
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of polyaspartamide-based adhesive hydrogels via Schiff base reaction with aldehyde-functionalized dextran†
Hend A.
Hegazy‡
 ac,
Hwi Hyun
Moon‡
a,
Dong-Hyun
Lee
b,
Suk Ho
Bhang
b,
Youn-Chul
Kim
*b,
Changsik
Song
*a and
Ji-Heung
Kim
*b
ac,
Hwi Hyun
Moon‡
a,
Dong-Hyun
Lee
b,
Suk Ho
Bhang
b,
Youn-Chul
Kim
*b,
Changsik
Song
*a and
Ji-Heung
Kim
*b
aDepartment of Chemistry, Sungkyunkwan University, Suwon, Gyeonggi 16419, Republic of Korea. E-mail: songcs@skku.edu
bDepartment of Chemical Engineering, Sungkyunkwan University, Suwon, Gyeonggi 16419, Republic of Korea. E-mail: younckim@skku.edu; kimjh@skku.edu
cDepartment of Chemistry, Faculty of Science, Zagazig University, Zagazig, Egypt
First published on 11th April 2023
Abstract
Tissue adhesives have become vital candidates for the treatment of injuries, working as hemostatic agents for wound-healing and tissue-sealing. The most accessible commercial adhesives are based on cyanoacrylate and fibrin glue; however, they suffer from drawbacks such as cytotoxicity and poor adhesive strength, which limit their bioapplications. Therefore, the development of an adhesive system that improves tissue repair and closure while exhibiting low cytotoxicity is an important but challenging task. Herein, to overcome the limitations of commercial adhesives, we fabricated biocompatible hydrogels based on amino-functionalized polyaspartamide crosslinked with naturally occurring dextran aldehyde via Schiff base chemistry and investigated their adhesive properties. The crosslink density is affected by the ratio between the amine and aldehyde functional groups of each polymer, as well as by the polymer concentrations. We evaluated the adhesive characteristics of the hydrogels using rheometer and lap shear tests. The polyaspartamide-based adhesive hydrogels showed good mechanical strength and self-healing properties and higher adhesive properties than fibrin glue, demonstrating their potential for tissue adhesion applications.
1. Introduction
Tissue adhesives have undergone substantial development with the expansion of the wound care industry.1,2 However, although several tissue adhesives have reached commercialization in clinical markets, they still have numerous limitations. For instance, despite its excellent tissue adherence, cyanoacrylate adhesive is harmful to cells and poorly biocompatible.3–5 Meanwhile, fibrin adhesives exhibit biocompatibility but poor mechanical strength and tissue adhesion.6–8 Ideal tissue adhesives should exhibit not only tissue adhesion properties and good biocompatibility but also mechanically robustness, manoeuvrability, and biodegradability to ensure an efficient removal of staples and sutures during clinical and surgical procedures.9,10Hydrogels are frequently employed as tissue adhesive materials because they physiologically resemble the natural extracellular matrix, which has both static and dynamic crosslinking networks.11–15 Natural-based hydrogels are promising candidates due to their great biocompatibility; however, their poor stability and mechanical properties limit their applications.16 Synthetic hydrogels also have an excellent prospect for biomedical applications due to the tunability and reproducibility of their physicochemical or mechanical properties. Unfortunately, they often suffer from poor biocompatibility.17 In this context, hydrogels consisting of biocompatible polymers, either natural or synthetic, and possessing mechanical and biological activity have emerged as potent platforms for a variety of biomedical applications, including tissue engineering and drug delivery.18
As a biocompatible, biodegradable, and low-cytotoxic polymer, polysuccinimide (PSI) has found a wide range of biomedical applications.19,20 PSI is typically produced by the acid-catalyzed polycondensation of aspartic acid.21 Because of the labile succinimidyl ring groups in its backbone, PSI undergoes nucleophilic substitution with amine-based molecules, forming polyaspartamide bearing a variety of functional groups with biomedical applications.22 Due to its protein-like structure, this polymer has a low level of inherent toxicity and degrades into amino acids or small peptides by the action of lysosomal enzymes.23 Several reports on polyaspartamide-based hydrogels have appeared in recent years, yet they mostly utilized polyaspartamide as the gel-forming macromers with crosslinkable functional groups such as catechol and methacrylate.24 In other studies, bis-amino compounds were crosslinked with polyaspartamide chains through unopened succinimidyl groups.25 Additionally, some studies have focused on the synthesis of polyaspartamide-based hydrogels; however, only the mechanical strength and degradability dependence on the degree of substitution have been investigated.26
In this study, with the aim of overcoming the drawbacks of commercial adhesives, we fabricated hydrogels using biocompatible polyaspartamide and oxidized dextran (Odex) and investigated their adhesive properties. As shown in Scheme 1, the in situ hydrogel was synthesized by coupling an amino-functionalized polyaspartamide (PHyAm) polymer with Odex using a Schiff base reaction. The polyaspartamide polymer was synthesized via the ring-opening aminolysis of PSI using ethanolamine (EA) and ethylenediamine (EDA). When aqueous solutions of polyaspartamide and dextran aldehyde polymers were combined at room temperature, gelation occurred at different gelation times depending on the polymer concentration and ratio. Finally, we conducted lap shear and rheometer tests to evaluate the adhesive properties of the synthesized hydrogels. Interestingly, the polyaspartamide-based adhesive hydrogels showed good mechanical strength, self-healing properties, and low cytotoxicity. The adhesive properties of the polyaspartamide-based hydrogels are higher than those of commercial fibrin glue, suggesting its potential use as novel materials in biomedical and wound-healing applications.
