
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanotechnology based therapeutic approaches: an advanced strategy to target the biofilm of ESKAPE pathogens
Arpita
Mukherjee
ab,
Somashree
Bose
ab,
Anirban
Shaoo
a and
Sujoy K.
Das
 *ab
*ab
aInfectious Diseases and Immunology Division, Council of Scientific and Industrial Research (CSIR)-Indian Institute of Chemical Biology, Kolkata-700032, India. E-mail: sujoydas@iicb.res.in; sujoydasiacs@gmail.com; Tel: +91 33 24995 905
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 12th April 2023
Abstract
Bacterial infection by ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp.) pathogens are one of the major health concerns and has caused a global crisis in the healthcare sectors, leading to mortality, morbidity, and socioeconomic loss. The overuse and abuse of antibiotics has led to an increased number of MDR bacteria. Thus, conventional antibiotics have failed to show notable improvement in bacterial infections. Biofilm formations make pathogens more recalcitrant than their planktonic form, and are becoming more challenging to treat with conventional antibiotics. To overcome the challenges of the biofilm-associated chronic infections of ESKAPE pathogens, a new therapeutic strategy is urgently needed. Recently, nanomaterial-based therapies have emerged as a novel approach to combat the biofilm infection of ESKAPE pathogens. The size, shape and other physicochemical properties of nanomaterials play a significant role in targeting the biofilm and overcoming the recalcitrant bacterial infection. Herein, we give a brief review about the formation and structure of biofilms of ESKAPE pathogens and the quorum-sensing (QS) mechanism in biofilm formation. Subsequently, we discuss the conventional methods and strategies for the treatment of biofilms and their limitations. Later, we highlight different strategies for the fabrication of nanoparticles, which include solid nanoparticles, conjugated nanoparticles and nanocarrier systems that can be used to target the bacterial biofilm. We also discuss different interaction mechanisms through which nanoparticles disrupt the biofilm and kill the sessile and persister cells. Therapeutic applications of nanomaterials in biomedical fields are also systematically reviewed. Lastly, we discuss the current status and future perspectives of nanotechnology. We believe that this article provides insights into the advancement of nanotechnology, and offers an alternative therapeutic strategy to treat biofilm-associated infections.
 Arpita Mukherjee | Arpita Mukherjee is presently pursuing her PhD at the Infectious Diseases and Immunology Division, Council of Scientific and Industrial Research- Indian Institute of Chemical Biology (CSIR-IICB), Kolkata, India under the supervision of Dr Sujoy K. Das, Principal Scientist at CSIR-IICB. She completed her graduation from Hooghly Women's College, The University of Burdwan, Bardhaman, West Bengal, India, majoring in Microbiology in 2017. She received her MSc degree from The University of Burdwan, Bardhaman, West Bengal, India, majoring in Microbiology in 2019. Her research focus is on the development of surface-engineered nanoparticles for application in the eradication of bacterial biofilms. |
 Somashree Bose | Somashree Bose received her BSc degree from Acharya Prafulla Chandra Roy Government College, University of North Bengal in 2015, majoring in Zoology. Then, she completed her MSc studies from Ranchi University in 2017, majoring in Zoology. She is currently pursuing her PhD degree at the Infectious Diseases and Immunology Division, CSIR-IICB, Kolkata, India under the supervision of Dr Sujoy K. Das. Her research focus is on the development and application of smart transdermal scaffolds as novel drug delivery systems to combat bacterial infections and accelerate wound healing. She is a coauthor of one research paper in an international journal of high repute, and has also filed a patent from India. |
 Anirban Shaoo | Anirban Shaoo received his BSc degree from Asutosh College, University of Calcutta, Kolkata, West Bengal, India in 2017, majoring in Biochemistry. Subsequently, he successfully completed his post-graduate studies in 2019 at Ballygunge Science College, University of Calcutta, Kolkata, West Bengal, India, majoring in Biochemistry. Currently, he is working as a Project JRF under the supervision of Dr Sujoy K. Das at the Infectious Diseases and Immunology Division, CSIR-IICB, Kolkata, India. His research focus is on the photothermal ablation of bacterial infections with the help of 3D hydrogels. |
 Sujoy K. Das | Dr Sujoy K. Das, Principal Scientist, CSIR-Indian Institute of Chemical Biology, India and Associate Professor of Academy of Scientific and Innovative Research (AcSIR), New Delhi, India. He is a Fellow of the Royal Society of Chemistry (FRSC), UK, and a Fellow of the Academy of Sciences Chennai (FASCh), India. Dr Das obtained his PhD degree from the Indian Association for the Cultivation of Science. He pursued his postdoctoral research studies at Dublin City University, Ireland and Technical University, Dresden, Germany. His research interests are broadly focused on the design and synthesis of bioinspired nanomaterials, delivery of antimicrobial peptides for the inhibition and disruption of bacterial biofilms, and smart hydrogel fabrication and photodynamic therapy for wound infection management. He has published more than 50 research articles (>5200 citations, h-index 36) in peer-reviewed journals, and has two patents in his credit. He also filed two patents and contributed to two book chapters. He is an Editorial Board Member of Enzyme and Microbial Technology, and edited a special issue as a Guest Editor. |
1. Introduction
ESKAPE pathogens are one of the major causes of nosocomial infections, and account for 2 million illnesses and 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year as per the reports of Centers for Disease Control and Prevention (CDC).1 Bacterial colonization and successive biofilm formation by ESKAPE pathogens are a major threat in the biomedical sector. The bacterial cells in the biofilm are surrounded by extracellular polymeric substances (EPS), which provide structural stability and protect the cells from harsh environmental conditions.3,4
000 deaths per year as per the reports of Centers for Disease Control and Prevention (CDC).1 Bacterial colonization and successive biofilm formation by ESKAPE pathogens are a major threat in the biomedical sector. The bacterial cells in the biofilm are surrounded by extracellular polymeric substances (EPS), which provide structural stability and protect the cells from harsh environmental conditions.3,4
It has been reported that the formation of a biofilm makes the bacteria 1000 times more antibiotic-resistant compared to their planktonic ones because EPS hinders the penetration of antibiotics.2,5 EPS accounts for 50%–90% of the total biomass of biofilms, thereby inhibiting the penetration of antimicrobials.6 QS7 plays a pivotal role in the initiation, development, and maturation of biofilms, as well as the release of planktonic bacteria from it. Autoinducers secreted by the pathogens help in cell-to-cell communication, which is further involved in biofilm formation, expression of pathogenic factors and evading host immunity. Besides that, the altered pH of the biofilm inactivates the antibiotics and helps the bacteria to survive in harsh environmental conditions.2,3,7
The abbreviation ESKAPE represents six groups of pathogens, i.e., Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.4 The ESKAPE pathogens are named due to their escaping strategies from known antibiotics, including the penicillin group of drugs, carbapenemase, vancomycin, and even colistin, making them multi-drug resistant. Many conventional approaches, such as physical and chemical-based strategies, exist for the treatment of biofilm-related infections by ESKAPE pathogens. Different therapeutic strategies, such as antibiotics, antimicrobial peptides, polymers and polysaccharides, are also used to treat the bacterial biofilm-related infections. Extensive utilization of antibiotics leads to the generation of MDR bacteria, which reduces the efficacy of the antibiotics.1,2 Bacteria inside the biofilm are prone to high mutation rates and interchangeable resistant genes on transposable elements, leading to the overall resistance to the cells of biofilm. Moreover, the abovementioned strategies are expensive, requiring multiple doses with uncertain and adverse outcomes.4,7
Recently, nanotechnology has emerged as a promising alternative to the conventional approaches for the effective treatment of bacterial biofilms. Nanomaterials having dimensions in the range of 1 to 100 nm possess unique physicochemical properties, such as small size, increased surface area-to-volume ratio, and increased thermal, electrical, optical and magnetic properties.8 Metal and metal oxide nanomaterials often act as antimicrobial agents due to their biotic interaction with microbes, which inhibits and disrupts the biofilm. Apart from that, nanomaterials like lipids, polymers, and micelles, also act as vehicles to deliver antimicrobial agents owing to their very small size and better penetration property within the biofilm matrix.9–11 In this review, we give a summary of the formation and structure of the biofilms of ESKAPE pathogens and their QS mechanism associated with the biofilm formation. Next, we highlight the efficacy of various nanomaterials based on different approaches to inhibit bacterial biofilm formation and disruption of the mature biofilm. Different treatment strategies use nanoparticles such as metal-based, carbon-based, 2D nanoparticles, nanoemulsions, lipid-based and several polymeric nanoparticles for removing biofilm. We also discuss the application of various nanomaterials such as metals, carbon, lipids and polymers in various biomedical fields to combat biofilm-related bacterial infections. Finally, we illustrate the current status and future perspective of nanotechnology for the effective treatment of ESKAPE pathogens. We believe that this review will give a brief idea to understand the development of nanomaterials as a promising antibacterial and antibiofouling agent to fight against chronic biofilm-related infections.
2. Biofilm formation and QS system of ESKAPE pathogens
Biofilms are heterogenous, multicellular surface-adhered sessile microbial communities surrounded by an EPS matrix, which consist of less than 10% dry weight of the microorganism and 90% dry weight of its matrix.12 The EPS of the biofilm contains exopolysaccharides, eDNA, lipids, proteins and other macromolecules. The EPS of the biofilm acts as a protective barrier, serving as the storage of nutrients. Biofilm formation takes place in four stages, such as surface attachment, biofilm formation, maturation and dispersal (Scheme 1).13–15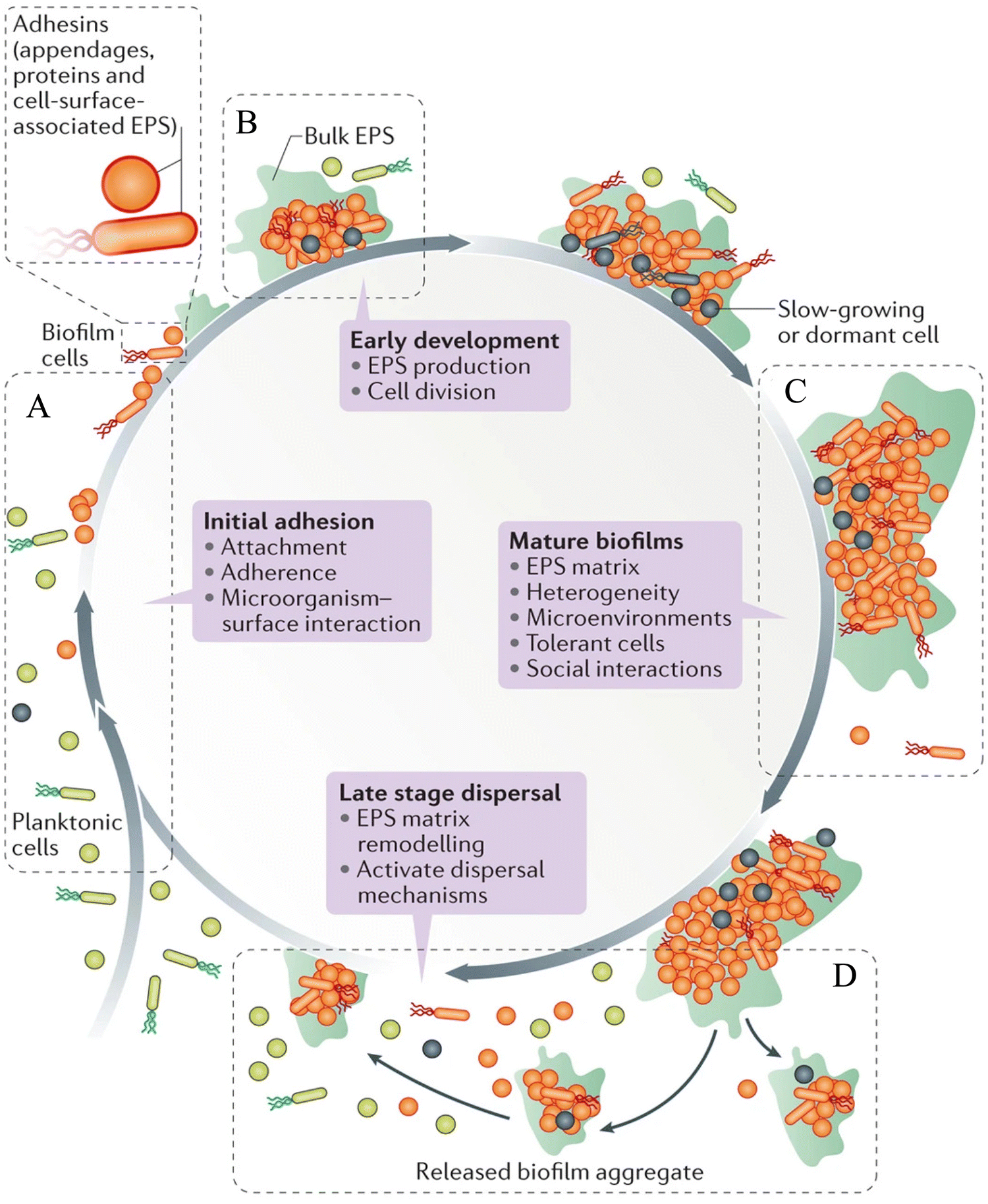 | ||
| Scheme 1 Various stages of the biofilm life-cycle comprising (A) surface attachment, (B) biofilm formation, (C) biofilm maturation and (D) dispersal of biofilm. Reprinted with permission from ref. 46. | ||
(1) Surface attachment
Microbial cells attach to the surfaces through various interactions, such as van der Waals, electrostatic and hydrophobic interactions, and become sessile.16 Capsular polysaccharide/adhesion (PS/A), fimbriae, pilus-like structures play a key role in the initial attachment of the bacteria to the surfaces.13(2) Biofilm formation
The surface-attached cells rapidly divide to form micro-colonies and secrete EPS, leading to the formation of a biofilm matrix.17(3) Biofilm maturation
In this stage, the biofilm forms a three-dimensional (3D) structure that provides structural stability and protects the cells from chemicals, host defense and harsh environmental conditions.17 Matured biofilms have dormant bacterial subpopulations called persisters, and show high tolerance to antibiotics.18(4) Dispersal of biofilm
The cells inside the biofilm detach from the substratum and go to a different location.13During biofilm formation, cell-to-cell communication takes place by a signaling process called QS. It plays a vital role in maintaining pathogenicity of the bacteria by controlling different virulent genes, which are required for the formation of the biofilm.7 The Gram-positive bacteria release small peptides named autoinducing peptides (AIP), while Gram-negative bacteria release a chemical called acyl-homoserine lactone (acyl-HSL) into the extracellular environment, and communicate with each other.19 After reaching a threshold concentration, these QS molecules (AIP and acyl-HSL) are recognized by the surrounding bacterial cells. These QS molecules bind to the specific receptor kinase of the cells, which further activates other sensory kinases and regulates the expression of different genes related to biofilm formation and pathogenesis.19,20
E. faecium is one of the common nosocomial pathogens that causes biofilm-mediated infection and becomes difficult to treat. Autolysin in E. faecium, named AtlAEfm, is involved in the stability of the biofilm. Among several virulence genes, enterococcal surface protein (Esp) and enterococcal biofilm regulator B (EbrB) take part in biofilm formation.21,22 Secretion of these virulence factors in E. faecium is regulated by the Fsr QS system.23S. aureus secretes glycocalyx-containing materials, which form a multilayered biofilm where heterogeneous protein expression occurs throughout the biofilm. EPS of S. aureus contains polysaccharide intracellular antigen (PIA), which is involved in the regulation of Staphylococcal biofilm formation.24 Biofilm-associated protein (Bap), surface-associated proteins such as protein A, fibrinogen binding proteins (FnBPA and FnBPB), S. aureus surface protein (SasG), and clumping factor B (ClfB) are important components for the attachment and development of the biofilm matrix. There are some secreted proteins like extracellular adherence protein (Eap) and beta toxin (Hlb), which help in the maturity of the Staphylococcal biofilm.25,26 The accessory gene regulator (agr) locus regulates the QS system, and is thus involved in the biofilm formation in S. aureus (Scheme 2A). AgrA and AgrC comprise the two component regulatory system, which senses the autoinducing cyclic octapeptide. The peptide is processed by AgrB, which is the product of agrD.27,28 Like other Gram-negative bacteria, K. pneumoniae (Scheme 2B) does not secrete acyl-HSLs AHL. Rather, it encodes SdiA (suppressor of the cell division inhibition), an orphan LuxR type receptor that responds to acyl-HSLs AHLs of other species. K. pneumoniae secretes different virulence factors like capsule polysaccharide, lipopolysaccharide (LPS), type 1 and type 3 fimbriae, which help to establish infection by evading the host immune system and biofilm formation.29–31 Type 3 fimbriae and capsular polysaccharide are the most important factors in biofilm establishment, where fimbriae mediate stable adherence and capsular polysaccharide is involved in cell-to-cell communication and biofilm structure formation.32 The biofilm formation of A. baumannii involves outer membrane protein A (OmpA), chaperon-usher pilus (Csu), biofilm associated protein (Bap), EPS, and poly-β-(1,6)-N-acetyl glucosamine (PNAG). OmpA facilitates A. baumannii to attach and invade the epithelial cell, and contribute to drug resistance and biofilm formation.33 Bap is a large surface exposed protein homologous to S. aureus, which is involved in cell–cell interaction and helps in adherence.34 The luxR/luxI type QS locus is called abaR/abaI in A. baumannii (Scheme 2C). The abaI gene encodes a 183 amino acid protein, which is involved in signal transduction, whereas abaR encodes a 238 amino acid protein, which acts as the autoinducer synthase receptor.35–37 Psl (polysaccharide synthesis locus), Pel (pellicle) and alginate are the three exopolysaccharides secreted by P. aeruginosa, where Psl helps to attach the cells to the surfaces, and is involved in cell–cell communication. It also helps to maintain the structural stability of the biofilm by presenting the periphery of the biofilm matrix. Pel is an important matrix component responsible for the surface attachment and structural stability of the biofilm, providing resistance against aminoglycoside antibiotics to the biofilm. Alginate plays a vital role in biofilm maturation, evading phagocytosis, opsonization and reducing antibiotic diffusion through the biofilm.38–40 Two complete acyl homoserine lactone systems are found in P. aeruginosa (Scheme 2D), named as the LasR-LasI and RhlR-rhlI system. LasI produces N-3-oxo-dodecanoyl-homoserine lactone (3OC12-HSL), while RhlI produces N-butanoyl-homoserine lactone (C4-HSL). LasR and RhlR are two transcription factors that are activated by 3OC12-HSL and C4-HSL, respectively, and regulate the transcription of different genes related to bacterial pathogenesis. The expression of several virulence factors like pyocyanin, elastase and exotoxin help in the development of biofilms in P. aeruginosa.41–43 Among Enterobacter spp., Enterbacter cloacae is mainly involved in biofilm-related infections. T6SS-2 of E. cloacae exhibits a vital role in the formation of biofilms, and also helps in attaching the bacteria to eukaryotic cells.44 This QS pathway can be disrupted by different mechanisms, such as (i) reducing the activity of receptors, (ii) inhibiting the synthesis of QS molecules (acyl-HSL, AIP), (iii) degradation of QS molecules, and (iv) mimicking of the signaling molecules.45
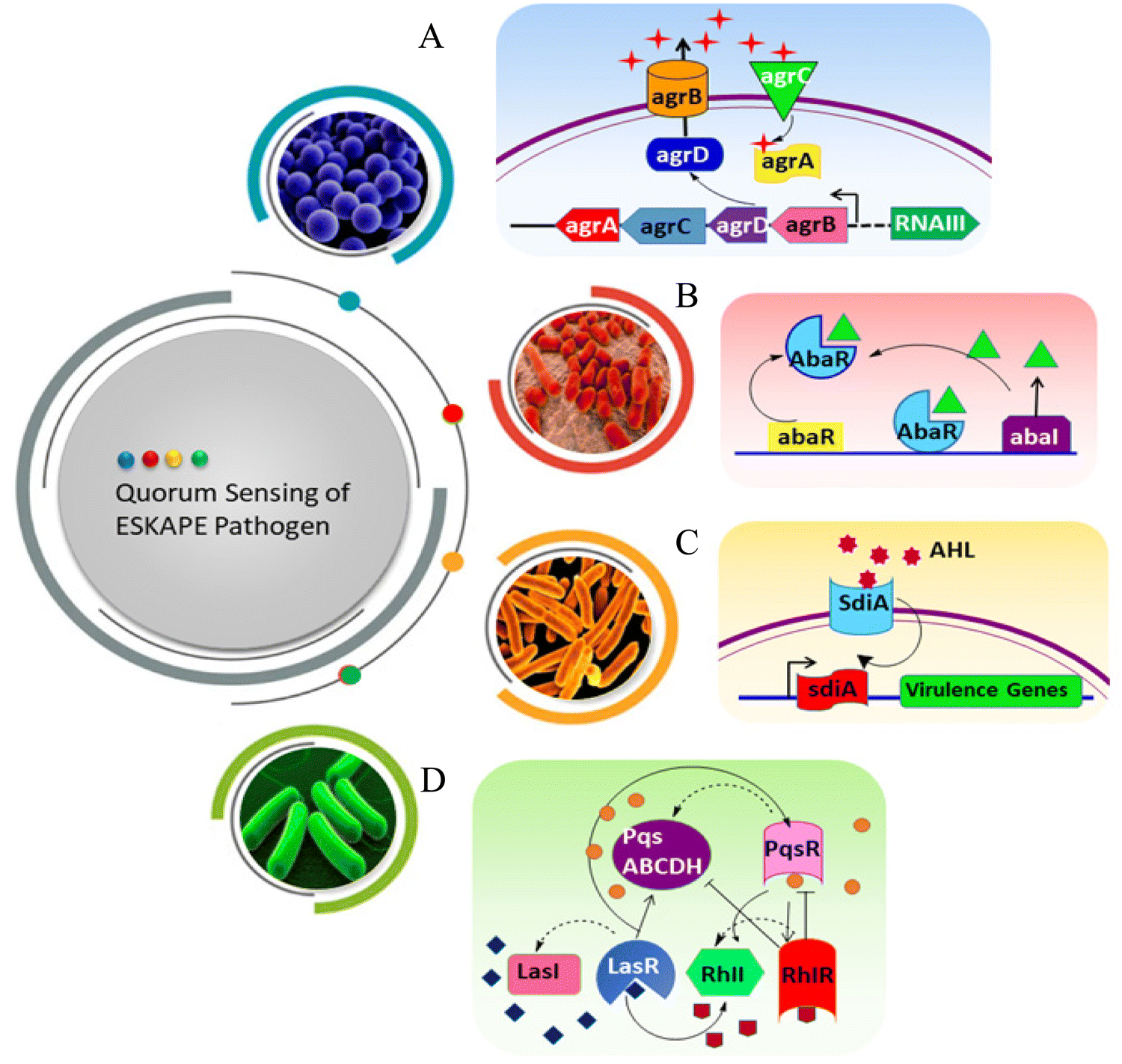 | ||
| Scheme 2 QS pathway of ESKAPE pathogens. (A) S. aureus, (B) A. baumanni, (C) K. pneumoniae and (D) P. aeruginosa. | ||
3. Existing strategies for biofilm inhibition and disruption
There are many existing strategies implemented for biofilm management in health care sectors, which include physical, chemical, drugs and other molecules. Scheme 3 illustrates three types of existing strategiesm and their limitations in biofilm inhibition and disruption. The physical method includes mechanical disruption of the biofilm by scrubbing, shear stress like jet spray, brushes, electric waves including ultrasound, laser shock, and UV rays, leading to the removal of the biofilm layer from the surgical instruments, hospital floors, bed, and others.46,47 On the other hand, different sprays and water-based jets are mostly used to eradicate the dental and surgical biofilms, and thus remove the dead necrotic tissue and exudates often referred to as debridement. The combination therapy of antibiotics along with debridement of the dead necrotic tissue could also be applied as a therapeutic strategy for curing bacterial infection.48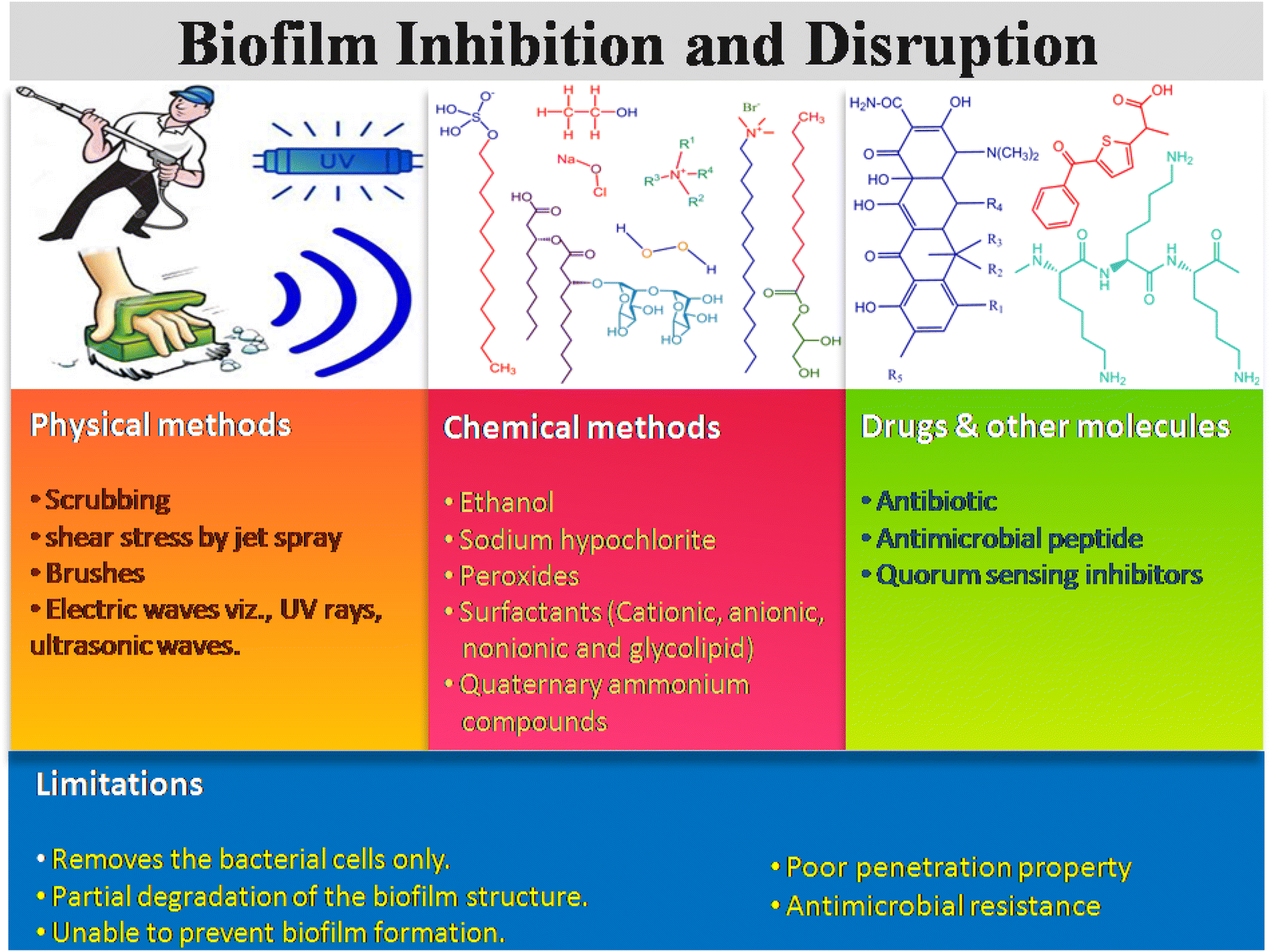 | ||
| Scheme 3 Conventional approaches for biofilm inhibition and disruption. | ||
The chemical-based antibiofilm agents involve the utilization of antimicrobial disinfectants, namely hydrogen peroxide (H2O2),49,50 chlorine-based chemicals (sodium hypochlorite, hypochlorous acid), surfactant-based chemicals, quaternary ammonium compounds, povidine-iodine and chlorhexidine gluconate.46 Among all these antimicrobial agents, H2O2, HOCl and HOBr are strong oxidants that attack the cellular membrane lipids, DNA and other cellular contents, and effectively kill the microorganism. In the presence of hydrogen peroxide and chloride/bromide ions, vanadium haloperoxidases have been reported to form HOCl/HOBr, which acts as a strong bactericidal and oxidizing agent against P. aeruginosa biofilms.51 QACs kill the microbes by cleaving their cell membrane, leading to nutrient efflux and hence cell lysis.52,53 Gemini QAC shows antibacterial and antibiofouling activities against P. aeruginosa.54 QACs are used in the cleaning of chronic wound infections. Different ionic and non-ionic surfactants are also conventionally used to inhibit and disrupt the biofilm formation.55–57 Surfactants usually weaken the EPS matrix, which causes surface detachment, leading to biofilm disruption. Apart from this, phage therapies (KPP10 against P. aeruginosa,58 AB-SA01 phage cocktail against MRSA,59 pIsf-AB02 against MDR A. baumannii60) against bacterial biofilms have also been developed.
Different small molecules, like antibiotics, antimicrobial peptides, and QS inhibitors with known antimicrobial activities, are used for treating the biofilm infections caused by ESKAPE pathogens.61,62 Antibiotics are still considered the best therapeutic agent to kill the bacterial cells through different modes of action, such as inhibition of DNA replication, cell wall synthesis, protein synthesis and various metabolic processes. Antibiotics like aztreonam, tobramycin and vancomycin serve as an antibiofouling agent and disrupt the biofilm formation through inhibition of filamentation, peptidoglycan biosynthesis, and affecting the bacterial cell permeability.63,64 The antimicrobial peptides (AMPs) have been explored as an alternative antibiofouling agent in the biomedical field. It has been reported that TAT-RasGAP317-326 with TAT HIV 48-57 sequence65 and AMP 1018-K666 exhibit antibiofilm properties, and inhibit the biofilm formation of P. aeruginosa, A. baumannii, S. aureus and methicillin resistant S. aureus (MRSA), respectively. Moreover, there are several limitations of these conventional techniques. Physical strategy causes the physical removal of the biofilm through aggressive debridement, but a few populations of sessile bacteria still remain on the surfaces. After completion of the treatment, the remaining bacterial cells act as a nucleation point and the biofilm repopulation takes place. For example, shear stress disturbs the biofilm integrity, but it fails to kill the persister cells.46 The antibiotics, antimicrobial peptides, QS inhibitors also have strong limitations, such as their reduced uptake into the biofilm matrix due to the protective EPS layer, inactivation by low pH, and anoxic condition of the biofilm, enzymatic degradation and/or their modification inside the biofilm microenvironment.2,67 Therefore, it is necessary to explore an alternative process and/or materials for effective dispersal of the bacterial biofilm.
4. Nanotechnology-based approach to combat biofilm
The emergence of nanoscience and nanotechnology provides an opportunity to deal with the biofilm of ESKAPE pathogens in an effective manner. Owing to their small size, large surface area and high surface-active groups, nanomaterials exhibit unique physicochemical properties. These physicochemical properties can be further tuned by varying the morphology (size and shape of the particles) and surface modifications of the nanoparticles.68 Nanoparticles act either as an antibacterial and antibiofouling agent, or as a smart carrier for the effective delivery of the antimicrobial agent/drugs inside the biofilm matrix.69 The nanotechnology-based approaches can be classified into three categories (Scheme 4): (1) solid nanoparticles, (2) conjugated nanomaterials and (3) nanocarrier system. The solid nanoparticles mainly focus on the utilization of inorganic nanoparticles, carbon-based, dendrimer and polymeric nanoparticles to inhibit and disrupt the biofilm formation.70–72 In the conjugated nanomaterials system, the solid nanoparticles are further functionalized with polymers, peptides and different bioactive molecules to increase the efficacy of the nanoparticles to disrupt the biofilm formation.73,74 In the nanocarrier system, nanomaterials have been explored to deliver the drugs, antibiotics, peptides and other small molecules to inhibit and disrupt the biofilm formation. Nanoparticles are also reported to play a role in breaking the QS system. Interference with the QS activity is an excellent strategy to inhibit bacterial pathogenicity without increasing the resistance among the bacteria. Some nanoparticles like AgNPs,75 SeNPs,76 and ZnO NPs77 show anti-QS activity against ESKAPE pathogens. They interfere with the AHL production, downregulate luxA and luxR, and attenuate the secretion of several virulence factors mediated by the QS system.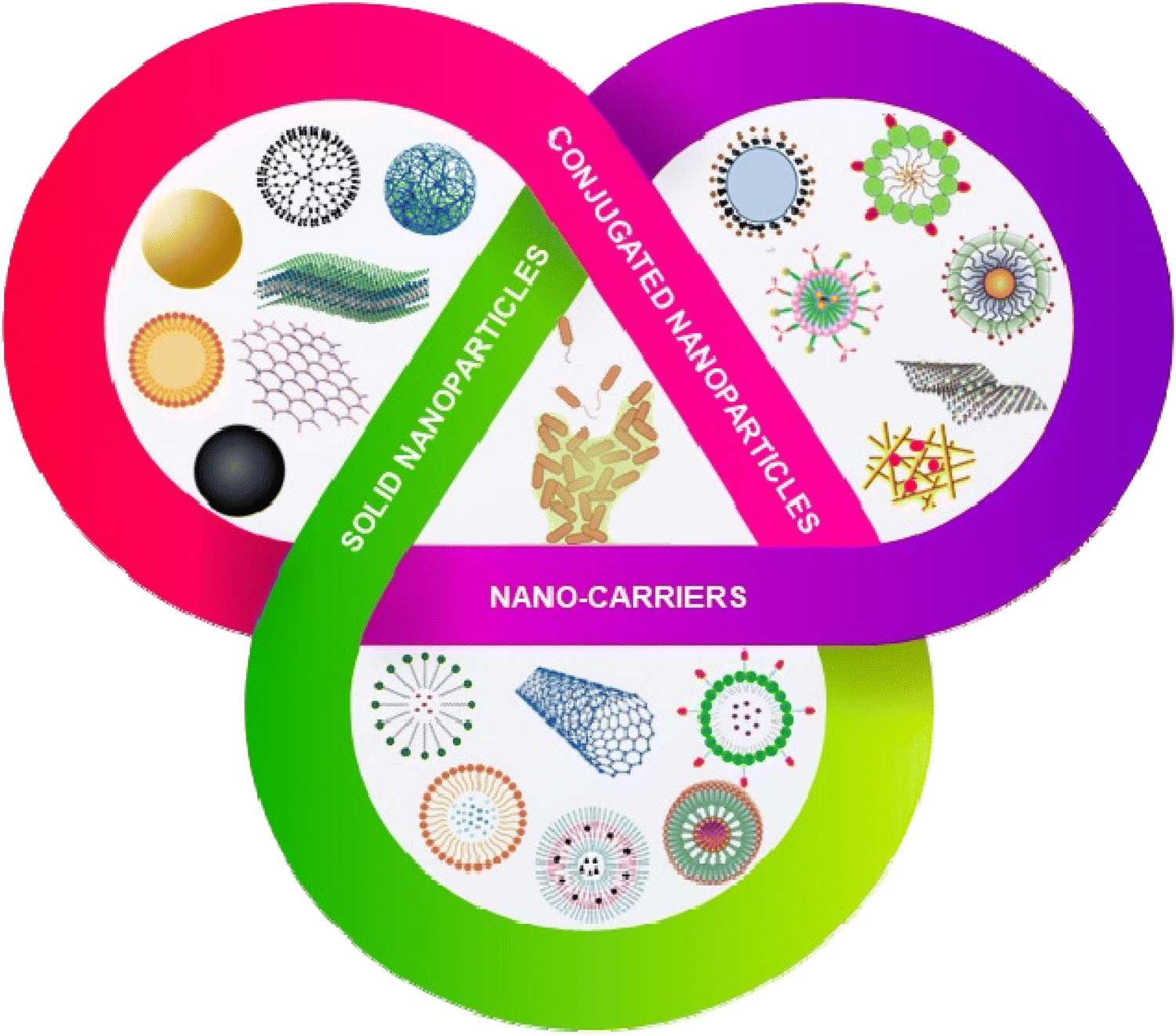 | ||
| Scheme 4 Nanotechnology based approaches to eradicate biofilms. Nanomaterials can be classified as solid nanoparticles, conjugated nanoparticles and nanocarrier systems. | ||
4.1. Inorganic nanoparticles and nanomaterials
Inorganic nanoparticles include a wide range of materials; namely, elemental metals, metal oxides, dichalcogenide, and others.78 Owing to their unique physicochemical properties, inorganic nanomaterials are widely used as antimicrobial and antibiofilm agents. The bactericidal action of the inorganic nanoparticles involves the alteration of the membrane potential, cell membrane damage, leakage of the cytosolic contents, generation of reactive oxygen species (ROS), oxidative stress and metabolic inactivation.79 Inorganic nanoparticles have also been modified by functionalization with different types of target specific molecules, like antimicrobial peptides and/or polymers. Some inorganic nanoparticles have also been explored as a nanocarrier to deliver different antibiotics and antimicrobial agents.Among different nanoparticles, silver nanoparticles (AgNPs) are considered one of the best antimicrobial metal nanoparticles.81 AgNPs have strong antibacterial and antibiofilm activity through binding with the cell wall of bacteria and damaging the cell membrane, causing leakage of the intracellular components, including proteins and nucleic acids.82 A 23%–86% biofilm inhibition of the K. pneumoniae strain was observed in the presence of different concentrations of AgNPs. Some other studies demonstrated that AgNPs disrupt the microbial membrane by leaching the Ag+ ions.83 To explore the antibiofilm property of gold nanoparticles (AuNPs), Qiao et al.84 functionalized gold nanorods (AuNRs) with NIR light-responsive and pH-activated surface charge changeable polymethacrylate having carboxyl betaine groups by reversible addition-fragmentation chain transfer polymerization reaction. The pH-responsive charge transformations led to the killing of the planktonic cells. Photothermal NIR irradiation causes local hyperthermia, leading to the death of S. aureus and consequently biofilm disruption (Fig. 1A). Khan et al.85 functionalized antibacterial fucoidan to stabilize AuNPs, which hampered the preliminary adhesion of the planktonic cells, thus inhibiting the biofilm formation. This conjugate also disintegrated the mature P. aeruginosa biofilm. Metal nanoparticles also act as a novel delivery system, wherein the P. aeruginosa PAO1 biofilm was disrupted and eliminated from the lungs of mice by a silver nanosystem (Ag@MON-AE) carrying AgNPs and a mesoporous organosilica layer for the delivery of alginate lyase and ceftazidime (Fig. 1B).86 Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images of Ag@MON-AE showed an alginate lyase-loaded Ag nanocomposite. The Ag@MON-AE nanocomposite showed 73.71% biofilm degradation at pH 6.4. The acidic environment of the biofilm increased the activity of alginate lyase, and was responsible for the release of ceftazidime. The synergistic activity of silver, alginate lyase and ceftazidime demonstrated the high biofilm degradation of PAO1. In another study, Wang et al.87 fabricated a multilayered film that consisted of gentamicin, tannic acid and silver nanoparticles coated on magnetic nanoparticles. The magnetic nanoparticles were further coated with hyaluronic acid to make it biocompatible. The nanocomposite could permeate through the S. aureus and E. coli biofilm by their magnetic field navigation, and exhibited strong antibiofilm and antibacterial efficacy. High ROS production in the presence of the magnetic field caused decomposition of the biofilm matrix, and finally led to the removal of the biofilm. Fig. 1C shows the confocal images of the control S. aureus biofilm, and the biofilm with and without a magnetic field. After the application of the magnetic field, the biofilm structure started to disintegrate and bacterial cells started to die. A copper nanoparticles (CuNPs)-encapsulated liposomal formulation was prepared to increase the effectiveness of the lipopeptide biosurfactant.88 The as-prepared nanoformulation reduces the cellular metabolism, and also influences the secretion of different virulence factors of MRSA and P. aeruginosa. High content screening (HCS) imaging of the biofilm using SYTO 9 and PI demonstrated that EL-LP-CuNP had immense killing efficiency at ½ MIC concentrations against both P. aeruginosa and MRSA (Fig. 1D).
