
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical fabrication of mesoporous metal-alloy films
Javeria
Bashir†
 ab,
Md. Belal
Chowdhury†
c,
Rahanuma Raihanu
Kathak†
c,
Shuvashis
Dey
b,
Atiya Tahira
Tasnim
c,
Mohammed A.
Amin
d,
Yusuf Valentino
Kaneti
b,
Mostafa Kamal
Masud
*bce and
Md. Shahriar A
Hossain
*ab
ab,
Md. Belal
Chowdhury†
c,
Rahanuma Raihanu
Kathak†
c,
Shuvashis
Dey
b,
Atiya Tahira
Tasnim
c,
Mohammed A.
Amin
d,
Yusuf Valentino
Kaneti
b,
Mostafa Kamal
Masud
*bce and
Md. Shahriar A
Hossain
*ab
aSchool of Mechanical and Mining Engineering, Faculty of Engineering, Architecture and Information Technology (EAIT), The University of Queensland, Brisbane, QLD 40721, Australia. E-mail: md.hossainn@uq.edu.au
bAustralian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: m.masud@uq.edu.au
cDepartment of Biochemistry and Molecular Biology, School of Life Sciences, Shahjalal University of Science & Technology, Sylhet 3114, Bangladesh
dDepartment of Chemistry, College of Science, Taif University, P.O. Box 11099, Taif, 21944, Saudi Arabia
eJST-ERATO Yamauchi Materials Space-Tectonics Project and International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science, Tsukuba, Ibaraki 305-0044, Japan
First published on 24th November 2022
Abstract
Nanoarchitectured mesoporous metal alloy films offer immense potential by integrating the intrinsic catalytic capabilities of their constituent metals to create suitable surface morphologies, different signal transductions and catalytic phenomena. These unique properties and functionalities have led to the development of several strategies for fabricating mesoporous alloy films, including template-based (e.g., lyotropic liquid crystalline (LLC) and di-block polymer-based templating) and template-free approaches (e.g., post-synthetic dealloying). More recently, the utilization of electrochemical methods for metallic mesoporous alloy films has ushered in a new way of fabrication and attracted much attention due to their simplicity, environmental friendliness, and cost-effectiveness. In this review article, we comprehensively reviewed the key electrochemical approaches for fabricating various types of metallic mesoporous alloy films (a wide range of geometries, pore sizes, form and tunneling structures, and metal compositions). With an emphasis on the fabrication of mesoporous bimetallic and trimetallic alloy films, we discuss their specific application for developing advanced technologies spanning from hydrogen production to fuel cell and sensor development. Furthermore, we also discuss the bottlenecks in the fabrication of mesoporous alloy films and provide a future perspective on the techniques. We envisage that this literature will guide the researcher towards research exploring engineered alloying of earth-abundant elements and translating them for industrial-scale electrocatalysis and nanomedicine application.
 Javeria Bashir | Javeria Bashir is a PhD student at the Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland (UQ), Australia. She received her master's degree in physics (2020) from the University of Lahore, Pakistan. Her research focuses on the synthesis and manipulation of magneto-plasmonic biocompatible mesoporous nanostructures for SERS based biosensing of clinically relevant biomarkers. |
 Md. Belal Chowdhury | Md. Belal Chowdhury graduated from the Department of Biochemistry and Molecular Biology from Shahjalal University of Science and Technology, Sylhet, Bangladesh. His research interest centers on the development of nanoarchitectured nanoparticles for early detection of disease-specific biomarkers with high sensitivity, specificity, and reliability along with faster detection time compared to the conventional diagnostic solutions. He has received several honors and distinctions for his excellent track record in academic study and published 4 peer-reviewed original research articles. |
 Rahanuma Raihanu Kathak | Rahanuma Raihanu Kathak is a lecturer of Biochemistry and Molecular Biology (BMB) at Primeasia University, Bangladesh. She has received her Master of Science and Bachelor of Science degrees in BMB from Shahjalal University of Science and Technology (SUST), Sylhet-3114, Bangladesh, with an academic record of 3.99 and 3.85 (out of 4.00) respectively. Her research interest lies in the field of medical diagnostics, especially in the emerging application of nano-architectonics for early disease detection. |
 Shuvashis Dey | Dr Shuvashis Dey is a Postdoctoral Research Fellow at the Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland (UQ). Prior to joining his current position, he received his PhD degree in biomedical engineering from UQ and developed microfluidic biosensors for early diagnosis and personalized treatment for better patient management. His research interest includes design and fabrication of micro/nanofluidic platforms and utilization of plasmonic nanomaterials for biosensing purpose. |
 Atiya Tahira Tasnim | Atiya Tahira Tasnim is a graduate student at the Department of Biochemistry and Molecular Biology under the School of Life Sciences at Shahjalal University of Science and Technology, Bangladesh. Her research interest focuses on the scope of biomolecule-based cancer nanomedicine, developing therapeutics in silico with a combination of wet-lab techniques, and exploring cutting-edge biotechnological methods ranging from single-cell genomics to nanotechnology. |
 Yusuf Valentino Kaneti | Dr Yusuf Valentino Kaneti received his PhD from the University of New South Wales in 2014. He has conducted postdoctoral research at Monash University (Australia), Graduate School at Shenzhen, Tsinghua University (China), and National Institute for Materials Science (Japan). Currently, he is working as an Advance Queensland Industry Research Fellow at the Australian Institute for Bioengineering and Nanotechnology, The University of Queensland. His research focuses on the rational design and fabrication of functional nanoporous materials and hybrid materials for energy and environmental applications. |
 Mostafa Kamal Masud | Dr Mostafa Kamal Masud is a Postdoctoral Research Fellow at Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland. Prior to joining AIBN, he has worked as a JSPS postdoctoral fellow at National Institute for Materials Science, Japan. He obtained his PhD from The University of Queensland, Australia, in 2020. His research focuses on the design and translation of bio-favourable engineered mesoporous nanostructures for the development of nanobiosensor to address critical issues in medical diagnosis. |
 Md. Shahriar A Hossain | Dr Shahriar Hossain is an Associate Professor at the School of Mechanical and Mining Engineering at The University of Queensland, Australia. He obtained his PhD degree in Materials Science and Engineering from the Institute for Superconducting and Electronic Materials (ISEM) at the University of Wollongong (UoW), Australia, in 2008. Dr Hossain was then employed as a Postdoctoral Research Fellow at the University of Geneva (Switzerland). He was awarded the prestigious DECRA Fellowship from ARC in 2013. His research expertise is on the fabrication and characterization of superconducting materials and magnetically triggered nanoparticles for medical, power, and energy applications. |
1. Introduction
Mesoporous nanomaterials including nanoparticles, films and building blocks have gained immense interest in recent years owing to their porous architecture that provides an exceptionally large surface area, mesochannels, and enhanced mass and ion transfer inside a nano-level material. These unique and fascinating features make them highly favorable and suitable for widespread applications including adsorption,1 isolation and separation of large molecules including chemicals and biomolecules,2 catalysis,3,4 targeted drug and nucleic acid delivery,3,5–9 sensor fabrication,10 advanced functional devices11,12 and highly efficient fuel cells.13 As a result, extensive research has been conducted to develop highly efficient mesoporous materials based on the target application, leading to the exploration of a wide range of mesoporous nanostructures of various chemical nature (e.g., carbon, metals, silica, aluminosilicates, metal oxides, metal carbides, metal sulphides, metal nitrides, polymers, etc.) as well as various frameworks (e.g., nanomaterials, nanocomposites and films).14 Among these materials, mesoporous metal architectures, particularly mesoporous metal films, are fascinating due to their intriguing physiochemical properties, such as ultra-large surface area, numerous exposed catalytic active sites, a widely open and interconnected porous structure that provides fast mass transport, high electrical conductivity, high mechanical stability and the ability to functionalize the pore surface in a variety of ways.15–18 Simultaneously, the ever-increasing demand for new materials with desirable properties and functionalities has fueled research into the synthesis of metallic alloys and fabrication of relevant devices.19–24Mesoporous metal alloy architectures combine the intrinsic catalytic characteristics of their constituent metals with the unique properties of mesoporous structures to provide distinctive signal transduction and catalytic properties. Mesoporous alloy films made up of multiple metals with varied reactivities and standard redox potentials (E0) can provide several advantages for electrocatalysis.21,23 Compared to the traditional nonporous noble metal based electrocatalysts, mesoporous metal alloy nanoarchitectures with a combination of noble metals can enhance electrocatalytic performance significantly because they produce a more favorable electrical structure and reduce the electrode fabrication cost. For instance, mesoporous Au–Cu films exhibit excellent performance in developing nonenzymatic glucose sensors with high sensitivity and selectivity, and reduce the cost involved in device fabrication due to the synergism between electronic response and the catalytic benefits from both components compared to that of monometallic Au based devices.23 Such superior electrocatalytic performances have also been reported for other mesoporous films, e.g., Pt–Cu,25 Au–Ni,26 Rh–Ni,21etc. Moreover, mesoporous noble metal–metal mixtures (e.g., Au–Pt, Pt–Pd,24,25 Pt–Ru,27 Au–Ag, Au–Ni,26etc.) have resulted in the development of high-performance heterogeneous electrocatalysts with better durability. For instance, the catalytic activity and electrochemical stability of mesoporous Pt–Au alloys or Pt–Ru alloys towards methanol oxidation (methanol fuel cell) and/or glucose oxidation (glucose detection) are substantially greater than that of mesoporous Pt films, which can be attributed to the well-developed mesoporous structure with a large surface area and the synergistic characteristics of Pt and Au.24,27
To date, several strategies for fabricating mesoporous alloy films have been reported, which can be classified as the hard-templating approach (i.e., nanocasting),29 the soft-templating approach (e.g., lyotropic liquid crystalline (LLC) and di-block polymer based templating)30,31 and the template-free approach (e.g., post-synthetic dealloying).32 Hard templates made of inorganic materials such as mesoporous silica are commonly used to guide and control the building of mesoporous metals. The generated replicas reverse the template's morphology; however, the dissolution process is typically slow and requires repeated exposure to dangerous chemicals. Although hard-templates provide a consistent and intuitive method for producing mesoporous metals, the complexity of the technique, lack of morphology control, and use of harsh chemicals have confined their use in wider applications.9,33 Soft-templating methods are promising alternatives that use organic surfactants (e.g., block polymers, lyotropic liquid crystals, etc.) as molecular templates to direct the formation of mesopores in metallic structures.31 Though soft-templating methods offer several distinctive benefits (e.g., cost-effectiveness, a wider porous structure with designed pore size and morphology, and customization), the high viscosity of the formed micellar system limits their applications.34 Mesoporous metal nanostructures have also been synthesized using a dealloying technique that selectively removes the most chemically active element.35 The chemical treatment preferentially dissolves a non-targeted element, leaving a porous metal or alloy surface behind. This technique is commonly seen in technologically significant alloy systems, such as stainless steel, brass, and different alloys especially Cu–Al alloys.36 However, the mechanical and physical properties of a porous overlayer differ greatly from those of the bulk alloy on which it sits, resulting in stress corrosion cracking, brittle crack propagation as well as other unwanted material failure.37 Even though these approaches are widely used for fabricating alloyed mesoporous metal architectures, these methods are complex and involve multistep processes, limiting their potential applicability, flexibility, and reproducibility. To address these issues, research has concentrated on the development of new strategies for the synthesis of more effective mesoporous materials in terms of fabrication time, flexibility, electrical properties, catalytic performance, and expense. In this regard, electrochemistry has opened an excellent and important window for the mesoporous world in terms of both electrochemical fabrication and characterization of mesoporous metal-based electrode structures.17,38,39 Compared to other methods, electrochemical synthesis of mesoporous alloy films is considered as a competitive and versatile synthetic method, in terms of simplicity, environmental friendliness and cost-effectiveness.14,22–26,40 The basic working principle of electrochemical techniques involves the application of an external potential on an appropriate electrode (as a working electrode) to convert the soluble precursor chemicals present in the electrolyte solution into solid mesoporous films. Metallic alloys have frequently been fabricated on conductive (as an electrode) surfaces using only one operational step, that is the simultaneous reduction of the precursor ions contained in the electrolyte by adjusting the potential/current density.41 A distinguishing feature of electrodeposition is that several reactions can occur simultaneously at the same working electrode that can be stimulated based on the external potential used.16 Because the applied potential is the driving force for depositing metal precursors dissolved in the electrolyte to form alloy films, the thickness of electrodeposited films can be controlled by varying the deposition time and metal precursor composition present in the electrolyte. Furthermore, because the films are deposited on conductive substrates, the as-prepared film can be immediately used for application purposes.20,21,24,42,43 Even though a plethora of research has been done in this field, no reviews have yet been published on the fabrication of mesoporous metal alloys utilizing both direct and indirect strategies, as well as on their applications. To address the research gap and to lead future science in mesoporous alloy films and their application in catalysis, energy, chemical sensor and biosensor applications, reports are sought in which both the fabrication and film-based targeted application are comprehensively discussed.
In this review we have comprehensively focused on the various electrochemical approaches for synthesizing different types (a wide range of geometries, pore sizes, shape and tunneling structures, and metal compositions) of metallic mesoporous alloy films. With an emphasis on the fabrication of mesoporous bimetallic and trimetallic alloy films, we have discussed their specific application for developing advanced technologies spanning from hydrogen production to fuel cell and sensor development. Following a thorough discussion in each section, this review also chronicles the challenges and needs implicated in the fabrication of mesoporous alloy films and provides a future perspective on the strategies presented.