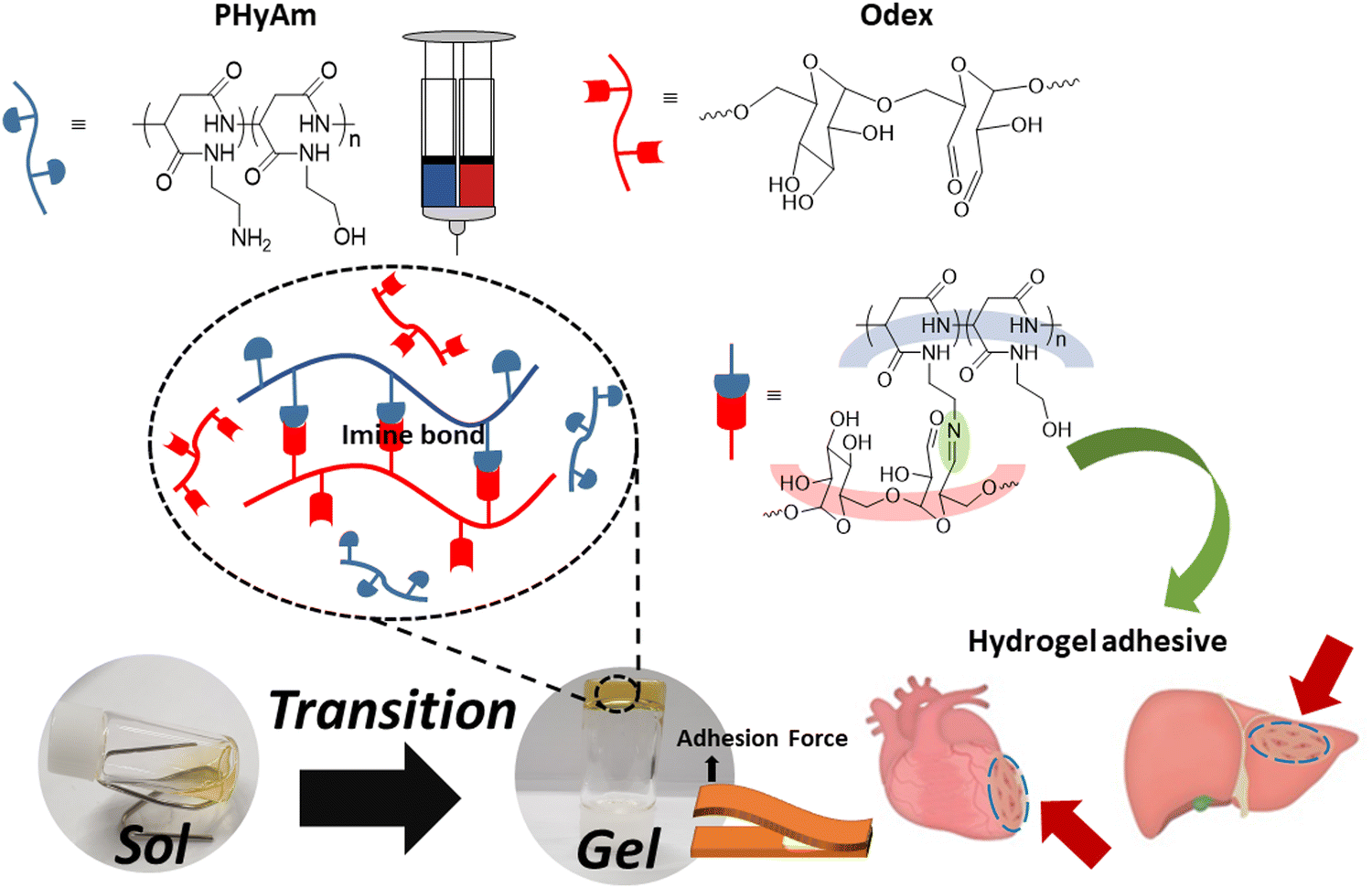 | ||
| Scheme 1 Schematic diagram of hydrogel formation based on amino-functionalized polyaspartamide (PHyAm) and aldehyde-functionalized dextran (Odex). | ||
2. Materials and methods
2.1. Materials and measurements
Alfa Aesar (MA, USA) supplied L-aspartic acid (98%) and EDA (99%). Phosphoric acid (98%) and EA (99%) were purchased from Acros Organics (MA, USA). Dextran (Mr ≈ 40 kDa) was provided by Leuconostoc spp. Fibrin glue was purchased from Green Cross (Yongin, Korea). Dialysis tubes with a molecular weight cut off (MWCO) of 3.5–14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and sodium periodate (NaIO4, 99.8%) were obtained from Sigma–Aldrich (MO, USA Dimethyl sulfoxide (DMSO) was purchased from Samchun chemicals (Seoul, Korea). Distilled water was produced using a Mili-Q water purification system.
000 Da and sodium periodate (NaIO4, 99.8%) were obtained from Sigma–Aldrich (MO, USA Dimethyl sulfoxide (DMSO) was purchased from Samchun chemicals (Seoul, Korea). Distilled water was produced using a Mili-Q water purification system.
1H Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker 500 MHz spectrometer. Fourier transform infrared (FTIR) spectra were recorded on a Vertex70 spectrometer (Bruker Optics, MA, USA) equipped with a diamond attenuated total reflection unit. Scanning electron microscopy (SEM) images were captured on a JEOL 7100 field emission SEM (JEOL, Tokyo, Japan) operated at an accelerating voltage of 15 kV. The rheological properties were investigated using an MCR 302e rheometer (Anton Paar, Graz, Austria). The average molecular weight and polydispersity index of the polymers were determined via gel permeation chromatography (GPC) using an Agilent Technology 1260 Infinity equipment (Agilent, CA, USA) with dimethylformamide (DMF) as the eluent and polystyrene as the standard. Lap shear tests were conducted using a universal testing machine (QC-508E, Cometech) with a load cell of 500 N. Cell viability was evaluated using a cell counting kit-8 (CCK-8, Dojindo Molecular Technologies, Inc., Kumamoto, Japan).
2.2. Methods
000 Da in the dark (water was changed 12 times) and maintained at 4 °C until further need.
Where V is the volume of NaOH (mL), M the molarity of NaOH (mol L−1), Mwt the molecular mass of dextran repeats unit (g mol−1), and wt the weight of Odex (g).
