
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRelation among absorbance shifts, mineralization morphology, and electronic conductivity of π-peptide aggregates with different amino acid residues†
Taein
Lee
a,
Sayak Subhra
Panda
b,
Grant E. K.
Hall
b,
Yunjia
Song
a,
John D.
Tovar
 ab and
Howard E.
Katz
*ab
ab and
Howard E.
Katz
*ab
aDepartment of Materials Science and Engineering, Johns Hopkins University, 3400 North Charles Street, Baltimore, 21218, MD, USA. E-mail: hekatz@jhu.edu
bDepartment of Chemistry, Johns Hopkins University, 3400 North Charles Street, Baltimore, 21218, MD, USA
First published on 29th March 2023
Abstract
Previously, the structures of self-assembled π-peptide aggregates, upon shifting from basic to acidic medium, have been shown computationally to vary greatly when the closest amino acid to the π-cores are varied between glycine and alanine. The absorbance spectra of π-peptide aggregates with different self-assembled structures showed different spectral shifts between acidic and basic medium. The relation of the absorbance shifts to various material properties of solid-state thin films made from the π-peptides by exposing basic medium to acid vapor were investigated for two types of π-cores: α-quaterthiophene (4T) and perylene diimide (PDI). π-Peptide nanostructure-templated mineralization of KCl was used as an indicator of structural morphology, which was further connected to electronic properties of the thin films. Thin films of π-peptides with 4T cores showed higher conductivity for samples with conspicuous KCl XRD peaks, which corresponded to samples with greater absorbance spectra shift from basic to acidic medium. Thin films of π-peptides with PDI cores that have glycine amino acids closest to the π-cores were previously shown by computational molecular dynamics simulation to exhibit co-facial stacking, while those with alanine amino acids closest to the π-cores were shown to exhibit rotationally shifted stacking. Self-assembled thin films of co-facially stacked π-peptides exhibited templated mineralization of dendritic KCl structures and good surface adhesion between thin film and the underlying substrate. On the other hand, those of rotationally shifted stacking π-peptides exhibited sporadic spots of bulk mineralized KCl and extreme brittleness of thin film due to poor adhesion with the underlying substrate. The observed brittleness in all rotationally shifted stacking π-peptide thin films made it impossible to accurately measure electrical properties, while co-facially stacked π-peptide thin films showed notable electrical conductivity.
Introduction
π-Conjugated moieties flanked with peptide wings can be self-assembled using a pH trigger to create supramolecular electronic materials.1–7 These peptide-flanked cores can stack to form supramolecular structures that assemble further to form aggregates with well-defined hierarchical structures based on multiple noncovalent interactions.8–15 Intermolecular electrostatic interactions (hydrogen bonding, van der Waals forces, etc.) have a profound effect on these supramolecular architectures.1,4,5,15 These interactions can be tuned at the molecular level either by varying the peptide sequences or the π chromophores to control the stacking properties of the π-peptide molecules.13–23 The π–π stacking properties, along with other noncovalent interactions, are integral to the supramolecular structure of the π-peptides, dictating nucleation and growth, and charge transport.17,18,20,21,24–26 Optoelectronic signals in solution and in solid forms,27,28 also tuned by the π–chromophore and peptide side chains, are shown to be related to the structures of these π–peptides.15,22,23,25,29,30 Electronic materials made of these supramolecular architectures are easily tunable, with small changes in π-peptide building blocks leading to significant changes in macrostructure and electronic properties.14,15,17,19,20,22,23,29,31,32 Therefore, controlling the stacking properties of these π-peptides may enable highly efficient and stable π-conjugated peptide materials to be used to address biological systems with electronic or optical signals, or to stimulate templated mineral growth.16–18,33 As will be seen below, the mineralization phenomenon was useful in the present work as an indicator of stacking morphology that also relates to conductivity in the π-peptide aggregates.Solid-phase peptide synthesis (SPPS) makes altering of the peptide side chains facile in order to control acidity, hydrophobicity, and polarity within the resulting macromolecule. Similar to varying peptide chains to alter properties of peptide nanomaterial thin films, modifying the alkyl spacer length in between the amino acid and central π-chromophores has led to variations in supramolecular aggregation.31 The chirality of the amino acids is also known to be another factor to tune the supramolecular arrangement of the cores to affect its electrical properties.23,32,34 Altering the central π-chromophores can also lead to changes in the stacking parameters, resulting in different material properties.8,25,35,36 In this manuscript, we focus on the change in morphological and electrical properties of solid-state thin film devices by altering the amino acids on side chains of two central π-chromophores. We focus on α-quaterthiophene (4T), a hole-transporting subunit, as the principal core of interest, and show that similar connections among absorbance spectroscopy, assembly morphology, and electrical conductivity are also obtained in a companion pair of nominally electron-transporting perylene diimide (PDI) peptides. The 4T π–chromophore was chosen for its demonstrated capability to form hole-transporting and fluorescent solids, as well as previous measurements of relatively high charge mobilities in oligopeptides with 4T conjugates.18,37,38 The PDI π chromophores were chosen for their high electron affinity and relatively efficient charge transport properties useful in OFETs, solar cells and light-emitting diodes.29,39 Electronic conductivity behavior of π-peptide aggregates may also have potential applications in electromagnetic nanocomposites.30,40,41
Computational guidance
Previously, Shmilovich et al.42 described how active learning computational modeling was used to track the changes in supramolecular structures of the cores for varying amino acid side chains with four p-type thiophene oligomers43 and two n-type arylenediimide cores for amino acid side chains with four different spacer units. Also, the molecular simulation trajectories for pH shifts from basic to acidic medium of 4T with 28 π-peptide side chains were acquired, yielding an average number of contacts (κ) and a radius of gyration (Rg) for each molecule.43 The same 28 π-peptide molecules’ self-assembly via pH shift from basic to acidic were tested experimentally for the spectral blue shift of the absorption maxima (λ) via UV-vis spectroscopy.43 Shmilovich et al.42 reported that, consistent with previous analysis,29 small non-polar residues adjacent to the π-core promote good regular and tightly packed stacking properties (large number of intermolecular contacts, larger blue shifts of UV-vis spectra on association) of the cores. Further, larger hydrophobic and bulky residues in any position of the amino acid side chains resulted in poor stacking properties from computational simulations as well as optoelectronic measurements. 4T was also associated with a high transfer integral in assemblies.42 Based on these findings, three π-peptide side chains were chosen from the pool of 28 molecules with 4T central π-chromophores: DGG-4T, DGA-4T, and DLAG-4T (Fig. 1). These molecules were chosen because they all had small non-polar residues adjacent to the π-core, while DLAG-4T contained a bulky hydrophobic aromatic residue in the amino acid side chain. Solid-state thin films of these π-peptides were explored to connect optoelectronic properties to morphologies and electrical conductivities.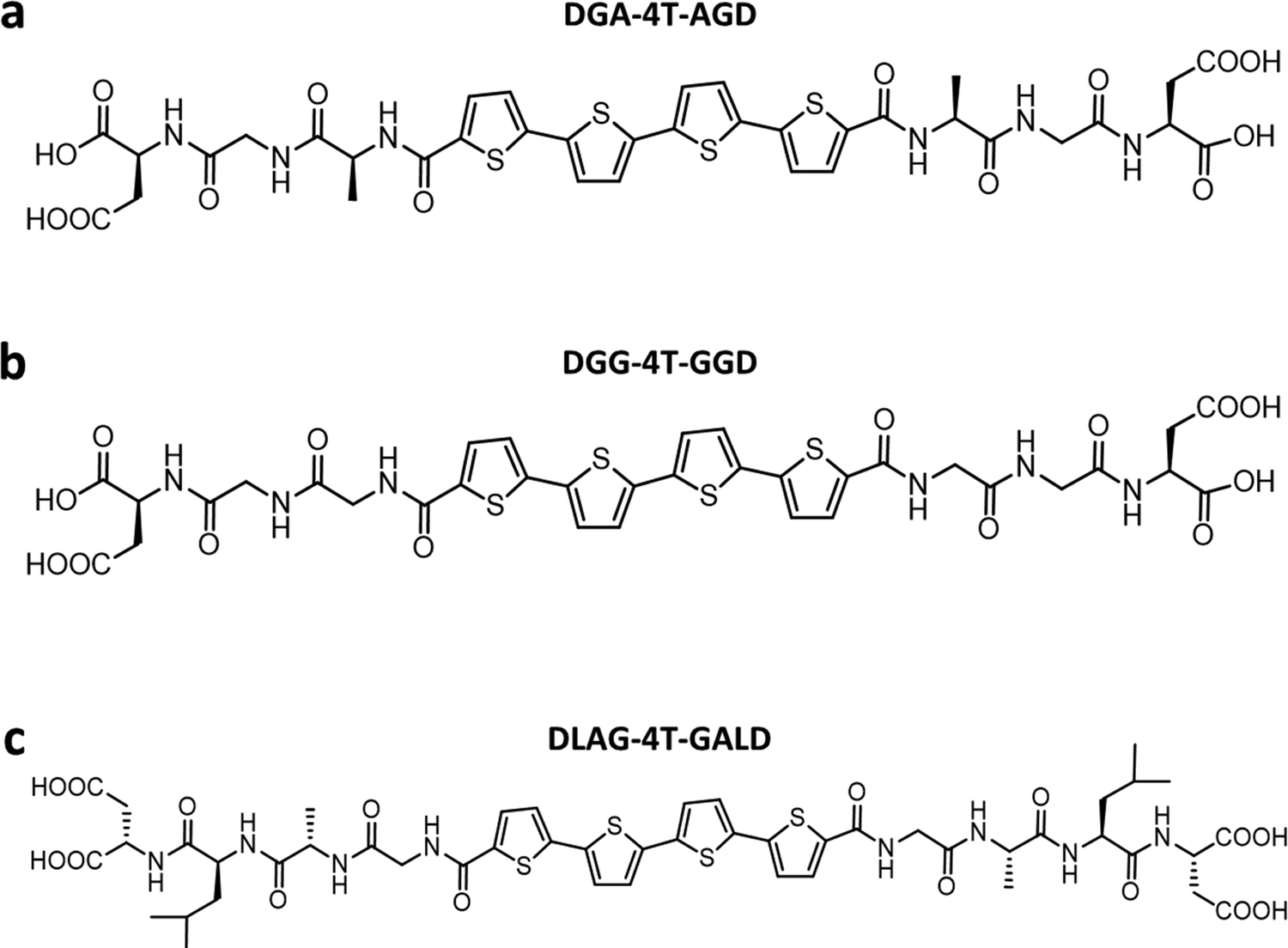 | ||
| Fig. 1 Chemical structures of (a) DGA-4T, (b) DGG-4T, and (c) DLAG-4T. | ||
We took advantage of a related investigation on the n-type PDI conjugated core system, which showed low reorganization energy compared to the naphthalenediimide core,42 to demonstrate the generality of the observed trends. Panda et al. reported computationally guided tuning of the peptidic perylene diimide (PDI) self-assembly.29 Two different self-assembly architectures were identified via computational modeling for the peptide-PDIs in acidic environments.29 The self-assembly architectures varied according to a change in the closest amino acid to the π core, especially by alternating between glycine and alanine.29,43 The different architectures anticipated by computational modeling were verified in solution via UV-vis, photoluminescence, and circular dichroism spectroscopies.43 Based on computational molecular dynamics simulation, the smaller size of glycine adjacent to the PDI π core prompted promotion of co-facial stacking, while the slightly larger alanine adjacent to the PDI π core prompted rotationally shifted stacking.29 Computational models anticipating the different stacking sequences showed different twist angles between DAVG and DAIA amino acid side chains with PDI cores as well as between VEVAG and VEVAA amino acid side chains with PDI cores (Fig. 2).29 Thus, altering the amino acid closest to the central PDI chromophores resulted in different twist angles, leading to different stacking parameters of the central π-chromophores.
