Tough tetrazine-functionalized styrene–butadiene rubber with self-adhesion through zinc–nitrogen coordination†
Received
14th July 2023
, Accepted 11th September 2023
First published on 12th September 2023
Abstract
We report a feasible system for the direct adhesion of cross-linked rubbers, based on incorporating tetrazine ligands into a styrene–butadiene rubber (SBR) followed by the addition of zinc dimethacrylate (ZDMA) with the aim of forming reversible coordination cross-links. The modification of SBR is achieved using an inverse-electron-demand Diels–Alder (IEDDA) click reaction with 3,6-di(2-pyridyl)-1,2,4,5-tetrazine (DPT) during the rubber-kneading process. The complexation capability of the bound DPT in the SBR with ZDMA is demonstrated using a small model molecule including a DPT unit via UV-vis spectra. The obtained DPT–ZDMA cross-linked rubbers show unique temperature dependence in dynamic mechanical analysis (DMA), demonstrating the dissociation and reassociation of the DPT–ZDMA cross-linking. Strain-sweep DMA tests exhibited a typical Payne effect with increasing amount of bound DPT units in the SBR, supporting the formation of ZDMA–ZDMA filler interactions. Furthermore, the reversible nature of DPT–ZDMA cross-linking endows the resulting composites with high strength. A tensile strength of up to 30.5 MPa at 579% elongation was achieved when 2.6 mol% of DPT was incorporated into SBR with 40 phr of ZDMA. Hysteresis measurements revealed that the amount of bound DPT in the SBR and the ZDMA content significantly impact the hysteresis loss due to the dissociation of the DPT–ZDMA cross-linking and ZDMA–ZDMA filler interactions, leading to high energy dissipation and high toughness. Finally, a direct adhesion application was demonstrated using T-peel tests, where the adhesion-peeling force reaches up to 5.72 N mm−1 when 2.6 mol% of DPT was incorporated into SBR with 60 phr of ZDMA. The maximum peeling force showed a good correlation with the difference between the E′ value at 25 °C and that at 145 °C as obtained from the DMA tests, clearly demonstrating that the degree of DPT–ZDMA cross-linking is a main factor in determining the adhesion strength in the DPT–ZDMA cross-linking system.
Introduction
Adhesion between cross-linked rubbers is one of the most important processes for engineering the manufacture of rubber products.1 However, it is commonly known that direct adhesion between cross-linked rubbers without treatment of the rubber surfaces is quite difficult due to the restricted polymer-chain mobility on account of cross-linking,1–5 and therefore, it is not surprising that pretreatment methods for rubber-surface modifications and adhesive technology have been widely studied.1,6–13 Nonetheless, repair processes, such as tyre retreading, for cured rubber products that involve an adhesion process between cross-linked rubbers are still limited given several issues with the processes, including curing temperature and time.14–18
To develop a more efficient adhesion system, it is important to induce the interdiffusion of polymer chains between two cured rubber samples during an adhesion process, such as heat-pressing, followed by interlinkage between the two rubber samples after cooling. Hence, the introduction of reversible linkages,1,19–22 such as hydrogen bonds,23–26 ionic interactions,27–33 host–guest interactions,34,35 dynamic covalent bonds36–45 and metal–ligand coordination bonds,25,46–55 given that introducing cross-linking bonds into cured rubber is a potential strategy to achieve direct adhesion between cross-linked rubbers, as reversible linkages often show a temperature dependence that is different from that of conventional covalent bonds, which results in good malleability at high temperature. Metal–ligand coordination bonds are particularly promising cross-linkages on account of their bonding energy, which can be controlled by changing the type of metal salt and the design of the ligands on the polymer chains. For example, Bao et al.47 have designed a highly stretchable autonomously self-healing poly(dimethylsiloxane) (PDMS) by introducing the 2,6-butylpyridinedicarboxyamide ligand and cross-linking with FeCl3. The resulting cross-linked PDMS samples show a high recovered-fracture strain due to the metal–ligand exchange and the low Tg (below −90 °C) of the PDMS, giving high mobility to the polymer chain. Weder et al.52 have demonstrated that cross-linkages that consist of the 2,6-bis(1′-methylbenzimidazolyl)-pyridine ligand, incorporated into a styrene–butadiene rubber (SBR) via a thiol–ene reaction, and Zn(OTf)2 can act as reversible bonds and show a unique tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ peak at ca. 90 °C due to energy dissipation upon activation of the metal–ligand cross-linkages at this temperature in dynamic-mechanical-analysis (DMA) measurements. Das et al.53 have reported that a mixture of epoxidized natural rubber (ENR), FeCl3, zinc acetate dihydrate, and 2,6-diaminopyridine (DAP) forms reversible cross-links and exhibits self-healing behaviour at 60 °C. In this system, Fe3+ ions form bonds with the oxygen atom of the epoxy group in the ENR, while DAP not only acts as a catalyst for the ring-opening of the epoxide moiety, but also kinetically controls the release of Fe3+.
δ peak at ca. 90 °C due to energy dissipation upon activation of the metal–ligand cross-linkages at this temperature in dynamic-mechanical-analysis (DMA) measurements. Das et al.53 have reported that a mixture of epoxidized natural rubber (ENR), FeCl3, zinc acetate dihydrate, and 2,6-diaminopyridine (DAP) forms reversible cross-links and exhibits self-healing behaviour at 60 °C. In this system, Fe3+ ions form bonds with the oxygen atom of the epoxy group in the ENR, while DAP not only acts as a catalyst for the ring-opening of the epoxide moiety, but also kinetically controls the release of Fe3+.
Meanwhile, the incorporation of ligand molecules into the conventional rubber polymer chains is of great interest from a manufacturing perspective. One of the most promising methods in this context is click chemistry,56 which is characterized by high atom-efficiency and performed under relatively mild conditions. In particular, inverse-electron-demand Diels–Alder (IEDDA) reactions between tetrazine moieties and unsaturated bonds57–60 is a powerful tool to incorporate functional groups into polymer chains.54,55,61,62 Recently, Wu et al. have reported a route for the modification of NBR54 and SBR55 using the IEDDA reaction with 3,6-di(2-pyridyl)-1,2,4,5-tetrazine (DPT) during the rubber-mixing process. The resulting polymers form reversible cross-links with metal salts (ZnCl2, CoCl2 and CuSO4), and show self-healing behaviour upon thermal treatment at 170–190 °C. However, the metal salts used in the aforementioned studies are not easy to handle in realistic industrial rubber-mixing processes due to their hygroscopicity and the risks they present to human health and/or the environment. Thus, we were interested to create a more feasible system, especially from the viewpoint of material handling in the rubber industry.
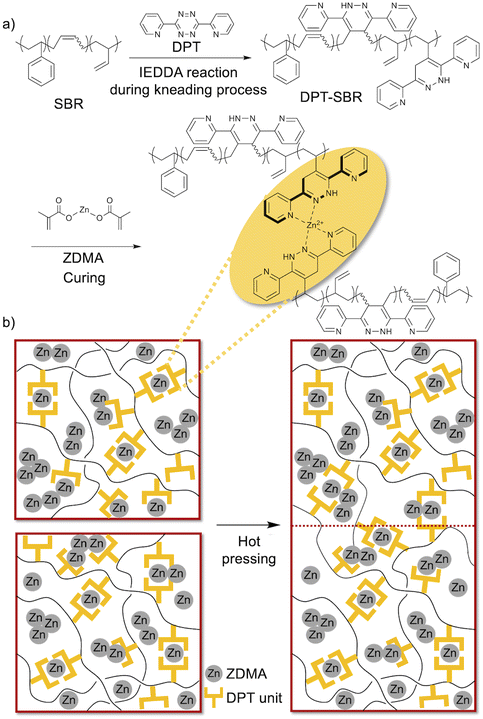
In this study, zinc dimethacrylate (ZDMA) was selected for the formation of the reversible metal–ligand cross-links with DPT-modified SBRs. ZDMA is not hygroscopic and less toxic than the aforementioned metal salts, and is thus widely used in the rubber industry as a reinforcing cross-linking coagent, mainly with peroxide curing systems.29,31–33,63–68 The objective of this study was to design novel cross-linked SBR composites that exhibit direct adhesion behaviour without the need for pretreatment (Fig. 1). For this purpose, we prepared DPT-modified SBRs using a rubber-kneading process with reference to a previous study,55 and investigated the coordination capability of the DPT unit towards ZDMA. Subsequently, the DPT-modified SBRs were compounded with ZDMA, which acted not only as a cross-linking agent, but also as a reinforcing filler. Finally, the physical properties of the resulting cross-linked samples, including their dynamic viscoelasticity, tensile behaviour and adhesive strength, were examined in detail. The obtained results demonstrate that the DPT–ZDMA cross-linking network is successfully formed to endow the resulting SBR composites with unique physical properties, including high tensile strength and improved adhesion strength.
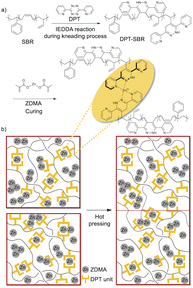 |
| | Fig. 1 Conceptual outline of this study: (a) preparation of DPT-modified SBRs by IEDDA reactions during the kneading process, followed by the formation of DPP–ZDMA cross-linkages and (b) schematic description of the adhesion process via rearrangement of the DPT–ZDMA cross-linkages. | |
Experimental
Materials
A random solution styrene–butadiene copolymer (SBR; 25 wt% styrene and 75 wt% butadiene; grade: TUFDENE™ 2000R) was purchased from Asahi Kasei Corp. Zinc dimethacrylate (ZDMA, Dymalink® 708) was obtained from Cray Valley Co., Ltd. Dichloroacetic acid, cis-cyclooctene and 3,6-di(2-pyridyl)-1,2,4,5-tetrazine (DPT) were acquired from Tokyo Chemical Industry Co., Ltd. Dicumyl peroxide and chloroform-d1 were purchased from Sigma Aldrich Co. LLC. Tetrahydrofuran (THF), toluene, dichloromethane, xylene, methanol, n-hexane, n-heptane and silica gel 60 N (spherical, neutral) were acquired from Kanto Chemical Co., Ltd. 1,1,2,2-Tetrachloroethane-d2 was purchased from Fujifilm Wako Pure Chemical Corp.
Measurements
Gel-permeation chromatography (GPC) measurements were carried out at 40 °C using a TOSOH HLC-8320 GPC system equipped with a guard column (TOSOH TSK guard column HXL-H), two separation columns (TOSOH TSK gel GMHXL), a differential refractive-index (RI) detector and a UV detector. Tetrahydrofuran (THF) was used as the eluent at a flow rate of 0.6 mL min−1. Polystyrene (PS) standards (Mw = 8420000–1010 g mol−1) were used to calibrate the GPC system. Differential-scanning-calorimetry (DSC) measurements were conducted using a DSC2500 (TA Instruments Inc.) instrument under an atmosphere of nitrogen at a heating rate of 20 °C min−1. High-resolution mass spectra (HRMS) were obtained using an Acquity UPLC system coupled to a Xevo G2-XS QTOF mass spectrometer (Waters) using MeOH as the eluent. Fourier-transform infrared (FT-IR) spectra were recorded at room temperature on a Nicolet iS5 FT-IR spectrometer using a germanium-crystal attenuated-total-reflectance (ATR) attachment (Thermo Scientific Inc.). 1H and 13C NMR spectra were measured in chloroform-d1 (CDCl3) with tetramethylsilane (TMS) as the internal standard or 1,1,2,2-tetrachloroethane-d2 (C2D2Cl4) at 300 K on a Bruker AVANCE III HD 600 MHz spectrometer. UV-vis spectra were measured at room temperature in CH2Cl2 using a V-680 (Jasco Corp.) spectrophotometer. Rotorless rheometer measurements were carried out at 145 °C using an RLR-4 (Toyo Seiki Manufacturing Co. Ltd) instrument under a pressure of 10 MPa with a fixed amplitude angle of ±1° and a frequency of 100 cpm. Dynamic mechanical analysis (DMA) tests were conducted on a GABO EPLEXOR (Netzsch) apparatus equipped with a 500 N load cell, using standard JIS-K6251 strip-shaped specimens (length: 40.0 mm; width: 4.7 mm). Temperature-sweep DMA tests were carried out under tensile mode with a fixed frequency of 15 Hz, a static strain of 6%, a dynamic strain of 0.4%, and a heating rate of 2 °C min−1 from −75 to 150 °C. The strain-sweep tests were conducted at 25 °C under tensile mode with a fixed frequency of 15 Hz, a static strain of 10%, and a dynamic strain range from 0.1 to 10%. Tensile tests were carried out at room temperature using an Instron 5965 (Instron) instrument equipped with a 5 kN load cell at a crosshead speed of 100 mm min−1, using dumbbell-shaped ISO 37-4 specimens. Three measurements were performed for each sample. The fracture energy, W, was calculated using the equation 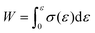 . Hysteresis tests were performed at room temperature using an Instron 5965 (Instron) apparatus equipped with a 5 kN load cell at a crosshead speed of 100 mm min−1, using dumbbell-shaped ISO 37-4 specimens. The hysteresis loss was calculated using the equation, Hysteresis loss = Wstored − Wunload, where
. Hysteresis tests were performed at room temperature using an Instron 5965 (Instron) apparatus equipped with a 5 kN load cell at a crosshead speed of 100 mm min−1, using dumbbell-shaped ISO 37-4 specimens. The hysteresis loss was calculated using the equation, Hysteresis loss = Wstored − Wunload, where 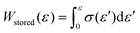 and Wunload were calculated using the stress–strain relationship for the unloading process. T-peel adhesion tests were conducted at room temperature on an Instron 5965 (Instron) instrument equipped with a 5 kN load cell at a crosshead speed of 300 mm min−1 using laminated stripes (length: 75 mm; width: 10 mm). Two measurements were performed for each sample.
and Wunload were calculated using the stress–strain relationship for the unloading process. T-peel adhesion tests were conducted at room temperature on an Instron 5965 (Instron) instrument equipped with a 5 kN load cell at a crosshead speed of 300 mm min−1 using laminated stripes (length: 75 mm; width: 10 mm). Two measurements were performed for each sample.