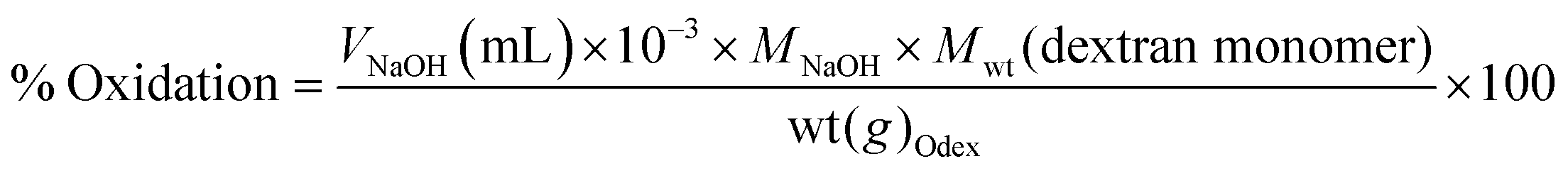
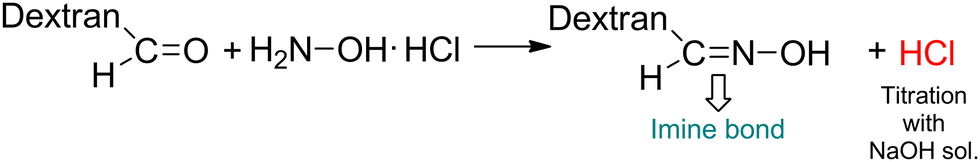 | ||
| Scheme 2 Determination of the oxidation degree of oxidized dextran (Odex) using the hydroxylamine hydrochloride method. | ||
000 g mol−1 by means of GPC using polystyrene standards and DMF as the eluent.
:0.5, 1:1, and 1:1.5 (P1O0.5, P1O1, and P1O1.5, respectively), and gelation was then achieved at room temperature to obtain the hydrogels. Both the tube inversion method and time sweep analysis of rheometer were used to calculate the gelation times. In the tube inversion method, the gelation time was determined by pouring a hydrogel sample into a vial and inverting it for 5 seconds. Time sweep analysis was performed by mixing PHyAm (20, 30, and 40 wt%) with Odex (20, 30, and 40 wt%) solutions at the rheometer's base plate, where moduli were recorded as a function of time at a 1 rad s−1 frequency and 1%. Strain.
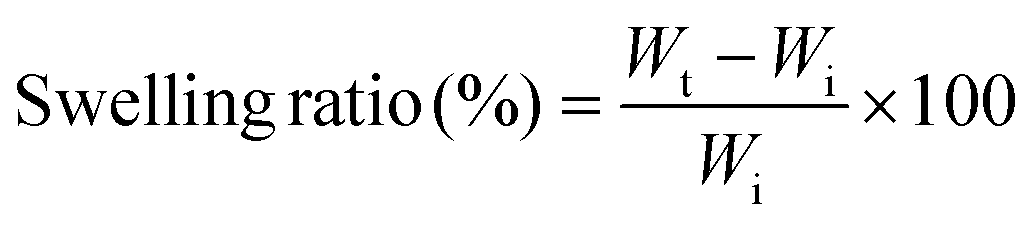
3. Results and discussion
3.1. Synthesis and characterization of the PHyAm/Odex hydrogels
For a variety of injuries, tissue adhesives have emerged as a promising contender, acting as hemostatic, tissue-sealing, and wound-healing agents. Thus, developing an adhesive system that can enhance tissue repair and closure with low cytotoxicity is highly desirable.31 Here, a hydrogel composed of PHyAm and Odex polymers was used to synthesize an injectable and self-healing hydrogel dressing with excellent adhesive properties.First, as shown in Scheme 3a, PSI was synthesized using the amino acid L-aspartic acid. Then, the PHyAm polymer was synthesized via an aminolysis reaction of PSI using EA and EDA (Scheme 3b). Subsequent experiments were conducted with an EA:EDA ratio of 3:7 because polymers having a higher EDA content may react with Odex. The structures and compositions of PSI and the PHyAm polymers were confirmed by 1H NMR spectroscopy using D2O as the solvent (Fig. S1, ESI†).
 | ||
| Scheme 3 Synthetic route to (a) PSI, (b) PHyAm, (c) Odex, and (d) the PHyAm/Odex hydrogel. | ||
Second, Odex was synthesized by oxidizing vicinal diols using NaIO4 as the oxidizing agent (Scheme 3c). As depicted in Fig. S2 (ESI†), the 1H NMR spectrum of Odex showed multiple new peaks at 5.0–6.0 ppm belonging to the hemiacetal structures of the aldehyde groups, which are absent in the 1H NMR spectrum of dextran, demonstrating the successful synthesis of Odex. Moreover, the FTIR spectrum showed a significant peak at 1748 cm−1 characteristic of a carbonyl group, confirming the chemical structure (Fig. S3, ESI†). Importantly, the oxidization degree of dextran aldehyde was found to be 30% using the hydroxylamine hydrochloride method.
The PHyAm/Odex hydrogels were easily synthesized by mixing an aqueous solution of PHyAm with an Odex solution at room temperature (Scheme 3d). Interestingly, a pale-yellow gel was formed within a few seconds without any external stimulus. This in situ gelation of the PHyAm/Odex mixture occurred via a Schiff base reaction, which resulted in the formation of imine bonds, as confirmed by FTIR analysis which showed a distinct peak at 1647 cm−1 corresponding to imine bond (Fig. S3, ESI†).