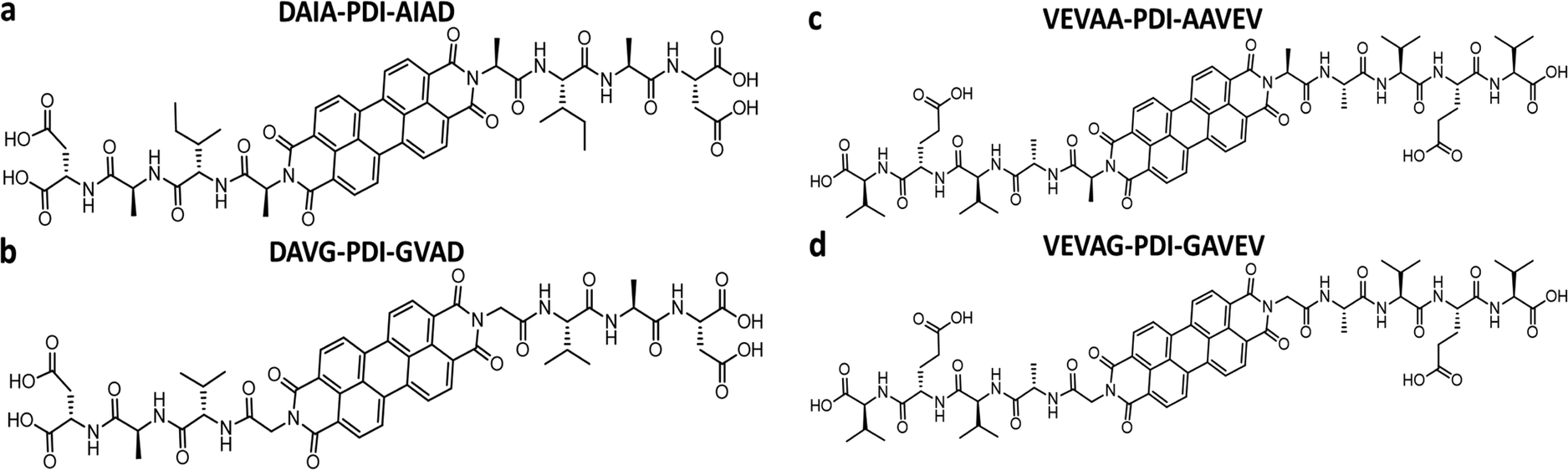 | ||
| Fig. 2 Chemical structures of (a) DAIA-PDI, (b) DAVG-PDI, (c) VEVAA-PDI, and (d) VEVAG-PDI. | ||
Experimental methods
Peptide synthesis
DLAG-4T, DGA-4T, DGG-4T peptides were prepared according to previously reported protocols using SPPS.43 VEVAG-PDI, VEVAA-PDI, DAVG-PDI, and DAIA-PDI were also prepared according to previously reported protocols using SPPS.29UV-vis spectroscopy
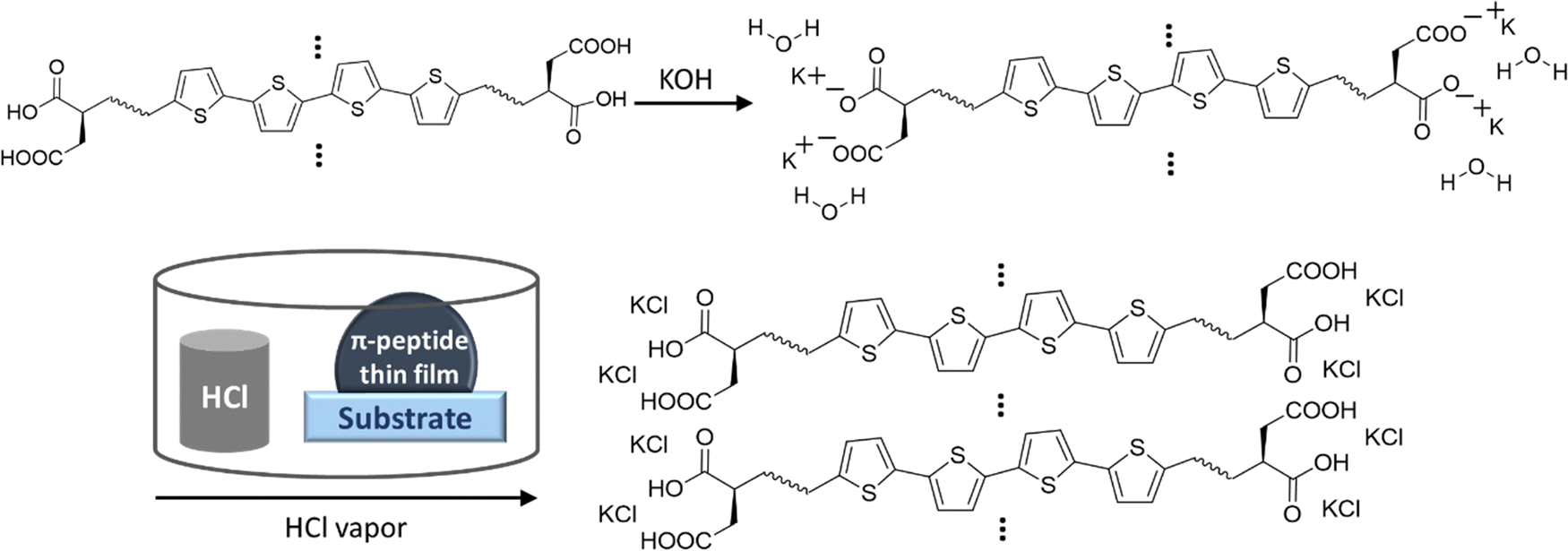
UV-vis spectra were obtained using a Varian Cary 50 Bio UV-vis spectrophotometer. Spectroscopic samples were prepared by diluting the peptide solution to the appropriate concentration in Millipore water to achieve an optical density near 0.1 in the monomeric/basic solution. In the case of 4T peptides, the pH was then adjusted by adding 15 μL of 1 M KOH (basic) followed by addition of 30 μL of 1 M HCl (acidic). The approximate concentration of the peptides was ∼2.25 μM for 4T peptides. In the case of PDI peptides, pH was adjusted by adding 15 μL of 1 M KOH for basic solutions and 40 μL of 1 M HCl for acidic solutions. The approximate concentration of the PDI peptides was in the range of 5–7.5 μM. The pH of the basic solutions was ∼12, whereas pH of the acidic solutions was ∼2.Solid-state device fabrication. Si/SiO2 substrates from University Wafers with a diameter of 150 mm with 300 nm SiO2 were cleaned using the following procedures: immersion in piranha solution with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of sulfuric acid to hydrogen peroxide for 1 day (Caution—Highly Corrosive and Toxic), rinsing in DI running water for 10 minutes, sonication in acetone for 10 minutes, rinsing in DI running water for 10 minutes, and sonication in propanol for 10 minutes. We dissolved the peptides in Millipore water to the concentration of 10 mg mL−1 (1 wt%) based on our previously published reports,44,45 and we also prepared 1 M KOH solution in Millipore water. We added the appropriate amount of KOH from the 1 M KOH solution to the 10 mg mL−1 peptide solutions to form the final molar ratio of ∼1:1.5 for peptide:KOH. The peptide solution presumably turned basic due to the addition of KOH. We drop-casted the basic peptide solution onto the cleaned substrates and immediately exposed it to HCl vapor in an inverted cylindrical glass beaker (170 mm diameter, 90 mm height) for 5 minutes. After HCl vapor chamber exposure, the π-peptide solutions were left to dry into solid-state films while undergoing self-assembly and templating mineralization of KCl overnight for 1 day. The drop-casting and drying of the samples were performed at room temperature in a well-ventilated space. 50 nm of Au was thermally evaporated in an Edwards 306 auto evaporator at a rate of 0.4 Å s−1 over the solids using a 200 mesh TEM grid patterned mask to make top gold electrodes. Note that this vacuum deposition would also complete the removal of any residual water not already removed by the air-drying. These procedures are outlined in Fig. 3.