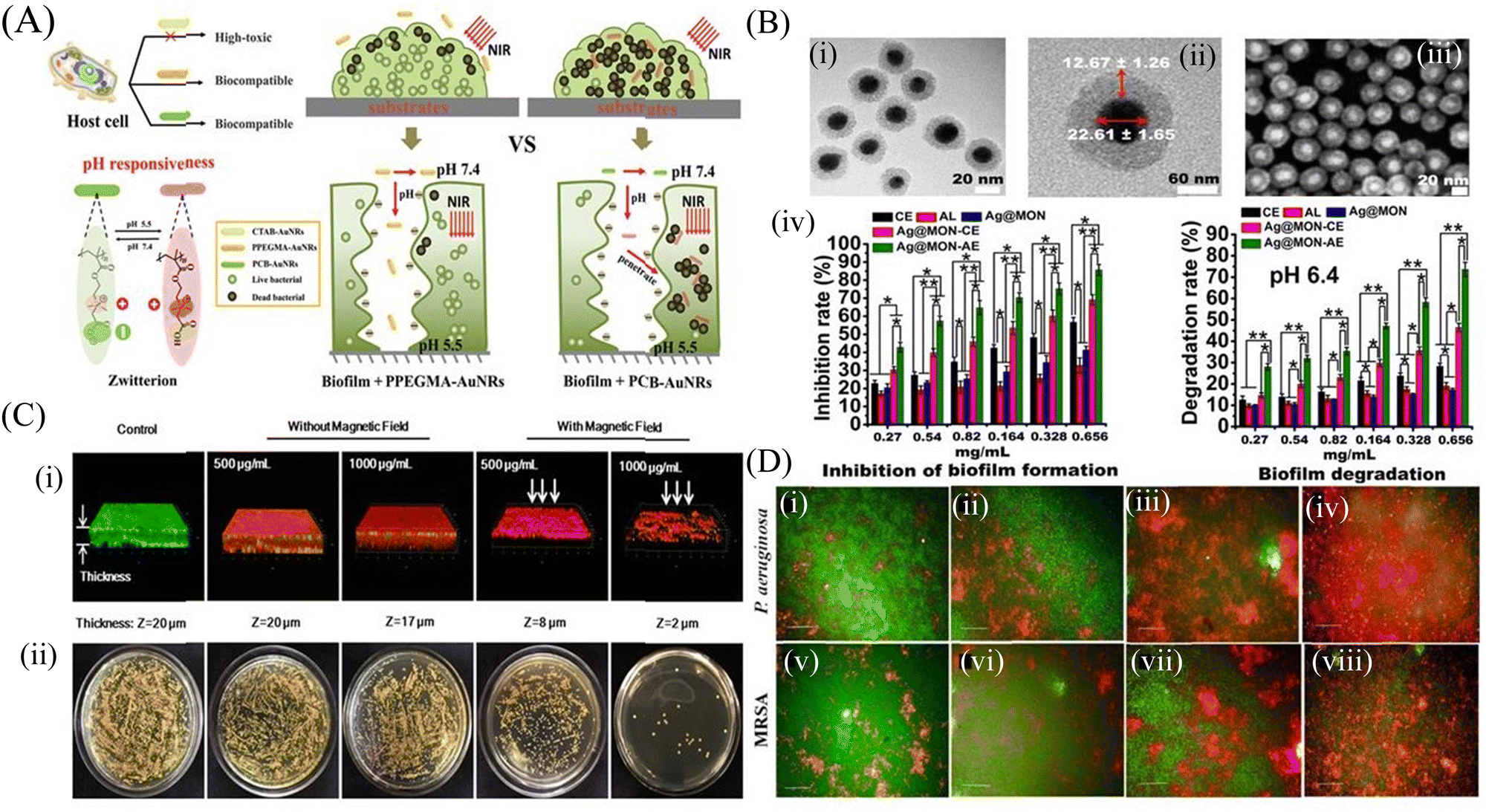 | ||
| Fig. 1 Inorganic nanomaterials: (A) Illustration of gold nanorods with charge-switchable properties and their interactions with mammalian cells and biofilms. (B) Characterization of the Ag@MON nanoparticles: (i) TEM image of the silver nanoparticles coated with mesoporous organosilica, (ii) Structural characteristics of Ag@MON at TEM magnification, (iii) SEM image of Ag@MON, (iv) Restraining and degrading the biofilm formed by P. aeruginosa PAO1. P. aeruginosa PAO1 was incubated with different concentrations of PBS, AL (alginate lyase), CE (ceftazidime), Ag@MON, Ag@MON-CE, and Ag@MON-AE for 36 h at 37 °C in a 96 well plate. The biofilm formed by the bacteria was determined. In addition, the ability of the compounds to degrade the biofilm formed by P. aeruginosa PAO1 after culturing for 72 h was analyzed under a pH 6.4 condition. (C) (i) Live/Dead staining of 3-D reconstructions of z-stacks collected across the S. aureus biofilms, and (ii) Bacterial colonies of surviving S. aureus in biofilms after treatment with MNPs@Ag@HA without and with an applied magnetic field (NdFeB, 2000 Gauss), respectively. (D) Membrane integrity assessment of preformed biofilms live and dead cells differentiated SYTO 9 and PI-stained P. aeruginosa and MRSA biofilms with treatment combinations: (i and v) Control biofilms without treatment, (ii and vi) Biofilms treated with LP alone, (iii and vii) Biofilms treated with CuNPs alone, (iv and viii) Biofilms treated with LP + CuNPs (50 μM). (A) Reprinted with permission from ref. 84. (B) Adapted with permission from ref. 86. (C) Reprinted with permission from ref. 87. (D) Reprinted with permission from ref. 88. | ||
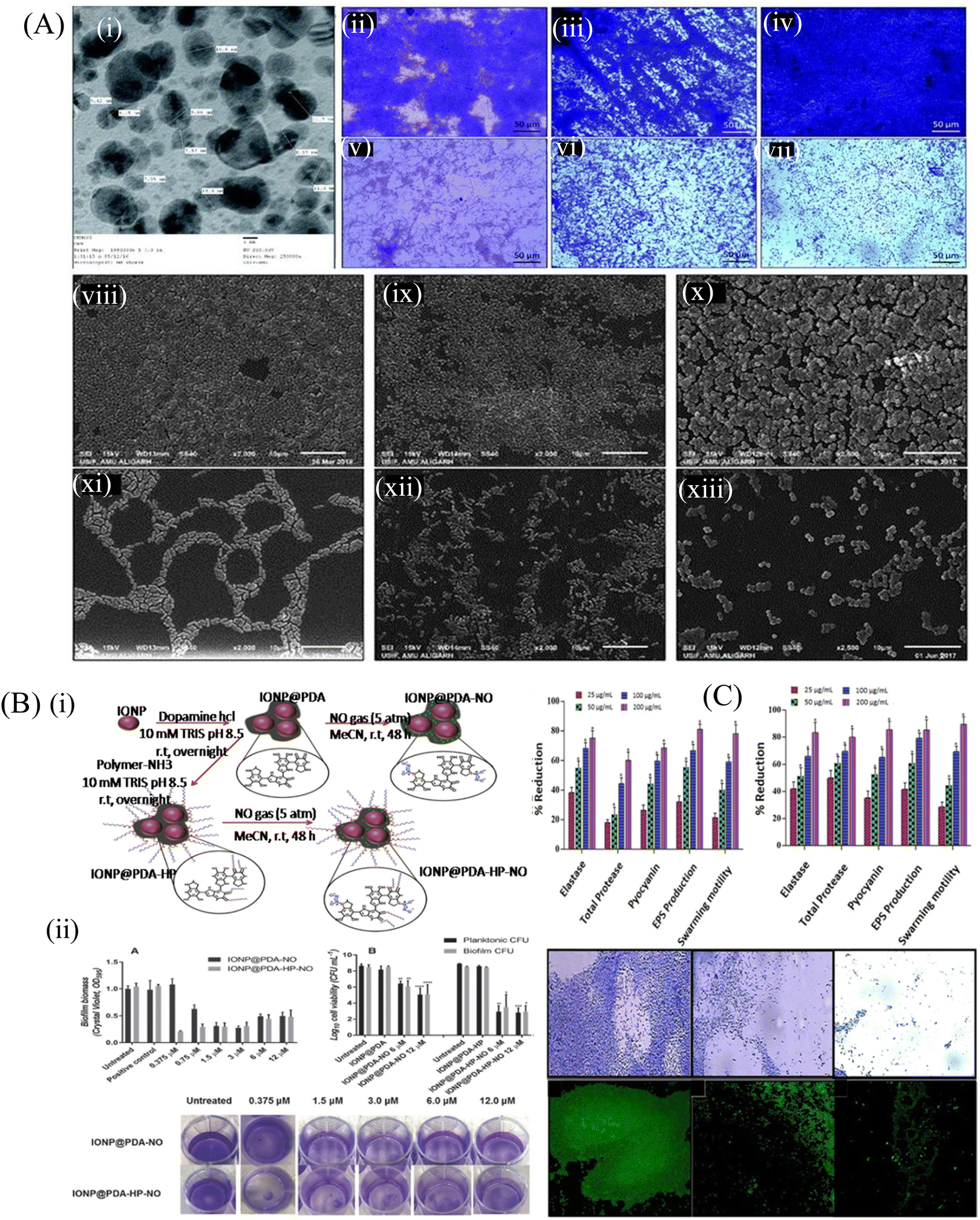 | ||
| Fig. 2 (A) (i) TEM of TiO2 NPs recorded at 250000 magnifications and 200 kV. Light microscopic images showing the effect of TiO2 NPs on the biofilm development of test bacteria, (ii) control E. coli ATCC 25922, (iii) control P. aeruginosa PAO1, (iv) control S. aureus MTCC 3160, (v) E. coli ATCC 25922 treated with 64 mg mL−1 TiO2 NPs, (vi) P. aeruginosa PAO1 treated with 64 mg mL−1 TiO2 NPs, (vii) S. aureus MTCC 3160 treated with 64 mg mL−1 TiO2 NPs. SEM images showing the effect of TiO2 NPs on the biofilm development of test bacteria, (viii) control E. coli ATCC 25922, (ix) control P. aeruginosa PAO1, (x) control S. aureus MTCC 3160, (xi) E. coli ATCC 25922 treated with 64 mg mL−1 TiO2 NPs, (xii) P. aeruginosa PAO1 treated with 64 mg mL−1 TiO2 NPs, (xiii) S. aureus MTCC 3160 treated with 64 mg mL−1 TiO2 NPs. All images were captured at 2500 magnification. (B) (i) Schematic illustration of the synthesis of the polydopamine-coated iron oxide nanoparticles (IONP@PDA) and the subsequent reaction with nitric oxide (NO), forming N-diazeniumdiolates (NONOates)-functionalized IONP@PDA (IONP@PDA-NO). Another route of synthesis involves the reaction of IONP@PDA with P(OEGMA)-b-P(ABA) by the Michael addition reaction, forming P(OEGMA)-functionalized IONP@PDA, IONP@PDAHP. NO was conjugated onto the IONP@PDA-HP to form NONOate-functionalized IONP@PDA-HP, hereby denoted as IONP@PDA-HP-NO, (ii) antibacterial activity of NO-functionalized nanoparticles on P. aeruginosa. Bacterial biofilms were grown in multiwell plates for 6.5 h in the absence of any treatment before being treated for a further 30 min with IONP@PDA-NO and IONP@PDA-HP-NO based on various concentrations of NO. The biofilm biomass was analyzed by crystal violet staining. Error bars represent standard error (n ≥ 2). (B) Effect of the NO-functionalized nanoparticles on P. aeruginosa viability after the release of NO. P. aeruginosa biofilms were grown in multiwell plates for 6.5 h in the absence of any treatments before being treated for further 30 min in the presence of the nanoparticles at 6 and 12 × 10−6 of NO. CFU analysis was used to determine the planktonic and biofilm viability. Error bars represent standard error (n ≥ 2). Asterisk indicate statistically significance difference of NO-loaded nanoparticles versus non-NO-loaded nanoparticles (*, P < 0.1; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). Stained biofilms treated with the indicated concentrations of NO. (C) QS-regulated virulence traits of PAO1: effects of sub-MICs of (i) ZN1 and (ii) ZN2 on inhibition of the QS-regulated virulence factor. Data are expressed as mean % reduction ± SD. Antibiofilm activity of ZNs: images of P. aeruginosa biofilm, (iii–v) crystal violet staining under a light microscope. Acridine orange staining under a CLSM, (vi) control, untreated, (vii) 200 μg mL−1 of ZN1, and (viii) 200 μg mL−1 of ZN2. (A) Reprinted with permission from ref. 93. (B) Adapted with permission from ref. 96. (C) Reprinted with permission from ref. 97. | ||
Some of the nanoparticles are reported to inhibit QS within the bacterial population. Khan et al.97 explored the antibacterial and antibiofilm efficacy of zinc oxide nanospikes (ZNs), i.e., ZN1 and ZN2, against the biofilm of P. aeruginosa (PAO1). ZNs prevented the QS signaling controlled by N-acyl homoserine lactone. Fig. 2C shows the QS inhibiting activity of both ZN1 and ZN2 at their sub-MIC concentration by interfering with the production of different virulence factors in P. aeruginosa. At a concentration of 25–200 μg mL−1, ZN1 demonstrated a dose-dependent decrease in the elastase (35%–74%), total protease (17%–58%) and pyocyanin (25%–67%) production, whereas ZN2 showed more efficacy than ZN1 at the same concentration. Microscopic analysis of the treated biofilm showed a significant decrease in the micro-colonies of the biofilm. The light microscopy result was further confirmed by confocal laser scanning microscopy (CLSM), which showed reduced biofilm biomass after the treatment with 200 μg mL−1 ZN1 concentration. This study highlighted the QS inhibiting activity of ZN, which further reduced the production of different virulence factors, and thus controlled the formation of biofilm. Tian et al.98 fabricated chitosan and polyethylene glycol (PEG)-coated Fe3O4 nanoparticles to load an antibiotic gentamicin, which showed a strong antibiofilm efficacy at a concentration of 500 μg mL−1 against the S. aureus biofilm. Dwivedi et al.95 observed that ZnO-NPs synthesized by a soft chemical/solution process were able to eradicate the biofilm and kill P. aeruginosa by generating intracellular ROS, which further affected the bacterial DNA and membrane. The low-resolution TEM image of ZnO-NPs demonstrated the smooth spherical morphology with the dimension of the NPs in the scale range of 10–15 nm. The TEM image of the NPs demonstrates the lattice fringes, separated by 0.265 nm, and this was similar to the lattice constant of the wurtzite ZnO phase.
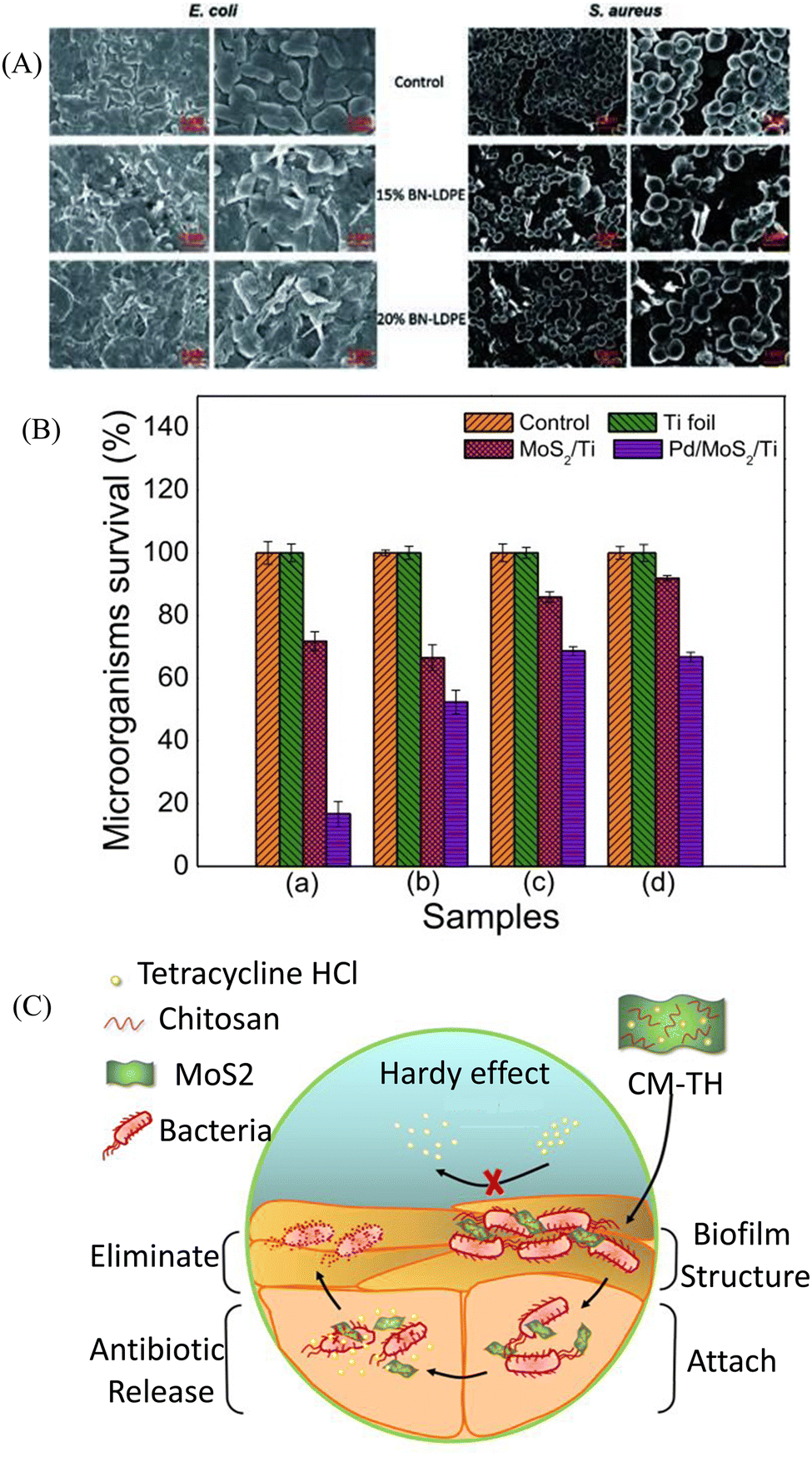 | ||
| Fig. 3 (A) Representative SEM images of E. coli and S. aureus. E. coli and S. aureus were grown for 24 h on LDPE (control) and LDPE composites with 15% and 20% of BN–LDPE composites, and grown bacterial cells were fixed and dehydrated before SEM imaging. (B) MoS2/Ti and Pd@MoS2/Ti coatings incubated in liquid nutrient broth and sabouraud media in the presence of P. aeruginosa (a), M. luteus (b), C. parapsilosis (c) and C. krusei (d) microorganisms. (C) Scheme of the CM-TH for anti-biofilm applications. Histogram obtained from quantitative analysis. (A) Reprinted with permission from ref. 101. (B) Adapted with permission from ref. 103. (C) Reprinted with permission from ref. 104. | ||
4.2. Carbon-based nanomaterials
Carbon-based nanomaterials are formed from the carbon material with a variety of nanostructures including carbon dots, carbon quantum dots, carbon nanotubes (CNT) and 2D nanomaterials, such as graphene oxide (GO), reduced graphene oxide (rGO) and others. Carbon-based nanostructures have been evaluated as an antibiofilm agent owing to their strong physicochemical and mechanical properties involving physical and chemical damage, including cell membrane damage, metabolic inactivation and ROS-mediated oxidative damage to the bacterial cells.105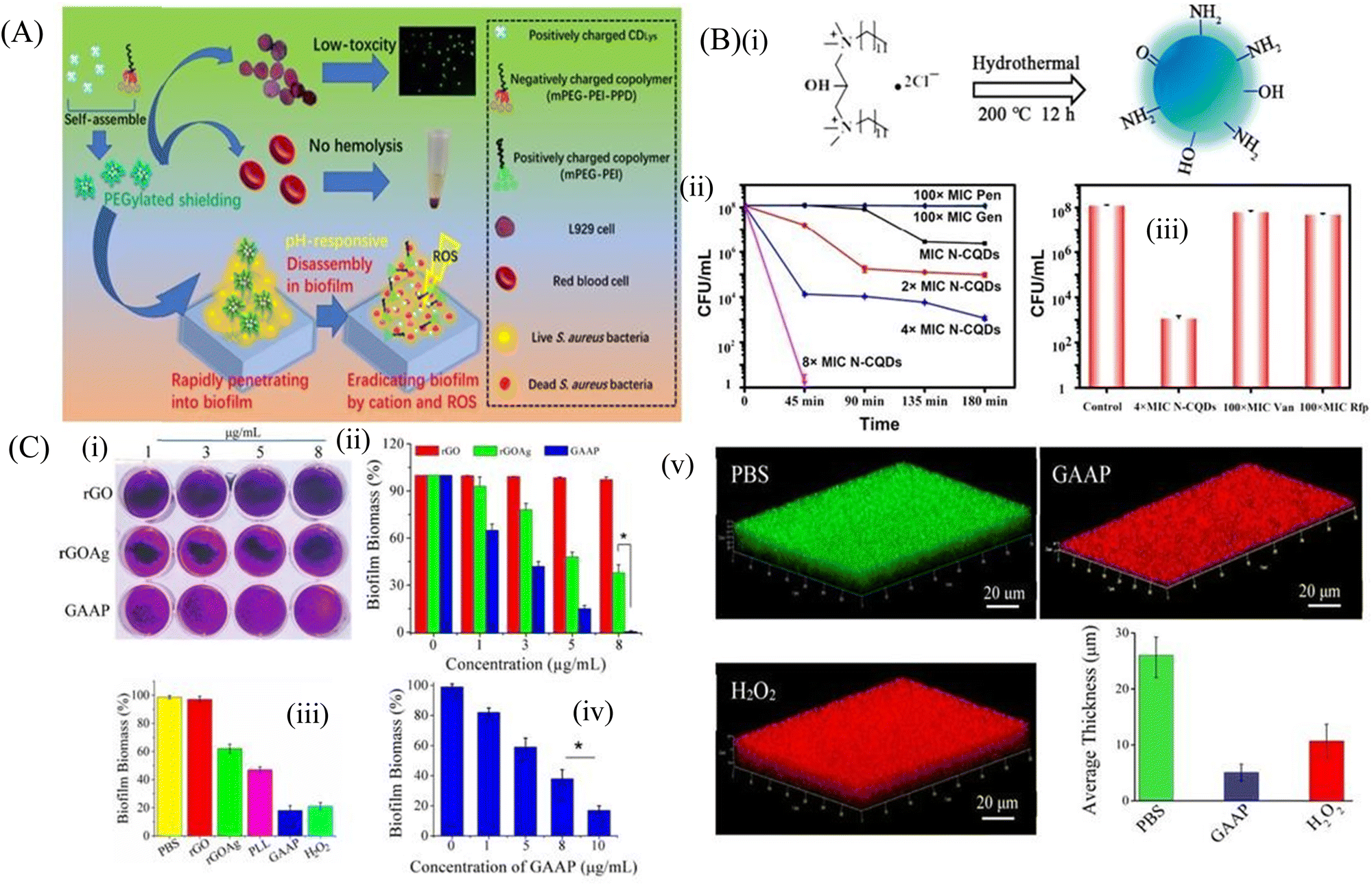 | ||
| Fig. 4 (A) Schematic representation of carbon dots. (B) (i) Synthesis route of N-CQDS, (ii) N-CQDs, Pen, and Gen were used to fight against persisters. Stationary-phase MRSA cells were treated with different concentrations of N-CQDs (MIC, 2 × MIC, 4 × MIC, and 8 × MIC) and 100 × MIC conventional antibiotics, (iii) Viability of stationary-phase MRSA cells treated with the indicated concentrations of each material for 3 h. The error bars indicate means ± SD (n = 3). (C) Crystal violet assay (i) to measure biofilm formation on rGO, rGOAg, and GAAP after incubation of S. aureus for 120 h. (ii) Concentration-dependent biofilm inhibition activity of rGO, rGOAg, and GAAP against S. aureus, (iii) activity of rGO, rGOAg, PLL, and GAAP measured by crystal violet staining, (iv) concentration-dependent biofilm disruption by GAAP. (v) Confocal microscopic images of the S. aureus biofilm treated with PBS (pH 7.2), GAAP (10 μg mL−1), and H2O2 (100 mM). Scale bar, 20 μm. The biofilm was grown for 72 h and treated with GAAP for 5 h at 37 °C; H2O2 was used as a positive control and PBS was used as a negative control. (A) Reprinted with permission from ref. 107. (B) Reprinted with permission from ref. 110. (C) Reprinted with permission from ref. 119. | ||
Carbon quantum dots (CQDs) have physicochemical properties similar to those of C-dots, and are used as a great antibiofouling and antibacterial agent. Their small size helps them to easily pass through the biofilm matrix and eradicate the persister cells inside the biofilm. They can be easily formulated by chemical ablation, electrochemical carbonization, microwave irradiation and hydrothermal treatment.109 Wang et al.110 used bis-quaternary ammonium salt as a carbon and nitrogen source to synthesize nitrogen-doped carbon quantum dots (N-CQDs) nanobiotics (Fig. 4B). The image also demonstrated that the high doses of antibiotics like penicillin and gentamicin (100 × MIC) at different time intervals showed no inhibition of the biofilm even after 4 h of treatment. In contrast, N-CQDs completely eradicated the persister cells of the biofilms in 45 min at a concentration of 8 × MIC. It was also shown that at 4 × MIC of N-CQDs, 90% of the persister cell population was destroyed. On the other hand, 100 × MIC of vancomycin and rifampin were able to eradicate only a small amount of the persister cells of biofilm. This N-CQDs nanosystem also showed strong in vitro antibacterial and antibiofilm activity against both MRSA and ampicillin-resistant E. coli bacteria without exhibiting any resistance. In addition, it restricted the biofilm formation and disrupted the matured biofilm, and finally accelerated the wound healing cascade when applied on infected wounds. The positively charged N-CQDs attached to the negatively charged bacterial membrane by electrostatic interaction. This led to the membrane damage and increased cellular permeability, which accelerated the insertion of N-CQDs into the cell and finally damaged the DNA through intracellular ROS production. The N-CQDs nanosystem also inhibited the intracellular metabolic pathways of MRSA. Peili et al.111 synthesized a series of CQDGents from gentamicin sulfate by direct calcinations. The CQDs exhibited antibacterial efficacy as compared to the individual antibiotics. Consequently, the efficacy of these CQDs on the fully matured S. aureus biofilm was assessed. The biofilm disrupting capacity of CQDs at 80 μg mL−1 was more than 99% as compared to individual antibiotics, which could only disrupt the biofilm by 10%.
4.3. Lipid-based nanomaterials
Lipid-based nanoparticles like liposome, solid lipid nanoparticle and nanostructured lipid carrier also draw great attention in the biomedical field. Their versatility, biocompatibility, solubility and high drug loading capacity makes them potential biofilm-targeted nanosystems.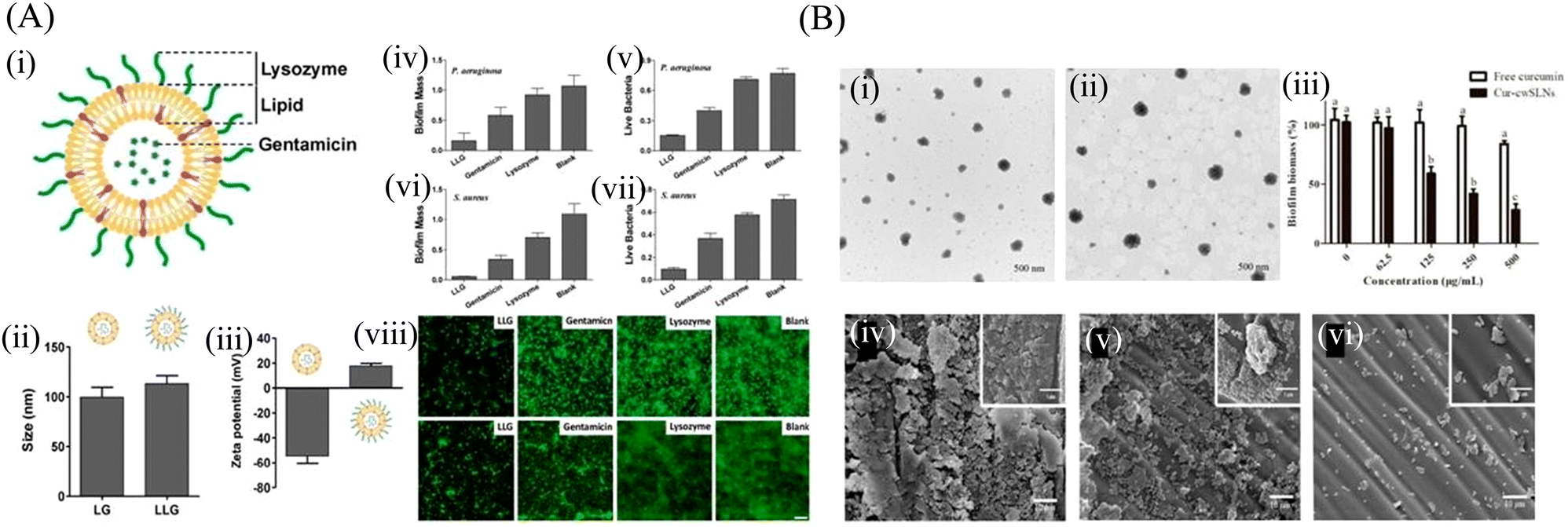 | ||
| Fig. 5 (A) (i) Schematic structure of lysozyme-associated liposomal gentamicin (LLG), (ii) hydrodynamic size, and (iii) surface zeta potential of liposome (without Lysozyme) and LLG, Crystal violet assay and 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay to assess the antibiofilm activity of LLG against P. aeruginosa biofilm (iv and v) and S. aureus (vi and vii). Fluorescence microscopy of P. aeruginosa (viii) and S. aureus (ix) biofilms. Biofilms incubated with tryptic soy broth (TSB) are used as the control. Scale bars were 10 μm. (B) The TEM of the SLNs (i) cwSLN, (ii) Cur-cwSLN, (iii) the cytotoxicity of cwSLNs and Cur-cwSLNs in 24 h. Data are presented as the mean ± SD, n = 3, (iv) SEM image of S. aureus biofilms, (v and vi) SEM image of inhibitory effects of pure curcumin and Cur-cwSLNs against biofilms produced by S. aureus, respectively. (A) Reprinted with permission from ref. 123. (B) Adapted with permission from ref. 131. | ||
4.4. Polymeric nanomaterials
Polymeric nanomaterials range from 10 to 1000 nm in size. There are different kinds of polymeric nanomaterials like polymeric shells or polymeric matrices. They are extensively used to deliver antimicrobial agents due to their high solubility index.132 They can be modified easily by different antimicrobial agents or peptides to achieve the desirable target-oriented removal of biofilms.:8 ratios of DSPE-PEG-HnMc and PLGA-PEG demonstrated better antibacterial efficacy than the HnMc prepared by 1:9 ratios at their critical micelle concentration (CMC). A 90% growth reduction of the drug-resistant P. aeruginosa CCARM 2073 and S. aureus CCARM 3078 was observed in the case of HnMc (2:8) at a concentration of 0.05 mg mL−1. Moreover, HnMc was able to inhibit biofilm formation of the above pathogens at a concentration of 0.1 mg mL−1, which established it as an excellent peptide-based nanomedicine for the treatment of bacterial infections. The micelle can be modified to make it stimuli-responsive to increase specificity towards the site of infections. Guo et al.138 synthesized the pH-sensitive micelle to target the biofilms, whereas Chen et al.139 formulated a pH/lipase-sensitive micelle to deliver ciprofloxacin to the P. aeruginosa biofilm. Guo et al.138 had synthesized three different types of pH sensitive copolymers named PAE–PLA–mPEG (A–L–E), PLA–PAE–mPEG (L–A–E) and PLA–PEG–PAE (L–E–A) composed of poly(β-amino ester) (PAE), poly(lactic-co-glycolic acid) (PLA) and polyethylene glycol (PEG), respectively, which were self-assembled in aqueous medium. The pH-sensitive chains that are PLA–PAE–mPEG, PAE–PLA–mPEG and PLA–PEG–PAE were self-assembled into micelles ML–A–E, MA–L–E and ML–E–A, respectively, where PLA was a hydrophobic core, PEG was a stable shell and PAE presented as pH-sensitive hydrophobic core moieties. They observed that at the triggering pH (pH at which the surface charge becomes reversed, pHt) on the biofilm surface, both the ML–A–E and ML–E–A were able to regulate the charge switching at the pH of 5.5 and 6.0, respectively (Fig. 6A). It is shown that ML–A–E effectively eradicates the implant-related biofilm infections, which can efficiently remove biofilms from the catheter. Chen et al.139 designed a vancomycin-functionalized micelle encapsulated with ciprofloxacin (CIP). Vancomycin-mediated targeting and pH/lipase-dependent release of CIP from the micelle shows higher survival of P. aeruginosa-infected mice. At pH 7.4 and 6.0, 20% release of CIP was observed, while in the presence of lipase enzyme, around 39 to 56% CIP release was found. A single dose of Van-hyd-PECL/Cip micelles reduced the bacterial load in lung tissues by around 3.1-log10 fold in comparison to the free Van-CIP. These micelles provided a workable infection microenvironment-responsive strategy to combat bacteria.
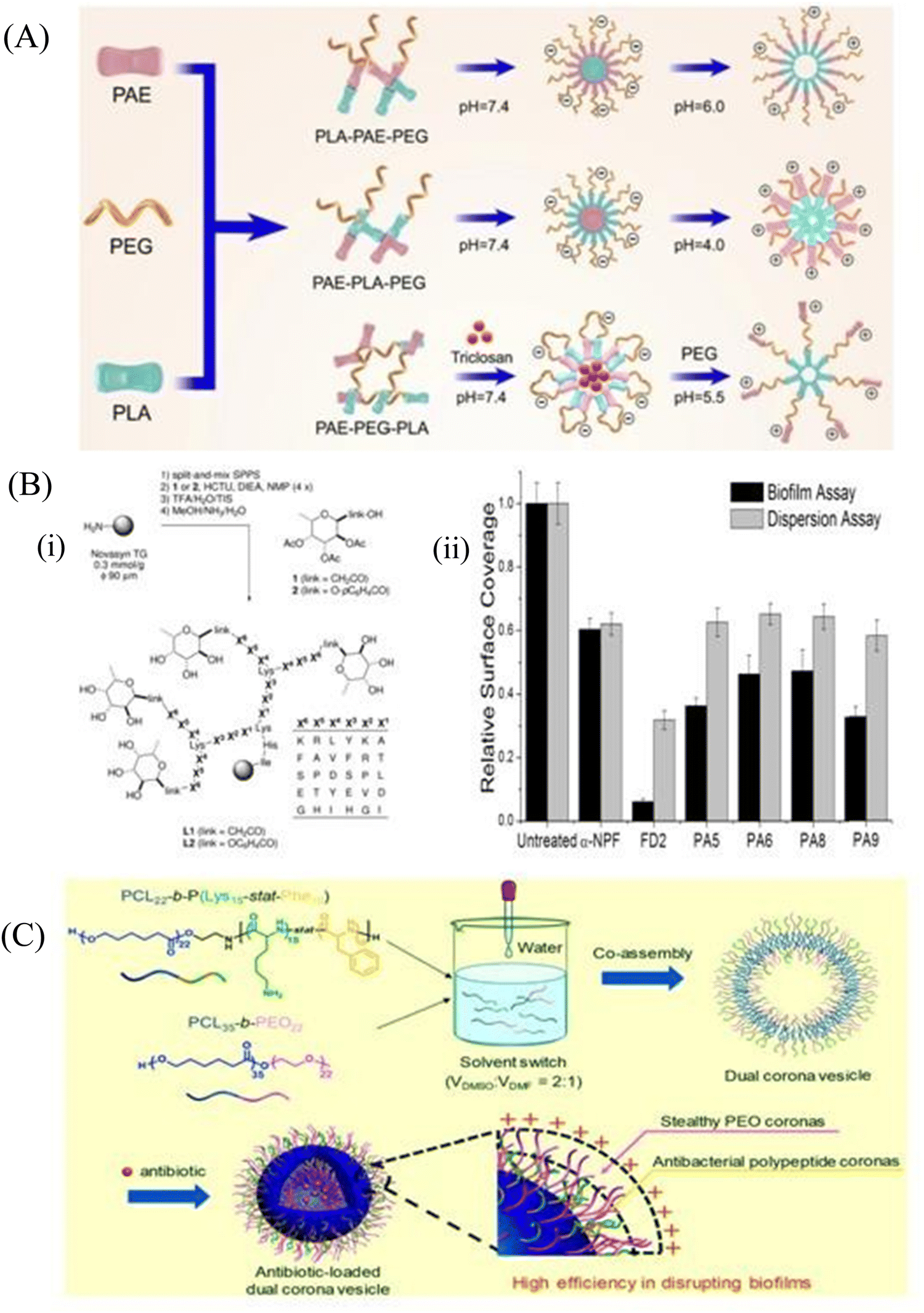 | ||
| Fig. 6 (A) Elaboration on the architecture of pH-sensitive copolymers with proper pH. (B) (i) Synthesis and structure of the C-fucosyl peptide dendrimer libraries, displayed on tentagel resin as solid support. The ‘link’ between amino acid X6 and the fucose is either an acetyl spacer in the C-fucosyl library L1 or phenoxyacyl in the O-fucoside library L2, obtained by coupling the corresponding building blocks 1 and 2. The positions X1–X6 display various amino acids according to the table at right (one-letter codes of L-amino acids) as a ‘one-bead-one-compound’ library obtained by the split-and-mix SPPS protocol, (ii) biofilm assays with P. aeruginosa and dendrimers. Biofilm inhibition and dispersion were tested with a-NPF (0.5 mM), dendrimers (0.05 mM), and wild-type strain PAO1. The values are normalized so that growth without any inhibitor is set to 1. A background control (no bacteria) gave reading values similar to the ones shown in the graph for the FD2 biofilm assay (0.05 mM). Mean values of three independent readings ± standard deviation are plotted. (C) PCL22-b-P (Lys15-stat-Phe10) and PEO22-b-PCL35 copolymers are co-assembled into a dual corona vesicle that can further encapsulate ciprofloxacin. (A) Reprinted with permission from ref. 138. (B) Reprinted with permission from ref. 143. (C) Reprinted with permission from ref. 147. | ||
4.5. Nanoemulsion
Emulsions are thermodynamically stable colloidal dispersions of two immiscible liquids. Emulsions can be macro, micro and nano size droplets depending upon their size. Nanoemulsions are a type of nanosized emulsions consisting of interdispersed hydrophobic and hydrophilic layers forming a nanocarrier system. Nanoemulsions are synthesized by high pressure homogenizer, high sheer stirring, ultrasound generator, and microfluidization.151 Owing to their small size along with solubilization properties, nanoemulsions can overcome the barrier of the biofilm matrix and deliver the antimicrobial agents to eradicate bacterial biofilms. Prateeksha et al.152 developed a nanoemulsion with the bioactive compounds of Gaultheria fragrantissima wall essential oil (EO), viz., eugenol (E-NE) and methyl salicylate (MS-NE) to inhibit the virulence and biofilm growth by E. coli O157:H7 (ECOH). COMSTAT analysis (Fig. 7A) demonstrated that the nanoemulsions made up of eugenol and methyl-salicylate prevented biofilm formation, and the substratum coverage and mean thickness were reduced by more than 80%. Nanoemulsion-loaded hydrogel coatings are preferably used to combat E. coli infestation on solid surfaces by their antibiofouling strategy. In another study, Ramalingam et al.153 synthesized a nanoemulsion containing EDTA with a mean diameter of 269 nm to show its inhibitory effect on the biofilm formation by A. baumannii. The nanoemulsion particularly prevented the adherence of the bacterial cells on the glass surface and restricted biofilm formation. For further stabilization of the nanoemulsion, various crosslinked nanoemulsions were prepared. Oz et al.154 synthesized a novel formulation based on a nanoemulsion, which was stabilized by cross-linked PLA (Fig. 7B). This polymer scaffold was highly antibacterial and exhibited strong antibiofilm efficacy against matured E. coli, A. baumannii, and MRSA biofilm. This stable nanoemulsion was made by the cross-linking of maleimide-functionalized PLA (MA-PLA) with the help of a dithiol cross-linker via in situ maleimide-thiol Michael addition. To increase its solubility, a dithiol crosslinker was developed. The nanoemulsion was loaded with carvacrol to disrupt the matured biofilms. Li et al.155 prepared a gelatin photo-crosslinked carvacrol nanoemulsion using a riboflavin photocatalyst. This antimicrobial nanoemulsion exhibited antibiofilm efficacy, and subsequently accelerated the wound healing by eradicating the wound biofilm of P. aeruginosa and MRSA (Fig. 7C). In spite of this progress, there are many drawbacks associated, such as poor stability due to the continuous fusion of the droplets, causing their aggregation. In addition, they are non-specific to the bacterial cell membrane.154 To overcome these limitations, Song et al.156 fabricated an antibacterial and antibiofilm chlorhexidine acetate nanoemulsion (CNE) for MRSA-infected skin burn wounds. CNE mostly targets and damages the cell wall and cell membranes of MRSA, leading to the release of alkaline phosphates, K+, Mg2+, DNA and protein, causing increased relative electrical conductivity. Hwang et al.157 demonstrated that ananoemulsion formulated with cetylpyridinium chloride (CPC) possessed antibacterial activity against planktonic and biofilm-forming A. baumannii. The CPC nanoemulsion helps in killing the planktonic bacterial cells, while the emulsified oil and detergent fractions degrade the biofilm structure. Recently, Das et al.158 developed a novel cationic nanoparticle conjugated oil in a water nanoemulsion to impart positive charges to the nanoemulsion for effective antibacterial and antibiofilm activity against S. aureus biofilms. The conjugated formulation penetrated the biofilm matrix and killed the persister cells through electrostatic interaction. In this manner, the matured biofilm was disrupted and new cells were restricted from adhesion, i.e., biofilm inhibition.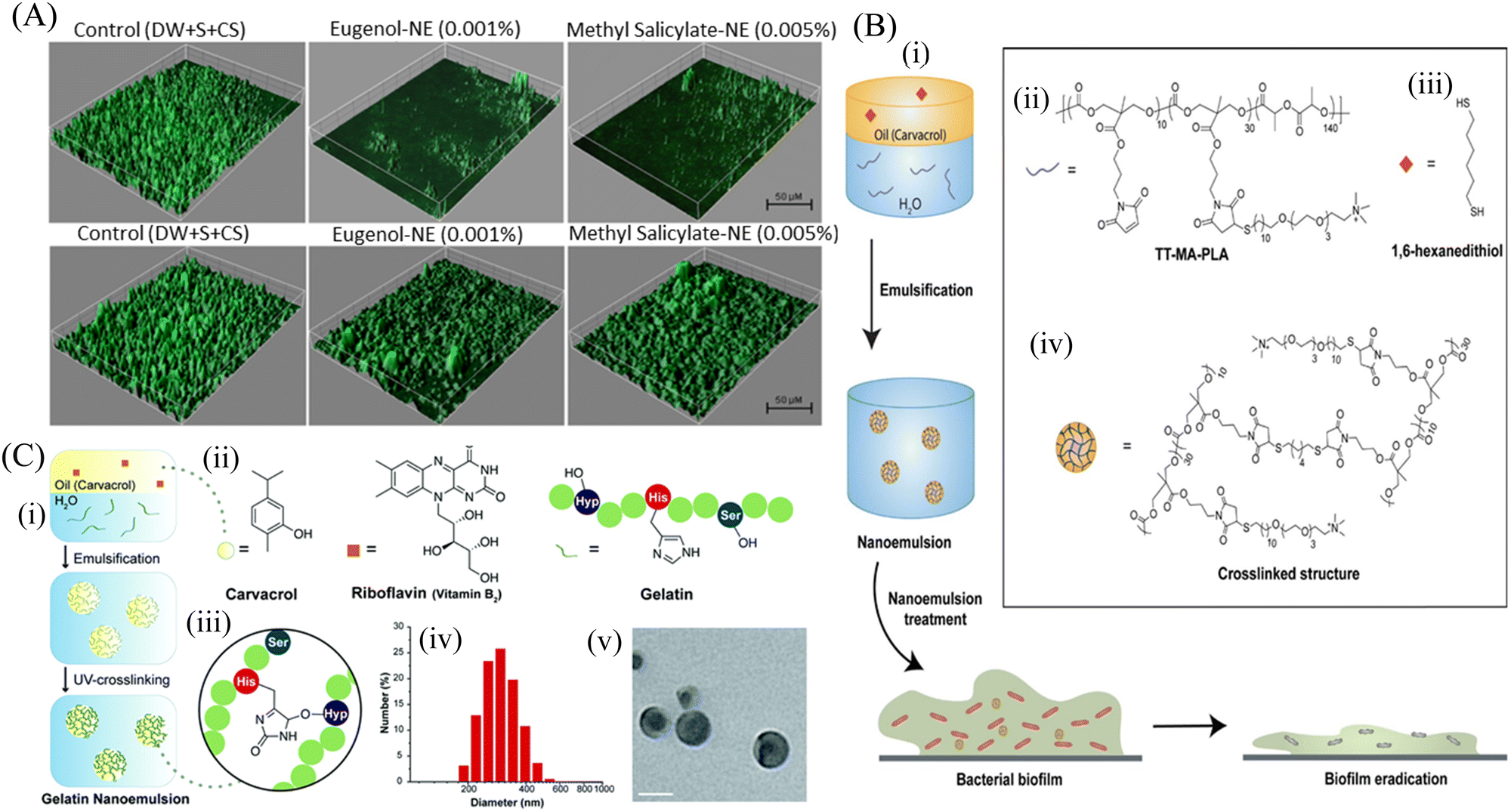 | ||
| Fig. 7 (A) Anti-biofilm activity of hydrogel coatings containing eugenol-NE and methyl salicylate-NE, eugenol and methyl salicylate against ECOH on glass surfaces, measured by CLSM. The mixture of DW, surfactant (S; Tween 80) and co-surfactant (CS; propylene glycol) in the ratio of 94.7:0.2:0.1 was used as a solvent control. (B) Schematic representation of the strategy utilized to generate carvacrol-loaded nanoemulsions, (i) resulting nanoemulsions showed improved antimicrobial activity against MDR bacterial biofilms, (ii) chemical structure of TT-MAPLA, (iii) chemical structure of 1,6-hexanedithiol, and (iv) Cross-linked structure of X-NEs. (C) Fabrication and characterization of gelatin nanoemulsions, (i) Riboflavin (UV cross-linking initiator) was dissolved in carvacrol. The oil mixture was then emulsified in an aqueous gelatin solution and cross-linked using long wavelength UV-A light (365 nm) to fabricate gelatin nanoemulsions, (ii) chemical structures of carvacrol, riboflavin, and the functional groups of gelatin participating in the cross-linking reaction, (iii) proposed cross-linked structure of gelatin nanoemulsions, (iv) dynamic light scattering histogram of nanoemulsions in PBS (150 mM), (v) TEM images of nanoemulsions. Scale bar is 100 nm. (A) Reprinted with permission from ref. 152. (B) Adapted with permission from ref. 154. (C) Adapted with permission from ref. 155. | ||
5. Antibiofouling mechanism of nanomaterials
The antibiofouling mechanism could be divided into two major activities, as follows: (1) inhibition of biofilm formation and (2) disruption of the existing biofilm (Scheme 5). Surface modification or the use of an agent that can interfere with the cell attachment to the surface can prevent biofilm formation. On the other hand, the preformed biofilm can be eradicated by the destruction of the EPS matrix and killing of the dispersed cells.159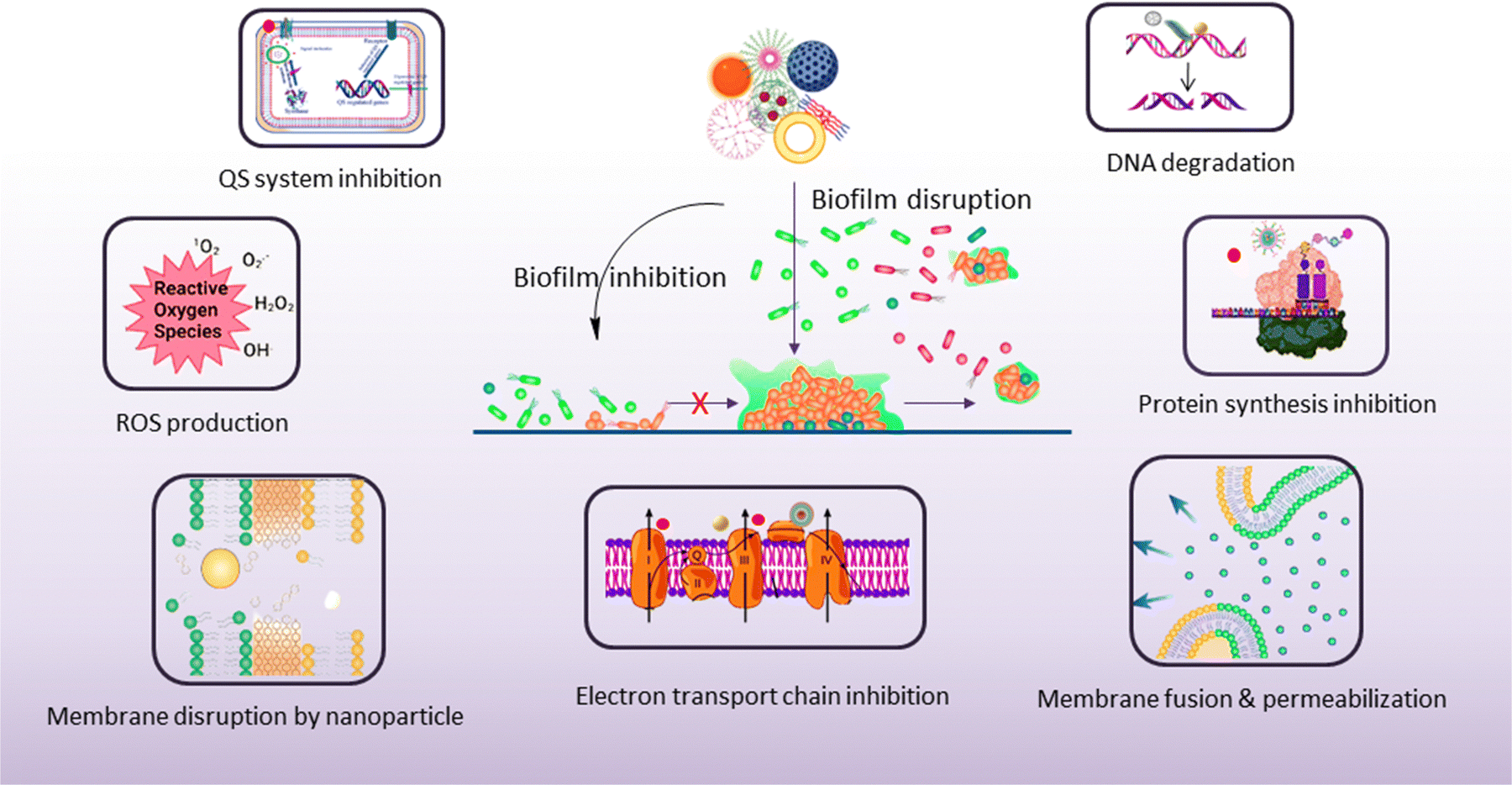 | ||
| Scheme 5 Biofilm inhibition and disruption mechanism of nanomaterials. | ||
5.1. Biofilm inhibition
Biofilm formation occurs in three consecutive stages, as follows: (i) attachment, (ii) colonization and proliferation of bacterial cells, and (3) maturation of the biofilms. Nanoparticles play an important role in the prevention of biofilm formation by inhibiting the initial attachment of planktonic cells on the substratum owing to its strong antibacterial activity. Nanomaterials inhibit bacterial adhesion because of their mobility creating an unstable substratum for the bacteria inhibiting the formation of a biofilm.112 Nanoparticles can reduce adhesion, maturation and proliferation of the biofilm by the formation of ROS, such as H2O2, hydroxyl radical (OH−) and superoxide ions (O2−). Nanomaterials interact with the bacterial cells and exhibit antibacterial activity through multiple pathways, which include membrane depolarization, ROS generation, inhibition of respiratory chain enzymes, damage of the intracellular components, inhibition of DNA replication and protein synthesis.158,160 Oxidative stress is an important factor mediated by metal nanoparticles that causes cytotoxicity and genotoxicity to the bacterial cells.161 Oxidative stress-related damage is of two types: ROS-dependent and ROS independent. The ROS-dependent pathway involves the overproduction of OH−, O2− and H2O2 to damage the cell membrane, destroy proteins, inhibit DNA replication and causes the leakage of intracellular components of the bacterial cells. Metal nanoparticles like AgNPs, AuNPs, and CuNPs bind with the bacterial cell membrane, resulting in the loss of cellular integrity, membrane damage, alteration of membrane permeability and exudation of cytoplasmic contents.80,90 In addition, ROS generation takes place, which leads to oxidative damage of the cellular components. Metal ions leached out from nanoparticles also contribute to antibacterial activity through the oligodynamic effect by destroying the proteins of the bacterial cell wall. Metal ions binding with the cell wall cause inhibition of respiratory chain dehydrogenase, uncoupling of oxidative phosphorylation, and metabolic arrest in the cells.162 Nanoparticles like AuNPs, AgNPs, and CuNPs also show peroxidase-like activities by converting H2O2 into toxic OH radicals to kill bacteria.163–165 The size, shape and surface properties of the nanoparticles play a pivotal role in the inhibition of biofilm formation. Smaller size nanoparticles show high antibacterial activity due to the high surface area-to-mass ratio and large interaction with the bacterial surface. It results in the disintegration of the bacterial cell wall, alteration of the membrane potential, destabilization of the outer membrane and leakage of the cytoplasmic contents.166,167 The shape of the nanoparticles also plays a significant role in the contact killing of bacterial cells. Sharp and pointed nanoparticles rupture the cell membrane of bacteria, leading to the leakage of cytoplasmic contents. Surface functionalization of the nanoparticle is another important factor, which enables multiple interactions with the bacterial cells and enhances the biofilm inhibition properties of nanomaterials. Surface modification of nanoparticles with different ligands and chemical groups, such as antimicrobial peptides, different polymers, and aptamers, strengthens interactions such as hydrophobic, electrostatic and van der Waals interactions between bacteria and nanoparticles. Cationic nanoparticles bearing a net positive charge interact with the negatively charged bacterial cell surface, leading to enhanced uptake of nanoparticles into the cells.168 This causes surface charge alteration, depolarization of the membrane potential, intracellular ROS generation and membrane pore formation.Nanomaterials can also be modified in such a way that it can interfere with the intracellular signaling pathways and disturb the cellular homeostasis. These disruptions include alteration in gene expression, DNA damage and protein synthesis. AuNPs functionalized with 4,6-diamino-2-pyrimidinethiol (DAPT) are able to inhibit the synthesis of bacterial tRNA.169 Au-DAPT binds to the genetic material, ribosomes, and inhibits protein synthesis of MDR strains of E. coli and P. aeruginosa. Au-DAPT also chelates Mg2+ ions, which results in destabilization of the bacterial cell membrane, followed by cell death. Nanoparticles can also act as carriers for loading antimicrobial drugs to increase the effectiveness, biocompatibility and bioavailability of antibiotics.99,121 Antibiotics, drugs and different therapeutics can be easily delivered either by encapsulating or conjugating with the nanomaterials.127,139 These nanomaterials protect them from enzymatic degradation, enabling prolonged circulation by increasing their half-life and sustainable targeted drug release, thus improving the pharmacokinetics efficacy.132,170 The utilization of the delivery system increases the stability, solubility and biocompatibility of challenging therapeutics. Targeted drug delivery using nanomaterials minimizes the selection of resistant strains, and allows target organ accumulation by functionalized surface modification, thereby limiting systemic side effects and immunosuppression. Nanoparticles can also respond in the presence of certain exogenous stimuli, such as pH, enzymes and chemicals, leading to effective delivery of therapeutics in the bacterial microenvironment.171,172
5.2. Biofilm disruption
Biofilm disruption is the main therapeutic challenge because the EPS provides a barrier against antimicrobial agents. The deeper layers of the biofilm have low concentrations of oxygen and nutrients, promoting the growth of persister cells, thereby resulting in increased antimicrobial tolerance and resistance. Nanomaterials have a unique size that enables them to penetrate the EPS matrix of the biofilm. The smaller size and higher aspect ratio have greater penetration properties, which is most effective in the eradication of biofilms.173,174 The surface charge properties of nanoparticles help in the efficient eradication of biofilm. The surface chemistry should be properly identified so that it leads to effective penetration into the biofilm. Stealth properties and biocompatibility through surface modifications make the nanoparticles more effective towards sufficient accumulation of nanomaterials inside the biofilm. Modifying the surface makes the nanoparticles more specific towards the biofilm. Anionic and zwitterionic nanoparticles have low EPS matrix penetration ability, whereas cationic nanoparticles with proper hydrophobicity can penetrate the EPS efficiently. Cationic nanoparticles can efficiently bind to the negatively charged matrix components due to the strong electrostatic force of attraction between the nanoparticles and the matrix.175 The shape of the nanoparticles plays an important role in biofilm disruption. Spherical nanoparticles can penetrate the biofilm very easily, so it can be used to deliver therapeutics inside the biofilm.176 Due to its small size, the diffusion of the nanocarrier carrying the therapeutic agent favors the killing of persister cells inside the biofilm matrix.177 The shape also plays an important role in generating heat upon NIR irradiation due to localized plasmon resonance. When the temperature of such nanoparticles is raised inside the biofilm, it is sufficient to kill the bacteria inside the biofilm.178 Nanomaterial penetration profiles are directly related to the success of biofilm elimination. After entering the EPS matrix of the biofilm, nanomaterials kill the sessile and persister cells by their own antibacterial effect or by release of therapeutic agents in a similar fashion, as described earlier in biofilm inhibition. Polymeric and lipid nanoparticles loaded with antimicrobial agents effectively pass and disrupt the biofilms, resulting in the killing of the pathogens, whereas traditional antibiotics fail to perform.146 Due to the distressing intramolecular forces, the AgNPs perturb the biofilm matrix.179 Although AuNPs have very little or no antibacterial activity, functionalized AuNPs enhance the activity of antimicrobial agents by delivering the agents into the biofilm.169 Micelles with surface charge can efficiently accumulate into the biofilm, showing significant destruction of the biofilm and bacteria within it.139 In addition, nanomaterials deliver matrix-degrading enzymes, nuclease, and other small molecules inside the biofilm and disperse the EPS matrix.180 Nanomaterials also inhibit the biofilm growth through interrupting the bacterial communication system, more specifically, the QS signaling pathway. Khan et al.97 showed that ZnO nanospikes had the ability to inhibit the QS of P. aeruginosa and prevent biofilm formation. These nanospikes inhibit the QS signaling and also affect the production of virulence factors, which is necessary to form the biofilm. Altogether, the nanomaterials provide a unique way to combat the deadly bacterial infection, showing that several mechanisms of action, low therapeutic doses, and targeted delivery of drugs minimize bacterial resistance against nanoparticles.6. Biomedical application of nanomaterials
Biofilm formation has been a major concern in the biomedical field since it may develop on surgical equipment, hospital beds, different medical implants, drug delivery devices, and most commonly on catheters. Besides the formation of the biofilm on the external substrates, the growth of biofilms also occurs on peripheral surfaces such as the wound bed. Biofilms may also form in internal organs, such as cardiovascular, dental, digestive, reproductive, respiratory, integumentary organs and the urinary system. According to the global report on sepsis by WHO, 24.4% mortality was reported among patients infected with health care-associated sepsis, which was increased to 52.3% among patients treated in ICUs.181 To treat the biofilm-linked morbidity and mortality, replacement of the contaminated apparatus and treatment with antibiofilm agent remain mandatory. Surface coating on medical devices, including metal nanoparticles,182,183 CNT,184 GO and rGO,185–188 has been one of the growing strategies to prevent biofilm colonization. Tran et al.183 synthesized a formulation of silver nanoparticles conjugated with polydopamine (DA) and a biofilm-lysing enzyme (α-Amylase) to prevent the biofilm colonization on titanium substrates (Fig. 8A). The antibacterial effect of AgNPs denatured the bacterial cell wall and disintegrated the extracellular matrix of the S. aureus biofilm, thus reducing the biofilm biomass on the titanium substrate. Periprosthetic joint infections (PJI) associated with MRSA are caused due to orthopedic implant surgeries. MRSA biofilms in PJI are difficult to remove due to the poor penetration property of antibiotics. To overcome this limitation, CNT-coated medical devices were fabricated by Morco et al.115 Following treatment of the MRSA biofilm with CICNT coatings for 48 h, there were 60–80% reductions of MRSA biofilm growth observed. Furthermore, FESEM images of the biofilm depicted that the CICNT treatment significantly reduced the biofilm biomass as compared to the untreated biofilm, and thus CICNT restricted the bacterial proliferation and bacterial growth (Fig. 8B). In another study, Kumar et al.186 fabricated PCL composites consisting of graphene for orthopedic applications. PCL composites were fabricated with GO, rGO and amine-functionalized GO (AGO) with varied filler contents (1%–5%). Among all composites, AGO composites inhibited the biofilm growth. It was suggested that AGO with the synergistic role of oxygen and amine containing functional groups demonstrated improved modulus, stem cell proliferation, and biofilm inhibition suitable for orthopedic applications. The SEM image (Fig. 8C) demonstrated that PCL/GO_5, PCL/rGO_5, and PCL/AGO_5 nanocomposites prevented the biofilm formation. A thick bacterial biofilm was seen on the PCL surface. However, very little adhered bacteria were visible on the composite surfaces. Furthermore, Gundel et al.189 synthesized a nanoemulsion consisting of Lemongrass oil having potential antimicrobial efficacy, antioxidant properties and antibiofouling efficacy. The essential oil nanoemulsion inhibited the biofilm formation of P. aeruginosa and S. aureus (Fig. 8D). Due to its efficacy as a strong antimicrobial agent, this nanoemulsion was found to be a better alternative to the conventional antibiofilm treatment. Worley et al.190 developed dual-action NO-releasing alkyl chain-modified poly(amidoamine) dendrimers to study its antibiofilm efficacy against S. aureus, MRSA and P. aeruginosa biofilms. This alkyl chain-modified poly(amidoamine) dendrimer was fabricated with different generations (G1–G4), and was modified with butyl (short) or hexyl (medium) chains, along with their further modification with N-diazeniumdiolate NO donors. With the increase in the dendrimer size and functional group density, the antibiofilm efficacy of the NO-releasing dendrimers was enhanced. A combination of short and long alkyl chain modifications in the dendrimers had less toxicity at therapeutic levels with enhanced anti-biofilm activity along with NO release, implying synergistic advantage of conjugating multiple biocides on a single macromolecular scaffold. G1 hexyl and G4 hexyl dendrimers have shown excellent biofilm penetration in comparison to either G1 butyl or G4 butyl dendrimers in the S. aureus biofilm (Fig. 8E). Therefore, the hexyl-modified dendrimer exhibited better antibiofilm efficacies than the butyl-altered dendrimer due to their increased biofilm penetration, membrane intercalation and cell membrane damage. With the increase in the cellular membrane damage, there was more intracellular NO build up, leading to further increased antibiofilm activity without altering the cell membrane. Meeker et al.191 synthesized gold nanocages (AuNC) and gold nanocages-coated polydopamine (AuNC@PDA). Furthermore, it was loaded with daptomycin (AuNC@Dap/PDA), and then finally conjugated with surface specific antibodies against staphylococcal protein A (aSpa) (AuNC@Dap/PDA-aSpa), which made it a highly photoactive and selective nanodrug against S. aureus (UAMS-1 and LAC strains). AuNC@PDA and AuNC@Dap/PDA exhibited a 3log reduction of S. aureus upon irradiation of 808 nm laser due to the synergistic effect of both photothermal and antibiotics (Fig. 9A). Mai et al.192 designed a cationic liposome-based delivery system augmented with photodynamic therapy for the effective treatment of burn infections. They developed a cationic lipid-mediated nano-sinoporphyrin sodium (DVDMS) delivery system to examine the antimicrobial activity of cationic DVDMS-liposome (CDL)-conjugated with photodynamic antimicrobial chemotherapy (PACT) in both P. aeruginosa and its multidrug-resistant strain. The positively charged modified liposome accelerated the penetration of DVDMS inside P. aeruginosa. CDL-PACT caused ROS-mediated bacterial cell death, along with reduced virulence factor-related genes expression upon irradiation with a laser at 100 J cm−2. Fluorescence imaging of CSDP hydrogen was seen both in vitro and in vivo (Fig. 9B). This nanosystem led to the complete removal of the bacterial infection by membrane permeabilization, damage, and then accelerated the wound healing cascade by decreasing the cytokines level in the presence of different growth factors, such as the bFGF, VEGF, TGF-β1 and Hyp. Ashbaugh et al.193 developed an excellent tunable antibiotic-loaded electrospun composite coating on the implant surface. This coating consists of poly(lactic-coglycolic acid) (PLGA) nanofibers embedded in a poly(ε-caprolactone) film to release combinatorial antibiotics (Van/Rif, Lin/Lin + Rif, and Dap/Dap + Rif) locally, and prevent chronic biofilm infection caused by MRSA. Van/Rif, Lin/Lin + Rif, and Dap/Dap + Rif combinations of antibiotic-loaded coatings were found to have strong antibiofouling efficacy to prevent biofilm formation in orthopedic implants (Fig. 9C). Patel et al.194 fabricated a silver sulfadiazine-loaded solid lipid nanoparticles (SSD-SLNs) formulation, and supplemented it with chitosan gel and DNase-I for the effective treatment of P. aeruginosa biofilm-associated burn wounds. DNase-I accelerated the antibiotic susceptibility against P. aeruginosa biofilms by cleaving the eDNA, and thus eliminated the biofilm. Pure SSD and SSD-SLN were able to eradicate only 58.1% and 78.7% of biofilm after 72 h, respectively (Fig. 10A and B). After the addition of DNase I-incorporated SSD-SLN, there was an inhibition of around 96.8% of the P. aeruginosa biofilm. SSD-SLNs along with DNase-I from the in vivo wound healing study exhibited absolute wound healing after 21 days, but formulations of SSD and SSD-LSNs showed incomplete healing after 21 days. Wang et al.195 fabricated a pH-switchable antimicrobial hydrogel by self-assembling of an octapeptide (Ac-Leu-Lys-Phe-Gln-Phe-His-Phe-Asp-NH2, IKFQFHFD) with nanofiber networks encapsulated with cypate (photothermal agent) and proline (procollagen component) for the disruption of the biofilm of MRSA and acceleration of the wound healing process. This hydrogel was destabilized at an acidic pH value of 5.5, which led to the release of the peptide, cypate and proline. The released peptide IKFQFHFD demonstrated antibacterial efficacy by the cell wall and membrane disintegration, and cypate under NIR light irradiation (808 nm, power density of 0.5 W cm−2) caused biofilm disruption by the photothermal activity. Furthermore, proline led to the acceleration of cell proliferation at the wound bed. The synergistic activity of the IKFQFHFD peptide and cypate of the hydrogel thus demonstrated efficient antibacterial and antibiofilm activity in infected wound care management. The in vivo studies suggested that this composite could heal diabetic mice within 20 days by complete removal of the MRSA biofilm. The results showed that the MRSA biofilm incubated
with hydrogel-cypate system under NIR laser irradiation was completely eradicated, while irradiation alone or cypate with irradiation were not able to eradicate the biofilm completely (Fig. 10C). Fumakiya et al.196 synthesized a topical nanomedicine for the controlled sustained release of LL37 (endogenous host defense peptide) and Serpin A1 (A1), an elastase inhibitor for chronic wound healing treatment. The combination of LL37 and A1 significantly promoted wound healing in BJ fibroblast cells and keratinocytes, eradicated bacterial contamination and inhibited biofilm formation by S. aureus and E. coli, and enhanced anti-inflammatory activity. For the first time, Thorn et al.197 developed lipid liquid crystal nanoparticles (LCNPs) to enhance the antimicrobial and penetration property of tobramycin against P. aeruginosa in full-thickness wounds in mice. Tobramycin-LCNPs significantly reduced the bacterial colonization in the wound by 1000-fold more than individual tobramycin, which showed similar results as the untreated bacterial cells. SEM images (Fig. 10D) showed that red-labeled LCNP is coated over the GFP PAO1 biofilm.
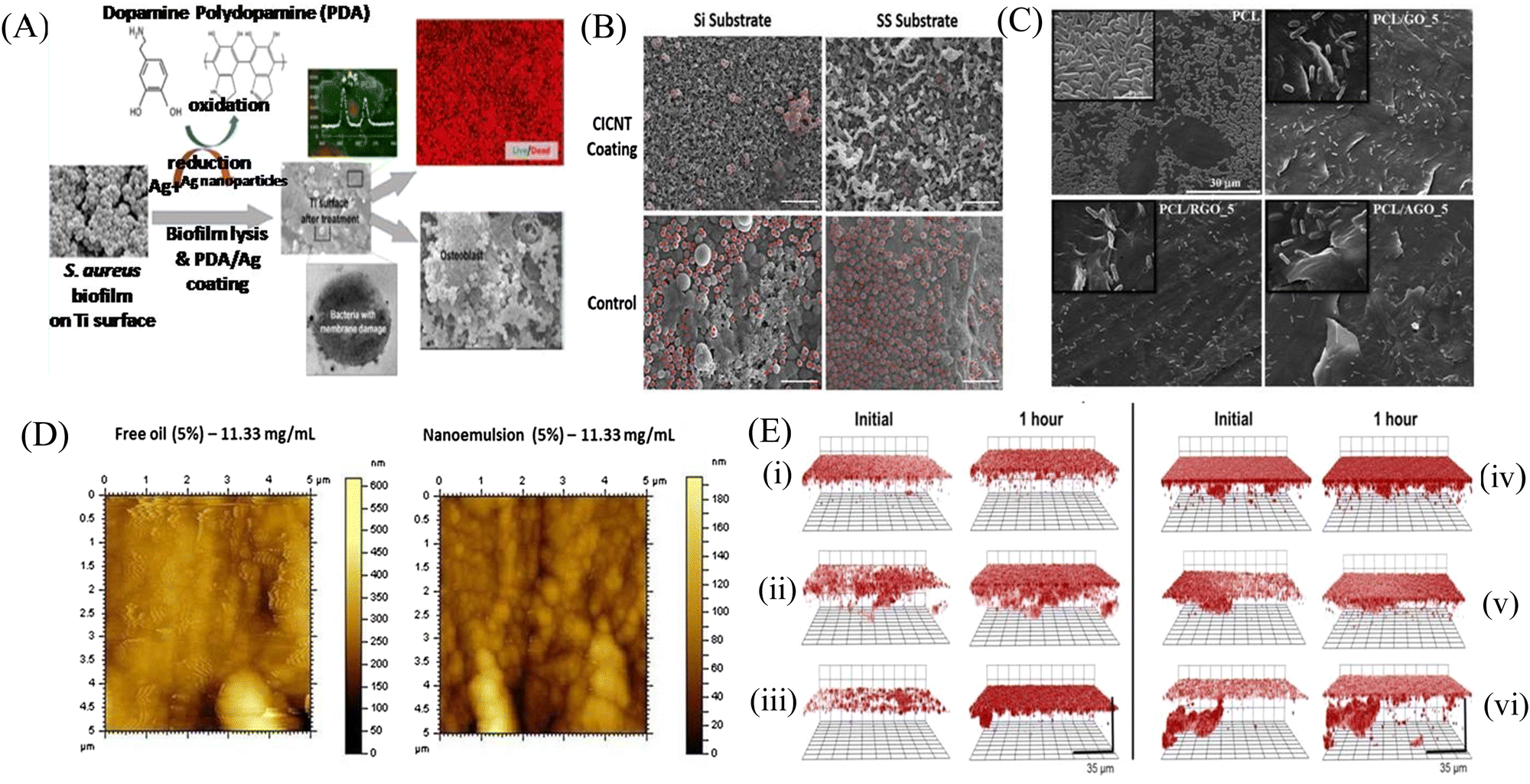 | ||
| Fig. 8 (A) Schematic representation of the silver-based nanosystem. (B) MRSA cells marked in red for each material: CICNT-Si, CICNT-SS, carbon control, and SS control. Scale bars are 5 μm. CICNT-Si, carbon-infiltrated carbon nanotube-silicon; CICNT-SS, carbon-infiltrated carbon nanotube-stainless steel, MRSA. (C) SEM micrographs of biofilm formation on neat PCL, PCL/GO-5, PCL/RGO-5, and PCL/AGO-5 composite surfaces. (D) Influence of free oil and nanoemulsion treatments against the formation of biofilms of P. aeruginosa. (E) Confocal microscopy images of S. aureus biofilms exposed to 50 μg mL−1 RITC-tagged (i) G1 butyl, (ii) G2 butyl, (iii) G4 butyl, (iv) G1 hexyl, (v) G2 hexyl, and (vi) G4 hexyl dendrimers for 1 h. Initial images obtained approximately 5 min after dendrimer exposure. Threshold inverted for clarity. (A) Reprinted with permission from ref. 183. (B) Reprinted with permission from ref. 115. (C) Adapted with permission from ref. 186. (D) Reprinted with permission from ref. 189. (E) Reprinted with permission from ref. 190. | ||
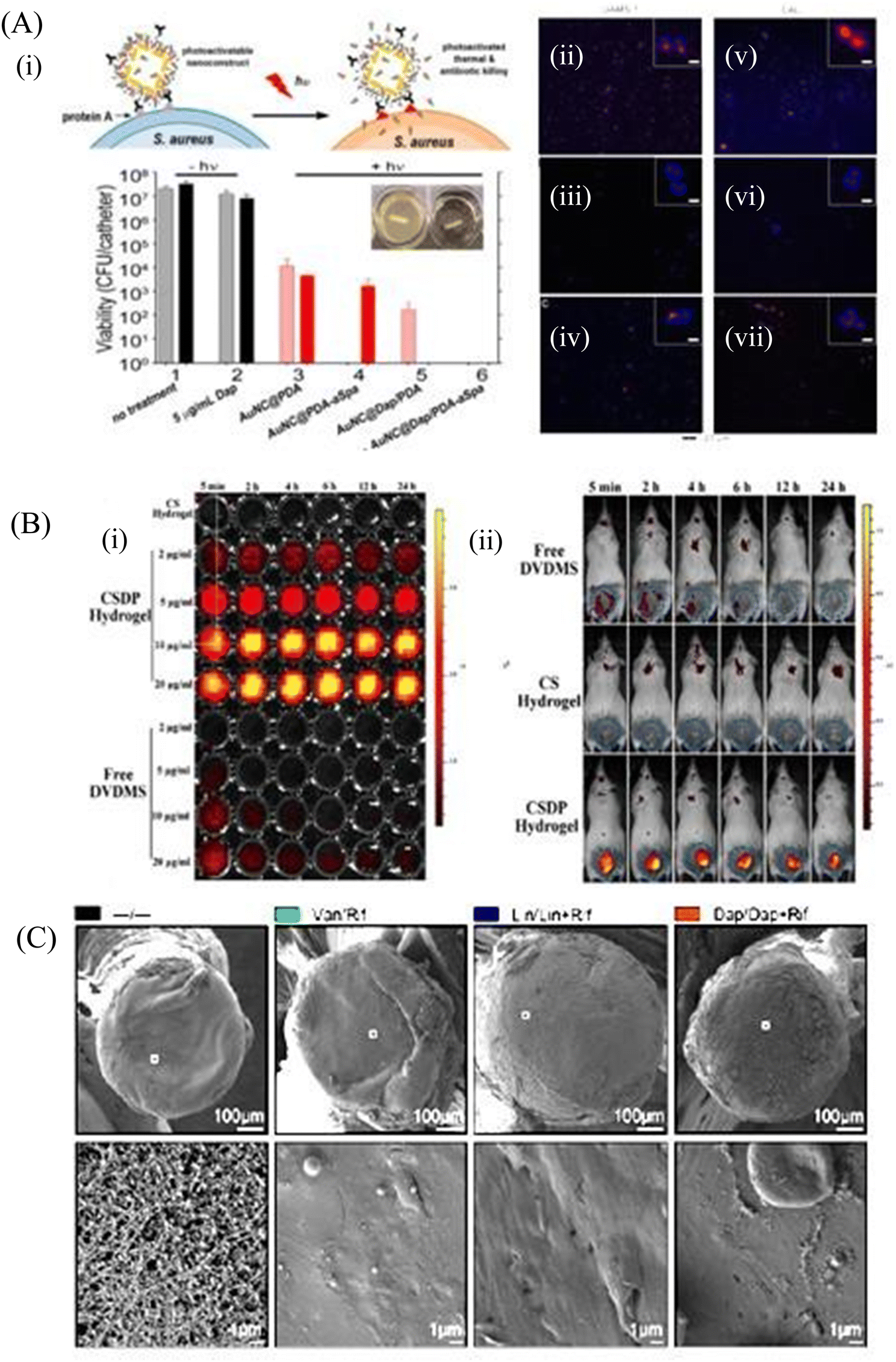 | ||
| Fig. 9 (A) (i) Schematic illustration of the working mechanism of the targeted photoactivatable nanoconstruct for synergistic photothermal and antibiotic treatment of S. aureus. Two-photon fluorescence images of S. aureus cells treated at different conditions, (ii) UAMS-1 exposed to AuNC@PDA–aSpa, (iii) UAMS-1 exposed to AuNC@PDA, (iv) UAMS-1 spa mutant exposed to AuNC@PDA–aSpa, (v) LAC exposed to AuNC@PDA–aSpa, (vi) LAC treated with AuNC@PDA, (vii) LAC spa mutant exposed to AuNC@PDA–aSpa. Cells were stained with DAPI colored in blue. Luminescence of AuNCs was colored in red. (B) Fluorescence imaging of CSDP hydrogel (C) In vivo efficacy on eradication of infection and biofilm formation. Biofilm formation was assessed by SEM on ex vivo implants. (A) Reprinted with permission from ref. 191. (B) Adapted with permission from ref. 192. (C) Reprinted with permission from ref. 193. | ||
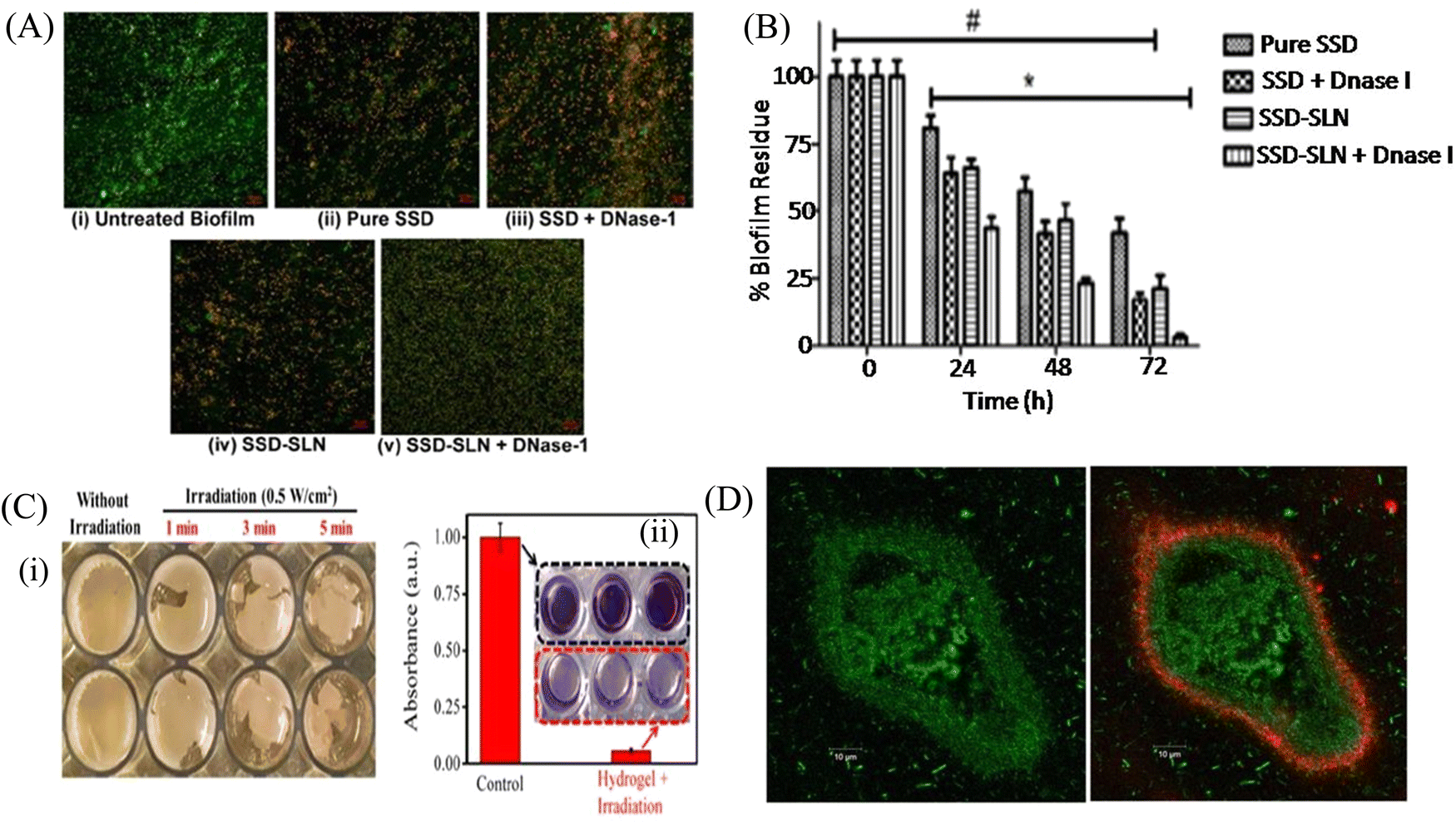 | ||
| Fig. 10 (A) Laser scanning microscopy images of the P. aeruginosa biofilm treated with different test samples (40× magnification). Green fluorescent (SYTO 9) cells indicate the live bacterial cells, and red fluorescent (PI) cells indicate dead cells. (i) Untreated P. aeruginosa biofilm, (ii) Pure SSD, (iii) SSD + DNase-I, (iv) SSD-SLNs, and (v) SSD-SLNs + DNase-I (SSD equivalent to 18.75 μg mL−1 and DNase-I equivalent to 20 μg mL−1). (B) Graph indicates the comparative inhibition of the established P. aeruginosa biofilm after three consecutive doses (once a day) of pure SSD or encapsulated SSD (18.75 μg mL−1) with or without DNase-I (20 μg mL−1). A repeated-measure two-way ANOVA was performed (*, P < 0.05, versus nontreated biofilms (0 h); #, P < 0.05, versus SSD-SLNs + DNase-I on 72 h). The viable count 2.3 × 109 ± 9.1 × 107 CFU mL−1 in the untreated biofilm was taken as one hundred percent. (C) (i) Representative photographs of integrated MRSA biofilm incubated with cypate released from the hydrogel-Cy system at scheduled time points of 4 h under a NIR laser (808 nm, 0.5 W cm−2) irradiation for different times, (ii) crystal violet staining image and its corresponding absorbance for the integrated MRSA biofilm incubated with the hydrogel-Cy system (under pH 5.5) for 4 h followed by NIR laser irradiation (808 nm, 0.5 W cm−2, 5 min). The biofilm under NIR laser irradiation without incubation with the hydrogel-Cy system was used as the control. (D) PAO1-GFP-tagged biofilm (green) grown in a flow cell (left) treated with octadecyl rhodamine B Chloride-tagged LCNPs (red). (A and B) Reprinted with permission from ref. 194. (C) Adapted with permission from ref. 195. (D) Reprinted with permission from ref. 197. | ||
7. Conclusion and future perspective
Biofilm formation by ESKAPE pathogens in the biomedical field has become difficult to treat due to the lack of effective conventional approaches. This is due to low penetration, enzymatic inactivation and degradation of antimicrobials in the microenvironment of biofilm. Nanomaterials have emerged as a novel strategy for the precise and effective treatment of bacterial biofilms. Because of their small size, nanomaterials can easily diffuse the EPS matrix of the biofilm and kill the persister and sessile cells through their intrinsic antibacterial activity and/or by delivering therapeutics. Nanomaterials interact with the bacterial cells and exhibit antibacterial activity through multiple pathways, which include ROS generation, damage of membrane and intracellular components, inhibition of protein synthesis and DNA replication. In addition, nanoparticles inhibit and disrupt biofilm formation through EPS and eDNA degradation, and interfere with the QS pathway. Even though nanotechnology has several advantages, it still has some limitations such as the cytotoxicity, effects on metabolism, poor renal clearance and aggregation with proteins and blood cells, causing health hazards. Dissolution of metal ions from nanoparticles waste also causes adverse health effects. Furthermore, the crossing of biological barriers and avoiding the immune system to achieve targeted delivery are huge challenges. Many nanomaterials have been developed to combat the biofilm of ESKAPE pathogens, but the majority of them are confined to cell line and animal models. Clinical trials in humans are still limited. The undetermined side effects and expensive clinical trials need to be identified and taken care of. Development of appropriate antimicrobial nanomaterials need proper in vitro and in vivo models in respect to antibacterial, antibiofouling, biocompatibility and nanotoxicology. More specifically, an extensive study regarding the fundamental, pharmacological and biological properties of nanoparticles and its interaction with pathogens is needed for clinical development of nanomaterials. Few studies have been conducted in vitro, out of which few proceeded for animal trials with some of them chosen for human testing. Two nanoformulated liposomes are already in phase III trials, namely, Amikacin (ArikaceTM), for the treatment of chronic infection due to P. aeruginosa infection in cystic fibrosis patients,198 and a nanoliposome-based formulation like Pulmaquin was prepared for the fast and sustained release of CIP.199 All of these shortcomings of nanomaterials could be addressed by future investigation. In the coming years, the in vivo toxicity assays, animal studies and clinical trials may pave the way for combating the nanomaterial synthesis, stability and discard related issues. Thus, we conclude that the nanomaterials may provide an excellent opportunity to combat biofilm-related infections caused by ESKAPE pathogens by minimizing the selection of resistance through the delivery of therapeutics with sub-inhibitory doses and killing of bacteria by multiple bactericidal pathways. The development of nanomaterials as antimicrobials and delivery agents requires the collaboration from scientists of different fields, such as biomedical researchers, microbiologists, chemists and engineers. This will lead to the successful generation and commercialization of nanomaterials, which will bring a revolutionary impact to treat biofilm-related infections.Author contributions
Arpita Mukherjee: investigation, formal analysis, writing – original draft, writing – review & editing. Somashree Bose: resources, data curation, visualization, and writing – original draft. Anirban Shaoo: resources, visualization, Sujoy K. Das: conceptualization, supervision, validation, funding acquisition, project administration, writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The financial support received from Science and Engineering Research Board (SERB), Govt. of India under Core Research Grant (CRG/2018/002307) is gratefully acknowledged. A. M. and S. B. gratefully acknowledge UGC, Govt. of India for fellowships.References
- https://emerypharma.com/blog/eskape-pathogens-explained. .
- D. Sharma, L. Misba and A. U. Khan, Antimicrob. Res. Infect. Control, 2019, 8, 76 CrossRef PubMed.
- Z. Jia, Antifouling Strategies-Interference with Bacterial Adhesion, IntechOpen, United Kingdom, 2022, vol. 14, p. 392 Search PubMed.
- D. M. P. De Oliveira, B. M. Forde, T. J. Kidd, N. A. H. Patrick, M. A. Schembri, S. A. Beatson, D. L. Paterson and M. J. Walker, Clin. Microbiol. Rev., 2020, 33, 181 CrossRef PubMed.
- O. Ciofu and T. Tolker-Nielsen, Front. Microbiol., 2019, 10, 913 CrossRef PubMed.
- R. M. Donlan, Emerging Infect. Dis., 2002, 8, 881 CrossRef PubMed.
- V. G. Preda and O. Săndulescu, Discoveries, 2019, 7, 100 CrossRef PubMed.
- N. Baig, I. Kammakakam and W. Falath, Mater. Adv., 2021, 2, 1821 RSC.
- Y. N. Slavin, J. Asnis, U. O. Häfeli and H. Bach, J. Nanobiotechnol., 2017, 15, 65 CrossRef PubMed.
- Q. L. Feng, J. Wu, G. Q. Chen, F. Z. Cui, T. N. Kim and J. O. Kim, J. Biomed. Mater. Res., 2000, 52, 662 CrossRef CAS PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623 CrossRef CAS PubMed.
- S. Veerachamy, T. Yarlagadda, G. Manivasagam and P. K. Yarlagadda, Proc. Inst. Mech. Eng., Part H, 2014, 228, 1083 CrossRef PubMed.
- G. Reid, Int. J. Antimicrob. Agents, 1999, 11, 223 CrossRef CAS PubMed.
- N. K. Archer, M. J. Mazaitis, J. W. Costerton, J. G. Leid, M. E. Powers and M. E. Shirtliff, Virulence, 2011, 2, 445 CrossRef PubMed.
- L. D. Blackman, Y. Qu, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2021, 50, 1587 RSC.
- C. G. Kumar and S. K. Anand, Int. J. Food Microbiol., 1998, 42, 9 CrossRef CAS PubMed.
- S. Park, M. Lee, J. Kim, H. Kim, M. Jung, M. Shin, W. Lee, G. Cheong, J. R. Lee and M. Jang, Molecules, 2019, 24, 4560 CrossRef CAS PubMed.
- M. Thoendel and A. R. Horswill, Adv. Appl. Microbiol., 2010, 71, 91 CAS.
- K. Papenfort and B. L. Bassler, Nat. Rev. Microbiol., 2016, 14, 576 CrossRef CAS PubMed.
- F. L. Paganelli, R. J. Willems, P. Jansen, A. Hendrickx, X. Zhang, M. J. M. Bonten and H. L. Leavis, mBio, 2013, 4, e00154 CrossRef PubMed.
- J. Top, F. L. Paganelli, X. Zhang, W. V. Schaik, H. L. Leavis, M. V. Luit-Asbroek, T. V. D. Poll, M. Leendertse, M. J. M. Bonten and R. J. L. Willems, PLoS One, 2013, 8, 65224 CrossRef PubMed.
- D. I. Johnson, Bacterial Pathogens and Their Virulence Factors. Enterococcus spp., Springer, Cham, 2018, ch. 5, p. 81 Search PubMed.
- C. R. Arciola, D. Campoccia, S. Ravaioli and L. Montanaro, Front. Cell. Infect. Microbiol., 2015, 5, 7 CrossRef PubMed.
- A. M. S. Figueiredo, F. A. Ferreira, C. O. Beltrame and M. F. Côrtes, Crit. Rev. Microbiol., 2017, 43, 602 CrossRef CAS PubMed.
- J. L. Lister and A. R. Horswill, Front. Cell. Infect. Microbiol., 2014, 4, 178 Search PubMed.
- J. M. Yarwood, D. J. Bartels, E. M. Volper and E. P. Greenberg, J. Bacteriol., 2004, 186, 1838 CrossRef CAS PubMed.
- J. Pratten, S. J. Foster, P. F. Chan, M. Wilson and S. P. Nair, Microbes Infect., 2001, 3, 633 CrossRef CAS PubMed.
- M. Habash and G. Reid, J. Clin. Pharmacol., 1999, 39, 887 CrossRef CAS PubMed.
- J. X. Zheng, Z. W. Lin, C. Chen, Z. Chen, F. J. Lin, Y. Wu, S. Y. Yang, X. Sun, W. M. Yao, D. Y. Li, Z. J. Yu, J. L. Jin, D. Qu and Q. W. Deng, Front. Cell. Infect. Microbiol., 2018, 8, 21 CrossRef PubMed.
- T. Pacheco, A. É. I. Gomes, N. M. G. Siqueira, L. Assoni, M. Darrieux, H. Venter and L. F. C. Ferraz, Front. Microbiol., 2021, 12, 597735 CrossRef PubMed.
- H. Nirwati, K. Sinanjung, F. Fahrunissa, F. Wijaya, S. Napitupulu, V. P. Hati, M. S. Hakim, A. Meliala, A. T. Aman and T. Nuryastuti, BMC Proc., 2019, 13, 20 CrossRef CAS PubMed.
- H. Zeighami, F. Valadkhani, R. Shapouri, E. Samadi and F. Haghi, BMC Infect. Dis., 2019, 19, 629 CrossRef PubMed.
- C. Cucarella, C. Solano, J. Valle, B. Amorena, I. Lasa and J. R. Penadés, J. Bacteriol., 2001, 183, 2888 CrossRef CAS PubMed.
- A. M. Asaad, S. Ansari, S. E. Ajlan and S. M. Awad, Infect. Drug Resist., 2021, 14, 709 CrossRef CAS PubMed.
- X. Sun, Z. Ni, J. Tang, Y. Ding, X. Wang and F. Li, Front. Microbiol., 2021, 12, 679241 CrossRef PubMed.
- M. López-Martín, J. F. Dubern, M. R. Alexander and P. Williams, J. Bacteriol., 2021, 203, 635 CrossRef PubMed.
- M. T. T. Thi, D. Wibowo and B. H. A. Rehm, Int. J. Mol. Sci., 2020, 21, 8671 CrossRef CAS PubMed.
- N. B. Cabra, B. Paetzold, T. Ferrar, R. Mazzolini, E. Torrents, L. Serrano and M. L. Luch-Sena, Sci. Rep., 2020, 10, 9390 CrossRef PubMed.
- P. Owlia, R. Nosrati, R. Alaghehbandan and A. R. Lari, GMS Hyg. Infect. Control, 2014, 9, Doc13, DOI:10.3205/dgkh000233.
- R. S. Smith, S. G. Harris, R. Phipps and B. Iglewski, J. Bacteriol., 2002, 184, 1132 CrossRef CAS PubMed.
- J. Tang, Y. Chen, X. Wang, Y. Ding, X. Sun and Z. Ni, Infect. Drug Resist., 2020, 13, 4273 CrossRef CAS PubMed.
- K. Oinuma and E. P. Greenberg, J. Bacteriol., 2011, 193, 421 CrossRef CAS PubMed.
- J. S. Bustos, M. A. Ares, C. A. G. Aldapa, J. A. G. Y. Merchand, J. A. Girón and M. A. D. L. Cruz, Front. Microbiol., 2020, 11, 560488 CrossRef PubMed.
- V. C. Kalia, Biotechnol. Adv., 2013, 31, 224 CrossRef CAS PubMed.
- H. Koo, R. N. Allan, R. P. Howlin, P. Stoodley and L. Hall-Stoodley, Nat. Rev. Microbiol., 2017, 15, 740 CrossRef CAS PubMed.
- X. Luo, B. Zhang, Y. Lu, Y. Mei and L. Shen, J. Hazard. Mater., 2022, 421, 126682 CrossRef CAS PubMed.
- S. L. Munasur, E. B. Turawa, C. U. M. E. Chikte and A. Musekiwa, Int. J. Environ. Res. Public Health, 2020, 17, 5601 CrossRef PubMed.
- C. B. Lineback, C. A. Nkemngong, S. T. Wu, X. Li, P. J. Teska and H. F. Oliver, Antimicrob. Res. Infect. Control, 2018, 7, 154 CrossRef PubMed.
- G. Orrù, S. D. Nero, E. Tuveri, M. L. Ciusa, F. Pilia, M. Erriu, G. Orrù, M. Liciardi, V. Piras and G. Denotti, Open Dent. J., 2010, 4, 140 CrossRef PubMed.
- F. Natalio, R. André, A. F. Hartog, B. Stoll, K. P. Jochum, R. Wever and W. Tremel, Nat. Nanotechnol., 2012, 7, 530 CrossRef CAS PubMed.
- S. Nadar, T. Khan, S. G. Patching and A. Omri, Microorganisms, 2022, 10, 303 CrossRef CAS PubMed.
- N. Ooi, K. Miller, C. Randall, W. Rhys-Williams, W. Love and I. Chopra, J. Antimicrob. Chemother., 2010, 65, 72 CrossRef CAS PubMed.
- K. Murakami, H. Yumoto, A. Murakami, T. Amoh, D. Viducic, K. Hirota, A. Tabat, H. Nagamune, H. Kourai, T. Matsuo and Y. Miyake, J. Appl. Microbiol., 2017, 122, 893 CrossRef CAS PubMed.
- C. Wu, J. Y. Lim, G. G. Fuller and L. Cegelski, Langmuir, 2013, 29, 920 CrossRef CAS PubMed.
- C. M. Toutain-Kidd, S. C. Kadivar, C. T. Bramante, S. A. Bobin and M. E. Zegans, Antimicrob. Agents Chemother., 2009, 53, 136 CrossRef CAS PubMed.
- B. V. G. Nguyen, T. Nagakubo, M. Toyofuku, N. Nomura and A. S. Utada, Langmuir, 2020, 36, 6411 CrossRef CAS PubMed.
- R. Watanabe, T. Matsumoto, G. Sano, Y. Ishii, K. Tateda, Y. Sumiyama, J. Uchiyama, S. Sakurai, S. Matsuzaki, S. Imai and K. Yamaguchi, Antimicrob. Agents Chemother., 2007, 51, 446 CrossRef CAS PubMed.
- L. G. Kifelew, M. S. Warner, S. Morales, L. Vaughan, R. Woodman, R. Fitridge, J. G. Mitchell and P. Speck, BMC Microbiol., 2020, 20, 204 CrossRef CAS PubMed.
- B. Sisakhtpour, A. Mirzaei, V. Karbasizadeh, N. Hosseini, M. Shabani and S. Moghim, Ann. Clin. Microbiol. Antimicrob., 2022, 21, 1 CrossRef CAS PubMed.
- M. M. Quinn, P. K. Henneberger, B. Braun, G. L. Delclos, K. Fagan, V. Huang, J. L. Knaack, L. Kusek, S. J. Lee, N. Le Moual, K. A. Maher, S. H. Mc Crone, A. H. Mitchell, E. Pechter, K. Rosenman, L. Sehulster, A. C. Stephens, S. Wilburn and J. P. Zock, Am. J. Infect. Control, 2015, 43, 424 CrossRef PubMed.
- A. Bridier, R. Briandet, V. Thomas and F. Dubois-Brissonnet, Biofouling, 2011, 27, 1017 CrossRef CAS PubMed.
- E. Rojo-Molinero, M. D. Macia, R. Rubio, B. Moya, G. Cabot, C. Lopez-Causap, J. L. Pérez, R. Cantón and A. Oliver, Antimicrob. Agents Chemother., 2016, 60, 2912 CrossRef CAS PubMed.
- C. Watanakunakorn, Rev. Infect. Dis., 1981, 3, 210 CrossRef CAS.
- T. Heinonen, S. Hargraves, M. Georgieva, C. Widmann, N. Jacquier and J. Glob, Antimicrob. Resist., 2021, 25, 227 CrossRef CAS PubMed.
- R. Festa, R. L. Ambrosio, A. Lamas, L. Gratino, G. Palmieri, C. M. Franco, A. Cepedaa and A. Anastasio, Foods, 2021, 10, 1372 CrossRef CAS PubMed.
- https://www.cdc.gov/drugresistance/about/how-resistance-happens.html. .
- T. Parandhaman, M. D. Dey and S. K. Das, Green Chem., 2019, 21, 5469 RSC.
- S. A. A. Rizvi and A. M. Saleh, Saudi Pharm. J., 2018, 26, 64 CrossRef PubMed.
- L. Wang, C. Hu and L. Shao, Int. J. Nanomed., 2017, 12, 1227 CrossRef CAS PubMed.
- H. Lan and D. Ph, The Use of Nanoparticles to Prevent and Eliminate Bacterial Biofilms, 2017, 344.
- S. N. Kłodzińska, F. Wan, H. Jumaa, C. Sternberg, T. Rades and H. M. Nielsen, J. Colloid Interface Sci., 2019, 555, 595 CrossRef PubMed.
- G. Sanità, B. Carrese and A. Lamberti, Front. Mol. Biosci., 2020, 7, 381 Search PubMed.
- A. Bahrami, R. Delshadi and S. M. Jafari, Trends Food Sci. Technol., 2020, 99, 217 CrossRef CAS.
- S. A. Masurkar, P. R. Chaudhari, V. B. Shidore and S. P. Kamble, IET Nanobiotechnol., 2012, 6, 110 CrossRef CAS PubMed.
- Prateeksha, B. R. Singh, M. Shoeb, S. Sharma, A. H. Naqvi, V. K. Gupta and B. N. Singh, Front. Cell. Infect. Microbiol., 2017, 7, 93 CrossRef CAS PubMed.
- N. A. Al-Shabib, F. M. Husain, F. Ahmed, R. A. Khan, I. Ahmad, E. Alsharaeh, M. S. Khan, A. Hussain, Md. T. Rehman, M. Yusuf, I. Hassan, J. M. Khan, G. Md Ashraf, A. Alsalme, M. F. Al-Ajmi, V. V. Tarasov and G. Aliev, Sci. Rep., 2016, 6, 36761 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, Z. Zhang, W. Niu, C. Li, N. Yang, B. Chen and H. Zhang, Chem. Rev., 2018, 118, 6409 CrossRef CAS PubMed.
- L. Wang, C. Hu and L. Shao, Int. J. Nanomed., 2017, 12, 1227 CrossRef CAS PubMed.
- E. S. López, D. Gomes, G. Esteruelas, L. Bonilla, A. L. L. Machado, R. Galindo, A. Cano, M. Espina, M. Ettcheto, A. Camins, A. M. Silva, A. Durazzo, A. Santini, M. L. Garcia and E. B. Souto, Nanomaterials, 2020, 10, 292 CrossRef PubMed.
- T. Ameh, M. Gibb, D. Stevens, S. H. Pradhan, E. Braswell and C. M. Sayes, Nanomaterials, 2022, 12, 2402 CrossRef CAS PubMed.
- J. M. V. Makabenta, A. Nabawy, C. H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19, 23 CrossRef CAS PubMed.
- M. H. Siddique, B. Aslam, M. Imran, A. Ashraf, H. Nadeem, S. Hayat, M. Khurshid, M. Afzal, I. R. Malik, M. Shahzad, U. Qureshi, Z. U. H. Khan and S. Muzammi, BioMed Res. Int., 2020, 6398165 CAS.
- Z. Qiao, Y. Yao, S. Song, M. Yin, M. Yang, D. Yan, L. Yang and J. Luo, J. Mater. Chem. B, 2020, 8, 3138 RSC.
- F. Khan, P. Manivasagan, J. W. Lee, D. T. N. Pham, J. Oh and Y. M. Kim, Mar. Drugs, 2019, 17, 208 CrossRef CAS PubMed.
- B. Wan, Y. Zhu, J. Tao, F. Zhu, J. Chen, L. Li, J. Zhao, L. Wang, S. Sun, Y. Yang, X. Zhang and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 9050 CrossRef CAS PubMed.
- X. Wang, J. Wu, P. Li, L. Wang, J. Zhou, G. Zhang, X. Li, B. Hu and X. Xing, ACS Appl. Mater. Interfaces, 2018, 10, 34905 CrossRef CAS PubMed.
- S. Kannan, A. Solomon, G. Krishnamoorthy and M. Marudhamuthu, Sci. Rep., 2021, 11, 1102 CrossRef CAS PubMed.
- M. S. Usman, M. E. E. Zowalaty, K. Shameli, N. Zainuddin, M. Salama and N. A. Ibrahim, Int. J. Nanomed., 2013, 8, 4467 Search PubMed.
- T. Amna, M. S. Hassan, N. A. Barakat, D. R. Pandeya, S. T. Hong, M. S. Khil and H. Y. Kim, Appl. Microbiol. Biotechnol., 2012, 93, 743 CrossRef CAS PubMed.
- S. V. Gudkov, D. E. Burmistrov, V. V. Smirnova, A. A. Semenova and A. B. Lisitsyn, Nanomaterials, 2022, 12, 2635 CrossRef CAS PubMed.
- J. H. Kim, H. Cho, S. E. Ryu and M. U. Choi, Arch. Biochem. Biophys., 2000, 382, 72 CrossRef CAS PubMed.
- M. Altaf, M. T. Zeyad, M. A. Hashmi, S. Manoharadas, S. A. Hussain, M. S. Ali Abuhasil and M. A. M. Almuzaini, RSC Adv., 2021, 11, 19248 RSC.
- N. Tran, A. Mir, D. Mallik, A. Sinha, S. Nayar and T. J. Webster, Int. J. Nanomed., 2010, 5, 277 CAS.
- S. R. Dwivedi, F. Wahab Khan, Y. K. Mishra, J. Musarrat and A. A. Al-Khedhairy, PLoS One, 2014, 9, 111289 CrossRef PubMed.
- N. N. M. Adnan, Z. Sadrearhami, A. Bagheri, T. K. Nguyen, E. H. H. Wong, K. K. K. Ho, M. Lim, N. Kumar and C. Boyer, Macromol. Rapid Commun., 2018, 39, 1800159 CrossRef PubMed.
- M. F. Khan, F. M. Husain, Q. Zia, E. Ahmad, A. Jamal, M. Alaidarous, S. Banawas, M. M. Alam, B. A. Alshehri, M. Jameel, P. Alam, M. I. Ahamed, A. H. Ansari and I. Ahmad, ACS Omega, 2020, 5, 32203 CrossRef CAS PubMed.
- B. Tian, S. Tang, C. Wang, W. Wang, C. Wu, Y. Guo, Y. Guo and Z. Zhu, Colloids Surf., B, 2014, 123, 403 CrossRef CAS PubMed.
- S. Begum, A. Pramanik, D. Davis, S. Patibandla, K. Gates, Y. Gao and P. C. Ray, ACS Omega, 2020, 5, 3116 CrossRef CAS PubMed.
- Z. L. Shaw, S. Kuriakose, S. Cheeseman, M. D. Dickey, J. Genzer, A. J. Christofferson, R. J. Crawford, C. F. McConville, J. Chapman, V. K. Truong, A. Elbourne and S. Walia, Nat. Commun., 2021, 12, 3897 CrossRef CAS PubMed.
- S. Pandit, K. Gaska, V. R. S. S. Mokkapati, S. Forsberg, M. Svensson, R. Kádár and I. Mijakovic, RSC Adv., 2019, 9, 33454 RSC.
- N. Qureshi, R. Patil, M. Shinde, G. Umarji, V. Causin, W. Gade, U. Mulik, A. Bhalerao and D. P. Amalnerkar, Appl. Nanosci., 2015, 5, 331 CrossRef CAS.
- R. Žalnėravičius, V. Klimas, A. Paškevičius, G. Grincienė, R. Karpicz, A. Jagminas and A. Ramanavičius, J. Colloid Interface, 2021, 591, 115 CrossRef PubMed.
- X. Zhang, W. Zhang, L. Liu, M. Yang, L. Huang, K. Chen, R. Wang, B. Yang, D. Zhang and J. Wang, Nanotechnology, 2017, 28, 225101 CrossRef PubMed.
- P. Choudhary, B. Ramalingam and S. K. Das, ACS Biomater. Sci. Eng., 2020, 6, 5911 CrossRef CAS PubMed.
- F. Lin, C. Li and Z. Chen, Front. Microbiol., 2018, 9, 259 CrossRef PubMed.
- P. Li, S. Liu, G. Zhang, X. Yang, W. Cao, X. Gong and X. Xing, ACS Appl. Bio Mater., 2020, 3, 1105 CrossRef CAS PubMed.
- M. Ritenberg, S. Nandi, S. Kolusheva, R. Dandela, M. M. Meijler and R. Jelinek, ACS Chem. Biol., 2016, 11, 1265 CrossRef CAS PubMed.
- G. Liang, H. Shi, Y. Qi, J. Li, A. Jing, Q. Liu, W. Feng, G. Li and S. Gao, Int. J. Nanomed., 2020, 15, 5473 CrossRef CAS PubMed.
- H. Wang, Z. Song, J. Gu, S. Li, Y. Wu and H. Han, ACS Biomater. Sci. Eng., 2019, 5, 4739 CrossRef CAS PubMed.
- P. Li, S. Liu, W. Cao, G. Zhang, X. Yang, X. Gong and X. Xing, Chem. Commun., 2020, 56, 2316 RSC.
- I. Malek, C. F. Schaber, T. Heinlein, J. J. Schneider, S. N. Gorb and R. A. Schmitz, J. Mater. Chem. B, 2016, 4, 5228 RSC.
- R. T. Santos, M. Gomes, L. C. Gomes and F. J. Mergulhao, iScience, 2020, 24, 102001 CrossRef PubMed.
- S. Kang, M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7528 CrossRef CAS PubMed.
- S. R. Morco, D. L. Williams, B. D. Jensen and A. E. Bowden, J. Orthop. Res., 2022, 40, 1953 CrossRef CAS PubMed.
- H. Fallatah, M. Elhaneid, H. Ali-Boucetta, T. W. Overton, H. El Kadri and K. Gkatzionis, Environ. Sci. Pollut. Res. Int., 2019, 26, 25057 CrossRef CAS PubMed.
- X. Dai, Y. Zhao, Y. Yu, X. Chen, X. Wei, X. Zhang and C. Li, Nanoscale, 2018, 10, 18520 RSC.
- K. Yuan, B. Jurado-Sánchez and A. Escarpa, Angew. Chem., Int. Ed., 2021, 60, 4915 CrossRef CAS PubMed.
- T. Parandhaman, P. Choudhary, B. Ramalingam, M. Schmidt, S. Janardhanam and S. K. Das, ACS Biomater. Sci. Eng., 2021, 7, 5899 CrossRef CAS PubMed.
- P. Choudhary, B. Ramalingam and S. K. Das, ACS Biomater. Sci. Eng., 2020, 6, 5911 CrossRef CAS PubMed.
- R. Banerjee, J. Biomater. Appl., 2001, 16, 3 CrossRef CAS PubMed.
- Z. Drulis-Kawa and A. Dorotkiewicz-Jach, Int. J. Pharm., 2010, 387, 187 CrossRef CAS PubMed.
- Y. Hou, Z. Wang, P. Zhang, H. Bai, Y. Sun, J. Duan and H. Mu, Int. J. Mol. Sci., 2017, 9, 784 CrossRef PubMed.
- D. Dong, N. Thomas, B. Thierry, S. Vreugde, C. A. Prestidge and P. J. Wormald, PLoS One, 2015, 10, 131806 Search PubMed.
- Y. Zhao, X. Dai, X. Wei, Y. Yu, X. Chen, X. Zhang and C. Li, ACS Appl. Mater. Interfaces, 2018, 10, 14426 CrossRef CAS PubMed.
- G. Mi, D. Shi, M. Wang and T. J. Webster, Adv. Healthcare Mater., 2018, 7, 1800103 CrossRef PubMed.
- E. B. Souto, I. Baldim, W. P. Oliveira, R. Rao, N. Yadav, F. M. Gama and S. Mahant, Expert Opin. Drug Delivery, 2020, 17, 357 CrossRef CAS PubMed.
- B. Rodenak-Kladniew, S. Scioli Montoto, M. L. Sbaraglini, M. Di Ianni, M. E. Ruiz, A. Talevi, V. A. Alvarez, N. Durán, G. R. Castro and G. A. Islan, Int. J. Pharm., 2019, 569, 118575 CrossRef CAS PubMed.
- A. Alalaiwe, P. W. Wang, P. Lu, Y. Chen, J. Fang and S. Yan, Front. Microbiol., 2018, 9, 1493 CrossRef PubMed.
- Md. M. Anjum, K. K. Patel, D. Dehari, N. Pandey, R. Tilak, A. K. Agrawal and S. Singh, Drug Delivery Transl. Res., 2021, 11, 305 CrossRef CAS PubMed.
- L. Luan, Z. Chi and C. Liu, Nanomaterials, 2019, 9, 763 CrossRef CAS PubMed.
- C. Sahli, S. E. Moya, J. S. Lomas, C. Gravier-Pelletier, R. Briandet and M. Hémadi, Theranostics, 2022, 12, 2383 CrossRef CAS PubMed.
- S. Perumal, R. Atchudan and W. Lee, Polymers, 2022, 14, 2510 CrossRef CAS PubMed.
- Y. Xi, T. Song, S. Tang, N. Wang and J. Du, Biomacromolecules, 2016, 17, 3922 CrossRef CAS PubMed.
- B. Hisey, P. J. Ragogna and E. R. Gillies, Biomacromolecules, 2017, 18, 914 CrossRef CAS PubMed.
- J. Qiao, M. Purro, Z. Liu and M. P. Xiong, ACS Infect. Dis., 2018, 4, 1346 CrossRef CAS PubMed.
- S. C. Park, C. Ko, H. Hyeon, M. K. Jang and D. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 54306 CrossRef CAS PubMed.
- R. Guo, K. Li, B. Tian, C. Wang, X. Chen, X. Jiang, H. He and W. Hong, J. Nanobiotechnol., 2021, 19, 232 CrossRef CAS PubMed.
- M. Chen, S. Xie, J. Wei, X. Song, Z. Ding and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36814 CrossRef CAS PubMed.
- F. Najafi, M. Salami-Kalajahi and H. Roghani-Mamaqani, J. Iran. Chem. Soc., 2021, 18, 503 CrossRef CAS.
- A. Felczak, N. Wrońska, A. Janaszewska, B. Klajnert, M. Bryszewska, D. Appelhans, B. Voit, S. Rozalskaa and K. Lisowska, New J. Chem., 2012, 36, 2215 RSC.
- A. A. Bahar, Z. Liu, F. Totsingan, C. Buitrago, N. Kallenbach and D. Ren, Appl. Microbiol. Biotechnol., 2015, 99, 8125 CrossRef CAS PubMed.
- E. M. V. Johansson, S. A. Crusz, E. Kolomiets, L. Buts, R. U. Kadam, M. Cacciarini, K. Bartels, S. P. Diggle, M. Cámara, P. Williams, R. Loris, C. Nativi, F. Rosenau, K. Jaeger, T. Darbre and J. Reymond, Chem. Biol., 2008, 15, 1249 CrossRef CAS PubMed.
- J. Fernandez, Á. Martin-Serrano, N. Gómez-Casanova, A. Falanga, S. Galdiero, F. J. de la Mata, I. Heredero-Bermejo and P. Ortega, Polymers, 2021, 13, 2127 CrossRef CAS PubMed.
- K. Winnicka, M. Wroblewska, P. Wieczorek, P. T. Sacha and E. A. Tryniszewska, Molecules, 2013, 18, 8607 CrossRef CAS PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323 CrossRef CAS PubMed.
- Y. Xi, Y. Wang, J. Gao, Y. Xiao and J. Du, ACS Nano, 2019, 13, 13645 CrossRef CAS PubMed.
- Y. Hong, Y. Xi, J. Zhang, D. Wang, H. Zhang, N. Yan, S. He and J. Du, J. Mater. Chem. B, 2018, 6, 6311 RSC.
- P. Walvekar, R. Gannimani, M. Salih, S. Makhathini, C. Mocktar and T. Govender, Colloids Surf., B, 2019, 182, 110388 CrossRef CAS PubMed.
- H. Wang, X. Nie, W. You, W. Huang, G. Chen, F. Gao, L. Xia, L. Zhang, L. Wang, A. Shen, K. Wu, S. Ding and Y. You, ACS Appl. Mater. Interfaces, 2021, 13, 56838 CrossRef CAS PubMed.
- A. Gupta, A. Z. Md Badruddoza and P. S. Doyle, Langmuir, 2017, 33, 7118 CrossRef CAS PubMed.
- P. Prateeksha, S. K. Barik and B. N. Singh, Sci. Rep., 2019, 9, 6520 CrossRef PubMed.
- K. Ramalingam and V. A. Lee, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 737 CrossRef PubMed.
- Y. Oz, A. Nabawy, S. Fedeli, A. Gupta, R. Huang, A. Sanyal and V. M. Rotello, ACS Appl. Mater. Interfaces, 2021, 13, 40325 CrossRef CAS PubMed.
- C. Li, R. F. Landis, J. M. Makabenta, A. Nabawy, T. Tronchet, D. Archambault, Y. Liu, R. Huang, M. Golan, W. Cui, J. Mager, A. Gupta, S. Schmidt-Malan, R. Patel and V. M. Rotello, Mater. Horiz., 2021, 8, 1776 RSC.
- Z. Song, H. Sun, Y. Yang, H. Jing, L. Yang, Y. Tong, C. Wei, Z. Wang, Q. Zou and H. Zeng, Nanomedicine, 2016, 12, 1543 CrossRef CAS PubMed.
- Y. Y. Hwang, K. Ramalingam, D. R. Bienek, V. Lee, T. You and R. Alvarez, Antimicrob. Agents Chemother., 2013, 57, 3568 CrossRef CAS PubMed.
- S. K. Das, D. Kumar, S. Swarnakar, S. Bose and Y. Dahat, A Novel Composition of Cationic Metal Nanoparticle Conjugated Nanoemulsion and a Process for the Preparation Thereof, India Pat., 202111045225, 2021 Search PubMed.
- F. Hizal, I. Zhuk, S. Sukhishvili, H. J. Busscher, H. C. van der Mei and C. H. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 20304 CrossRef CAS PubMed.
- O. Bondarenko, A. Ivask, A. Käkinen, I. Kurvet and A. Kahru, PLoS One, 2013, 8, 64060 CrossRef PubMed.
- J. S. Kim, E. Kuk, K. N. Yu, J. H. Kim, S. J. Park, H. J. Lee, S. H. Kim, Y. K. Park, Y. H. Park, C. Y. Hwang, Y. K. Kim, Y. S. Lee, D. H. Jeong and M. H. Cho, Nanomedicine, 2007, 3, 95 CrossRef CAS PubMed.
- O. Gordon, T. V. Slenters, P. S. Brunetto, A. E. Villaruz, D. E. Sturdevant, M. Otto, R. Landmann and K. M. Fromm, Antimicrob. Agents Chemother., 2010, 54, 4208 CrossRef CAS PubMed.
- W. Hu, M. R. Younis, Y. Zhou, C. Wang and X. H. Xia, Small, 2020, 16, 2000553 CrossRef CAS PubMed.
- Z. Chen, J. J. Yin, Y. T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu and N. Gu, ACS Nano, 2012, 6, 4001 CrossRef CAS PubMed.
- S. Singh, Front. Chem., 2019, 7, 46 CrossRef CAS PubMed.
- C. N. Lok, C. M. Ho, R. Chen, Q. He, W. Yu, H. Sun, P. K. Tam, J. Chiu and C. Che, J. Biol. Inorg. Chem., 2007, 12, 527 CrossRef CAS PubMed.
- S. Kang, M. Herzberg, D. F. Rodrigues and M. Elimelech, Langmuir, 2008, 24, 6409 CrossRef CAS PubMed.
- G. E. Valenti, S. Alfei, D. Caviglia, C. Domenicotti and B. Marengo, Int. J. Mol. Sci., 2022, 23, 6108 CrossRef CAS PubMed.
- Y. Zhao, Y. Tian, Y. Cui, W. Liu, W. Ma and X. Jiang, J. Am. Chem. Soc., 2010, 132, 12349 CrossRef CAS PubMed.
- S. A. A. Rizvi and A. M. Saleh, Saudi Pharm J., 2018, 26, 64 CrossRef PubMed.
- L. L. Li, J. H. Xu, G. B. Qi, X. Zhao, F. Yu and H. Wang, ACS Nano, 2014, 8, 4975 CrossRef CAS PubMed.
- A. F. Radovic-Moreno, T. K. Lu, V. A. Puscasu, C. J. Yoon, R. Langer and O. C. Farokhzad, ACS Nano, 2012, 6, 4279 CrossRef CAS PubMed.
- D. L. Slomberg, Y. Lu, A. D. Broadnax, R. A. Hunter, A. W. Carpenter and M. H. Schoenfisch, ACS Appl. Mater. Interfaces, 2013, 5, 9322 CrossRef CAS PubMed.
- K. Ikuma, A. W. Decho and B. L. Lau, Front. Microbiol., 2015, 6, 591 Search PubMed.
- J. M. V. Makabenta, A. Nabawy, C. H. Li, S. S. chmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19, 23 CrossRef CAS PubMed.
- T. O. Peulen and K. J. Wilkinson, Environ. Sci. Technol., 2011, 45, 3367 CrossRef CAS PubMed.
- P. Meers, M. Neville, V. Malinin, A. W. Scotto, G. Sardaryan, R. Kurumunda, C. Mackinson, G. James, S. Fisher and W. R. Perkins, J. Antimicrob. Chemother., 2008, 61, 859 CrossRef CAS PubMed.
- D. Hu, H. Li, B. Wang, Z. Ye, W. Lei, F. Jia, Q. Jin, K. F. Ren and J. Ji, ACS Nano, 2017, 11, 9330 CrossRef CAS PubMed.
- E. O. Mikhailova, J. Funct. Biomater., 2020, 11, 84 CrossRef CAS PubMed.
- C. R. Thorn, P. L. Howell, D. J. Wozniak, C. A. Prestidge and N. Thomas, Adv. Drug Delivery Rev., 2021, 179, 113916 CrossRef CAS PubMed.
- https://apps.who.int/iris/bitstream/handle/10665/334216/9789240010789-eng.pdf .
- Q. Wan and T. J. Webster, J. Biomed. Mater. Res., 2012, 100, 3205 Search PubMed.
- H. A. Tran and P. A. Tran, ACS Appl. Mater. Interfaces, 2021, 13, 41435 CrossRef CAS PubMed.
- R. Teixeira-Santos, M. Gomes, L. C. Gomes and F. J. Mergulhão, iScience, 2020, 24, 102001 CrossRef PubMed.
- H. Mohammed, A. Kumar, E. Bekyarova, Y. Al Hadeethi, X. Zhang, M. Chen, M. S. Ansari, A. Cochis and L. Rimondini, Front. Bioeng. Biotechnol., 2020, 8, 465 CrossRef PubMed.
- S. Kumar, S. Raj, E. Kolanthai, A. K. Sood, S. Sampath and K. Chatterjee, ACS Appl. Mater. Interfaces, 2015, 7, 3237 CrossRef CAS PubMed.
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei, R. Jiang, J. Kong and Y. Chen, ACS Nano, 2011, 5, 6971 CrossRef CAS PubMed.
- O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731 CrossRef CAS PubMed.
- S. Gündel, M. E. de Souza, P. M. Quatrin, B. Klein, R. Wagner, A. Gündel, R. A. Vaucher, R. C. V. Santos and A. F. Ourique, Microb. Pathog., 2018, 118, 268 CrossRef PubMed.
- B. V. Worley, K. M. Schilly and M. H. Schoenfisch, Mol. Pharmacol., 2015, 12, 1573 CrossRef CAS PubMed.
- D. G. Meeker, S. V. Jenkins, E. K. Miller, K. E. Beenken, A. J. Loughran, A. Powless, T. J. Muldoon, E. I. Galanzha, V. P. Zharov, M. S. Smeltzer and J. Chen, ACS Infect. Dis., 2016, 2, 241 CrossRef CAS PubMed.
- B. Mai, M. Jia, S. Liu, Z. Sheng, M. Li, Y. Gao, X. Wang, Q. Liu and P. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 10156 CrossRef CAS PubMed.
- A. G. Ashbaugh, X. Jiang, J. Zheng, A. S. Tsai, W. Kim, J. M. Thompson, R. J. Miller, J. H. Shahbazian, Y. Wang, C. A. Dillen, A. A. Ordonez, Y. S. Chang, S. K. Jain, L. C. Jones, R. S. Sterling, H. Mao and L. S. Miller, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6919 CrossRef PubMed.
- K. K. Patel, D. B. Surekha, M. Tripathi, Md. M. Anjum, M. S. Muthu, R. Tilak, A. K. Agrawal and S. Singh, Mol. Pharmacol., 2019, 16, 3916 CrossRef CAS PubMed.
- J. Wang, X. Y. Chen, Y. Zhao, Y. Yang, W. Wang, C. Wu, B. Yang, Z. Zhang, L. Zhang, Y. Liu, X. Du, W. Li, L. Qiu, P. Jiang, X. Mou and Y. Li, ACS Nano, 2019, 13, 11686 CrossRef CAS PubMed.
- M. Fumakia and E. A. Ho, Mol. Pharmacol., 2016, 13, 2318 CrossRef CAS PubMed.
- C. R. Thorn, A. Wignall, Z. Kopecki, A. Kral, C. A. Prestidge and N. Thomas, ACS Infect. Dis., 2022, 8, 841 CrossRef CAS PubMed.
- US National Library of Medicine. http://ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01315691(2018).
- US National Library of Medicine. http://ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02104245 (2018).
| This journal is © The Royal Society of Chemistry 2023 |