3.2. Analysis of the physical properties of the hydrogels
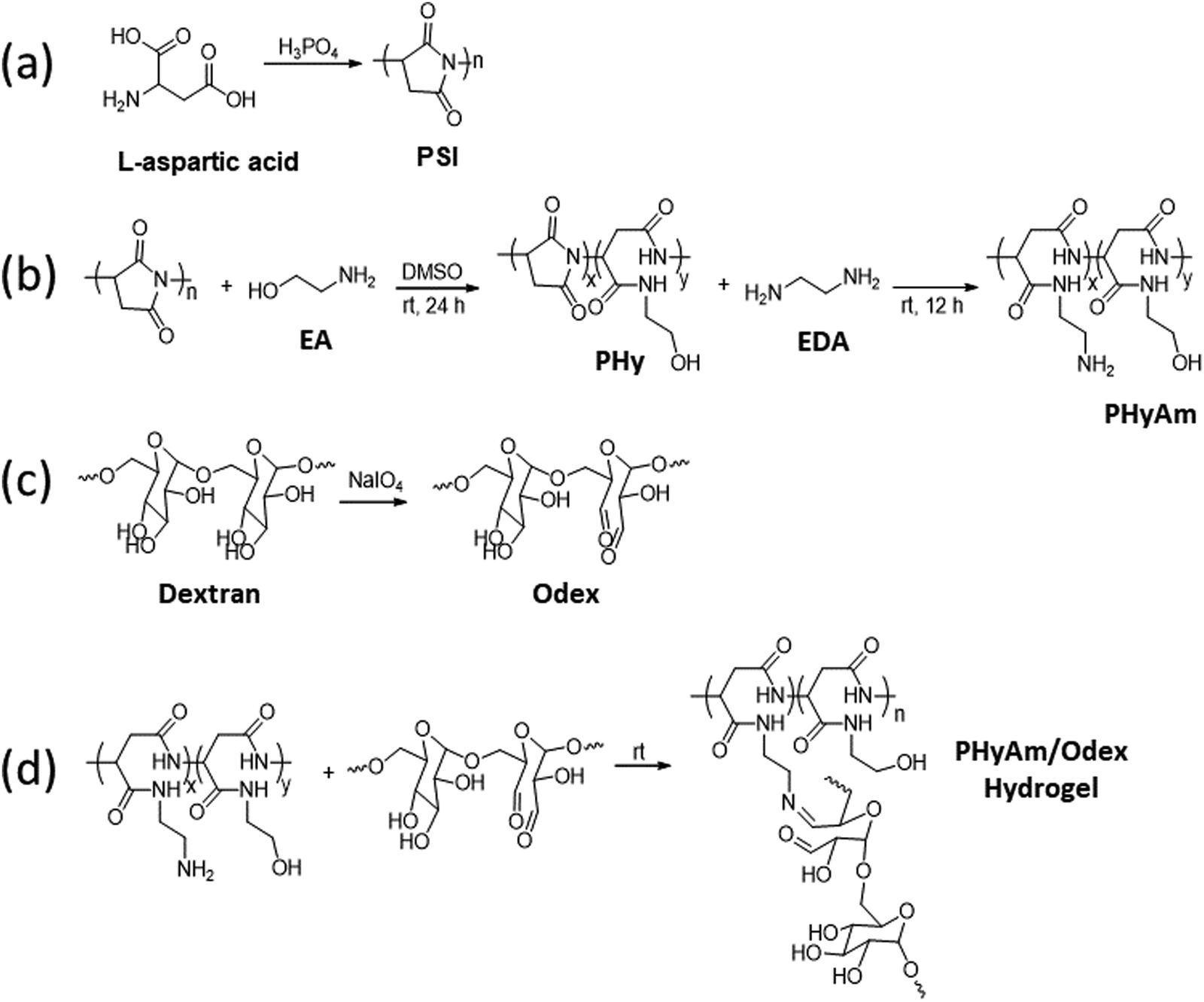
The gelation time of hydrogels is crucial to their practical biomedical application. Since surgery requires a definite time, a short gelation time is undesirable, whereas a long gelation time hinders closing promptly the wound site.32 In this study, the gelation time was determined by the tube inversion method33,34 and rheological measurements.33 It was observed in both methods that the gelation time increased with decreasing crosslinker concentration. As shown in (Fig. 1a), the gelation time decreased from near 180 s to 20 s by increasing the crosslinker concentration from 20 to 40 wt% for P1O1.5 hydrogel because of the gradually enhanced crosslinking density of the hydrogels. The extremely quick setting of the 40% hydrogel could be advantageous for preventing the gel from spreading to nearby tissues. Similar results were observed in rheological measurements using oscillation time sweep test, in which the gelation point was determined as the time at which G′ and G′′ crossed each other (Fig. S4, ESI†). The mechanical strength of hydrogels is also important to shield the wound from further injuries.35 To assess the rheological characteristics of the hydrogels, the frequency dependence of the storage (G′) and loss (G′′) modulus was observed at angular frequencies between 1 and 100 rad s−1 and 1% strain. In all samples, G′ was much greater than G′′ throughout the frequency range, which suggested that the hydrogels were highly stable and elastic. Additionally, the precursor concentrations and ratios were found to influence the final stiffness of the hydrogels. Thus, the 40 wt% hydrogel (33 kPa) exhibited higher final moduli and faster gelation time than the 30 wt% hydrogel (23 kPa) and the 20 wt% hydrogel (7 kPa) due to the increase in the crosslinking densities with increasing the PHyAm and Odex concentration, hence increasing the stiffness of the hydrogel (Fig. 1b and Fig. S5a–c, ESI†). The damping factor of the hydrogels was found to be less than 1, demonstrating their elastic nature (Fig. S5d, ESI†).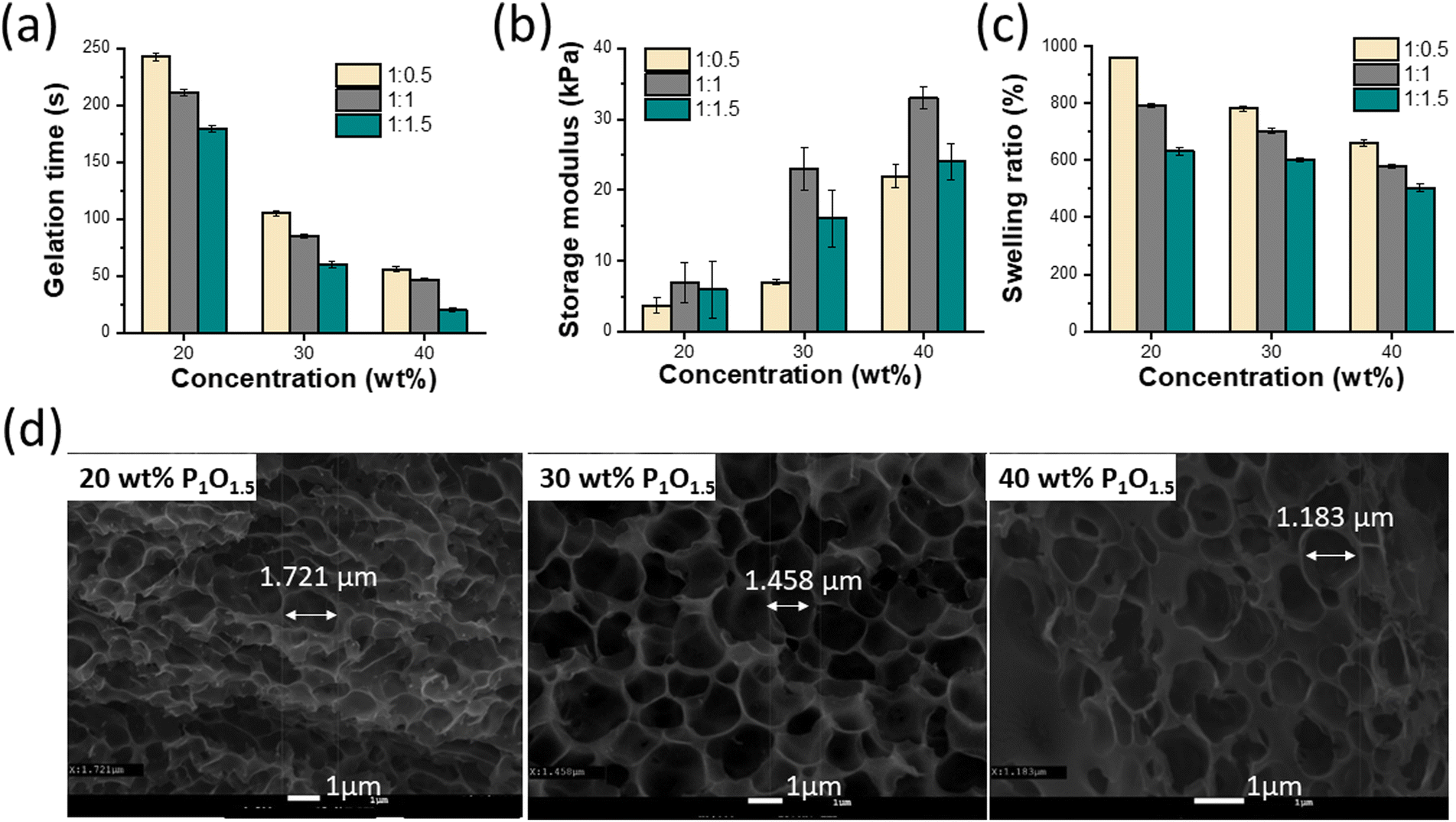 | ||
| Fig. 1 (a) Gelation time, (b) frequency dependence of the storage (G′) and loss (G′′) modulus, (c) swelling ratio of the hydrogels formed at concentrations of 20, 30, and 40 wt%. (d) SEM images of the P1O1.5 hydrogels formed at concentrations of 20, 30, and 40 wt%. | ||
Furthermore, as revealed by an SEM analysis, all the hydrogels showed interconnected porous structures (Fig. 1d and Fig. S6a, b, ESI†). The corresponding mean pore diameter significantly decreased from 1.721 to 1.183 μm with increasing the concentration of the hydrogels from 20 to 40 wt% for P1O1.5. It is well known that the pore volume and pore size of porous materials can be mostly controlled by the crosslink density in the hydrogel network.
3.3. Swelling studies
Swelling is a frequent characteristic of hydrophilic polymer adhesives and vital for hydrogels with prospective applications in biomedical fields because they are exposed to biological fluids and can be harmful to adhesion and to adjacent tissues.36 When water is absorbed into the adhesive matrix, the density of the polymer decreases, which might result in the volumetric expansion of adhesives during swelling, which in turn compresses the nearby tissue mechanically. Basically, many factors affect the swelling process, such as the nature of the polymer, the rigidity of the polymer chain, its ability to expand, and the degree of crosslinking. The swelling properties of the proposed hydrogels at different ratios and concentrations are shown in Fig. 1c. These results demonstrated that the equilibrium swelling ratios decreased with increasing concentration (20–40 wt%) and ratio (1:05–1:1.5), indicating increase in the crosslinking density accordingly.
3.4. Self-healing properties of the hydrogels
Hydrogel wound dressings can withstand external mechanical strain after being applied to the wound site. Therefore, using self-healing hydrogels as wound dressings can extend their durability.37 To evaluate the self-healing ability of the hydrogels, a hydrogel sample formed into a cylindrical shape with a diameter of 8 mm and a thickness of roughly 3 mm was cut into two pieces, which were then brought into contact and allowed to self-heal. After 5–10 min, the rebuilt hydrogel disk was lifted using tweezers to confirm its ability to keep integrity under gravity (Fig. 2a), which can be attributed to the reversible dynamic covalent bonds (imine bonds) within the hydrogel network migrating to the interfacial chains to undergo recrosslinking. The self-healing properties of the hydrogels were further confirmed by means of a dynamic rheological analysis. The critical strain point near which the hydrogel changes from solid to liquid state was determined using a strain amplitude sweep test. As shown in Fig. 2b, at low strain, G′ and G′′ had constant values. Then, upon increasing the strain, the G′ and G′′ curves intersected at about 3.8% strain, which is the critical point. Further increasing the strain up to 10% led to a sharp decrease in the G′ value below the G′′ value, demonstrating the collapse of the hydrogel network. After 5 min, the strain amplitude sweep test was repeated on the same sample to check the self-healing ability, which was about 60% of the value obtained in the first measurement. These findings showed that the PHyAm/Odex hydrogels have considerable self-healing abilities due to the formation of imine bonds between the amine groups of PHyAm and the aldehyde groups of Odex.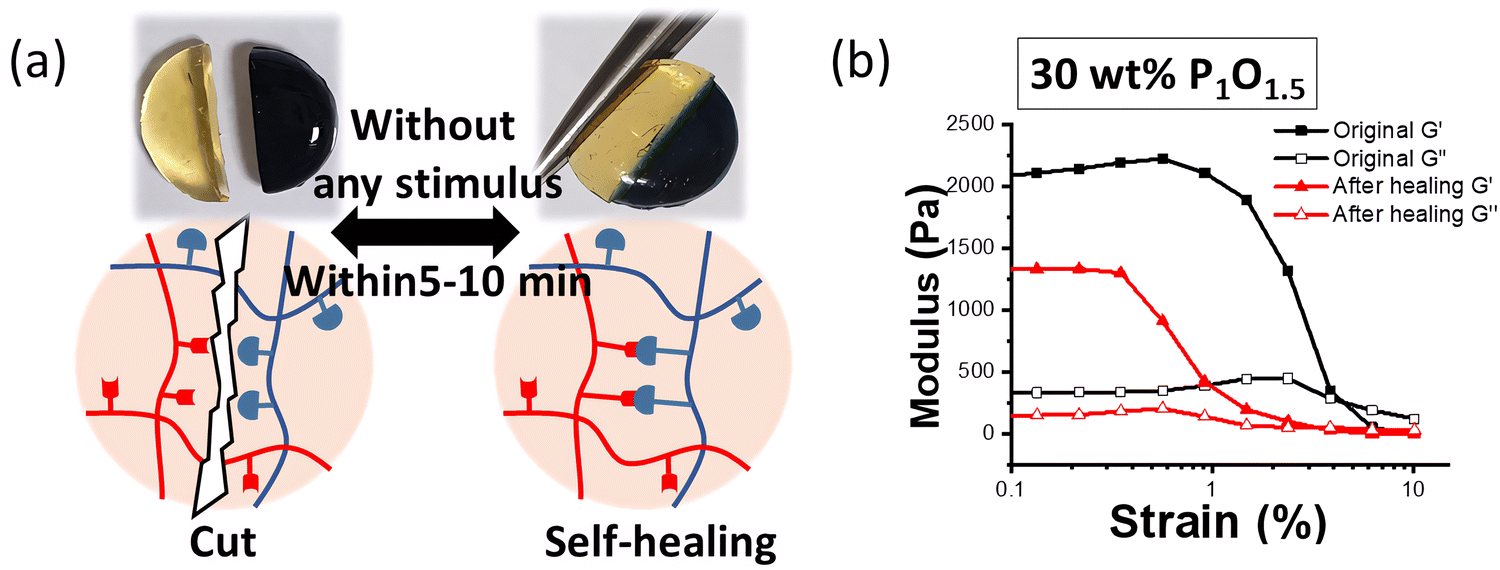 | ||
| Fig. 2 (a) Diagram of macroscopic analysis of the self-healing property of the hydrogels. (b) rheological analysis of self-healing recovery after 5 min. | ||
3.5. Adhesive properties of the hydrogels
The adhesion properties of hydrogel dressings are critical in the wound-healing process because it controls bleeding and prevents gas or fluid leakage from injured sites.38–40 Tissue adhesive polymers containing aldehyde groups have been utilized extensively. In the case of Odex, the aldehyde groups can react with the amino groups in tissues to create chemical linkages for adhesion.38 To quantify the adhesion behaviour of the PHyAm/Odex hydrogels, a widely used lap shear test method was employed. Among the many parameters that affect adhesion, the substrate to which the material is adhered is one of the critical factors.41Fig. 3a shows the preparation of the sample for lap shear testing on different substrates. In general, substrates can be divided into low-surface-energy plastics and high-surface-energy metals. Typically, a strong adhesion is more favoured on high-energy, rough surfaces.41Fig. 3b shows the immediate bulk adhesive strengths for various substrates commonly used as daily-life materials (aluminum foil, glass, and plastic). The highest adhesive strength values of 131, 74, 65, and 58 kPa for glass, PMMA, aluminum, and PET plastic substrate, respectively, were obtained at a 40 wt% P1O1 hydrogel. The best adherence was observed on glass substrates, which might be due to the formation of strong hydrogen bonds between the amine groups of the hydrogel and the hydroxyl groups of glass (Si–OH).42 For PET and PMMA plastics, attractive interactions caused by dipole–dipole interactions and hydrogen bonds between polyaspartamide and the ester groups (hydrogen acceptors) of PET and PMMA may be responsible for the observed adhesive properties.43 | ||