Preparation of DPT-modified SBRs
SBR (65.0 g) and varying amounts of DPT were kneaded at 70 rpm at 120 °C for 3 min in an internal mixer (Toyo Seiki Manufacturing Co. Ltd). The resulting DPT–SBRs were denoted as DPT–SBR1, DPT–SBR2 and DPT–SBR3, for DPT loading amounts of 1, 2 and 4 mol% relative to the butadiene unit in the SBR, respectively.
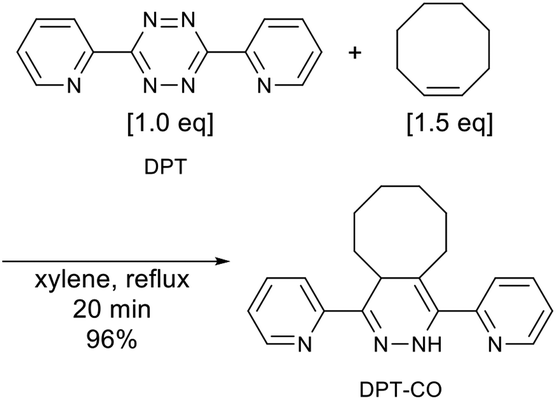
Synthesis of DPT-CO57
A round-bottom flask was charged with 3,6-di(2-pyridyl)-1,2,4,5-tetrazine (0.472 g, 2.00 mmol) and xylene (20 mL), before the resulting solution was sparged with nitrogen gas. Subsequently, cis-cyclooctene (0.331 g, 3.00 mmol) was added to the solution, which was subsequently stirred under reflux conditions at a setting temperature of 160 °C. After 20 min, the red solution turned pale yellow, indicating that the reaction had reached completion. The solution was then concentrated under reduced pressure, before the thus obtained residue was subjected to column chromatography on silica gel using acetone/n-hexane 5/95 (v/v) to give 0.612 g (1.92 mmol, 96%) of DPT-CO as a yellow solid. 1H NMR (CDCl3, 600 MHz, δ): 8.81 (s, 1H), 8.63 (ddd, J = 4.8, 1.7, 1.1 Hz, 1H), 8.59 (ddd, J = 4.8, 1.7, 1.1 Hz, 1H), 8.08 (dt, J = 8.0, 1.0 Hz, 1H), 7.72 (td, J = 7.7, 1.4 Hz, 1H), 7.63 (m, 1H), 7.54 (m, 1H), 7.21 (ddd, J = 7.5, 4.8, 1.0 Hz, 1H), 7.16 (ddd, J = 7.4, 4.8, 1.1 Hz, 1H), 4.40 (m, 1H), 2.90 (ddd, J = 15.2, 9.0, 3.3 Hz, 1H), 2.15 (m, 1H), 2.07 (m, 1H), 1.88 (m, 1H), 1.83–1.65 (m, 5H), 1.63–1.56 (m, 2H), 1.53 (m, 1H). 13C NMR (CDCl3, 150 MHz, δ): 154.8, 152.6, 149.4, 148.6, 143.2, 136.3, 135.8, 133.8, 124.0, 122.5, 122.4, 121.0, 112.4, 35.3, 31.4, 28.8, 27.6, 26.9, 24.7, 22.8. IR (ATR, cm−1) 3265, 2917, 2860, 1580, 1564, 1488, 1471, 1427, 1353, 1152. HRMS (ESI) [M + Na] calcd for C20H22N4Na 341.1742; found 341.1790. UV-vis (6.4 × 10−5 M in CH2Cl2, nm) λmax 255, 325.
Mixing and curing conditions for the uncured rubber samples
SBR or DPT–SBRs (15 g, 100 phr) were mixed with different amounts of ZDMA for 10 min on a two-roll open mill with a roller speed of 15 rpm at 65 °C. The SBR/ZDMA mixtures were further mixed with varying amount of dicumyl peroxide (DCP) for 2 min on the two-roll open mill with a roller speed of 15 rpm at 65 °C. The resulting samples were hot-pressed at 145 °C and 15 MPa for 30 min to give sheets with a thickness of ∼1 mm.
Dissolution study of cross-linked samples
The cross-linked sheets were cut into disc-shaped specimens with a diameter of 8.0 mm. The cut samples were immersed in 10 mL of toluene/dichloroacetic acid (v/v = 95/5)31 and kept at room temperature for 4 days, followed by heating (80 °C for 6 h).
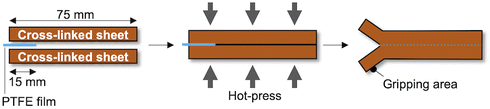
Preparation of T-peel test samples
Two sheets of each of the prepared cross-linked rubber samples were attached to each other without any pretreatment or adhesive (Fig. 2; left). To ensure a sufficient gripping area on the specimens for T-peel tests, a thin piece of poly(tetrafluoroethylene) (PTFE) film (length: 15–20 mm) was placed between the two sheets at the edge of the specimens (Fig. 2; middle and right). Then, the laminates were hot-pressed at 145 °C under 15 MPa for 30 min to give specimens of approximately 2 mm.
 |
| | Fig. 2 Schematic description of the preparation of the T-peel test specimens. | |
Results and discussion
Preparation of DPT–SBRs and a small model molecule (DPT-CO)
To prepare the DPT–SBRs, DPT and SBR were kneaded using an internal mixer. After the kneading process, the reaction between red DPT and colourless SBR was visually confirmed to have proceeded by the disappearance of the red colour of DPT and the appearance of a yellow colour (Fig. 3).
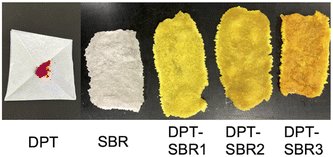 |
| | Fig. 3 Photographs of the DPT, SBR and DPT–SBR samples. | |
For a detailed DPT–SBR characterization, a small model molecule (DPT-CO) was synthesized for comparison. DPT-CO was obtained from the IEDDA reaction between cis-cyclooctene and DPT (Scheme 1). Compared to previously reported synthesis conditions, which use trans-cyclooctene in THF at room temperature to reach completion immediately,57 a much higher temperature was required for the reaction with cis-cyclooctene, most likely due to its lower strain energy compared to that of trans-cyclooctene.69 Nevertheless, the targeted DPT-CO was obtained in 96% yield.
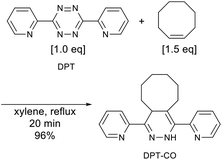 |
| | Scheme 1 Synthesis of DPT-CO from DPT and cis-cyclooctene. | |
Characterization of SBR and DPT–SBRs
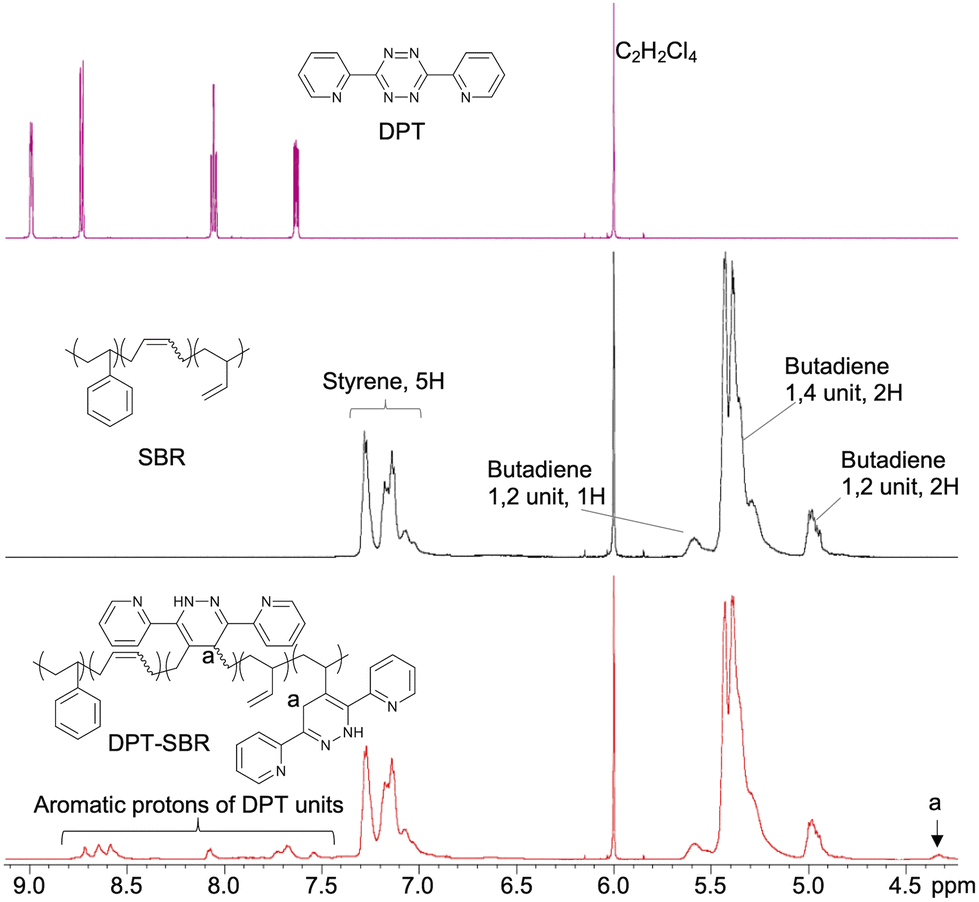
To characterize the original SBR and obtained DPT–SBRs, 1H NMR, GPC and DSC measurements were carried out and the characteristic values are summarized in Table 1. 1H NMR measurements were performed to characterize the obtained DPT–SBRs in detail (Fig. 4 and Fig. S1 as well as Table S1†). The initial signals for DPT disappeared in the spectrum of the DPT–SBR samples, and new distinctive signals corresponding to the aromatic protons in the DPT unit in the aromatic region and C–H protons (a) at δ 4.33 ppm appeared. In a previous study,55 the signal at 4.33 ppm was assigned to the N–H protons of the DPT units, but this interpretation contradicts the assignment of the 1H NMR spectrum for a DPT unit in a model small molecule (DPT-CO), which was supported by several NMR techniques, including 2D NMR experiments (Fig. S2–S5†). A comparison of the NMR spectra of the DPT–SBR samples with those of DPT-CO suggested that the signal at δ 4.33 ppm for the DPT–SBRs should be assigned to the C–H proton (a) of the DPT units. A close observation of the signals of the butadiene units (δ 5.70–4.70 ppm) revealed that both the content of 1,4 and 1,2 units per mole SBR decrease upon DPT loading, indicating that DPT reacts with both units in the butadiene segments. The modification-efficiency was calculated based on the ratio of the aromatic protons of the DPT units (8H, δ 8.81–7.46 ppm) relative to the aromatic protons of the styrene units in the SBR as well as the protons of the of butadiene 1,4 unit (2H) and the butadiene 1,2 unit (3H, 5.70–4.70 ppm) (eqn (S1)–(S6)†). In their entirety, these results indicate modification-efficiency values of 75.7, 76.4 and 75.8% for DPT–SBR1, DPT–SBR2 and DPT–SBR3, respectively. The unreacted residual DPT in each sample was considered to have further reacted with the SBR during the subsequent curing process and increased the modification efficiency.55
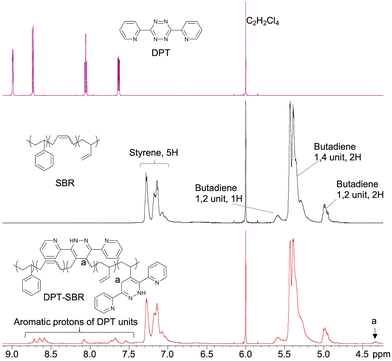 |
| | Fig. 4
1H NMR spectra of DPT, SBR and DPT–SBR3. | |
Table 1 Characterization of SBR and DPT–SBRs
| Polymer |
Butadiene (1,4 unit:1,2 unit):styrene:DPTa (mol%) |
M
n
|
M
w/Mnb |
T
g
(°C) |
|
Determined by 1H NMR spectroscopy.
Determined by GPC based on polystyrene standards (eluent: THF).