2. Strategies for fabricating mesoporous metal alloy films
With the growing interest in mesoporous metal alloys with controllable porosity and pore morphology (size, shape, distribution and tunneling) as well as unique chemical and physical properties, several strategies have been developed to obtain the porous alloy film of interest. The cutting-edge nanotechnology is capable of developing porous structures with pore sizes ranging from 2 nm to 100 nm and spacing widths ranging from 50 to 100 nm with desired geometry.44 The most used strategies adapted for fabricating such alloys include template-based strategies (soft and hard template), potential-controlled surfactant assembly, colloidal assembly, interfacial surfactant templating, deposition from lyotropic liquid crystalline phases of surfactants, sol–gel assembly, electro-assisted self-assembly method, etc. Each strategy is specific and provides films with defined shape, quality, elemental composition, architecture, and pore definition. The pore size, shape, and morphology are also affected by the substrate (e.g., silicon wafer, glass, stainless steel, etc.) and template used to develop porous structures. For instance, synthesis of mesoporous titania films on freshly ground steel produced extensively fractured films with distorted porosity, whereas films produced on pre-calcined steel demonstrated good integrity, high substrate coverage, and a narrow pore size distribution with ordered and interconnected pores.45 Though the above mentioned strategies have their own advantages and superiority, each methodology also suffers from drawbacks.27,46,47 Among all reported strategies, the template based strategies (e.g. endo-template or soft-templating approach, exo-template or hard templating approach, etc.)48–51 are popular where an engineered template is used to direct the geometry of pores in the alloy films. Herein, we have divided the above-mentioned strategies into two broad categories: (i) direct electrodeposition of metals by electroreduction of metal cations, and (ii) indirect precipitation of metal hydroxides through electrogenerated hydroxide ions.2.1. Direct electrodeposition of metals by electroreduction of metal cations
This is one of the most common deposition processes that involves the electrochemical reduction of metal precursors to their solid metal states, where electrons are directly transferred by the working electrode.39,50 In the direct electrodeposition, the metal ions interact with the polar part of polymeric micelles. The micelles’ geometry depends on the nature of both free ions and the polymer. These ions as well as the ion-capped polymeric micelles become positively charged even though initially they are non-ionic. If some paramagnetic ions like Ni2+ are present in the micellar system, they have no significant effect on the process in the absence of a magnetic field. However, in the presence of an electric field, the capped micelles move to the negative electrode where the ion precursors are reduced. Therefore, the applied electric field is a critical factor for obtaining the desired porous structure. The reduction potential is also a determining factor of the porous structure as during the deposition process, a higher reduction rate is achieved through a large negative potential. If two precursors with different potential are present in the system, the precursor having higher redox potential (value vs. the standard electrode) will be favored for reduction. For example, at a deposition potential of −0.7 V (vs. Ag/AgCl), the reduction of the Au precursor in micelles containing both Au and Ni ions is higher due to the large difference between the standard redox potentials of Au and Ni (i.e., the standard redox potential of [AuCl4]−/Au is 1.00 V vs. standard calomel electrode (SCE), while the standard redox potential of Ni2+/Ni is −0.25 V vs. SCE). Within a certain range of applied potential the metal alloy film can be fabricated but no porosity can be observed due to too fast or too slow reduction of ions. Therefore, to get highly uniform and porous metal alloy films, the voltage always needs to be optimized.28 Overall, the electrical potential applied to electrodes regulates the rate of electron generation and, as a result, nucleation and crystal growth. Temperature also plays a big role as higher temperature leads to large ligament of the mesoporous structure having large size nanocrystals. These variables are critical in determining the morphology of the nanoporous film. These electrodeposition techniques have grown in popularity over the last two decades as a means of developing efficient and reliable processes for creating mesoporous metals.16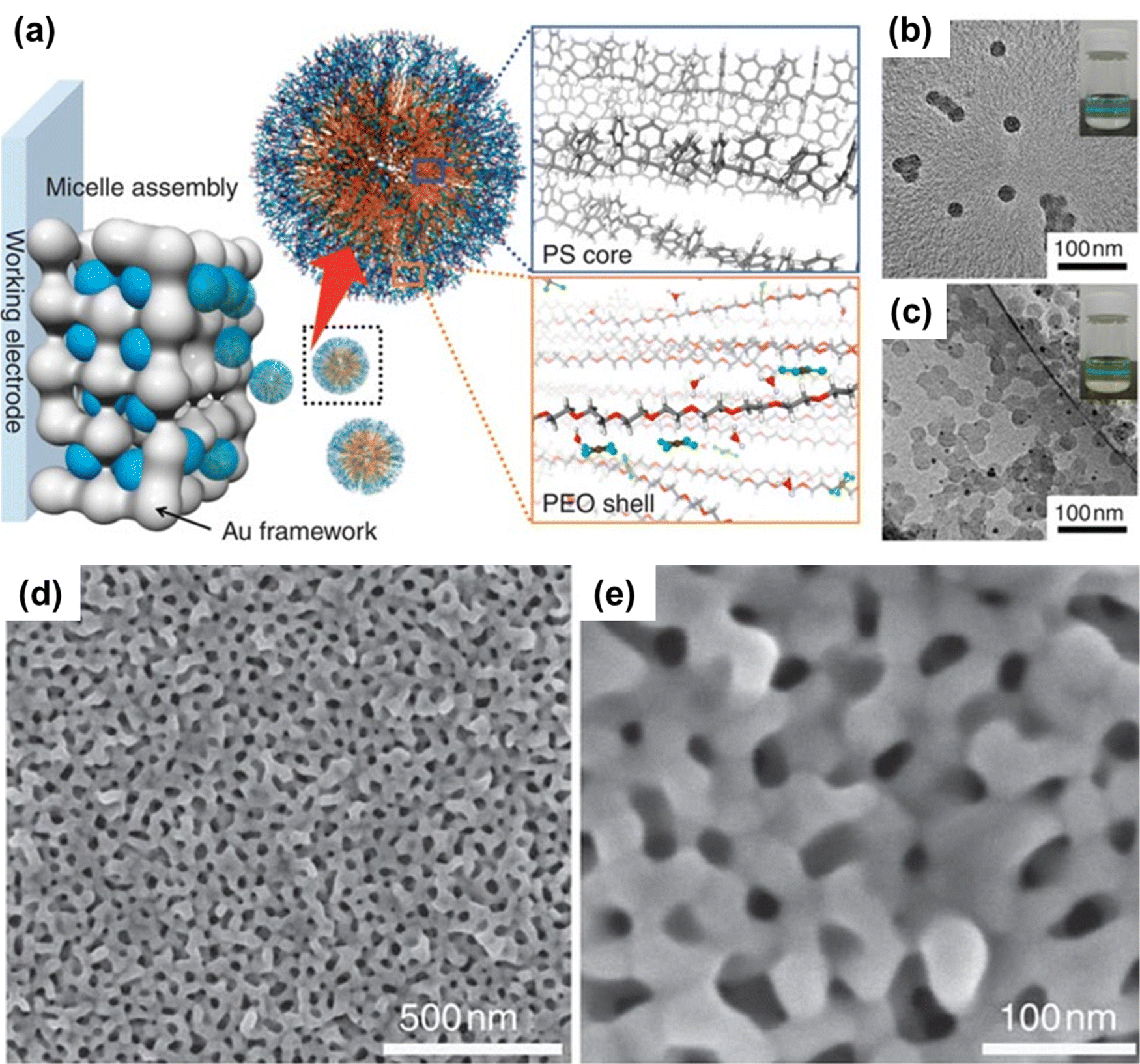 | ||
Fig. 1 (a) Schematic illustration of the mechanism involved in the formation of PS-b-PEO micelles with a gold precursor (HAuCl4). Under a suitable composition, the PS-b-PEO block polymer reacts with Au ions to form a spherical micelle. (b) The TEM images reveal the formation of spherical micelles containing the PS18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-b-PEO7500 block polymer without Au ions. (c) The formation of micelles with Au ions. The black dots represent Au nanoparticles formed on the surface of micelles by electron beam irradiation. The inset image depicts the Tyndall effect. (d and e) Top-surface SEM images of the mesoporous Au film prepared at 1000s deposition time. Reproduced with permission.70 Copyright 2015 Springer Nature. 000-b-PEO7500 block polymer without Au ions. (c) The formation of micelles with Au ions. The black dots represent Au nanoparticles formed on the surface of micelles by electron beam irradiation. The inset image depicts the Tyndall effect. (d and e) Top-surface SEM images of the mesoporous Au film prepared at 1000s deposition time. Reproduced with permission.70 Copyright 2015 Springer Nature. | ||
2.2 Indirect precipitation of metal hydroxides through electrogenerated hydroxide ions
3. Fabrication of mesoporous metallic alloys
3.1 Gold–silver (Au–Ag) alloy
The electrochemical fabrication of a mesoporous Au–Ag alloy can be achieved by several electrochemical methods. These distinctive methods have a significant impact on the variable properties of the synthesized alloy such as pore size and shape, pore diameter, surface area, and structure–activity relationship. The soft and hard template approach along with the electrochemical dealloying method has been extensively employed to fabricate mesoporous metals. Although a fine control over the size and structure of mesoporous metals can be achieved by the soft and hard template approach, irregular morphology of the resulting film can be a major drawback. The challenge of fabricating monodispersed mesoporous Au–Ag by the hard template approach is attributed to the faster deposition rate in mesoporous templates and formation of bulk particles due to the preferential migration to the outer layer of the mesoporous silica during the reduction process.115 To prevent the migration of metal species outside of the mesoporous support, Fang and colleagues developed a method using a soft-enveloping phase transfer mechanism.116 The phase transfer mechanism is comprised of three distinct phases: solid-liquid-solution (SLS) interface where the solvent (liquid) layer acts as a barrier to block the migration of metal species to the template (solid) and cannot dissolve the used precursor molecules. The solution phase contains the reducing agent, and the final reduction of the metal precursors occurs at the interface of the solid and solution phase facilitated by the spontaneous phase transfer at the interface of the solution and liquid phase. Based on this strategy and using hexane and 1,1,3,3-tetramethyldisiloxane (TMDS) as a barrier layer and reducing agent respectively on a KIT-6 hard template, monodispersed Au–Ag NPs with highly ordered uniform pores with an average diameter of ∼100 nm were synthesized. The resulting Au–Ag networks exhibited higher catalytic properties in the methanol oxidation reaction (MOR) producing higher peak current up to 19.2 μA μg−1 which is 3.1- and 24-fold higher than that of the Au-4 (nm) NPs and Au-70 NPs respectively. They also exhibited lower electro-oxidation potentials than the control Au NPs and 3- and 24-times higher mass activity and 7 and 5 times improved specific activity compared to Au-4 and Au-70 NPs respectively.The Au–Ag alloy film can also be fabricated by using a block copolymer as a soft template that directs the pore formation and pore morphology. Such an electrochemical process of fabricating mesoporous Au–Ag alloy films has been reported by Park and colleagues.117 In this work, a block copolymer templated approach was used to generate mesoporous Au–Ag alloy films with a high surface area to volume ratio and enhanced biosensing capacity. A sacrificial soft-template, PS-b-PEO block copolymer, has been utilized as a pore directing agent.
The PS-b-PEO block polymer (dissolved in THF) forms spherical micelles upon addition of ethanol and an aqueous solution of HAuCl4 or (and) AgNO3 (as Au and Ag precursors, respectively). Upon the addition of the aqueous salt, the hydrophobic part of the block polymer (PS) aggregated as a core, leaving the hydrophilic EO as a shell to interact with the bimetallic species (Au and Ag ions) at the outer layer of the formed micelles. The electrodeposition of a micellar solution containing 50:50 Au3+:Ag+ at −0.5 V resulted in a homogeneously dispersed mesoporous film (Fig. 2). The as-synthesized Au–Ag alloy films were reported to contain uniformly sized pores of 12 nm throughout the alloy surface and a wider pore size of 23 nm was also found with the increase of Ag content. The resultant alloy also exhibited higher catalytic activity towards the [Fe(CN)6]3−/4− redox molecule. The homogeneous mesopores with a large surface area and the higher electrocatalytic activity of the synthesized alloy enable the construction of a highly sensitive miRNA biosensor as a transduction surface (working electrode) without requiring any enzymatic and amplification process. The magnetically isolated and purified miRNAs were absorbed onto the surface of the mesoporous Au–Ag film that contributed significantly to achieve a higher current response of 144.21 μA cm−2 in differential pulse voltammetry (DPV) interrogation which is 18 and 2-fold higher compared to the mesoporous Ag and Au electrodes respectively. This rapid, amplification-free miRNA biosensing assay using the cost-effective Au–Ag alloy film is highly sensitive to attomolar levels (100 aM LOD) which is multiple times higher than amplification-based molecular techniques and commensurate with the electrochemical biosensor-based detection techniques.118,119 In terms of stability, the mesoporous Au–Ag films sustained more than 40 runs measured using cyclic voltammetry in both reaction media ([Fe(CN)6]3−/4−) and acidic media (0.5 M H2SO4).
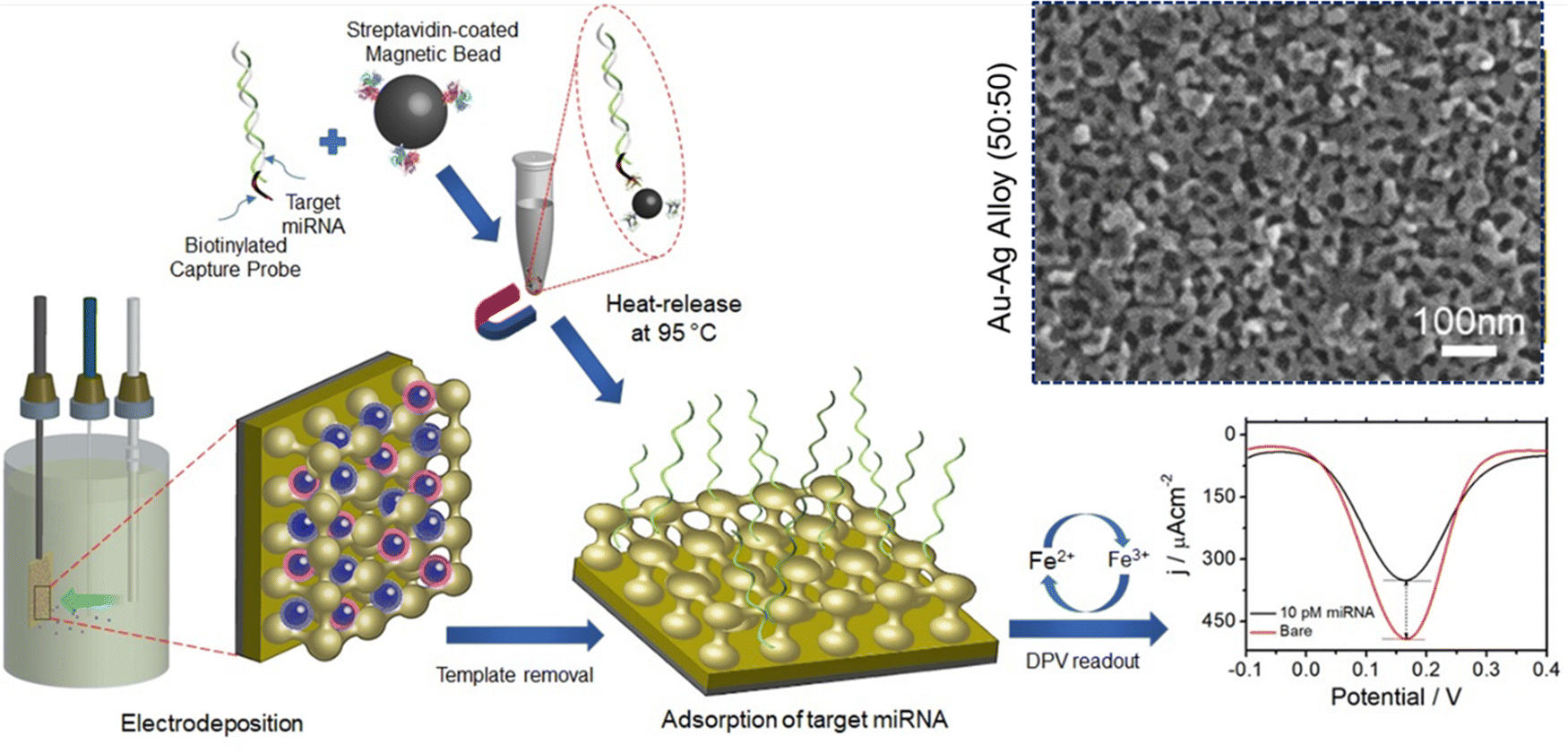 | ||
| Fig. 2 Schematic diagram of mesoporous Au–Ag alloy film synthesis via the electrodeposition process. Metal precursors were deposited electrochemically onto the PS-b-PEO derived diblock-copolymer micelles followed by template removal and adsorption of magnetically isolated and purified miRNA on the Au–Ag film. Changes in the DPV current in the [Fe(CN)6]3−/4− system before and after the target miRNA adsorption were recorded as a part of evaluating mesoporous Au–Ag films as a single step non-enzymatic amplification free miRNA sensor. Inset: Top-surface SEM images of the mesoporous Au–Ag alloy prepared from a 50:50 precursor ratio. Reproduced with permission.117 Copyright 2020 Royal Society of Chemistry. | ||
In another report, Lv et al. fabricated mesoporous Au–Ag nanospheres including mesoporous Au–Ag alloy, hollow mesoporous Au–Ag, and core–shell Ag-mesoporous Au.120 A seedless, one-pot, surfactant-templating approach was utilized to fabricate the well-controlled nanostructures. A synthetic surfactant template C22H45N+(CH3)2–C3H6-SH (Cl−) (C22N-SH) along with AuCl4− and Ag+ as metal precursors and N2H4 as the reducing agent was used to fabricate the alloys. The covalently stable intermediate generated by thiolate-metal linkages between C22N-S-Au(I) and C22N-S-Ag(I) precluded quick nucleation and high mobility of Au/Ag precursors.
3.2 Platinum–gold (Pt–Au) alloy
Although Au and Pt are referred to as the most chemically stable metals due to their resistance to oxidation reaction, synthesis of a Pt–Au bimetallic alloy is a challenging task. The imposed challenge is attributed to the different reduction kinetics of Au and Pt ions leading to phase segregation.121–123 Massive attention has been directed towards the effective synthesis of bimetallic Pt–Au alloys due to their high catalytic properties than their pure counterparts. Moreover, the surface area of Pt–Au binary alloys largely impacts the electrocatalytic and biosensing performance since the presence of Au prevents the adsorption of poisoning intermediates such as CO by structure-induced promotional effects.124 The challenge of fabricating a Pt–Au bimetallic alloy has been overcome by Yamauchi and co-workers, who electrochemically synthesized a mesoporous Pt–Au alloy by employing a soft-templating strategy (Fig. 3).40 Herein, the LLCs prepared from concentrated surfactant solution by solvent evaporation have been used as the soft-template. The use of LLCs prevented phase separation of Au and Pt ions thereby resulting in a uniform distribution despite varying deposition potentials of independent ions. They have also shown that the framework composition of the mesoporous structure can be readily controlled by tuning the ratio of the precursor solution. Shifting the Au content from low to high yielded distorted mesoporous structures (Pt62Au38) compared to the well-ordered 2D hexagonal mesostructures (Pt90Au10 and Pt80Au20) which in turn reduced the electrochemically active surface area. In SEM analysis, pores were reported to be distributed over the entire surface of mesoporous alloy films in a honeycomb arrangement with a pore diameter of around 3 nm. When compared to nonporous Pt sheets and mesoporous Pt, the resulting mesoporous alloy performed admirably as a catalytic electrode in electrochemical methanol oxidation reactions, exhibiting substantial current density for a longer period. This durable feature was achieved due to the prevention of electrode deactivation by poisoning in the presence of Au which is absent in nonporous and mesoporous Pt.125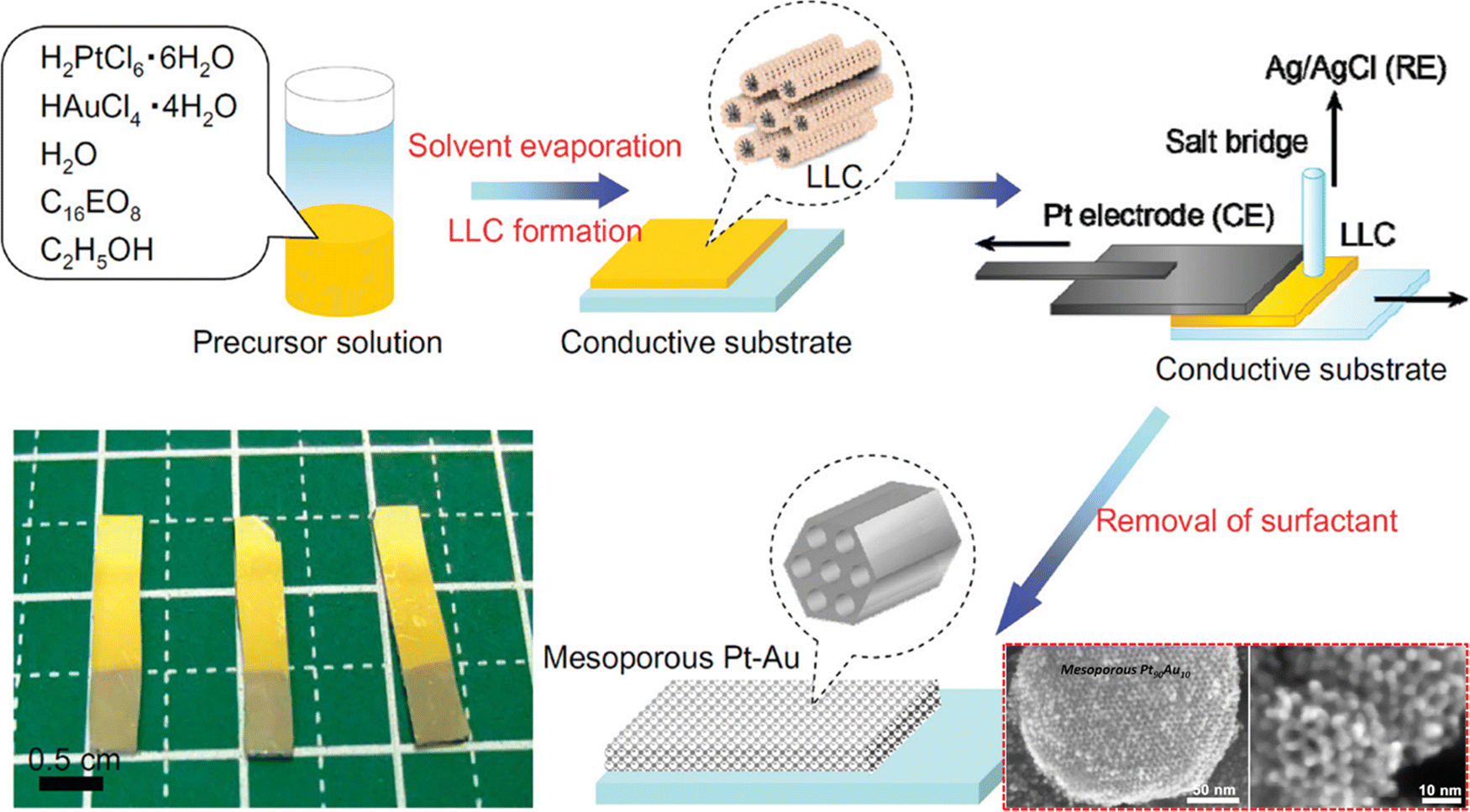 | ||
| Fig. 3 Illustration of the experimental steps involved in the fabrication of the mesoporous Pt–Au alloy film. The highly ordered two-dimensional LLC template was prepared on a conductive surface by a drop-coating method from a surfactant solution containing ethanol, water, nonionic surfactant, and the corresponding metal species. The resulting LLC film comprising Pt and Au species was used to perform metal electrodeposition utilizing a galvanostat and a Pt substrate as the counter electrode and Ag/AgCl as the reference electrode, followed by removal of surfactants and undeposited metal species. Inset: SEM images of mesoporous Pt90Au10. Reproduced with permission.40 Copyright 2012 American Chemical Society. | ||
Another surfactant based templating strategy using poly(ethylene glycol)–poly(propylene glycol)–poly(ethylene glycol) (P123) as a template was reported for the fabrication of Pt–Au mesoporous alloy films by electrodeposition on indium–tin oxide (ITO) coated glass.125 The P123 template contributed to the surface morphology by affecting the growth of particles. The fabricated Pt–Au alloy films also exhibited higher catalytic activity in the methanol oxidation reaction and induced a peak area increase and peak shift to lower potential in the I–V curve (at 50 mV s−1 scan rate) compared to the fabricated nonporous and mesoporous Pt electrode. In the performance characteristics test of the synthesized film in the methanol oxidation reaction, the Pt–Au film displayed a higher current density for a longer period of time compared to the nonporous Pt and mesoporous Pt indicating the alloy's significant stability.