| Fig. 3 (a) Schematic illustration of adhesion strength measurements. (b) Adhesion strength of a 40 wt% P1O1.5 hydrogel measured using various substrates under wet condition. (c) Adhesion strength of 20, 30, and 40 wt% P1O1 hydrogels on glass substrates under wet and dry conditions. (d) Image of the lap shear test using porcine skin. (e) Wet adhesion strength of 20, 30, and 40 wt% P1O1.5 hydrogels on porcine skin. (f) Wet adhesion strength of hydrogels with various hydrogels (P1O0.5, P1O1, P1O1.5) and fibrin glue. | ||
The adhesion strength of the hydrogels as a function of the aging time on a glass slide is depicted in Fig. S7a (ESI†). As the aging time increased from 10 min to 24 h, the adhesive strength increased up to 1.26 MPa after 24 h. Fig. 3c shows the adhesive strength of PHyAm/Odex hydrogels with different polymer concentrations on a glass substrate after 10 min and 12 h. After 10 min, the adhesive strength increased from 57 kPa to 150 kPa upon increasing the concentration from 20 to 40 wt%. Whereas it decreased from 668 kPa to 363 kPa by increasing the concentration from 20 to 40 wt% after 12 h, which probably due to the dehydration of the hydrogel with time. Moreover, the adhesion properties of different ratios of the hydrogel after 10 min and after 12 h (Fig. S7b and c, ESI†), respectively showed that P1O1 has the highest adhesion properties. The results of the lap shear adhesion tests and the G′ value of the hydrogel derived from rheological measurements, which evaluate the cohesion strength of materials, were in good agreement.
The adhesion ability of the hydrogels was then compared with clinically administered fibrin glue as a positive control using PS, which is commonly used for adhesive testing in biomedical applications owing to its similarity to the human dermis.44Fig. 3d depicts a schematic illustration and a typical image for the lap shear strength test. After spreading the hydrogels on the PS surface, the samples were overlaid and compressed by a weight of 2000 g for 30 min. With increasing the PHyAm/Odex concentration from 20 to 40 wt%, the adhesive strength increased from about 5 to 16 kPa (Fig. 3e). The adhesion to PS might result from the formation of hydrogen bonds between the polymer and the hydroxyl, amine, and carboxyl functional groups present on PS.45 As shown in Fig. 3f, the tissue adhesion was measured using different PHyAm:Odex mole ratios (P1O0.5, P1O1, and P1O1.5), finding that the hydrogel with a P1O1.5 exhibited much higher adhesive properties. This could be attributed to the following factors: (1) the higher Odex ratio provides more aldehyde groups for the reaction with the amino groups of the PS tissue, resulting in bonding;46 (2) the robust cohesion of the hydrogel network improves the adhesion strength on PS. Additionally, in order to evaluate the feasibility and applicability of our adhesive polymers, wet and dry adhesion strength properties have been investigated using porcine skin tissue and the results showed that there are no noticeable differences in both cases (Fig. S7d, ESI†), indicating the real applicability of the prepared polymers in real biofields. In these measurements, all the hydrogels showed stronger adherence than the commercial adhesive fibrin glue (4.95 kPa).