Determined by DSC.
|
| SBR |
85.7 (77.3:8.4):14.3:— |
141606 |
2.45 |
−68.4 |
| DPT–SBR1 |
84.7 (76.6:8.1):14.6:0.7 |
81800 |
1.78 |
−65.1 |
| DPT–SBR2 |
83.6 (75.9:7.7):15.1:1.3 |
75096 |
1.76 |
−53.5 |
| DPT–SBR3 |
81.7 (74.1:7.6):15.7:2.6 |
60303 |
1.76 |
−52.0 |
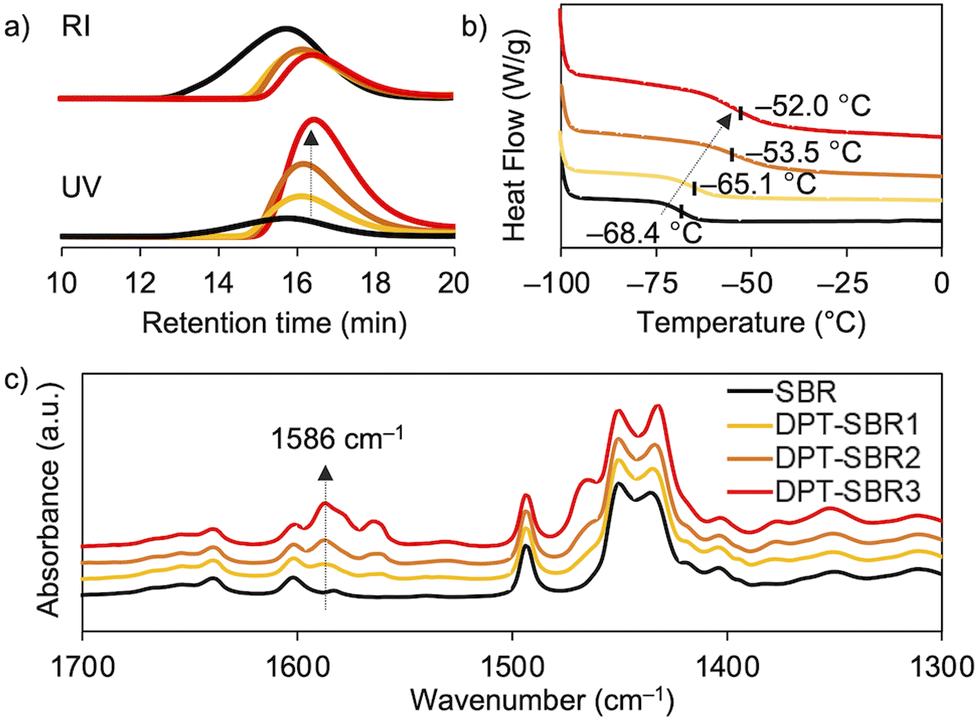
The Mn and Mw/Mn values of the SBR and DPT–SBR samples were determined using GPC. The Mn values of the resulting DPT–SBR samples were lower than those of the starting SBR samples (Fig. 5a). The decrease in Mn upon DPT modification is most likely predominantly due to the main-chain scission caused by the oxidative decomposition of the DPT units70 induced by heat and/or mechanical shear forces71 during the kneading process. Furthermore, in the GPC profiles monitored using a UV detector, the measured peak intensities of the DPT–SBR samples increased with increasing loading amount of DPT, while the peak intensities measured using an RI detector decreased slightly compared to the unmodified SBR. The increased UV absorbance intensity in the GPC measurements is consistent with the strong UV absorbance of the DPT unit (Fig. S6†), which suggests that the degree of DPT incorporation into the SBR is increased by increasing the amount of DPT used. DSC measurements were performed to determine the glass-transition temperature (Tg) of the samples, and the obtained Tg values of the unmodified SBR, DPT–SBR1, DPT–SBR2 and DPT–SBR3 are −68.4, −65.1, −53.5 and −52.0 °C, respectively (Fig. 5b). The increase in Tg with increased DPT loading is probably due to a decrease in polymer-chain mobility72 upon incorporation of the bulky DPT units, indicating the successful modification of the SBR with DPT.55 The DPT-modification of SBR is also supported by FT-IR spectroscopy, in which the distinctive absorbance of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bending vibration55 is observed at 1586 cm−1. The intensity of this peak increases with increasing DPT loading (Fig. 5c) and almost identical absorption peaks were observed for the original DPT-CO (Fig. S7†).
N bending vibration55 is observed at 1586 cm−1. The intensity of this peak increases with increasing DPT loading (Fig. 5c) and almost identical absorption peaks were observed for the original DPT-CO (Fig. S7†).
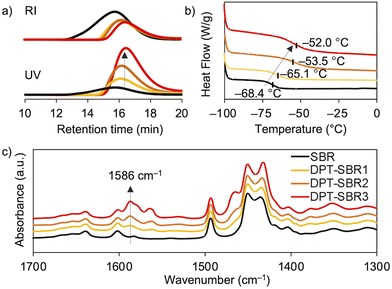 |
| | Fig. 5 Characterization of SBR (black), DPT–SBR1 (yellow), DPT–SBR2 (orange) and DPT–SBR3 (red): (a) GPC curves obtained using an RI detector (top) or UV detector (bottom), (b) DSC curves and (c) ATR FT-IR spectra. | |
ZDMA complexation study
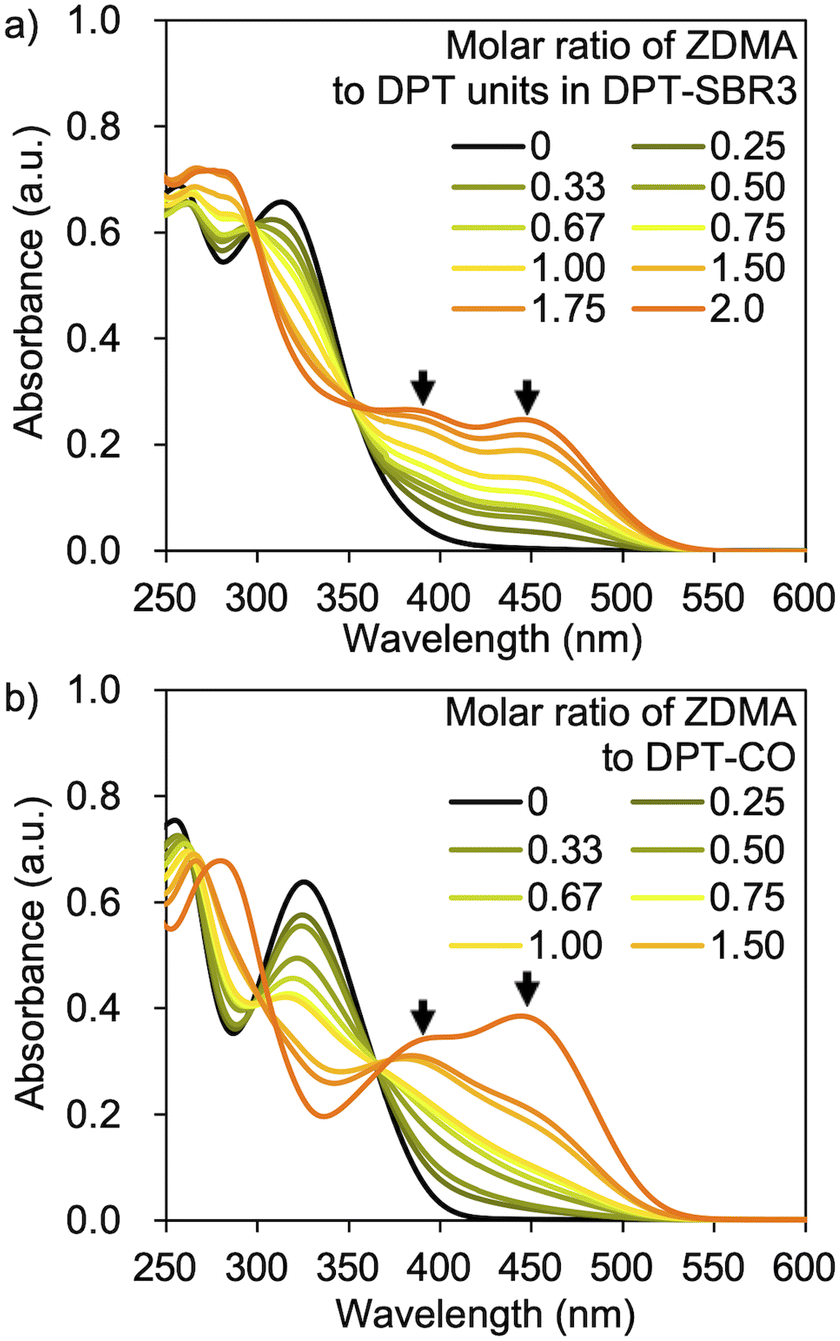
UV-vis measurements were conducted in order to examine the complexation capability of the DPT units in the SBRs with ZDMA, as well as the complexation of DPT-CO with ZDMA. The UV-vis measurements were performed using DPT–SBR3 or DPT-CO in CH2Cl2 with a constant DPT-unit concentration of 6.4 × 10−5 M and a gradually varying molar ratio of ZDMA to DPT units from 0 to 2.0 (Fig. 6). Distinctive absorbances at approximately 380 and 450 nm appeared with increasing ZDMA ratio for both DPT–SBR3 and DPT-CO. The latter absorbance is presumably due to the complexation of one DPT unit to a Zn2+ ion, given that this absorbance appeared strongly in the presence of an excess of ZDMA relative to the DPT unit. On the other hand, the absorbance at approximately 380 nm appeared first, suggesting that it is due to complexation of two or more DPT units to a Zn2+ ion. Furthermore, TOF-MS study using the mixture of DPT-CO and ZDMA was also performed and demonstrated the formation of a complex consisting of a Zn2+ ion coordinated to two DPT-CO ligands (Fig. S8†).
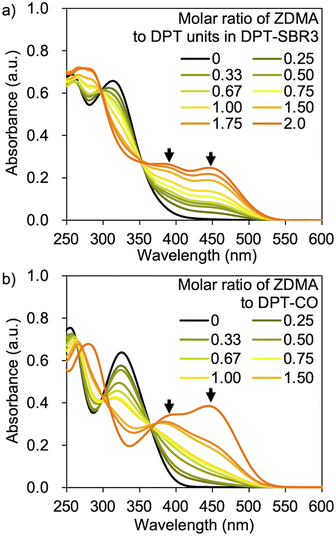 |
| | Fig. 6 UV-vis spectra for (a) DPT–SBR3 with ZDMA and (b) DPT-CO with ZDMA. | |
Preparation of cross-linked rubber samples
In the next step, cross-linked rubber composites were prepared using unmodified SBR or DPT–SBR samples; the formulation of all samples is summarized in Table 2 and Table S2.† To achieve cross-linking in the unmodified SBR as a control sample, 40 phr of ZDMA and 0.2 phr of dicumyl peroxide (DCP) were mixed (run 1). The control samples with varying degrees of cross-linking density were also prepared by mixing the unmodified SBR with 40 phr of ZDMA and 0.05 or 0.4 phr of DCP (runs S1 and S2†). The samples for runs 2, 3 and 4 were prepared using 40 phr of ZDMA with DPT–SBR1, DPT–SBR2 and DPT–SBR3, respectively, in order to clarify the effects of the amount of bound DPT in the SBR on the physical properties. Further samples were synthesized using 30, 50 and 60 phr of ZDMA, in order to gain further insight into the impact of the amount of incorporated ZDMA on the mechanical behaviour (runs 5, 6 and 7). All samples were mixed with an open roll and hot-pressed with an ordinary condition (145 °C; 30 min) into sheets with an approximate thickness of 1 mm. The hot-pressing condition was set to adjust the cross-linking density indicated by the swelling experiment using n-heptane of SBR/Z40/DCP and DPT–SBR2/Z40 within the comparable level (Fig. S9†).
Table 2 Formulation of prepared cross-linked samples. The unit of the values (phr) refers to weight fraction of the specified rubber component per 100 units of the base rubber
| Run |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
| Sample code |
SBR/Z40/DCP |
DPT–SBR1/Z40 |
DPT–SBR2/Z40 |
DPT–SBR3/Z40 |
DPT–SBR3/Z30 |
DPT–SBR3/Z50 |
DPT–SBR3/Z60 |
| SBR |
100 |
|
|
|
|
|
|
| DPT–SBR1 |
|
100 |
|
|
|
|
|
| DPT–SBR2 |
|
|
100 |
|
|
|
|
| DPT–SBR3 |
|
|
|
100 |
100 |
100 |
100 |
| ZDMA |
40 |
40 |
40 |
40 |
30 |
50 |
60 |
| DCP |
0.2 |
|
|
|
|
|
|
Demonstration of DPT/ZDMA cross-linking
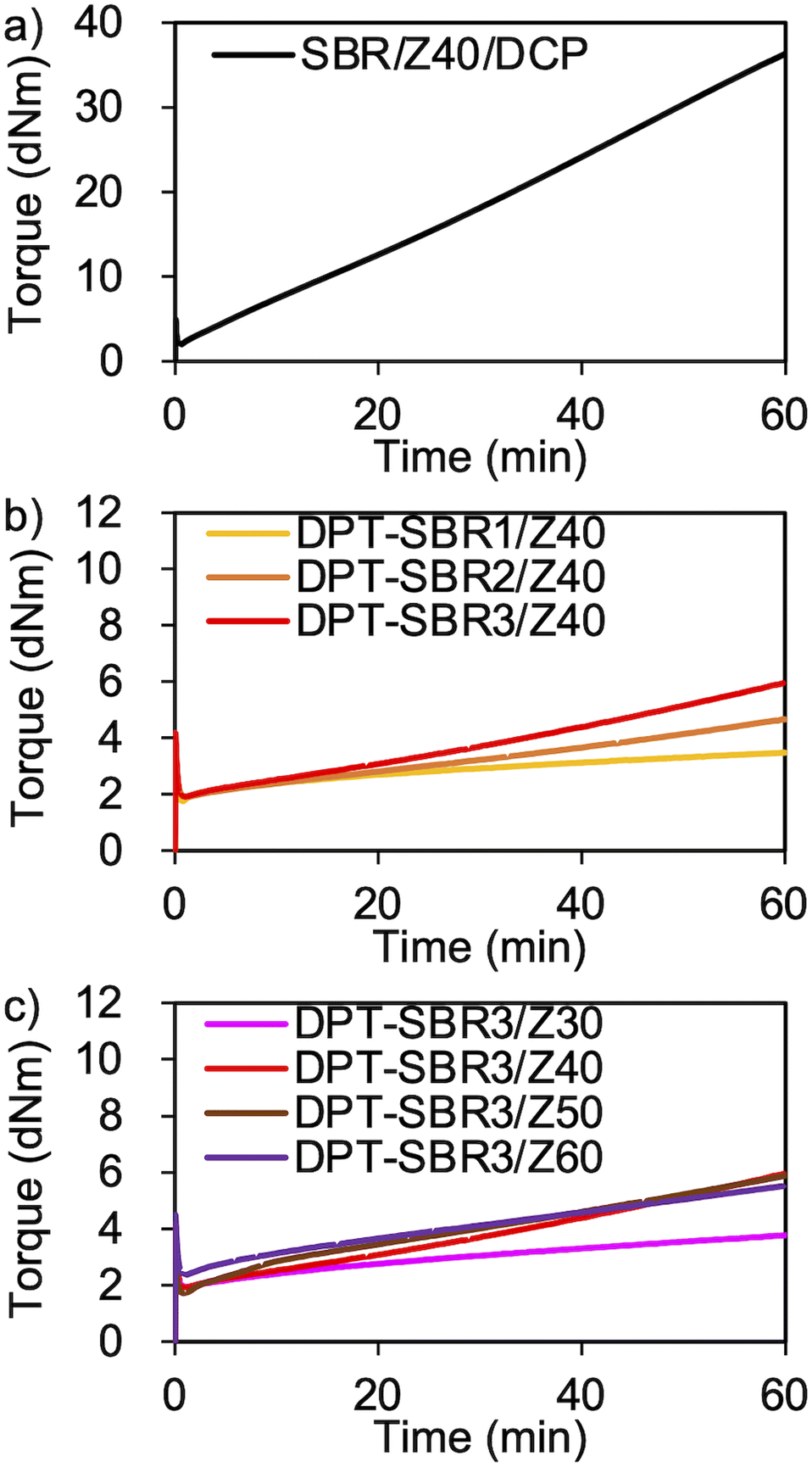
To demonstrate the successful cross-linking of DPT–ZDMA, the elastic torque of the samples was analysed using a rheometer at 145 °C (Fig. 7 and S10†). As shown in Fig. 7a, the control SBR sample using DCP based on a conventional peroxide and ZDMA-based curing system showed a rapid and significant increase in its elastic torque.63 In Fig. 7b, the increase in the elastic torque observed for the DPT–SBR samples loaded with 40 phr of ZDMA indicates the formation of a cross-linking network due to the complexation of DPT with ZDMA. With increasing amount of bound DPT in the SBR samples, the elastic torque increased, suggesting that a higher degree of DPT–ZDMA cross-linking was achieved. The enhanced DPT–ZDMA cross-linking density was confirmed through a swelling experiment with n-heptane, which revealed that the swelling degrees of DPT–SBR1/Z40, DPT–SBR2/Z40 and DPT–SBR3/Z40 are consistent with the order of their torque values (Fig. S11†). In Fig. 7c, the impact of ZDMA loading on the elastic torque is presented using DPT–SBR3. The maximum torque for the sample with a 30 phr ZDMA loading is significantly decreased compared to those of the other DPT–SBR3 composites, indicating a lower degree of DPT–ZDMA complexation. On the other hand, the DPT–SBR3 samples with 50 and 60 phr of ZDMA show slightly higher torque values than DPT–SBR3/Z40 at the initial stage of the test, albeit that the maximum torque values are almost the same for these samples. These results suggest that DPT–SBR3/Z40, DPT–SBR3/Z50 and DPT–SBR3/Z60 has almost the same degree of DPT/ZDMA cross-linking at 145 °C.
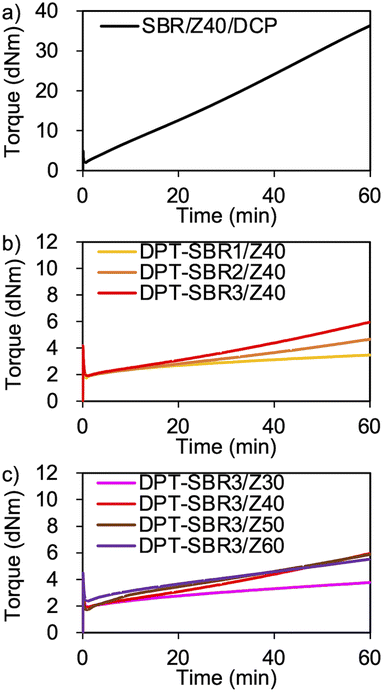 |
| | Fig. 7 Elastic torque curves of rubber composites measured using a moving die rheometer at 145 °C: (a) control sample (SBR/Z40/DCP), (b) effect of the amount of bound DPT in the SBRs and (c) effect of the ZDMA content. | |
To further examine the formation of DPT–ZDMA cross-linkages, dissolution and swelling tests were conducted for all composites using toluene/dichloroacetic acid (v/v = 95/5) referring to a previous study31 (Fig. 8). For that purpose, disc-shaped specimens with a diameter of 8.0 mm (Fig. 8a) were immersed in the solvent (Fig. 8b) and kept at room temperature for 4 days (Fig. 8c). At that point, the solutions of the DPT–ZDMA cross-linked samples turned yellow, indicating that some fraction of the DPT–ZDMA cross-links were dissociated by the acid. Especially DPT–SBR1/Z40 was completely dissolved, while the other DPT–ZDMA cross-linked samples remained partially undissolved, suggesting that the DPT–ZDMA cross-linking density of DPT–SBR1/Z40 is lower than that of the other samples. These samples were further treated at 80 °C for 6 h, which resulted in complete dissolution of all DPT–ZDMA cross-linked samples (Fig. 8d). Interestingly, SBR/Z40/DCP did not show dissolution behaviour, but exhibited swelling instead. These results demonstrate that the SBR/Z40/DCP contains direct C–C covalent cross-links among the SBR main chains, while the DPT–SBRs with ZDMA composites without DCP are formed mainly by DPT–ZDMA coordination cross-linking.
 |
| | Fig. 8 Photographs documenting the dissolution and swelling behaviour of the cured rubber composites in toluene/dichloroacetic acid (v/v = 95/5): (a) prepared specimens, (b) beginning of the test, (c) after soaking at room temperature for 4 days and (d) after further treatment at 80 °C for 6 h. | |
DMA measurements
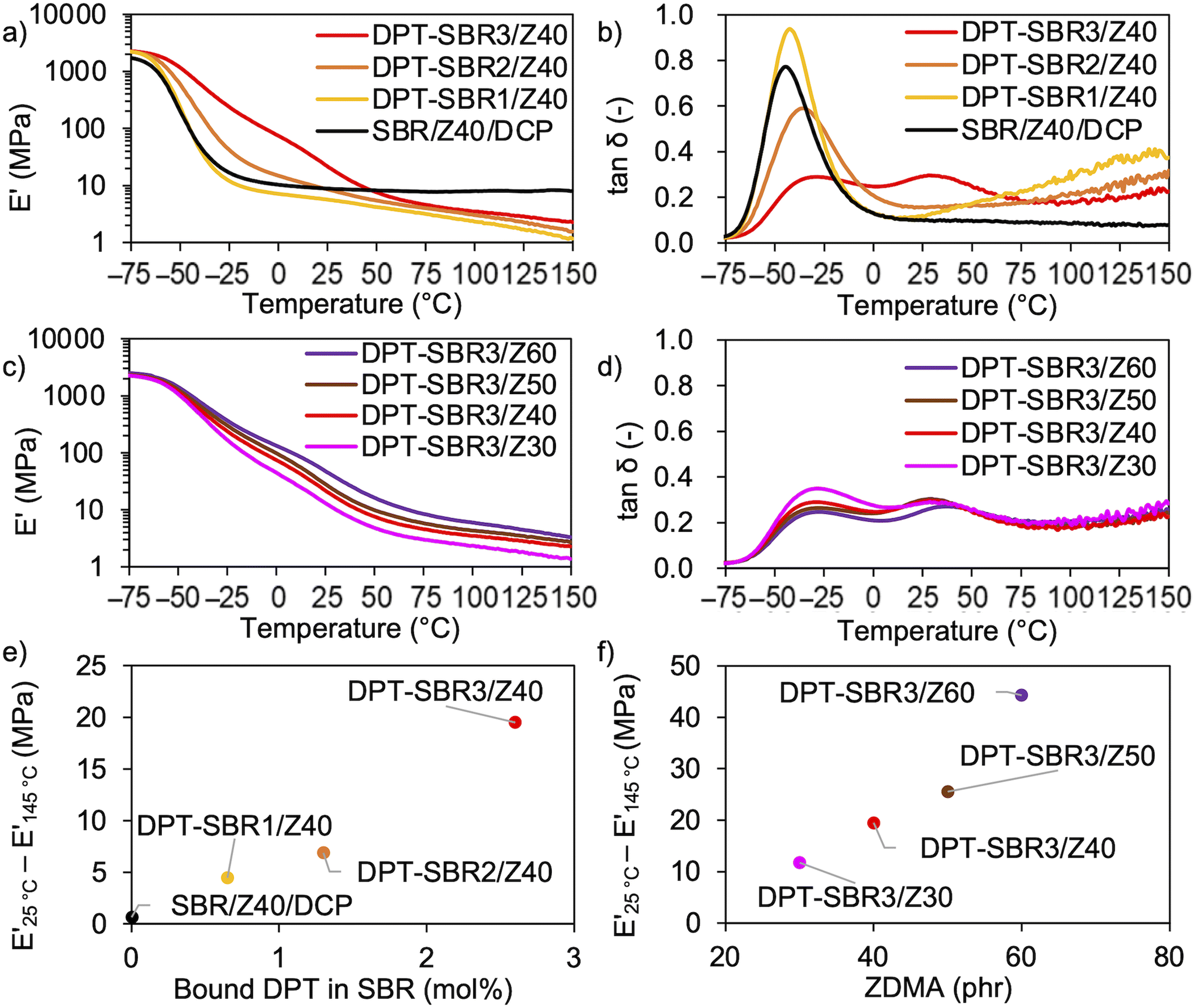
To investigate the temperature dependence of the prepared samples, temperature-sweep DMA tests were carried out at temperatures ranging from −75 to 150 °C (Fig. 9). In Fig. 9a and b, the effects of the amount of bound DPT in the SBRs on E′ and tanδ are described. The results show that decrease in E′ induced upon raising the testing temperature becomes slower with increasing the amount of bound DPT in the SBRs. Furthermore, the peaks of the tanδ curves due to the glass transition (Fig. S12†) at approximately −40 °C are broader for DPT–SBR2/Z40 and DPT–SBR3/Z40. These results suggest that ZDMA acts as a reinforcing filler and forms bound rubber73via coordination linkages. Notably, the tanδ curve of SBR-DPT3/Z40 shows a second transition peak at approximately 30 °C. This peak is most likely due to the dynamic dissociation/reassociation of the DPT/ZDMA coordination cross-linkages and ionic multiplets74 of ZDMA, and possibly including the effect of hydrogen bonding via N–H bonds in DPT units with the pyridine groups or the ester groups in ZDMA. The same type of ionic transition peak has been observed in a previous study.27 In Fig. 9c and d, the impact of the amount of ZDMA on E′ and tanδ is described. Upon increasing the ZDMA content from 30 to 60 phr, the shoulders of the E′ curves over 0 °C become larger and the peaks in the tanδ curves due to the glass transition become broader, strongly supporting the idea that ZDMA acts not only as a coordination cross-linking point, but also as a reinforcing filler,73,75via the bound DPT in the SBRs. According to the above discussion, the gap between E′ at high and low temperatures seems to reflect the degree of coordination cross-linking, given that the slower decrease in E′ observed for the DPT–SBR and ZDMA composites with increasing temperature is most likely due to the dissociation of DPT–ZDMA coordination cross-links. In other words, the difference between the values of E′ at high and low temperatures is expected to be a predictor of the degree of DPT–ZDMA cross-linking. As expected, the difference between the values of E′ at 25 °C and 145 °C, denoted as E′25 °C − E′145 °C, shows a good correlation with the degree of bound DPT in the SBRs and the ZDMA loading (Fig. 9e and f). Based on these results, we consider the difference in the value of E′ to be a useful tool to interpret the hot-pressed adhesion behaviour, given that the interdiffusion and interlinking behaviour prompted by the dissociation and reassociation of cross-linking are important for hot-pressing adhesion, as mentioned in the introduction.
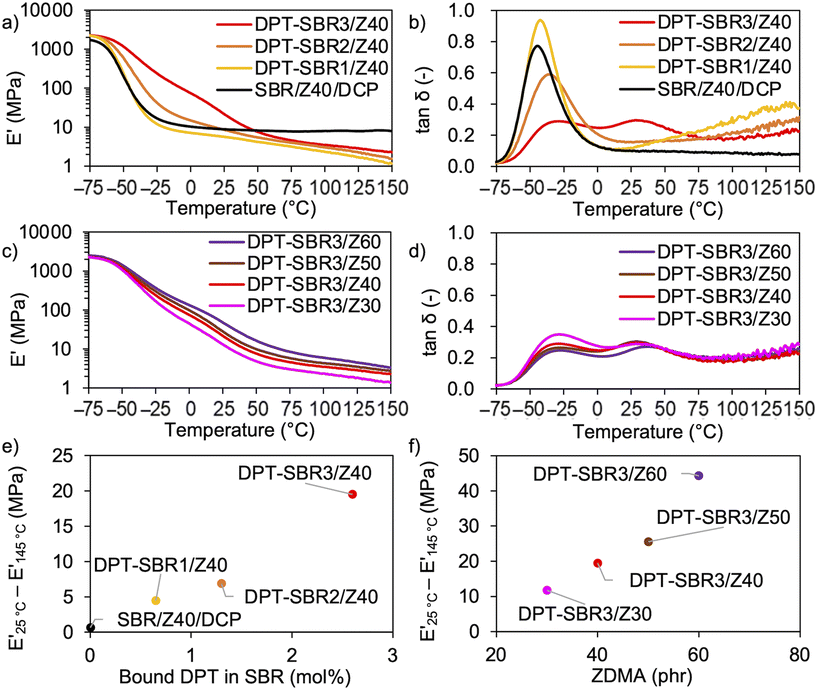 |
| | Fig. 9 Temperature dependence of the viscoelasticity: (a) effect of the amount of bound DPT in the SBR on E′, (b) effect of the amount of bound DPT in the SBR on tanδ, (c) effect of ZDMA content on E′, (d) effect of the loaded ZDMA content on tanδ, (e) effect of the amount of bound DPT in the SBR on the difference between E′ at 25 °C and E′ at 145 °C and (f) effect of ZDMA content on the difference between E′ at 25 °C and E′ at 145 °C. | |
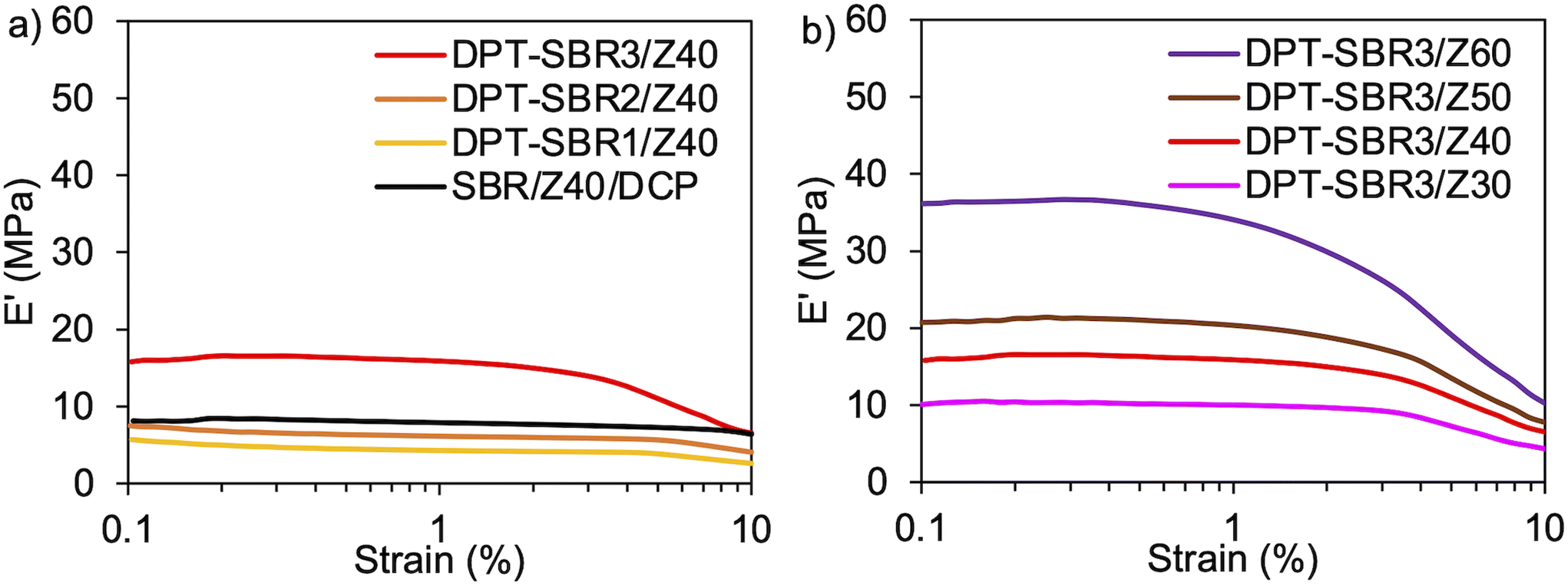
To further investigate the impact of DPT/ZDMA cross-linking on the dynamic mechanical behaviour, strain-sweep DMA tests were conducted at strains ranging from 0.1 to 10% (Fig. 10). In Fig. 10a, the impact of the amount of bound DPT in the SBRs on the strain dependence of E′ are depicted. The control, SBR/Z40/DCP, shows almost no decrease in E′ (Payne effect)76 with increasing strain, indicating that a significant network of ZDMA particles is not formed. On the other hand, DPT–SBR1/Z40 and DPT–SBR2/Z40 show a slight decrease in E′ at strains ranging from 5 to 10%, and importantly, DPT–SBR3/Z40 reveals an obvious Payne effect, demonstrating that the bound DPT in the SBRs enhanced the ZDMA–ZDMA filler interaction66,67via DPT–ZDMA linkages. Fig. 10b depicts the effects of the ZDMA content on the strain dependence of E′; the Payne effect is significantly amplified with increasing ZDMA content, demonstrating that strong ZDMA–ZDMA filler interactions are formed.65 Based on these results, it seems obvious that ZDMA plays important roles as both a cross-linking point and a reinforcing filler in the DPT–ZDMA cross-linking system.
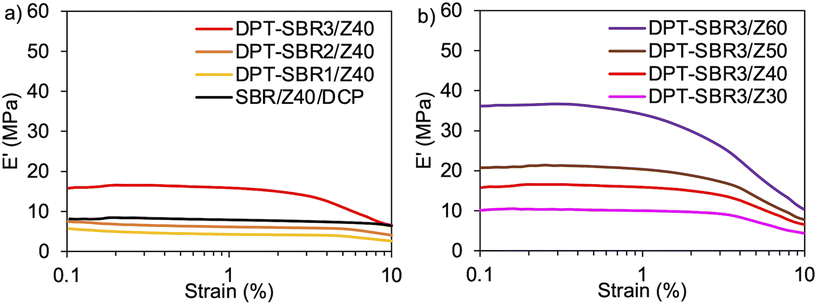 |
| | Fig. 10 Strain dependence of E′: (a) effect of the amount of bound DPT in the SBR on E′ and (b) effect of loaded ZDMA content on E′. | |
Tensile-test and hysteresis-test results
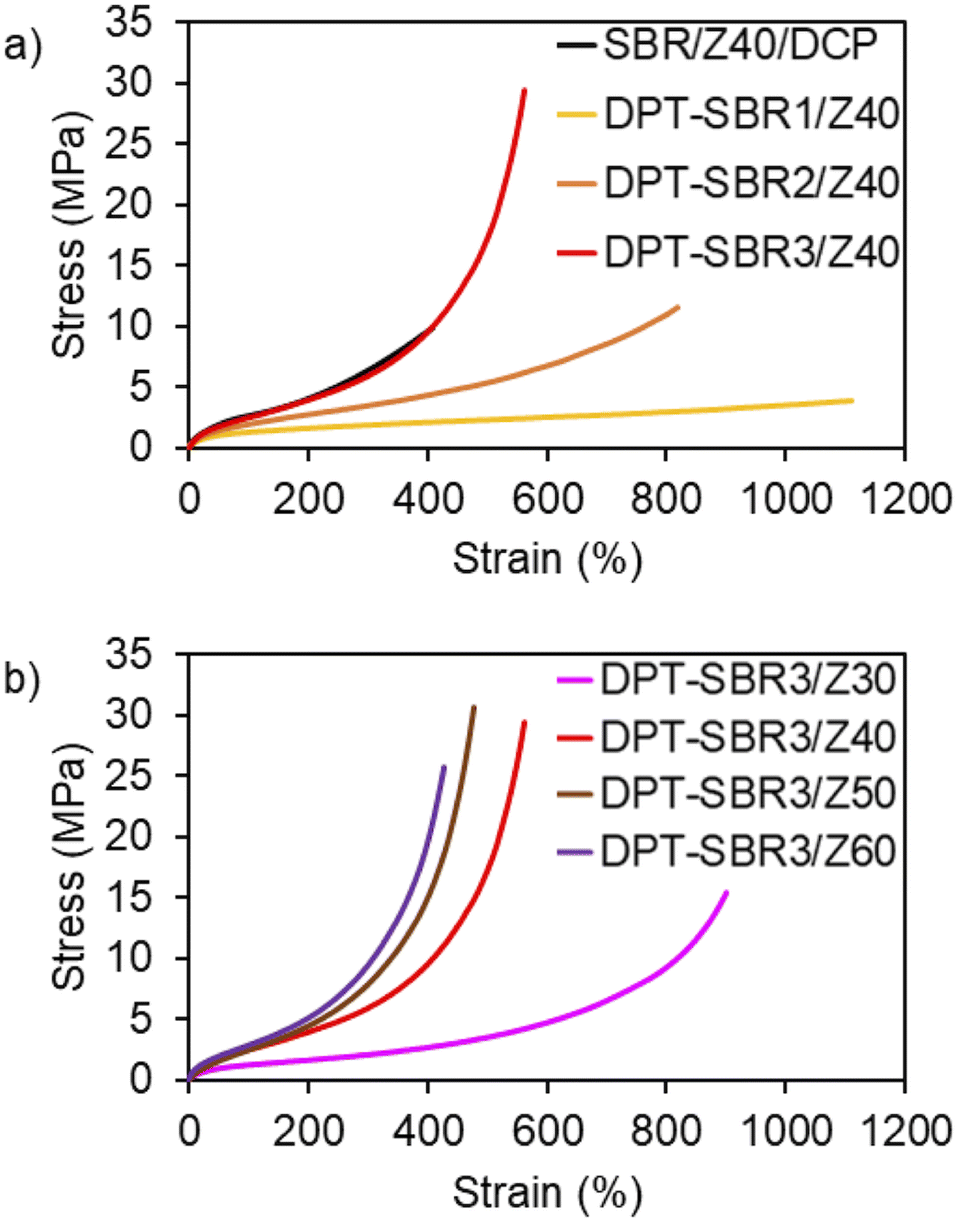
To investigate the impact of the DPT/ZDMA linkages on the mechanical strength, tensile tests were conducted, and engineering stress–strain curves were obtained (Fig. 11 and S13†); the mechanical properties are summarized in Table 3 and Table S3.† In Fig. 11a, the effect of the amount of bound DPT in the SBRs on the stress–strain curve is depicted. While the control SBR/Z40/DCP shows a relatively low elongation at break (Eb) of 365%, the DPT–SBR composites with 40 phr of ZDMA, DPT–SBR1/Z40, DPT–SBR2/Z40 and DPT–SBR3/Z40, give higher Eb values of 1048, 746 and 512%, respectively. Furthermore, the tensile strength at break (Tb) and the fracture energy of the DPT–SBR composites increases significantly with increasing amount of bound DPT in the SBRs, providing Tb values of 3.99, 9.50 and 30.5 MPa for DPT–SBR1/Z40, DPT–SBR2/Z40 and DPT–SBR3/Z40, respectively. The values of fracture energy of each DPT–SBR composite are higher than the control samples with varying cross-linking density indicated by swelling rate (Fig. S14†). Notably, the Tb value of DPT–SBR3/Z40 is higher than previous reported studies using SBRs with ligand–metal coordination systems.52,55 The dramatically improved Tb of DPT–SBR3/Z40 is appears to be derived from the strong reinforcement by ZDMA, which is consistent with the results of the DMA measurements. This interpretation is supported by the well-known fact that composites of ZDMA with nitrile–butadiene rubber (NBR) or hydrogenated nitrile–butadiene rubber (HNBR) show high Tb values due to strong reinforcement by ZDMA via coordination of the nitrile groups.63,67 In Fig. 11b, the impact of the level of ZDMA loading on the stress–strain curve is summarized; Tb values of 14.5, 30.5, 29.1 and 25.6 MPa were found for DPT–SBR3/Z30, DPT–SBR3/Z40, DPT–SBR3/Z50 and DPT–SBR3/Z60, respectively, indicating that 40–50 phr is the optimum ZDMA content in terms of tensile strength in this study. The decrease of the Tb of DPT–SBR3/Z30 in comparison with that of DPT–SBR3/Z40 is drastic, which suggests that 30 phr of ZDMA is insufficient to form a ZDMA-reinforced network via the bound DPT in the SBR. In contrast, DPT–SBR3/Z50 and DPT–SBR3/Z60 show higher moduli than DPT–SBR3/Z40 over a wide strain range, while the Tb values of DPT–SBR3/Z60 decreased. The increased moduli indicate an increased cross-linking density due to more efficient DPT–ZDMA coordination in addition to the conventional filler-induced hydrodynamic effects and strain-amplification effects.77 The decreased values of Tb for DPT–SBR3/Z60 suggest that excessive loading of ZDMA probably increases the degree of initial flaws.78
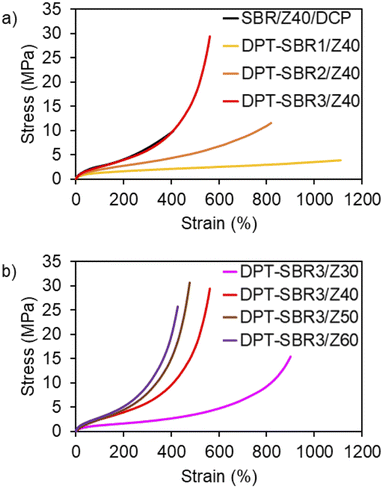 |
| | Fig. 11 Engineering stress–strain curves: (a) effect of the amount of bound DPT in the SBRs and (b) effect of loaded ZDMA content. | |
Table 3 Moduli and fracture parameters of the cross-linked samples shown in Fig. 12. Values in parentheses refer to the standard deviation
| Run/sample code |
Tensile stress at 100% elongation (MPa) |
Tensile stress at 300% elongation (MPa) |
Elongation at break (Eb) (%) |
Tensile strength at break (Tb) (MPa) |
Fracture energy (MJ m−3) |
| 1/SBR/Z40/DCP |
2.64 (0.03) |
6.44 (0.32) |
365 (32) |
8.50 (0.98) |
14.7 (2.91) |
| 2/DPT–SBR1/Z40 |
1.28 (0.02) |
1.89 (0.04) |
1048 (46) |
3.99 (0.09) |
25.2 (0.95) |
| 3/DPT–SBR2/Z40 |
1.91 (0.01) |
3.43 (0.04) |
746 (75) |
9.50 (1.60) |
33.4 (6.01) |
| 4/DPT–SBR3/Z40 |
2.57 (0.09) |
5.83 (0.09) |
512 (11) |
30.5 (3.01) |
46.3 (2.38) |
| 5/DPT–SBR3/Z30 |
1.25 (0.01) |
2.09 (0.03) |
893 (4.9) |
14.5 (1.31) |
36.0 (1.61) |
| 6/DPT–SBR3/Z50 |
2.62 (0.17) |
8.65 (0.97) |
455 (24) |
29.1 (1.13) |
36.0 (2.20) |
| 7/DPT–SBR3/Z60 |
2.80 (0.07) |
10.16 (0.81) |
408 (20) |
25.6 (0.30) |
30.1 (2.26) |
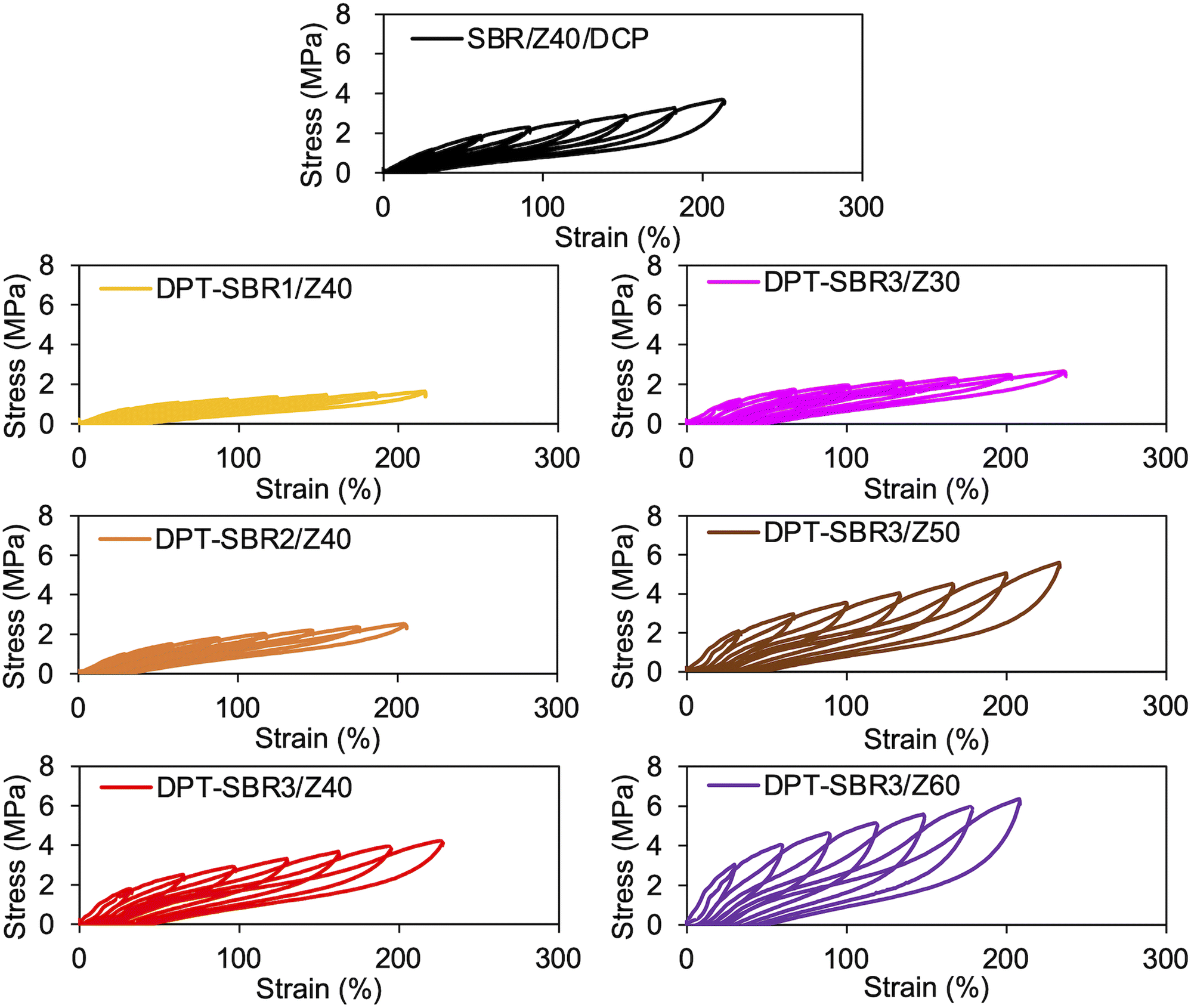
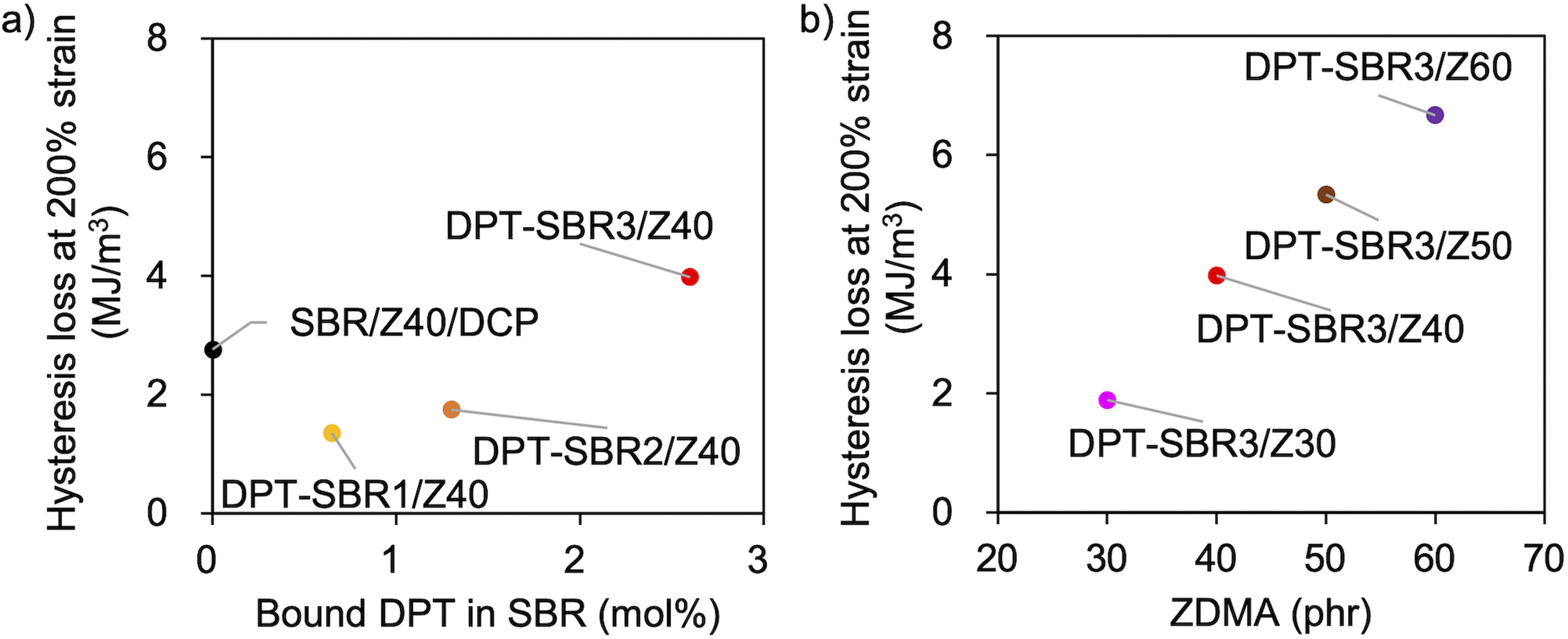
To further investigate the mechanical strength of the DPT–SBR and ZDMA composites, the strain dependence of the hysteresis loss was determined using hysteresis tests (Fig. 12 and S15†). The effect of the amount of bound DPT in the SBRs and the ZDMA content on the hysteresis loss at 200% as a representative value of hysteresis loss at large strain is summarized in Fig. 13. Fig. 13a shows that the hysteresis loss at 200% strain increases significantly with increasing amount of bound DPT in the SBRs, showing the same trend as the relationship between the amount of bound DPT in the SBRs and the Tb values. These results suggest that DPT–ZDMA cross-linking and ZDMA–ZDMA ionic multiplets74 are dissociated at large strain, accompanied by high energy dissipation, resulting in high Tb values with increasing amount of bound DPT in the SBRs. Notably, DPT–SBR3/Z40 shows comparable hysteresis loss with SBR/Z40/DCP, but the Tb value of the former is much higher than the latter, suggesting that the inherent flaw size79 in the former composite is smaller probably due to the strong reinforcement with ZDMA via coordination bonds with DPT units. Fig. 13b presents the impact of the ZDMA content on the hysteresis loss at 200% strain; these values are significantly improved by increasing the ZDMA content. This trend is in line with previous results67 for NBR and ZDMA composites.
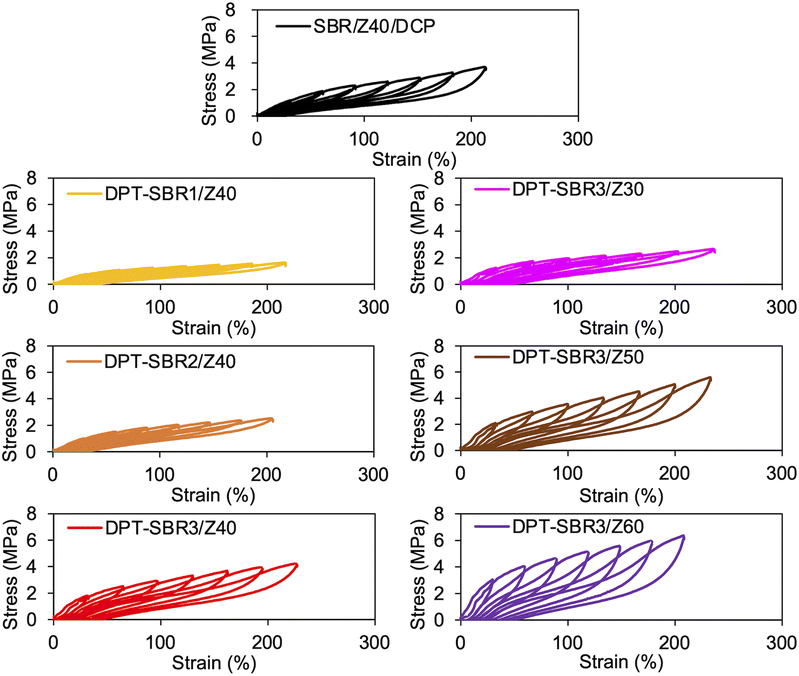 |
| | Fig. 12 Hysteresis test results of SBR composites. | |
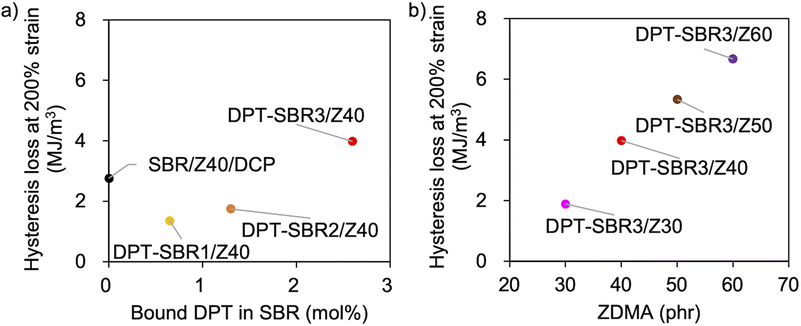 |
| | Fig. 13 Hysteresis loss at 200% strain: (a) effect of the amount of bound DPT in the SBR and (b) effect of the amount of loaded ZDMA. | |
Adhesion performance
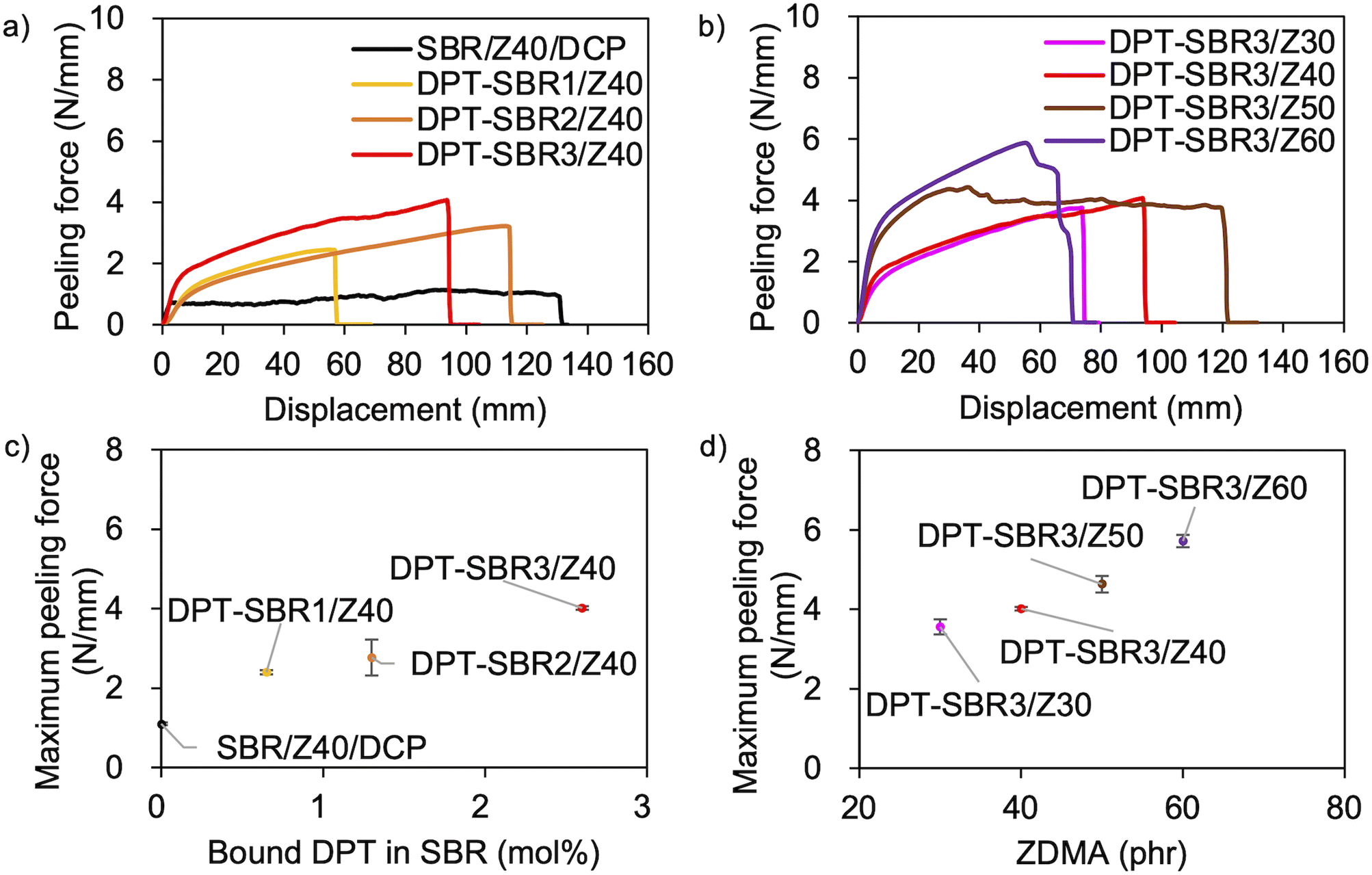
The above discussion demonstrates that the DPT–ZDMA cross-links obviously act as reversible bonds, and accordingly, malleability can be expected at high temperatures. Thus, we speculated that the direct adhesion of DPT–ZDMA cross-linked SBRs could be achieved using a hot press. To confirm such a direct adhesion capability, two cross-linked sheets of each sample were stacked and remoulded at 145 °C under 15 MPa for 30 min, before T-peel tests were conducted using the prepared adhered samples (Fig. 14 and S16†). The condition of the adhesion sample preparation was determined by several experimental results with varying temperature and time, and the condition providing the highest adhesion performance was selected (Fig. S17†). Fig. 14a shows the effect of the amount of bound DPT in the SBRs on the peeling force; the composites of DPT–SBR with ZDMA clearly show higher peeling forces than the control sample cured with DCP. Fig. 14b depicts the impact of the ZDMA content on the peeling force; the maximum peeling force values gradually increase with increasing amount of ZDMA. The relationships between the resulting maximum peeling force and the amount of bound DPT in the SBR as well as the ZDMA content are summarized in Fig. 14c and d, respectively. These results demonstrate that the maximum peeling force is dramatically increased with increasing both the amount of bound DPT units in the SBRs and the loaded ZDMA content. Furthermore, the values of maximum peeling force of each DPT–SBR composite are higher than the control samples with varying cross-linking density indicated by swelling rate (Fig. S18†).
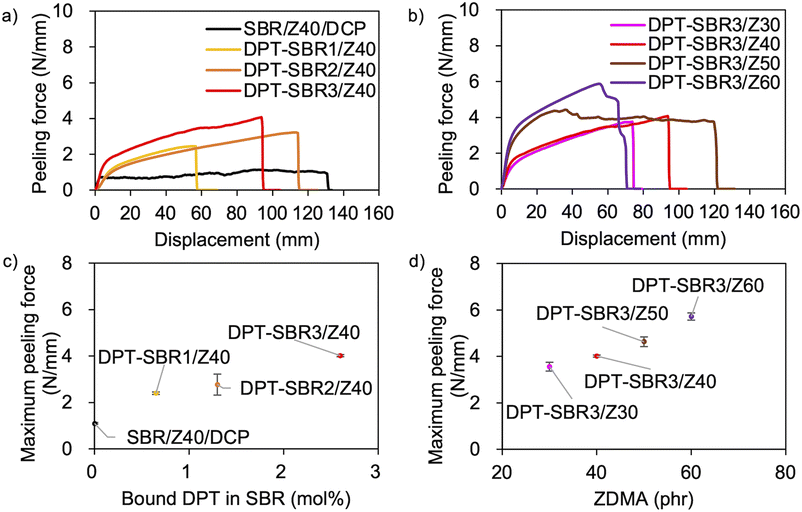 |
| | Fig. 14 T-peel test results: (a) effect of the amount of bound DPT in the SBRs, (b) effect of the loaded ZDMA content. Relationship between the maximum peeling force and (c) the amount of bound DPT in the polymer or (d) the loaded ZDMA content. | |
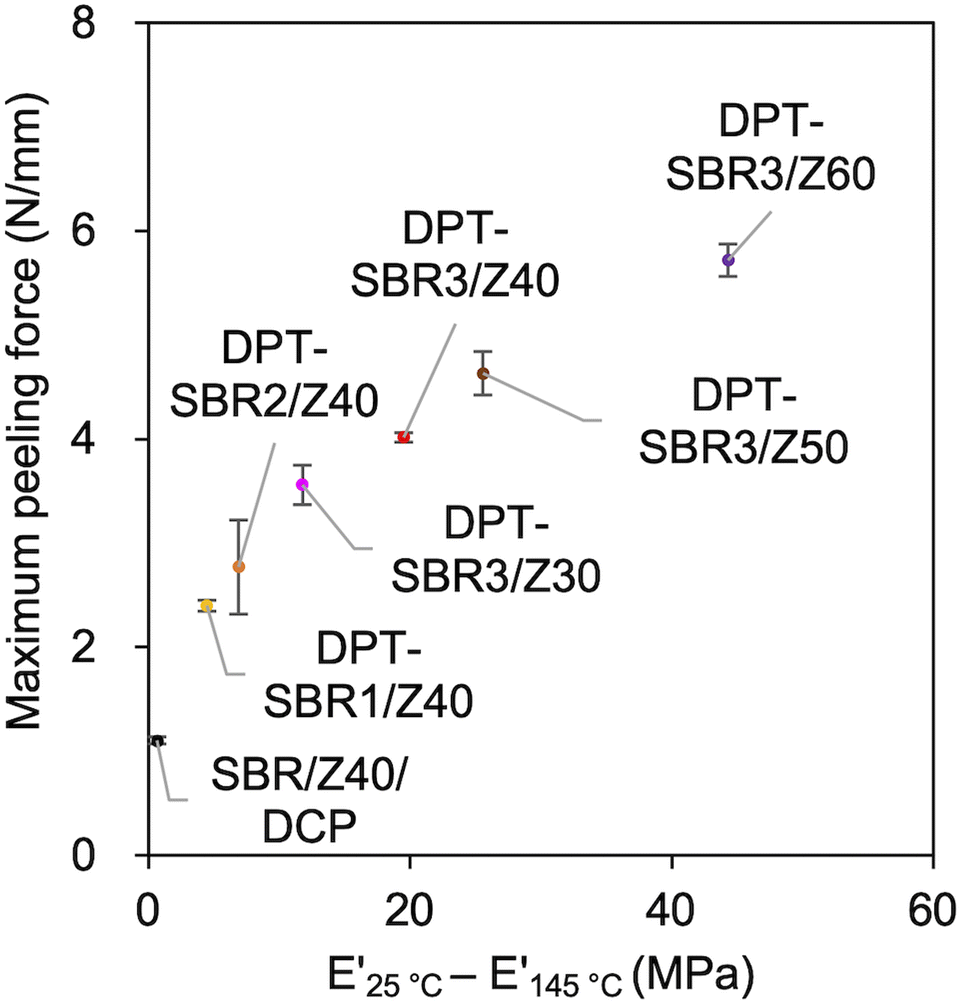
In this adhesion process, the degree of DPT–ZDMA cross-linking seems to be one of the most important factors for controlling the adhesion capability. Based on this assumption, the relationship between the maximum peeling force and the predictor of the degree of DPT–ZDMA cross-linking (E′25 °C − E′145 °C) mentioned in the discussion of the temperature sweep DMA results (vide supra) was investigated (Fig. 15). The value of the maximum peeling force was found to improve with increasing DPT–ZDMA cross-linking predictor value (E′25 °C − E′145 °C), demonstrating that the direct adhesion strength of the DPT–ZDMA cross-linking system can be explained by the degree of DPT–ZDMA cross-linking.
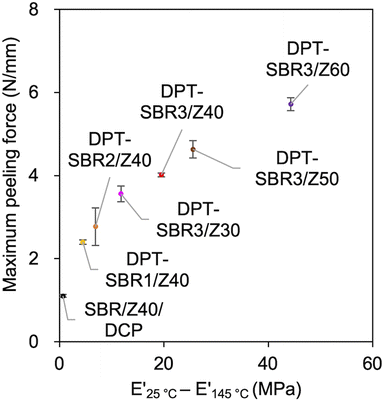 |
| | Fig. 15 Relationship between the maximum peeling force and the difference between E′ at 25 °C and E′ at 145 °C as a predictor of the degree of DPT/ZDMA coordination cross-linking. | |
Conclusions
To achieve direct adhesion between cross-linked rubbers without any pretreatment of the rubber surfaces or the use of adhesives, we designed novel modified SBR composites that contain zinc dimethacrylate (ZDMA)–nitrogen coordination linkages. These composites were prepared via tetrazine click reactions with 3,6-di(2-pyridyl)-1,2,4,5-tetrazine (DPT) during the rubber-kneading process, followed by the addition of ZDMA and a thermal curing process. The DPT modification was confirmed via GPC and DSC as well as FT-IR and 1H NMR spectroscopy. The complexation capability of the DPT unit with ZDMA was investigated in detail by conducting a study using a model small molecule (DPT-CO). The UV-vis spectra revealed that both DPT-CO and the DPT units in the SBR showed almost the same absorbance wavelength due to complexation with ZDMA, indicating that similar types of complexation occurred.
In the compounding study, the formation of DPT–ZDMA cross-linking was confirmed by the increase in the elastic torque monitored using a rheometer and the dissolution behaviour in toluene/dichloroacetic acid. The resulting DPT–ZDMA cross-linked SBRs showed a unique decrease in E′ and increase in tanδ with increasing temperature in temperature-sweep DMA measurements, most likely due to dynamic dissociation/reassociation of DPT–ZDMA linkages, and the parameter E′25 °C − E′145 °C was proposed as a predictor of the degree of DPT–ZDMA cross-linking. It was clarified that this predictor value can be controlled by the amount of bound DPT in the SBR and the ZDMA loading amount. Strain-sweep DMA measurements were also carried out, and a typical Payne effect was observed for DPT–ZDMA cross-linked samples, demonstrating that ZDMA plays the role of both cross-linking point and reinforcing filler. Notably, DPT–ZDMA cross-linking also led to unique stress–strain behaviour. Hysteresis tests revealed improved hysteresis loss with a larger amount of bound DPT in the samples of SBR with ZDMA, resulting in a much higher Tb than that of the conventional peroxide-cured sample. Finally, direct adhesion between cross-linked rubbers was demonstrated, and DPT–ZDMA cross-linked samples showed improved adhesion strength, consistent with the increase in the predictor of the degree of DPT–ZDMA cross-linking (E′25 °C − E′145 °C). In conclusion, we have successfully established a novel system for direct adhesion between cross-linked rubbers. We are convinced that this technology not only represents a new approach to rubber bonding, but can also be expected to contribute to the improvement of rubber repair processes, which would lead to a reduction in CO2 emissions and rubber waste.
Author contributions
K. K., K. T. and H. O. conceived the concept and designed the experiments. K. K. performed the experiments and analysed the data. K. K., K. T. and H. O. wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JST-Mirai Program Grant Number JPMJMI18A2, Japan.
References
-
D. K. Kotnee and A. K. Bhowmick, Rubber to Rubber Adhesion, Scrivener Publishing LLC, Beverly, 2021 Search PubMed.
- R.-J. Chang and A. N. Gent, J. Polym. Sci., Polym. Phys. Ed., 1981, 19, 1619–1633 CrossRef CAS.
- A. K. Bhowmick and A. N. Gent, Rubber Chem. Technol., 1984, 57, 216–226 CrossRef CAS.
- H. Chun and A. N. Gent, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2223–2229 CrossRef CAS.
- F. Ruch, M. O. David and M. F. Vallat, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 3189–3199 CrossRef CAS.
- D. Oldfield and T. E. F. Symes, J. Adhes., 1983, 16, 77–96 CrossRef CAS.
- J. M. Martín-Martínez, J. C. Fernández-García, F. Huerta and A. C. Orgilés-Barceló, Rubber Chem. Technol., 1991, 64, 510–521 CrossRef.
- M. S. Sánchez-Adsuar, M. M. Pastor-Blas, R. Torregrosa-Maciá and J. M. Martín-Martínez, Int. J. Adhes. Adhes., 1994, 14, 193–200 CrossRef.
- M. D. Romero-Sánchez, M. M. Pastor-Blas and J. M. Martín-Martínez, Int. J. Adhes. Adhes., 2001, 21, 325–337 CrossRef.
- M. M. Pastor-Blas, J. M. Martín-Martínez and J. G. Dillard, Surf. Interface Anal., 1998, 26, 385–399 CrossRef CAS.
- H. Hirahara, K. Mori and Y. Oishi, J. Adhes. Sci. Technol., 1997, 11, 1459–1474 CrossRef CAS.
- J. Tyczkowski, I. Krawczyk and B. Wożniak, Surf. Coat. Technol., 2003, 174–175, 849–853 CrossRef CAS.
- T. F. Petrova, A. A. Shcherbina and A. E. Chalykh, Polym. Sci., Ser. C, 2007, 49, 89–94 CrossRef.
- S. Nakane, Nippon Gomu Kyokaishi, 2012, 85, 204–207 CrossRef CAS.
- Indriasari, J. Noordermeer and W. Dierkes, Appl. Sci., 2021, 11, 9834 CrossRef CAS.
- G. Ferrer, Resour., Conserv. Recycl., 1997, 19, 221–255 CrossRef.
- D. Dobrotă, G. Dobrotă and T. Dobrescu, J. Cleaner Prod., 2020, 260, 121141 CrossRef.
- J. Gaidhane, I. Ullah and A. Khalatkar, Mater. Today: Proc., 2022, 60, 2257–2261 CrossRef.
- P. K. Behera, S. Mohanty and V. K. Gupta, Polym. Chem., 2021, 12, 1598–1621 RSC.
- J. Xu, L. Zhu, Y. Nie, Y. Li, S. Wei, X. Chen, W. Zhao and S. Yan, Materials, 2022, 15, 5993 CrossRef CAS PubMed.
- B. Li, P.-F. Cao, T. Saito and A. P. Sokolov, Chem. Rev., 2023, 123, 701–735 CrossRef CAS.
- M. Das, A. R. Parathodika, P. Maji and K. Naskar, Eur. Polym. J., 2023, 186, 111844 CrossRef CAS.
- K. Chino and M. Ashiura, Macromolecules, 2001, 34, 9201–9204 CrossRef CAS.
- C.-C. Peng and V. A. Abetz, Macromolecules, 2005, 38, 5575–5580 CrossRef CAS.
- J. Liu, S. Wang, Z. Tang, J. Huang, B. Guo and G. Huang, Macromolecules, 2016, 49, 8593–8604 CrossRef CAS.
- Y. Shoda, D. Aoki, K. Tsunoda and H. Otsuka, Polymer, 2020, 202, 122700 CrossRef CAS.
- I. Mora-Barrantes, M. A. Malmierca, J. L. Valentin, A. Rodriguez and L. Ibarra, Soft Matter, 2012, 8, 5201–5213 RSC.
- A. Das, A. Sallat, F. Böhme, M. Suckow, D. Basu, S. Wießner, K. W. Stöckelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS PubMed.
- C. Xu, L. Cao, X. Huang, Y. Chen, B. Lin and L. Fu, ACS Appl. Mater. Interfaces, 2017, 9, 29363–29373 CrossRef CAS PubMed.
- Y. Miwa, J. Kurachi, Y. Kohbara and S. Kutsumizu, Commun. Chem., 2018, 1, 5 CrossRef.
- C. Li, Z. Yuan and L. Ye, Composites, Part A, 2019, 126, 105580 CrossRef CAS.
- J. Liu, C. Xiao, J. Tang, Y. Liu and J. Hua, Ind. Eng. Chem. Res., 2020, 59, 12755–12765 CrossRef CAS.
- A. M. Wemyss, A. Marathianos, E. L. Heeley, J. Ekeocha, Y. Morishita, R. di Ronza, M. M. Bernal, D. M. Haddleton and C. Wan, ACS Appl. Polym. Mater., 2022, 4, 7868–7877 CrossRef CAS.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef CAS PubMed.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2020, 56, 4381–4395 RSC.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- N. V. Herck and F. E. Du Prez, Macromolecules, 2018, 51, 3405–3414 CrossRef.
- P. Tanasi, M. Hernández Santana, J. Carretero-González, R. Verdejo and M. A. López-Manchado, Polymer, 2019, 175, 15–24 CrossRef CAS.
- A. L. Dobson, N. J. Bongiardina and C. N. Bowman, ACS Appl. Polym. Mater., 2020, 2, 1053–1060 CrossRef CAS PubMed.
- B. T. Michal, E. J. Spencer and S. J. Rowan, ACS Appl. Mater. Interfaces, 2016, 8, 11041–11049 CrossRef CAS PubMed.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- A. Takahashi, R. Goseki, K. Ito and H. Otsuka, ACS Macro Lett., 2017, 6, 1280–1284 CrossRef CAS PubMed.
- A. Tsuruoka, A. Takahashi, D. Aoki and H. Otsuka, Angew. Chem., Int. Ed., 2020, 59, 4294–4298 CrossRef CAS PubMed.
- S. Kataoka, A. Tsuruoka, D. Aoki and H. Otsuka, ACS Appl. Polym. Mater., 2021, 3, 888–895 CrossRef CAS.
- L. L. Robinson, E. S. Taddese, J. L. Self, C. M. Bates, J. R. de Alaniz, Z. Geng and C. J. Hawker, Macromolecules, 2022, 55, 9780–9789 CrossRef CAS.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. Guan, J. Am. Chem. Soc., 2014, 136, 16128–16131 CrossRef CAS.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. A. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- X. Zhang, Z. Tang, B. Guo and L. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 32520–32527 CrossRef CAS PubMed.
- Y.-L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y.-C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. Gu, C. Wang, M. He, J. W. Chung and Z. Bao, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS PubMed.
- Z. Tang, J. Huang, B. Guo, L. Zhang and F. Liu, Macromolecules, 2016, 49, 1781–1789 CrossRef CAS.
- M. Das, S. Pal and K. Naskar, eXPRESS Polym. Lett., 2020, 14, 860–880 CrossRef CAS.
- M. Mareliati, L. Tadiello, S. Guerra, L. Giannini, S. Schrettl and C. Weder, Macromolecules, 2022, 55, 5164–5175 CrossRef CAS.
- S. Mandal, F. Simon, S. S. Banerjee, L. B. Tunnicliffe, C. Nakason, C. Das, M. Das, K. Naskar, S. Wiessner, G. Heinrich and A. Das, ACS Appl. Polym. Mater., 2021, 3, 1190–1202 CrossRef CAS.
- Q. Wang, Y. Shi, Q. Li and C. Wu, Eur. Polym. J., 2021, 150, 110415 CrossRef CAS.
- Q. Wang, W. Wang, Q. Li and C. Wu, Ind. Eng. Chem. Res., 2021, 60, 2163–2177 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- K. Kang, J. Park and E. Kim, Proteome Sci., 2016, 15, 15 CrossRef PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- I. A. Barker, D. J. Hall, C. F. Hansell, F. E. Du Prez, R. K. O'Reilly and A. P. Dove, Macromol. Rapid Commun., 2011, 32, 1362–1366 CrossRef CAS PubMed.
- S. Jain, K. Neumann, Y. Zhang, J. Geng and M. Bradley, Macromolecules, 2016, 49, 5438–5443 CrossRef CAS.
- A. Nomura, J. Takano, A. Toyoda and T. Saito, Nippon Gomu Kyokaishi, 1993, 66, 830–838 CrossRef CAS.
- R. Costin, W. Nagel and R. Ekwall, Rubber Chem. Technol., 1991, 64, 152–161 CrossRef CAS.
- Y. Lu, L. Liu, M. Tian, H. Geng and L. Zhang, Eur. Polym. J., 2005, 41, 589–598 CrossRef CAS.
- Y. Chen and C. Xu, Polym. Compos., 2011, 32, 1593–1600 CrossRef CAS.
- Y. Chen, C. Xu and Y. Wang, J. Reinf. Plast. Compos., 2012, 31, 705–716 CrossRef CAS.
- J. Chen, L. Liao, F. Zhang, S. Hu, J. Huang, S. Shuyun, H. Dongning and X. Mingyue, Polym. Eng. Sci., 2023, 63, 1388–1400 CrossRef CAS.
- P. v. R. Schleyer, J. E. Williams and K. R. Blanchard, J. Am. Chem. Soc., 1970, 92, 2377–2386 CrossRef CAS.
- F. Karaki, T. Kiguchi, K. Itoh, N. Sato, K. Konishi and H. Fujii, Tetrahedron, 2021, 97, 132411 CrossRef CAS.
- F. Bueche, J. Appl. Polym. Sci., 1960, 4, 101–106 CrossRef CAS.
- E. A. DiMarzio and J. H. Gibbs, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 1089–1472 CrossRef.
- I. Pliskin and N. Tokita, J. Appl. Polym. Sci., 1972, 16, 473–492 CrossRef CAS.
- A. Eisenberg, B. Hird and R. B. Moore, Macromolecules, 1990, 23, 4098–4107 CrossRef CAS.
- Y. Fukahori, Rubber Chem. Technol., 2007, 80, 701–725 CrossRef CAS.
- A. R. Payne, J. Appl. Polym. Sci., 1965, 9, 2273–2284 CrossRef CAS.
- M. Klüppel and J. Schramm, Macromol. Theory Simul., 2000, 9, 742–754 CrossRef.
-
A. N. Gent, in Science and Technology of Rubber, ed. F. R. Eirich, Academic Press, Inc., New York, 1st edn, 1978, ch. 10, pp. 419–454 Search PubMed.
- G. R. Hamed, Rubber Chem. Technol., 1983, 56, 244–251 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b
*b
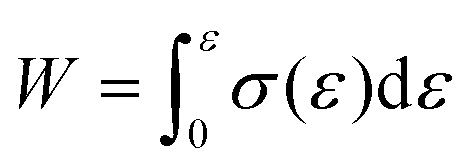 . Hysteresis tests were performed at room temperature using an Instron 5965 (Instron) apparatus equipped with a 5 kN load cell at a crosshead speed of 100 mm min−1, using dumbbell-shaped ISO 37-4 specimens. The hysteresis loss was calculated using the equation, Hysteresis loss = Wstored − Wunload, where
. Hysteresis tests were performed at room temperature using an Instron 5965 (Instron) apparatus equipped with a 5 kN load cell at a crosshead speed of 100 mm min−1, using dumbbell-shaped ISO 37-4 specimens. The hysteresis loss was calculated using the equation, Hysteresis loss = Wstored − Wunload, where 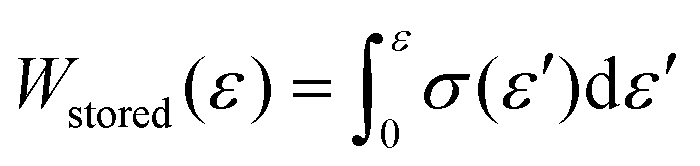 and Wunload were calculated using the stress–strain relationship for the unloading process. T-peel adhesion tests were conducted at room temperature on an Instron 5965 (Instron) instrument equipped with a 5 kN load cell at a crosshead speed of 300 mm min−1 using laminated stripes (length: 75 mm; width: 10 mm). Two measurements were performed for each sample.
and Wunload were calculated using the stress–strain relationship for the unloading process. T-peel adhesion tests were conducted at room temperature on an Instron 5965 (Instron) instrument equipped with a 5 kN load cell at a crosshead speed of 300 mm min−1 using laminated stripes (length: 75 mm; width: 10 mm). Two measurements were performed for each sample.