In 2013, Cuiling Li and colleagues reported another approach of electrochemical synthesis of a mesoporous Pt–Au alloy film by a soft-templating method.22 In their work, they were able to fabricate a mesoporous Pt–Au alloy by electrochemical deposition on a Au-coated Si substrate. Rather than applying constant potential deposition, a square-wave potential program was applied in the process. Application of a square-wave potential facilitated the uniform reduction and constant concentration of metallic ions on the substrate surface resulting in the formation of a Pt–Au alloy with a pore size of approximately 7–8 nm and without any phase separation. The square-wave potential also permitted the retention of a constant mesoporous structure with up to 50% atomic content of Au whereas a distorted mesoporous structure in the Pt62Au38 alloy was noticed in the absence of a square-wave potential. The fabricated Pt–Au alloy films were found to be a highly effective glucose sensor owing to their high catalytic activity towards glucose oxidation. This is due to the synergy of the large surface area of the Pt–Au alloy and mesoporous structures. As an amperometric glucose sensor, the fabricated mesoporous Pt51Au49 alloy was reported to be highly sensitive (352 μA cm−2 mM−1) with a lower detection limit (LOD) of 6 μM and a detection range of 6 μM – 11 mM which is within the normal physiological level of glucose (8–11 mM).126,127 Moreover, after their catalytic activity, the native state of the Pt–Au electrodes can be achieved with a simple rinse with distilled water and the electrode structure can sustain at least 10 cycles of glucose detection. Electrochemically fabricated mesoporous Pt–Au binary alloy films (Pt51Au49) featuring high sensitivity, low LOD, wider detection range, high fabrication reproducibility, and anti-poisoning effects enable the application of the Pt–Au alloy as a non-enzymatic glucose sensor for a wide range of clinical purposes.
3.3 Gold–copper (Au–Cu) alloy
Highly viscous and concentrated surfactants used in a typical liquid crystal-based synthesis of mesoporous alloys interfere with the pore size tuning of the fabricating alloy.34 To get around the problem, researchers have employed a combination of electrochemical deposition, polymeric micelle assembly, and easy solvent extraction to create a mesoporous Au–Cu alloy with variable composition. Nugraha et al. have reported the synthesis of Au–Cu mesoporous alloy films by electrodeposition on Au–Si substrates using spherical polymeric micelles as the electrolyte solution.23 In this strategy, the PS-b-PEO polymeric micelle was used as a sacrificial soft-template. Fine control over the architecture of mesopores was achieved by tuning the compositional ratio of precursor species (Au and Cu) present in the electrolyte solution. Uniform distribution of the mesopores was reported in Au94Cu6, Au87Cu13, Au78Cu22, Au49Cu51, and Au41Cu59 alloy samples with an average pore diameter of 28 nm (Fig. 4). Interestingly, it was found that the final structures of the alloy mimic the soft template used as a support. Au94Cu6 and Au87Cu13 alloys which were low in Cu content appeared to replicate the cage-like mesopores present in the parent micellar template. In contrast, the higher Cu content in Au78Cu22 and Au49Cu51 alloys mediated the fusion of mesopores resulting in one-dimensional vertically aligned mesochannels. These vertical channels provide lesser diffusion resistance which in turn facilitates mass transfer within the mesoporous channels. Electrochemically synthesized Au–Cu bimetallic mesoporous alloy films were also found to be highly active in the electro-oxidation of glucose. In general, the performance of a non-enzymatic glucose sensor heavily depends upon two critical parameters: electroconductivity and the large surface area of electrodes that are crucial for electro-oxidation of glucose.23 All of the Au–Cu alloys possessed a high volume-normalized electrochemically active Au surface area (ECSA) compared to the Au films. Moreover, the higher current response and more positive potential of bimetallic Au–Cu mesoporous alloys compared to the mesoporous Au films in the electro-oxidation of glucose characterize the alloys to be more catalytically active. The mesoporous Au49Cu51 film was also found to be highly sensitive (643.6 μA cm−2 mM−1) than the nonporous Au49Cu51 film and mesoporous Au film over a broad range of glucose concentration (0.01 – 19 mM) with a LOD of 1.5 μM. To date, there are only two Au- and Cu-based nanomaterial-based glucose sensors available that possess higher sensitivity than the mesoporous Au–Cu film.128,129 | ||
| Fig. 4 The top-surface SEM images of Au–Cu alloy films with different compositions of both Au and Cu, fabricated by using the PS-b-PEO polymeric micelle. The presence of mesopores in the Au–Cu alloy samples confirms the formation of mesoporous films. Reproduced with permission.23 Copyright 2018 American Chemical Society. | ||
3.4 Gold–nickel (Au–Ni) alloy
Application of electrochemical deposition to synthesize mesoporous alloy films can be extended further to fabricate Au–Ni bimetallic alloy films with mesoporous morphology. Such an approach was reported by Nugraha and colleagues where a block-copolymer template was utilized to synthesize mesoporous Au–Ni alloy films with different compositions.26 In this soft-template mediated electrodeposition, Au and Ni precursors were reduced electrochemically in the presence of PS-b-PEO diblock copolymer micelles. The resultant Au–Ni films consist of mesopores distributed uniformly throughout the entire surface with an average pore size of 28 nm. By adjusting the Au:Ni ratio in the precursor solution, several Au–Ni alloy films with varying compositions (Au100, Au81Ni19, Au76Ni24, Au60Ni40) were produced. A negative correlation between alloy thickness and Ni2+ content in electrolyte solution was observed. The well-defined mesopores are generated throughout the film, from the outermost to the innermost parts (Fig. 5). By enhancing accessibility to active sites and lowering mass and ion transport resistance, such a mesoporous architecture can be of considerable benefits for electrocatalytic applications, allowing for an optimum contact area between the catalyst and the analyte. Mesoporous Au100, Au81Ni19, Au76Ni24, and Au60Ni40 had a Au mass-normalized ECSA-Au of 113.9, 38.2, 94.2, and 53.4 cm2 mg−1_Au, respectively. In the cyclic voltammetry (CV) test to measure the electrocatalytic performance toward the methanol oxidation reaction (MOR), mesoporous alloy films exhibited two characteristic anodic peaks during positive and negative-going scans; the former is due to the oxidation of methanol whereas the latter is linked to the removal of the leftover carbonaceous intermediates.130,131 The catalytic current density of alloy compositions increased gradually with the increase of Ni content until reaching the optimum for Au76Ni24 (410 mA mg−1_Au) and then declined. This obtained current density is the highest among other reported catalysts used for MOR.132–135 In durability check of the binary Au–Ni alloy for MOR by chronoamperometry, Au76Ni24 exhibited stable current density and the lowest deterioration rate of current density for 1200 seconds among other compositions owing to the prevention of poisoning by intermediate carbonaceous species.136 The simple preparation reinforced by excellent electrocatalytic capability and stability underscores the practical implementation of the mesoporous Au76Ni24 bimetallic alloy film in direct methanol fuel cells (DMFCs).
 | ||
| Fig. 5 The cross-sectional SEM images of mesoporous bimetallic Au–Ni films obtained from precursor solutions using PS-b-PEO diblock copolymer micelles containing different molar ratios of Au and Ni (a) 100:0, (b) 75:25, (c) 50:50, and (d) 25:75 at an applied potential of −0.7 V and temperature of 40 °C. Reproduced with permission.26 Copyright 2019 Elsevier Ltd. | ||
3.5 Platinum-based (Pt–Pd/Ru/Cu) alloy
Unlike other bimetallic alloys, platinum–palladium alloys are in extensive use as catalysts in several application fields including fuel-cell technology, hydrogenation reactions, hydrocracking, hydrotreatment, hydro-isomerization, and so on.137 But the synthesis of Pt–Pd mesoporous alloys requires a lot of complex processes with poor control over the framework composition. Wang and Yamauchi demonstrated in 2012 that electrochemical plating in an aqueous surfactant solution may be used to make mesoporous Pt–Pd alloy films (Fig. 6).24 The aqueous electroplating solution contained K2PtCl4 and Na2PdCl4 along with a non-ionic surfactant Brij 58. Brij 58 was taken at a concentration of 1.0 wt%, which is above its CMC. By changing the compositional ratio of precursor species, compositional control over the mesopores of the synthesized alloy (Pt93Pd7, Pt82Pd18, Pt69Pd31, and Pt57Pd43) was achieved. The resulting film contained mesopores distributed uniformly throughout the surface with a pore size of 7 nm and a wall thickness of 3 nm. From the SEM image, it was evident that the absence of the platinum precursor induced the palladium to develop as a branch without forming any mesopore whereas an increased content of palladium caused visible cracks and an uneven alloy surface. The same effect was also reflected upon the alloys’ catalytic performance in the methanol oxidation reaction where Pt69Pd31 and Pt57Pd43 exhibited lower catalytic activity than mesoporous platinum and Pt93Pd7 and Pt82Pd18 alloy films. Pt93Pd7 and Pt82Pd18 achieved increased catalytic activity of 30% and 60% respectively than the mesoporous platinum film. This enhanced performance is due to the high palladium content which reduces the energy for methanol adsorption and facilitates dehydrogenation of methanol to adsorbed CO.138,139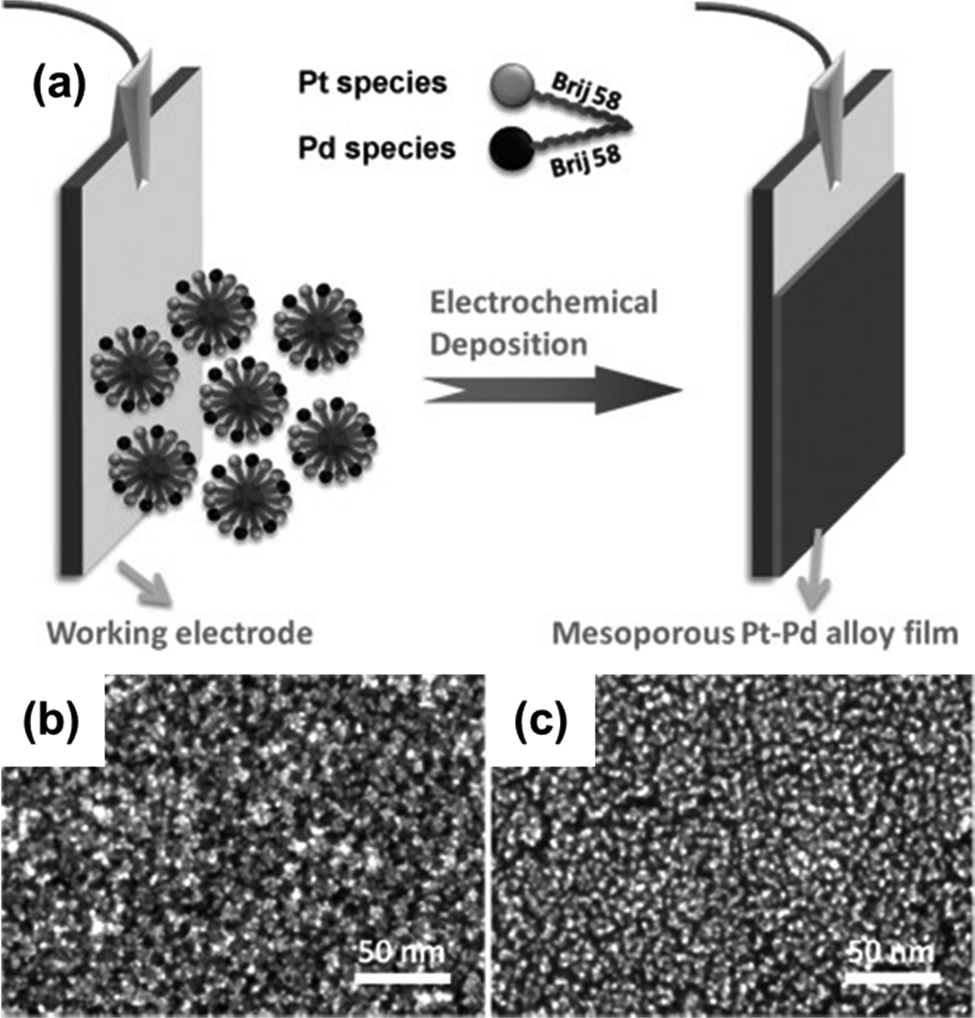 | ||
| Fig. 6 (a) Pictorial representation of the mesoporous bimetallic Pt–Pd alloy film synthesis by an electrochemical plating approach where Pt and Pd metal species are electrodeposited into the working electrode from a precursor solution containing K2PtCl4, Na2PdCl4, and non-ionic aqueous surfactant Brij 58. High-resolution SEM images of mesoporous Pt (b) and Pt57–Pd43 alloy films (c). (b) Reproduced with permission.24 Copyright 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In a regular platinum catalyst, CO can trigger the poisoning of the platinum surface. But the synergy between platinum and palladium in the Pt–Pd alloy film alleviates CO poisoning. Moreover, the presence of platinum nanoparticles in the wall of mesopores also boosts up the methanol oxidation reaction.63 Alongside catalytic performance, the Pt–Pd film also possessed significant stability in CV analysis as it achieved the highest current density for 2000 seconds compared to the mesoporous Pt and Pt catalyst. Similar to the Pt–Pd alloy, mesoporous Pt–Ru alloy film synthesis of different compositions has been reported by Wang and colleagues.66 In their work, they have used Brij58 as a non-ionic surfactant solution and K2PtCl4 and RuCl3 as metal precursors to perform electroplating on gold (Au) surfaces (Fig. 7). Different compositions of Pt and Ru in the precursor solution (Pt2+/Ru3+) such as 100:0, 75:25, 50:50, and 25:75 resulted in alloys of Pt100, Pt95Ru5, Pt93Ru7, and Pt87Ru13 respectively. Surprisingly, a substantial increase of Ru species in the precursor solution did not correspond to the amount deposited on the films (50% in the precursor to mere 7% in the product). This unique result could be attributed to the lower reduction tendency of Ru3+ ions than [PtCl4]2− in the reduction process during electrodeposition leading to the preference of [PtCl4]2− ions over Ru3+ ions.140 The as-synthesized alloy films possessed uniformly distributed mesopores with a diameter of 7 nm along with inter-connected nanoparticles in the pore wall. These connected nanoparticles contributed greatly to attain a concave surface offering numerous rich edges and corner atoms.141 In the methanol oxidation reaction, the mesoporous Pt95Ru5 alloy exhibited increased catalytic activity compared to the Pt nanoparticles, mesoporous Pt films, and the commercially available Pt catalyst. The chronoamperometric curve of the Pt–Ru alloy against the mesoporous Pt and Pt catalyst in the methanol dehydrogenation reaction showed considerably higher current density indicating good stability of the alloy film.
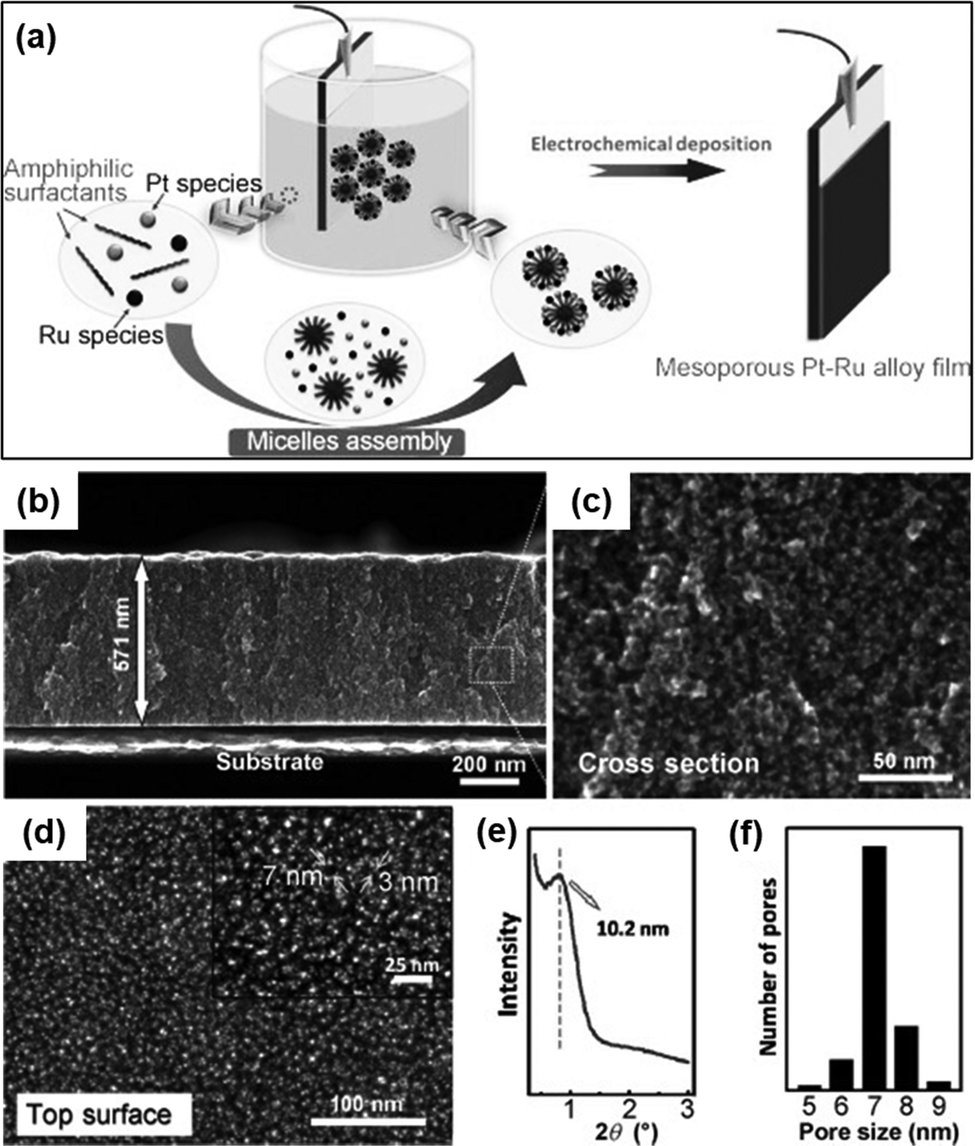 | ||
| Fig. 7 (a) Fabrication process of the mesoporous Pt–Ru alloy film by using electrochemical reduction using Brij58 as an amphiphilic surfactant and K2PtCl4 and RuCl3 as Pt and Ru precursors respectively. (b) Cross-sectional SEM image at low magnification. (c) Corresponding highly magnified SEM image. (d) Top-surface SEM image. Inset: Highly magnified image of the top surface. (e) Low-angle XRD profile. (f) Pore-size distribution curve in the film. Reproduced with permission.66 Copyright 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Another study focusing on the electrochemical method to synthesize mesoporous Pt–Cu alloy films has been reported by Li and Yamauchi in 2014.25 Similar to the Pt–Pd and Pt–Ru, mesoporous Pt–Cu films were electrochemically deposited on a gold-coated silicon substrate (as a working electrode) at a reduction potential of −0.2 V in an electrolyte solution containing metal precursors and non-ionic surfactant Brij58. A linear relationship between the metal content in the precursor solution and the resulting films of various Pt and Cu compositions was reflected. However, a shift of surface morphology from flat to a cauliflower-like structure with the increase of Cu content was reported from SEM images. From the synthesized alloy films of different compositions (Pt100, Pt86Cu14, Pt73Cu27, Pt57Cu43, Pt39Cu61 and Pt30Cu70) the thickness of Pt86Cu14 and Pt57Cu43 was reported to be similar around 550 nm. In the catalytic performance test towards the methanol oxidation reaction (MOR), Pt57Cu43 generated the highest current density of 1.02 mA cm−2 Pt compared to 0.98 mA cm−2 Pt and 0.96 mA cm−2 Pt for Pt86Cu14 and Pt100 respectively. Moreover, a negative shift of the onset potential and peak potential in mesoporous Pt–Cu films was evident compared to the mesoporous Pt film. A comparatively higher current density of Pt57Cu43 than Pt86Cu14 despite sharing other properties was due to the fine balance between Pt and Cu content where Cu scavenges poisonous intermediates generated on the Pt electrodes suppressing CO poisoning.142 However, the mesoporous Pt–Au alloy possess relatively less stability (long-term) towards the MOR than that of mesoporous Pt. This is probably due to the less stability of individual Cu for MOR, that also shown by Pt NPs catalyst for MOR.143
3.6 Rhodium–nickel (Rh–Ni) alloy
Nanomaterials based on rhodium (Rh) have been used substantially in various applied fields such as fuel cell technology, synthesis of refined chemicals, etc., owing to their high electrocatalytic activity.144,145 However, several associated factors such as complexity in the construction of high-surface-area structures due to high surface energy, less control over the final product, and high cost of Rh metal render the traditional wet chemical synthesis of Rh nanoparticles an inefficient process.146 To overcome the issue, Kani and colleagues undertook an alloying approach of Rh with inexpensive transition metal Ni to fabricate mesoporous RhNi alloy films via an electrochemical method (Fig. 8).21 Metal precursors of Rh and Ni were co-deposited on Au-coated Si substrates by a self-assembled PS-b-PEO block copolymer micelle. To identify the optimal electrochemical deposition potential for RhNi films, linear sweep voltammetry (LSV) analysis was performed, and Rh deposition started at −0.2 V irrespective of the presence of Ni precursors in solutions. Since Ni has a much lower redox potential (E0) than Rh (−0.26 V and 0.76 V respectively), it required a much higher energy and low potential (−1.0 V) and a lowered pH of 2.4. But at low potential, fast deposition of Rh occurs that hinders incorporation into the micelles. Hence the co-deposition of Ni on the Rh layers occurs to generate mesoporous RhNi alloys at −0.7 V deposition potential. Though the Ni content in the final alloy materials corresponds linearly to the Ni concentration in the precursor solution, the final alloy films did not exhibit more than 50% of Ni content, supporting the co-deposition phenomena. By tuning the composition in precursor solutions, Rh100, Rh76Ni24, Rh58Ni42, and Rh49Ni51 films with uniformly distributed pore diameters of 17.0, 16.5, 16.0, and 16.6 nm respectively were produced.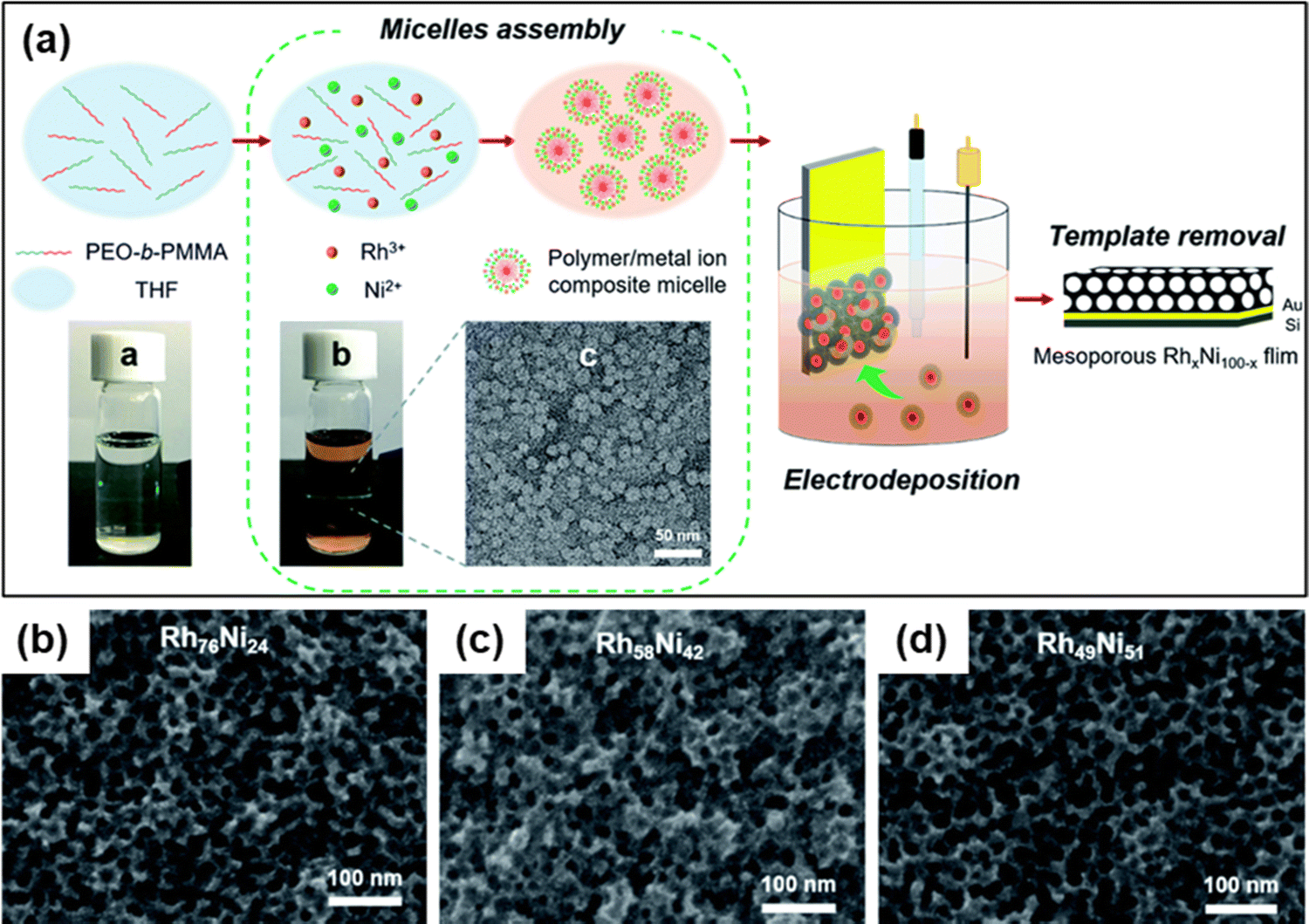 | ||
| Fig. 8 (a) Schematic representation of the electrochemical fabrication of mesoporous Rh–Ni alloy films. A PEO-b-PMMA block copolymer template was utilized to electrochemically deposit Rh and Ni metal species on a gold (Au)-coated silicon (Si) substrate followed by the template removal step to generate a mesoporous bimetallic Rh–Ni alloy film. Top-surface SEM images of mesoporous (b) Rh76Ni24, (c) Rh58Ni42, and (d) Rh49Ni51 films deposited at −0.7 V for 270, 240, and 180 s, respectively. Reproduced with permission.21 Copyright 2021 Royal Society of Chemistry. | ||
The high-angle annular dark-field scanning TEM (HAADF-STEM) revealed that the pore walls consisted an array of tiny nanomaterials contributing to the increase of surface area. Formation of alloy crystals with Ni and Rh was also proved by peak shift towards larger angles in XRD with the increase of Ni concentration. The catalytic performance of the synthesized alloys was evaluated by alkali-mediated hydrogen evolution reaction (HER), which serves as a model reaction for the performance test. The ECSA_Rh of Rh100, Rh76Ni24, Rh58Ni42, and Rh49Ni51 was reported as 39.9, 40.6, 44.0, and 34.3 m2 g−1 respectively. RhNi films also exhibited a positively shifted polarization curve compared to the Ni100 film (>−100 mV vs. −276 mV) indicating higher production of hydrogen. The performance in HER was reported to increase with the increase of Ni concentration in the alloy, supported by current densities of −72, −65, and −59 mV @ 10 mA cm−2 for Rh76Ni24, Rh58Ni42 and Rh49Ni51 alloys respectively. Tafel slopes, an indicative measure for the HER performance (the lower the better), also indicate the high-performance rating of the mesoporous Rh49Ni51 alloy film (67 mV dec−1 compared to 69, 74, 91, and 100 mV dec−1 for Rh58Ni42, Rh76Ni24, Rh100, and Ni100) among other alloys of varying compositions.147–149 In chronopotentiometry examination of the Rh–Ni alloy film for 10 straight hours, the catalytic activity decreased over time compared to the pure Rh film. Although pore morphology and Rh–Ni composition remained unchanged, the properties of catalytically active sites were reported to be affected. It is due to the common characteristics of Ni and its alloys to produce Ni hydrides (NiHx) on the electrode surface during prolonged activity thus lowering the performance in HER.150
3.7 Mesoporous chalcogenide semiconductors (Cd–Se and Cd–Te)
In recent years, a new avenue of research has opened up focusing on mesoporous semiconductors, a new class of materials with potential applications in optoelectronics and solar cells. The adjustable morphological aspects of mesoporous semiconductors (particle size, pore size, etc.) that directly influence the light scattering pattern inside the nanoporous structures make them more promising. Though there are several methods currently available for synthesizing chalcogenide semiconductors, the reports about mesoporous CdX (X = chalcogenide) are very limited. In 2020, Nagaura and colleagues reported for the first time an electrochemical method for fabricating CdSe and CdTe films (Fig. 9).151 A soft-template directed electrodeposition was performed using PS-b-PEO block copolymer micelles. The pore size of the fabricated films was found to be substantially dependent on the molecular weight of the block copolymer, with pore diameters of 9–9.5 nm and 17.7–18.0 nm for PS(5000)-b-PEO(2000) and PS(10000)-b-PEO(4100) respectively. From the XRD pattern, it was proved that the CdSe and CdTe films posed as a hexagonal/cubic mixture and cubic structure respectively. By using this as-prepared mesoporous CdSe film and non-porous CdSe film as the photo-absorber, they also developed highly sensitive photodetectors. The photo-to-dark current ration (PDCR) which measures the photodetection sensitivity of the non-porous CdSe film was reported as 0.059 at 0.05 mW cm−2 and the optical sensitivity (S) was 1.18 mW−1 cm2 compared to 10 and S = 204 for the mesoporous CdSe based photodetector. Massive improvements of the mesoporous CdSe based photosensor are due to two important factors. Firstly, the low dark current of the mesoporous CdSe than that of the non-porous samples, and smaller grain and pore size (∼9 nm) than the mean free path of electrons of n-type CdSe (≈20 nm) result in a larger PDCR value.151 Secondly, numerous electrical semiconducting nano-junctions inside of mesoporous architecture can be directly exposed to the light as light can penetrate and scatter through the nanopore networks, substantially enhances the quantum efficiency of the photosensors.152,153
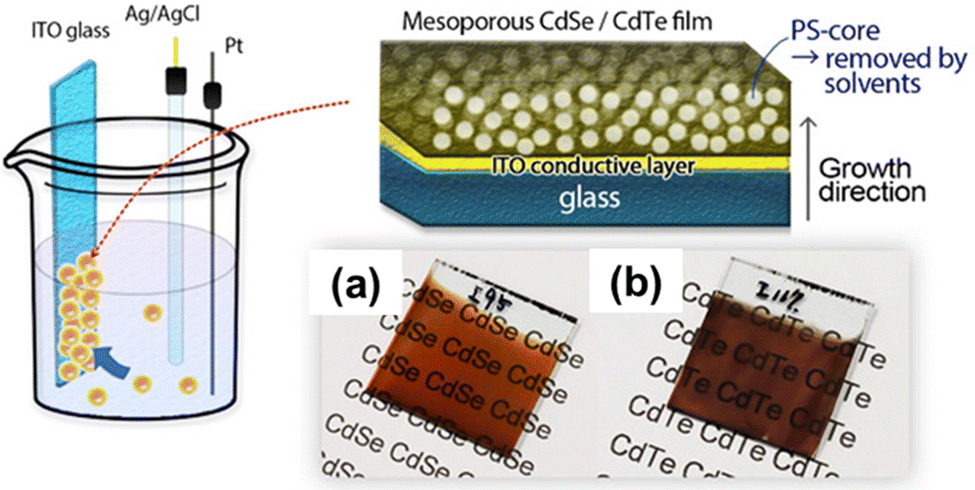 | ||
| Fig. 9 Schematic representation of the fabrication of mesoporous CdTe and CdSe films, and their morphological structure: (a) CdSe and (b) CdTe film. The films were grown onto an ITO glass substrate using PS-b-PEO block copolymers. Reproduced with permission.151 Copyright 2020 Wiley-VCH GmbH. | ||
3.8 Mesoporous trimetallic alloys
Alloying of metals by electrochemical fabrication methods with tunable compositions can also be extended to trimetallic alloys. Along with lowering the production cost, electrochemically synthesized trimetallic alloys offer enhanced catalytic performance with a high degree of reaction stability.154–156 Electrochemical fabrication methods also offer a multitude of options to optimize alloy composition to adjust the catalytic performance of the resultant ternary alloys. Such a study has been reported by Nugraha et al. in 2018, where the group synthesized mesoporous AuCuNi films by electrochemical deposition (Fig. 10).157 The co-electrodeposition of Au, Cu, and Ni (HAuCl4, CuSO4, and NiCl2 as precursors respectively) on a conductive Au-coated Si surface using PS-b-PEO polymeric micelles as a sacrificial template resulted in mesoporous AuCuNi films having a uniform distribution of mesopores of 28 nm throughout the entire surface. The synthesized films were also reported to exhibit a strong dependency on the metal precursors present in the micellar (electrolyte) solution. By adjusting the precursor solution's composition, mesoporous Au37Cu48Ni15, Au41Cu46Ni13, and Au60Cu35Ni5 films were obtained. Interestingly, SEM, TEM, EDS, and XRD analysis of Au60Cu35Ni5 films revealed that the mesoporous structures were not confined only to the surface area of the film, rather they expanded across the entire cross-sectional area. CV analysis of the synthesized alloy films showed an ECSA of 43.2, 45.8, and 18.1 m2 g−1 for Au37Cu48Ni15, Au41Cu46Ni13, and Au60Cu35Ni5 respectively indicating a larger contact area and easily accessible pores available for welcoming guest species to undergo electrocatalytic reaction.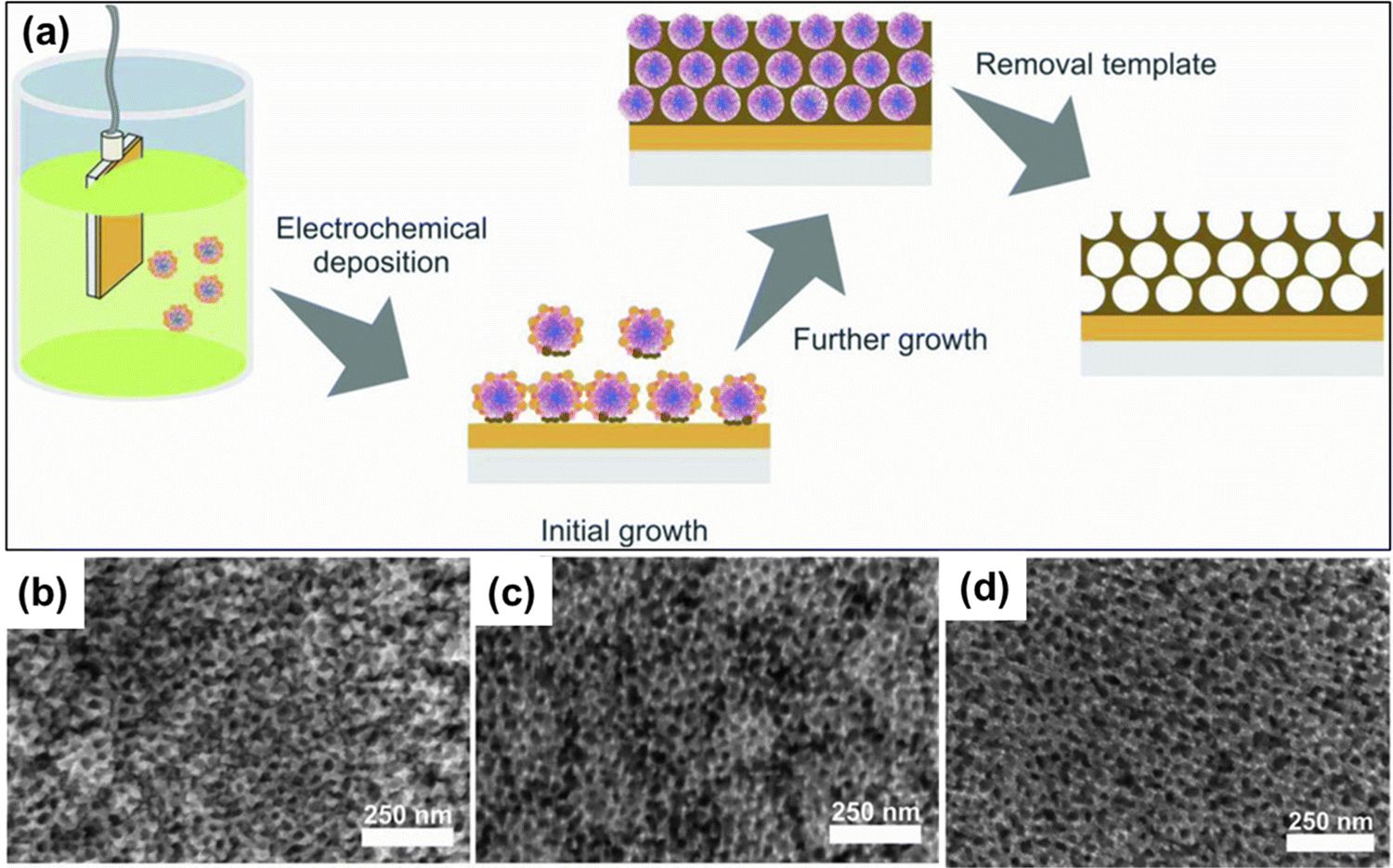 | ||
| Fig. 10 Mesoporous trimetallic alloy synthesis process by electrodeposition involving the polymeric micelle assembly strategy. The ternary AuCuNi alloy with well-organized mesopores was synthesized by co-electrodeposition of Au, Cu, and Ni precursors on an Au-coated Si surface in the presence of PS-b-PEO polymeric micelles. SEM images of the mesoporous AuCuNi films synthesized from a Au:Cu:Ni precursor ratio of 3:1:1 at an applied potential of (b) −0.70 V, (c) −0.80 V, and (d) −0.90 V (vs. Ag/AgCl) at 40 °C for 1000 s. Reproduced with permission.157 Copyright 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Electrochemically fabricated mesoporous AuCuNi films have displayed intensified activity in the electrocatalysis of glucose compared to the pure (non-porous) Au film.72 Mesoporous Au60Cu35Ni5 was able to detect glucose at concentrations ranging from 0.1 mM to 8.0 mM with a detection sensitivity of 1823 μA mM−1 cm−2 and a detection limit of 0.82 μM, which is relatively higher than most of the non-enzymatic glucose sensing electrodes. The fabricated ternary alloy film is also reported to be immune to the naturally occurring interfering agents such as maltose, uric acid, fructose, and ascorbic acid in glucose sensing warranting its high selectivity. Moreover, in the methanol oxidation reaction, all of the AuCuNi alloys exhibited increased peak current density where the highest current density was achieved with Au41Cu46Ni13 (3.8 mA cm−2), which is also notably higher than that achieved with Pt-based electrocatalysts.158,159 The corresponding ternary alloys also presented a great deal of stability in retaining their activity in glucose and methanol oxidation reactions by minimizing the loss of ECSA in respective reactions. The ECSA of the ternary alloy decreased merely 19% after 6500 seconds of electro-oxidation of glucose and the alloy showed a high catalytic activity in the methanol oxidation reaction evaluated by the amperometric response for 1200 seconds. Another insightful study by Nugraha and colleagues undertook an exploratory approach by Bayesian optimization to find the ternary PtPdAu alloy with optimum catalytic performance in the methanol oxidation reaction.42 This exploratory search curtailed the need for time-consuming and exhaustive experimentation to filter out the chemical composition of the most efficient mesoporous PtPdAu alloys. To put the numbers in perspective, possible compositions of individual metal species to fabricate trimetallic alloys can be 5151 which makes it very challenging to determine the proper composition for obtaining trimetallic alloys with highest electrocatalytic activity. Mesoporous trimetallic PtPdAu alloys with varying compositions were electrochemically synthesized by the micelle assembly approach using the Pluronic P123 EO20-PO70-EO20 triblock polymer and K2PtCl4, Na2PdCl4, and HAuCl4 as metal precursors. Structures with well-defined and well-distributed mesopores with 9 nm pore size devoid of any cracks were reported, which mimic the mesopores of the micelle template. The synthesized alloys also manifested a higher catalytic activity in the methanol oxidation reaction generating an oxidation peak in the forward scan associated with methanol oxidation and a peak in the backward scan associated with removal of reaction intermediates (CO for example) generated in the forward scan.160–162 The obtained current responses were reported to be higher than those of the bimetallic alloy of PtPd and PtAu supporting the synergistic effects of the third metal species in electrocatalytic performance.162,163 Of the various compositional ratio of each metal species in the precursor solution, the one with the lowest Au content (Pt:Pd:Au ratio of 59:40:1) generated the best current density in methanol oxidation, which was also the composition derived from the Bayesian optimization. This might be attributed to the change of surface adsorption of Pt by Au leading to better CO removal and catalytic performance.164,165
4. Application of mesoporous metallic alloy films
Mesoporous metallic alloy films provide significant promise for advanced technology development in response to the most pressing global concerns (e.g., energy generation, storage, water decontamination, wettability, automated device fabrication, photosensing, biosensing and nanomedicine).34,166–168 These materials are excellent candidates for developing advanced technological applications which range from hydrogen production to fuel cells and sensors, not only because they possess special physiochemical properties, but also because of the high flexibility of their synthesis.4.1. Hydrogen evolution reaction (HER)
Fuel cell development is accelerating in response to current environmental concerns and the growing demand for more efficient power sources.169 The fuel cell, which transforms the chemical energy of a fuel directly to electrical energy, has numerous advantages over traditional combustion-based technologies.170,171 Hydrogen has emerged as a possible successor for fossil fuels, as a clean and sustainable energy with the highest energy density. When used as a fuel in a fuel cell, it not only converts energy efficiently, but also creates no pollution because it only emits water as a byproduct. However, yet it is very challenging to generate a large quantity of hydrogen that can serve commercial purpose. Water splitting electrolysis is a viable technique for achieving efficient hydrogen synthesis in terms of energy conversion and storage, in which catalysis or electrocatalysis plays a vital role. The hydrogen evolution reaction (HER) produces hydrogen from water electrolysis. Owing to the excellent intrinsic activity, Pt-group metals such as Pt, Pd, and Ru have attracted increased study interest in recent decades as excellent HER catalysts.172 However, expensive synthesis procedures and scarcity of these metals limit their widespread use for HER.173 As a result, developing effective, robust, and inexpensive catalysts or electrocatalysts is a must if meaningful electrocatalytic hydrogen production from water splitting is to be achieved. Mesoporous Rh–Ni alloy films fabricated by an alloying approach of Rh with inexpensive transition metal Ni via an electrochemical method resulted in improved catalytic performance as an electrocatalyst in HER.21 The performance of the alloy in HER improves with the increase of Ni concentration in the alloy, and the mesoporous Rh49Ni51 alloy film is reported to be the best supported by a current density of −59 mV @ 10 mA cm−2 (Fig. 11). Tafel slopes, another indicative measure for the HER performance (the lower the better), also indicate the high-performance rating of the mesoporous Rh49Ni51 alloy film. The alloy's mesoporous structure and favourable synergistic electronic characteristics are credited with the improved HER performance.21 Again mesoporous Pt with ΔGH ≈ 0 is well known as the best-performing catalyst for the HER, which requires a negligible over-potential to achieve high reaction rates in acidic solutions.174,175 However, its functionality as the HER catalyst in practical use is limited by the high cost of this metal. Mesoporous Ni-rich Ni–Pt thin films were produced by alloying Pt with the transition (relatively inexpensive) metal Ni which significantly reduced the cost of the whole catalyst without compromising catalytic activity. Also, the secondary metal heteroatoms optimized the d-band center and ΔGH of Pt which resulted in enhanced performance and excellent stability of the entire bimetallic system. The HER activity is found to be increased by almost one order of magnitude compared to Pt/C.176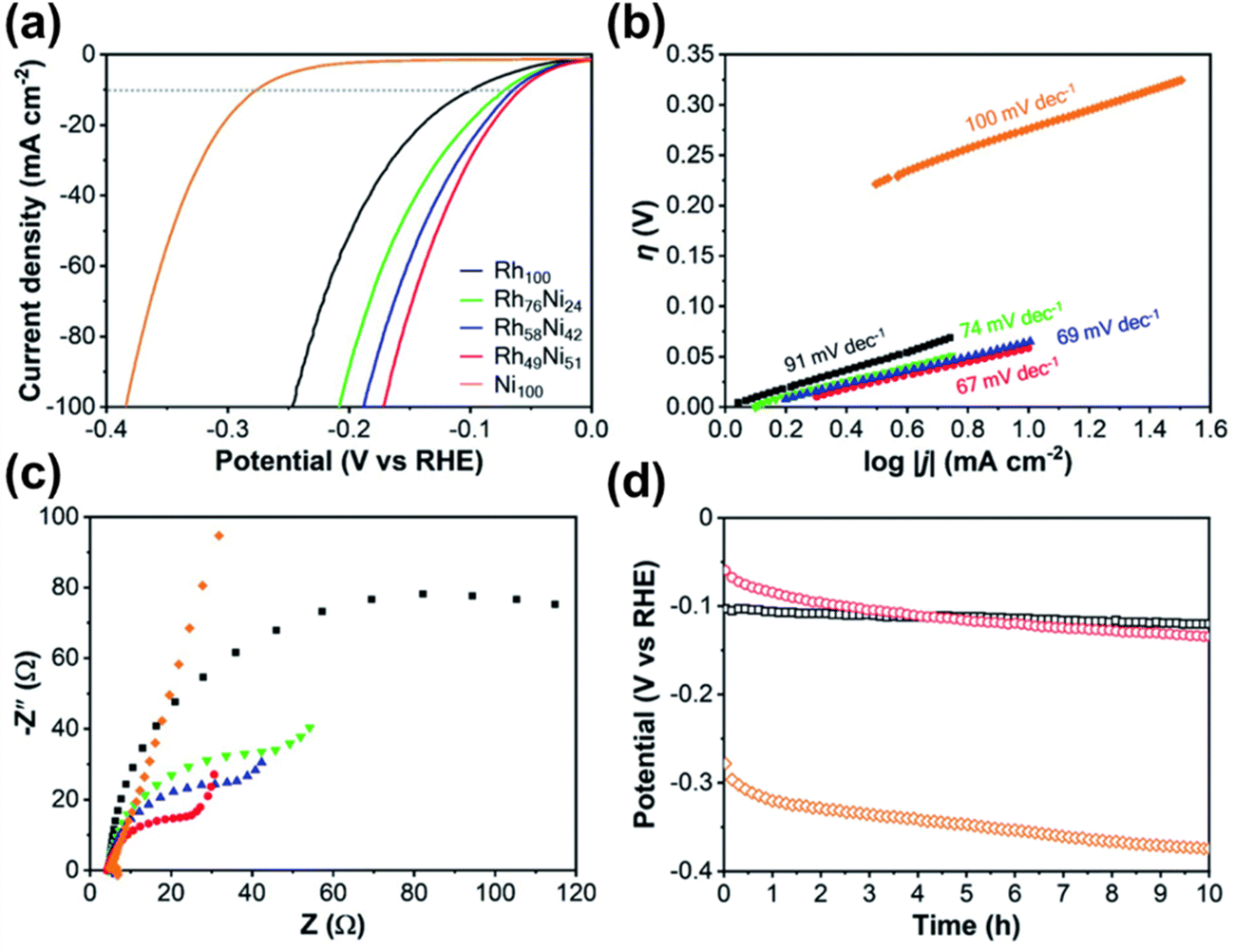 | ||
| Fig. 11 Demonstration of the alkali-mediated HER performance of the Rh–Ni alloy in 1.0 M KOH solution. (a) The polarization curves of mesoporous Rh100, Rh76Ni24, Rh58Ni42, Rh49Ni51, and Ni100 films recorded at a scan rate of 2 mV s−1. (b) Tafel plots and (c) EIS of the corresponding samples. (d) The HER stability of mesoporous Rh100 (black), Rh49Ni51 (red), and Ni100 (orange) films demonstrated using chronopotentiometric V–t graphs obtained at a current density of 10 mA cm2. Reproduced with permission.21 Copyright 2021 Royal Society of Chemistry. | ||
4.2. Methanol oxidation reaction (MOR)
When compared to hydrogen, methanol is a very appealing fuel because of its availability, low cost, and high energy density, as well as its ease of storage and transportation.177–179 Because of their portability and promise of high energy density, DMFCs have sparked a lot of scientific interest.177,178,180 Without reforming the fuel into hydrogen, DMFCs may generate power from methanol and water at low temperatures.181 While DMFCs are expected to be one of the future energy sources in a hypothetical carbon-neutral economy, there is still a significant gap in the development of a suitable electrocatalyst, which necessitates further research.179 Though mesoporous Pt with a high surface area demonstrates significant activity in the electrocatalytic oxidation of methanol, which is a half-reaction of a DMFC, the adsorbed CO molecules formed as a reaction intermediate hamper the reaction by obstructing the active sites of Pt.182 As a result, tremendous work has gone into preventing CO poisoning. Yamauchi and colleagues undertook an alloying approach of Pt with Au to fabricate a Pt–Au mesoporous film which tends to exhibit an excellent electrocatalytic performance for methanol oxidation reactions. The presence of Au suppresses the adsorption of CO onto the Pt surface, to alleviate its poisoning effects (Fig. 12).40 In terms of electrochemical stability toward methanol oxidation, the stability of mesoporous Pt–Au alloy films exceeds that of nonporous Pt and mesoporous Pt films as well as the commercial Pt black powders.40 Similar to the mesoporous Pt–Au alloy film, other Pt-based mesoporous alloy films such as Pt–Ru and Pt–Cu films also showed strong potential for use as an electrocatalyst in direct methanol fuel cells.25,66 Likewise, the excellent electrocatalytic capability and stability of the mesoporous Au–Ni bimetallic alloy film fabricated by electrochemical deposition underscores the practical implementation in DMFCs.26 Furthermore, compared to binary alloys, mesoporous tri-metallic alloy catalysts such as mesoporous Au–Cu–Ni films and Pt–Pd–Au films have been observed to exhibit greater overall catalytic activity.42,157,183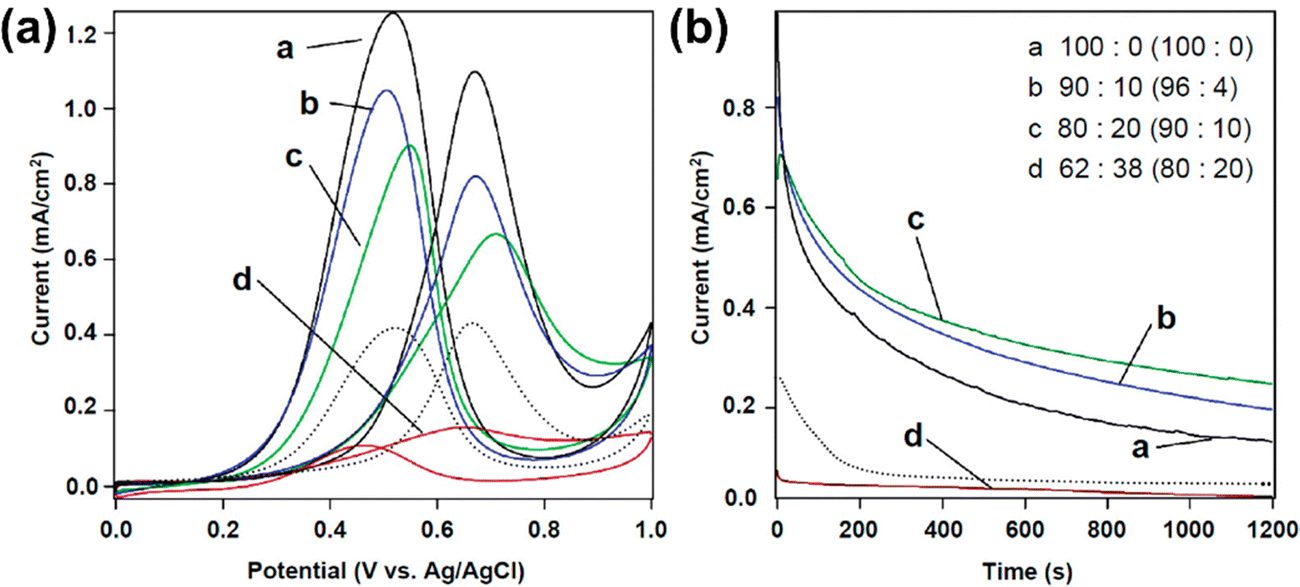 | ||
| Fig. 12 (a) Cyclic voltammetry and (b) chronoamperometric curves of the Pt–Au mesoporous film at a potential of 0.6 V in the presence of 0.5 M H2SO4 containing 0.5 M methanol. The currents were normalized to the ECSAs obtained from CV curves in 0.1 M NaOH solution. The currents were compared to the ECSAs calculated from CV curves in 0.1 M NaOH. The product compositions (Pt:Au) are given in parentheses, as are the Pt4+:Au3+ compositional ratios in the precursor solutions. A bulk Pt film (shown by a dotted line) is used as a comparison to the other samples. Electrochemical deposition at a constant voltage (0.04 V vs. Ag/AgCl) was used to make the bulk Pt film. All the samples had a film thickness of less than 100 nanometers. Reproduced with permission.40 Copyright 2012 American Chemical Society. | ||
4.3. Glucose sensor
Glucose sensors have received increased attention in the medical use for blood glucose measurement and monitoring over the last few decades. The accurate monitoring and careful control of the amount of glucose in human blood are essential.184,185 However, the last few decades have experienced an obvious rise in diabetes cases worldwide. This has resulted in a surge in demand for non-enzymatic amperometric glucose sensors that are extremely efficient, stable, selective and sensitive. Noble metals, such as Pt and Au, have been identified as the most promising candidates for glucose electro-oxidation.23,186,187 These metals as electrodes, on the other hand, have limited sensitivity and poor selectivity, and are poisoned by reaction intermediates. Some of these flaws were addressed by combining various noble metals. Mesoporous Pt–Au alloy films, for example, have shown great promise as an electrode in glucose sensing devices.22 Because of the synergistic effect of mesoporous structures and Pt–Au binary-alloy surfaces, they have good catalytic activity for glucose oxidation. The amperometric glucose sensors made from mesoporous Pt–Au alloy films have a good anti-poisoning effect, as well as good stability and reproducibility. These films can retain their activity towards the detection of glucose at least for ten cycles. The film was able to detect up to 352 μA cm−2 mM−1 glucose with a LOD of 6.0 μM (S/N = 3) (Fig. 13).22 When it comes to industrial manufacturing, however, the cost-effectiveness of using precious metals for electrode fabrication is becoming an issue that needs to be addressed. This drawback can be minimized by introducing cheap metals along with the noble metal. For instance, Au and Cu were coupled to form mesoporous Au–Cu alloy sheets, which reduced the manufacturing costs as compared to monometallic Au-based devices. Moreover, this bimetallic alloy boosted both selectivity and sensitivity due to the synergistic electrical response and catalytic benefits from both components.23 With great selectivity, sensitivity, and good linearity over a broad range of glucose concentrations with a low detection limit, this mesoporous Au–Cu film exhibits outstanding performance at a reasonably strong negative potential, indicating the removal of interferents.188 Besides these mesoporous bimetallic alloys, ternary alloys, mesoporous Au–Cu–Ni films, also displayed intensified activity in the electrocatalysis of glucose compared to the pure Au film.72,157 These trimetallic alloys presented a great deal of stability in retaining their activity in glucose oxidation reactions by minimizing the loss of ECSA in respective reactions.157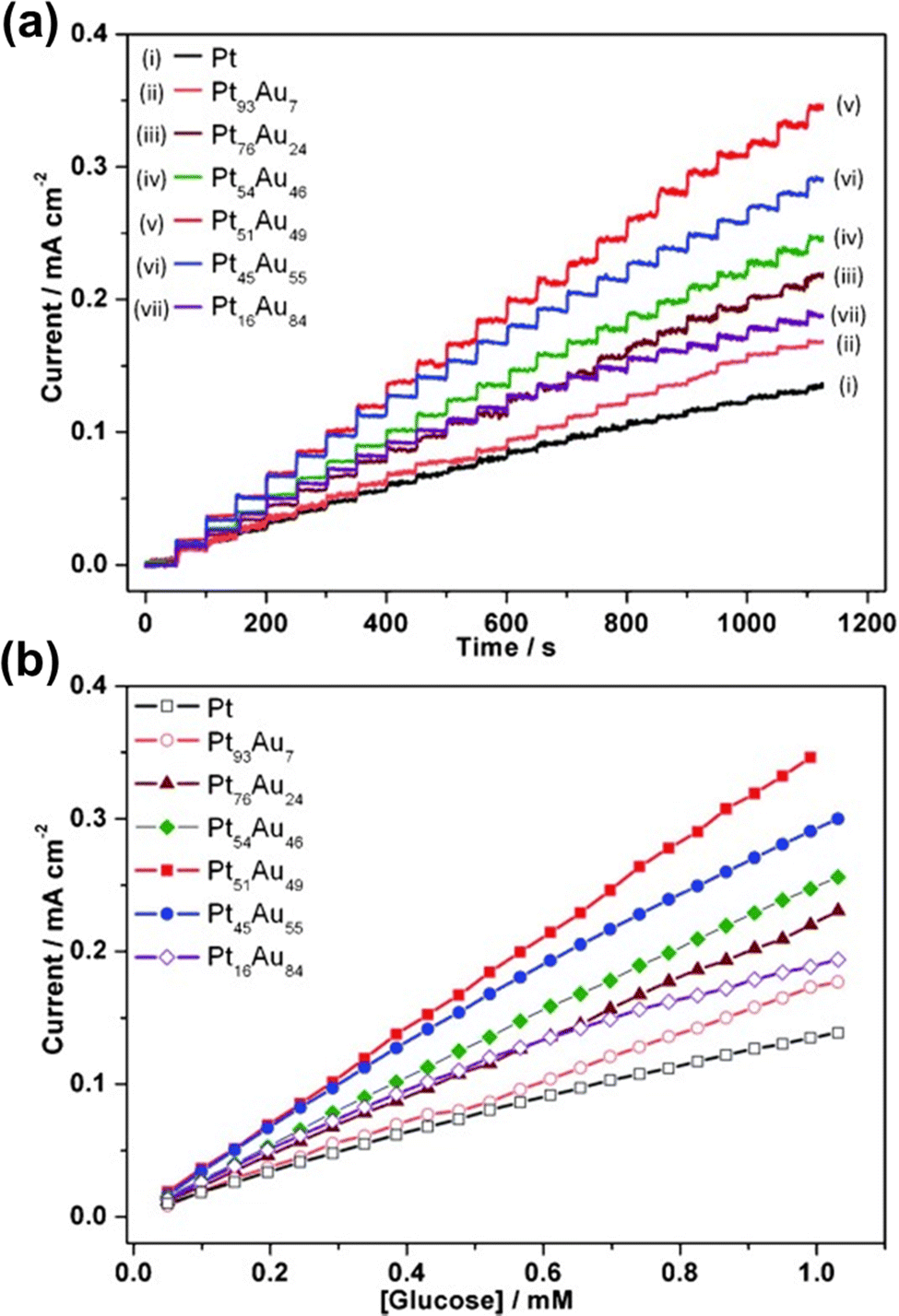 | ||
| Fig. 13 (a) The dynamic amperometric response of a Pt–Au alloy for the successive addition of various amounts of glucose, ranging from 0 to 1.05 mM at 0.3 V. (b) The corresponding curves of those concentrations. The average values are obtained by using five independent measurements from five different film electrodes. Reproduced with permission.22 Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
4.4. Biosensors
Apart from use in glucose sensing devices, mesoporous metallic alloys also have application in ultrasensitive detection of nucleic acids such as microRNA (miRNA). miRNA, a kind of small non-coding RNA (21–24 nucleotides) that regulates over 30% of human genome, is involved extensively in almost every cellular process including cell differentiation, genetic expression, and biological development.189–191 The expression of miRNA changes according to the type and stage of disease, especially cancer, and has been reported to be imbalanced in many disease conditions, either upregulated or downregulated relative to the healthy state.192 As a result, miRNA expression has been extensively studied as a potential biomarker for the diagnosis and prognosis of a wide range of illnesses, including cancer.193–195 Mesoporous Au–Ag alloy films have a lot of potential for fabricating portable, high-performance miRNA sensors.117 These DPV sensors enable ultrasensitive detection of miRNA with an LOD of 100 aM and possess activity in the dynamic range of 100 pM to 100 aM with good reproducibility (RSD, of <5%, for S/N = 3) (Fig. 14). These bimetallic alloys provide several benefits over the current miRNA assays with different cascade signal amplification including high stability, increased adsorption sites for miRNA, enhanced catalytic effect, and therefore, superior sensitivity.196 Furthermore, the fabrication technique is both cost-effective and reproducible and re-usable. This assay requires a single-step electrochemical detection of isolated target miRNA from the source, requiring less than 1 hour for miRNA detection, making it a good candidate for a portable miRNA sensor for clinical use. It could be used in biological studies, bioanalysis, and clinical diagnosis. Although the potential of the mesoporous nanoarchitecture is utilized for electrochemical biosensing, it could be also applicable for many other types of biosensing applications. For example, the unique properties of the metallic nanostructures might potentially enhance plasmonic phenomena which in turn could be utilized in surface plasmon resonance (SPR) based biosensing platforms.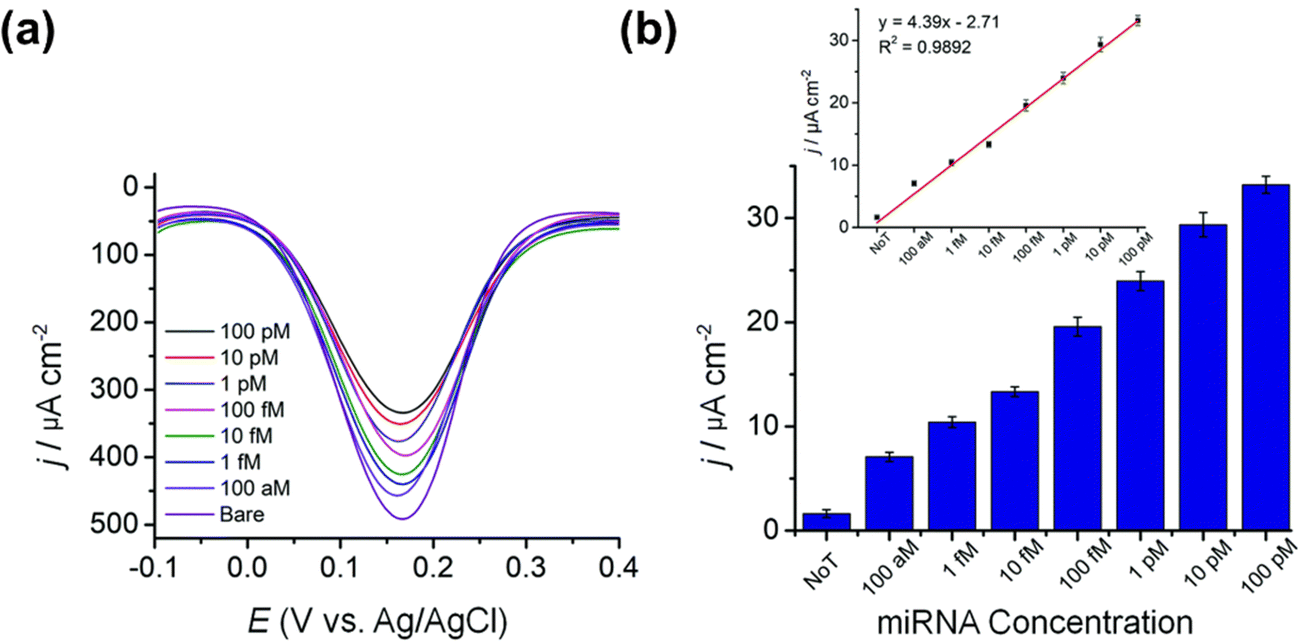 | ||
| Fig. 14 Assay sensitivity for the Au–Ag bimetallic alloy-based sensor for miRNA. (a) The DPV responses for the Au–Ag film (50:50) sensor for a range of microRNA (miR-9-2) ranging from 100 aM to 100 pM. (b) The relative current is determined by using the bare (non-porous) mesoporous Au–Ag film as a reference. Reproduced with permission.117 Copyright 2020 Royal Society of Chemistry. | ||
5. Conclusions and perspectives
In this comprehensive review, we have comprehensively reviewed the current strategies available for the synthesis of mesoporous alloy films. We have discussed the electrochemical procedures to fabricate various bimetallic and trimetallic alloy films and their corresponding application. To sum up, alloying of noble metals with inexpensive metal species contributes positively to both procedural and functional aspects. Alloying of metals in a bimetallic and trimetallic fashion reduces the overall production cost significantly compared to that of single metal-based catalysts. Alloying also offers highly diversified explorable options to fabricate multi-component alloys with tunable metal constituents. Moreover, the fabrication of binary and ternary alloys by electrochemical methods is highly reliable, cost-effective, and easily reproducible. From functional aspects, electrochemically synthesized multi-metallic mesoporous alloy films exhibit substantial performance boosts in catalytic conversion reactions. On top of that, electrochemically fabricated mesoporous alloy films show high stability, re-usability, and durability in participating reactions alongside notable efficiency in resisting catalytic poisoning effects. The high functionality and catalytic performance of electrochemically synthesized mesoporous alloy films (binary and ternary) can be attributed to several factors including highly ordered mesopores across the film, evenly distributed mesopores of the same size throughout the entire cross-sectional area of the alloy film, absence of any cracks and voids, larger surface area provided by constituent metal species, synergistic effects of participating metal species, availability of abundant multifunctional active sites for the guest species, variability in the electronic structure and strain effects, and lastly high stability and durability. Electrochemical methods also offer customizability during the fabrication process to tailor the final film composition and subsequent catalytic performance of the resulting alloys. Adjustment parameters include the compositional ratio of participating metal precursors, size and type of the micellar templates, duration of electrodeposition, etc. Mesoporous metallic alloys fabricated by electrochemical methods are suitable for a wide range of applications including electrocatalytic activity in HER, MOR and electrocatalytic signal amplifications for glucose, protein and nucleic acid sensor. Bi- and tri-metallic mesoporous alloys have been reported to function as non-enzymatic glucose sensors with high sensitivity, accuracy, and low detection limit compared to the monometallic counterparts and conventional glucose sensors. The versatility and applicability of mesoporous binary and ternary alloy films explored to date are just the tips of the iceberg. Despite the fact that mesoporous metal alloys have been extensively studied in HER and MOR, the application of these films in the avenue of biological sciences such as detection of various types of nucleic acids, biosensing of epigenetic modifications (DNA methylation and RNA methylation), detection of post-translational protein modification, protein purification, biomolecule detection, and so on is still unanswered. We envisage that more focus should be directed towards research exploring alloying of earth-abundant elements and translating their application in biological studies and industrial-scale electrocatalysis.Author contributions
J. Bashir, B. Chowdhury and R. R. Kathak contributed equally.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study is supported by the JSPS fellowship (Grant Number P20039) to M. K. M. The authors are also grateful to the Taif University Researchers Supporting Project number (TURSP-2020/03), Taif University, Taif, KSA. The authors are also grateful to the Queensland node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and micro-fabrication facilities for Australian researchers.References
- Y. Deng, D. Qi, C. Deng, X. Zhang and D. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS PubMed.
- A. Yamaguchi, F. Uejo, T. Yoda, T. Uchida, Y. Tanamura, T. Yamashita and N. Teramae, Nat. Mater., 2004, 3, 337–341 CrossRef CAS PubMed.
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- G. Ma, X. Yan, Y. Li, L. Xiao, Z. Huang, Y. Lu and J. Fan, J. Am. Chem. Soc., 2010, 132, 9596–9597 CrossRef CAS PubMed.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- R. Liu, Y. Zhang, X. Zhao, A. Agarwal, L. J. Mueller and P. Feng, J. Am. Chem. Soc., 2010, 132, 1500–1501 CrossRef CAS PubMed.
- D. R. Radu, C.-Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216–13217 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- P.-K. Chen, N.-C. Lai, C.-H. Ho, Y.-W. Hu, J.-F. Lee and C.-M. Yang, Chem. Mater., 2013, 25, 4269–4277 CrossRef CAS.
- F. Jiao and P. G. Bruce, Adv. Mater., 2007, 19, 657–660 CrossRef CAS.
- E. Rossinyol, A. Prim, E. Pellicer, J. Arbiol, F. Hernández-Ramírez, F. Peiró, A. Cornet, J. R. Morante, L. A. Solovyov, B. Tian, T. Bo and D. Zhao, Adv. Funct. Mater., 2007, 17, 1801–1806 CrossRef CAS.
- Y. Shi, B. Guo, S. A. Corr, Q. Shi, Y.-S. Hu, K. R. Heier, L. Chen, R. Seshadri and G. D. Stucky, Nano Lett., 2009, 9, 4215–4220 CrossRef CAS PubMed.
- F. Pereira, K. Vallé, P. Belleville, A. Morin, S. Lambert and C. Sanchez, Chem. Mater., 2008, 20, 1710–1718 CrossRef CAS.
- A. Serrà and E. Vallés, Catalysts, 2018, 8, 395 CrossRef.
- P. Innocenzi and L. Malfatti, Chem. Soc. Rev., 2013, 42, 4198–4216 RSC.
- C. Li, M. Iqbal, J. Lin, X. Luo, B. Jiang, V. Malgras, K. C. W. Wu, J. Kim and Y. Yamauchi, Acc. Chem. Res., 2018, 51, 1764–1773 CrossRef CAS PubMed.
- A. Walcarius, Chem. Soc. Rev., 2013, 42, 4098–4140 RSC.
- D. Baba, J. Kim, J. Henzie, C. Li, B. Jiang, Ö. Dag, Y. Yamauchi and T. Asahi, Chem. Commun., 2018, 54, 10347–10350 RSC.
- K.-H. Huynh, X.-H. Pham, J. Kim, S. H. Lee, H. Chang, W.-Y. Rho and B.-H. Jun, Int. J. Mol. Sci., 2020, 21, 5174 CrossRef CAS PubMed.
- B. Jiang, C. Li, H. Qian, M. S. A. Hossain, V. Malgras and Y. Yamauchi, Angew. Chem., Int. Ed., 2017, 56, 7836–7841 CrossRef CAS PubMed.
- K. Kani, H. Lim, A. E. Whitten, K. Wood, A. J. E. Yago, M. S. A. Hossain, J. Henzie, J. Na and Y. Yamauchi, J. Mater. Chem. A, 2021, 9, 2754–2763 RSC.
- C. Li, H. Wang and Y. Yamauchi, Chem. – Eur. J., 2013, 19, 2242–2246 CrossRef CAS PubMed.
- A. S. Nugraha, V. Malgras, M. Iqbal, B. Jiang, C. Li, Y. Bando, A. Alshehri, J. Kim, Y. Yamauchi and T. Asahi, ACS Appl. Mater. Interfaces, 2018, 10, 23783–23791 CrossRef CAS PubMed.
- H. Wang and Y. Yamauchi, Chem. – Asian J., 2012, 7, 2133–2138 CrossRef CAS.
- C. Li and Y. Yamauchi, Chem. – Eur. J., 2014, 20, 729–733 CrossRef CAS PubMed.
- A. S. Nugraha, J. Na, Md. S. A. Hossain, J. Lin, Y. V. Kaneti, M. Iqbal, B. Jiang, Y. Bando, T. Asahi and Y. Yamauchi, Appl. Mater. Today, 2020, 18, 100526 CrossRef.
- W. Xu, Z. Wu and S. Tao, J. Mater. Chem. A, 2016, 4, 16272–16287 RSC.
- A. S. Nugraha, G. Lambard, J. Na, M. S. A. Hossain, T. Asahi, W. Chaikittisilp and Y. Yamauchi, J. Mater. Chem. A, 2020, 8, 13532–13540 RSC.
- X. Gao, X. Pei, D. W. Gardner, C. S. Diercks, S. Lee, B. Rungtaweevoranit, M. S. Prevot, C. Zhu, S. Fakra and R. Maboudian, Adv. Mater., 2019, 31, 1807553 CrossRef PubMed.
- Y. Yamauchi, A. Sugiyama, R. Morimoto, A. Takai and K. Kuroda, Angew. Chem., Int. Ed., 2008, 47, 5371–5373 CrossRef CAS PubMed.
- G. S. Attard, J. M. Corker, C. G. Göltner, S. Henke and R. H. Templer, Angew. Chem., Int. Ed. Engl., 1997, 36, 1315–1317 CrossRef CAS.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- J. Wang, Q. Chen, X. Liu, W. Qiao, D. Long and L. Ling, Mater. Chem. Phys., 2011, 129, 1035–1041 CrossRef CAS.
- V. Malgras, H. Ataee-Esfahani, H. Wang, B. Jiang, C. Li, K. C.-W. Wu, J. H. Kim and Y. Yamauchi, Adv. Mater., 2016, 28, 993–1010 CrossRef CAS PubMed.
- S. Cherevko, N. Kulyk and C.-H. Chung, Nanoscale, 2012, 4, 568–575 RSC.
- D. E. Williams, R. C. Newman, Q. Song and R. G. Kelly, Nature, 1991, 350, 216–219 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450–453 CrossRef CAS PubMed.
- D. H. Nagaraju and V. Lakshminarayanan, J. Phys. Chem. C, 2009, 113, 14922–14926 CrossRef CAS.
- M. K. Masud, M. Umer, M. S. A. Hossain, Y. Yamauchi, N.-T. Nguyen and M. J. A. Shiddiky, Trends Biochem. Sci., 2019, 44, 433–452 CrossRef CAS PubMed.
- Y. Yamauchi, A. Tonegawa, M. Komatsu, H. Wang, L. Wang, Y. Nemoto, N. Suzuki and K. Kuroda, J. Am. Chem. Soc., 2012, 134, 5100–5109 CrossRef CAS PubMed.
- L. P. Bicelli, B. Bozzini, C. Mele and L. D’Urzo, Int. J. Electrochem. Sci., 2008, 3, 53 Search PubMed.
- A. S. Nugraha, G. Lambard, J. Na, M. S. A. Hossain, T. Asahi, W. Chaikittisilp and Y. Yamauchi, J. Mater. Chem. A, 2020, 8, 13532–13540 RSC.
- Y. Yamauchi and K. Kuroda, Chem. – Asian J., 2008, 3, 664–676 CrossRef CAS PubMed.
- J. Jiang and C. Wang, J. Electrochem. Soc., 2019, 167, 037521 CrossRef.
- S. Sokolov, E. Ortel and R. Kraehnert, Mater. Res. Bull., 2009, 44, 2222–2227 CrossRef CAS.
- S. L. Suib, Chem. Rec., 2017, 17, 1169–1183 CrossRef CAS PubMed.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- J. Cai, W. Wei, X. Hu and D. A. Wood, Earth-Sci. Rev., 2017, 171, 419–433 CrossRef.
- X. Fei, W. Li, Z. Shao, S. Seeger, D. Zhao and X. Chen, J. Am. Chem. Soc., 2014, 136, 15781–15786 CrossRef CAS PubMed.
- K. Jüttner, Encyclopedia of Electrochemistry, John Wiley & Sons, Ltd, 2007 Search PubMed.
- W. Li, Y. Deng, Z. Wu, X. Qian, J. Yang, Y. Wang, D. Gu, F. Zhang, B. Tu and D. Zhao, J. Am. Chem. Soc., 2011, 133, 15830–15833 CrossRef CAS PubMed.
- V. Malgras, Q. Ji, Y. Kamachi, T. Mori, F.-K. Shieh, K. C.-W. Wu, K. Ariga and Y. Yamauchi, Bull. Chem. Soc. Jpn., 2015, 88, 1171–1200 CrossRef CAS.
- B. Guo, X. Wang, P. F. Fulvio, M. Chi, S. M. Mahurin, X.-G. Sun and S. Dai, Adv. Mater., 2011, 23, 4661–4666 CrossRef CAS PubMed.
- N. Pal and A. Bhaumik, Adv. Colloid Interface Sci., 2013, 189–190, 21–41 CrossRef CAS PubMed.
- C. Li, B. Jiang, Z. Wang, Y. Li, M. S. A. Hossain, J. H. Kim, T. Takei, J. Henzie, Ö. Dag, Y. Bando and Y. Yamauchi, Angew. Chem., Int. Ed., 2016, 55, 12746–12750 CrossRef CAS PubMed.
- H. Yan, Chem. Commun., 2012, 48, 3430–3432 RSC.
- S. R. Gajjela, K. Ananthanarayanan, C. Yap, M. Grätzel and P. Balaya, Energy Environ. Sci., 2010, 3, 838–845 RSC.
- W.-C. Li, A.-H. Lu, C. Weidenthaler and F. Schüth, Chem. Mater., 2004, 16, 5676–5681 CrossRef CAS.
- Z. Xia, L. Liao and S. Zhao, Mater. Res. Bull., 2009, 44, 1626–1629 CrossRef CAS.
- R. Ding, L. Lv, L. Qi, M. Jia and H. Wang, RSC Adv., 2013, 4, 1754–1760 RSC.
- N. D. Petkovich and A. Stein, Chem. Soc. Rev., 2013, 42, 3721–3739 RSC.
- C. Li, T. Sato and Y. Yamauchi, Angew. Chem., Int. Ed., 2013, 52, 8050–8053 CrossRef CAS PubMed.
- H. Wang, L. Wang, T. Sato, Y. Sakamoto, S. Tominaka, K. Miyasaka, N. Miyamoto, Y. Nemoto, O. Terasaki and Y. Yamauchi, Chem. Mater., 2012, 24, 1591–1598 CrossRef CAS.
- M. Iqbal, C. Li, K. Wood, B. Jiang, T. Takei, Ö. Dag, D. Baba, A. S. Nugraha, T. Asahi, A. E. Whitten, M. S. A. Hossain, V. Malgras and Y. Yamauchi, Chem. Mater., 2017, 29, 6405–6413 CrossRef CAS.
- C. Li, V. Malgras, A. Aldalbahi and Y. Yamauchi, Chem. – Asian J., 2015, 10, 316–320 CrossRef CAS PubMed.
- H. Wang, M. Imura, Y. Nemoto, L. Wang, H. Y. Jeong, T. Yokoshima, O. Terasaki and Y. Yamauchi, Chem. – Eur. J., 2012, 18, 13142–13148 CrossRef CAS PubMed.
- C. Li, V. Malgras, S. M. Alshehri, J. H. Kim and Y. Yamauchi, Electrochim. Acta, 2015, 183, 107–111 CrossRef CAS.
- B. P. Bastakoti, Y. Li, T. Kimura and Y. Yamauchi, Small, 2015, 11, 1992–2002 CrossRef CAS PubMed.
- Y. Li, B. P. Bastakoti and Y. Yamauchi, APL Mater., 2016, 4, 040703 CrossRef.
- C. Li, Ö. Dag, T. D. Dao, T. Nagao, Y. Sakamoto, T. Kimura, O. Terasaki and Y. Yamauchi, Nat. Commun., 2015, 6, 6608 CrossRef CAS PubMed.
- C. Li, B. Jiang, H. Chen, M. Imura, L. Sang, V. Malgras, Y. Bando, T. Ahamad, S. M. Alshehri, S. Tominaka and Y. Yamauchi, Nano Res., 2016, 9, 1752–1762 CrossRef CAS.
- A. S. Nugraha, C. Li, J. Bo, M. Iqbal, S. M. Alshehri, T. Ahamad, V. Malgras, Y. Yamauchi and T. Asahi, ChemElectroChem, 2017, 4, 2571–2576 CrossRef CAS.
- B. Jiang, C. Li, J. Tang, T. Takei, J. H. Kim, Y. Ide, J. Henzie, S. Tominaka and Y. Yamauchi, Angew. Chem., 2016, 128, 10191–10195 CrossRef.
- K.-S. Choi, H. C. Lichtenegger, G. D. Stucky and E. W. McFarland, J. Am. Chem. Soc., 2002, 124, 12402–12403 CrossRef CAS PubMed.
- S. Manne, J. P. Cleveland, H. E. Gaub, G. D. Stucky and P. K. Hansma, Langmuir, 1994, 10, 4409–4413 CrossRef CAS.
- K. Hu and A. J. Bard, Langmuir, 1997, 13, 5418–5425 CrossRef CAS.
- E. J. Wanless and W. A. Ducker, J. Phys. Chem., 1996, 100, 3207–3214 CrossRef CAS.
- H. N. Patrick, G. G. Warr, S. Manne and I. A. Aksay, Langmuir, 1999, 15, 1685–1692 CrossRef CAS.
- J.-F. Liu and W. A. Ducker, J. Phys. Chem. B, 1999, 103, 8558–8567 CrossRef CAS.
- I. Burgess, C. A. Jeffrey, X. Cai, G. Szymanski, Z. Galus and J. Lipkowski, Langmuir, 1999, 15, 2607–2616 CrossRef CAS.
- Y. Tan, E. M. P. Steinmiller and K.-S. Choi, Langmuir, 2005, 21, 9618–9624 CrossRef CAS PubMed.
- Y. Yamauchi, T. Momma, H. Kitoh, T. Osaka and K. Kuroda, Electrochem. Commun., 2005, 7, 1364–1370 CrossRef CAS.
- Y. Yamauchi, T. Ohsuna and K. Kuroda, Chem. Mater., 2007, 19, 1335–1342 CrossRef CAS.
- X. Zhang, W. Lu, J. Dai, L. Bourgeois, N. Hao, H. Wang, D. Zhao and P. A. Webley, Angew. Chem., Int. Ed., 2010, 49, 10101–10105 CrossRef CAS PubMed.
- K. Luo, C. T. Walker and K. J. Edler, Adv. Mater., 2007, 19, 1506–1509 CrossRef CAS.
- K. Tanaka, N. Moritake and H. Uchiki, Sol. Energy Mater. Sol. Cells, 2007, 91, 1199–1201 CrossRef CAS.
- O. Lev, Z. Wu, S. Bharathi, V. Glezer, A. Modestov, J. Gun, L. Rabinovich and S. Sampath, Chem. Mater., 1997, 9, 2354–2375 CrossRef CAS.
- A. Walcarius, D. Mandler, J. A. Cox, M. Collinson and O. Lev, J. Mater. Chem., 2005, 15, 3663–3689 RSC.
- S. V. Aurobind, K. P. Amirthalingam and H. Gomathi, Adv. Colloid Interface Sci., 2006, 121, 1–7 CrossRef CAS PubMed.
- J. Wang, Anal. Chim. Acta, 1999, 399, 21–27 CrossRef CAS.
- D. B. Mitzi, Chem. Mater., 2001, 13, 3283–3298 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. de, A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- R. Shacham, D. Avnir and D. Mandler, Adv. Mater., 1999, 11, 384–388 CrossRef CAS.
- P. N. Deepa, M. Kanungo, G. Claycomb, P. M. A. Sherwood and M. M. Collinson, Anal. Chem., 2003, 75, 5399–5405 CrossRef CAS PubMed.
- M. M. Collinson, Acc. Chem. Res., 2007, 40, 777–783 CrossRef CAS PubMed.
- S. Sayen and A. Walcarius, Electrochem. Commun., 2003, 5, 341–348 CrossRef CAS.
- E. Sibottier, S. Sayen, F. Gaboriaud and A. Walcarius, Langmuir, 2006, 22, 8366–8373 CrossRef CAS PubMed.
- R. Toledano, R. Shacham, D. Avnir and D. Mandler, Chem. Mater., 2008, 20, 4276–4283 CrossRef CAS.
- M. Raveh, L. Liu and D. Mandler, Phys. Chem. Chem. Phys., 2013, 15, 10876–10884 RSC.