3.6. Cytotoxicity of the hydrogels
Biocompatibility studies were conducted in 24-well transwell plates using different amounts (5, 10, 20, 40, and 80 mg) of hydrogel in the transwell and 400 μL of medium in the peripheral well. The results depicted in Fig. 4 were obtained by measuring the cell viability after 24 h of incubation. Even high amounts of hydrogel showed 85% or more viability compared with the blank experiment (no treatment). Reducing cell viability by more than 30% is defined as a cytotoxic effect by International Standard ISO 10993-5 (2009),47 demonstrating that the hydrogel did not exert a severe cytotoxic effect on hDF cell.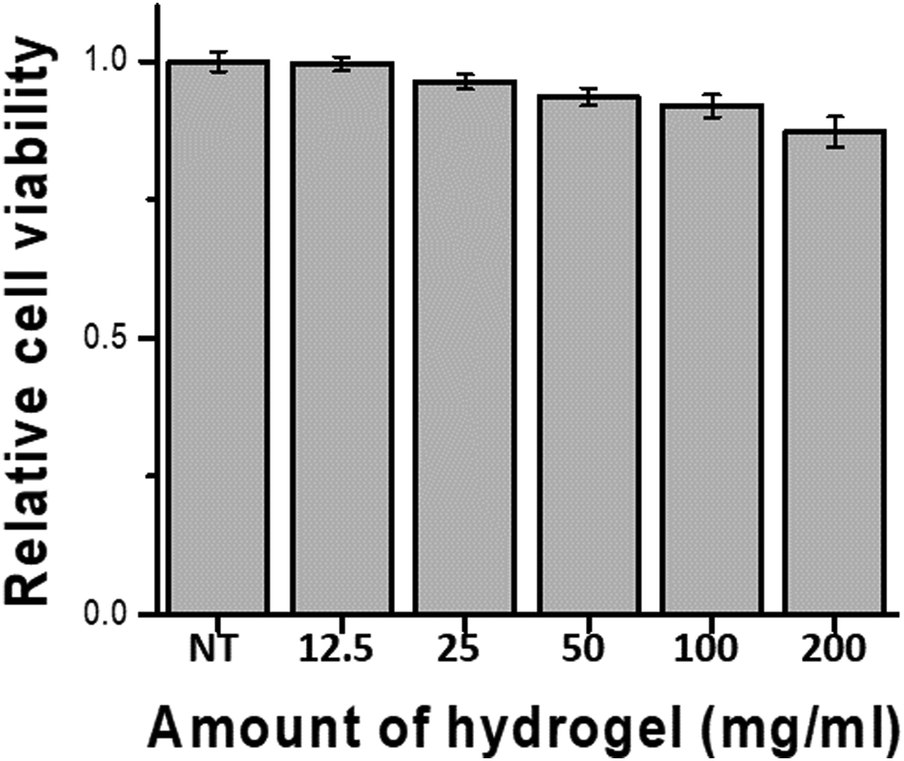 | ||
| Fig. 4 Cell viability test of the 30 wt% P1O1.5 hydrogel (NT: no treatment). | ||
Conclusion
In this work, adhesive hydrogels were synthesized by forming imine bonds via a Schiff base reaction between the amine functional groups of polyaspartamide and the aldehyde groups of dextran. The rheological and adhesive properties of hydrogels synthesized at various concentrations and ratios of amines and aldehydes were analyzed, finding that the 20 wt% hydrogel had high wet adhesion and the 40% hydrogel exhibited high dry adhesion. Among the various surfaces evaluated, glass showed the highest adhesion when the P1O0.5 hydrogel, while PS showed the highest adhesion when the P1O1.5 hydrogel. The synthesized hydrogels exhibited self-healing properties within 5–10 min without any external stimuli, as well as low cytotoxicity. Notably, the synthesized hydrogels exhibited higher adhesive strength on PS than fibrin glue, which is currently used as a skin adhesive. Therefore, this work demonstrates the potential of polyaspartamide-based adhesive hydrogels as biomedical adhesives in clinical applications.Author contributions
H. A. Hegazy: conceptualization, investigation, formal analysis, writing – original draft; H. H. Moon: conceptualization, investigation, data curation, writing – original draft; D. H. Lee: data curation; S. H. Bhang: supervision; Y. C. Kim: conceptualization, investigation, writing – review & editing, supervision; C. S. Song: conceptualization, investigation, writing – review & editing, supervision; J. H. Kim: conceptualization, investigation, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Materials and Components Technology Development Program (Project No. 20013223) and the Technology Innovation Program (Project No. 20013794, Center for Composite Materials and Concurrent Design) funded by the Ministry of Trade, Industry & Energy (MOTIE, KOREA).References
- S. Nam and D. Mooney, Chem. Rev., 2021, 121, 11336–11384 CrossRef CAS PubMed.
- M. B. Detweiler, J. G. Detweiler and J. Fenton, J. Invest. Surg., 1999, 12, 245–262 CrossRef CAS PubMed.
- G. Pascual, S. Sotomayor, M. Rodriguez, B. Perez-Kohler, A. Kuhnhardt, M. Fernandez-Gutierrez, J. San Roman and J. M. Bellon, PLoS One, 2016, 11, e0157920 CrossRef PubMed.
- B. Mizrahi, C. F. Stefanescu, C. Yang, M. W. Lawlor, D. Ko, R. Langer and D. S. Kohane, Acta Biomater., 2011, 7, 3150–3157 CrossRef CAS PubMed.
- B. J. Vote and M. J. Elder, Clin. Exp. Ophthalmol., 2000, 28, 437–442 CrossRef CAS PubMed.
- G. Lopezcarasa-Hernandez, J.-F. Perez-Vazquez, J.-L. Guerrero-Naranjo and M. A. Martinez-Castellanos, Int. J. Retina Vitreous, 2021, 7, 33 CrossRef PubMed.
- M. Radosevich, H. I. Goubran and T. Burnouf, Vox Sang., 1997, 72, 133–143 CrossRef CAS PubMed.
- K. Conrad and A. Yoskovitch, Arch. Facial Plast. Surg., 2003, 5, 522–527 CrossRef PubMed.
- X. Zhang, Y. Jiang, L. Han and X. Lu, Biosurface and Biotribology, 2021, 7, 163–179 CrossRef.
- R. Yu, M. Li, Z. Li, G. Pan, Y. Liang and B. Guo, Adv. Healthcare Mater., 2022, 11, 2102749 CrossRef CAS PubMed.
- Y. Yi, C. Xie, J. Liu, Y. Zheng, J. Wang and X. Lu, J. Mater. Chem. B, 2021, 9, 8739–8767 RSC.
- H. Geckil, F. Xu, X. Zhang, S. Moon and U. Demirci, Nanomedicine, 2010, 5, 469–484 CrossRef CAS PubMed.
- M. Okawa, A. Tanabe, S. Ohta, S. Nagatoishi, K. Tsumoto and T. Ito, Commun. Mater., 2022, 3, 81 CrossRef CAS.
- A. Sigen, Q. Xu, M. Johnson, J. Creagh-Flynn, M. Venet, D. Zhou, I. Lara-Sáez, H. Tai and W. Wang, Appl. Mater. Today, 2021, 22, 100967 CrossRef.
- Y. Liang, M. Li, Y. Yang, L. Qiao, H. Xu and B. Guo, ACS Nano, 2022, 16, 3194–3207 CrossRef CAS PubMed.
- M. Klein and E. Poverenov, J. Sci. Food Agric., 2020, 100, 2337–2347 CrossRef CAS PubMed.
- U. S. K. Madduma-Bandarage and S. V. Madihally, J. Appl. Polym. Sci., 2021, 138, 50376 CrossRef CAS.
- J. Jang and C. Cha, Biomacromolecules, 2018, 19, 691–700 CrossRef CAS PubMed.
- H. Adelnia, I. Blakey, P. J. Little and H. T. Ta, Front. chem., 2019, 7, 755 CrossRef CAS PubMed.
- H. Adelnia, H. D. N. Tran, P. J. Little, I. Blakey and H. T. Ta, ACS Biomater. Sci. Eng., 2021, 7, 2083–2105 CrossRef CAS PubMed.
- T. Nakato, A. Kusuno and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 117–122 CrossRef CAS.
- E. Jalalvandi and A. Shavandi, Eur. Polym. J., 2018, 109, 43–54 CrossRef CAS.
- G. Pitarresi, F. S. Palumbo, G. Giammona, M. A. Casadei and F. Micheletti Moracci, Biomaterials, 2003, 24, 4301–4313 CrossRef CAS PubMed.
- G. Pitarresi, P. Pierro, G. Tripodo, D. Mandracchia and G. Giammona, J. Drug Delivery Sci. Technol., 2005, 15, 377–382 CrossRef CAS.
- J. R. Moon and J.-H. Kim, Macromol. Res., 2008, 16, 489–491 CrossRef CAS.
- M. Kim and C. Cha, Biomacromolecules, 2020, 21, 3693–3703 CrossRef CAS PubMed.
- J. Maia, R. A. Carvalho, J. F. J. Coelho, P. N. Simões and M. H. Gil, Polymer, 2011, 52, 258–265 CrossRef CAS.
- H. Zhao and N. D. Heindel, Pharm. Res., 1991, 8, 400–402 CrossRef CAS PubMed.
- D. Lu, H. Wang, T. Li, Y. Li, F. Dou, S. Sun, H. Guo, S. Liao, Z. Yang, Q. Wei and Z. Lei, ACS Appl. Mater. Interfaces, 2017, 9, 16756–16766 CrossRef CAS PubMed.
- Y.-z Bu, G.-f Sun, L.-c Zhang, J.-h Liu, F. Yang, P.-f Tang and D.-c Wu, Chin. J. Polym. Sci., 2017, 35, 1231–1242 CrossRef CAS.
- A. Bal-Ozturk, B. Cecen, M. Avci-Adali, S. N. Topkaya, E. Alarcin, G. Yasayan, Y.-C. Ethan, B. Bulkurcuoglu, A. Akpek, H. Avci, K. Shi, S. R. Shin and S. Hassan, Nano Today, 2021, 36, 101049 CrossRef CAS PubMed.
- X. Yang, G. Liu, L. Peng, J. Guo, L. Tao, J. Yuan, C. Chang, Y. Wei and L. Zhang, Adv. Funct. Mater., 2017, 27, 1703174 CrossRef.
- C. T. Huynh, Z. Zheng, M. K. Nguyen, A. McMillan, G. Yesilbag Tonga, V. M. Rotello and E. Alsberg, ACS Biomater. Sci. Eng., 2017, 3, 2011–2023 CrossRef CAS PubMed.
- M. K. Nguyen, O. Jeon, M. D. Krebs, D. Schapira and E. Alsberg, Biomaterials, 2014, 35, 6278–6286 CrossRef CAS PubMed.
- X. Du, Y. Liu, X. Wang, H. Yan, L. Wang, L. Qu, D. Kong, M. Qiao and L. Wang, Mater. Sci. Eng., C, 2019, 104, 109930 CrossRef CAS PubMed.
- Q. Chai, Y. Jiao and X. Yu, Gels, 2017, 3, 6 CrossRef PubMed.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS PubMed.
- C. Ghobril and M. W. Grinstaff, Chem. Soc. Rev., 2015, 44, 1820–1835 RSC.
- N. Annabi, K. Yue, A. Tamayol and A. Khademhosseini, Eur. J. Pharm. Biopharm., 2015, 95, 27–39 CrossRef CAS PubMed.
- E. Y. Jeon, B. H. Hwang, Y. J. Yang, B. J. Kim, B. H. Choi, G. Y. Jung and H. J. Cha, Biomaterials, 2015, 67, 11–19 CrossRef CAS PubMed.
- M. Guvendiren, D. A. Brass, P. B. Messersmith and K. R. Shull, J. Adhes., 2009, 85, 631–645 CrossRef CAS PubMed.
- G. Bovone, O. Y. Dudaryeva, B. Marco-Dufort and M. W. Tibbitt, ACS Biomater. Sci. Eng., 2021, 7, 4048–4076 CrossRef CAS PubMed.
- M. A. Sibeko, M. L. Saladino, A. S. Luyt and E. Caponetti, J. Mater. Sci., 2016, 51, 3957–3970 CrossRef CAS.
- H. Chung and R. H. Grubbs, Macromolecules, 2012, 45, 9666–9673 CrossRef CAS.
- X. Pei, J. Wang, Y. Cong and J. Fu, J. Polym. Sci., 2021, 59, 1312–1337 CrossRef CAS.
- J. Hoque, R. G. Prakash, K. Paramanandham, B. R. Shome and J. Haldar, Mol. Pharmaceutics, 2017, 14, 1218–1230 CrossRef CAS PubMed.
- D. Zhou, S. Li, M. Pei, H. Yang, S. Gu, Y. Tao, D. Ye, Y. Zhou, W. Xu and P. Xiao, ACS Appl. Mater. Interfaces, 2020, 12, 18225–18234 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00032j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |