
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect engineering in carbon materials for electrochemical energy storage and catalytic conversion
Zhiqiang
Zhao
a,
Huan
Chen
a,
Wanyu
Zhang
a,
Shan
Yi
 a,
Hongli
Chen
a,
Zhe
Su
a,
Bo
Niu
a,
Yayun
Zhang
*ab and
Donghui
Long
ab
a,
Hongli
Chen
a,
Zhe
Su
a,
Bo
Niu
a,
Yayun
Zhang
*ab and
Donghui
Long
ab
aState Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: yy.zhang@ecust.edu.cn
bKey Laboratory of Specially Functional Polymeric Materials and Related Technology (Ministry of Education), School of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 9th January 2023
Abstract
Carbon, featured by its distinct physical, chemical, and electronic properties, has been considered a significant functional material for electrochemical energy storage and conversion systems. Significant improvements in the configuration, electron distribution, and chemical environment of the carbon matrix by unavoidable defect sites have hastened parturition in the study of carbon defect engineering which has become a vital research subject in defect chemistry. Compared to pure carbon with natural inertness, the defect-rich carbon matrix can modulate the electronic properties, increase exposed active sites, and further accelerate the electrochemical redox reaction, thus ultimately improving battery performance and electrocatalyst activity. This review covers recent advances in understanding, designing, and exploring defects in carbon materials toward energy-related applications. In particular, the role and active origin of defects have been comprehensively discussed on the basis of both experimental results and theoretical calculations. This article aims to provide reference and guidance for large-scale, diversified, and industrial applications of defect-rich carbon functional materials.
1. Introduction
Rapid advancement in urbanization and continuous development of industrialization have greatly exacerbated the excessive use of non-renewable fossil sources (e.g., coal, oil, natural gas, etc.), and further highlighted the serious energy crisis and environmental problems.1–3 Developing efficient, green, safe and continuable electrochemical energy storage as well as conversion technologies has become an imminent task in the process of global energy transformation and “decarbonization”.4–7 In this field, metal-ion batteries (MIBs), metal–sulfur batteries (MSBs) and electrocatalysts have attracted extensive attention as high-performance electrochemical energy storage and conversion systems. Both MIBs and MSBs have been at the forefront of energy storage devices thanks to their high capacity and fast charge–discharge rate.8–10 Electrocatalysis, as an important conversion pathway for renewable energy, has been widely reported and won inspiring success.11–16 Even though there are different structures and operating mechanisms, these energy storage and conversion devices have one thing in common: carbon materials are extensively adopted. Carbon, as an important part of battery functional components and catalytic materials, has received considerable attention because of its low cost, broad source, light weight, good electrical conductivity, and physical and chemical versatility.17–21 Unfortunately, the reactivity of pure carbon materials is unsatisfactory, restricted by their natural inertness. To obtain higher performance energy storage and conversion systems, developing higher reactive carbon has become particularly significant.22–24 Encouragingly, defect engineering can provide plentiful catalytic sites for electrochemical redox reactions and is considered to be one of the most promising strategies for carbon material modification.25–27The term “defect” tends to have a negative connotation, meaning something lacking or imperfect in a material. However, with the deepening of research, the impact of defects on material properties has gradually been understood by researchers, and this impact may bring great changes. Therefore, defect structures have started being deliberately introduced or intentionally suppressed to customize the properties of the material.28,29 Meanwhile, some theories of defect chemistry have been gradually established and perfected, and defect engineering was widely used in the research studies of advanced functional materials.30 In recent years, defect-rich carbon has been broadly applied to the field of energy storage and conversion, and its positive role in improving the performance of rechargeable batteries and electrocatalysts has been approbated.31,32 We employ the term “defect” in this article as a technical term to contain both intrinsic defects (topological defects, vacancy defects, and edge defects) and extrinsic defects. Defect engineering can tune the geometry and electron distribution of the carbon matrix, provide defective catalytic sites, and further accelerate electrochemical redox reactions.33 For MIBs, defect engineering of carbon not only induces the formation of sites with high charge and spin densities, but also lowers the migration energy and diffusion barriers during metal ion intercalation/adsorption, contributing to high capacity and fast storage.34,35 Furthermore, the defect sites endow the carbon material with strong chemisorption and efficient catalytic activity, which is beneficial for the anchoring and catalytic conversion of polysulfides, and subsequently enhances the all-around electrochemical performance of LSBs.36,37 In the field of electrocatalysis, the introduction of defects can tune the electron distribution and surface properties of electrocatalysts, endow them with excellent catalytic activity, and serve to overcome the problem of slow redox kinetics.15,38,39 It should be heeded that the defect density is strictly controlled. Excessive defect concentration will destroy the C–C sp2 conjugated structure and make the conductivity worse, thereby impairing the cycling stability of the battery and the activity of electrochemistry.40
Recently, great progress has been made with regard to carbon defect engineering for energy storage and conversion, while revealing the active origin and precisely customizing defects in the carbon matrix currently remain a challenge. An in-depth understanding of the role of carbon defects in electrochemical redox reactions is crucial, which can guide the rational design of high-performance carbon defect sites. In this review, we summarize the recent advances in carbon defect engineering applied to energy storage and catalytic conversion as well as review the efforts to unravel the role of defects via experimental and computational methods (Fig. 1). Specifically, we describe the classification of carbon defects, reveal the structure–activity relationship between the defect sites and reactivity, and briefly introduce the commonly used defect construction strategies. Then, the recent progress in carbon defect engineering in the field of MIBs, MSBs and electrocatalysts is presented in detail, emphasizing the role and activity origin of the defect sites. Finally, some perspectives are provided for further research on carbon defect engineering. This paper aims to elucidate the significant impact of defect engineering on the properties of carbon materials and to provide some guidance for the future development of this promising research direction. We hope that through the descriptions in this paper, readers can gain a better understanding of carbon defect engineering and inspire some new research in this field.
 | ||
| Fig. 1 An overview of defect engineering in carbon for energy conversion and storage. | ||
2. Fundamentals of carbon defect engineering
Defect structures are widespread in solid materials. With the promotion of high-precision and high-sensitivity research strategies such as in situ characterization, density functional theory (DFT) calculations, etc., the effects of defect sites on the atomic structure and electron distribution of materials are gradually clear. Advanced characterization techniques are essential to identify defect types, assess defect density, and gain insight into the reactivity of defect sites. Exhaustive defect characterization methods have been introduced in many reports and reviews,41–45 which provide basic support for further detailed analysis of the defect sites. To identify the active sources of defect sites in different reactions, it is quite essential to master the effects of specific defect structures on carbon materials. At the same time, understanding the relationship between defect structure and material performance can furnish a new idea for the development and design of carbon defect engineering. We briefly outline the development of carbon defect engineering in the field of electrochemical energy storage and catalytic conversion as a way to provide readers with important information that captures the important history of this field (Fig. 2). In this section, we will introduce the classification of defects, the common construction strategies of defects, elaborate on the structure–activity relationship of defective carbon materials, and clarify the active sites of defective structures.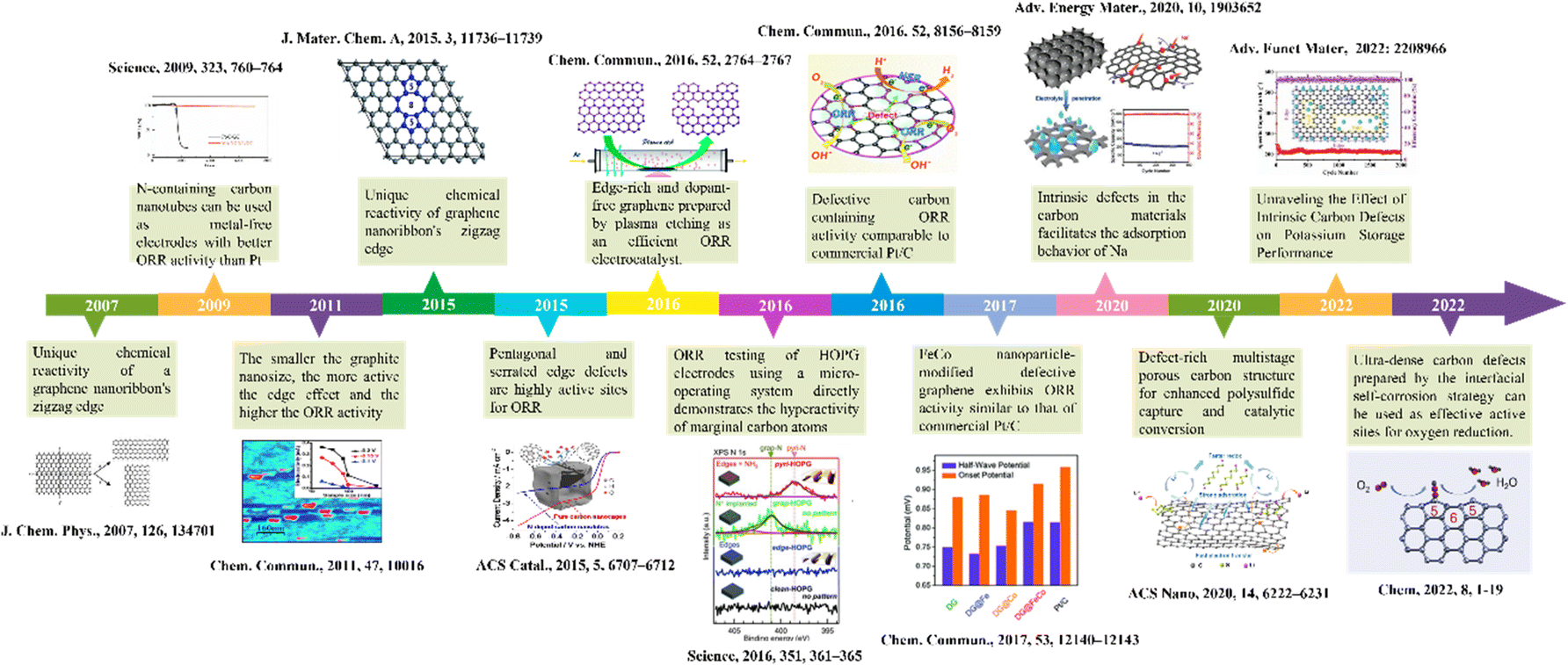 | ||
| Fig. 2 Outline of the history of carbon defect engineering in the field of electrochemical energy storage and catalytic conversion.12,46–57 | ||
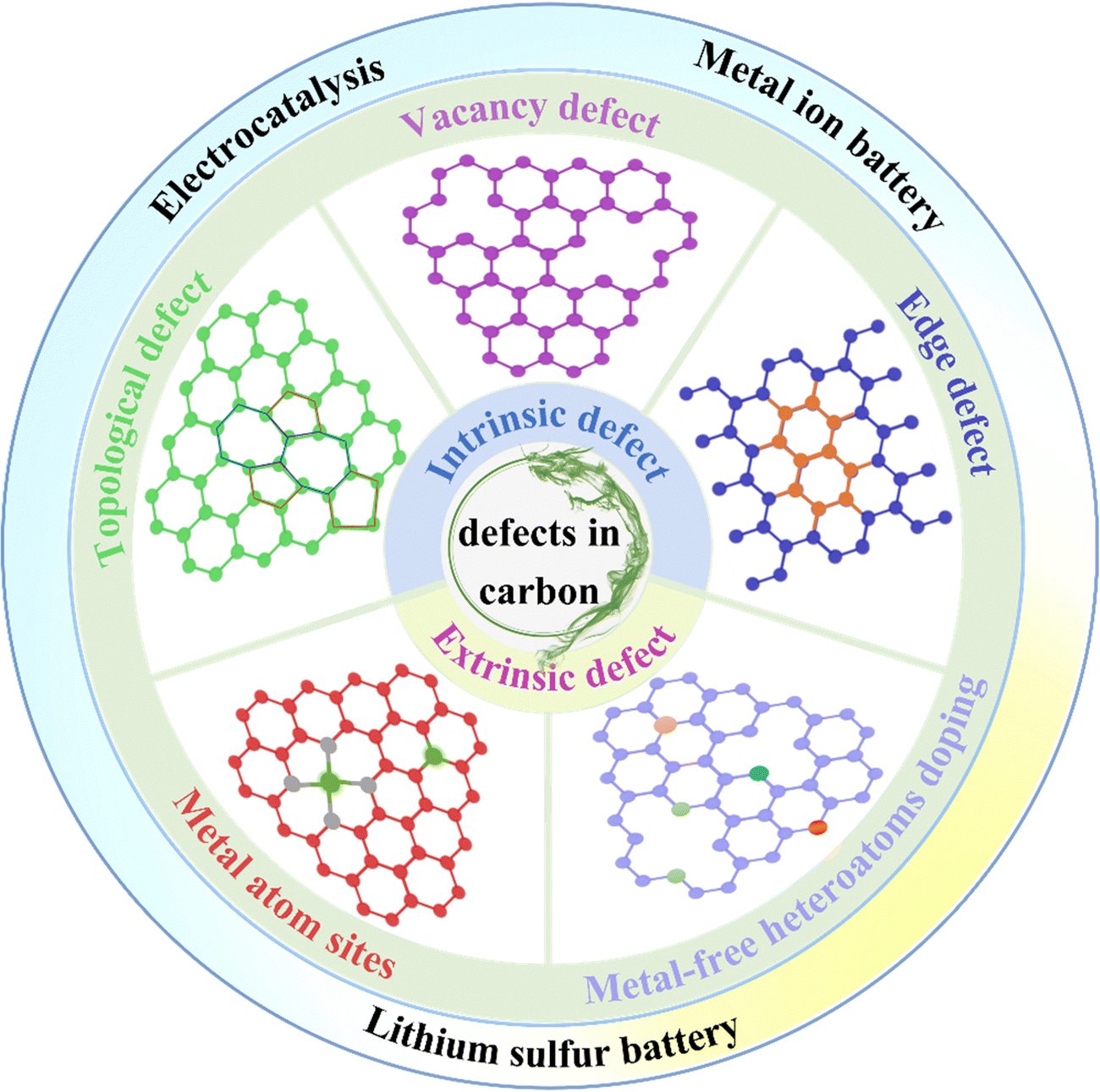
2.1. Defect classification
The multiplex types and complex structure of materials endow the defect with highly intricate manifestations. Therefore, the reasonable classification of defects is of great significance and affirmatively affects the rational design and application of defect engineering.Defect in solid materials can be compartmentalized into four main types according to their dimensions, including (1) zero-dimensional (0D) point defects, such as vacancies, interstitials, substitutions, heteroatom doping defects; (2) one-dimensional (1D) line defects, involving steps, dislocations, etc. (3) two-dimensional (2D) planar defects, mainly referring to grain boundaries, phase boundaries, etc. (4) three-dimensional (3D) volume defects, having precipitates, voids, etc. This is the most extensively employed classification method, and the applications of defect engineering based on this sorting in electrocatalysis,58 electrode materials,22 thermoelectric energy conversion44 and other fields have been summarized and reviewed. Focusing on past reports, it has been demonstrated that point defect engineering can make materials more active, and related studies are reported in a much greater volume than other types.50 Point defects can be further divided into intrinsic defects and non-intrinsic defects. Intrinsic defects appear as anomalous arrangements of crystal structures in nodes or adjacent microscopic regions, involving Schottky defects and Frenkel defects.59,60 Non-intrinsic defects, also known as doping defects, are formed by the intercalation of dopant atoms or ions into the crystal lattice.
Beyond that, for a clearer understanding, some researchers are interested in classifying the defect of solid materials on the basis of their source. Defects are assorted into intrinsic defects and impurity defects. The intrinsic defects described here are not limited to point defects, but also include line defects (e.g., steps and dislocations) and bulk defects (e.g., hole defects). Moreover, on the basis of the formation mechanism of defects, they can be partitioned into atom vacancies, gap defects, substitution defects and high-dimensional defects (dislocations, grain boundary, precipitates, etc.).61 In addition, defects can be divided into in situ defects and post-processing defects according to different construction strategies. The former is the direct formation of specific defects during material preparation, and the latter is the construction of defects through various methods on pre-prepared materials.
Carbon, as a member of the solid materials family, adopts the systematization mentioned above strategies without any inappropriateness. Defect engineering of carbon materials aims to design rational defect structures to increase their activity.54,62 Related research chiefly emphasizes point defects, which can be partitioned into intrinsic defects and extrinsic defects. Intrinsic defects have the following three common forms in carbon materials: lattice distortion (topological defect), carbon vacancy defects and sp3 hybrid carbon defects (edge defects). In terms of extrinsic defects, including metal-free heteroatoms (N, B, S, etc.) doped defects, and metal atom (Fe, Co, Ni, etc.) site induced defects.
2.2. Intrinsic defects
Early studies on the modification of carbon materials mainly emphasized surface functionalization and heteroatom doping, while intrinsic carbon defects were often overlooked. With the continuous consummation of related research, multitudinous researchers have certified that intrinsic defects have better electrochemical activity than heteroatom doping. The catalytic function of intrinsic defect sites has been extensively accepted and has aroused great interest.63–66Topological defects describe the distortion of the carbon lattice into one or more unconventional polygonal forms or even dangling bonds with the redistribution of asymmetric electrons, and common non-hexagonal forms formed include pentagon (C5), pentagon–heptagon–pentagon–heptagon (Stone–Wales defects, SW, C5-7-5-7, Fig. 3a), etc.39,67 Vacancy defects are easy to understand and are characterized by the absence of carbon atoms in the hexagonal carbon lattice. According to the number of missing atoms, it can be divided into monovacancy (MV), divacancy (DV), and multivacancy (losing three or more carbon atoms) (Fig. 3b). The formation of a single vacancy can be explained by the absence of a carbon atom resulting in the placement of three electrons in three dangling σ-orbitals, which further leads to the deformation of the three carbon atoms next to the vacancy and the formation of a new bond between two carbon atoms. Changes in the interatomic distances provide the most intuitive evidence for the appearance of an intrinsic defect in hexagonal lattices. As early as 2003, Hawelek et al.65 calculated through molecular dynamics modeling that the bond lengths of carbon atoms in SW defect, monovacancy, and DV are about 1.36–1.46 Å, 1.34–1.74 Å, and 1.33–1.81 Å, respectively, while in the conventional hexagonal lattice they are about 1.42 Å. A few years later, Kudur et al.68 reported that in SW defect graphene, the distance between rotating carbon atoms (C5–C6) can be reduced to 1.30 Å (Fig. 3c and d). Structures leading to changes in the carbon–carbon bond length of defect regions are closely related to the reactivity of carbon materials. A recent theoretical study has shown that defective sites affect the band structure of the carbon lattice.69 Usually, in a perfect graphene, the Fermi level (EF) is very consistent with the Dirac point, and the π and π* bands in the vicinity of the EF exhibit double degeneracy (Fig. 3e).70Fig. 3f shows the band structure of MV graphene. It can be seen that the EF is below the Dirac point, indicating the role of MV as a hole dopant. In contrast, the energy band structure of graphene with DV (Fig. 3g) and SW (C5775) defects (Fig. 3h) indicates that the defective π state lies above the EF, i.e., is indicative of the n-type doping behavior of DV and SW defects. Moreover, for the SW structure, the Fermi level coincides with the Dirac point and no additional carriers are introduced into the graphene.
 | ||
| Fig. 3 (a) Schematic images of defect sites of a topological defect, and (b) vacancy defect. (a and b) Reproduced with permission.71 Copyright 2015, Wiley-VCH. (c) Carbon atom arrangement of SW defect and DV defect at C96H24. (d) C–C bond lengths at different graphene models. Reproduced with permission.68 Copyright 2016. American Chemical Society. The band structures of (e) perfect graphene, (f) graphene with MV, (g) graphene with 5-8-5 DV, (h) graphene with SW. Reproduced with permission.69 Copyright 2013. (i) Schematic images of defect sites of an edge defect in graphene. Reproduced with permission.72 Copyright 2019, Wiley-VCH. | ||
On the one hand, edge defects are edge carbon atoms with high spin and charge density exposed by missing carbon atoms in a fixed orientation. On the other hand, no perfect material can achieve a borderless state, and carbon materials themselves have boundaries. The local charge redistribution induced by the edge carbon atoms enriches the edge sites with charge, thus conferring rich electrocatalytic reactivity on the edge sites. According to the difference in the structure index, the hexagonal carbon skeleton edge can be compartmentalized into the zigzag edge and the armchair edge (Fig. 3i)73,74 where the serrated edge positions are filled with a large number of unpaired π-electrons, and the armchair edges, the unpaired π-electrons on two adjacent carbon atoms tend to form covalent bonds. Interestingly, carbon-based materials rich in porous structures, especially microporous structures, have the potential to exhibit better activity. Furthermore, graphene nanoribbons (GNRs) have been developed as one of the promising metal-free catalysts by virtue of their large aspect ratio and numerous defects along their edges. Graphene quantum dots (GQD) have extremely small material dimensions that provide extremely rich edges and thus exhibit high edge activity.
Defect engineering in carbon materials can alter their characteristics, including mechanical properties, conductivity, catalytic activity, etc. The improvement in mechanical properties was confirmed by Rajasekaran et al. through molecular dynamics studies, suggesting that topological defects contribute to the intrinsic strength and fracture toughness of graphene carbon sheets.75 Moreover, Bocko and Pavol reported that the increase in the defect content reduces the critical buckling force of single-walled carbon nanotubes.76 As for conductivity, Zhu et al.39 calculated the energy gap values of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) in C6 and C5 by DFT. Compared with C6, the narrower HOMO–LUMO energy gap of C5 is more favorable for electron transfer.
Intrinsic defects caused by interference with the carbon lattice electron distribution can serve as active sites to enhance the activity of carbon materials. Kudur et al.68 compared the redox reactivity of pristine and defective graphene lattices using an analytical Fukui function based on density functional theory (DFT). It was found that both Stone–Wales defects and divacancy defects enhanced the redox reactivity. Moreover, the carbon atoms exposed by the defect structure have high charge density and high spin, which greatly improves the electrical conductivity of carbon materials.54,77 Jiang et al.46 demonstrated that zigzag edges possess unique local electronic states and chemical reactivity. Patel et al.78 proposed that structural defects may increase the adhesion of other atoms and molecules to carbon materials (CNTs). Wang et al.79 found that the intrinsic carbon defect has the ability to destroy the integrity of π conjugation and activate lone pairs of π electrons, enhancing the π–π electron donor–acceptor interactions between carbon-based catalytic materials and other molecules. All of these properties endow intrinsic defects with better electrochemical reactivity compared to regular hexagonal configurations.
It is noteworthy that porous carbon materials are extensively used in the field of electrochemical energy storage and conversion, especially for various electrode materials, because of their rich pore structure, high electronic conductivity, good chemical and thermal stability, easily adjustable physicochemical properties and low production cost.80,81 The large specific surface area provided by porous nanoarrays is believed to play an important role in loading a high number of defect sites on the electrodes, thus providing a great number of reactive sites for electrochemical redox reactions.82 The porous structure of the electrode increases the electrolyte infiltration rate, which helps to improve the utilization of active materials, reduce the electrode inhomogeneous polarization and improve the electrode stability. The association of physically acting porous structures with chemically acting active sites (intrinsically defective sites, heteroatoms, metal single atom and other catalytic materials) will effectively improve the electrode performance.83–85 Varying pore sizes confer a wide variety of functions on carbon-based materials, making them shine in electrodes. The microporous pores facilitate increase ion transport paths and are rich in edge-activated carbon, but the diffusion rate of lithium ions in them is hindered, making the multiplicative performance poor. Although large holes can relieve stresses in electrodes with large volume changes like sulfur electrodes, thus improving electrode stability, large holes are not conducive to the increase of electrode energy density. Mesopores are preferred by researchers because they can balance energy density and multiplicity performance as much as possible. Meanwhile, carbon-based electrodes with hierarchical pores have attracted much attention in recent years.86–89 In terms of the types of porous carbon materials used in porous electrodes, porous activated carbon,90,91 porous carbon nanotubes,92,93 porous graphene94–96 and porous conductive polymers show great potential for energy storage and conversion applications. In general, the research related to porous electrodes has made great progress and the applications will become more and more widespread.
2.3. Extrinsic defects
The intrinsic carbon defect sites have high compatibility and can serve as anchor sites and heteroatoms/clusters to form coordination active centers. Exotic atoms with different electronegativities and electronic structures can supersede the atoms in the carbon skeleton, be incorporated into the intrinsic defect of carbon, or be firmly adsorbed by carbon, which will induce intramolecular charge transfer or spin redistribution and generate the advancement of electrochemical activity. The research on improving the activity of carbon materials employing metal-free heteroatoms develops earliest and becomes increasingly mature. Relevant studies have been vastly reported,42,47,97–99 and the concept of metal-free heteroatom doped defects has won support among researchers. Metal atoms, including noble metal atoms and transition metal atoms, have high dispersion and atomic utilization. More importantly, they can be anchored on carbon materials to regulate the electronic distribution and chemical environment.| Element | C | N | F | I | Si | B | P | S |
|---|---|---|---|---|---|---|---|---|
| Electronegativity (χ, Pauling scale) | 2.55 | 3.04 | 3.98 | 2.66 | 1.98 | 2.04 | 2.19 | 2.58 |
| Δχ (vs. C) | +0.49 | +1.43 | +0.11 | −0.57 | −0.51 | −0.36 | +0.03 | |
| Atomic radius (Å) | 0.91 | 0.75 | 0.57 | 1.32 | 1.46 | 1.17 | 1.23 | 1.09 |
| Atomic volume (cm3 mol−1) | 4.58 | 17.3 | 17.1 | 25.74 | 12.1 | 4.6 | 17.0 | 15.5 |
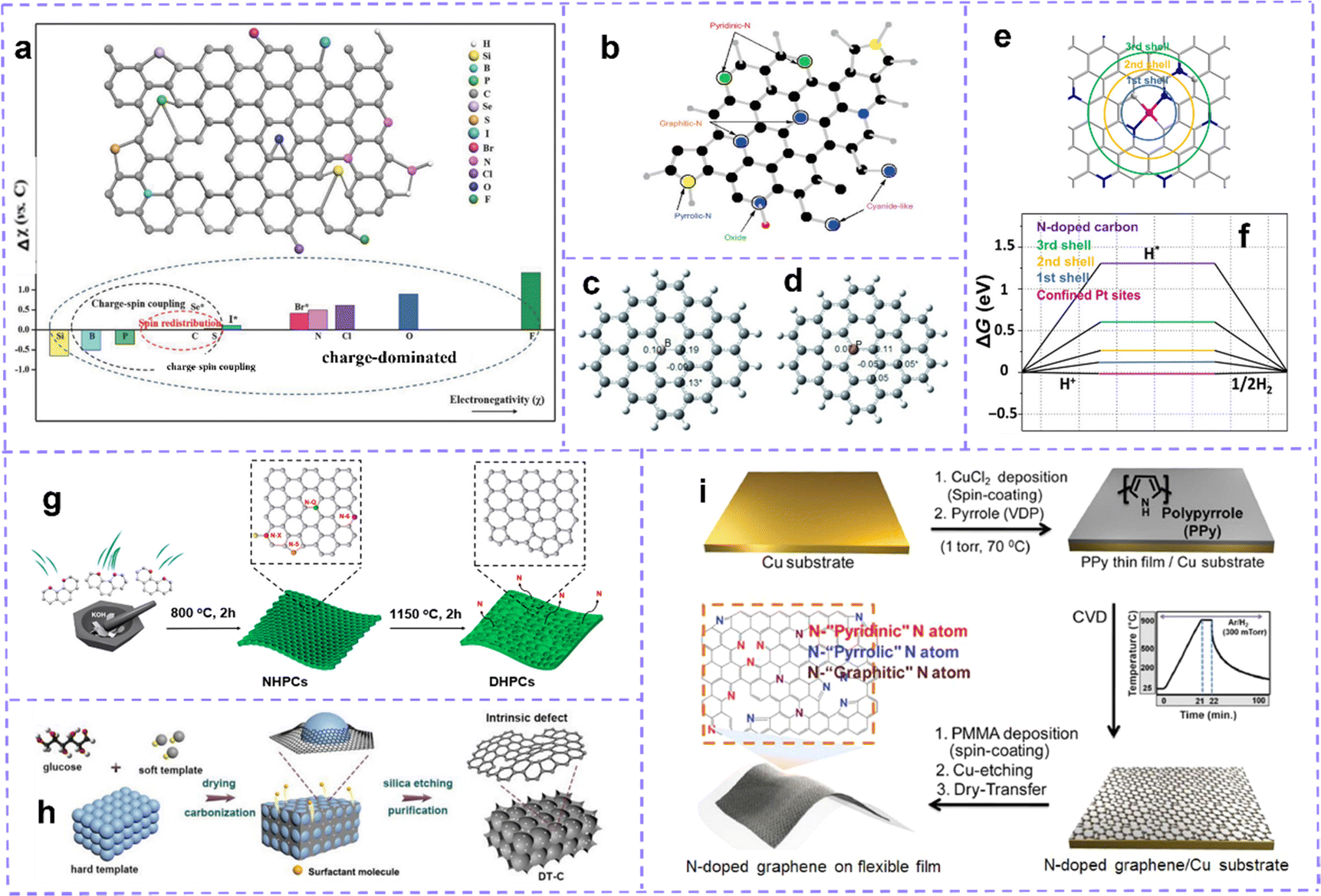 | ||
| Fig. 4 (a) The lattice structure of carbon doped with different heteroatoms and the electronegativity of non-metallic heteroatoms compared to carbon. According to the relative electronegativity of heteroatom dopants, the origin of doping effect is classified into charge redistribution, spin redistribution and charge spin coupling. The superscript of Se, Br and I marked with * indicates that these heteroatoms are too large to be incorporated into carbon crystals. Reproduced with permission.109 Copyright 2018, Wiley-VCH. (b) Different N-doping configurations. Reproduced with permission.112 (c) Mulliken spin density distribution of B-substituted graphene and (d) P-substituted graphene. Reproduced with permission.113 Copyright 2013, Wiley-VCH. (e) Schematic description for the coordination shells for the isolated Pt over the graphene. (f) ΔGH* on pure and Pt-decorated graphene in different coordination shells. Reproduced with permission.114 Copyright 2018, Science publishing group. (g) Schematic illustration of the synthetic process of EC, POC, PC by N Removal. Reproduced with permission.115 Copyright 2020, American Chemical Society. (h) Schematic illustrations of the synthesis process of the DT-C. Reproduced with permission.54 Copyright 2020, Wiley-VCH. (i) Schematic illustration of the synthesis of N-doped graphene on a flexible substrate by CVD. Reproduced with permission.116 Copyright 2018, Wiley-VCH. | ||
Nitrogen (N)-doping and halogen atom doping belong to the typical charge-dominant mechanism. N is the most frequently used heteroatom dopant in carbon material doping modification. N (χ = 3.04) has higher electronegativity than C (χ = 2.55). The difference in electronegativity between C atoms and N atoms enables the C atoms around the N-doped site to carry a positive charge density, allowing the carbon atoms to build a higher electron spin density. The valence electron structure and void orbital of N-doped carbon are both close to the Fermi level, resulting in more electronic transitions being provided.117 As a result, these effects enable N-doped carbon to exhibit desirable electron transfer ability and electrochemical reactivity.
Generally, the N atoms in nitrogen-containing carbon materials are in the following forms: graphitic-N (quaternary N), pyridinic-N, and pyrrolic-N (Fig. 4b). Each configuration is associated with an accompanying intrinsic defect structure. Graphitic N, also known as quaternary N, can donate electrons to π-conjugated systems (n-type doping), increasing the nucleophilic strength of the surrounding carbon rings. The graphitic N–C bond length is 1.3927 Å. Pyridine N (p-type doping) usually refers to an N atom bonded to two C atoms and is often found in MV, DV, and SW. Near the MV, pyridine N is the most stable configuration with an N–C bond length of 1.33 Å.118 The N atom is placed in the heptagonal ring of the SW defect as pyridine N, while in the pentagon of the SW defect, it is labeled as pyrrolic N (1.372 Å N–C bond length).119
Doping with halogen atoms, including fluorine (F), chlorine (Cl), bromide (Br), and iodine (I), is another effective strategy to construct doping sites. Halogen atoms with high electronegativity (χ: from 2.66 to 3.98), can form C–X bonds (X: F, Cl, Br, or I) and acts as an electron-acceptor. Halogen atoms form doped structures by breaking the π bonds between carbon atoms and their adjacent carbon atoms. Due to the larger radius of the halogen atoms, doping at the edges is easier and more stable.120 Halogen atoms are connected to sp2 carbon by replacing H atoms and changing the hybridization state of C atoms to sp3, which in turn affects the electronic properties and local structure production of carbon materials. Besides the fact that the C–F bond (1.378 Å) length is slightly shorter than the C–C bond (1.425 Å) in pristine graphene, the distances between the other halogen atoms and the adjacent carbon atoms are all greater than 1.425 Å; the detailed data are reported by Caio.64
The substitution of halogen atoms significantly affects the electron distribution and framework structure of carbon materials. Doping with halogen atoms increases the repulsive interactions within the carbon lattice while preserving the hexagonal lattice symmetry of the graphene structure. This substitution structure results in the loss of π-conjugated electron clouds above and below the graphene plane.121 The degree of halogenation can control the electronic properties. In suitable amounts, C–X bonds tend to enhance the polarization of highly electronegative halogen functional groups (Cδ+–Xδ−), thereby improving electronic conductivity and transport properties.122 But when the degree of halogenation is too high, the material tends to be insulating. For example, the charge carrier mobility of fully substituted graphene with F atoms (F atoms attached to each carbon atom) was shown to be three orders of magnitude smaller than that of pristine graphene. Karlicky et al.123 systematically summarized the structure, synthesis, and electrical, mechanical, optical, vibrational and thermodynamic properties of graphene halides.
In the periodic table of elements, C and silicon (Si) are in the same family, so they have similar structures and electronic configurations as well as the progressive covalent atomic radius (77 pm vs. 111 pm). The electronegativity of Si (χ = 1.98) is much lower than that of carbon so that the Si doping effect can be grasped by a charge-dominant mechanism. The reactivity of Si-doped carbon materials has long been demonstrated.44,45
Boron (B) and phosphorus (P) doping are classified as charge–spin coupling mechanisms. B atoms have a size close to that of C atoms and can be easily incorporated into the carbon framework. A longer B–B bond strains the C–C bond, which represents an increase in reactivity. Although B (χ = 2.04) has a lower electronegativity than C, it often requires electrons to partially fill empty orbitals in order to achieve more favorable energies, so it tends to act as an electron acceptor near the C–B bond.124 The incorporation of B atoms endows the surrounding C atoms with a positive charge density, which effectively promotes the transfer of charges, thereby promoting electrical conductivity.125,126 Moreover, unpaired pz orbitals of B atoms are vacancies, inducing relatively high spin densities (Fig. 4c).113 The most active sites in B-doped carbon materials are the edge carbon atoms with high electron-donating ability.127 B-doped carbon materials exhibit interesting reactivity. P atoms have slightly less electronegativity (χ = 2.19) than carbon, which can donate electrons to form the P–C bond and show a positive charge density.128 The atomic radius of P is larger than that of C, causing the P atoms to protrude in the C plane, the C–P bond is relatively long, and doping is difficult.129,130 Due to the larger structural deformation caused by the larger radius of the P atom, its induced spin density is relatively low. (Fig. 4d).113
Sulfur (S) is also a common dopant for lattice modification of sp2 carbon. However, because the electronegativity values of S (χ = 2.58) and C are similar, S doping has little effect on the charge distribution. The active sites of S-doped carbon materials can be explained by the “electron spin” contribution mechanism.131 The results of theoretical calculation show that compared with the original graphene and surface S-doped graphene, the edge-selectively sulfurized graphene has higher activity. Although the charge densities of the three types of graphene are similar, the edge sulfur oxide (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO) doping produces a high spin density (0.39 vs. 0 vs. 0).132
SO) doping produces a high spin density (0.39 vs. 0 vs. 0).132
Metal atom modification greatly enhances the activity of carbon materials. In recent literature reports, coordinating monatomic metals with N-doped carbon materials (M–N–C) to stabilize metal species has been an efficient strategy to obtain metal site induced defects. The 2p orbitals of N can be hybridized with the d orbitals of metals, and various forms of N functional groups furnish plentiful coordination sites for metal atoms. In addition to doping defect, the intrinsic defect sites in the carbon matrix are also conducive to capturing metal species.135,136 Guan et al.137 proposed that the adsorption environment coupling between the mononuclear Mn site and four N atoms is the source of the catalytic activity for water oxidation. Similarly, Wen et al.138 explained that the atomic Co corresponding to the N atom should be the active center of the HER and ORR. Analysis of noble metals also showed similar conclusions. Zhang et al.114 investigated the catalytic activity of different coordination shell sites around Pt, and the results showed that the reactivity increased with the clustering towards the metal atom center sites, and the activity of inert carbon could be induced by decorative metal atoms (Fig. 4e and f).
Both intrinsic and extrinsic defects have been demonstrated to effectively alter the charge/spin distribution on the sp2 lattice, thereby imparting activity to carbon materials. Different configurations of the intrinsic defect and extrinsic defect have different effects on the change of carbon lattice structure and the improvement of the activity. In the heteroatom doping process, it is inevitable to cause more or less changes in the carbon skeleton, and even induce a large number of intrinsic defects. It is encouraging that this may also be an important reason for the improved doping activity. The synergy of various defects induces comprehensive effects.21,114,139–141 and involves multiple defect structures that are expected to improve the electronic properties, stability, electrochemical performance and catalytic activity, all of which are conducive to their applications in electrochemical energy storage systems and energy conversion systems.
2.4. Strategies to construct defects
Various synthetic strategies have been employed to fabricate defect-rich carbon materials. Some of the most commonly used methods are discussed below, including mechanical ball milling, heat treatment, chemical vapor deposition (CVD), etc.Mechanical ball milling is a simple, effective, low-cost, and environmentally friendly strategy for constructing intrinsic and extrinsic defects. The ball milling process can grind large-sized powders into fine particles by shearing. Segmental cleavage of C–C bonds under a large shear induces the formation of intrinsic defects. As the grinding time increases, the lattice structure shows more defects. In the presence of a source of metal-free and/or metal heteroatoms in the grinding system, the heteroatoms form chemical bonds with carbon atoms, resulting in doped carbon.67,142 In addition, studies have shown that it is possible to harvest carbon materials with a controllable degree of defects by regulating the parameters of the ball milling process.143,144
Heat treatment is a common method for mass production of defect structures under a specified reaction atmosphere and reaction temperature. The degree of defect can be adjusted by changing the heat treatment temperature and the amount of gas introduced. It generally does not alter the shape and overall chemical composition of the matrix. At high temperatures, N2, H2S and other gases introduced into the system can be used as a source of heteroatom, or a source of heteroatom placed upstream of the carbon substrate can be gasified at high temperatures to induce doping. Studies have shown that a certain amount of heteroatoms can diffuse into the carbon lattice at high temperatures.145,146 Furthermore, after heat treatment, the heteroatoms in the doped carbon are removed to form MV defects, which can drive the reorganization of peripheral carbon atoms to form various topological defects at high temperatures.57,147 Gan et al.115 prepared edge-rich carbon (EC), pentagon–octagon-rich carbon (POC), and pentagon-rich carbon (PC) by removing N heteroatoms at 1150 °C for 120 minutes (Fig. 4g). Recombination has irreversibility, the thermal motion of carbon atoms is limited, and the carbon skeleton that has formed defect is difficult to recover. Adjusting the ratio of intrinsic defect introduced by the removed atoms to the retained atoms in the carbon matrix is also beneficial for synergistic effects.148
A template assisted method, which can assist in the preparation of defect in the heat treatment process, has been developed into a mature method of defect preparation. Carbon atoms around the template are stretched or squeezed to induce lots of intrinsic defects in the carbon lattice. In the case of containing dopants, doped carbon can be obtained. Templates can be divided into hard templates and soft templates. The removal of most external hard stencils requires complex etching or washing procedures. Guo et al.54 used silica as a hard template and a surfactant as a soft template to prepare porous interconnected intrinsic defect-rich carbon (DT-C) (Fig. 4h). Yuan et al.116 prepared N, P-doped carbon using polystyrene nanospheres (PS), polyvinylpyrrolidone (PVP), and phosphoric acid as templates, carbon/N sources, and P sources. Peng et al.149 prepared Fe, N-doped carbon employing Fe2O3 nanoparticles as the template and iron source. Moreover, Chen et al.150 prepared nanosheets by pyrolyzing rhodamine and releasing gas as a soft template.
Chemical vapor deposition (CVD) is a common in situ method for introducing heteroatoms into carbon matrices. Using metals and their compounds as substrates and catalysts, copper foil is the most commonly used, and then carbon precursors and heteroatom precursors are introduced, which will decompose and release carbon atoms and heteroatoms to be deposited on the substrate under high temperatures. After cooling, doped graphene forms on the surface of the substrate (Fig. 4i). The doping quality and quantity are affected by the precursor ratio and deposition conditions. This process shows obvious advantages in the preparation of high levels of heteroatom-doped carbon.151–154 It is worth noting that we only briefly introduce the construction strategy for defects here, and there are many excellent reviews and research articles that summarize this aspect in detail, and readers can refer to them as needed.115,155–159
In current reports, most defective carbon materials exhibit good stability, which ensures that the overall cell performance does not suffer from rapid degradation during service. However, most catalyst materials, including defective carbon materials, suffer from significant stability challenges. Defective carbon-based catalytic sites can exhibit some reversible or irreversible structural damage during service, especially in harsh environments. First, to ensure better activity, the stability of the defect sites needs to be improved. On the other hand, the dynamic evolution of the defect sites may have unexpected effects on the performance, which requires more accurate characterization techniques.
3. Recent advances in carbon defect engineering for MIBs
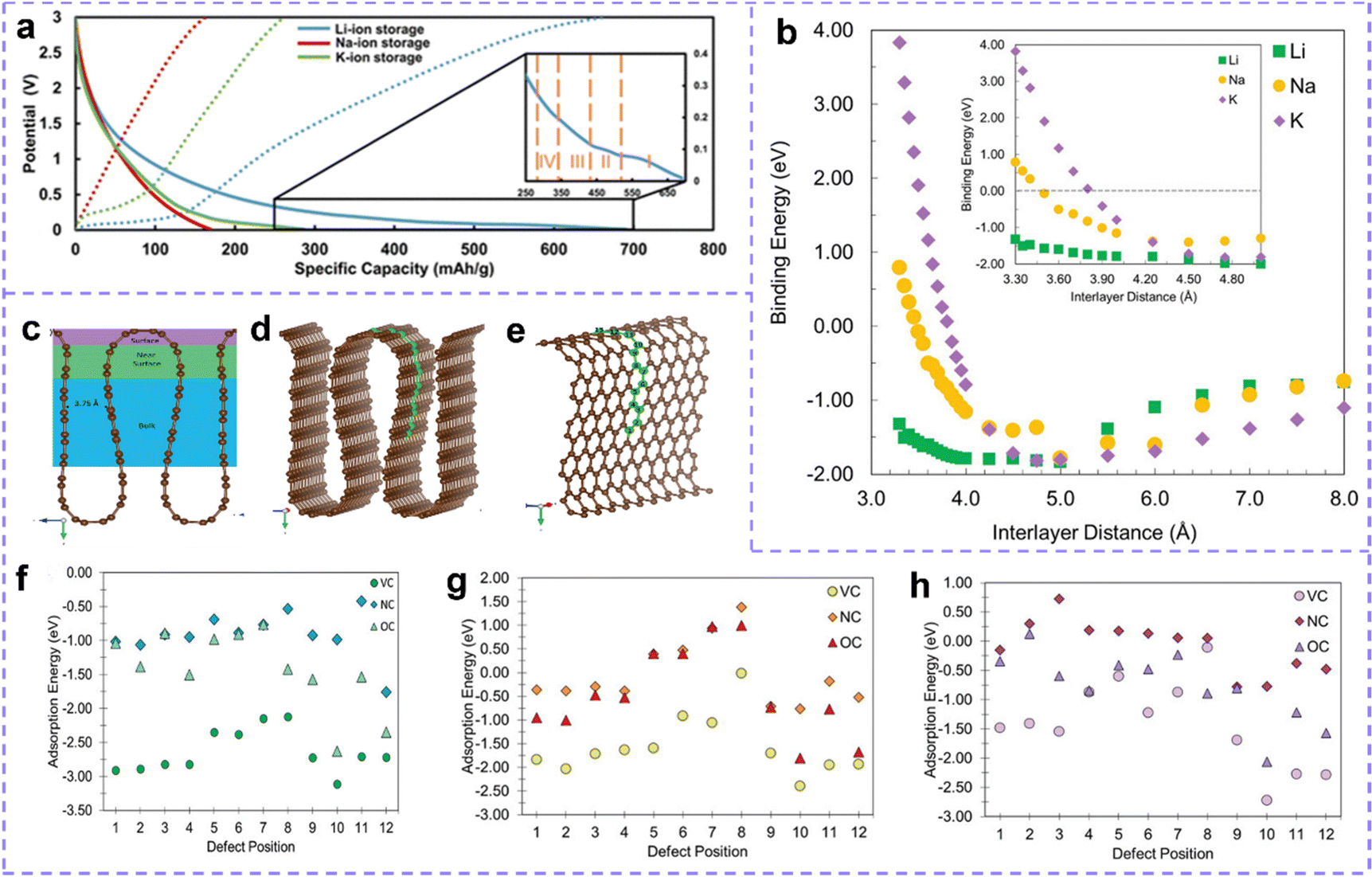
Rechargeable metal ion batteries (MIBs), including Li+, Na+, K+, Mg2+, Zn2+, Al3+, etc., have the preponderance of good safety, high capacity, energy-saving and environmental protection, and is regarded as a foreground energy storage system. The battery has four main components: anode, cathode, electrolyte and separator. In the internal circuit, metal ions as a shuttling matter are exchanged between the cathode and the anode, and the external circuit relies on electron transfer to provide electric energy. Take the lithium-ion battery (LIBs) as an example. During charging, Li ions deintercalate from the cathode, enter the electrolyte, pass through the electrolyte and the separator, and are finally inserted into the anode. When discharging, the reverse process occurs, and Li ions are stored on the cathode side. As we all know, electrode materials, as one of the most critical components, have the control force which determines the capacity, efficiency, stability, rate capability and safety of MIBs.160–162 Among many electrode materials, carbon materials have received extensive solicitude due to their good electrical conductivity, stability and low cost. Carbon has both redox properties, and common storage ions include cationic (Li+, Na+, K+, Ca2+, etc.) and anionic (AlCl4−, BF4−, etc.). The storage behaviors of metal ions in carbon materials include adsorption, intercalation/deintercalation, diffusion, etc. Numerous findings suggest that defect sites can participate in the storage of metal ions and render distinctive functions in the electrode.6,163,1643.1. Investigations of the relationship between carbon defect engineering and MIB performance
A tremendous number of studies have manifested that active sites, including intrinsic defect sites and extrinsic atom doping sites, can improve the electrochemical activity of carbon materials and the capacity of batteries.165,166 However, the impact of defective spots on performance is not always as desirable as expected, and the actual response mechanism induced by defective spots remains controversial.Some views that carbon defect sites negatively affect the performance of MIBs have been reported.167–171 Chen et al.171 shared a view in 2017: The fewer the defects, the better the electrochemical manifestation. Graphene aerogels with rich defects (GA-2000), fewer defects and no defect were prepared by controlling the annealing temperature. After testing, it is clear that GA-2000 with ample vacancy and tortilla carbon network shows a lower capacity and worse rate performance. The blue shift of the G band in the Raman spectrum during charging and redshift during discharging confirmed the insertion and extraction of AlCl4− in the graphene layer. However, the D band related to the defect reveals no deviation, which indicates that the AlCl4− anion has not been embedded into the defect site (Fig. 5a and b). Furthermore, the defect also hinders the rapid insertion of AlCl4− into the graphene layer and lowers the conductivity of the electrode. At present, there is no consensus on whether this negative effect is a special case or a universal one, which shows that there is still some confusion about the actual function and mechanism of defect engineering in MIBs.
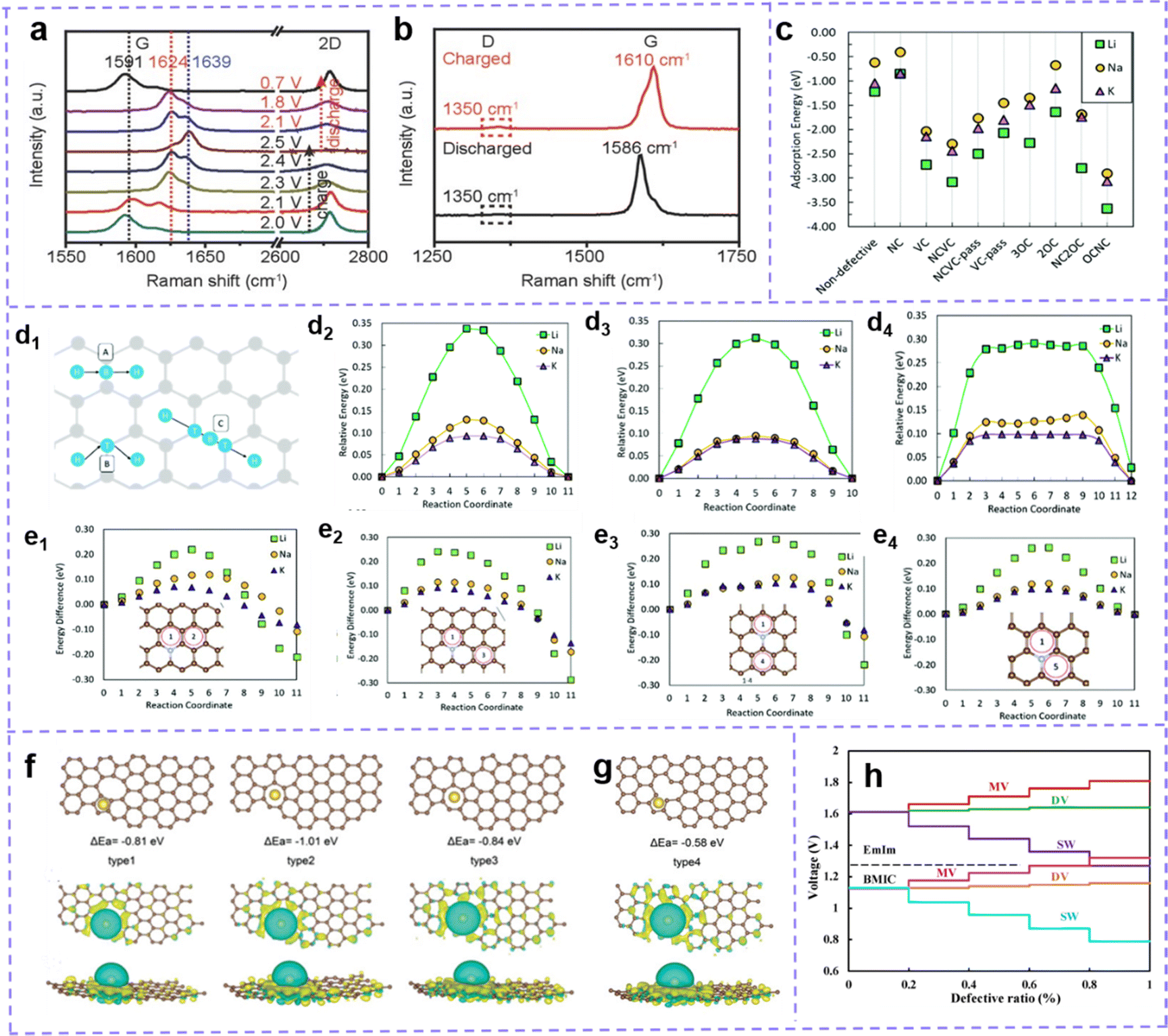 | ||
| Fig. 5 (a) In situ and (b) Ex situ Raman spectra of GA-2000 during the cyclic process. Reproduced with permission.171 Copyright 2017, Wiley-VCH. (c) Li, Na, and K adsorption energy on a carbon surface with defects. (d) Schematic of the three considered metal diffusion paths, and their respective migration barriers for Li, Na, and K on pure graphene, (d2) H–B–H, (d3) H–T–H, and (d4) H–T–B–T–H using the climbing image nudged elastic band method. (d1) Grey circles are carbon atoms, and blue the migrating metal atom moving in the direction of the arrows. B denotes the bridge site, H the hole site, and T the top site. (e) Metal migration activation energies (in eV) on N-doped graphene (NC) following metal migration paths (e1) 1–2 across a C–C bond, (e2) 1–3 along a C–C bond, (e3) 1–4 along a C–N bond, and (e4) 1–5 across the C–N bond. (c and e) Reproduced with permission.172 Copyright 2019, Royal Society of Chemistry. (f) Configuration, adsorption energy, and top and side views of the electron density of Na atoms adsorbed on carbon structure with divacancy defects and (g) monovacancy defects. The brown and yellow balls represent the C and Na atoms, respectively. Reproduced with permission.54 Copyright 2020, Wiley-VCH. (h) Voltage profile of intrinsic defects varies with different concentration ratio for EMIm and BMIC ionic liquids. Reproduced with permission.173 Copyright 2020, Elsevier. | ||
Theoretical capacity and operating voltage are two clutch factors for the electrochemical performance of MIBs. The allegro charge transfer with the metal and matrix implies the increase of capacity. Charge transfer directly pertained to the bonding between metal ions and electrodes as well as the migration of metal ions. A moderately high bonding energy means resultful adsorption of metals, which is beneficial to the stability of electrochemical properties. If the bonding energy is too high, the metal ions will be trapped in the defect sites. These trapped ions will generate electrostatic repulsion of subsequent ions, resulting in low ion concentration between graphite layers. In addition, the high bonding strength is associated with the reduction of battery voltage.168,173,174 When the binding energy is low, it is difficult for the metal to intercalate or adsorb, which is not conducive to the increase of the metal ion concentration. Although the bonding strength affects the migration, it is also deeply influenced by the metal migration barrier around the metal adsorption or insertion site. Even the strong adsorption of metals may adversely affect the diffusion.172 This may be related to the types of metals, the properties of adsorption sites, and so on, which are discussed below.
The advantages and disadvantages of defect engineering to carbon-based electrode materials should be evaluated by the comprehensive effects of bonding and migration. Zhang et al.172 systematically studied the adsorption and migration of Li, Na and K on graphene. Compared with the defect-free system, most defects show a stronger adsorption force (lower negative adsorption energy) (Fig. 5c). The energy barrier of metal migration/diffusion consists of metal-defect interaction energy and activation energy of metal migration. The data of metal–defect interaction energy show that the defect structure does not have unfavorable high metal defect bonding. However, in most cases, the calculated migration energy barrier is higher than that of pure graphene. Taking the common N-doped defect as an example, the doped sites are not suitable for the strong adsorption sites of metals. However, according to the calculation of migration energy, compared with pure graphene (Fig. 5d), the existence of an N-site reduces the energy barrier of alkali metal migration (Fig. 5e). So N doping defect engineering benefits from its positive side and can also be used as an effective strategy to upgrade MIBs (M: Li, Na, and K).
Besides, there are also reported that monovacancy in graphene has the effect of trapping Li+, but it resists the migration of Li+ (migration barrier increases from 0.32 to 0.87 eV).175 Yang et al.176 discussed the influence of defect structure on the specific capacity and migration energy barrier of Na. When the voltage is appropriate (>0.5 V), bilayer graphene (BLG) with a monovacancy defect improves the intercalation and adsorption of Na, and the capacity is increased (from 123.97 mA h g−1 to 382.54 mA h g−1) compared with pure BLG. The monovacancy defect of carbon is also unfavorable for Na migration, and its energy barrier (0.59 eV on the surface and 0.56 eV between layers) is larger than that in the original BLG (0.15 eV on the surface and 0.32 eV between layers).
The effect of carbon defect sites on the performance of MIBs is highly dependent on the defect type. Guo et al.54 calculated the adsorption energy and charge density of defect sites through DFT, and the results showed that the divacancy defect was more helpful to improve the adsorption of Na on carbon materials than monovacancy defects (Fig. 5f and g). The trend of charge density accumulation is more obvious in the structures with divacancy defects. Defects that reduce the surrounding charge density facilitate the transfer of charge from Li, Na, and K atoms to carbon structures, which are more suitable for introduction in anodic carbon materials. And those defect structures that facilitate the transfer of charge from carbon skeleton to metal ions are more favorable for the cathode performance. Moreover, Debnath et al.174 calculated that the binding energy between graphene and AlCl4− is −2.21 eV. When carbon atoms are replaced by N, B and Si atoms, the values become −3.14 eV, −2.11 eV and −4.23 eV, respectively. B atoms weaken the bonding, while Si doping will produce such strong bonding that AlCl4− may not be able to achieve intercalation. Considering the binding energy, N doping is a suitable choice in AlIBs. The influence of N-doped configuration was further studied. Compared with primitive graphene, the existence of pyridine N and pyrolytic N reduces the bonding strength; Graphite N can obviously improve the bonding strength between Al and adsorption sites. In alkali metal batteries, because the intercalation ion is a cation, the situation is the opposite. Yang et al.177 reported that the adsorption of K by pyrrole and pyridine N-doping is stronger than that by graphite N-doping. This may be attributed to the electron enrichment of graphite doping while pyrrole N-doping, pyridine N-doping and carbon vacancy defects can cause electron destitution and the inclination for extracting electrons from K atoms, thus facilitating adsorption.
Zamri et al.173 investigated the role of different intrinsic defects in AlIBs. The binding energies after intercalation of AlCl4− of bilayer graphene with pure, monovacancy (MV), divacancy (DV) and Stone–Wales (SW) defects are −1.78, −1.74, −1.83 and −2.30 eV, respectively. Therefore, the SW defects endow bilayer graphene with favorable adsorption. Besides, an AlCl4− molecule receives 0.8074, 0.6251, 0.7955 and 0.9343 |e| from the adjacent carbon atoms of pristine, MV, DV and SW, respectively. This means that SW defect sites can enhance the charge transfer of the Al ion storage positive electrode. By calculating the AlCl4− diffusivity rates in pristine (8.01 ×× 10−6 cm2 s−1), DV (8.87× 10−6 cm2 s−1) and SW (1.03 × 10−5 cm2 s−1) bilayer graphene, it can be seen that SW shows obvious improvement in ion diffusion. Based on this, bilayer graphene with SW defects should be able to achieve satisfactory theoretical capacity, but it is disappointing that the voltage fluctuates greatly with the proportion of SW defects, which makes it difficult to match the working voltage of AlIBs (Fig. 5h).
The influence of the same defect structure on the storage performance of metal ions has many aspects, and the final effect is superimposed by multiple factors. Debnath et al.174 concluded that in the graphene network, two AlCl4− groups began to agglomerate when they were placed in the 43.85 Å2 regions, while in C11N and C4N networks, this value was reduced to 27.792 Å2 and 34.812 Å2, respectively. The introduction of N sites can accommodate more AlCl4− without agglomeration in the same space. Once they aggregate, two AlCl4− can easily form one Al2Cl7− and release one Cl atom, which could integrate with other Cl atoms to produce Cl2 molecules. From the point of view of preventing agglomeration, N-doped sites are beneficial to Al ion intercalation. However, Childress et al.178 proposed that a N dopant would hinder the intercalation of AlCl4−. On the one hand, N-doped graphene has enhanced redox catalytic performance, resulting in AlCl4− reduction to generate chlorine molecules; On the other hand, the repulsion between N-doped sites and highly electronegative Cl will prevent intercalation. Then, finding out the influence of N doping on AlCl4− intercalation may require further all-round research by researchers.
From the above discussion, it can be seen that the defect engineering of carbon-based electrode materials in MIBs is very complicated, which is also true for other electrochemical systems. Sophistication comes from many aspects. First, the diversity of carbon materials and the complexity of defective structures have set up obstacles for the comparison and popularization of the results. And at present, the intercalation mechanism of some metal ions has not been fully grasped, which is not conducive to studying the adsorption or intercalation of metal ions at defect sites. Besides, the defects induced in the carbon layer are usually random, and there may be differences between theoretical analysis and experimental results.
Every coin has two sides. We know that some defective structures can play a positive role in specific types of batteries, while some defects have more obvious negative effects on the specific batteries. The former is worth popularizing to realize high-performance carbon-based electrode materials, while the latter requires defect shielding to improve efficiency and cycle stability. Generally, defect structures that can promote charge transfer between the metal species (Li, Na, K, Al, etc.) and the carbon matrix to achieve strong bonding, and defect structures that can offer more migration paths and faster migration rates for metal species are preferred, because they are conducive to the adsorption and embedding of metal species, and realize high capacity, high cycle stability and high rate capability of MIBs. Anyway, it is undeniable that abundant defects will definitely have a significant impact on the capacity and rate capability of carbon-based electrode materials, which has endowed defect engineering of carbon materials with strong persuasiveness.
3.2. Carbon-based electrode materials for MIBs
Generally, large interlayer spacing and clipping kinetics are beneficial to the anode, while platitudinous active sites and big specific surface areas are the requirements of the cathode. Obviously, the characteristics of carbon materials met application demands perfectly. At present, the most widely employed carbon-based electrode materials include graphite, graphene, graphite alkyne, hard carbon, soft carbon, and so on.As we know, graphite has a layered structure. This structure is connected by a strong covalent bond in the plane and a weak van der Waals force in the vertical direction, with a layer spacing of 3.35 Å, which creates the possibility for ion intercalation.179,180 As can be seen from the discharge curve, for graphite, an obvious low voltage plateau can be observed in the discharge curves of Li and K (Fig. 6a), which means that charge intercalation will occur at a low voltage before electroplating metal. However, there is no low voltage plateau on the Na discharge curve, which indicates that Na is not intercalated into graphite.168,181 The argument that Na cannot be inserted into graphite is repeatedly mentioned in the literature, which may be related to thermodynamic factors rather than the limitation of only size effect.182
 | ||
| Fig. 6 (a) Galvanostatic charge–discharge profiles of graphite electrode measured at a current density of 1 A g−1. Reproduced with permission.181 Copyright 2021, American Chemical Society. (b) The curves of binding energies of Li, Na and K in planar graphite with the distance between layers. Reproduced with permission.183 Copyright 2020, Wiley-VCH. (c) Schematic diagram of different areas (volumetric, near-surface and surface). (d and e) Simulation unit, brown sphere represents carbon atom, green sphere represents carbon atom general defect location. The red arrow represents the x direction, the green represents the y direction, and the blue represents the z direction. Metal adsorption energies for (f) Li, (g) Na, and (h) K in the VC, NC, and OC defect systems. (c–h) Reproduced with permission.184 Copyright 2021, Wiley-VCH. | ||
However, the advancement of high-performance MIBs is hindered by the small distance between graphite layers (0.335 nm), few ion intercalation sites, high sensitivity to electrolyte and long diffusion range between graphite layers.185 In fact, there are many types of carbon materials with unique graphitization and layer spacing available. For example, non-graphitic carbon (hard carbon and soft carbon) with a disordered structure, its binding mechanism with metal ions may include intercalation, adsorption, pore filling, deposition and so on.170,186 The complexity and diversity of storage mechanisms have aroused great interest in researchers. Perfect graphite is idealized, and disordered parts (grain boundaries, edges, and non-graphite areas) in graphite will also have an impact on ion storage. In recent years, multifarious full-fledged carbon-based electrode materials have been widely designed and developed to equip high-performance MIBs.187 In particular, defect-rich carbon materials will expose auxiliary surface intercalation/adsorption sites to improve the insertion of metal ions and charge transfer, which will lead to higher metal storage.188
Metal adsorption or intercalation is strongly associated with the distance between layers and defect position.23,189 Take the planar graphitic layer as an example, lithium is always remunerative for insertion at 3.35–8.0 Å interlayer distance, while the binding energy of Na and K becomes negative only when the interlayer distance is greater than 3.49 Å and 3.85 Å respectively, which means favorable insertion (Fig. 6b).183 The presence of defect sites improves the intercalation and adsorption of metal ions, and the location of defect is crucial. Olsson et al.184 studied the effects of defective structure on the storage capacity of metal ions (Li+, Na+, and K+) at different positions in hard carbon anode materials. The analysis shows that the intercalation capacity of metal strongly depends on the defect position (Fig. 6c–h). Taking Li+ as an example (Fig. 6f), for vacancy carbon defect (VC), N substitution (NC), and O substitution (OC) defect, Li has similar adsorption strengths at defect positions 1–4 and defect positions 9–12. The former may be due to strong intercalation binding, while the latter represents strong adsorption binding. Positions 5–8 represent the transition from surface adsorption to intercalation and thus weaker adsorption/intercalation.
The defect structure produces strong chemical bonds between metal and matrix, and then affects the intercalation and adsorption of metal ions. The difference in adsorption energy leads to the difference in metal migration behavior. Reports from Liu and co-workers confirm that alkali metals and alkaline earth metals have strong binding to carbon materials, especially N-doped carbon. In addition, it is found that Na and Mg generally have the weakest binding to the matrix among alkali and alkaline earth metals due to the competition between trends in the ionization energy and the ion–substrate coupling.190 So, the low sodium capacity of commercial graphite was also identified.
Intrinsic defect sites in carbon materials can upgrade the metal storage capability and contribute to the electrochemical reaction of batteries. Zhou et al.191 proposed that the carbon vacancy is beneficial to Li storage, and the isolated carbon vacancy in bilayer graphene can capture three Li ions between two graphene sheets. Ou et al.25 introduced carbon vacancy defects on graphene (BMG) substrates using mechanical ball milling, and the milling time was controlled to tailor the vacancy density (Fig. 7a). BMG-30 h with a relatively high defect density exhibits excellent Li and Na storage properties. Wang et al.192 reported that the exposed intrinsic defect in carbon nanosheets promoted Na ion adsorption, thereby enhancing the capacity storage and the rate performance of SIBs. Um et al.193 confirmed that the reduction of K ions in defect-rich graphite is easier than that of first hand graphite, which means that it has higher K ion storage capacity and more stable adsorption. Wang et al. synthesized carbon materials with different intrinsic defects using a template-assisted method and investigated the effect of intrinsic defects on the potassium storage performance.53 It was found that higher levels of intrinsic defects were more beneficial to improve the capacitive behavior by providing sufficient potassium adsorption sites. The researchers revealed a strong positive correlation between the level of intrinsic defects and the capacity/capacity retention, while the wettability of the carbon materials increased with the increase of the defect concentration, resulting in the rapid penetration of electrolyte ions. Furthermore, Ha et al.194 calculated the binding energy of the perfect graphene model and topological defect model (C585, C55777) to Al2Cl7− and Al. The results show a forceful interaction between Al ions and defective carbon sites, and the defect sites are conducive to reducing Al3+ to Al atoms through electron transfer on the electrode surface (Fig. 7b and c).
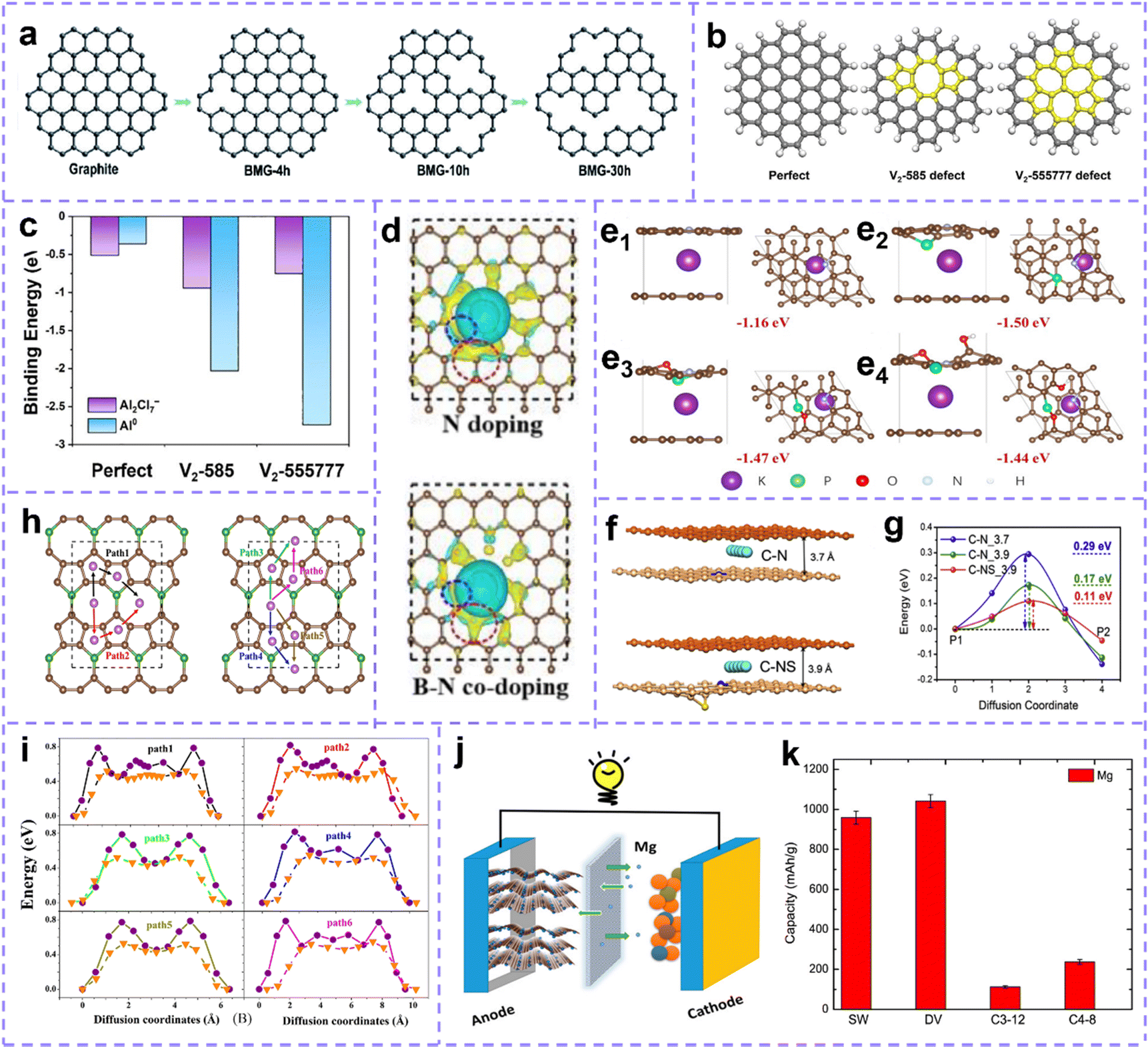 | ||
| Fig. 7 (a) Schematic of the structural transformation of BGMs. Reproduced with permission.25 Copyright 2021, Royal Society of Chemistry. (b) Structures of perfect and defective carbon networks. (c) The calculated binding energy of Al species and each matrix. (b and c) Reproduced with permission.194 Copyright 2022, Elsevier. (d) The charge density difference between pyridine-N doped and B, pyridine-N dual-doped carbon. The blue and red circles reflect the charge difference around the doped N and B centers, respectively. Reproduced with permission.195 Copyright 2022, Elsevier. (e) Side and top views of K+ adsorption on (e1) pyridine N-doped hard carbon (HC), (e2) pyridine N, P, doped HC, (e3) pyridine N, P, O doped HC, and (e4) pyridine N, P, O, OH doped HC. Reproduced with permission.196 Copyright 2022, Wiley-VCH. (f) The interlayer spacing of N-doped carbon before and after S-doping changes. (g) Diffusion barrier energies for the C–N and C–NS models. Reproduced with permission.186 Copyright 2020, Elsevier. (h) Top and side views of the atomic structure of popC5B monolayer and Li/Na migration paths. (i) The corresponding diffusion barrier distributions for Li and Na ions. (h and i) Reproduced with permission.197 Copyright 2021, Wiley-VCH. (j) Schematic model of a defective 2D carbon-based structure as a high-capacity negative electrode for magnesium ion batteries. (k) Maximum capacity of magnesium on graphene (allotropes) with different defect. (j and k) Reproduced with permission.198 Copyright 2016, American Chemical Society. | ||
Furthermore, the construction of extrinsic defect is treated as a simple and effective strategy to increase the electrochemical activity of carbon materials, enhance the capacity, promote charge transfer and accelerate the reaction kinetics. Hu et al.195 studied the adsorption behavior of graphite, pyridinic-N and pyrrolic-N doped carbon layer to K+, and the data showed that N doping is helpful to K+ adsorption. Then, the adsorption behavior of B, N dual-doping was compared, which revealed that the addition of B enhanced the adsorption performance of K. And the accumulated electrons around B (red circle) increased significantly, which signifies that the B site has a stronger attraction to K than the N site (Fig. 7d). Recently, Huang et al.163 proved that increasing S doping content and defect level can enhance the insertion depth of K ions and enrich additional active sites. Moreover, Chen et al.199 sorted out the application progress of heteroatom-doped carbon anode in SIBs in their review article.
Both intrinsic and extrinsic defects can positively affect the electrochemical activity of carbon. Then the synergistic effect of various defects is of great benefit to the application of carbon materials in MIBs. Recently, Yang et al.196 studied the adsorption energy (ΔE) of K by N-doped defect sites (N-5), O-doped and P-doped. The results show that ΔE increases to −1.50 eV after N and P dual doping compared with pyrrole N doping (−1.16 eV). On this basis, O and OH were introduced, but ΔE decreased slightly (Fig. 7e). The existence of synergistic effects or hindrance between dopant atoms is confirmed by this case. Pei et al.186 reported the effects of carbon vacancy, N and S doping on the diffusion and storage of Na. With the addition of N, the carbon layer spacing (3.7 Å to 3.9 Å) is increased and the diffusion barrier energy is decreased (Fig. 7f and g). More importantly, the interaction between dopants and the intrinsic defect has been born out. The synergy between N, S dopants and carbon vacancy profoundly affects the storage performance of Na. Moreover, Wei et al.197 designed graphene (popC5B) with B doping and pentagonal–octagonal–pentagonal ring structure (Fig. 7h). Electron transfer is enhanced by substituted B atoms. Carbon–boron vacancy–vacancy pair structure further strengthens the delocalization of charge. The maximum migration energy barrier of Li and Na atoms in this structure is 0.77–0.81 eV, 0.52–0.55 eV, which implies that popC5B can own the same charge/discharge rate as LIBs and SIBs (Fig. 7i). When used as the anode in LIBs and SIBs, the maximum storage capacity reaches 1891 and 1135 mA h g−1 respectively. The synergy of heteroatoms with metal compounds, metal nanoparticles also exhibit a strong metal ion storage capacity. High content of heteroatom (N and O) doped Co/CoOx nanoparticles anchored porous carbon rectangles were designed by Sun et al.200 The carbon material exhibits a hyperpolar surface with high hydrophilicity and abundant surface defects due to high heteroatom doping (both N-/O-doping above 10 atomic %) and layered pore system.
Defect-rich carbon tends to have high specific metal storage capacities and excellent reversibility. For graphene with the divacancy defect density of 25%, the maximum Li storage capacity is as high as 1675 mA h g−1,6 which is 4.5 times that of graphite (372 mA h g−1). For the Stone–Wales defect, the maximum Li storage capacity can reach 1100 mA h g−1 and the maximum capacity of Ca is 2142 mA h g−1.7,201 B-doped graphene (BC5) can form the Li6BC5 compound after Li+ adsorption, with a Li storage capacity of 2271 mA h g−1.202 In beryllium (Be) doped graphene, sixteen Li+ can be easily connected around a Be center (divacancy case), and the theoretical storage capacity of Li increases to 2303.295 mA h g−1 due to the appearance of Li8BeC7.203 The schematic model of a defective 2D carbon-based structure as a high-capacity negative electrode for magnesium ion batteries is shown in Fig. 7j. The maximum magnesium (Mg) storage capacity on graphene with a different defect shown in Fig. 7k illustrates the discharge capacity of 1042 mA h g−1 with 25% divacancy defects.198 Not only the theoretical capacity is satisfactory, but the actual capacity that can be achieved in the application process is also distinctly improved.6,204,205
This subsection highlights recent advances in the application of carbon materials with defect sites in metal ion batteries. The contribution of defect sites to the improvement of carbon-based electrode performance is reported. The introduction of defect sites is a powerful means to improve the intercalation capability of carbon materials, which can lead to substantial improvements in energy storage applications.
4. Recent advances in carbon defect engineering for MSBs
Although LIBs have achieved overwhelming applications, their specific capacity, energy density and safety are still strenuous to content the future energy storage needs. No discoveries without challenges. Numerous researchers are inspiringly seeking alternative solutions, hoping to develop a new type of secondary battery with considerable performance advantages and cost superiority. MSBs stand out with advantages of economy, stability, ecological compatibility and high theoretical energy density. Lithium–sulfur batteries (LSBs) in particular have attracted great interest from researchers and are regarded as promising candidates for high-energy-density systems.Rechargeable LSB consists of a Li metal anode, sulfur cathode, separator and organic lithium-based electrolyte. Unlike the LIB intercalation mechanism, the operation of LSBs relies on Li metal electroplating and stripping on the anode side and sulfur species conversion on the cathode side.206,207 The reduction of S8 molecules is to first transform into high-order polysulfides Li2Sn (4 ≤ n ≤ 8), then reduce to low-order polysulfides Li2Sn (1 ≤ n < 4, Li2S2/Li2S), and finally generate lithium sulfide, and the oxidation process is reversed. During the reaction, if no protective measures are taken, polysulfides prefer to move back and forth between the anode and the cathode. The shuttle effect will bring serious consequences, such as anode corrosion, irreversible capacity loss, the low utilization rate of active materials, self-discharge, etc. Moreover, the challenges to be conquered for sulfur cathodes include poor electrical conductivity and severe volume change during the lithiation process.208–211
In 2009, Nazar et al.212 encapsulated sulfur in mesoporous carbon and obtained a reversible capacity as high as 1320 mA h g−1. Apparently, embedding sulfur into a carbon matrix to make a carbon–sulfur composite cathode is an effective strategy, and the proposal of this scheme opens a new window for the design of high-performance sulfur cathodes. For practical carbon materials, effective adsorption and catalytic conversion of polysulfides are requisite. The dissolution of polysulfides is affected by the adsorption of anchoring materials and solubility in electrolytes. When the adsorption of polysulfides by anchoring materials exceeds the tendency of its dissolution in electrolytes, the dissolution is inhibited.213 In addition, if the adsorption of carbon materials is violent enough, a large amount of polysulfides adsorbed on the surface will reduce the conductivity of carbon and prevent the subsequent adsorption of polysulfides. Therefore, the adsorption energy between carbon matrix and polysulfide should be neither too strong nor too weak.214,215 Enhancing the kinetics of the oxidation–reduction reaction of polysulfides is also a core issue. On the one hand, slow conversion will lead to the saturation of polysulfide adsorption, limiting subsequent adsorption and reducing the utilization of active sulfur; on the other hand, fast conversion is favorable for the adsorption of polysulfides and has a positive effect on the overall performance of LSBs.
Over the past few years, some pioneering studies have shown that defect-rich carbon materials with adsorption–catalytic-conversion function can be widely used in LSBs to overcome the above challenges. The defective carbon materials will expose coordinated unsaturated sites, thus inducing feasible adsorption and enhancing transformation kinetics184,190,191 In this part, the research progress of defect-rich carbon materials as the sulfur storage host and separator modification materials is summarized, emphasizing the structure–function relationship between defect sites and adsorption, catalysis as well as the conversion of polysulfides.
4.1. Carbon-based sulfur hosts
Carbon materials with the defective structures are identified as promising sulfur host for LSBs. Defect engineering plays a crucial role in inducing active site exposure and causing electron redistribution, which is expected to enhance the all-around electrochemical performance of the LSBs.216Intrinsic defect of carbon materials has great potential for efficient capture and catalytic conversion of polysulfides. To further reveal the catalytic reaction abilities of intrinsic defect sites, Wang et al.217 used DFT calculations to analyze the intrinsic defect-rich carbon (IDC) and the extrinsic defect-rich carbon (EIDC, O-doping). The theoretical models of the two kinds of carbon before being loaded with sulfur, before and after the cycle are shown in Fig. 8a and b. It can be seen that after loading sulfur, sulfur forms C–S bonds with graphitic carbon atoms in EIDC/S and defect carbon atoms in IDC/S, respectively. The O atoms in EIDC are highly electronegative, and easily lose electrons forming a negative charge with strong adsorption. After circulation, the S atom is more willing to combine with the O atom, and this relatively weak C–S bond brings more losses to polysulfides. While in IDC, electron transfer occurs directly between sulfur species and C atoms, forming stable C–S bonds, which can enhance the adsorption capacity of IDC and accelerate the conversion of polysulfides. After the cycle, sulfur can be trapped again by defective C atoms, the reaction diagram is shown in Fig. 8c. Such cyclic charging and discharging processes result in the IDC/S electrode being better than the EIDC/S electrode.
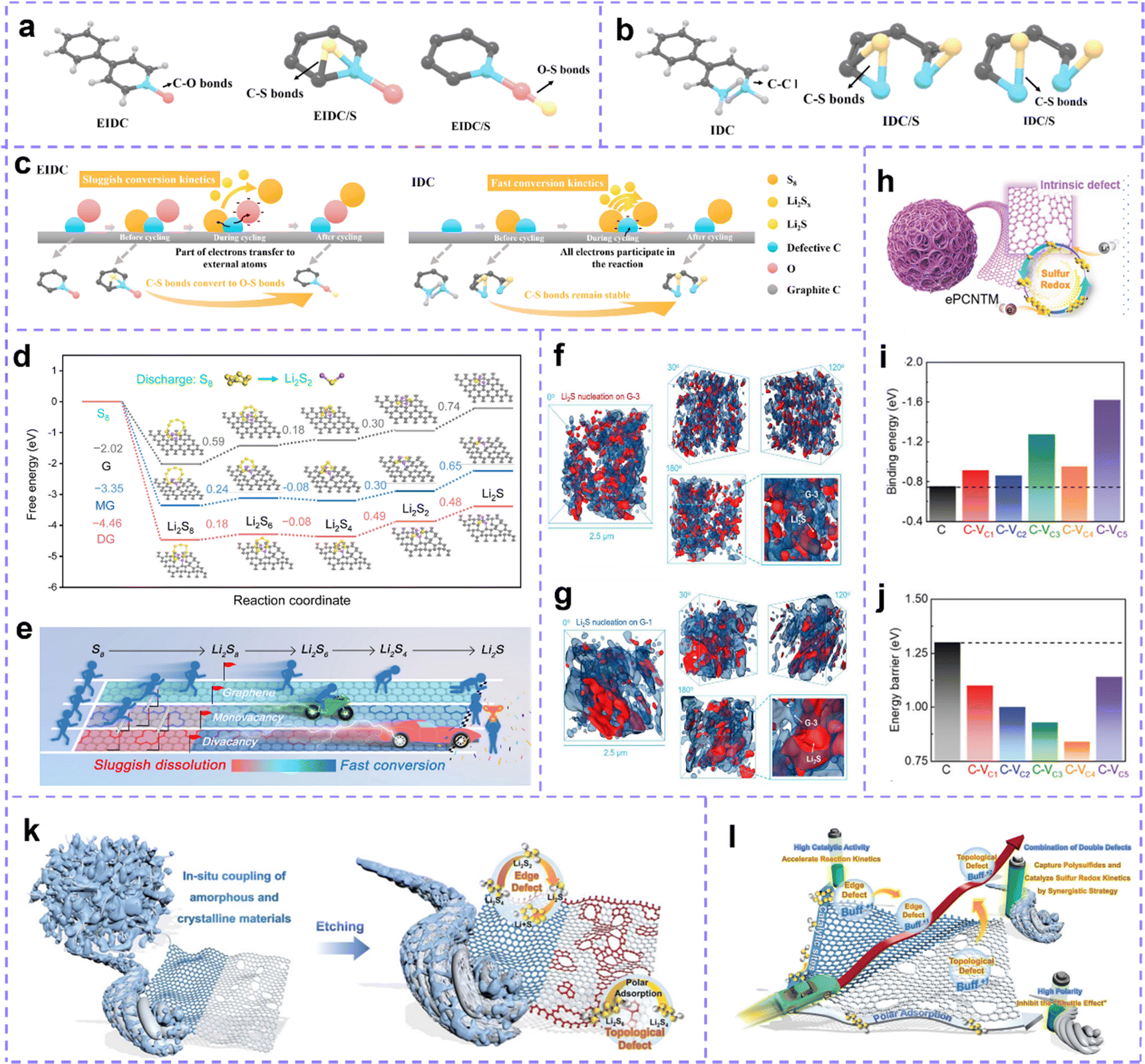 | ||
| Fig. 8 (a) From left to right are the theoretical models of EIDC (EIDC/S) and (b) IDC (IDC/S) before sulfur loading, before cycling, and after cycle. (c) C–S bond stabilities of two defective carbon materials in the reaction process. Reproduced with permission.217 Copyright 2021, Elsevier. (d) Gibbs free energy curves of sulfur conversion on pristine graphene (G), MG and DG hosts. (e) A brief schematic diagram of the effect of various substrates on the conversion process of sulfur species. (f) X-ray 3D Nano-CT images of Li2S deposited on defect-rich samples and (g) perfect graphene substrates at different rotation angles, respectively. (d–g) Reproduced with permission.218 Copyright 2022, Wiley-VCH. (h) Schematic diagram of ePCNTM. (i) Binding energies for Li2S6 and (j) Li2S adsorption on a series of defective carbon sites. (h–j) Reproduced with permission.219 Copyright 2021, Wiley-VCH. (k) Schematic diagram of the synthesis of eCPAC materials. (l) Schematic diagram of a synergistic strategy to regulate the kinetics of multiphase sulfur redox reactions through a composite carbon structure and dual intrinsic defects. (k and l) Reproduced with permission.220 Copyright 2022, Wiley-VCH. | ||
Understanding the effect of the intrinsic defect on the transformation of sulfur species at each step is consequential. Song et al.218 calculated the adsorption energies of monovacancy (MV), divacancy (DV) and pure graphene to Li2S4 were −1.70, −2.80 and −0.95 eV, respectively, which indicated that the intrinsic defective carbon had stronger interaction with polysulfides to prevent their dissolution. Meanwhile, compared with pure graphene (−0.36 eV), the adsorption energy of MV (−0.49 eV) and DV (−0.57 eV) for S8 is larger, which means that the defect site slows down the formation of Li2S8. Through the calculation of the energy barrier, it is further confirmed that the defect site decelerates the emergence of Li2S8, but catalyzes its transformation to low-order polysulfides (Fig. 8d and e). Besides, the catalytic kinetics of Li2S nucleation on graphene is accelerated by the defect sites. The deposition behavior of Li2S was studied by the synchrotron X-ray 3D nano-CT technique. It shows that the size of Li2S in the defect-rich sample is small and well-distributed (Fig. 8f), while the Li2S lump deposits appear in the sample without defects (Fig. 8g).
Intrinsic defects in carbon materials can help address the thorny issues of the shuttle effect and slow kinetics in LSBs. Zhang et al.219 fabricated intrinsic defect-rich porous carbon nanotube microspheres (ePCNTM) (Fig. 8h). Spectral characterization confirmed the presence of a large number of intrinsic carbon defects. Adsorption energy calculations for Li2S6 for samples with 0–5 carbon atom vacancies indicate that the binding energy of the defective surface for Li2S6 exceeds that of the perfect counterparts (Fig. 8i). Energy barrier calculations for the decomposition of Li2S on the carbon surface with 0–5 carbon atom vacancies show that on defective carbon, the barrier is lowered (Fig. 8j). These computational results vigorously suggest that intrinsic carbon defect have stronger polysulfide fixation capacity and higher catalytic activity. Electrochemical test data certify that the developed ePCNTM, acting as a sulfur host, renders meritorious service to the advancement of LSBs. Zhang et al. demonstrated a composite carbon host material (eCPAC) with edge defects and topological defects to capture LiPS and catalyze sulfur redox kinetics through a synergistic strategy (Fig. 8k and l).220 The researchers coupled in situ with a large amount of edge-defective carbon obtained by chemical activation and carbon materials with numerous topological defects in biomass-based carbon structures into composite carbon materials. The composite carbon materials have ultra-high specific surface area and multi-level porous structures, which accelerate the bidirectional catalysis of solid-phase LiPSs through physical confinement and chemisorption. Benefiting from these structural advantages and electrocatalytic activity, the S@eCPAC cathode material exhibits excellent electrochemical performance. This S@eCPAC cell has ultra-high cyclability and multiplicative performance, which can be stably cycled more than 1000 times at 0.5C with an average capacity decay rate as low as 0.05% and a high coulombic efficiency of 99.95%. Even after 600 cycles at 5C, the average decay rate is still as low as 0.032%. Moreover, Pint et al. investigated the effect of the proportion of defective carbon nanotubes (CNTs) with good mechanical and electrical conductivity on the electrode performance of lithium–sulfur batteries by coupling them to carbon nanohorns (CNHs) with chemical defect properties.221 The best defect-engineered cathode was demonstrated with a reversible capacity of more than 1200 mA h g−1 at 0.1C, more than twice that of the one-component structure of carbon nanotubes and carbon nanohorns. This study provides carefully optimized performance for binder-free lithium–sulfur battery cathodes.
Building extrinsic defect is also an attractive and effective strategy to improve sulfur host properties. Zhang et al.122 prepared N, F dual-doped hollow carbon frameworks (NFHCs), the heteroatoms strongly chemisorbed with Li, which promoted the anchoring of polysulfides. Moreover, heteroatom doping can enhance the polarity of the carbon matrix, endow it with ultrafast electrolyte wetting, and the wetting angle is quickly invisible once exposed to the electrolyte (Fig. 9a). Benefiting from the structural advantages, doping defect and fleet electrolyte wetting ability, NFHC possesses fast Li ion diffusion and sulfur conversion kinetics (Fig. 9b). Recently, Wang et al.222 proposed that the active lithium–nitrogen (Li–N) bond (LNB) can act as a catalytic site, which can lower the conversion barrier of Li2S during the discharge process and enhance the mobility of Li ions. The existence of the Li–N bridge effectively reduces the Gibbs free energy of sulfur species reduction and improves the electrochemical performance of LSB during discharge (Fig. 9c). Moreover, Chen et al. developed a porous 2D-defective ZIF-7 with abundant active edges as the host material for sulfur.223 In addition, the pores expose abundant active sites for enhanced anchoring to polysulfides. The introduction of the active edge formed by N defects at the pore edge can further accelerate the redox kinetics of active sulfur.
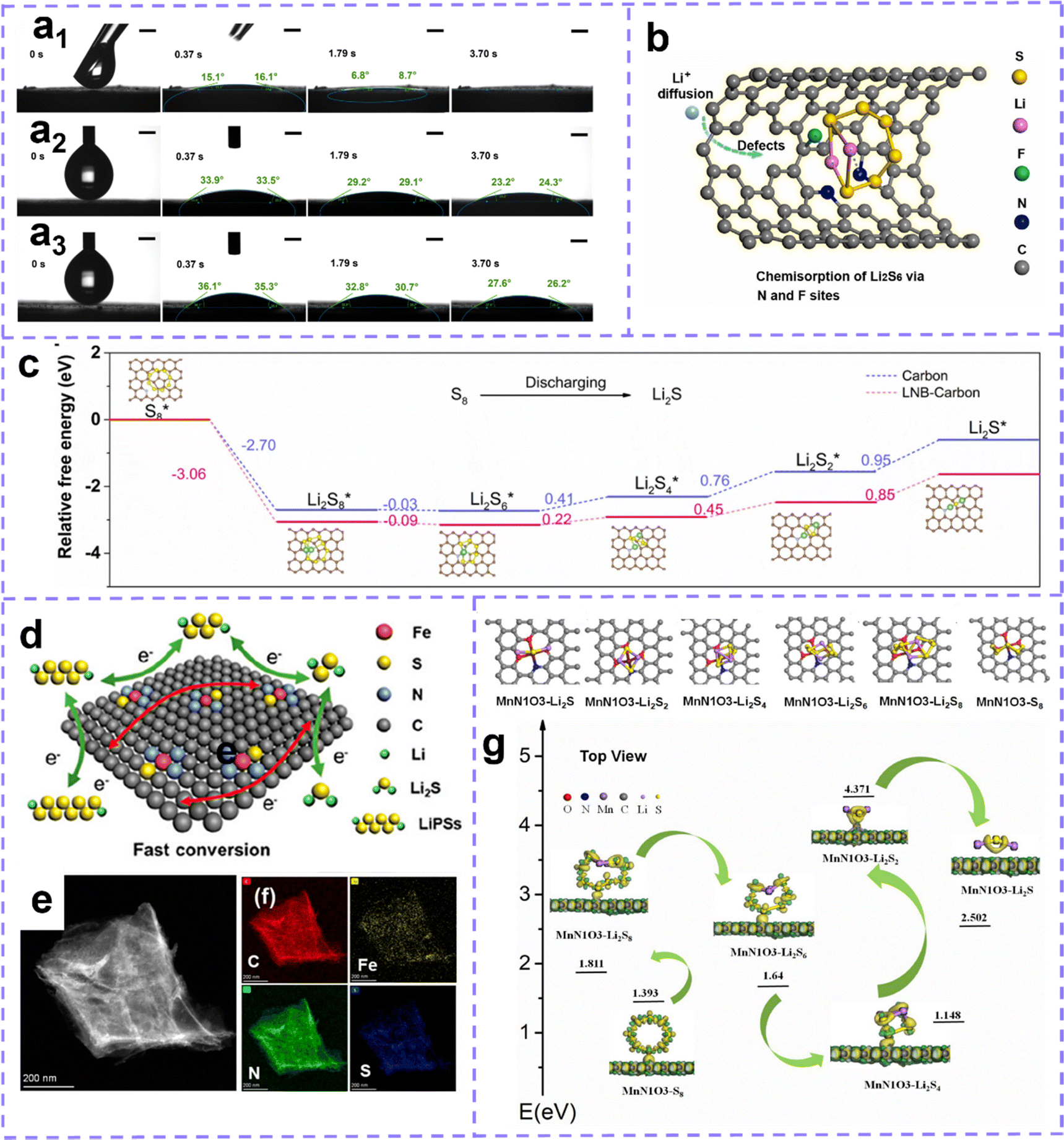 | ||
| Fig. 9 (a) Contact angle tests for (a1) NFHC/S, (a2) NC/S and (a3) CNT/S samples. The scale bar is 1 mm. (b) Schematic diagram of polysulfide capture and Li+ diffusion behavior in NFHC/S with N, F doping. Reproduced with permission.122 Copyright 2022, Elsevier. (c) Relative free energy profiles for the reduction of sulfur species on pristine nanocarbon and nanocarbon with Li–N bridge. Reproduced with permission.222 Copyright 2022, Elsevier. (d) Schematic diagram of electrocatalytic porous carbon promoting polysulfide conversion. (e and f) TEM-EDX elemental mapping images of C, Fe, N, and S for FeNSC. (d–f) Reproduced with permission.224 Copyright 2021, American Chemical Society. (g) Adsorption structures of polysulfides and the calculated energy diagram. Reproduced with permission.225 Copyright 2021, Elsevier. | ||
Metal atom-induced defect sites with excellent catalytic activity can be served as anchoring and catalytic conversion centers of polysulfides.226–228 Inducing metal defect sites can not only enhance electrical conductivity, but also produce the synergistic effect with doped metal-free heteroatoms.135 Zhao et al.224 constructed Fe active sites on N, S dual-doped porous carbon (FeNSCs). Calculation of adsorption energy and decomposition energy barrier, stating that Fe and N, S heteroatoms exhibit a synergistic effect during polysulfide anchoring and accelerate the reaction kinetics (Fig. 9d–f). Recently, Liu et al.225 introduced Mn atoms into O, N doped hollow carbon spheres, O and N atoms can play a role in stabilizing Mn atoms, and enhance their catalytic reactivity (Fig. 9g). Mn atoms introduced into the composite cathode can significantly inhibit the shuttle and catalyze the conversion of polysulfides. So far, many studies have also certificated that the Co atom,229–231 Ni atom,232 and Zn atom37,233 embedded in the carbon skeleton can act as defect sites to improve the performance of carbon–sulfur cathode. Recently, Liu et al.234 summarized the latest progress in improving the performance of the sulfur cathode with a single metal atom dopant in a review journal.
The promoting effects of intrinsic and extrinsic defects on polysulfide conversion and the diffusion of Li ions have been recognized, as well as their superposition tend to significantly affect the activity of carbon catalytic materials. Wang et al. found that there are both synergies and differences between intrinsic defects and extrinsic defects.235 Both extrinsic (oxygen-containing defect) and intrinsic defects are conducive to the rapid formation of polysulfides. However, when the content of oxygen-containing defects is high, the capture of polysulfides is more efficient, while the content of intrinsic defect increases and the conversion of polysulfides is faster. Therefore, the best performance can be obtained by adjusting the relative content of the two defects. When the content of two defect in the carbon-based host is equal, excellent rate performance (299.4 mA h·g−1 at 20C) can be obtained at a high sulfur loading of 80 wt%.
To briefly summarize, porous carbon materials act as sulfur hosts with great advantages. And defective sites in carbon can improve its electrical conductivity by changing the electron distribution, which is a good remedy for the electrical insulation of sulfur electrodes. In addition, porous carbon materials have huge spaces to accommodate active sulfur and physically limit the irreversible loss of sulfur, and defective sites exhibit strong chemisorption to polysulfides. The exposed defective catalytic sites are able to modulate the electronic structure and induce charge redistribution, thus promoting diffusion kinetics and accelerating sulfur redox reactions in the lithium–sulfur battery system.
4.2. Carbon-based multifunctional separators
To mitigate a series of problems caused by the dissolution and diffusion of polysulfides, scientists from all over the world have put in a lot of effort. In the early research, the emphasis was mainly on the preparation and modification of composite sulfur cathodes. Now many reports have proved that constructing multi-purpose modified separators is also an effective strategy.8,236,237 A commercial polypropylene (PP) separator was employed in the early LSBs, which not only allowed Li ions to traverse but also let polysulfides pass through, resulting in a serious shuttle effect. If the existing separator can be retrofitted, the shuttle effect can be effectively subsided. A deeper understanding proves that carbon materials can be employed as multifunctional embedded layers and defect-rich carbon is widely accepted for its good adsorption and catalytic conversion of polysulfides. The application of defect-rich carbon-based modified separators in LSBs will be discussed in the following sections.Integration of defect-rich carbon and a commercial separator is an efficacious and practicable strategy. Wei et al.238 prepared N-doped graphitized carbon (NGC) derived from coconut shell to modify the PP separator. NGC has evenly distributed N elements of about 4.7 wt%, which offers strong adsorption and catalytic site for polysulfides. Kong et al.128 attained P-doped carbon nanosheet array (MPC) and employed it as the separator coating. The doping of P into the carbon skeleton disturbs the electron distribution of the hexagonal carbon lattice and endows MPC with polarity. In the process of polysulfide adsorption, the P–Li bond and the P-S bond are formed, which effectively enhances the anchoring of polysulfides and inhibits their dissolution and diffusion (Fig. 10a and b).
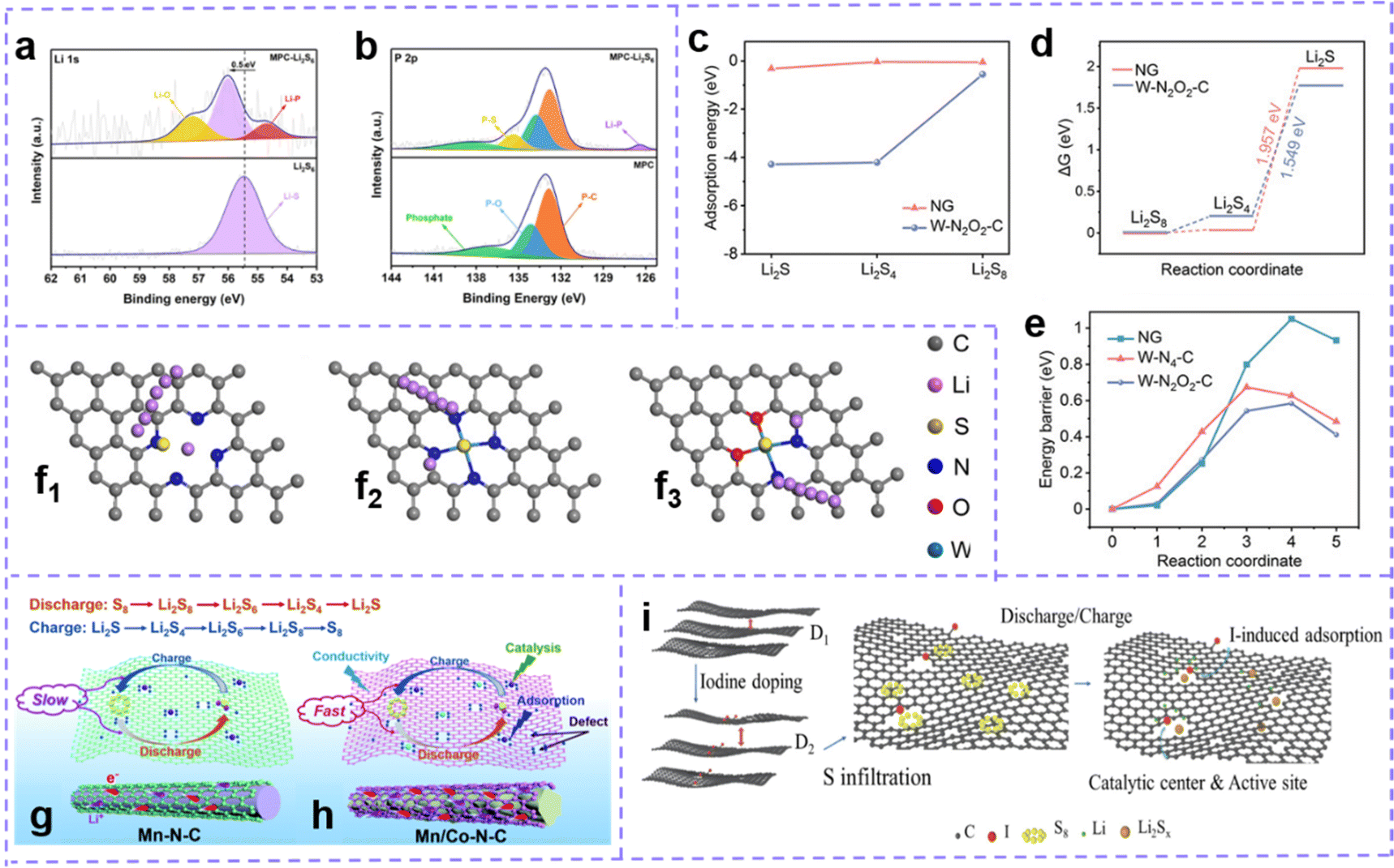 | ||
| Fig. 10 (a) Li 1s XPS spectra for pristine Li2S6 and MPC-Li2S6. (b) P 2p XPS spectra for MPC and MPC-Li2S6. (a and b) Reproduced with permission.128 Copyright 2022, Elsevier. (c) Adsorption energies and (d) Gibbs free energy changes of Li2S8, Li2S4, and Li2S on NG and W/NG. (e) Energy profiles for the decomposition of Li2S on NG, W–N4–C and W–O2N2–C. (f) Decomposition pathways of Li2S on (f1) NG, (f2) W–N4–C and (f3) W–O2N2–C. (c–f) Reproduced with permission.239 Copyright 2021, Wiley-VCH. (g) Schematic illustration of the working mechanism of Mn–N–C and (h) Mn/Co–N–C catalytic materials in LSBs. Reproduced with permission.36 Copyright 2022, Royal Society of Chemistry. (i) Schematic illustration of the mechanism of a separator with abundant I-doped active sites. Reproduced with permission.240 Copyright 2022, Wiley-VCH. | ||
Metal-induced defect sites have been testified to be an effective method to construct high-performance modified separators. Wang et al.239 reported novel tungsten (W)-embedded N-doped graphene oxide (W/NG) to fit out a separator. The local coordination environment of W–O2N2–C is diagnosed as the active center. Compared with the other carbon (NG, W–N4–C), W–O2N2–C has stronger adsorption of polysulfides and lower decomposition energy barrier of Li2S, thus giving the combined separator higher catalytic activity and faster conversion (Fig. 10c–f). Moreover, Qiao et al.26 made a porous carbon doped with Mn and N (Mn–N–C) for the modification of separators. Mn atoms in Mn–N–C are beneficial to improve the adsorption performance and enhance the conductivity of the carbon matrix. This team also manufactured an N-doped carbon (Mn/Co–N–C) embedded in a Mn and Co bimetal to modify the separator.36 Compared with Mn–N–C, the higher catalytic activity of Mn/Co–N–C can be attributed to the synergistic effect of Mn–N and Co–N sites. Benefiting from the Mn/Co–N–C separator to inhibit shuttle and catalyze the conversion of polysulfides, LSB shows bonzer performance. As a result, the LSB achieved a high initial specific capacity of 1662 mA h g−1 at 0.1C, and excellent stability of only 0.036% capacity decay per cycle at 2C over 1000 cycles (Fig. 10g and h).
The dual-function configuration of the cathode and the separator improves the performance of LSBs. Zhang et al.240 prepared I-doped soft carbon for sulfur storage, and an I-modified separator to inhibit the shuttle of polysulfides and allow the passage of Li ions. The I doping increases the layer spacing, which is conducive to the rapid ion transport, the anchoring of polysulfides and the acceleration of redox reaction kinetics (Fig. 10i). Moreover, Song et al.241 obtained an N/P dual-doped conductive carbon framework (NPPC) and acted as a sulfur host as well as the free-standing interlayer. The assembled LSB has relatively ideal performance, with an approving initial capacity of 1412.6 mA h g−1 at 0.1C and well-pleasing stability with 0.032% degradation rate per cycle for 500 cycles at 1.0C.
It is obvious that the defect-site rich carbon modified separator or self-supporting interlayer is beneficial to suppress polysulfide shuttling and enhancing the overall performance of Li–sulfur batteries. Although the research on the defect engineering of carbon-based materials in the field of lithium–sulfur batteries started relatively late, its development has made remarkable progress. The defective catalytic sites exhibit strong adsorption and catalytic conversion of polysulfides, which can efficiently trap polysulfides, improve active sulfur utilization, and accelerate electrochemical redox reactions. Therefore, it is very meaningful to deeply explore the mechanism of carbon material defect engineering on the electrochemical performance of lithium–sulfur batteries. It is hoped that more researchers will devote themselves to this research direction and contribute to the development of lithium–sulfur batteries.
5. Recent advances in carbon defect engineering for electrocatalysis
The oxygen reduction reaction (ORR), oxygen evolution reaction (OER), hydrogen evolution reaction (HER), and carbon dioxide reduction reaction (CO2RR) are the core steps in various electrochemical energy conversion systems.133,242 An electrocatalytic reaction is usually a multiphase interface transformation process, including diffusion, reactant/intermediate adsorption, charge transfer, product desorption and other essential procedures. Like other chemical reactions, electrocatalytic conversion also has energy barriers (overpotential) to surmount. High overpotentials lead to ultralow kinetics of the electrocatalytic reactions. Therefore, it is crucial to develop suitable catalytic materials to reduce the overpotential and accelerate the reaction kinetics.243–245Common electrocatalysts include noble metals, non-noble metal materials, and carbon materials. Among them, with its advantages of economy, environmental protection and electrochemical stability, etc. carbon-based electrocatalytic materials have aroused great interest.27,246,247 Thanks to the hard work of scientists around the world, great achievements have been made in the design and application of carbon-based catalytic materials with abundant electrocatalytic active sites. Benefiting from their high efficiency, the research on defect engineering of carbon-based electrocatalysts has become a hot topic day by day, and the related research flourish well. The following section denotes solicitude for the origins of electrocatalytic activity of defect-rich carbon materials and their application progress in ORR, HER and CO2RR.
5.1. ORR
The oxygen reduction reaction (ORR), the cathode reaction in rechargeable metal–air batteries and fuel cells, is the cornerstone of sustainable energy storage and conversion technologies. Because the breaking of the OO bond requires high energy to achieve, the ORR is kinetically rather slow. Therefore, catalysts play a pivotal role in accelerating the ORR. Over the past few decades, a great deal of work has been devoted to exploring efficient and low-cost ORR catalytic materials. The catalytic activity of defect-rich carbon-based electrocatalytic materials in the ORR has been demonstrated, and related research has developed in a promising direction.38,66,246,248,249
To improve the ORR performance of intrinsic defect-rich carbon materials, Yao and co-workers published a series of seminal and important reports both in theory and in experiment.12,51,58,70,98,250,251 For example, they prepared a metal-free N-doped carbon-based electrocatalytic material in 2014, which can improve the behavior of carbon materials for the ORR.250 They also demonstrated a new defect mechanism for carbon in the ORR. First-principles calculations predict that a C585 topological defect on graphene (G585) is more efficient than N doping, achieving commensurate activity to Pt in all steps of the ORR (Fig. 11a).12 In addition, they published a review paper in 2018 summarizing the mechanism of carbon defect catalyzing the ORR, strongly supporting the idea that defects can serve as active sites for electrochemical reactions.70 Moreover, in 2020, the catalysis effect of defect and heteroatoms for the ORR was systematically explored. Fourteen heteroatom-doped C5 defect models were constructed and compared with heteroatom-doped perfect graphene (Fig. 11b). The results show that C5 + N + S wins the best active site for the acidic ORR (Fig. 11c), and the heteroatom doping catalysis strongly depends on the specific inherent defects of graphene. It is elucidated that heteroatom modification can tune the electronic structure of defects, while the role of dopants themselves is very limited.251 Recently, they reported a strategy to build defect sites on carbon materials based on the thermal reduction reaction of metal oxides with carbon, which can both increase the carbon defect density and avoid the reconfiguration of carbon materials at high temperatures.40 Materials with higher defect densities exhibit better performance in the ORR than materials with lower defect densities, and the performance is positively correlated with defect density. This case illustrates that a quantitative change in the number of defects can lead to an increase in the corresponding catalytic activity.
 | ||
| Fig. 11 (a) Free energy diagrams of ideal monolayer graphene (G), N-doped graphene (N–G), G585-defective graphene (G585), and ideal catalyst for the ORR. Reproduced with permission.12 Copyright 2015, Royal Society of Chemistry. (b) Schematic summary of the 14 configurations of the single and dual heteroatom-tuned C5 defects; brown, blue, green, pink, and yellow represent C, N, B, P, and S atoms, respectively. (c) ORR volcano plot of overpotential versus adsorption energy of OH*. (b and c) Reproduced with permission.251 Copyright 2020, Elsevier. (d) Configuration map of carbon edge defect with or without N doping, including Cpeak (C1), Cvalley (C2), Npyridinic–Cvalley (C3), Ngraphiticvalley–Cpeak (C4), and Ngraphiticcenter–Cpeak (C5). (e) ORR free energy diagrams calculated for the five C defect sites. (d and e) Reproduced with permission.252 Copyright 2020, Wiley-VCH. (f) Schematic diagram of the nanotube structure: (f1) BC3NT, (f2) 7MR, and (f3) 8MR. (g) Free energy change of the ORR. (f and g) Reproduced with permission.253 Copyright 2022, Royal Society of Chemistry. (h) Free energy diagram for the ORR and (i) OER on FeN4C, FeN3SC and RuO2 at an electrode potential of U = 0 V. Reproduced with permission.254 Copyright 2022, Royal Society of Chemistry. | ||
Besides, Cheng et al.15 in 2016 demonstrated that edge and topological defect endow carbon materials with moderate adsorption and high activity for oxygen species, thereby enhancing electrocatalytic activity for the ORR. The edge-rich and dopant-free graphene, carbon nanotubes, and graphite exploited by Li et al. exhibited efficient ORR electrocatalytic performance.56 This example illustrates the excellent ORR activity of intrinsic defect-rich carbon-based electrocatalysts even in the absence of dopants. Zhu et al.39 prepared pentagonal defect-rich carbon nanomaterials (PD-C) by cutting fullerenes (C60). The electrochemical tests and DFT calculation results confirmed that intrinsic pentagons play a greater role in enhancing ORR activity, optimizing reaction pathways and achieving high stability compared to ordinary hexagons. Zhang et al. used DFT to reveal that the presence of edge defects can activate other defects and act synergistically with them to improve the overall oxygen catalytic activity.255 Moreover, in a review paper published in 2021, Zhang et al.249 summarized the latest progress in defect engineering of carbon materials used in ORR, focusing on the collaborative design and combined application of heteroatom doping with edge defects, vacancy defects and topological defects, revealing the relationship between defect/dopants and electrocatalytic ORR performance.
Heteroatom-doped carbon-based electrocatalytic materials exhibit high electrocatalytic activity for the ORR. As mentioned earlier, the introduction of N atoms increases the electronegativity, allowing the rearrangement of the electronic structure of adjacent carbon atoms. The rearranged electron distribution of carbon atoms results in stronger interactions with oxygen molecules and contributes to enhanced adsorption and dissociation of oxygen molecules. Unfortunately, the exhaustive real catalytic mechanism induced by heteroatom N doping for the ORR is still controversial. For example, in N-doped systems, the issue of ORR active sites generated by pyridinic-N or graphitic-N has been discussed. In their 2016 paper, Guo et al. mightily supported the Lewis base site created by pyridine N as the active site for the ORR, and detailed the possible mechanism of ORR on N-doped carbon.52 The adsorption of O2 molecules by the carbon atoms adjacent to the pyridinic-N is the initial step of the ORR, after which O2 is protonated, and two pathways may be followed. One is a “four-electron mechanism” that occurs at a single site, and two protons are attached to two oxygen atoms, causing the breaking of the O–OH bond and the formation of OH species, the additional protons then react with the adsorbed OH to form H2O. The other is the “2 + 2 electron mechanism”, where O–OH adsorbs one proton to form H2O2, which is subsequently reduced to two molecules of H2O.
Yang et al. demonstrated through experiments that the electron-donating graphitic-N site acts as the ORR active site, and the electron-withdrawing pyridine-N serves as the OER active site. Close to the graphitic-N and/or pyrolytic-N plus the nucleophilic strength (Cδ−) of the adjacent carbocyclic ring, which is energetically favorable for O2 adsorption, thus accelerates the ORR because O2 has a high density of O lone pair electrons (Oδ+). Pyridinic-N attracts electrons from surrounding C atoms, and makes oxidation intermediates (OH− and OOH−) more easily adsorbed, which is beneficial to the OER.256 For graphitic-N sites actually include graphitic center N (Fig. 11d, C5) and graphitic valley N (Fig. 11d, C4). And calculated the dependence between N-doped carbon defect configuration change and ORR activity by DFT. The carbon configuration and free energy change during the ORR are shown in Fig. 11d and e. For the C4 site with graphitic valley N doping in the carbon edge defect, the highest resistance to overcome in the ORR is only 0.56 eV (O* formation: ΔG = −2.41 eV, OOH* formation: ΔG = 0.06 eV, OH* formation, ΔG = 0.56 eV, OH* desorption: ΔG = 0.13 eV). However, in the C1, C2, C3 and C5 configurations, the ΔG for this rate-determining step is 1.49, 1.38, 1.43 and 0.99 eV, respectively, larger than the C4 site. Thus, the graphite valley N-doped carbon defect creates the best active site for ORR electrocatalysis in alkaline surroundings. Liang et al. developed a novel molecular design strategy to prepare 2D porous vortex-layered carbon nanowebs with abundant carbon edge defects and N-doping sites.252 The optimized electrocatalytic oxygen reduction reaction catalysts exhibited ORR activity comparable to that of commercially available 30% Pt/C in alkaline media. Guan et al. synthesized N-doped carbon nanotubes with controlled defect sites and investigated the activity of different N-doped configurations on the selectivity towards the ORR pathway.257 It was found that the ORR on N-doped defect sites passes through a 2e− electron pathway and the formed H2O2 can be further reduced to H2O at pyridine and pyrrole N sites, while on the other hand, the ORR process passes through a 4e− pathway at graphitic nitrogen sites. This study provides an insightful guide to elucidate the catalytic selectivity of nitrogen species in nitrogen-doped carbon catalysts.
The ORR performance of intrinsic defects and heteroatom-doped defects is much better than that of pure carbon but needs to be further improved as it still cannot outperform Pt/C. Fortunately, intrinsic defect and extrinsic defect co-catalyzed ORR exhibits high performance. Yao et al. have designed defective structures coupled with heteroatom doping for S–C defects and N–S–C active sites.258 By introducing topological defects into the S-doped structure, it contributes to the reduction of the activation energy barrier, leading to its ORR activity in acidic electrolytes. By introducing N atoms into the S–C defect, the active structure can confer higher ORR activity of the catalyst in acidic versus basic electrolytes. High-content N-doped porous carbon nanosheets (PNCNs) were synthesized via a facile template-assisted method.259 DFT calculations confirmed that carbon sites near graphitic-N in PNCNs-900 are highly active sites for the ORR, and the increased electrocatalytic activity is due to the coupling effect of a large number of defects and graphitic-N. Moreover, N-doped graphitic carbon nanosheets (d-pGCS) with abundant structural defects were prepared by a simple two-step thermal treatment route and applied as ORR electrocatalysts.260 After testing, it was found that d-pGCS-1000 exhibits surpassing ORR activity and stability, even comparable to Pt/C. Recently, Wang et al.253 compared the performance of B-doped carbon nanotubes (BC3NTs), seven-membered ring (7MR) and eight-membered ring (8MR) B-doped topological defect structures for catalyzing the ORR and OER (Fig. 11f). The data show that 8MR is more favorable for the adsorption of O2, followed by 7MR, and the perfect BC3NT is inferior. Topological defects effectively reduce the ORR and OER overpotential, and the larger the topological defect ring, the smaller the overpotential (Fig. 11g).
Besides, the coupling of metal-free and metal-heteroatoms shows high catalytic activity for the ORR. To understand the origin of their activity, Zhang et al. studied the catalytic mechanism of S-doped Fe–N–C materials through DFT theoretical calculations, and proposed that the key to bifunctional activity is the stabilization effect of OOH*.254 For ORRs, S-doping reduces the energy barrier by the stabilization of OH* and OOH* species, especially for the difficult potential determining step (PDS) in Pt (O* → OH*) and FeN4C (O2* → OOH*) (Fig. 11h). Regarding the OER, S-doping lowers the barrier of PDS (O* → OOH*) by stabilizing OOH* species (Fig. 11i). The S doping stabilization effect sourced from the enhanced interaction between Fe and oxygen species, the improved interatomic coupling of Fe–N3S active sites and the moving of the d band to the Fermi level. Compared with N and S atoms, other heteroatoms have different electronegativity and atomic orbitals, which will produce different coordination structures and electronic properties, and then cause different ORR and OER reactivities. This study may be beneficial for designing activated carbon-based catalytic materials with metal-induced defect sites in coordination with heteroatoms for the ORR and the OER.
Tuning the atomic coordination environment of metal atoms and metal-free heteroatoms on carbon substrates can efficaciously enhance catalytic activity. Zhang et al. investigated the promotion of ORR performance by edge Co–N4 sites, and tuned Co–N4 site-doped carbon with different edge volume ratios.261 The results show that edge-loaded Co–N4 sites are more favorable for 2e− ORR than in-plane sites. When Co–N4 sites are anchored on multistage porous carbon (HPC), the resulting Co–N/HPC catalysts are rich in edge-loaded active sites and have a 2e− high selectivity of ≈95% ORR in alkaline media. Wang et al.262 obtained r-Fe–N–C catalytic material (r refers to reduced Fe) containing the FeN4 site. Compared with the conventional FeN4 sites (Fe–NC catalysts), the Fe–N bond distance in r-Fe–N–C is slightly extended and the electron density on the Fe atom is higher (i.e., the Fe valence is lower). Experiments show that r-Fe–N–C exhibits better ORR performance than Fe–NC. Furthermore, metal-induced defect sites of two-component metal can better meet the requirements of high ORR activity. Recently, Mao et al.245 fabricated Ni-doped Co–N/C mesoporous dodecahedron (Co&Ni@N/C) for accelerating ORR kinetics. The results of in situ enhanced Raman scattering (SERS) spectra show that there are superoxide ions (O−2) on the surface of Co&Ni@N/C and they are quickly converted into OH−.
The above summarizes in detail the progress of the research on carbon material defect engineering for ORR catalysis. The study of the ORR catalyzed by carbon-based defect sites is relatively early and well developed. It has been demonstrated that defect engineering provides a feasible and effective method to improve the intrinsic activity and increase the number of active sites in carbon-based electrocatalysts.
5.2. HER
Hydrogen energy is being actively pursued as a green, clean and sustainable energy. Hydrogen (H2) can be generated in a water electrolysis system via the hydrogen evolution reaction (HER), which is an economical and feasible strategy. However, the HER has the disadvantages of slow kinetics and high overpotential, resulting in poor electrocatalytic performance. The high activity of metal-free carbon-based electrocatalytic materials for HER has been demonstrated, and in particular, defect-rich carbon is increasingly developed as the most promising metal-free alternative to commercial Pt/C catalysts.70,246,263Heteroatoms doping endow carbon-based electrocatalytic materials with high activity, which can be used not only in the ORR, but also in the HER. For example, Zhang et al.264 introduced N atoms into dopamine to generate pyridine-based molecules, resulting in high-level N-doped defect-rich porous carbon (DRPC). DRPC in an alkaline medium exhibits an ultralow overpotential of 217 mV to achieve 10 mA cm−2 HER current density, which were superb or equivalent to those of most metal-free carbon catalysts and non-noble metal HER catalysts. DFT was employed to model the adsorption of hydrogen on graphitic-N, pyrrolic-N and pyridinic-N sites, as well as N-adjacent carbon by Li et al.265 The corresponding free energy (ΔG) of H+ adsorption on different sites is shown in Fig. 12a. When hydrogen is adsorbed on the N-site, the ΔG of the pyridinic N-site (−0.74 eV) is smaller than that of the bare carbon (1.74 eV) and graphitic N (1.87 eV), while the ΔG of the pyrrolic-N (−1.79 eV) is smaller, which is difficult to desorb. Therefore, in general, pyridinic-N is more suitable for hydrogen adsorption/desorption. But when it comes to the N-adjacent carbon, the ΔG of pyridine N–C sites (1.65 eV) is much higher than that of graphitic N–C (0.59 eV) and pyrrolic N–C sites (0.17 eV). This case illustrates that three types of N dopants enhance HER activity through different mechanisms. Not only N doping but also other heteroatoms are introduced to enhance HER activity. Zheng et al.266 developed N, P dual-doped graphene (N, P–G) as a non-metallic electrocatalyst for HER. The N and P heteroatoms synergistically induce the catalytic activity of the surrounding carbon atoms, thus exhibiting higher electrocatalytic HER activity than single-doped graphene (Fig. 12b and c), whether in an acidic or basic environment.
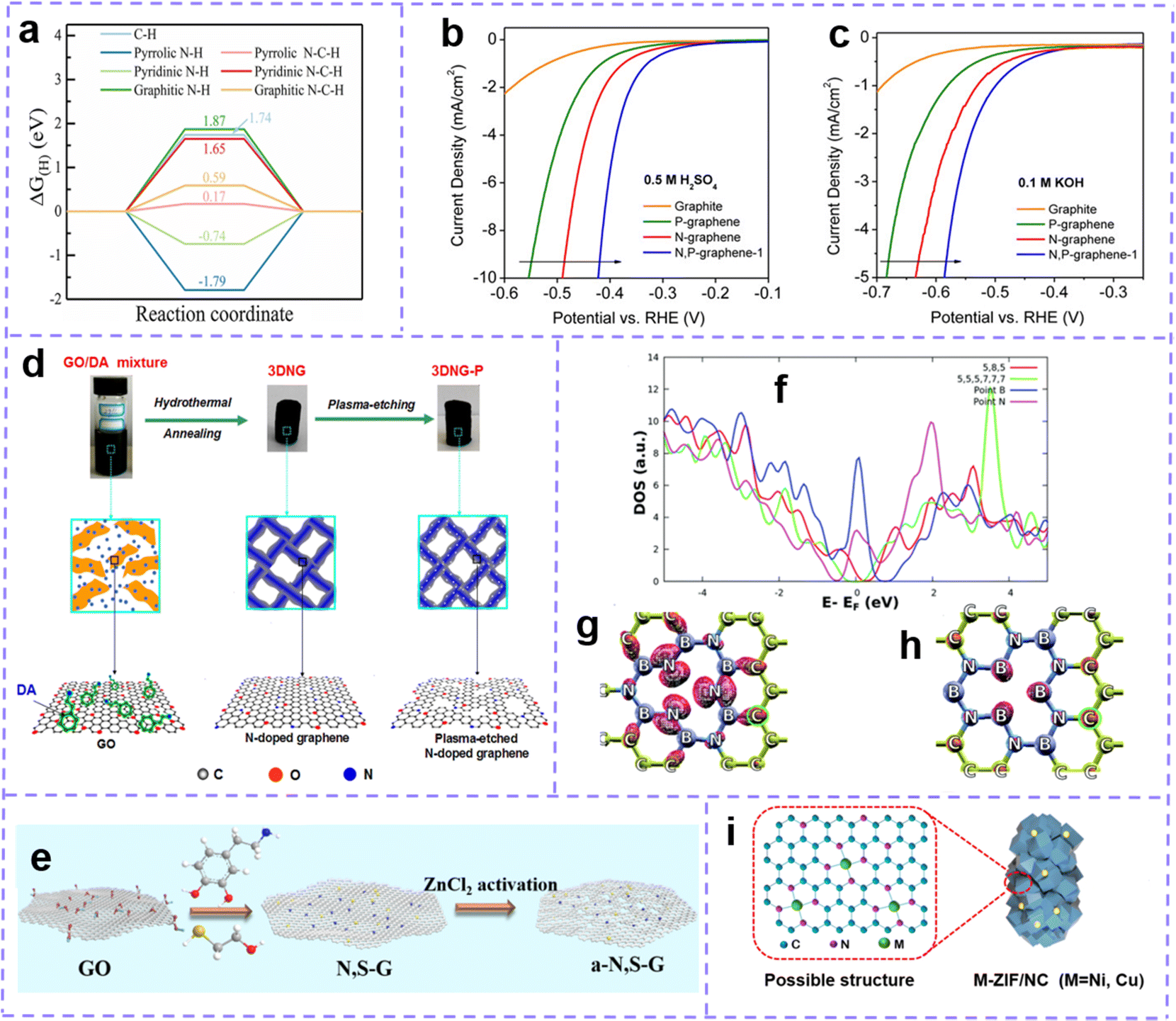 | ||
| Fig. 12 (a) Gibbs free energy of H+ adsorption on different sites. Reproduced with permission.265 Copyright 2021, Elsevier. (b) HER polarization curves of N and/or P doped graphene in (b) 0.5 M H2SO4 (pH = 0) and (c) 0.1 M KOH (pH = 13). Reproduced with permission.266 Copyright 2014, American Chemical Society. (d) Schematic illustration of the synthesis process of 3DNG-P. Reproduced with permission.267 Copyright 2017, Elsevier. (e) Schematic diagram of the activation process of N, S–G. Reproduced with permission.268 Copyright 2019, Elsevier. (f) Density of states (DOS) for defective B, N doped graphene, with (5,8,5), (5,5,5,7,7,7) and defect of B and N doping. (g) Local density of states (LDOS) near the Fermi energy for B doped and (h) N doped graphene. (f–h) Reproduced with permission.263 Copyright 2019, Royal Society of Chemistry. (i) Schematic diagram of the structure of M–ZIF/NC. Reproduced with permission.269 Copyright 2021, Elsevier. | ||
So far, the brilliant catalytic performance of intrinsic defects and heteroatom dopants has been demonstrated in the HER. The coupling effect between them is crucial to implementing preeminent HER performance. Tian et al.267 obtained a 3D N-doped graphene (3DNG-P) by plasma etching (Fig. 12d). Structural defect engraved on graphene by plasma etching couple with high content of N-dopants to strengthen the HER activity of 3DNG-P, showing low overpotentials of 128 mV to achieve 10 mA cm−2 in acidic media. Moreover, activation of N, S-doped carbon (NS–G) by ZnCl2 produces abundant defect (Fig. 12e). Compared with pristine NS–G without chemical activation, the catalytic performance of the activated carbon for both the HER and OER is tellingly improved. Except for the synergistic catalysis by N, S heteroatom dopants, the structural defect caused by the activation process to carbon materials was also shown to be active sites for HER.268 Yoshikazu et al.270 confirmed by DFT calculations that the cooperation of S and N dopants to intrinsic defect in the graphene carbon lattice can tune the Gibbs free energy of H* absorption to improve HER activity. Ozden et al.263 calculated the density of states (DOS) of intrinsic defect-rich carbon (C585, C555777) and B and N doped C59 by DFT. It can be seen from Fig. 12f that the DOS of C585 and C555777 is not distributed near the Fermi level without doping, but with heteroatom doping, the high DOC is shown at the Fermi level, which means that electrons on the HOMO can be easily transferred to the LOMO, thereby promoting electrochemical reactions. In the case of N-doped C59, there is nice occupancy on nearby B and C (Fig. 12g). When B doped C59, there is sizeable occupancy on nearby N and C (Fig. 12h), and B doping shows a better effect. This study points out that the joint effect of extrinsic dopant atoms and intrinsic defect enhance the catalytic activity for the HER, and the coupling of different heteroatoms and different defect breeds different catalytic activities. It is worth mentioning that, drawing on the synergistic effect of metal catalyst defect sites and heterojunctions, the coupling between the intrinsic defect sites of the carbon matrix and the heterojunction interface between the carbon matrix and polar substances has also received attention from researchers.271,272 Heterojunction interfaces and intrinsic carbon defects have somewhat similar principles of action on the carbon skeleton. They both effectively change the electronic structure of the bureau of the carbon matrix, altering the adsorption energy of the reacting material and lowering the reaction energy barrier, thus reducing the overpotential required for the reaction. Activated carbon sites are often considered as potential active sites for different electrocatalytic reactions.273,274
Besides, the synergistic effect defective sites also enhance the activity of carbon-based electrocatalytic materials for the HER. Liu et al. prepared single-atom Pt, Co-anchored catalysts using ZIF-8-derived defect-rich N-doped carbon material as a carrier.275 It was confirmed that the carbon defects were favorable for the single-atom anchoring and the catalysts showed ultra-high HER activity, which was up to 54 times higher than that of commercial 20wt% Pt/C in acidic media. Zhang et al.269 introduced metal sites and N-doping sites into zeolitic imidazolate zinc framework (ZIF-8)-derived carbon, and obtained heteroatom-modified carbon-based catalysts Ni–ZIF/NC and Cu–ZIF/NC for the HER (Fig. 12i). The coordination of metal atoms with N atoms to form M–Nx serves as catalytically active sites in the HER, and the incorporation of metal atoms significantly improves the catalytic activity of ZIF/NC. Ni–ZIF/NC showed superior catalytic activity compared with Cu–ZIF/NC and ZIF/NC, especially in an alkaline environment, with an overpotential of 163.0 mV and Tafel slope of 85.0 mV dec−1, which is markedly better than the results in other literature reports, and the gap with Pt/C is narrowed (37.8 mV and 34.2 mV dec−1). MOFs-derived Co-modified Ni, N co-doped carbon (Co–Ni@NC) was synthesized by a simple hydrothermal and high-temperature pyrolysis.276 Metal species are considered important HER active sites. The HER and OER activities of Co–Ni/NC electrocatalysts are much higher than those of Ni/NC and NC. The Co content has a great influence on the electrocatalytic performance, and Co-1-Ni-0.5/NC exhibits better HER and OER activities with an HER overpotential of only 179 mV.
5.3. CO2RR
From the perspective of green chemistry, catalytic conversion of carbon dioxide (CO2) is of great significance, which not only helps to solve the energy crisis, but also relieves the environmental pressure. Electrocatalytic CO2 reduction is a low-cost and high-efficiency catalytic conversion technology, through which many value-added chemicals can be harvested, including carbon monoxide (CO), methane, ethanol, etc. If the application of CO2 reduction can be realized, the current situation of CO production, which is still produced by coal gasification or steam methane reforming, will be greatly improved.13,16,42,277 Carbon-based catalysts have attracted much attention due to their high catalytic activity for the CO2RR. After recent development, they have become promising alternatives to metal-based catalysts for CO2RR. Defect-rich carbon materials, especially intrinsic defect-rich carbon, have made great progress as efficient metal-free carbon-based catalysts for CO2RR.Intrinsic carbon defect has irreplaceable effects on CO2RR performance. Although the related research started relatively late, it has been widely reported in recent years. Jiang et al. developed a K-assisted strategy to achieve the fabrication of defective structures in dopant-free carbon materials.278 K@Bio-MOF-1 was pyrolyzed at 1100 °C to generate defect-rich porous carbon with 12 adjacent carbon atom vacancies (K-defect-C-1100). The catalyst exhibited an ultra-high CO faradaic efficiency (FE) of up to 99% at −0.45 V, which was much better than for N-doped carbon in electrocatalytic CO (N–C-1100). Theoretical calculations indicate that the excellent performance is attributed to the tendency of defects in K-defect-C-1100 to adsorb electrophilic CO2 molecules and significantly accelerate the formation of COOH* intermediates. Wang et al.72 compared the CO2RR performance of two intrinsic defect-rich carbon materials with and without N. The results showed that carbon without N atoms also manifested excellent CO2RR catalytic performance, and the CO2RR activity was positively correlated with the intrinsic defect concentration. This proves that intrinsic carbon defect, rather than dopants, are possible active sites for CO2RR. Then, modeling and calculation are carried out for zigzag and armchair edge defects as well as pentagonal and octagonal topological defects. The change of free energy in CO2RR is shown in Fig. 13a. When COOH* is formed, pure carbon, zigzag and armchair edges and octagonal topological defects all show an increase in free energy, while a decrease in the pentagonal defect is observed. However, the free energy of COOH* to CO* increases on pentagonal carbon, while the other four types of carbon are opposite. On the whole, octagonal and pentagonal defect won the best performance. Near edge X-ray absorption fine structure (NEXAFS) spectroscopy further verifies that topological defects are more beneficial to CO2RR than edge defects.
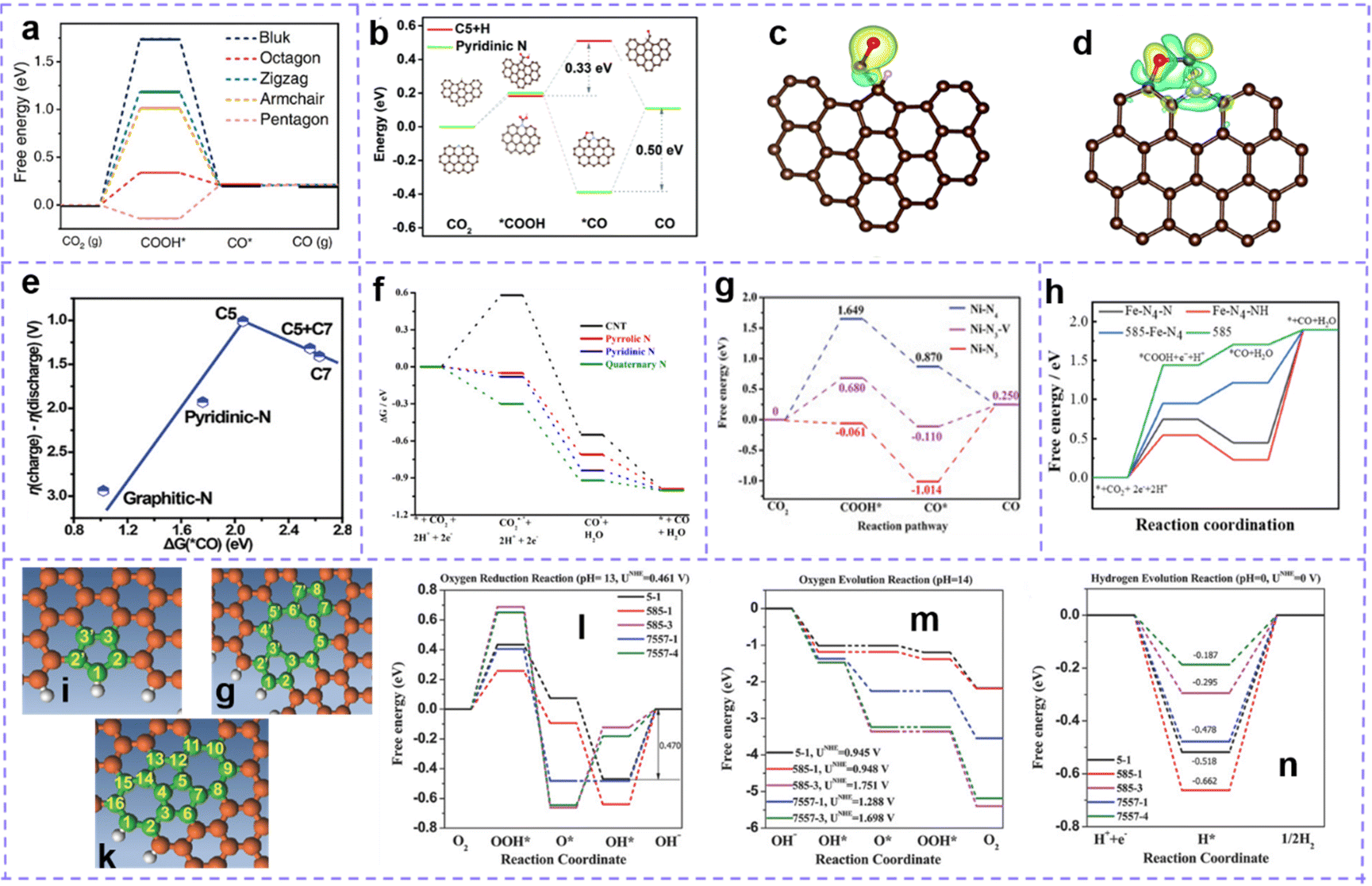 | ||
| Fig. 13 (a) DFT calculations for CO2RR activities of different defects. Reproduced with permission.72 Copyright 2019, Wiley-VCH. (b) Free energy diagram for CO2RR over the C5 + H defect site and the pyridinic N site. (c) Differential charge density of CO molecules adsorbed at the C5 + H site and (d) pyridine N site. (b–d) Reproduced with permission.244 Copyright 2020, Royal Society of Chemistry. (e) The relationship between the overpotential and the adsorption energy of the *CO (ΔG*CO) intermediate. Reproduced with permission.16 Copyright 2021, Wiley-VCH. (f) Gibbs free energy for CO2 electrocatalytic reduction to CO on pristine CNT and different N sites at −0.40 V vs. RHE. Reproduced with permission.279 Copyright 2016, Wiley-VCH. (g) Calculated free-energy diagram for the conversion of CO2 to CO. Reproduced with permission.280 Copyright 2019, Wiley-VCH. (h) Gibbs free energy diagrams (in eV) of CO2RR over Fe–N4–N and 585-Fe–N4 site with different applied potentials. Reproduced with permission.281 Copyright 2021, Wiley-VCH. (i) Configuration diagrams of Edge pentagon, (j) 5-8-5 defect, (k) 7-55-7 defect. (l) Schematic energy profiles for the ORR pathway, (m) the OER pathway, and (n) the HER pathway on defective graphene in alkaline/acidic media. (i–n) Reproduced with permission.147 Copyright 2016, Wiley-VCH. | ||
According to the analysis of X-ray absorption spectroscopy (XAS) results and aberration-corrected transmission electron microscope (ACTEM) images, Wu et al.244 concluded that the pentagonal defect (C5 + H) located at the edge has a high activity for the CO2RR. The performances of the C5 + H edge, pyridine N, armchair and zigzag edge in CO2RR are analyzed. The calculation shows that the pyridine N model has tight interaction with *CO molecules, which leads to the difficult absorption of *CO (Fig. 13d). The zigzag edge sites strongly attract *COOH. The edge of the armchair powerfully absorbs *COOH and *CO. While C5 + H sites with properties of both topological defects and edge defects showed rapid kinetics (Fig. 13b and c). Topological defects, especially pentagons, are further revealed for high catalytic activity in CO2RR. Topological defect-rich graphene (TDG) was prepared by removing N dopants from the carbon framework and used as a cathode catalyst for the Li–CO2 battery.16 According to the relationship between the overpotential difference between charge and discharge (η = ηcharge − ηdischarge) and adsorption energy of the *CO (ΔG*CO), a “volcano-type” Fig. 13e was drawn, from which it can be found that C5 has the highest catalytic activity for CO2 reduction and precipitation. And N-doped materials perform poorly compared to intrinsic defects. Even the overpotential difference of 1.01 V for C5 is lower than that induced by the active site of metal atoms (FeN4). Several other researchers have also reported that the pentagonal topological defect structure serves as the main active site for CO2 reduction.282–284
Extrinsic defects, including heteroatoms doping and metal atom-induced defect, can induce activity sites on the carbon matrix. Han et al.285 obtained B, N, and P heteroatom-doped carbon. N with greater electronegativity induces a positive charge density on the surrounding C atoms, which promotes the adsorption of CO2; P and B have lower electronegativity and high electron-donating ability, which can enhance the electron delocalization of adjacent C atoms, enhancing electrical conductivity. The synergistic effect of the three heteroatoms vastly reduces the free energy barrier for the formation of *COOH, thereby enhancing CO2RR catalytic activity. Xu et al.279 synthesized a series of N-doped carbon nanotubes (NCNTs) for the CO2RR. DFT calculations suggest that the enhanced activity of the N-doped carbon catalyst is accomplished by stabilizing the CO2.−* (COOH*) intermediate. The catalytic activity of different N configurations is different, the most effective is quaternary N, followed by pyridinic-N and pyrrolic-N (Fig. 13f).
Intrinsic defect and extrinsic atom-induced defect in carbon materials have been found to enhance the electrocatalytic activity of CO2RR, and their combination is often able to create catalytic sites that are favorable for the CO2RR. Zhang et al.280 prepared Ni-atom-coordinated N-rich carbon (Ni–N3–V) containing vacancy defects for CO2 reduction by adjusting the coordination environment of Ni–N4. The changes of free energy during the CO2 reduction process for the three models of Ni–N4, Ni–N3–V, and Ni–N3 were calculated by DFT, as shown in Fig. 13g. From the two steps of CO2 → COOH* → CO*, Ni–N3 is the most favorable, but it is difficult for Ni–N3 to release CO from the reaction site during the CO* → CO reaction. Therefore, comprehensively considered, Ni–N3–V has the relatively best electrocatalytic activity because of its modest and medium ΔG values for CO2 activation and CO desorption. This report enables the coupling of vacancy defects with M–N–C to be demonstrated. In addition, Nie and co-workers intercalated Fe–N4-rich graphene-like porous carbon and calculated the free energy of CO2RR under the catalysis of Fe–N4–N, Fe–N4–NH, 585-Fe–N4, and 585-defects.281 It can be seen from Fig. 13h that Fe–N4–N and Fe–N4–NH sites have strong adsorption on *CO. For the defect site of 585-Fe–N4, the key step of the CO2RR is the formation of *COOH. The corresponding energy barrier is 0.94 eV, which is lower than that of the topological 585 defects (1.43 eV). This report shows that topological defects (C585) and Fe–N4 have a synergistic effect and this result has been verified by electrochemical tests.
Multifunctional electrocatalysts that can significantly promote the adsorption/desorption of two or more active species are suitable for a variety of electrocatalytic reactions. Yao et al.147 verified by DFT calculations that several sites 5-1, 585-1, 585-3, 7557-1 and 7557-4 have the highest catalytic activity among edge pentagons, 5-8-5 edge defects and 7-55-7 edge defects (Fig. 13i–k). The energy changes of ORR, OER and HER at these sites were calculated as shown in Fig. 13l–n, and superior activity of ORR and OER was highlighted on marginal carbon in 5-1, 585-1 and 7557-1defect atomic sites. The HER active sites are 7557-4 and 585-3 because hydrogen has the required binding energy at this site. Notably, when carbon-based electrocatalytic materials are introduced with a variety of extrinsic heteroatoms, including metal-free and metal atoms, they often exhibit trifunctional catalytic activity. For instance, Liu et al.286 developed N, O, P multi-doped porous carbon (GNP-900), which has a high specific surface area and huge defect sites. And multi-heteroatom doping can vigorously induce charge redistribution, inducing excellent performance for the HER, OER and ORR. A multi-doped porous carbon material with Cu, N and S atoms (Cu–N–SC-1100) was prepared and exhibits excellent electrocatalytic activity for the ORR, OER and HER.27 Besides, Br, Co, N multi-doped defect-rich porous carbon frameworks (BrHT@CoNC),287 Fe, Co, Ni embedded N-doped carbon nanorod frameworks,14etc. have also been known to exhibit high electrocatalytic activity for the ORR, OER and HER.
In this section, the progress of carbon defect engineering in electrocatalysis is summarized, as it has been robustly developed in recent years. There are many studies on electrocatalytic reactions, such as the ORR, OER, HER, nitrogen reduction reaction (NRR), CO2RR, etc., and only some of them are reported here. It is hoped that this study will be of interest to researchers and encourage more research on improving the catalytic performance of electrocatalysts.
6. Summaries and perspectives
Carbon materials have the virtues of environmental protection, low cost, stable performance, etc., and show distinctive advantages in energy storage and conversion systems. However, it is difficult for the activity of raw carbon to meet the requirements of future energy-related applications, especially smart grids and electric vehicles. Recently, defect electrochemistry has developed rapidly, and great progress has been made in both theoretical research and practical exploration. Defect engineering has become an effective and popular strategy to prepare carbon for high-capacity batteries and high-performance electrocatalysts. The intrinsic and extrinsic defects of carbon materials can adjust the characteristics of charge distribution, spin density, energy band structure, and adsorption energy, thereby improving the activity and increasing the reaction sites, which have a positive effect on the improvement of performance. This article reviews recent advances in defect engineering of carbon materials for energy storage and conversion systems, with a focus on applications in MIBs, LSBs and electrocatalysts.Defect engineering in carbon materials for MIB electrodes. Based on the analysis of defect engineering and MIB performance, the role of defect is further elucidated, which has been a controversial topic in the field of MIBs for many years. Despite the inconsistent correlation between active sites and electrochemical activity, it is undeniable that designated defect sites can trap metal ions, enhance the adsorption and intercalation of metal ions, increase the electrochemical activity of carbon materials and offer additional capacity. In addition, the introduction of heteroatoms makes the carbon interlayer spacing tend to increase, providing a large space for the intercalation of metal ions. Defective sites also enhance electrode wettability in electrolytes, facilitating charge transfer and electrode/electrolyte interaction.
Defect engineering in carbon materials for a LSB cathode and separator. As mentioned earlier, local electron reconstruction caused by defects can usually promote many electrochemical reactions. Oxidation–reduction of sulfur is the basic reaction of LSB, and carbon is an important part of a sulfur cathode and modified separator. Defect-rich carbon exposes abundant coordinated unsaturated sites. These sites can not only enhance the conductivity and wettability, but also weaken the shuttling effect. Defect sites with adsorption–catalytic-conversion function willing to adsorb polysulfides through electrostatic interaction and/or chemical bond, and furnish faster kinetics for the conversion of sulfur species.
Defect engineering in carbon materials for electrocatalysis. With the deepening of the research on the mechanisms related to defect electrocatalysis, the understanding of catalytic active sites is now becoming more and more clear. The intrinsic defect electrocatalytic activity has been demonstrated by numerous experimental results and theoretical simulations. The active sites doped by metal-free heteroatoms are modified carbon atoms around the dopant. The coordination coupling between metal atoms and the surrounding environment is the active source of metal atom-induced defect sites. Defect sites can improve adsorption energy, stabilize intermediates, and enhance the electrocatalytic activity of carbon materials in the ORR, HER, and CO2RR.
Numerous studies have confirmed that the development of defect engineering in carbon materials is extremely promising and that the activity and catalytic performance of electrochemical reactions can indeed be greatly improved. However, there are still some challenges to be overcome during the defect engineering research of carbon-based materials.
 | ||
| Fig. 14 Future direction of defect engineering in carbon materials. Cartoon cars and experimental operation pictures are provided by 51yuansu.com, the illustration of functional identification is from ref. 229, and the illustration of mechanism analysis is cited from ref. 254. Reproduced with permission. | ||
(1) The current research on defect engineering of carbon-based materials, including intrinsic and extrinsic defects, focuses on the fields of electrocatalysis and fuel cells, but there is a relative lack of research on electrochemical energy storage systems such as secondary batteries and supercapacitors.
(2) Currently, defective structures are manufactured by relatively traditional methods, many of which are pyrolysis and ball milling, and are difficult to innovatively design with high precision and number of defective sites.
(3) The scale of defect sites is at the atomic level, and advanced characterization techniques that are highly accurate and affordable, such as electron microscopy, which allows direct observation of defect structures, and spectroscopy, which determines defect types and densities, are not yet widely available. In addition, in situ characterization techniques to detect the stability and dynamic evolution of defect sites during electrochemical reactions are not commonly applied.
(4) The synergistic mechanism of intrinsic defect sites with non-metallic heteroatoms, and metal single-atom is debatable, and the mechanism of how they interact and jointly promote electrochemical reactivity is not yet understood.
(5) The practicality of defect engineering for carbon-based materials in practical electrochemical devices has not been explored, and metrics and standard protocols for defect site activity and durability analysis have not been established.
In the future road, more detailed and systematic research work should continue to be carried out. The future development of defect engineering in carbon materials may fasten on, but is not confined to, the following four aspects (Fig. 14).
(1) Controlled synthesis. It is essential to explore synthetic methods and optimize synthetic strategies to achieve accurate and controllable defect sites as well as quantitatively adjustable defect density. Light irradiation, plasma etching, arc discharge methods, electrochemical exfoliation, ion exchange, vaporized salt induction and other methods are feasible strategies for constructing defects, and how to achieve the desired defects in carbon materials is worthy of in-depth study.
(2) Function identification. The identification of defect types and the evaluation of defect density are the prerequisites for studying the activity of defect sites, and advanced characterization techniques (Kelvin probe force microscope, positron annihilation, electron paramagnetic resonance, electron energy loss spectroscopy, etc.) are essential to characterize defects. Furthermore, the effects of various defect sites on electrochemical activity are not consistent and clarifying the contribution of each defect to the performance is also important and challenging. This will accelerate the understanding of the active mechanism of different defect sites, facilitate the design of multifunctional materials, and broaden the application scope of defect materials.
(3) Mechanism analysis. Current mechanism exploration mainly relies on the strategy of combining theoretical calculation prediction and experimental result verification. Therefore, it is feasible to further increase the application of theoretical simulation methods including density functional calculation and machine learning in the understanding of electrochemical reactions. Furthermore, most catalyst materials, including defective electrocatalysts, face challenges related to the stability of catalytic reactions, despite the good stability exhibited by defective sites in the current report. Some reversible or irreversible structural rearrangement of defective catalytic materials during electrochemical reactions requires in situ characterization techniques with higher spatiotemporal resolution.
(4) Practical application. The large-scale production and application of defective carbon materials is the goal pursued. Unfortunately, the current preparation of defect-rich carbon is still stagnant at the laboratory scale, and therefore, it is vital and necessary to reduce the fabrication cost and improve the properties of defect-rich carbon to speed up their practical applications.
Undoubtedly, defect engineering in carbon materials has great application potential in the field of energy storage and conversion. We believe that through the hard work of scientists all over the world in the field of defect engineering, the related research will definitely bear fruitful results, and the extensive industrial application of defect-rich carbon will surely be realized. We hope this paper can provide some inspiration and ideas for the construction of defect engineering in carbon material and the understanding of catalytic sites, as well as help readers find out the application progress and broad prospects of defect engineering in the field of energy storage and conversion.
Author contributions
Zhiqiang Zhao participated in the investigation and wrote the manuscript. Huan Chen and Wanyu Zhang conducted a reference analysis. Shan Yi, Hongli Chen and Zhe Su contributed to the conceptualization. Bo Niu and Yayun Zhang helped write reviews and edits. Donghui Long helped funding acquisition, project management and oversight. All authors finally approved for publication.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22008073, No. 21878091, No. 22078100, and No. 52102098), the Shanghai Sailing Program (No. 20YF1410600), the Fundamental Research Funds for the Central Universities, and the Shanghai Talent Development Fund (No. 2021026).References
- R. R. Baidoo, F. Ferguson and H. Singh, in 2008 IEEE Energy 2030 Conference, IEEE, Atlanta, GA, USA, 2008, pp. 1–6.
- A. Benítez, J. Amaro-Gahete, Y.-C. Chien, Á. Caballero, J. Morales and D. Brandell, Renewable Sustainable Energy Rev., 2022, 154, 111783 CrossRef.
- Y. Liu, Z. Yu, J. Chen, C. Li, Z. Zhang, X. Yan, X. Liu and S. Yang, Chin. Chem. Lett., 2022, 33, 1817–1830 CrossRef CAS.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- Q. Lin, J. Zhang, W. Lv, J. Ma, Y. He, F. Kang and Q. Yang, Small, 2020, 16, 1902603 CrossRef CAS PubMed.
- R. Mukherjee, A. V. Thomas, D. Datta, E. Singh, J. Li, O. Eksik, V. B. Shenoy and N. Koratkar, Nat. Commun., 2014, 5, 3710 CrossRef CAS PubMed.
- D. Datta, J. Li, N. Koratkar and V. B. Shenoy, Carbon, 2014, 80, 305–310 CrossRef CAS.
- Y. Gao, Y. Bai, R. Sun, M. Qu, M. Wang, L. Peng, Z. Wang, W. Sun and K. Sun, Ind. Eng. Chem. Res., 2022, 61, 6957–6966 CrossRef CAS.
- C. Wei, Y. Han, H. Liu, R. Gan, W. Ma, H. Liu, Y. Song, X. Zhang, J. Shi and C. Ma, J. Colloid Interface Sci., 2022, S0021979722010207 Search PubMed.
- H. Li, M. Zhao, B. Jin, Z. Wen, H. K. Liu and Q. Jiang, Small, 2020, 16, 1907464 CrossRef CAS PubMed.
- Y. Li, H. Wang, C. Priest, S. Li, P. Xu and G. Wu, Adv. Mater., 2021, 33, 2000381 CrossRef CAS PubMed.
- H. Zhao, C. Sun, Z. Jin, D.-W. Wang, X. Yan, Z. Chen, G. Zhu and X. Yao, J. Mater. Chem. A, 2015, 3, 11736–11739 RSC.
- P. Han, X. Yu, D. Yuan, M. Kuang, Y. Wang, A. M. Al-Enizi and G. Zheng, J. Colloid Interface Sci., 2019, 534, 332–337 CrossRef CAS PubMed.
- Q. Jin, L. Xiao, W. He, H. Cui and C. Wang, Green Energy Environ., 2022, S2468025722000590 Search PubMed.
- C. Tang, H.-F. Wang, X. Chen, B.-Q. Li, T.-Z. Hou, B. Zhang, Q. Zhang, M.-M. Titirici and F. Wei, Adv. Mater., 2016, 28, 6845–6851 CrossRef CAS PubMed.
- F. Ye, L. Gong, Y. Long, S. N. Talapaneni, L. Zhang, Y. Xiao, D. Liu, C. Hu and L. Dai, Adv. Energy Mater., 2021, 11, 2101390 CrossRef CAS.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, Beilstein J. Nanotechnol., 2015, 6, 1016–1055 CrossRef CAS PubMed.
- F. Y. Fan, W. C. Carter and Y.-M. Chiang, Adv. Mater., 2015, 27, 5203–5209 CrossRef CAS PubMed.
- K. Kumaresan, Y. Mikhaylik and R. E. White, J. Electrochem. Soc., 2008, 155, A576 CrossRef CAS.
- X. Ren, Z. Liu, M. Zhang, D. Li, S. Yuan and C. Lu, ChemElectroChem, 2021, 8, 3457–3471 CrossRef CAS.
- L. Zhang, Y. Shi, Y. Wang and N. R. Shiju, Adv. Sci., 2020, 7, 1902126 CrossRef CAS PubMed.
- Y. Zhang, L. Tao, C. Xie, D. Wang, Y. Zou, R. Chen, Y. Wang, C. Jia and S. Wang, Adv. Mater., 2020, 32, 1905923 CrossRef CAS PubMed.
- M. S. Dresselhaus and G. Dresselhaus, Adv. Phys., 1981, 30, 139–326 CrossRef CAS.
- X. Wu, Y. Chen, Z. Xing, C. W. K. Lam, S. Pang, W. Zhang and Z. Ju, Adv. Energy Mater., 2019, 9, 1900343 CrossRef.
- M. Ou, S. Sun, Y. Liu, Y. Xu, C. Chen, P. Hu, C. Fang, Q. Li and J. Han, RSC Adv., 2021, 11, 22297–22304 RSC.
- S. Qiao, D. Lei, Q. Wang, X. Shi, Q. Zhang, C. Huang, A. Liu, G. He and F. Zhang, Chem. Eng. J., 2022, 442, 136258 CrossRef CAS.
- M. Wang, K. Su, M. Zhang, X. Du and Z. Li, ACS Sustainable Chem. Eng., 2021, 9, 13324–13336 CrossRef CAS.
- C. Xie, D. Yan, H. Li, S. Du, W. Chen, Y. Wang, Y. Zou, R. Chen and S. Wang, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- Y. Zhang, N. Afzal, L. Pan, X. Zhang and J. Zou, Adv. Sci., 2019, 6, 1900053 CrossRef PubMed.
- Z. Yu, Y. Pan, Y. Shen, Z. Wang, Z.-Y. Ong, T. Xu, R. Xin, L. Pan, B. Wang, L. Sun, J. Wang, G. Zhang, Y. W. Zhang, Y. Shi and X. Wang, Nat. Commun., 2014, 5, 5290 CrossRef CAS PubMed.
- J. Wang, H. Shi, W. Wang, Z. Xu, C. Hong, Y. Xue and F. Tian, Chem. Eng. J., 2022, 432, 133617 CrossRef CAS.
- C. Xie, D. Yan, W. Chen, Y. Zou, R. Chen, S. Zang, Y. Wang, X. Yao and S. Wang, Mater. Today, 2019, 31, 47–68 CrossRef CAS.
- E. Uchaker and G. Cao, Chem. – Asian J., 2015, 10, 1608–1617 CrossRef CAS PubMed.
- P. Luo, C. Zheng, J. He, X. Tu, W. Sun, H. Pan, Y. Zhou, X. Rui, B. Zhang and K. Huang, Adv. Funct. Mater., 2022, 32, 2107277 CrossRef CAS.
- S. Qiao, Q. Wang, D. Lei, X. Shi, Q. Zhang, C. Huang, A. Liu, G. He and F. Zhang, J. Mater. Chem. A, 2022, 10, 11702–11711 RSC.
- H. Liu, Z. Chen, L. Zhou, H. Man, Q. Zeng, K. Pei, G. Chen, Q. Wang, Y. Song, X. Li, Y. Zhou, F. Fang, J. Liu, D. Sun and R. Che, Energy Storage Mater., 2021, 41, 703–714 CrossRef.
- X. Yan and X. Yao, in Carbon-Based Metal-Free Catalysts, ed. L. Dai, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018, pp. 59–75 Search PubMed.
- J. Zhu, Y. Huang, W. Mei, C. Zhao, C. Zhang, J. Zhang, I. S. Amiinu and S. Mu, Angew. Chem., Int. Ed., 2019, 58, 3859–3864 CrossRef CAS PubMed.
- Q. Wu, Y. Jia, Q. Liu, F. Liu, A. Du and X. Yao, Chem, 2022, 8, 2715–2733 CAS.
- Z. Xiao, C. Xie, Y. Wang, R. Chen and S. Wang, J. Energy Chem., 2021, 53, 208–225 CrossRef CAS.
- D. Xue, H. Xia, W. Yan, J. Zhang and S. Mu, Nano-Micro Lett., 2021, 13, 5 CrossRef PubMed.
- Z. Shi, M. Li, J. Sun and Z. Chen, Adv. Energy Mater., 2021, 11, 2100332 CrossRef CAS.
- Y. Zheng, T. J. Slade, L. Hu, X. Y. Tan, Y. Luo, Z.-Z. Luo, J. Xu, Q. Yan and M. G. Kanatzidis, Chem. Soc. Rev., 2021, 50, 9022–9054 RSC.
- X. Luo, H. Zheng, W. Lai, P. Yuan, S. Li, D. Li and Y. Chen, Energy Environ. Mater., 2022, eem2.12402 CrossRef.
- D. Jiang, B. G. Sumpter and S. Dai, J. Chem. Phys., 2007, 126, 134701 CrossRef PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Pan, S. Wang, X. Chen, P. Hu, L. Sun and X. Bao, Chem. Commun., 2011, 47, 10016 RSC.
- Y. Jiang, L. Yang, T. Sun, J. Zhao, Z. Lyu, O. Zhuo, X. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 CrossRef CAS.
- X. Yan, Y. Jia, T. Odedairo, X. Zhao, Z. Jin, Z. Zhu and X. Yao, Chem. Commun., 2016, 52, 8156–8159 RSC.
- X. Yan, Y. Jia, L. Zhang, M. Teng Soo and X. Yao, Chem. Commun., 2017, 53, 12140–12143 RSC.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS PubMed.
- F. Yuan, C. Shi, Q. Li, J. Wang, D. Zhang, Q. Wang, H. Wang, Z. Li, W. Wang and B. Wang, Adv. Funct. Mater., 2022, 2208966 CrossRef CAS.
- R. Guo, C. Lv, W. Xu, J. Sun, Y. Zhu, X. Yang, J. Li, J. Sun, L. Zhang and D. Yang, Adv. Energy Mater., 2020, 10, 1903652 CrossRef CAS.
- Q. Wu, Y. Jia, Q. Liu, X. Mao, Q. Guo, X. Yan, J. Zhao, F. Liu, A. Du and X. Yao, Chem, 2022, S2451929422003187 Search PubMed.
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764–2767 RSC.
- L. Guan, H. Hu, L. Li, Y. Pan, Y. Zhu, Q. Li, H. Guo, K. Wang, Y. Huang, M. Zhang, Y. Yan, Z. Li, X. Teng, J. Yang, J. Xiao, Y. Zhang, X. Wang and M. Wu, ACS Nano, 2020, 14, 6222–6231 CrossRef CAS PubMed.
- X. Yan, L. Zhuang, Z. Zhu and X. Yao, Nanoscale, 2021, 13, 3327–3345 RSC.
- A. Walsh, J. Phys. Chem. Lett., 2010, 1, 1284–1287 CrossRef CAS.
- W.-F. Li, C.-M. Fang, M. Dijkstra and M. A. van Huis, J. Phys.: Condens. Matter, 2015, 27, 355801 CrossRef PubMed.
- T. Xiong, Y. Zhang, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS.
- Y. Cao, Z. Liu, Y. Tang, C. Huang, Z. Wang, F. Liu, Y. Wen, B. Shan and R. Chen, Carbon, 2021, 180, 1–9 CrossRef CAS.
- L. Hou, W. Yang, B. Jiang, P. Wang, L. Yan, C. Zhang, G. Huang, F. Yang and Y. Li, Carbon, 2021, 183, 176–186 CrossRef CAS.
- P. V. C. Medeiros, A. J. S. Mascarenhas, F. de Brito Mota and C. M. C. de Castilho, Nanotechnology, 2010, 21, 485701 CrossRef PubMed.
- L. Hawelek, W. Wrzalik, A. Brodka, J. C. Dore, A. C. Hannon, S. Iijima, M. Yudasaka, T. Ohba, K. Kaneko and A. Burian, Chem. Phys. Lett., 2011, 502, 87–91 CrossRef CAS.
- Y. Jia, L. Zhang, L. Zhuang, H. Liu, X. Yan, X. Wang, J. Liu, J. Wang, Y. Zheng, Z. Xiao, E. Taran, J. Chen, D. Yang, Z. Zhu, S. Wang, L. Dai and X. Yao, Nat. Catal., 2019, 2, 688–695 CrossRef CAS.
- M. Yi and Z. Shen, J. Mater. Chem. A, 2015, 3, 11700–11715 RSC.
- G. Kudur Jayaprakash, N. Casillas, P. D. Astudillo-Sánchez and R. Flores-Moreno, J. Phys. Chem. A, 2016, 120, 9101–9108 CrossRef CAS PubMed.
- Z. Hou, X. Wang, T. Ikeda, K. Terakura, M. Oshima and M. Kakimoto, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165401 CrossRef.
- Y. Jia, J. Chen and X. Yao, Mater. Chem. Front., 2018, 2, 1250–1268 RSC.
- Y. S. Yun, K.-Y. Park, B. Lee, S. Y. Cho, Y.-U. Park, S. J. Hong, B. H. Kim, H. Gwon, H. Kim, S. Lee, Y. W. Park, H.-J. Jin and K. Kang, Adv. Mater., 2015, 27, 6914–6921 CrossRef CAS PubMed.
- W. Wang, L. Shang, G. Chang, C. Yan, R. Shi, Y. Zhao, G. I. N. Waterhouse, D. Yang and T. Zhang, Adv. Mater., 2019, 31, 1808276 CrossRef PubMed.
- J. Tan, N. Tian, Z. Li, J. Li, X. Yao, M. Vakili, Y. Lu and T. Zhang, Chem. Eng. J., 2021, 421, 127729 CrossRef CAS.
- Q. Wu, X. Yan, Y. Jia and X. Yao, EnergyChem, 2021, 3, 100059 CrossRef CAS.
- G. Rajasekaran and A. Parashar, Mater. Today: Proc., 2018, 5, 6780–6788 CAS.
- J. Bocko and P. Lengvarský, J. Mech. Sci. Technol., 2017, 31, 1825–1833 CrossRef.
- Y. Gao, S. Zhang, X. Li, L. Li, L. Bao, N. Zhang, J. Peng and X. Li, Carbon, 2021, 181, 323–334 CrossRef CAS.
- A. M. Patel, N. Gosai and A. Y. Joshi, Appl. Mech. Mater., 2015, 813–814, 145–150 Search PubMed.
- T. Wang, L. Xue, Y. Liu, T. Fang, L. Zhang and B. Xing, Chem. Eng. J., 2022, 435, 134822 CrossRef CAS.
- Z. Zhao, Z. Su, H. Chen, S. Yi, W. Zhang, B. Niu, Y. Zhang and D. Long, Sustainable Energy Fuels, 2022, 6, 5211–5242 RSC.
- J. Wu, Z. Ju, X. Zhang, X. Xu, K. J. Takeuchi, A. C. Marschilok, E. S. Takeuchi and G. Yu, ACS Nano, 2022, 16, 4805–4812 CrossRef CAS PubMed.
- C. Li, X. Li, Q. Yang, P. Sun, L. Wu, B. Nie, H. Tian, Y. Wang, C. Wang, X. Chen and J. Shao, Adv. Sci., 2021, 8, 2004957 CrossRef CAS PubMed.
- J. Lai, A. Nsabimana, R. Luque and G. Xu, Joule, 2018, 2, 76–93 CrossRef CAS.
- J. C. Bui, E. W. Lees, L. M. Pant, I. V. Zenyuk, A. T. Bell and A. Z. Weber, Chem. Rev., 2022, 122, 11022–11084 CrossRef CAS PubMed.
- H. Hamed, S. Yari, J. D’Haen, F. U. Renner, N. Reddy, A. Hardy and M. Safari, Adv. Energy Mater., 2020, 10, 2002492 CrossRef CAS.
- E. Harimohan, K. Nanaji, B. V. A. Rao and T. N. Rao, Int. J. Energy Res., 2022, 46, 4339–4351 CrossRef CAS.
- X. Zhang, Z. Hui, S. King, L. Wang, Z. Ju, J. Wu, K. J. Takeuchi, A. C. Marschilok, A. C. West, E. S. Takeuchi and G. Yu, Nano Lett., 2021, 21, 5896–5904 CrossRef CAS PubMed.
- X. Zhang, Z. Hui, S. T. King, J. Wu, Z. Ju, K. J. Takeuchi, A. C. Marschilok, A. C. West, E. S. Takeuchi, L. Wang and G. Yu, Nano Lett., 2022, 22, 2521–2528 CrossRef CAS PubMed.
- W. Zhou, S. Yao, H. Wang, Q. Du, Y. Ma and Y. Zhu, ACS Nano, 2020, 14, 5798–5805 CrossRef CAS PubMed.
- W. Zhang, Z. Ren, Z. Ying, X. Liu and H. Wan, J. Alloys Compd., 2018, 743, 44–51 CrossRef CAS.
- Z. Liu, C. Duan, S. Dou, Q. Yuan, J. Xu, W. Liu and Y. Chen, Small, 2022, 18, 2200954 CrossRef CAS PubMed.
- L. Wan, E. Shamsaei, C. D. Easton, D. Yu, Y. Liang, X. Chen, Z. Abbasi, A. Akbari, X. Zhang and H. Wang, Carbon, 2017, 121, 330–336 CrossRef CAS.
- D. Fang, Y. Wang, X. Liu, J. Yu, C. Qian, S. Chen, X. Wang and S. Zhang, ACS Nano, 2019, acsnano.8b07491 CrossRef PubMed.
- H. Sun, L. Mei, J. Liang, Z. Zhao, C. Lee, H. Fei, M. Ding, J. Lau, M. Li, C. Wang, X. Xu, G. Hao, B. Papandrea, I. Shakir, B. Dunn, Y. Huang and X. Duan, Science, 2017, 356, 599–604 CrossRef CAS PubMed.
- D. Wang, R. Dai, X. Zhang, L. Liu, H. Zhuang, Y. Lu, Y. Wang, Y. Liao and Q. Nian, Carbon, 2020, 161, 880–891 CrossRef CAS.
- Z. Li, X. Zhang, H. Tan, W. Qi, L. Wang, M. C. Ali, H. Zhang, J. Chen, P. Hu, C. Fan and H. Qiu, Adv. Funct. Mater., 2018, 28, 1805026 CrossRef.
- J. Wang and W. Han, Adv. Funct. Mater., 2022, 32, 2107166 CrossRef CAS.
- X. Yan, Y. Jia and X. Yao, Chem. Soc. Rev., 2018, 47, 7628–7658 RSC.
- Y. Zhu, X. Yang, C. Peng, C. Priest, Y. Mei and G. Wu, Small, 2021, 17, 2005148 CrossRef CAS PubMed.
- D. Jana, C.-L. Sun, L.-C. Chen and K.-H. Chen, Prog. Mater. Sci., 2013, 58, 565–635 CrossRef CAS.
- M. Skorupska, A. Ilnicka and J. P. Lukaszewicz, Processes, 2022, 10, 643 CrossRef CAS.
- S. Ghosh, S. Barg, S. M. Jeong and K. (Ken) Ostrikov, Adv. Energy Mater., 2020, 10, 2001239 CrossRef CAS.
- Y. Wang, Z. Tang, S. Shen and J. Yang, New Carbon Mater., 2022, 37, 321–336 CrossRef.
- S. Gouse Peera, H.-J. Kwon, T. G. Lee and A. M. Hussain, Ionics, 2020, 26, 1563–1589 CrossRef CAS.
- D. Saurel, B. Orayech, B. Xiao, D. Carriazo, X. Li and T. Rojo, Adv. Energy Mater., 2018, 8, 1703268 CrossRef.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 RSC.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66–72 CrossRef CAS.
- K. Gao, B. Wang, L. Tao, B. V. Cunning, Z. Zhang, S. Wang, R. S. Ruoff and L. Qu, Adv. Mater., 2019, 31, 1805121 CrossRef PubMed.
- M. Rahm, T. Zeng and R. Hoffmann, J. Am. Chem. Soc., 2019, 141, 342–351 CrossRef CAS PubMed.
- C. Tantardini and A. R. Oganov, Nat. Commun., 2021, 12, 2087 CrossRef CAS PubMed.
- S. Majeed, J. Zhao, L. Zhang, S. Anjum, Z. Liu and G. Xu, Nanotechnol. Rev., 2013, 2, 615–635 CAS.
- X. Kong, Q. Chen and Z. Sun, Chem. Phys. Chem., 2013, 14, 514–519 CrossRef CAS PubMed.
- H. Zhang, P. An, W. Zhou, B. Y. Guan, P. Zhang, J. Dong and X. W. (David) Lou, Sci. Adv., 2018, 4, eaao6657 CrossRef PubMed.
- G. Gan, S. Fan, X. Li, J. Wang, C. Bai, X. Guo, M. Tade and S. Liu, ACS Catal., 2021, 11, 14284–14292 CrossRef CAS.
- F. Yuan, H. Sun, D. Zhang, Z. Li, J. Wang, H. Wang, Q. Wang, Y. Wu and B. Wang, J. Colloid Interface Sci., 2022, 611, 513–522 CrossRef CAS PubMed.
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef CAS PubMed.
- Y.-C. Lin, P.-Y. Teng, C.-H. Yeh, M. Koshino, P.-W. Chiu and K. Suenaga, Nano Lett., 2015, 15, 7408–7413 CrossRef CAS PubMed.
- Y. Nam, D. Cho and J. Y. Lee, J. Phys. Chem. C, 2016, 120, 11237–11244 CrossRef CAS.
- R. Saito, M. Yagi, T. Kimura, G. Dresselhaus and M. S. Dresselhaus, J. Phys. Chem. Solids, 1999, 60, 715–721 CrossRef CAS.
- H. Zhou, Y. Peng, H. B. Wu, F. Sun, H. Yu, F. Liu, Q. Xu and Y. Lu, Nano Energy, 2016, 21, 80–89 CrossRef CAS.
- S. Zhang, Z. Xu, Z. Jiang, Z. Xiao, A. Tang and H. Yang, Carbon, 2022, 195, 207–218 CrossRef CAS.
- F. Karlický, K. Kumara Ramanatha Datta, M. Otyepka and R. Zbořil, ACS Nano, 2013, 7, 6434–6464 CrossRef PubMed.
- V. V. Chaban and O. V. Prezhdo, Nanoscale, 2016, 8, 15521–15528 RSC.
- S. Huang, D.-D. Ma, X. Wang, Y. Shi, R. Xun, H. Chen, H. Guan and Y. Tong, J. Colloid Interface Sci., 2022, 608, 334–343 CrossRef CAS PubMed.
- S. Xue, J. Tan, X. Ma, Y. Xu, R. Wan and H. Tao, FlatChem, 2022, 34, 100390 CrossRef CAS.
- Z. Qu, F. Sun, J. Gao and G. Zhao, J. Colloid Interface Sci., 2022, 613, 47–56 CrossRef CAS PubMed.
- Z. Kong, Y. Lin, J. Hu, Y. Wang and L. Zhan, Chem. Eng. J., 2022, 436, 132719 CrossRef CAS.
- I. O. Maciel, J. Campos-Delgado, E. Cruz-Silva, M. A. Pimenta, B. G. Sumpter, V. Meunier, F. López-Urías, E. Muñoz-Sandoval, H. Terrones, M. Terrones and A. Jorio, Nano Lett., 2009, 9, 2267–2272 CrossRef CAS PubMed.
- V. V. Strelko, V. S. Kuts and P. A. Thrower, Carbon, 2000, 38, 1499–1503 CrossRef CAS.
- J. Zhu and S. Mu, Adv. Funct. Mater., 2020, 30, 2001097 CrossRef CAS.
- I.-Y. Jeon, S. Zhang, L. Zhang, H.-J. Choi, J.-M. Seo, Z. Xia, L. Dai and J.-B. Baek, Adv. Mater., 2013, 25, 6138–6145 CrossRef CAS PubMed.
- Y.-J. Wang, B. Fang, D. Zhang, A. Li, D. P. Wilkinson, A. Ignaszak, L. Zhang and J. Zhang, Electrochem. Energy Rev., 2018, 1, 1–34 CrossRef CAS.
- J. Zhang, C. You, H. Lin and J. Wang, Energy Environ. Mater., 2022, eem2.12250 Search PubMed.
- B. Bayatsarmadi, Y. Zheng, A. Vasileff and S. Qiao, Small, 2017, 13, 1700191 CrossRef PubMed.
- C. Rivera-Cárcamo and P. Serp, ChemCatChem, 2018, 10, 5058–5091 CrossRef.
- J. Guan, Z. Duan, F. Zhang, S. D. Kelly, R. Si, M. Dupuis, Q. Huang, J. Q. Chen, C. Tang and C. Li, Nat. Catal., 2018, 1, 870–877 CrossRef CAS.
- X. Wen, L. Bai, M. Li and J. Guan, ACS Sustainable Chem. Eng., 2019, 7, 9249–9256 CrossRef CAS.
- A. K. Thakur, K. Kurtyka, M. Majumder, X. Yang, H. Q. Ta, A. Bachmatiuk, L. Liu, B. Trzebicka and M. H. Rummeli, Adv. Mater. Interfaces, 2022, 9, 2101964 CrossRef CAS.
- M. Fan, J. Cui, J. Wu, R. Vajtai, D. Sun and P. M. Ajayan, Small, 2020, 16, 1906782 CrossRef CAS PubMed.
- Y. Huang, K.-D. Seo, K. A. Jannath, D.-S. Park and Y.-B. Shim, Carbon, 2022, 196, 621–632 CrossRef CAS.
- G. Ba, T. Huo, Q. Deng, H. Li and W. Hou, ACS Sustainable Chem. Eng., 2020, 8, 18606–18615 CrossRef CAS.
- Y. Dong, X. Lin, D. Wang, R. Yuan, S. Zhang, X. Chen, L. G. Bulusheva, A. V. Okotrub and H. Song, Energy Storage Mater., 2020, 30, 287–295 CrossRef.
- Y. Dong, S. Zhang, X. Du, S. Hong, S. Zhao, Y. Chen, X. Chen and H. Song, Adv. Funct. Mater., 2019, 29, 1901127 CrossRef.
- C. E. Lowell, J. Am. Ceram. Soc., 1967, 50, 142–144 CrossRef CAS.
- Y.-J. Lee and H. Hatori, Chem. Phys. Lett., 2002, 362, 326–330 CrossRef CAS.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- L. Chen, S. Yu, Y. Zhang, Y. Song and L. Song, J. Power Sources, 2021, 514, 230607 CrossRef CAS.
- P. Tian, J. Zang, S. Song, S. Zhou, H. Gao, H. Xu, X. Tian and Y. Wang, J. Power Sources, 2020, 448, 227443 CrossRef CAS.
- Y. Chen, B. Xi, M. Huang, L. Shi, S. Huang, N. Guo, D. Li, Z. Ju and S. Xiong, Adv. Mater., 2022, 34, 2108621 CrossRef CAS PubMed.
- L. G. Bulusheva, V. E. Arkhipov, K. M. Popov, V. I. Sysoev, A. A. Makarova and A. V. Okotrub, Materials, 2020, 13, 1173 CrossRef CAS PubMed.
- C. Wang and C. Xu, Coatings, 2021, 11, 523 CrossRef CAS.
- V. K. R. Kondapalli, X. He, M. Khosravifar, S. Khodabakhsh, B. Collins, S. Yarmolenko, A. Paz y Puente and V. Shanov, ACS Omega, 2021, 6, 29009–29021 CrossRef CAS PubMed.
- H. Xu, L. Ma and Z. Jin, J. Energy Chem., 2018, 27, 146–160 CrossRef.
- X. Zhang, X. Liu, W. Zhang and Y. Song, Green Energy Environ., 2022, S2468025722000577 Search PubMed.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef PubMed.
- Q. Zhang and J. Guan, J. Power Sources, 2020, 471, 228446 CrossRef CAS.
- Y. Yuan, Z. Chen, H. Yu, X. Zhang, T. Liu, M. Xia, R. Zheng, M. Shui and J. Shu, Energy Storage Mater., 2020, 32, 65–90 CrossRef.
- Y. Zhang, Y. Zhang, H. Zhang, L. Bai, L. Hao, T. Ma and H. Huang, Coord. Chem. Rev., 2021, 448, 214147 CrossRef CAS.
- Z. Xu, M. M. Rahman, L. Mu, Y. Liu and F. Lin, J. Mater. Chem. A, 2018, 6, 21859–21884 RSC.
- F. Wei, Q. Zhang, P. Zhang, W. Tian, K. Dai, L. Zhang, J. Mao and G. Shao, J. Electrochem. Soc., 2021, 168, 050524 CrossRef CAS.
- M. K. Shobana, J. Alloys Compd., 2019, 802, 477–487 CrossRef CAS.
- R. Huang, X. Zhang, Z. Qu, X. Zhang, J. Lin, F. Wu, R. Chen and L. Li, J. Mater. Chem. A, 2022, 10, 682–689 RSC.
- X. Yao, Y. Ke, W. Ren, X. Wang, F. Xiong, W. Yang, M. Qin, Q. Li and L. Mai, Adv. Energy Mater., 2019, 9, 1803260 CrossRef.
- W. Zhang, J. Ming, W. Zhao, X. Dong, M. N. Hedhili, P. M. F. J. Costa and H. N. Alshareef, Adv. Funct. Mater., 2019, 29, 1903641 CrossRef.
- N. Khossossi, D. Singh, A. Ainane and R. Ahuja, Chem. – Asian J., 2020, 15, 3390–3404 CrossRef CAS PubMed.
- L. Han, X. Zhu, F. Yang, Q. Liu and X. Jia, Powder Technol., 2021, 382, 40–47 CrossRef CAS.
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2018, 8, 1703238 CrossRef.
- X. Zhao, Y. Ding, Q. Xu, X. Yu, Y. Liu and H. Shen, Adv. Energy Mater., 2019, 9, 1803648 CrossRef.
- X.-X. He, J.-H. Zhao, W.-H. Lai, R. Li, Z. Yang, C. Xu, Y. Dai, Y. Gao, X.-H. Liu, L. Li, G. Xu, Y. Qiao, S.-L. Chou and M. Wu, ACS Appl. Mater. Interfaces, 2021, 13, 44358–44368 CrossRef CAS PubMed.
- H. Chen, F. Guo, Y. Liu, T. Huang, B. Zheng, N. Ananth, Z. Xu, W. Gao and C. Gao, Adv. Mater., 2017, 29, 1605958 CrossRef PubMed.
- E. Olsson, G. Chai, M. Dove and Q. Cai, Nanoscale, 2019, 11, 5274–5284 RSC.
- F. A. Zamri and M. H. Abu Bakar, Mater. Today Commun., 2020, 25, 101641 CrossRef CAS.
- S. Debnath, M. Horscheck-Diaz, D. J. Searles and M. Hankel, Carbon, 2021, 183, 546–559 CrossRef CAS.
- J. Zheng, Z. Ren, P. Guo, L. Fang and J. Fan, Appl. Surf. Sci., 2011, 258, 1651–1655 CrossRef CAS.
- S. Yang, S. Li, S. Tang, W. Dong, W. Sun, D. Shen and M. Wang, Theor. Chem. Acc., 2016, 135, 164 Search PubMed.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 Search PubMed.
- A. S. Childress, P. Parajuli, J. Zhu, R. Podila and A. M. Rao, Nano Energy, 2017, 39, 69–76 Search PubMed.
- H. Zhang, Y. Yang, D. Ren, L. Wang and X. He, Energy Storage Mater., 2021, 36, 147–170 Search PubMed.
- H. Zhang, G. Guo, H. Adenusi, B. Qin, H. Li, S. Passerini and W. Huang, Mater. Today, 2022, 53, 162–172 CrossRef CAS.
- Y. Rangom, R. R. Gaddam, T. T. Duignan, Y. Wu, Z. Hu and X. S. Zhao, ACS Appl. Energy Mater., 2021, 4, 13272–13278 CrossRef CAS.
- Y. Li, Y. Lu, P. Adelhelm, M.-M. Titirici and Y.-S. Hu, Chem. Soc. Rev., 2019, 48, 4655–4687 RSC.
- E. Olsson, J. Cottom, H. Au, Z. Guo, A. C. S. Jensen, H. Alptekin, A. J. Drew, M. Titirici and Q. Cai, Adv. Funct. Mater., 2020, 30, 1908209 CrossRef CAS.
- E. Olsson, J. Cottom and Q. Cai, Small, 2021, 17, 2007652 CrossRef CAS PubMed.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- Z. Pei, Q. Meng, L. Wei, J. Fan, Y. Chen and C. Zhi, Energy Storage Mater., 2020, 28, 55–63 CrossRef.
- M.-C. Lin, M. Gong, B. Lu, Y. Wu, D.-Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B.-J. Hwang and H. Dai, Nature, 2015, 520, 324–328 CrossRef CAS PubMed.
- Y. Hai, W. Cui, Y. Lin, P. Han, H. Chen, Z. Zhu, C. Li, B. Yang, C. Zhu and J. Xu, Appl. Surf. Sci., 2019, 484, 726–731 CrossRef CAS.
- M. Dahbi, M. Kiso, K. Kubota, T. Horiba, T. Chafik, K. Hida, T. Matsuyama and S. Komaba, J. Mater. Chem. A, 2017, 5, 9917–9928 RSC.
- Y. Liu, B. V. Merinov and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3735–3739 CrossRef CAS PubMed.
- J. Zhou, W. Zhou, C. Guan, J. Shen, C. Ouyang, M. Lei, S. Shi and W. Tang, Sci. China: Phys., Mech. Astron., 2012, 55, 1376–1382 CrossRef CAS.
- C. Wang, J. Huang, J. Li, L. Cao, Q. Xi and S. Chen, J. Electroanal. Chem., 2020, 860, 113924 CrossRef CAS.
- J. Um, S. U. Yoon, H. Kim, B. S. Youn, H.-J. Jin, H.-K. Lim and Y. S. Yun, J. Energy Chem., 2022, 67, 814–823 CrossRef CAS.
- S. Ha, J. C. Hyun, J. H. Kwak, H.-D. Lim, B. S. Youn, S. Cho, H.-J. Jin, H.-K. Lim, S. M. Lee and Y. S. Yun, Chem. Eng. J., 2022, 437, 135416 CrossRef CAS.
- Y. Hu, C. Tang, H. Li, A. Du, W. Luo, M. Wu and H. Zhang, Chin. Chem. Lett., 2022, 33, 480–485 CrossRef CAS.
- M. Yang, Q. Kong, W. Feng, W. Yao and Q. Wang, Carbon Energy, 2022, 4, 45–59 CrossRef CAS.
- Y. Wei, B. Yang, S. Wang, D. Wu, S. Zhang, H. Chen and E. Ruckenstein, EcoMat, 2022, 4(1), e12150 CrossRef CAS.
- D. Er, E. Detsi, H. Kumar and V. B. Shenoy, ACS Energy Lett., 2016, 1, 638–645 CrossRef CAS.
- W. Chen, M. Wan, Q. Liu, X. Xiong, F. Yu and Y. Huang, Small Methods, 2019, 3, 1800323 CrossRef CAS.
- X. Sun, G.-P. Hao, X. Lu, L. Xi, B. Liu, W. Si, C. Ma, Q. Liu, Q. Zhang, S. Kaskel and O. G. Schmidt, J. Mater. Chem. A, 2016, 4, 10166–10173 RSC.
- D. Datta, J. Li and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 1788–1795 CrossRef CAS PubMed.
- X. Wang, Z. Zeng, H. Ahn and G. Wang, Appl. Phys. Lett., 2009, 95, 183103 CrossRef.
- S. Ullah, P. A. Denis and F. Sato, Appl. Mater. Today, 2017, 9, 333–340 CrossRef.
- M. Riyaz, S. Garg, N. Kaur and N. Goel, Comput. Theor. Chem., 2022, 1214, 113757 CrossRef CAS.
- W. Zhou, Y. Liu, H. Dai, X. Yuan, Y. Peng, W. Huang, L. Fu, Y. Zhu, Y. Wu and X. Wang, J. Solid State Electrochem., 2021, 25, 457–464 CrossRef CAS.
- R. F. Service, Science, 2018, 359, 1080–1081 CrossRef CAS PubMed.
- D. Wang, J. Liu, X. Bao, C. Qing, T. Zhu and H.-E. Wang, ACS Appl. Energy Mater., 2022, 5, 2573–2579 CrossRef CAS.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- G. Li, Z. Chen and J. Lu, Chem, 2018, 4, 3–7 CAS.
- N. Angulakshmi, R. B. Dhanalakshmi, S. Sathya, J. Ahn and A. M. Stephan, Batteries Supercaps, 2021, 4, 1064–1095 CrossRef CAS.
- T. Tao, S. Lu, Y. Fan, W. Lei, S. Huang and Y. Chen, Adv. Mater., 2017, 29, 1700542 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- H. R. Jiang, W. Shyy, M. Liu, Y. X. Ren and T. S. Zhao, J. Mater. Chem. A, 2018, 6, 2107–2114 RSC.
- Y. Chen, X. Gao, D. Su, C. Wang and G. Wang, Trends Chem., 2020, 2, 1020–1033 CrossRef CAS.
- Z. Yuan, H.-J. Peng, T.-Z. Hou, J.-Q. Huang, C.-M. Chen, D.-W. Wang, X.-B. Cheng, F. Wei and Q. Zhang, Nano Lett., 2016, 16, 519–527 CrossRef CAS PubMed.
- S. Qiu, X. Liang, S. Niu, Q. Chen, G. Wang and M. Chen, Nano Res., 2022, 15, 7925–7932 CrossRef CAS.
- C. Wang, J. Huang, L. Cao, J. Li, C. He, H. Li, J. Tian and K. Kajiyoshi, Carbon, 2021, 183, 899–911 CrossRef CAS.
- Y. Song, H. Gao, M. Wang, L. Chen, X. Cao, L. Song, X. Liu, W. Cai, J. Sun and W. Zhang, EcoMat, 2022, 4(3), e12182 CAS.
- Y. Zhang, G. Li, J. Wang, D. Luo, Z. Sun, Y. Zhao, A. Yu, X. Wang and Z. Chen, Adv. Energy Mater., 2021, 11, 2100497 CrossRef CAS.
- Y. Cui, J. Li, Y. Cai, H. Zhang and S. Zhang, Small, 2022, 18, 2204183 CrossRef CAS PubMed.
- L. Oakes, R. Carter and C. L. Pint, Nanoscale, 2016, 8, 19368–19375 RSC.
- J. Wang, J. Zhang, S. Duan, T. Li, L. Jia, H. Liu, L. Li, S. Cheng, H. Hu, M. Huang, H. Hu, S. Zhang, Q. Xiao and H. Lin, Chem. Eng. J., 2022, 429, 132352 CrossRef CAS.
- X. Wang, C. Zhao, B. Liu, S. Zhao, Y. Zhang, L. Qian, Z. Chen, J. Wang, X. Wang and Z. Chen, Adv. Energy Mater., 2022, 12, 2201960 CrossRef CAS.
- H. Zhao, B. Tian, C. Su and Y. Li, ACS Appl. Mater. Interfaces, 2021, 13, 7171–7177 CrossRef CAS PubMed.
- Y. Liu, Z. Wei, B. Zhong, H. Wang, L. Xia, T. Zhang, X. Duan, D. Jia, Y. Zhou and X. Huang, Energy Storage Mater., 2021, 35, 12–18 CrossRef.
- L. Fang, Z. Feng, L. Cheng, R. E. Winans and T. Li, Small Methods, 2020, 4, 2000315 CrossRef CAS.
- J. Wang, B. Ding, X. Lu, H. Nara, Y. Sugahara and Y. Yamauchi, Adv. Mater. Interfaces, 2021, 8, 2002159 CrossRef CAS.
- J. Wang, W. Qiu, G. Li, J. Liu, D. Luo, Y. Zhang, Y. Zhao, G. Zhou, L. Shui, X. Wang and Z. Chen, Energy Storage Mater., 2022, 46, 269–277 CrossRef.
- Y. Li, J. Wu, B. Zhang, W. Wang, G. Zhang, Z. W. Seh, N. Zhang, J. Sun, L. Huang, J. Jiang, J. Zhou and Y. Sun, Energy Storage Mater., 2020, 30, 250–259 CrossRef.
- J. Wang, L. Jia, S. Duan, H. Liu, Q. Xiao, T. Li, H. Fan, K. Feng, J. Yang, Q. Wang, M. Liu, J. Zhong, W. Duan, H. Lin and Y. Zhang, Energy Storage Mater., 2020, 28, 375–382 CrossRef.
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L.-J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed.
- S. Zhang, X. Ao, J. Huang, B. Wei, Y. Zhai, D. Zhai, W. Deng, C. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 9691–9698 CrossRef CAS PubMed.
- H. Shi, X. Ren, J. Lu, C. Dong, J. Liu, Q. Yang, J. Chen and Z. Wu, Adv. Energy Mater., 2020, 10, 2002271 CrossRef CAS.
- Y. Miao, Y. Zheng, F. Tao, Z. Chen, Y. Xiong, F. Ren and Y. Liu, Chin. Chem. Lett., 2022, S1001841722000304 Search PubMed.
- C. Wang, J. Huang, J. Li, L. Cao, R. Lang and K. Kajiyoshi, J. Nanostruct. Chem., 2023, 13, 67–68 CrossRef CAS.
- G. Liu, Q. Zeng, Z. Fan, S. Tian, X. Li, X. Lv, W. Zhang, K. Tao, E. Xie and Z. Zhang, Chem. Eng. J., 2022, 137683 CrossRef CAS.
- J. Lei, T. Liu, J. Chen, M. Zheng, Q. Zhang, B. Mao and Q. Dong, Chem, 2020, 6, 2533–2557 CAS.
- B. Wei, Y. Xia, S. Chen and H. Wang, Electroanalysis, 2022, elan.202200003 Search PubMed.
- P. Wang, B. Xi, Z. Zhang, M. Huang, J. Feng and S. Xiong, Angew. Chem., Int. Ed., 2021, 60, 15563–15571 CrossRef CAS PubMed.
- Y. Zhang, S. Pal, Y. Yang, C. Huang, J. Wang, P. Apostol, Z. Liu, A. Hu, A. Vlad and X. Chen, Batteries Supercaps, 2022, 5(8), e202200124 CAS.
- Z. Song, X. Lu, Q. Hu, J. Ren, W. Zhang, Q. Zheng and D. Lin, J. Power Sources, 2019, 421, 23–31 CrossRef CAS.
- T. Ogasawara, A. Débart, M. Holzapfel, P. Novák and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390–1393 CrossRef CAS PubMed.
- A. Debart, J. Bao, G. Armstrong and P. G. Bruce, ECS Trans., 2007, 3, 225–232 CrossRef CAS.
- Q. Wu, J. Gao, J. Feng, Q. Liu, Y. Zhou, S. Zhang, M. Nie, Y. Liu, J. Zhao, F. Liu, J. Zhong and Z. Kang, J. Mater. Chem. A, 2020, 8, 1205–1211 RSC.
- J. Mao, P. Liu, J. Li, J. Yan, S. Ye and W. Song, J. Energy Chem., 2022, S2095495622002479 Search PubMed.
- S. Lu and Z. Zhuang, Sci. China Mater., 2016, 59, 217–238 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- D. Cao, Y. Bai, J. Zhang, G. Tan and C. Wu, Nano Energy, 2021, 89, 106464 CrossRef CAS.
- J. Zhang, J. Zhang, F. He, Y. Chen, J. Zhu, D. Wang, S. Mu and H. Y. Yang, Nano-Micro Lett., 2021, 13, 65 CrossRef PubMed.
- X. Zhao, H. Zhao, T. Zhang, X. Yan, Y. Yuan, H. Zhang, H. Zhao, D. Zhang, G. Zhu and X. Yao, J. Mater. Chem. A, 2014, 2, 11666–11671 RSC.
- X. Yan, H. Liu, Y. Jia, L. Zhang, W. Xu, X. Wang, J. Chen, D. Yang and X. Yao, Cell Rep. Phys. Sci., 2020, 1, 100083 CrossRef.
- Q. Lai, J. Zheng, Z. Tang, D. Bi, J. Zhao and Y. Liang, Angew. Chem., Int. Ed., 2020, 59, 11999–12006 CrossRef CAS PubMed.
- Y. Wang, M. Yu, T. Zhang, Z. Xue, Y. Ma and H. Sun, Catal. Sci. Technol., 2022, 12, 332–338 RSC.
- X. Zhang, Q. Zhang, J. Cui, J. Yan, J. Liu and Y. Wu, Nanoscale, 2022, 14, 3212–3223 RSC.
- L. Zhang, Q. Xu, J. Niu and Z. Xia, Phys. Chem. Chem. Phys., 2015, 17, 16733–16743 RSC.
- H. B. Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, e1501122 CrossRef PubMed.
- B. Huang, K. Hou, Y. Liu, R. Hu and L. Guan, J. Energy Chem., 2021, 63, 521–527 CrossRef CAS.
- D. Li, Y. Jia, G. Chang, J. Chen, H. Liu, J. Wang, Y. Hu, Y. Xia, D. Yang and X. Yao, Chem, 2018, 4, 2345–2356 CAS.
- J. Lei, K. Wang, B. Deng, Y. Li, S. Zhang and Y. Cao, J. Alloys Compd., 2022, 914, 165359 CrossRef CAS.
- J. Yu, Y. Dai, Z. Zhang, T. Liu, S. Zhao, Q. He, P. Tan, Z. Shao and M. Ni, Chem. Eng. Sci., 2022, 259, 117816 CrossRef CAS.
- Y. Tian, M. Li, Z. Wu, Q. Sun, D. Yuan, B. Johannessen, L. Xu, Y. Wang, Y. Dou, H. Zhao and S. Zhang, Angew. Chem., Int. Ed., 2022, 61(51), e202213296 CAS.
- Q. Wang, R. Lu, Y. Yang, X. Li, G. Chen, L. Shang, L. Peng, D. Sun-Waterhouse, B. C. C. Cowie, X. Meng, Y. Zhao, T. Zhang and G. I. N. Waterhouse, Sci. Bull., 2022, S2095927322001803 Search PubMed.
- S. Ozden, S. Bawari, S. Vinod, U. Martinez, S. Susarla, C. Narvaez, J. Joyner, C. S. Tiwary, T. N. Narayanan and P. M. Ajayan, Nanoscale, 2019, 11, 12489–12496 RSC.
- Z. Zhang, Z. Yi, J. Wang, X. Tian, P. Xu, G. Shi and S. Wang, J. Mater. Chem. A, 2017, 5, 17064–17072 RSC.
- Y. Li, C. Ai, S. Deng, Y. Wang, X. Tong, X. Wang, X. Xia and J. Tu, Mater. Res. Bull., 2021, 134, 111094 CrossRef CAS.
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed.
- Y. Tian, Y. Ye, X. Wang, S. Peng, Z. Wei, X. Zhang and W. Liu, Appl. Catal., A, 2017, 529, 127–133 CrossRef CAS.
- X. Li, X. Duan, C. Han, X. Fan, Y. Li, F. Zhang, G. Zhang, W. Peng and S. Wang, Carbon, 2019, 148, 540–549 CrossRef CAS.
- Y. Zhang, S. Yun, M. Sun, X. Wang, L. Zhang, J. Dang, C. Yang, J. Yang, C. Dang and S. Yuan, J. Colloid Interface Sci., 2021, 604, 441–457 CrossRef CAS PubMed.
- Y. Ito, W. Cong, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 2131–2136 CrossRef CAS PubMed.
- J. Lin, P. Wang, H. Wang, C. Li, X. Si, J. Qi, J. Cao, Z. Zhong, W. Fei and J. Feng, Adv. Sci., 2019, 6, 1900246 CrossRef PubMed.
- Y. Yan, J. Lin, T. Xu, B. Liu, K. Huang, L. Qiao, S. Liu, J. Cao, S. C. Jun, Y. Yamauchi and J. Qi, Adv. Energy Mater., 2022, 12, 2200434 CrossRef CAS.
- S. Liu, L. Kang and S. C. Jun, Adv. Mater., 2021, 33, 2004689 CrossRef CAS PubMed.
- S. Liu, L. Kang, J. Henzie, J. Zhang, J. Ha, M. A. Amin, M. S. A. Hossain, S. C. Jun and Y. Yamauchi, ACS Nano, 2021, 15, 18931–18973 CrossRef CAS PubMed.
- Y. Chen, R. Ding, J. Li and J. Liu, Appl. Catal., B, 2022, 301, 120830 CrossRef CAS.
- X. Shen, W. Tan, Z. Wei, X. Wang, H. Zhou, J. Lv, M. Zhao, M. Zhang, J. Yang, G. He and L. Yang, J. Mater. Sci.: Mater. Electron., 2021, 32, 22974–22983 CrossRef CAS.
- M. Liu, M. Liu, X. Wang, S. M. Kozlov, Z. Cao, P. De Luna, H. Li, X. Qiu, K. Liu, J. Hu, C. Jia, P. Wang, H. Zhou, J. He, M. Zhong, X. Lan, Y. Zhou, Z. Wang, J. Li, A. Seifitokaldani, C. T. Dinh, H. Liang, C. Zou, D. Zhang, Y. Yang, T.-S. Chan, Y. Han, L. Cavallo, T.-K. Sham, B.-J. Hwang and E. H. Sargent, Joule, 2019, 3, 1703–1718 CrossRef CAS.
- L. Ling, L. Jiao, X. Liu, Y. Dong, W. Yang, H. Zhang, B. Ye, J. Chen and H. Jiang, Adv. Mater., 2022, 34, 2205933 CrossRef CAS PubMed.
- J. Xu, Y. Kan, R. Huang, B. Zhang, B. Wang, K.-H. Wu, Y. Lin, X. Sun, Q. Li, G. Centi and D. Su, ChemSusChem, 2016, 9, 1085–1089 CrossRef CAS PubMed.
- X. Rong, H. Wang, X. Lu, R. Si and T. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CrossRef CAS PubMed.
- W. Ni, Z. Liu, Y. Zhang, C. Ma, H. Deng, S. Zhang and S. Wang, Adv. Mater., 2021, 33, 2003238 CrossRef CAS PubMed.
- M. Chen, S. Wang, H. Zhang, P. Zhang, Z. Tian, M. Lu, X. Xie, L. Huang and W. Huang, Nano Res., 2020, 13, 729–735 CrossRef CAS.
- P. Jiang, K. Jiang, D. Tranca, J. Zhu, F. Qiu, C. Ke, C. Lu, E. Kymakis and X. Zhuang, Adv. Mater. Interfaces, 2021, 8, 2100051 CrossRef CAS.
- Y. Dong, Q. Zhang, Z. Tian, B. Li, W. Yan, S. Wang, K. Jiang, J. Su, C. W. Oloman, E. L. Gyenge, R. Ge, Z. Lu, X. Ji and L. Chen, Adv. Mater., 2020, 32, 2001300 CrossRef CAS PubMed.
- J. Han, X. Deng, K. Chen, S. Imhanria, Y. Sun and W. Wang, Renewable Energy, 2021, 177, 636–642 CrossRef CAS.
- Z. Liu, M. Wang, X. Luo, S. Li, S. Li, Q. Zhou, W. Xu and R. Wu, Appl. Surf. Sci., 2021, 544, 148912 CrossRef CAS.
- S. Dilpazir, R. Liu, M. Yuan, M. Imran, Z. Liu, Y. Xie, H. Zhao and G. Zhang, J. Mater. Chem. A, 2020, 8, 10865–10874 RSC.
| This journal is © The Royal Society of Chemistry 2023 |