:1 ratio of sulfuric acid to hydrogen peroxide for 1 day (Caution—Highly Corrosive and Toxic), rinsing in DI running water for 10 minutes, sonication in acetone for 10 minutes, rinsing in DI running water for 10 minutes, and sonication in propanol for 10 minutes. We dissolved the peptides in Millipore water to the concentration of 10 mg mL−1 (1 wt%) based on our previously published reports,44,45 and we also prepared 1 M KOH solution in Millipore water. We added the appropriate amount of KOH from the 1 M KOH solution to the 10 mg mL−1 peptide solutions to form the final molar ratio of ∼1:1.5 for peptide:KOH. The peptide solution presumably turned basic due to the addition of KOH. We drop-casted the basic peptide solution onto the cleaned substrates and immediately exposed it to HCl vapor in an inverted cylindrical glass beaker (170 mm diameter, 90 mm height) for 5 minutes. After HCl vapor chamber exposure, the π-peptide solutions were left to dry into solid-state films while undergoing self-assembly and templating mineralization of KCl overnight for 1 day. The drop-casting and drying of the samples were performed at room temperature in a well-ventilated space. 50 nm of Au was thermally evaporated in an Edwards 306 auto evaporator at a rate of 0.4 Å s−1 over the solids using a 200 mesh TEM grid patterned mask to make top gold electrodes. Note that this vacuum deposition would also complete the removal of any residual water not already removed by the air-drying. These procedures are outlined in Fig. 3.
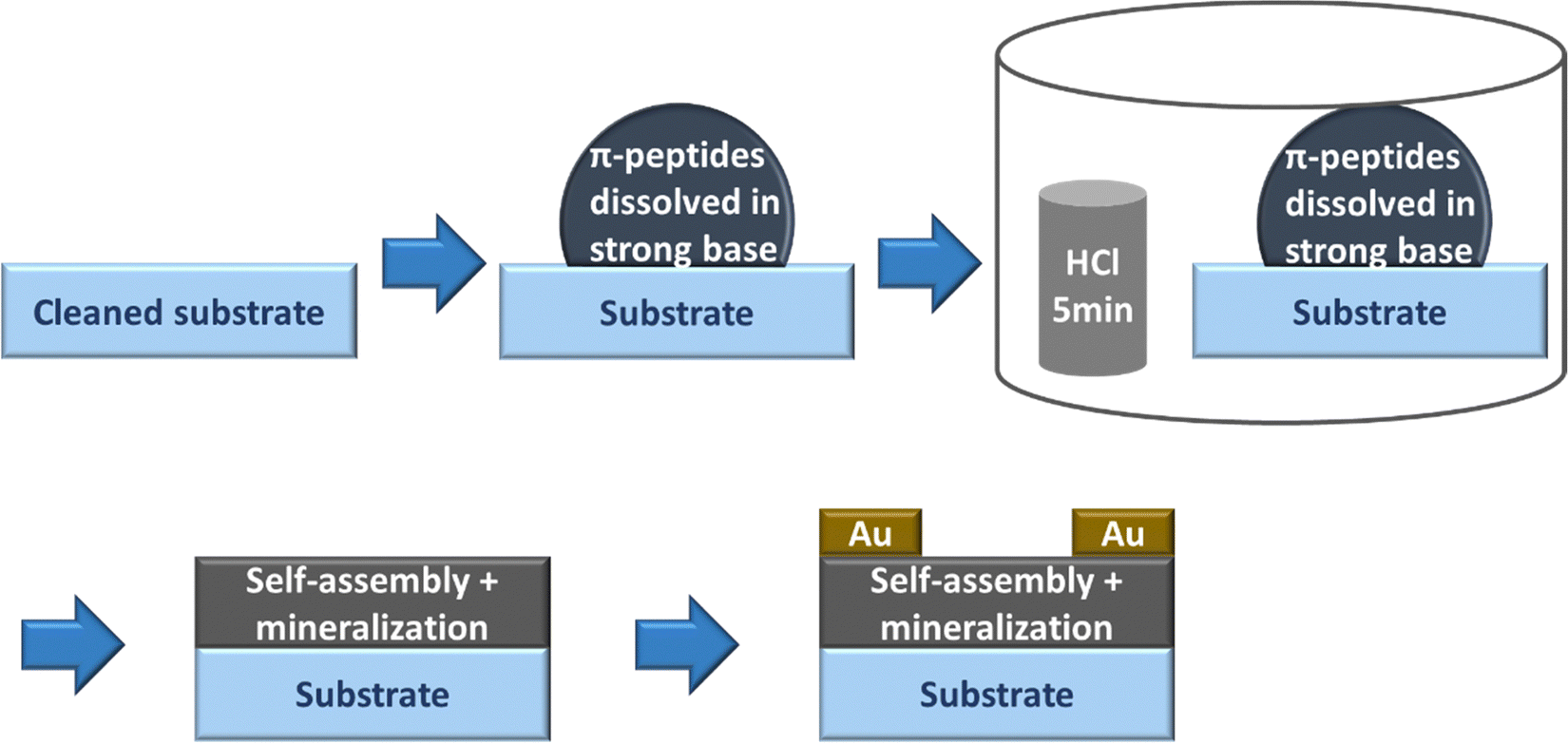 | ||
| Fig. 3 Schematic of the procedure to obtain π-peptide solid-state thin films on Si substrates. | ||
X-ray diffraction (XRD) characterization of the thin films was performed using a Bruker D8 Focus diffractometer (λ = 1.5424 Å) with a LynxEye detector. Both Cuk-α1 and 2 were used for the XRD with sample to detector distance 200.5 mm and a linear PSD. Scans were performed at 0.1 s per step in 0.01 degrees increments from 5 to 60 2 theta degrees for all XRD measurements. Separate sets of samples were stored for one month or three months in closed containers in air before measurement.
3D laser optical scanning microscopy
All optical, laser, and 3D microscope images as well as roughness profile measurements were acquired using a Keyence VK-X 100 laser microscope.Scanning electron microscopy/energy dispersive X-ray spectroscopy (SEM/EDS)
All samples were coated with 5–7 nm of Cr using a thermal evaporator before SEM/EDS to prevent burning of the organic solid-state thin films from the electron beam in the SEM. All Cr coated samples were observed under a Tescan MIRA 3 GMU for SEM images at 15–20 keV with working distances between 5 and 20 mm. All EDS data were acquired using an AMATEK EDAX Octane Plus.Atomic force microscopy (AFM)
A Dimension 3100 AFM (Bruker Nano, Santa Barbara, CA) was used for the AFM measurements. The measurements were run in tapping mode in air.Results and discussion
UV-vis absorbance spectra of peptides dissolved in basic solution by deprotonation of carboxylic acid groups and peptides self-assembled in acidic solution via re-protonation of carboxylic acid groups were measured. The absorbance changes observed between the different pH levels represent structural and orientational reorganization of the peptides upon self-assembly induced by noncovalent interactions promoted in acidic media. These noncovalent interactions include π–π stacking interactions between the central π chromophores coupled with interactions between amino acid side chains. Significant change in UV-vis spectroscopy was observed when varying the amino acid closest to the central π–chromophore leading to varying degrees of UV-vis peak shift or totally different peak positions resulting from different types of stacking parameters.The optoelectronic characteristics of each peptide were related to macroscopic structural morphologies obtained via 3D laser optical microscopy SEM/EDS, XRD, and to electrical properties, by fabricating devices comprising gold source/drain electrodes. Analysis of the π–chromophore assembly-templated KCl mineralization can be related to the morphologies of the deposited assemblies. Analysis of the templated growth of KCl minerals provides an innovative method to analyze and track peptide–π-peptide molecule self-assembly.
Relation among absorbance signatures, morphologies, and conductivities for 4T peptides
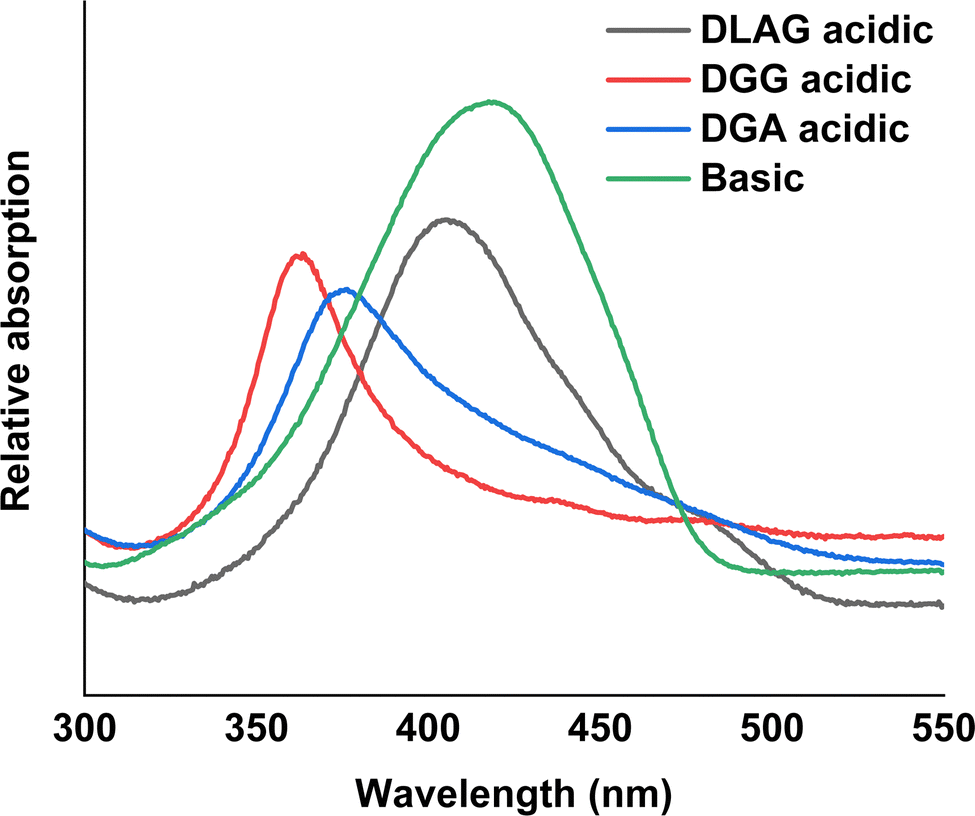 | ||
| Fig. 4 UV-vis spectroscopy measurements in basic and acidic medium for DLAG-4T, DGG-4T, and DGA-4T. | ||
It has been reported previously that larger peak shifts from the observed absorption maxima in basic conditions correlate to better co-facial stacking in self-assembled nanoaggregates.40 To investigate how optoelectrical signals evolve in solid-state assemblies, basic solutions were acidified in an HCl vapor-filled enclosed chamber (Fig. 3).
 | ||
| Fig. 5 Holistic view images via laser optical microscope for (a) DGG-4T, (b) DGA-4T, right DLAG-4T. The first two, DGG-4T and DGA-4T showed thicker, more definite aggregates. | ||
XRD showed KCl peaks for all three DGG-4T/DGA-4T/DLAG-4T molecules. The KCl was made upon exposure to HCl because KOH was used as the base additive to dissolve the molecules in distilled water. Since the KOH base is used to dissolve the peptides in solution, the K+ ions function as counter ions to the COO− groups in the deprotonated aspartic acid (D amino acid) and therefore are in close proximity to the π-peptides. Hence, the KCl crystal growth resulting from exposure to HCl vapor marks regions of self-assembly as shown in Scheme 1.
 | ||
| Scheme 1 Schematic of localization of K+ ions during dissolving of peptides and formation of KCl in close proximity to peptide aggregates upon HCl vapor exposure. | ||
KCl XRD peaks for the mineral grown on the aggregates were slightly shifted to a higher two-theta angle than that of pure KCl synthesized from dropping KOH solution on substrates and exposing it to HCl in the same enclosed vapor chamber as shown in Fig. 6. Pure KCl has an XRD peak around 28.2°, representing the (200) plane, while all DGG-4T/DGA-4T/DLAG-4T peptide assemblies had KCl peaks around 28.6°. The peak shifts in XRD for this case most likely occur because of changed lattice parameters due to lattice strain or change in d-spacing.46 Generally, introduction of lattice strain that decreases lattice parameter or d-spacing shifts XRD peaks of two-theta angles higher.46 In this case, it is likely that both occurred simultaneously as peptide self-assembly templated KCl mineralization resulted in strained KCl lattice parameters, as K+ functioned as counter ions in close proximity to the COO− groups in the aspartate amino acid residues. The peptide stacking during self-assembly therefore affected the K+ ions to be placed under influence of the stacking distance during mineralization to KCl. The degree of this influence would be related based on the difference in the peptidic supramolecular orientation and the rate of KCl mineralization.
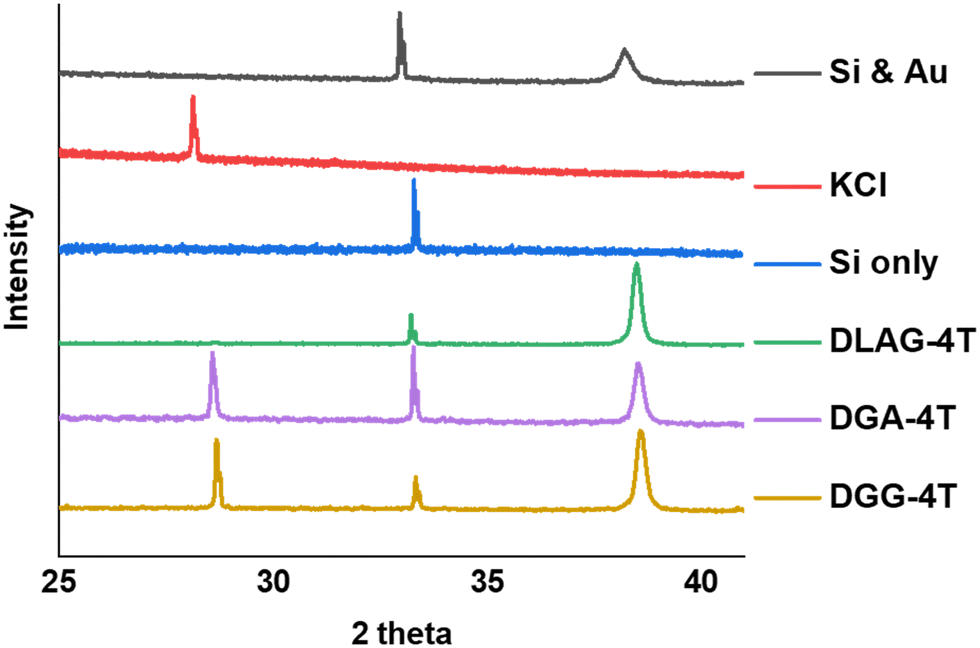 | ||
| Fig. 6 XRD measurements for DGG-4T, DGA-4T, DLAG-4T, Si only and KCl only. DLAG barely makes crystalline planes of KCl—there is a very small peak around 28 two-theta degrees (Fig. S7, ESI†). | ||
The degree of co-facial aggregation upon acidification was expected to be highest for DGG-4T, then DGA-4T, and much lower for DLAG-4T based on UV-vis spectroscopy discussed above. This is also portrayed in XRD data showing KCl peaks being more distinct for DGG-4T and DGA-4T samples whereas DLAG-4T showed almost no KCl peaks as there was negligible peptide aggregation in either basic or acidic media. These observations were made on samples stored for both one and three months before measurement. The strongest KCl XRD peak for DGG-4T and DGA-4T corresponded to most atoms in the (200) plane. Considering this is a specific peak for KCl and that DGG-4T does not generally self-assemble into distinctive crystal planes via 3D supramolecular assembly, the conspicuous (200) peak implies that more (200) plane KCl mineralized in DGG-4T and DGA-4T relative to DLAG-4T. This means that because DGG-4T/DGA-4T peptides undergo greater degree of aggregation via self-assembly upon exposure to HCl, more K+ coupled to COO− concentrated in the aggregation sites compared to DLAG-4T, leading to more concentrated templated growth of KCl. The opposite phenomenon happens for DLAG-4T, which had smaller and more distributed KCl crystal growth whose (200) plane reflections were too small to be detected significantly by X-rays from the XRD as shown in Fig. S8 (ESI†). Aspartic acid (D) being slightly further from the 4T central chromophores for DLAG-4T compared to DGA-4T and DGG-4T may have additionally impacted the structural orientation of peptides as well as the ensuing electrical properties.
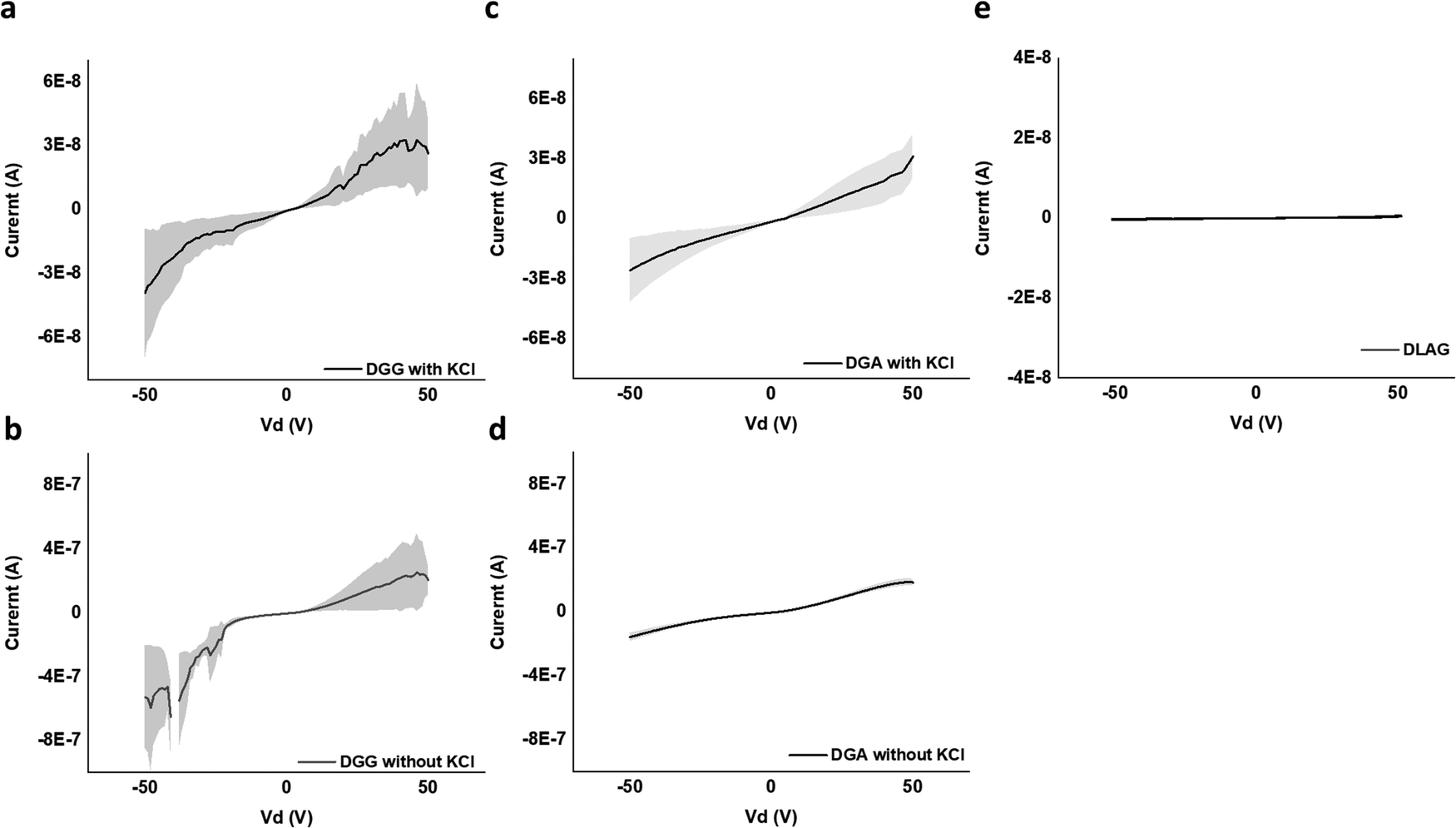 | ||
| Fig. 7 2 terminal IV curve for regions of (a) DGG-4T with KCl at that device, (b) DGG-4T without KCl at that device, (c) DGA-4T with KCl at that device, (d) DGA-4T without KCl at that device, and (e) DLAG-4T—no current whether or not KCl was present. | ||
Ohm's Law was used to calculate conductivity from linear regions of IV plots from electrode pairs contacting aggregates without KCl as follows (eqn (1))
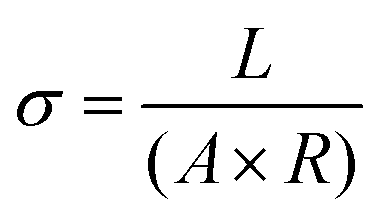 | (1) |
| V (V) | I (A) | R (Ohm) | L (cm) | A (cm2) | σ (S cm−1) | |
|---|---|---|---|---|---|---|
| DGG-4T | 50 | 2.1 × 10−7 ± 9.3 × 10−8 | 2.4 × 108 | 3.8 × 10−3 | 9.0 × 10−7 | 1.8 × 10−5 ± 7.8 × 10−6 |
| DGA-4T | 50 | 1.9 × 10−7 ± 2.0 × 10−8 | 2.7 × 108 | 3.8 × 10−3 | 9.0 × 10−7 | 1.5 × 10−5 ± 1.7 × 10−6 |
| DLAG-4T | 50 | 5.1 × 10−10 ± 1.0 × 10−10 | 9.9 × 1010 | 3.8 × 10−3 | 9.0 × 10−7 | 4.2 × 10−8 ± 8.3 × 10−9 |
DGG-4T with the 55 nm UV-vis peak shift showed conductivity of 1.8 × 10−5 S cm−1, whereas DGA-4T with the 45 nm UV-vis peak shift showed nearly the same conductivity. DLAG-4T, with lowest spectral shift of 10 nm among the three 4T-conjugated peptides, showed conductivity of 4.2 × 10−8 S cm−1. The increase in conductivity values with increasing UV-vis spectral shifts suggests a possible correlation between the two factors. The stark difference in conductivity between DGG-4T, DGA-4T, and DLAG-4T implies that there is a certain degree of aggregation or UV-vis peak shift required for adequate π-stacking of oligomers to increase the overlap among assembled peptides to form conductive pathways.
Relation among absorbance signatures, morphologies, and conductivities of PDI peptides
UV-vis spectra of VEVAA-PDI/VEVAG-PDI & DAIA-PDI/DAVG-PDI were measured in basic and acidic medium to observe the changes in electronic structure (Fig. 8). Similar to what was previously reported,29 we observed two distinct peaks at 535 and 502 nm for all 4 peptides in basic condition, suggesting that these peaks are from isolated and essentially molecularly dissolved PDI cores. The broadening and extinction of peaks for VEVAG-PDI and DAVG-PDI in acidic medium have also been previously correlated to co-facial stacking due to smaller twist angles of stacking from computational modeling of dimer pairs.29 The dampening of the peaks shown in VEVAA-PDI/DAIA-PDI has been previously reported as rotationally shifted stacking of the internal PDI cores due to larger twist angles of stacking from the same modeling experiments.29 VEVAG-PDI and VEVAA-PDI UV-vis peak shifts from basic to acidic medium also show that the bulkiness of the amino acid closest to the π-core is critical to the self-assembly properties of these peptide systems.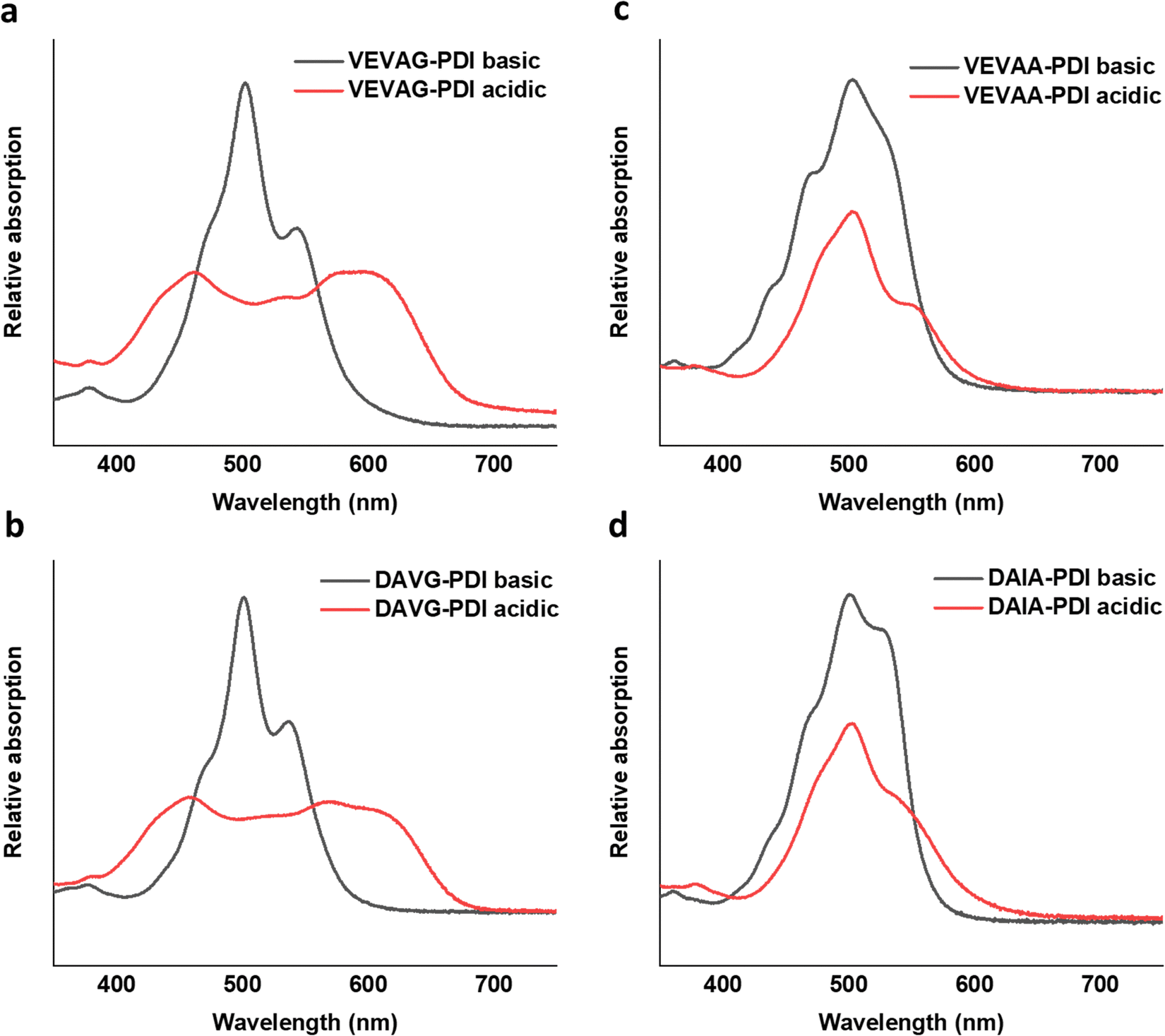 | ||
| Fig. 8 UV-vis spectra for basic (15 μL of 1 M KOH to 2.25 μM peptides) and acidic (40 μL of 1 M HCl to basic solution) peak shift for (a) VEVAG, (b) DAVG, (c) VEVAA, (d) DAIA PDI peptides. All spectra were recorded in the 5–7.5 μM concentration range. | ||
 | ||
| Fig. 9 (a) VEVAG (b) VEVAA (c) DAVG (d) DAIA PDI peptide thin films stitched 20× magnified optical microscopy images. Features in (a) and (c) are much more extended. | ||
Similarly, the crystallization of KCl mineralized on these peptide aggregates is also connected to spectroscopy of the aggregates in solution. Previous work by Panda et al.29 classified PDI peptides as having either co-facial for VEVAG-PDI and DAVG-PDI (Fig. 10a) or rotationally shifted stacking for VEVAA-PDI and DAIA-PDI (Fig. 10b and c) We reconfirmed that UV-vis spectroscopy from basic to acidic medium showed peak broadening for co-facially stacked peptides. Those peptides yielded dendritic macroscopic structures upon mineralization. On the other hand, peptides with rotationally shifted stacking showed dampening of the same peaks. The rotationally shifted stacking of those peptides may have contributed to forming spherical micellar structures upon mineralization (Fig. 10b) in some regions resulting in the observed circular dot aggregates of templated KCl mineralization in Fig. 9b and d. We anticipate that rotationally shifted stacking may have contributed to elongated stacks of spiraling peptides (Fig. 10c) that could not adhere properly to the surface of the Si/SiO2 substrate as well as the more co-facially stacked aggregates, perhaps leading to the brittle surface shown in Fig. S22 (ESI†).
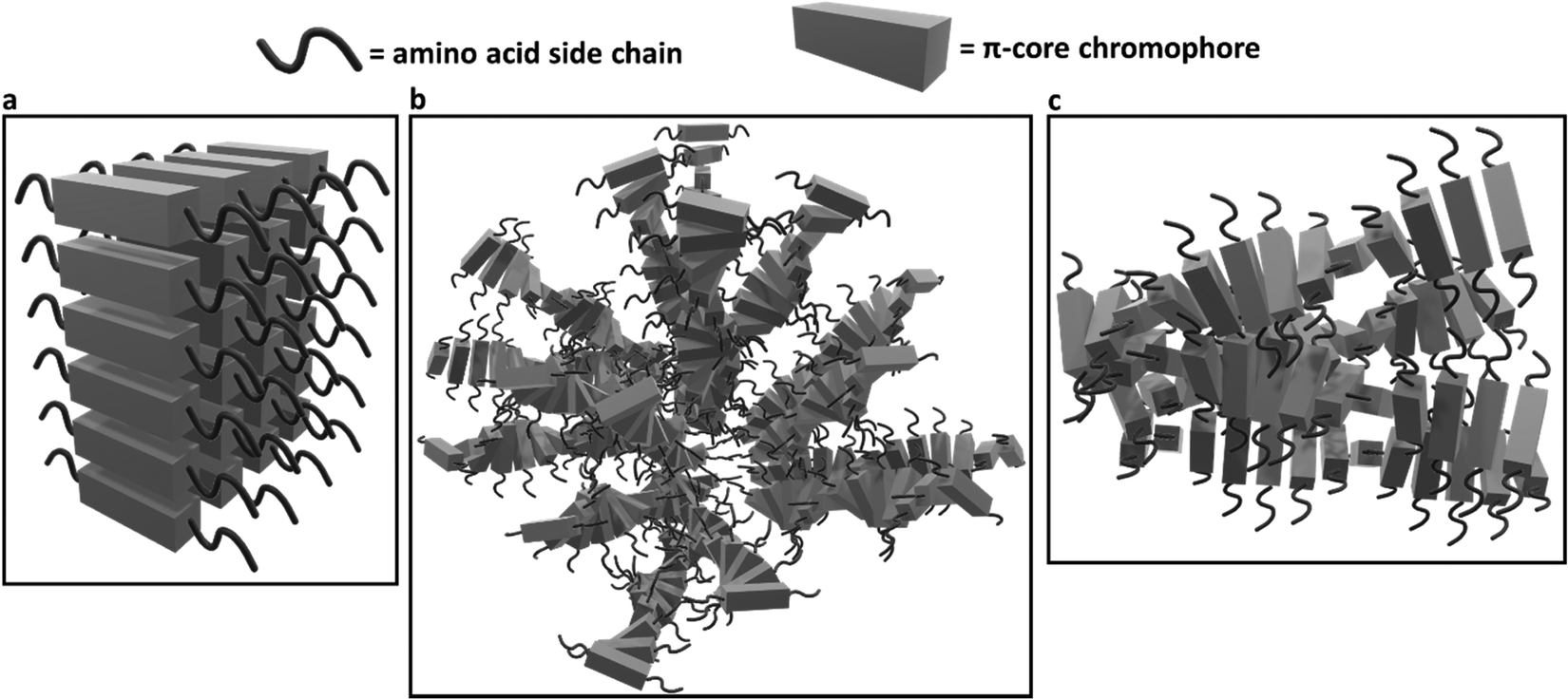 | ||
| Fig. 10 Schematic of different stacking structures deriving from change in amino acid sequence in peptide side chains based on evidence presented in Panda et al.29 (a) Estimated 3D structures of co-facially stacked π-peptide structures for VEVAG-PDI and DAVG-PDI with peak broadening and extinction for UV-vis spectroscopy from basic to acidic medium, (b) estimated 3D structures for rotationally shifted stacking of π-peptide structures anticipated to contribute to the spherical enveloping micellar structures that may template the formation of bulk KCl crystals for VEVAA-PDI and DAIA-PDI based on previously reported models.29 (c) Estimated 3D structures of elongated spiraling stacks of rotationally shifted stacking π-peptides may have prevented proper adhesion of the VEVAA-PDI and DAIA-PDI peptides to the surface of Si/SiO2 substrate, which turned out to be very brittle (Fig. S20, ESI†). | ||
These observations were also related to XRD structural data. Different macrostructures of KCl mineralization were templated by different peptide stacking sequence as shown in previous work.29 KCl peak shifts in the present work from solidified samples of VEVAA-PDI/VEVAG-PDI to DAIA-PDI/DAVG-PDI implied the presence of different KCl crystallite polymorphs due to lattice strain (Fig. 11). As discussed earlier, peptide self-assembly templated KCl mineralization can cause strain on the KCl lattice, increasing d-spacing.46 It appears that VEVAA-PDI, DAVG-PDI, and VEVAG-PDI peptides have at least some presence of KCl crystals with larger lattice parameter (28.6° two-theta) almost identical to the KCl peak observed for 4T based peptides. DAIA-PDI peptide assemblies, with the bulkiest single amino acid residue (isoleucine), had almost entirely the 29.2° two-theta, corresponding to the KCl with smaller lattice parameter, which was true after both one and three months storage.
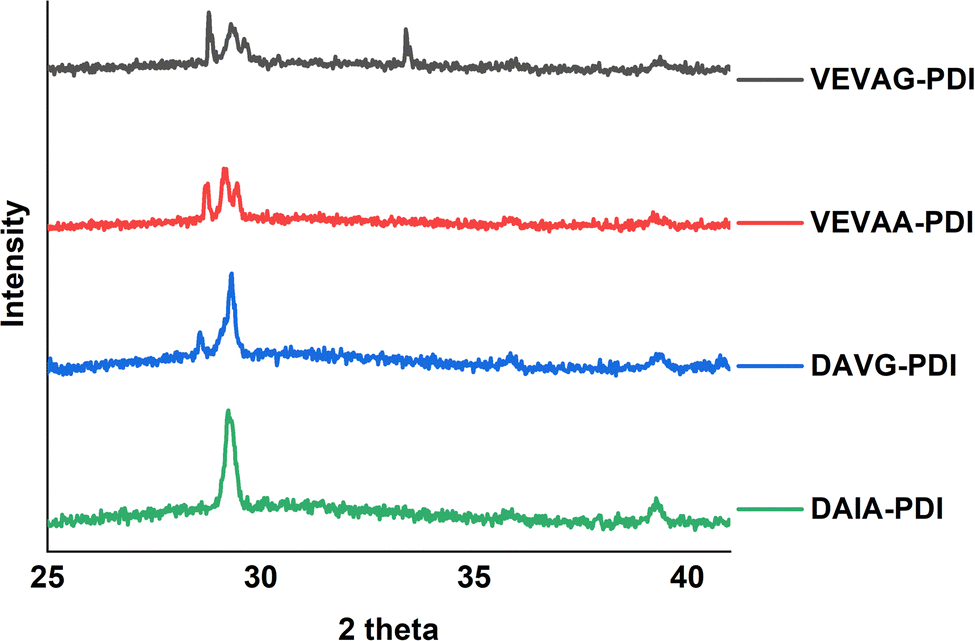 | ||
| Fig. 11 XRD graph for solid-state thin films of VEVAG-PDI, VEVAA-PDI, DAVG-PDI, and DAIA-PDIpi-peptides. Leftmost peaks are stronger for G versus A near the π system. Data shown are after three months storage; stronger relative contributions of the lower angle peaks were observed after one month storage. | ||
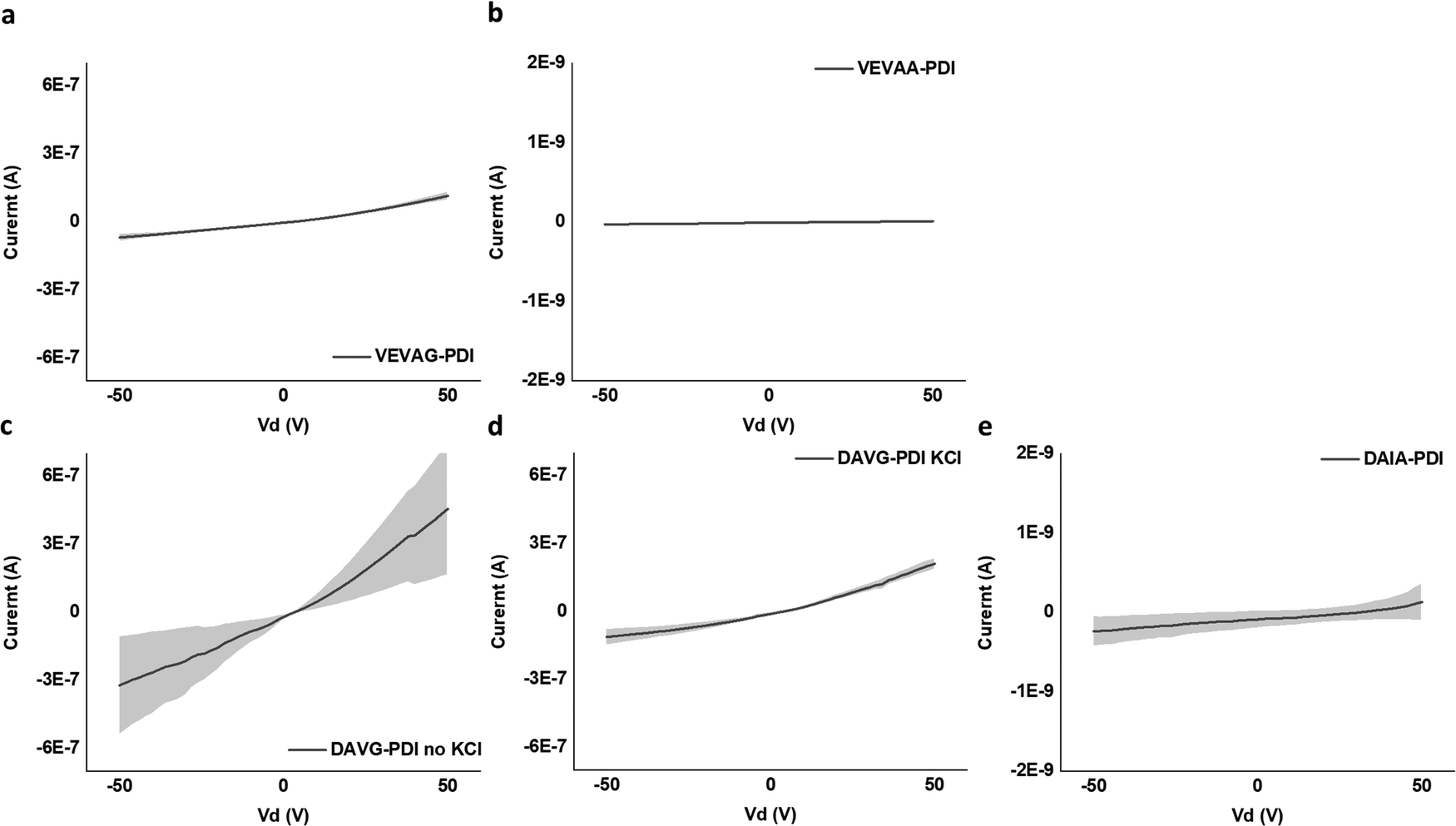 | ||
| Fig. 12 2 terminal IV curve for regions of (a) VEVAG-PDI, (b) VEVAA-PDI, (c) DAVG-PDI with no KCl at that device, (d) DAVG-PDI with KCl at that device, and (e) DAIA-PDI. VEVAG-PDI did not have samples without KCl. VEVAA-PDI and DAIA-PDI samples had spherical micellar structures dispersed throughout the samples. | ||
It is noted that the conductance/conductivity values of DAIA-PDI and VEVAA-PDI were not completely reliable as it was almost impossible to achieve stable probe tip contact with the electrodes due to the solid-state domains of these peptides being too brittle and shattering upon touch (Fig. S22, ESI†). The samples that did not shatter completely were measured and are discussed subsequently.
Only for DVAG-PDI could we obtain different IV curves for areas without KCl and areas with KCl on the solid-state thin films. The region with KCl structure had lower conductance than areas without KCl, again likely due to contact resistance. Thickness of around 1 μm was measured via 3D laser optical microscopy surface roughness scans, as shown in Fig. S23–S26 (ESI†). Eqn (1) was applied to IV curves for regions without KCl. Table 2. summarizes the values used to calculate the conductivity of DAIA-PDI, DAVG-PDI, VEVAA-PDI, and VEVAG-PDI peptides.
| V (V) | I (A) | R (Ohm) | L (cm) | A (cm2) | σ (S cm−1) | |
|---|---|---|---|---|---|---|
| VEVAG-PDI | 50 | 1.2 × 10−7 ± 1.6 × 10−8 | 4.4 × 108 | 3.8 × 10−3 | 9.0 × 10−7 | 9.6 × 10−6 ± 1.4 × 10−2 |
| VEVAA-PDI | 50 | 9.2 × 10−12 ± 3.0 × 10−14 | 5.5 × 1012 | 3.8 × 10−3 | 9.0 × 10−7 | 7.6 × 10−10 ± 2.5 × 10−12 |
| DAVG-PDI | 50 | 4.6 × 10−7 ± 2.9 × 10−7 | 1.1 × 108 | 3.8 × 10−3 | 9.0 × 10−7 | 3.8 × 10−5 ± 2.4 × 10−5 |
| DAIA-PDI | 50 | 1.3 × 10−10 ± 2.3 × 10−10 | 3.9 × 1011 | 3.8 × 10−3 | 9.0 × 10−7 | 1.1 × 10−8 ± 1.9 × 10−8 |
DAVG-PDI structural features included co-facial stacking and shorter stacking distance between peptides, resulting in the highest conductivity of 0.38 S cm−1 as anticipated previously from computational molecular simulations.48 VEVAG-PDI structures included co-facial stacking and relatively longer and multiple types of stacking distance between peptides than DAVG-PDI, resulting in the second highest conductivity of 9.58 × 10−6 S cm−1. DAIA-PDI structures included rotationally shifted stacking and shorter stacking distance between peptides, resulting in very low conductivity of 1.07 × 10−8 S cm−1. VEVAA-PDI structures included rotationally shifted stacking and longer stacking distance between peptides, resulting in much lower conductivity of 7.6 × 10−10 S cm−1.
Comparing the conductivities of DAIA-PDI and VEVAA-PDI with DAVG-PDI and VEVAG-PDI suggests that co-facial stacking creates an environment more favorable for the formation of connected macroscopic conduction pathways, while rotationally shifted stacking leads to unfavorable conditions for formation of connected macroscopic conduction pathways due to lack of proper interfacial contact with the underlying substrate. 50× optical and 3D laser microscope images suggest that rotationally shifted stacking may have resulted in either isolated micellar envelope structures (Fig. 10b. and Fig. S16a, b, e, f, ESI†) templating the growth of KCl blocks, or the formation of very brittle surfaces by helical twist structures leading to less adhesion to the surface (Fig. 10c) making it very difficult to measure electrical properties (Fig. S22, ESI†).
The comparison of DAVG-PDI to VEVAG-PDI is consistent with closer stacking distance between peptides facilitating the creation of more conductive pathways. Templated KCl crystals’ XRD peak for DAVG-PDI (29.2°) had smaller lattice parameter compared to the bulk of VEVAG-PDI peaks (28.6°) meaning closer stacking distance between peptides. It is well known that the electronic conduction pathway formed by peptide chromophores’ π stacking is closely related to the overlap of π-orbitals,49–53 which affect the bandgap energy levels. Closer peptide stacking is assumed to lead to decrease in the hopping activation energies, increasing conductivity.49–51
Conclusion
Three very different types of observations – absorption spectral shifts in solution, crystallinity and morphology of mineralized KCl on aggregates, and conductivity of solid aggregates – appeared to be grouped according to the bulkiness of π–chromophore peptide side chains being assembled. For one pair of π–chromophore peptides, a single methyl group associated with an alanine-for-glycine substitution was sufficient to alter all three observations in a manner consistent with weaker self-assembly. The innovative method of relating the templated growth of KCl minerals to the self-assembly characteristics of different types of π-peptides could be used to investigate different self-assembling systems in the future. It should also be possible in principle to grow electrically active minerals instead of insulating KCl crystals to form conduction pathways templated by the π–chromophore–peptide assemblies.A major goal of this research area is to establish defined conducting pathways in biological media and address those pathways with realistic contact strategies for neural stimulation and light-to-electricity energy conversion. The absolute conductivity magnitude is not the main variable being considered here. Our conductivities are largely in the middle of the range of values previously observed. Rather, it is the use of conductivity along with the spectral shifts as a marker of different pi-stacking geometries, and showing that those geometries influence the arrangements of COOH terminal groups on peptides that nucleate inorganic materials, that are the most important findings.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the National Science Foundation DMREF program, grant number DMR-1728947. We thank Professor Patricia McGuiggan for AFM measurements and images.References
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, About supramolecular assemblies of π-conjugated systems, Chem. Rev., 2005, 105(4), 1491–1546, DOI:10.1021/cr030070z.
- E. Moulin, J.-J. Cid and N. Giuseppone, Advances in supramolecular electronics—From randomly self-assembled nanostructures to addressable self-organized interconnects, Adv. Mater., 2013, 25(3), 477–487, DOI:10.1002/adma.201201949.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional supramolecular polymers, Science, 2012, 335(6070), 813–817, DOI:10.1126/science.1205962.
- P. Leclère, M. Surin, P. Viville, R. Lazzaroni, A. F. M. Kilbinger, O. Henze, W. J. Feast, M. Cavallini, F. Biscarini, A. P. H. J. Schenning and E. W. Meijer, About oligothiophene self-assembly: From aggregation in solution to solid-state nanostructures, Chem. Mater., 2004, 16(23), 4452–4466, DOI:10.1021/cm049673x.
- J. D. Tovar, Supramolecular construction of optoelectronic biomaterials, Acc. Chem. Res., 2013, 46(7), 1527–1537, DOI:10.1021/ar3002969.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Supramolecular biomaterials, Nat. Mater., 2016, 15(1), 13–26, DOI:10.1038/nmat4474.
- S. S. Panda, H. E. Katz and J. D. Tovar, Solid-state electrical applications of protein and peptide based nanomaterials, Chem. Soc. Rev., 2018, 47(10), 3640–3658, 10.1039/C7CS00817A.
- J. D. Tovar, Supramolecular construction of optoelectronic biomaterials, Acc. Chem. Res., 2013, 46(7), 1527–1537, DOI:10.1021/ar3002969.
- E. R. Draper and D. J. Adams, Controlling the Assembly and Properties of Low-Molecular-Weight Hydrogelators, Langmuir, 2019, 35(20), 6506–6521, DOI:10.1021/acs.langmuir.9b00716.
- M. Wehner, M. I. S. Röhr, V. Stepanenko and F. Würthner, Control of self-assembly pathways toward conglomerate and racemic supramolecular polymers, Nat. Commun., 2020, 11, 5460, DOI:10.1038/s41467-020-19189-8.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Amino-acid-encoded biocatalytic self-assembly enables the formation of transient conducting nanostructures, Nat. Chem., 2018, 10(7), 696–703, DOI:10.1038/s41557-018-0047-2.
- M. Amit, S. Yuran, E. Gazit, M. Reches and N. Ashkenasy, Tailor-made functional peptide self-assembling nanostructures, Adv. Mater., 2018, 30(41), 1707083, DOI:10.1002/adma.201707083.
- H. Shao, J. Seifert, N. C. Romano, M. Gao, J. J. Helmus, C. P. Jaroniec, D. A. Modarelli and J. R. Parquette, Amphiphilic self-assembly of an n-type nanotube, Angew. Chem., Int. Ed., 2010, 49(42), 7688–7691, DOI:10.1002/anie.201003415.
- E.-K. Schillinger, E. Mena-Osteritz, J. Hentschel, H. G. Börner and P. Bäuerle, Oligothiophene versus β-sheet peptide: Synthesis and self-assembly of an organic semiconductor–peptide hybrid, Adv. Mater., 2009, 21(16), 1562–1567, DOI:10.1002/adma.200803110.
- D. A. Stone, L. Hsu and S. I. Stupp, Self-assembling quinquethiophene–oligopeptide hydrogelators, Soft Matter, 2009, 5(10), 1990–1993, 10.1039/B904326H.
- J. E. Bullock, R. Carmieli, S. M. Mickley, J. Vura-Weis and M. R. Wasielewski, Photoinitiated charge transport through π-stacked electron conduits in supramolecular ordered assemblies of donor−acceptor triads, J. Am. Chem. Soc., 2009, 131(33), 11919–11929, DOI:10.1021/ja903903q.
- R. J. Kumar, J. M. MacDonald, Th. B. Singh, L. J. Waddington and A. B. Holmes, Hierarchical self-assembly of semiconductor functionalized peptide α-helices and optoelectronic properties, J. Am. Chem. Soc., 2011, 133(22), 8564–8573, DOI:10.1021/ja110858k.
- K. Besar, H. A. M. Ardoña, J. D. Tovar and H. E. Katz, Demonstration of hole transport and voltage equilibration in self-assembled π-conjugated peptide nanostructures using field-effect transistor architectures, ACS Nano, 2015, 9(12), 12401–12409, DOI:10.1021/acsnano.5b05752.
- E. T. Pashuck, H. Cui and S. I. Stupp, Tuning supramolecular rigidity of peptide fibers through molecular structure, J. Am. Chem. Soc., 2010, 132(17), 6041–6046, DOI:10.1021/ja908560n.
- B. D. Wall, A. E. Zacca, A. M. Sanders, W. L. Wilson, A. L. Ferguson and J. D. Tovar, Supramolecular polymorphism: Tunable electronic interactions within π-conjugated peptide nanostructures dictated by primary amino acid sequence, Langmuir, 2014, 30(20), 5946–5956, DOI:10.1021/la500222y.
- R. Marty, R. Nigon, D. Leite and H. Frauenrath, Two-fold odd–even effect in self-assembled nanowires from oligopeptide-polymer-substituted perylene bisimides, J. Am. Chem. Soc., 2014, 136(10), 3919–3927, DOI:10.1021/ja412384p.
- R. Matmour, I. De Cat, S. J. George, W. Adriaens, P. Leclère, P. H. H. Bomans, N. A. J. M. Sommerdijk, J. C. Gielen, P. C. M. Christianen, J. T. Heldens, J. C. M. van Hest, D. W. P. M. Löwik, S. De Feyter, E. W. Meijer and A. P. H. J. Schenning, Oligo(p-phenylenevinylene)−peptide conjugates: Synthesis and self-assembly in solution and at the solid−liquid interface, J. Am. Chem. Soc., 2008, 130(44), 14576–14583, DOI:10.1021/ja803026j.
- D. Ivnitski, M. Amit, B. Rubinov, R. Cohen-Luria, N. Ashkenasy and G. Ashkenasy, Introducing charge transfer functionality into prebiotically relevant β-sheet peptide fibrils, Chem. Commun., 2014, 50(51), 6733–6736, 10.1039/C4CC00717D.
- H. Shao, T. Nguyen, N. C. Romano, D. A. Modarelli and J. R. Parquette, Self-assembly of 1-d n-type nanostructures based on naphthalene diimide-appended dipeptides, J. Am. Chem. Soc., 2009, 131(45), 16374–16376, DOI:10.1021/ja906377q.
- A. S. Weingarten, A. J. Dannenhoffer, R. V. Kazantsev, H. Sai, D. Huang and S. I. Stupp, Chromophore dipole directs morphology and photocatalytic hydrogen generation, J. Am. Chem. Soc., 2018, 140(15), 4965–4968, DOI:10.1021/jacs.7b12641.
- E. P. Bruckner and S. I. Stupp, Designing supramolecular polymers with nucleation and growth processes, Polym. Int., 2022, 71(5), 590–595, DOI:10.1002/pi.6384.
- J. J. Walsh, J. R. Lee, E. R. Draper, S. M. King, F. Jäckel, M. A. Zwijnenburg, D. J. Adams and A. J. Cowan, Controlling Visible Light Driven Photoconductivity in Self-Assembled Perylene Bisimide Structures, J. Phys. Chem. C, 2016, 120, 18479–18486, DOI:10.1021/acs.jpcc.6b06222.
- M. R. Wasielewski, Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems, Acc. Chem. Res., 2009, 42, 1910–1921, DOI:10.1021/ar9001735.
- S. S. Panda, K. Shmilovich, N. S. M. Herringer, N. Marin, A. L. Ferguson and J. D. Tovar, Computationally Guided Tuning of Peptide-Conjugated Perylene Diimide Self-Assembly, Langmuir, 2021, 37(28), 8594–8606, DOI:10.1021/acs.langmuir.1c01213.
- P. Xie, Z. Shi, M. Feng, K. Sun, Y. Liu, K. Yan, C. Liu, T. A. A. Moussa, M. Huang, S. Meng, G. Liang, H. Hou, R. Fan and Z. Guo, Recent advances in radio-frequency negative dielectric metamaterials by designing heterogeneous composites, Adv. Compos. Hybrid Mater., 2022, 5, 679–695, DOI:10.1007/s42114-022-00479-2.
- S. S. Panda, K. Shmilovich, A. L. Ferguson and J. D. Tovar, Controlling supramolecular chirality in peptide–π–peptide networks by variation of the alkyl spacer length, Langmuir, 2019, 35(43), 14060–14073, DOI:10.1021/acs.langmuir.9b02683.
- S. S. Panda, K. Shmilovich, A. L. Ferguson and J. D. Tovar, Computationally guided tuning of amino acid configuration influences the chiroptical properties of supramolecular peptide–π–peptide nanostructures, Langmuir, 2020, 36(24), 6782–6792, DOI:10.1021/acs.langmuir.0c00961.
- M. A. Khalily, G. Bakan, B. Kucukoz, A. E. Topal, A. Karatay, H. G. Yaglioglu, A. Dana and M. O. Guler, Fabrication of supramolecular n/p-nanowires via coassembly of oppositely charged peptide-chromophore systems in aqueous media, ACS Nano, 2017, 11(7), 6881–6892, DOI:10.1021/acsnano.7b02025.
- R. Kundu, A. Chandra and A. Datta, Fluorescent chemical tools for tracking anionic phospholipids, Isr. J. Chem., 2021, 61(3–4), 199–216, DOI:10.1002/ijch.202100003.
- J. Cornil, D. A. dos Santos, X. Crispin, R. Silbey and J. L. Brédas, Influence of interchain interactions on the absorption and luminescence of conjugated oligomers and polymers: A quantum-chemical characterization, J. Am. Chem. Soc., 1998, 120(6), 1289–1299, DOI:10.1021/ja973761j.
- H. A. Bechtel, Z. H. Kim, J. P. Camden and R. N. Zare, Bond and mode selectivity in the reaction of atomic chlorine with vibrationally excited CH2D2, J. Chem. Phys., 2004, 120(2), 791–799, DOI:10.1063/1.1630961.
- B. X. Dong, Z. Liu, M. Misra, J. Strzalka, J. Niklas, O. G. Poluektov, F. A. Escobedo, C. K. Ober, P. F. Nealey and S. N. Patel, Structure control of a π-conjugated oligothiophene-based liquid crystal for enhanced mixed ion/electron transport characteristics, ACS Nano, 2019, 13(7), 7665–7675, DOI:10.1021/acsnano.9b01055.
- R. D. McCullough, The chemistry of conducting polythiophenes, Adv. Mater., 1998, 10(2), 93–116, DOI:10.1002/(SICI)1521-4095(199801)10:2.
- O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Supramolecular energy materials, Adv. Mater., 2020, 32(17), 1907247, DOI:10.1002/adma.201907247.
- Z. Zhang, M. Liu, M. M. Ibrahim, H. Wu, Y. Wu, Y. Li, G. A. M. Mersal, I. H. El Azab, S. M. El-Bahy, M. Huang, Y. Jiang, G. Liang, P. Xie and C. Liu, Flexible polystyrene/graphene composites with epsilon-near-zero properties, Adv. Compos. Hybrid Mater., 2022, 5, 1054–1066, DOI:10.1007/s42114-022-00486-3.
- M. Liu, H. Wu, Y. Wu, P. Xie, R. A. Pashameah, H. M. Abo-Dief, S. M. El-Bahy, Y. Wei, G. Li, W. Li, G. Liang, C. Liu, K. Sun and R. Fan, The weakly negative permittivity with low-frequency-dispersion behavior in percolative carbon nanotubes/epoxy nanocomposites at radio-frequency range, Adv. Compos. Hybrid Mater., 2022, 5, 2021–2030, DOI:10.1007/s42114-022-00541-z.
- K. Shmilovich, Y. Yao, J. D. Tovar, H. E. Katz, A. Schleife and A. L. Ferguson, Computational discovery of high charge mobility self-assembling π-conjugated peptides, Mol. Syst. Des. Eng., 2022, 7(5), 447–459, 10.1039/D2ME00017B.
- K. Shmilovich, S. S. Panda, A. Stouffer, J. Tovar and A. L. Ferguson, Hybrid computational-experimental data-driven design of self-assembling pi-conjugated peptides, Digital Discovery, 2022, 1, 448–462, 10.1039/d1dd00047k.
- K. Besar, H. A. M. Ardona, J. D. Tovar and H. E. Katz, Demonstration of Hole Transport and Voltage Equilibration in Self-Assembled π-Conjugated Peptide Nanostructures Using Field-Effect Transistor Architectures, ACS Nano, 2011, 9, 12401–12409, DOI:10.1021/acsnano.5b05752.
- T. Lee, S. S. Panda, J. D. Tovar and H. E. Katz, Unusually Conductive Organic–Inorganic Hybrid Nanostructures Derived from Bio-Inspired Mineralization of Peptide/Pi-Electron Assemblies, ACS Nano, 2020, 14(2), 1846–1855, DOI:10.1021/acsnano.9b07911.
- P. Scherrer, Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, Mathematisch-Physikalische Klasse, 1918, 1918, 98–100 Search PubMed.
- C. Videlot-Ackermann, J. Ackermann, H. Brisset, K. Kawamura, N. Yoshimoto, P. Raynal, A. El Kassmi and F. Fages, A,ω-distyryl oligothiophenes: High mobility semiconductors for environmentally stable organic thin film transistors, J. Am. Chem. Soc., 2005, 127(47), 16346–16347, DOI:10.1021/ja054358c.
- B. A. Thurston and A. L. Ferguson, Machine learning and molecular design of self-assembling-conjugated oligopeptides, Mol. Simul., 2018, 44(11), 930–945, DOI:10.1080/08927022.2018.1469754.
- B. C. Streifel, J. F. M. Hardigree, H. E. Katz and J. D. Tovar, Heteroaromatic variation in amorphous 1,6-methano[10]annulene-based charge-transporting organic semiconductors, J. Mater. Chem. C, 2014, 2(37), 7851–7858, 10.1039/C4TC01326C.
- W. Bao, L. Jing, J. Velasco, Y. Lee, G. Liu, D. Tran, B. Standley, M. Aykol, S. B. Cronin, D. Smirnov, M. Koshino, E. McCann, M. Bockrath and C. N. Lau, Stacking-dependent band gap and quantum transport in trilayer graphene, Nat. Phys., 2011, 7(12), 948–952, DOI:10.1038/nphys2103.
- Y. Ma, D. Cai, S. Wan, P. Yin, P. Wang, W. Lin and Q. Zheng, Control over π–π stacking of heteroheptacene-based nonfullerene acceptors for 16% efficiency polymer solar cells, Natl. Sci. Rev., 2020, 7(12), 1886–1895, DOI:10.1093/nsr/nwaa189.
- S. Ehrlich, J. Moellmann and S. Grimme, Dispersion-Corrected Density Functional Theory for Aromatic Interactions in Complex Systems, Acc. Chem. Res., 2013, 46(4), 916–926, DOI:10.1021/ar3000844.
- B. Liu, Y. Vonhausen, A. Schulz, C. Hobartner and F. Wurthner, Peptide Backbone Directed Self-Assembly of Merocyanine Oligomers into Duplex Structures, Angew. Chem., Int. Ed., 2022, 61(21), e202200120, DOI:10.1002/anie.202200120.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00979j |
| This journal is © The Royal Society of Chemistry 2023 |