- O. Nadzhafova, M. Etienne and A. Walcarius, Electrochem. Commun., 2007, 9, 1189–1195 CrossRef CAS.
- Z. Wang, M. Etienne, G.-W. Kohring, Y. Bon-Saint-Côme, A. Kuhn and A. Walcarius, Electrochim. Acta, 2011, 56, 9032 CrossRef CAS.
- M. Etienne, A. Goux, E. Sibottier and A. Walcarius, J. Nanosci. Nanotechnol., 2009, 9, 2398–2406 CrossRef CAS PubMed.
- A. Maghear, M. Etienne, M. Tertiş, R. Săndulescu and A. Walcarius, Electrochim. Acta, 2013, 112, 333 CrossRef CAS.
- F. Qu, R. Nasraoui, M. Etienne, Y. B. S. Côme, A. Kuhn, J. Lenz, J. Gajdzik, R. Hempelmann and A. Walcarius, Electrochem. Commun., 2011, 13, 138–142 CrossRef CAS.
- I. Mazurenko, M. Etienne, R. Ostermann, B. Smarsly, O. Tananaiko, V. Zaitsev and A. Walcarius, Langmuir, 2011, 27, 7140–7147 CrossRef CAS PubMed.
- R. Shacham, D. Mandler and D. Avnir, C. R. Chim., 2010, 13, 237–241 CrossRef CAS.
- L. Liu, R. Toledano, T. Danieli, J.-Q. Zhang, J.-M. Hu and D. Mandler, Chem. Commun., 2011, 47, 6909–6911 RSC.
- Y. Guillemin, M. Etienne, E. Sibottier and A. Walcarius, Chem. Mater., 2011, 23, 5313–5322 CrossRef CAS.
- H. Lim, J. Kim, K. Kani, M. K. Masud, H. Park, M. Kim, S. M. Alsheri, T. Ahamad, N. Alhokbany, J. Na, V. Malgras, Y. Bando and Y. Yamauchi, Small, 2020, 16, 1902934 CrossRef CAS PubMed.
- M. Etienne, S. Sallard, M. Schröder, Y. Guillemin, S. Mascotto, B. M. Smarsly and A. Walcarius, Chem. Mater., 2010, 22, 3426–3432 CrossRef CAS.
- W. Ghach, M. Etienne, P. Billard, F. P. A. Jorand and A. Walcarius, J. Mater. Chem. B, 2013, 1, 1052–1059 RSC.
- A. Walcarius, E. Sibottier, M. Etienne and J. Ghanbaja, Nat. Mater., 2007, 6, 602–608 CrossRef CAS PubMed.
- A. Goux, M. Etienne, E. Aubert, C. Lecomte, J. Ghanbaja and A. Walcarius, Chem. Mater., 2009, 21, 731–741 CrossRef CAS.
- S. L. Z. Jiokeng, I. Tonle and A. Walcarius, Sens. Actuators, B, 2019, 287, 296–305 CrossRef CAS.
- A. Takai, Y. Doi, Y. Yamauchi and K. Kuroda, J. Phys. Chem. C, 2010, 114, 7586–7593 CrossRef CAS.
- J. Fang, L. Zhang, J. Li, L. Lu, C. Ma, S. Cheng, Z. Li, Q. Xiong and H. You, Nat. Commun., 2018, 9, 521 CrossRef PubMed.
- H. Park, M. K. Masud, J. Na, H. Lim, H. P. Phan, Y. V. Kaneti, A. A. Alothman, C. Salomon, N. T. Nguyen, M. S. A. Hossain and Y. Yamauchi, J. Mater. Chem. B, 2020, 8, 9512–9523 RSC.
- M. El Aamri, G. Yammouri, H. Mohammadi, A. Amine and H. Korri-Youssoufi, Biosensors, 2020, 10, 186 CrossRef CAS PubMed.
- B. N. Johnson and R. Mutharasan, Analyst, 2014, 139, 1576–1588 RSC.
- H. Lv, L. Sun, J. Feng, J. Na, D. Xu, Y. Yamauchi and B. Liu, Chem. Commun., 2020, 56, 9679–9682 RSC.
- H. Ataee-Esfahani, L. Wang, Y. Nemoto and Y. Yamauchi, Chem. Mater., 2010, 22, 6310–6318 CrossRef CAS.
- P. Hernández-Fernández, S. Rojas, P. Ocón, J. L. Gómez de la Fuente, J. San Fabián, J. Sanza, M. A. Peña, F. J. García-García, P. Terreros and J. L. G. Fierro, J. Phys. Chem. C, 2007, 111, 2913–2923 CrossRef.
- L. Wang and Y. Yamauchi, Chem. Mater., 2011, 23, 2457–2465 CrossRef CAS.
- J. Luo, M. M. Maye, V. Petkov, N. N. Kariuki, L. Wang, P. Njoki, D. Mott, Y. Lin and C.-J. Zhong, Chem. Mater., 2005, 17, 3086–3091 CrossRef CAS.
- E.-K. Park, J.-K. Lee, Y.-S. Kim, G.-P. Kim and S.-H. Baeck, J. Phys. Chem. Solids, 2008, 69, 1284–1287 CrossRef CAS.
- C. Li, Y. Su, X. Lv, H. Xia, H. Shi, X. Yang, J. Zhang and Y. Wang, Biosens. Bioelectron., 2012, 38, 402–406 CrossRef CAS PubMed.
- C. Li, Y. Su, S. Zhang, X. Lv, H. Xia and Y. Wang, Biosens. Bioelectron., 2010, 26, 903–907 CrossRef CAS PubMed.
- J. Huang, Z. Dong, Y. Li, J. Li, J. Wang, H. Yang, S. Li, S. Guo, J. Jin and R. Li, Sens. Actuators, B, 2013, 618–624 CrossRef CAS.
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver and J. K. Nørskov, J. Mol. Catal. A: Chem., 1997, 3, 421–429 CrossRef.
- P. Kannan, C.-S. Yoon, S.-C. Yi, S. Y. Lee and D.-H. Kim, Mater. Chem. Phys., 2015, 156, 1–8 CrossRef CAS.
- Y. Wang, H. Ji, W. Peng, L. Liu, F. Gao and M. Li, Int. J. Hydrogen Energy, 2012, 37, 9324–9329 CrossRef CAS.
- P. N. Njoki, M. E. D. Roots and M. M. Maye, ACS Appl. Nano Mater., 2018, 1, 5640–5645 CrossRef CAS.
- S. Wang, J. Yin, H. Wei, K. Huang, L.-M. Liu and H. Wu, ACS Appl. Energy Mater., 2019, 2, 468–476 CrossRef CAS.
- S. Yan, L. Gao, S. Zhang, L. Gao, W. Zhang and Y. Li, Int. J. Hydrogen Energy, 2013, 38, 12838–12846 CrossRef CAS.
- S. Yan, S. Zhang, Y. Lin and G. Liu, J. Phys. Chem. C, 2011, 115, 6986–6993 CrossRef CAS.
- X. Cui, P. Xiao, J. Wang, M. Zhou, W. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang, Y. Zhang and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 4488–4493 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- Z.-C. Zhang, J.-F. Hui, Z.-G. Guo, Q.-Y. Yu, B. Xu, X. Zhang, Z.-C. Liu, C.-M. Xu, J.-S. Gao and X. Wang, Nanoscale, 2012, 4, 2633–2639 RSC.
- A.-X. Yin, X.-Q. Min, W. Zhu, H.-S. Wu, Y.-W. Zhang and C.-H. Yan, Chem. Commun., 2012, 48, 543–545 RSC.
- H. Strehlow, Ber. Bunsen-Ges. Phys. Chem., 1978, 82, 1114 CrossRef.
- A. Takai, H. Ataee-Esfahani, Y. Doi, M. Fuziwara, Y. Yamauchi and K. Kuroda, Chem. Commun., 2011, 47, 7701–7703 RSC.
- Z. Yin, W. Zhou, Y. Gao, D. Ma, C. J. Kiely and X. Bao, Chem. – Eur. J., 2012, 18, 4887–4893 CrossRef CAS PubMed.
- S. Guo, S. Zhang, X. Sun and S. Sun, J. Am. Chem. Soc., 2011, 133, 15354–15357 CrossRef CAS PubMed.
- H. Duan, N. Yan, R. Yu, C.-R. Chang, G. Zhou, H.-S. Hu, H. Rong, Z. Niu, J. Mao, H. Asakura, T. Tanaka, P. J. Dyson, J. Li and Y. Li, Nat. Commun., 2014, 5, 3093 CrossRef.
- Y. Jiang, J. Su, Y. Yang, Y. Jia, Q. Chen, Z. Xie and L. Zheng, Nano Res., 2016, 9, 849–856 CrossRef CAS.
- Y.-N. Wen and J.-M. Zhang, Solid State Commun., 2007, 144, 163–167 CrossRef CAS.
- Q.-Q. Chen, C.-C. Hou, C.-J. Wang, X. Yang, R. Shi and Y. Chen, Chem. Commun., 2018, 54, 6400–6403 RSC.
- X. Wang, C. Xu, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Nat. Commun., 2019, 10, 4876 CrossRef.
- Z. Xing, D. Wang, Q. Li, A. M. Asiri and X. Sun, Electrochim. Acta, 2016, 210, 729–733 CrossRef CAS.
- D. S. Hall, C. Bock and B. R. MacDougall, J. Electrochem. Soc., 2013, 160, F235 CrossRef CAS.
- T. Nagaura, H.-P. Phan, V. Malgras, T.-A. Pham, H. Lim, A. Ashok, J. Kim, J. You, N.-T. Nguyen, J. Na and Y. Yamauchi, Angew. Chem., Int. Ed., 2021, 60, 9660–9665 CrossRef CAS PubMed.
- M. C. Beard, G. M. Turner and C. A. Schmuttenmaer, Nano Lett., 2002, 2, 983–987 CrossRef CAS.
- Y. Qiu, W. Liu, W. Chen, W. Chen, G. Zhou, P.-C. Hsu, R. Zhang, Z. Liang, S. Fan, Y. Zhang and Y. Cui, Sci. Adv., 2016, 2, e1501764 CrossRef PubMed.
- A. Lu, Z. P. Wu, B. Chen, D. L. Peng, S. Yan, S. Shan, Z. Skeete, F. Chang, Y. Chen, H. Zheng, D. Zeng, L. Yang, A. Sharma, J. Luo, L. Wang, V. Petkov and C. J. Zhong, J. Mater. Chem. A, 2018, 6, 5143–5155 RSC.
- X. Sun, D. Li, Y. Ding, W. Zhu, S. Guo, Z. L. Wang and S. Sun, J. Am. Chem. Soc., 2014, 136, 5745–5749 CrossRef CAS PubMed.
- S. Zhang, Y. Shao, H. G. Liao, J. Liu, I. A. Aksay, G. Yin and Y. Lin, Chem. Mater., 2011, 23, 1079–1081 CrossRef CAS.
- A. S. Nugraha, V. Malgras, J. Kim, J. Bo, C. Li, M. Iqbal, Y. Yamauchi and T. Asahi, Small Methods, 2018, 2, 1800283 CrossRef.
- Y. Zhang, G. Chang, H. Shu, M. Oyama, X. Liu and Y. He, J. Power Sources, 2014, 262, 279–285 CrossRef CAS.
- Z. Wang, W. Xie, F. Zhang, J. Xia, S. Gong and Y. Xia, Electrochim. Acta, 2016, 192, 400–406 CrossRef CAS.
- Y. Liu, M. Chi, V. Mazumder, K. L. More, S. Soled, J. D. Henao and S. Sun, Chem. Mater., 2011, 23, 4199–4203 CrossRef CAS.
- S. J. Ye, H. T. Bui, Y. Y. Kim, K. Liao, K. M. Cho, H.-T. Jung, Y. Kang, D. Y. Kim and O. O. Park, Chemistry, 2017, 23, 17136–17143 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang and X. Wang, Nanoscale, 2011, 3, 1663–1674 RSC.
- C. Zhu, S. Guo and S. Dong, Adv. Mater., 2012, 24, 2326–2331 CrossRef CAS PubMed.
- K. Mikkelsen, B. Cassidy, N. Hofstetter, L. Bergquist, A. Taylor and D. A. Rider, Chem. Mater., 2014, 26, 6928–6940 CrossRef CAS.
- G. You, J. Jiang, M. Li, L. Li, D. Tang, J. Zhang, X. C. Zeng and R. He, ACS Catal., 2018, 8, 132–143 CrossRef CAS.
- Z. Sun, J. H. Kim, Y. Zhao, F. Bijarbooneh, V. Malgras and S. X. Dou, J. Mater. Chem., 2012, 22, 11711–11719 RSC.
- F. H. Bijarbooneh, Y. Zhao, Z. Sun, Y.-U. Heo, V. Malgras, J. H. Kim and S. X. Dou, APL Mater., 2013, 1, 032106 CrossRef.
- Z. Sun, T. Liao, K. Liu, L. Jiang, J. H. Kim and S. X. Dou, Nano Res., 2013, 6, 726–735 CrossRef CAS.
- M. P. Walsh, J. Power Sources, 1990, 29, 13–28 CrossRef CAS.
- K. Maheshwari, P. D. S. Sharma, D. A. K. Sharma and P. S. Verma, Int. J. Eng. Res. Sci. Technol., 2018, 7, 060019 Search PubMed.
- N. Sazali, W. N. Wan Salleh, A. S. Jamaludin and M. N. Mhd Razali, Membranes, 2020, 10, 99 CrossRef CAS PubMed.
- L. Zu, W. Zhang, L. Qu, L. Liu, W. Li, A. Yu and D. Zhao, Adv. Energy Mater., 2020, 10, 2002152 CrossRef CAS.
- Y. Yan, B. Yu Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- K. Eiler, S. Suriñach, J. Sort and E. Pellicer, Appl. Catal., B, 2020, 265, 118597 CrossRef CAS.
- X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736–2753 RSC.
- M. F. Sgroi, F. Zedde, O. Barbera, A. Stassi, D. Sebastián, F. Lufrano, V. Baglio, A. S. Aricò, J. L. Bonde and M. Schuster, Energies, 2016, 9, 1008 CrossRef.
- X. Wang, S. Xi, W. S. V. Lee, P. Huang, P. Cui, L. Zhao, W. Hao, X. Zhao, Z. Wang, H. Wu, H. Wang, C. Diao, A. Borgna, Y. Du, Z. G. Yu, S. Pennycook and J. Xue, Nat. Commun., 2020, 11, 4647 CrossRef CAS PubMed.
- X. Li and A. Faghri, J. Power Sources, 2013, 226, 223–240 CrossRef CAS.
- T. Shimizu, M. Mohamedi, T. Momma and T. Osaka, Electrochemistry, 2006, 74, 326–331 CrossRef CAS.
- X. Cheng, Z. Shi, N. Glass, L. Zhang, J. Zhang, D. Song, Z. S. Liu, H. Wang and J. Shen, J. Power Sources, 2007, 165, 739–756 CrossRef CAS.
- C. Li, H. Wang, Y. Li, H. Yu, S. Yin, H. Xue, X. Li, Y. Xu and L. Wang, Nanotechnology, 2018, 29, 255404 CrossRef PubMed.
- S. Y. Tee, C. P. Teng and E. Ye, Mater. Sci. Eng., C, 2017, 70, 1018–1030 CrossRef CAS PubMed.
- R. Li, X. Deng and L. Xia, Sci. Rep., 2020, 10, 16788 CrossRef CAS PubMed.
- K. Tian, M. Prestgard and A. Tiwari, Mater. Sci. Eng., C, 2014, 41, 100–118 CrossRef CAS PubMed.
- F. Kurniawan, V. Tsakova and V. M. Mirsky, Electroanalysis, 2006, 18, 1937–1942 CrossRef CAS.
- J. Wang, D. F. Thomas and A. Chen, Anal. Chem., 2008, 80, 997–1004 CrossRef CAS PubMed.
- K. U. Tüfekci, M. G. Öner, R. L. J. Meuwissen and Ş. Genç, Methods Mol. Biol., 2014, 1107, 33–50 CrossRef PubMed.
- J. O’Brien, H. Hayder, Y. Zayed and C. Peng, Front. Endocrinol., 2018, 9, 402 CrossRef PubMed.
- G. Fu, J. Brkić, H. Hayder and C. Peng, Int. J. Mol. Sci., 2013, 14, 5519–5544 CrossRef CAS PubMed.
- J. Hayes, P. P. Peruzzi and S. Lawler, Trends Mol. Med., 2014, 20, 460–469 CrossRef CAS PubMed.
- M. N. Islam, M. K. Masud, M. H. Haque, M. S. Al Hossain, Y. Yamauchi, N.-T. Nguyen and M. J. A. Shiddiky, Small Methods, 2017, 1, 1700131 CrossRef.
- L. Zhang, W. Su, S. Liu, C. Huang, B. Ghalandari, A. Divsalar and X. Ding, Phenomics, 2022, 2, 18–32 CrossRef.
- M. K. Masud, J. Na, M. Younus, M. S. A. Hossain, Y. Bando, M. J. A. Shiddiky and Y. Yamauchi, Chem. Soc. Rev., 2019, 48, 5717–5751 RSC.
- M. K. Masud, J. Na, T.-E. Lin, V. Malgras, A. Preet, A. A. Ibn Sina, K. Wood, M. Billah, J. Kim, J. You, K. Kani, A. E. Whitten, C. Salomon, N.-T. Nguyen, M. J. A. Shiddiky, M. Trau, M. S. A. Hossain and Y. Yamauchi, Biosens. Bioelectron., 2020, 168, 112429 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |