
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface modifications of COP-based microfluidic devices for improved immobilisation of hydrogel proteins: long-term 3D culture with contractile cell types and ischaemia model†
Sandra
González-Lana
 ac,
Teodora
Randelovic
abd,
Jesús
Ciriza
ab,
María
López-Valdeolivas
e,
Rosa
Monge
c,
Carlos
Sánchez-Somolinos
de and
Ignacio
Ochoa
*abd
ac,
Teodora
Randelovic
abd,
Jesús
Ciriza
ab,
María
López-Valdeolivas
e,
Rosa
Monge
c,
Carlos
Sánchez-Somolinos
de and
Ignacio
Ochoa
*abd
aTissue Microenvironment (TME) Lab. Aragón Institute of Engineering Research (I3A), University of Zaragoza, C/ Mariano Esquillor s/n, 500018 Zaragoza, Spain. E-mail: iochgar@unizar.es
bInstitute for Health Research Aragón (IIS Aragón), Paseo de Isabel La Católica 1-3, 50009 Zaragoza, Spain
cBEONCHIP S.L., CEMINEM, Campus Río Ebro. C/ Mariano Esquillor Gómez s/n, 50018 Zaragoza, Spain
dCIBER in Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Madrid, Spain
eAragón Institute of Nanoscience and Materials (INMA), Department of Condensed Matter Physics (Faculty of Science), CSIC-University of Zaragoza, C/ Pedro Cerbuna 12, 50009 Zaragoza, Spain
First published on 29th March 2023
Abstract
The tissue microenvironment plays a crucial role in tissue homeostasis and disease progression. However, the in vitro simulation has been limited by the lack of adequate biomimetic models in the last decades. Thanks to the advent of microfluidic technology for cell culture applications, these complex microenvironments can be recreated by combining hydrogels, cells and microfluidic devices. Nevertheless, this advance has several limitations. When cultured in three-dimensional (3D) hydrogels inside microfluidic devices, contractile cells may exert forces that eventually collapse the 3D structure. Disrupting the compartmentalisation creates an obstacle to long-term or highly cell-concentrated assays, which are extremely relevant for multiple applications such as fibrosis or ischaemia. Therefore, we tested surface treatments on cyclic-olefin polymer-based microfluidic devices (COP-MD) to promote the immobilisation of collagen as a 3D matrix protein. Thus, we compared three surface treatments in COP devices for culturing human cardiac fibroblasts (HCF) embedded in collagen hydrogels. We determined the immobilisation efficiency of collagen hydrogel by quantifying the hydrogel transversal area within the devices at the studied time points. Altogether, our results indicated that surface modification with polyacrylic acid photografting (PAA-PG) of COP-MD is the most effective treatment to avoid the quick collapse of collagen hydrogels. As a proof-of-concept experiment, and taking advantage of the low-gas permeability properties of COP-MD, we studied the application of PAA-PG pre-treatment to generate a self-induced ischaemia model. Different necrotic core sizes were developed depending on initial HCF density seeding with no noticeable gel collapse. We conclude that PAA-PG allows long-term culture, gradient generation and necrotic core formation of contractile cell types such as myofibroblasts. This novel approach will pave the way for new relevant in vitro co-culture models where fibroblasts play a key role such as wound healing, tumour microenvironment and ischaemia within microfluidic devices.
Introduction
Cell culture has been one of the most used preclinical models for decades. It has evolved from conventional and traditional bi-dimensional to the most advanced three-dimensional (3D) based models. The new models try to resemble, as much as possible, the pathophysiological in vivo cellular behaviour. In vivo, cells embedded or surrounded by a 3D architecture are influenced by different biochemical and mechanical stimuli, depending on the cell type and tissue. Thus, the 3D microenvironment enhances interactions among neighbouring cells and extracellular matrix (ECM), influencing pathophysiological cell motility, proliferation, migration, and morphology.1–3 In the last decades, several 3D culture systems have been developed4–6 trying to mimic the complex tissue microenvironment. However, although spheroids, organoids, scaffolds based on hydrogels and 3D bioprinted-based models provide closer in vivo scenarios than cell monolayers, they do not entirely resemble the microenvironmental characteristics.7–10 They present limitations such as lack of vasculature and mechanical stimulation, imprecise control over gradients, or limited medium exchange at discrete points.11 These disadvantages have been overcome by coupling the aforementioned 3D cultures with microfluidic devices (MD). The compartmentalisation of chambers and channels in a micrometric scale allows spatial control, resembling the in vivo microenvironmental cell distribution more closely.12,13 Moreover, microfluidic devices allow perfusing medium adjacent to or through a cell culture with a stable nutrient and oxygen supply, waste removal and generating chemical gradients. They also allow exerting mechanical forces such as shear stress and interstitial pressure, affecting the cellular morphology and gene expression.14–16 Optical monitoring in microfluidic devices also facilitates easy measurements due to the microfluidic channels defined heights that lead to a controlled focal distance between the sample and the optical objective. Matrices or scaffolds to support 3D cell cultures inside microfluidic device chambers should mimic the spatial complexity and mechanical properties of in vivo microenvironment. In natural protein matrices, cells can migrate through the fibrillar network by a motion of non-crosslinked collagen. Thus, cells bind to ECM through integrin and focal adhesion points, exerting active contractility and mechanosensing.17,18 The interaction with integrins is transduced to biochemical signals that lead to cell attachment, migration or ECM remodelling. If the hydrogel has insufficient resistance to the cell-applied force, it deforms, modifying its initial geometry that might eventually lead to a collapse (Fig. 1).19–21 This hydrogel collapse can be noticed as an X–Y hydrogel detachment (top view) from the microfluidic device walls (Fig. 1A), or just with a Z axis contraction (Fig. 1C, section view of Fig. 1B). Therefore, the analysis of the three axes is required to monitor the evolution of the gel integrity over time (Fig. 1D, non-contracted gel). Collagen-based hydrogel contraction and remodelling is a natural outcome of cellular expansion and interaction with the matrix.22–24 However, this phenomenon may ruin the hydrogel architecture within the microfluidic device,25 disrupting the compartmentalisation and affecting chemical gradients (ESI† Fig. S1).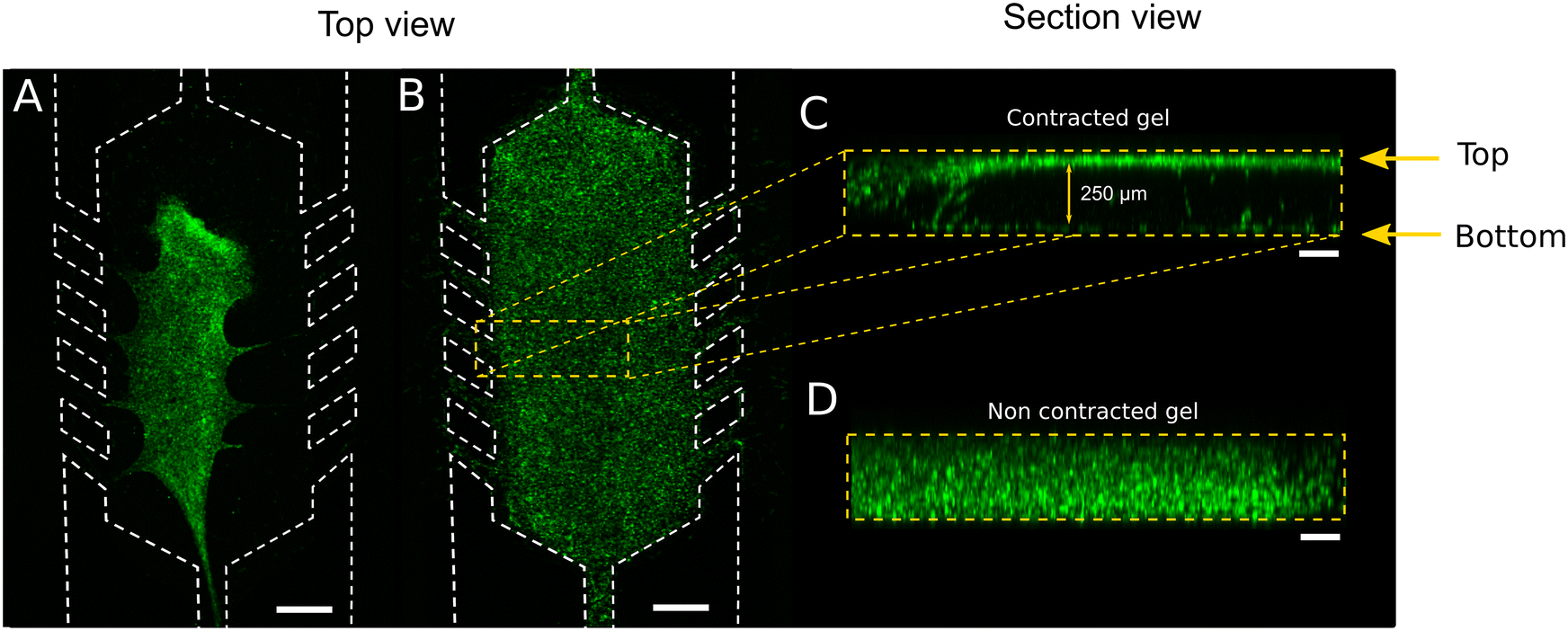 | ||
| Fig. 1 3D culture of green labelled fibroblasts embedded in collagen hydrogel in a microfluidic device. Confocal top view micrograph of (A) collapsed collagen embedded cells and (B) apparently conserving the 3D structure. (C) Section view of the culture chamber by confocal Z-stacks reconstruction images of the hydrogel from B showing a non-homogeneous cell distribution, that tends to localise on the walls of the device. (D) Section view of the culture chamber of a non-contracted gel with cell homogeneously distributed along the height of the chamber. Note: scale bar 500 μm (A and B), 100 μm (C and D). Height of the section in (C) and (D) 250 μm. | ||
The techniques to improve the bonding between the device and the ECM protein to prevent hydrogel contraction in microfluidic devices can be classified into three main groups: physical adsorption (physisorption), covalent bonding and their combination.26 In physical adsorption, proteins are adsorbed to the surface by intermolecular forces (electrostatic, hydrophobic, van der Waals, hydrogen bonding interactions or combination). Surface oxidation is the most common method used to condition the surface. For covalent protein bonding, among others,27 silanization and grafting, with alkoxy- or chloro-silanes, such as 3-aminopropyltriethoxysilane (APTES)28–30 that later is covalently bonded to the protein. Another treatment used for covalent immobilisation consists of the ultraviolet (UV)-graft polymerisation with benzophenone as photoinitiator and, for example, acrylic acid (AA) to produce polyacrylic acid (PAA) brushes on the surface26,31 to which the proteins are subsequently covalently linked. The combination of two or more immobilisation mechanisms is possible. Thus, physisorption and covalent bonding might be performed by adsorbing positively charged molecules such as poly-D-lysine (PDL) or poly-L-lysine (PLL) on activated surfaces, further activating them for covalent protein immobilisation.32,33
The aforementioned methods for protein immobilisation have been mainly validated for PDMS-based microfluidic devices. PDMS is widely used in microfluidics because of its high biocompatibility, optical transparency, gas permeability, malleability and easy prototyping. Nonetheless, thermoplastic materials such as cyclic olefin polymer (COP) and cyclic olefin copolymer (COC) are becoming materials of great interest. These low-cost materials can be massively produced with mould-based techniques, such as injection moulding or hot embossing. Moreover, they are considered medical-grade plastics, with very low gas permeability, resistance to solvents, acids and bases, heat resistance, excellent optical properties, and no unspecific adsorption issues.26 COP and COC are available from various vendors under different brand names (Topas®, Apel®, Zeonor® and Zeonex®) and grades. Depending on the monomers included, their ratios, molecular weight as well as the presence of additives, different material properties can be tuned to some extent adapting their performance to different applications.34 Although the hydrophobicity of native cyclic olefin (co)polymers represents a great challenge for the use of these materials in microfluidic applications and as cell culture platforms, their surface can be modified to increase hydrophilicity and create functional groups for the attachment of biological entities.35
Protein immobilisation by silanization36–38 and polyacrylic photografting have never been applied on COP-based microfluidic devices (COP-MD)39–42 to preserve the three-dimensionality nor has its effect to counteract cellular forces in 3D cultures been characterised. On the other hand, the adsorption of positively charged molecules such as PDL/PLL on activated surfaces43–46 and the subsequent activation of amine functional groups with GA for covalent immobilisation to proteins is barely described in PDMS devices.47 Nevertheless, this approach has not been previously reported on COC/COP surfaces. We were encouraged, therefore, to test several surface treatments (Fig. 2) on COP-MD to form stronger bonds with collagen hydrogel. We hypothesised that improved bonding between collagen hydrogel-embedded fibroblasts and the COP-MD surface should avoid and delay the collapse and detachment caused by fibroblast cell contractility. This would allow the development of in vitro models requiring contractile cells.
 | ||
Fig. 2 Schematic illustration of COP surface modification procedures involved in collagen gel immobilisation. A) Oxygen plasma activates the inert surface of the COP by oxidizing and inducing the formation of polar functional groups carboxyl (C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and hydroxyl (–OH). It increases the hydrophilicity of the material and the subsequent physisorption of biomolecules. B) APTES can bind to hydroxyl functional groups of the material surface providing an amine functional group to facilitate covalent bonding with proteins on the other end. That amine group can be activated by different covalent linking agents such as glutaraldehyde. C) UV-graft polymerization with benzophenone and AA produces PAA brushes on the surface. The carboxyl groups of grafted PAA can be covalently linked to ECM proteins via carbodiimide-mediated amide formation. D) Positive charged molecules as PDL/PLL are adsorbed by electrostatic interaction on the surface of activated surfaces (negatively charged). Amine groups on PDL can be activated with GA and proteins are covalently immobilised. O) and hydroxyl (–OH). It increases the hydrophilicity of the material and the subsequent physisorption of biomolecules. B) APTES can bind to hydroxyl functional groups of the material surface providing an amine functional group to facilitate covalent bonding with proteins on the other end. That amine group can be activated by different covalent linking agents such as glutaraldehyde. C) UV-graft polymerization with benzophenone and AA produces PAA brushes on the surface. The carboxyl groups of grafted PAA can be covalently linked to ECM proteins via carbodiimide-mediated amide formation. D) Positive charged molecules as PDL/PLL are adsorbed by electrostatic interaction on the surface of activated surfaces (negatively charged). Amine groups on PDL can be activated with GA and proteins are covalently immobilised. | ||
Experimental
Microdevice fabrication/characteristics
Microfluidic devices (MD) based on cyclic olefin polymers (ZEONOR, Zeon Corporation) were manufactured by injection moulding. The design (Fig. 3A) comprised a central chamber 2000 μm wide and 250 μm deep, flanked by two lateral channels (700 μm wide, 250 μm deep) connected to the central chamber by micropillars. The injected piece (ZEONOR 1420R) was attached to a sheet of COP (ZEONOR 14-100) by biocompatible adhesive (ARseal 8026) and cut out in the area of the central chamber to preserve the whole seeding area in contact with COP (free of adhesive) (Fig. 3B). After applying the surface treatment, the hydrogel seeding solution was injected through an inlet port into the central chamber by manual pipetting. The lateral microchannels remained hydrogel-free and filled with culture medium, simulating blood vessels. | ||
| Fig. 3 Microfluidic device appearance and components. (A) Magnified representation of a COP-MD with collagen hydrogel embedded cells (green) confined into the central chamber and culture medium perfused through the two lateral channels creating a gradient of medium. (B) Schematic illustration of the elements integrating the microfluidic device. | ||
Surface modification and immobilisation strategies
All devices were sterilised by exposure to a germicidal UV lamp for 30 minutes before cell-embedded collagen hydrogel seeding.
3D cell culture
Human cardiac fibroblasts (HCF) (ScienCell) and human dermal fibroblasts (HDF) (Gibco) were grown between passages 3 to 6. HCF were grown in DMEM (Lonza 12-707F) culture medium supplemented with 10% FBS (Sigma F7524) and 2.5 mM L-glutamine (Lonza 17-605c), 1% penicillin/streptomycin (100 μg mL−1) (Lonza DE 17-602E), 10 ng ml−1 FGF-2 (Peprotech 100-18B) and 1.5% HEPES (Lonza BE17-737C). HDF were grown in DMEM (Lonza 12-707F) culture media supplemented with 10% FBS (Sigma F7524) and 2.5 mM L-glutamine (Lonza 17-605c), 1% penicillin/streptomycin (100 μg mL−1) (Lonza DE 17-602E), 0.5 ng ml−1 FGF-2 (Peprotech 100-18B) and 1.5% HEPES (Lonza BE17-737C). Human glioblastoma cell line U-87 MG was purchased from Sigma Aldrich (Sigma 89081402 lot: 16L024). U-87 MG were grown in high glucose DMEM (Lonza H3BE12-614F) culture medium supplemented with 10% FBS (Sigma F7524) and 2.5 mM L-glutamine (Lonza 17-605c), 1% penicillin/streptomycin (100 μg mL−1) (Lonza DE 17-602E). Media was refreshed every other day, passaging cells every 3–4 days with trypsin/EDTA solution (Lonza, CC-5012). All cell lines were maintained within a humidified TEB-1000 incubator set at 5% CO2 and 37 °C (EBERS Medical Technology).Hydrogel mixture for 3D culture into COP-MD was composed of 50% (v/v) of the double desired cell density in growth media and 50% (v/v) collagen matrix. For collagen matrix, either 3.36 mg mL−1 (Corning 354236) or 10.98 mg mL−1 (Corning 354249) type I rat tail collagen, NaOH 1 N (Sigma 655104) at the proportion of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 (v/v of collagen volume) and DMEM 5X (Sigma D5523) at 1:5 (v/v of collagen matrix volume) were mixed to get a final collagen density of 1.2 mg mL−1 or 4 mg mL−1 respectively. Sterile water was added to reach the final collagen matrix volume. The collagen gel matrix was resuspended, homogenised and kept on ice until injection into the devices. The cell suspension at the desired cell density was homogenised with collagen matrix before injecting into the device. COP-MD were pre-warmed at 37 °C for 1 h before cell seeding. Once the cells-collagen gel suspension was confined in the central chamber, devices were turned up and down every 20 seconds for 3 minutes to obtain a homogeneous cell distribution before introducing into the incubator (37 °C, 5% CO2) for 15 minutes to promote collagen gel polymerisation. Afterwards, the pre-warmed culture medium was perfused through lateral channels and refreshed daily.52
:40 (v/v of collagen volume) and DMEM 5X (Sigma D5523) at 1:5 (v/v of collagen matrix volume) were mixed to get a final collagen density of 1.2 mg mL−1 or 4 mg mL−1 respectively. Sterile water was added to reach the final collagen matrix volume. The collagen gel matrix was resuspended, homogenised and kept on ice until injection into the devices. The cell suspension at the desired cell density was homogenised with collagen matrix before injecting into the device. COP-MD were pre-warmed at 37 °C for 1 h before cell seeding. Once the cells-collagen gel suspension was confined in the central chamber, devices were turned up and down every 20 seconds for 3 minutes to obtain a homogeneous cell distribution before introducing into the incubator (37 °C, 5% CO2) for 15 minutes to promote collagen gel polymerisation. Afterwards, the pre-warmed culture medium was perfused through lateral channels and refreshed daily.52
Cell tracking and viability assessment
Cells were fluorescently labelled before embedding into the hydrogel by adding 5 μl of lipophilic cell membrane dye Vybrant™ DiO (Invitrogen V22886) per 106 cells per mL suspension. Cells were incubated for 15 min, centrifuged and washed three times with a culture medium. Cell viability was assessed by perfusing through the lateral channels with 1 μg mL−1 calcein AM (CAM) (Sigma 17783) and 4 μg mL−1 propidium iodide (PI) (Sigma P4170). Both stainings were incubated at 37 °C in a humidified chamber with 5% CO2. Cells cultured into COP-MD were visualised by confocal microscopy (Nikon Ti-E coupled to a C1 modular confocal microscope), at 590/50 nm for Vybrant and CAM and at 650LP nm for PI. Two image types were taken from each device: the top and section view of the central chamber (Fig. 1). To obtain the hydrogel area occupied in the section view, confocal Z-stacks were taken at different time points. To acquire the entire height of the chamber, 300 μm thick confocal stacks were performed with images every 10 μm. Z-Stacks were performed in at least two regions of each device, evaluating at least three devices per condition. Images were analyzed with Fiji® software53 for area quantification (ESI† Fig. S2).The immobilisation efficiency of collagen hydrogel was related to the hydrogel resistance to collapse by cell traction forces and was assessed by measuring the section area occupied (ESI† Fig. S2). Hence, the greater the cell area occupied, the greater the resistance of the hydrogel to collapse and the better hydrogel immobilisation. For cell viability, fluorescence profile intensity across the chamber was quantified by selecting a rectangular region of the central chamber between the medium channels.
Statistical analysis
For statistical analysis, trend lines of transversal area quantification over time were computed using weighted linear regression with an R square ranging from 0.6 to 0.97, from which the slope was estimated. The slope was considered an immobilisation efficiency indicator, so the greater the slope, the greater the gel contraction and, therefore, less hydrogel immobilisation. The significant difference among the conditions was determined by a statistical comparison of the slope pairs using a two-tailed test. Cell viability analysis was determined by a statistical parametric unpaired Student t-test using the statistical software GraphPad Prism 6.01. All experiments were performed at least thrice, and a p-value < 0.05 was considered statistically significant.Results
Hydrogel immobilisation efficiency evaluation after different COP-MD surface treatments
Three surface treatments in COP devices were assessed prior to immobilisation of fibroblast embedded into 1.2 mg mL−1 collagen hydrogels. Thus, collagen I was immobilised to PAA-PG, APTES and PDL pre-treated COP-MD surfaces, taking a plasma-treated surface as control. The most representative changes of hydrogel integrity were monitored and observed in the section view, as the top view does not evidence the whole hydrogel integrity. The section area occupied by green-fluorescent DiO Vybrant™ labelled human cardiac fibroblasts (HCF) (107 cells per mL) embedded into collagen (1.2 mg mL−1) hydrogels was monitored over 13 days. Images were acquired until collagen hydrogels were completely collapsed (Fig. 4A). Silanization of the surface with APTES displayed a clear hydrogel area reduction after two days, being pronounced on the third day (slope −9.58 ± 0.86) (Fig. 4B). PDL-treated chips reduced their hydrogel area below 90% after four days in culture (slope −6.81 ± 1.64). Finally, PAA-photografting (PAA-PG) treatment showed the longest hydrogel structure preservation, keeping over 90% area after 8 days of culture (slope −1.69 ± 0.36). Then, the first fissures between the device surface and the hydrogel began to be displayed, showing statistically significant differences compared to the control (p < 0.001). No tendencies were observed between the top (47.5%) and the bottom (52.5%) when the gels detached from the COP-MD. | ||
| Fig. 4 Culture of HCF (107 cells per mL) embedded in 1.2 mg mL−1 collagen after device pre-treatments. A) Transversal area evolution overtime of HCF labelled with green-fluorescent DiO Vybrant™ dye. B) Area percentage of HCF embedded collagen hydrogels within the device chamber over time (dashed line) after treatment with different conditions and trend lines (continuous line). C) Statistical comparison among the slopes (absolute values) of trend lines from each condition. D) Micrographs and area percentage of calcein–propidium iodide stained cells from micrographs of collagen embedded HCF within treated devices after 13 days. Error bars SD. Note: **** p < 0.0001, *** p < 0.001, ** p < 0.01, * p < 0.05. Error bars SD. Scale bar 100 μm. | ||
Although all studied hydrogels were contracted over time except PAA-PG pre-treatment, we evaluated embedded cell viability after 13 days in all the conditions to determine any possible toxic effect from the chemicals applied on the device surface. Thus, we quantified the cell viability of collagen-embedded HCF and corroborated that all the surface treatments did not significantly affect the cell viability compared with the control (Fig. 4D).
The treatment showing the longest prevention of collagen hydrogel collapse was also tested with other high-contractile cell lines to avoid cell line-specific bias. Thus, PAA-PG was applied to devices seeded with human glioblastoma cell line U-87 MG previously embedded in 1.2 mg mL−1 collagen hydrogel. Consistently with the observations of HCF collagen-embedded hydrogels, the treatment displayed significant differences to control, with longer preservation of 3D structure (ESI† Fig. S3). Moreover, PAA-PG treatment exhibited also improved gel resistance to cell forces with collagen-embedded human dermal fibroblasts (HDF) for 3 days, at 2 and 5 × 106 cells per mL (ESI† Fig. S4).
Evaluation of collagen concentration effect on the hydrogel immobilisation efficiency
In order to test the outcomes of the studied surface treatments on collagen contraction within COP-MD at higher collagen concentrations, we tested them with 4 mg mL−1 collagen hydrogels (Fig. 5A–C). Thus, the section area of 107 HCF embedded in 4 mg mL−1 collagen hydrogels within treated COP-MD was monitored over time. In all the studied treatments the shrinkage of 4 mg mL−1 collagen hydrogels was delayed compared to that observed in 1.2 mg mL−1 collagen hydrogels, still following a similar tendency. This way, PAA-PG treatment displayed the least contracted collagen hydrogels for up to 20 days, followed by PDL-treated devices. Furthermore, in contrast to 1.2 mg mL−1 assays, PDL treatment kept uncontracted collagen hydrogels nearly as long as PAA-PG. This behaviour in PDL-treated devices was reflected by the slope of the trend lines from the cell area percentage occupied within the hydrogel in the device chamber, showing statistically significant differences to control and comparable to PAA-PG (p < 0.0001). In fact, the cell area percentage occupied in PDL-treated microdevices kept above 90% until day 20 of culture, similar to PAA-PG. Still, it decreased slightly at this point, with some gels detaching from the surface of the microdevice. Finally, silanization with APTES (slope −4.245 ± 1.146) did not show differences to control as described with 1.2 mg mL−1 collagen results, preserving cell area percentage occupied within devices above 90% up to the 6 day of culture. | ||
| Fig. 5 Culture of HCF (107 cells per mL) embedded in 4 mg mL−1 collagen after device pre-treatments. (A) Transversal area evolution overtime of HCF labelled with green-fluorescent DiO Vybrant™ dye. (B) Area percentage of HCF occupation of the collagen hydrogel within the device chamber over time (dashed line) and trend lines (continuous line). (C) Statistical comparison among the slopes of trend lines from each condition. Note: **** p < 0.0001, *** p < 0.001, ** p < 0.01 and * p < 0.05. Error bars SD. Scale bar 100 μm. | ||
To better understand the surface distribution of the treatment and surface attachment forces to collagen I, complementary assays with shear stress were performed (ESI† Fig. S5). The surface of rectangular COP-channels was treated and coated with fluorescently-labelled collagen I.
The distribution of functional groups, responsible for protein immobilisation, was indirectly quantified by analysing the collagen bonded to the COP surface for each condition before applying shear stress (0 dyn cm−2). Contrary to other types of proteins (ESI† Fig. S6A) collagen distributed in aggregates (ESI† Fig. S5A). This is probably caused by the variability in their size and structural properties. Regarding the quantity of immobilised collagen, significant differences were only observed between control and PDL surfaces (ESI† Fig. S5B).
To compare the adhesion forces of the collagen to the differently treated COP surfaces, collagen-coated channels were exposed to increasing shear stress forces (0.15, 25 and 150 dyn cm−2). Fluorescence microscopy revealed that the collagen presence varied before and after shear stress application. Consistently with 3D cell embedded assays, in control condition (plasma) collagen was almost completely detached from the surface after applying the lowest shear stress (0.15 dyn cm−2). Indeed, the remaining quantity of collagen was 12.5% of the initial state. In the other treated surfaces (APTES, PDL and PAA-PG) collagen quantity slightly decreased when shear forces of either 0.15 or 25 dyn cm−2 were applied (ESI† Fig. S5B). Lastly, more pronounced changes were observed with the maximum shear stress (150 dyn cm−2). For the APTES surface, the collagen quantity was reduced to approximately 28% of the initial state. In turn, for the PDL and PAA-PG channels, this parameter was reduced to 61% and 75%, respectively. These results agree with the previously described observations for collagen hydrogel resistance to cell contraction (Fig. 4 and 5).
PAA-PG was tested with other types of extracellular matrix proteins with a key role in cell adhesion such as fibronectin and laminin (ESI† Fig. S6). Plasma control and PAA-PG channels were coated with fluorescently-labelled fibronectin and laminin and exposed to 150 dyn cm−2. Although the initial distribution was very homogeneous for both laminin and fibronectin, the fluorescence intensity showed different values (ESI† Fig. S6B). Fibronectin control channels displayed a minor intensity than PAA-PG channels. Though in PAA-PG channels fluorescence intensity decreased after flow, it still maintained a higher intensity than control in static conditions. On the other hand, the intensity of laminin was greater in PAA-PG channels compared with the control ones before flow. Nevertheless, despite both conditions showed protein detachment, the intensity levels of control channels were better maintained than PAA-PG ones. These results suggested an improved immobilisation of fibronectin to PAA-PG surfaces in comparison with the laminin.
Evaluation of PAA-PG immobilisation technique for the in vitro simulation of the necrotic core formation
The presence of fibroblasts in co-cultures of different cell types is often needed to create more biomimetic in vitro models. However, fibroblasts are one of the main cell types affecting 3D hydrogel integrity. Moreover, some experiments require high cell densities inside the gel as the self-induced ischaemia models,52,54 in which the fibroblast role is extremely relevant. Thus, we tested if PAA-PG pre-treatment could overcome this technical obstacle, focusing on a necrotic core model with human cardiac fibroblasts in 1.2 mg mL−1 collagen hydrogels.First, we assessed the influence of the HCF cell density on contracting the hydrogel. Low-dense cultures (106 cells per mL) of HCF did not produce collagen hydrogel contraction when the device was not treated (control) (Fig. 6A). However, the hydrogel contraction was observed after 3 days with 5 × 106 cells per mL and only 24 hours with higher densities (107 and 2 × 107 cells per mL) (Fig. 6B). The PAA-PG pre-treated microdevices improved performance significantly, successfully preserving the hydrogel structure after 3 days in culture at all the densities studied (Fig. 6C). Vybrant DiO fluorescence intensity and cell morphology differences were observed in the central area of the culture chamber in the highest HCF concentration hydrogels (107 and 2 × 107 cells per mL). Fibroblasts located closer to the medium channels were more fluorescent and spread. At the same time, cell morphology in the central area was rounded and fluorescence intensity lower, suggesting cell death (Fig. 6D–H, S7 and ESI† video). Cell viability staining after 48 hours showed rounded central cells as dead and membrane compromised cells, generating a necrotic core in the innermost hydrogel region. The necrotic core generated after 48 hours by a density of 107 cells per mL occupied 500–600 μm of the central chamber (Fig. 6I). A higher cell density of 2 × 107 cells per mL provided a larger necrotic core, up to 1500 μm (Fig. 6J).
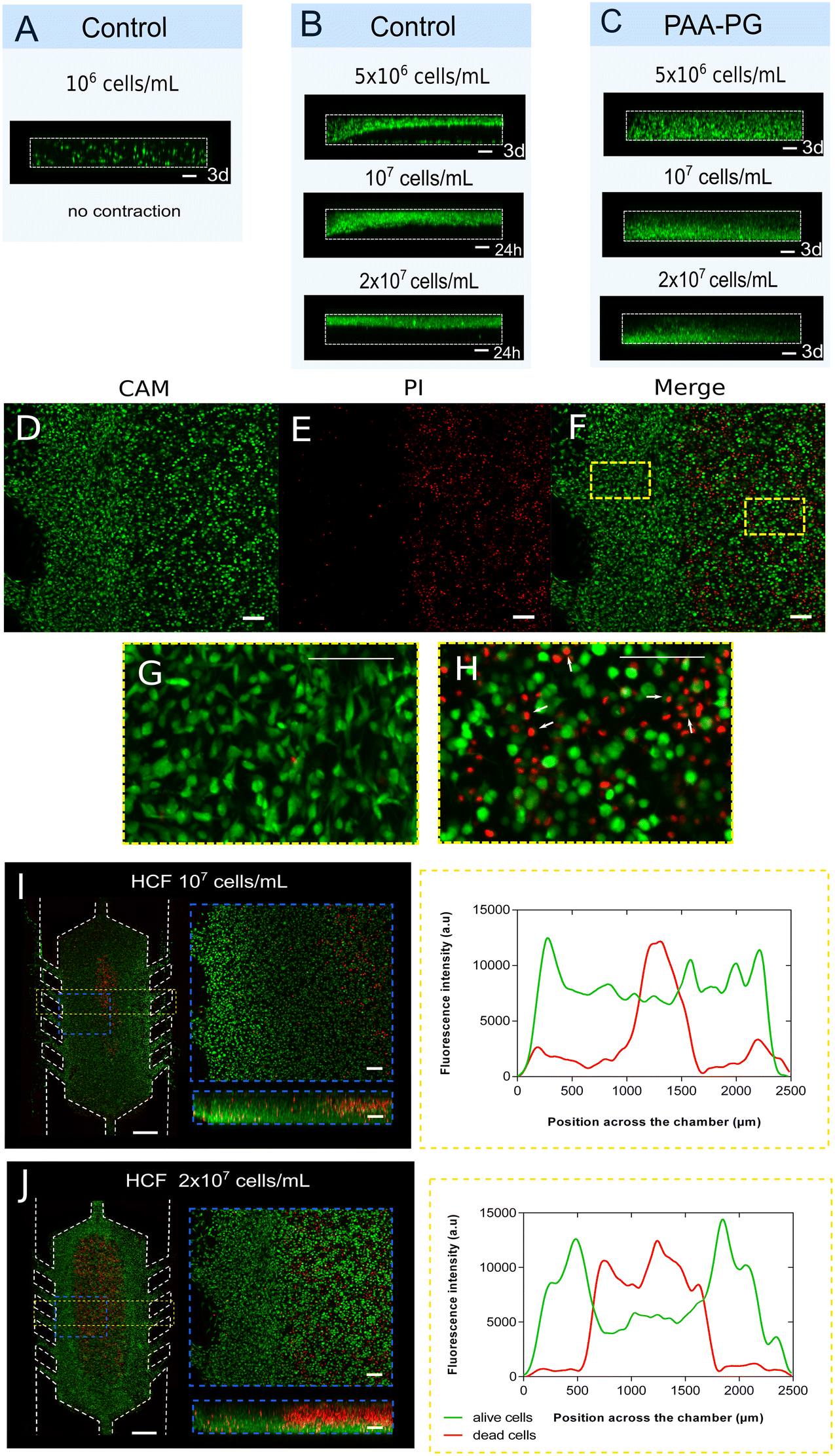 | ||
| Fig. 6 HCF 3D culture for necrotic core formation in PAA-PG treated devices. (A) Green labelled (DiO) HCF at 106 cells per mL in collagen (1.2 mg mL−1) hydrogel within non-treated microdevice after 3 days. Cardiac fibroblasts at 5 × 106, 107 and 2 × 107 cells per mL section view within non-treated (B) and PAA-PG treated (C) COP-MD. 2 × 107 HCF mL−1 after 48 hours culture labelled with (D) calcein (CAM) and (E) propidium iodide (PI) viability stain presented two different areas, being the area on the left densely populated whereas that the right area displayed less cells, separated from each other. PI binds to nucleic acids of dead cells, mainly grouped in the center of the chamber (right area in E), where gases and nutrients levels decrease. Merging CAM and PI micrographs (F) revealed the coincidence of the right area with the predominance of dead cells. Magnifying the image in the region closer to the medium channels (left in F) cells exhibited a spindle like form (G) while in the central region of the chamber (right in F) cells showed a rounded morphology (H). In addition, in the right area (marked with white arrows in H) red stained nuclei disclosed inside green fluorescent cells, suggesting that these cells have a compromised membrane. Top and section view (blue bordered regions) of alive/dead fibroblasts of 107 (I) and 2 × 107 (J) HCF mL−1 at 48 hours after CAM/PI staining. The graphs show quantification of CAM (green) and PI (red) fluorescence intensity profile along the chamber device (yellow bordered regions). Scale bar 100 μm except in (I) and (J) left image of the whole device where scale bar is 500 μm. | ||
Accordingly, the pre-treatment with PAA-PG allows the generation of a necrotic core with fibroblasts embedded in collagen hydrogel, which was impossible without collagen hydrogel immobilisation (Fig. 6B and C). The effectiveness of PAA-PG was even more evident when studying the human glioblastoma U87-MG cell line. Thus, comparing non-treated COP-MD with PAA-PG treated, we were able to prolong the culture of 4 × 107 U87-MG cells per mL embedded in collagen from 4 hours to 9 days. Moreover, a necrotic core was preserved 9 days after cell seeding (ESI† Fig. S8).
Discussion
Fibroblast concentration and ECM stiffness are parameters that influence physiological and non-physiological phenomena. For example, in fibrosis and wound healing processes, an acute injury triggers a cascade of events that lead to fibroblast migration and proliferation. These fibroblasts produce large amounts of ECM proteins, including collagen type I and III, fibronectin and hyaluronic acid, leading to increased mechanical tensions in combination with other chemical stimuli that induce transdifferentiation of fibroblasts into myofibroblasts. Myofibroblasts are characterised by the ability to exert strong contractile forces on the ECM, and produce fibrillar collagen bundles that stiffen the tissue. Similarly, tumours have been described as wounds that fail to heal, where the fibroblasts play an important role by producing and depositing ECM that enhances stiffness and strain.2,55 All these aspects make the in vitro biomimetic recreation of these processes, in which fibroblasts are involved, a matter of crucial importance. However, the generation of models based on microfluidic devices has been very limited by the contractile capacity of fibroblasts on the hydrogel structure in which they are embedded. These contractions cause gel collapse, altering the organisation and distribution of cells in microfluidic devices. Therefore, the need arises to develop surface treatments for the chip materials to avoid hydrogel collapse. In this work, several protein immobilisation methods have been compared in COP-MD.From the different methodologies reported (low-pressure plasma, oxygen plasma, UV/ozone, photografting of PEGMA/PEtOx, permanent PVA coating and dynamic HEC coating), oxygen plasma appears as a very effective technique in terms of reducing hydrophobicity and generating functional groups in COP/COC substrates.56,57 However, the oxygen plasma effect depends on the polymer type. The reduction of the WCA is an indicator of the increase in surface energy due to the introduction of polar groups on the surface of the polymer. For example, differences in the ethylene/norbornene content in COC (Topas®) are described to affect the WCA. An increased norbornene content lead to a lower WCA and more hydrophilic surface after oxygen plasma-activation.58 Haq et al.,59 applied a corona treatment to polycarbonate, COC and COP surfaces, obtaining a reduction from 80° to 38°, 96° to 39° and 93° to 27°, respectively. The WCA reduction was related to the increment of oxygen to carbon ratio in C–O, COH, COOH and CO forms. These results suggest a different surface activation of COP and COC under the same treatment conditions. In cell culture applications,60 COP (Zeonex®) and COC (Topas®) air plasma-treated substrates were seeded with HeLa cells (cervix carcinoma cells). COP was described as a suitable substrate for cell culture while COC did not provide an appropriate chemistry for cells. Despite COP and COC are chemically related, spectroscopy analysis showed different spectra. Authors suggested the presence of non-specified additives in the polymers to explain the differences observed in the cell culture studies.
Among all the treatments studied in this work, the physisorption treatment (control) has shown the worst response to counter hydrogel contraction. This was expected because the intramolecular forces involved are weak and unstable, needing additional treatments to enhance protein attachment.30,61,62 The short stability of plasma oxidation has been widely reported in both PDMS and COC materials36,63 extensive also to their ability to adsorb molecules on their surface. For example, AFM height images from collagen coating physically adsorbed onto PDMS after oxygen plasma treatment show lower stability after incubation in a growth medium for five days than stable covalent coating based on an aminosilane chemistry.64 Moreover, according to published studies of monolayer cultures on collagen coating65,66 and collagen-embedded cells46 within microfluidic devices, it seems that the physical adsorption of collagen I does not represent an ideal host surface for the long-term culture of cells. Although this lack of stability can be attributed to cell activity and homeostasis, the adsorbed collagen can also be exchanged by other proteins from the cell culture medium, known as the Vroman effect, making immobilisation less stable and resistant to cell remodelling.66
Positively charged molecules (PDL/PLL) have been commonly used as a cell adhesion molecule to enhance cell attachment, adhesion and differentiation of many cell types.67 Some works describe the immobilisation of PDL through APTES67 or Pluronic F12768 to stabilise and improve neuron adhesion with in long-term cultures. Direct PDL adsorption on plasma-activated surfaces prevents shrinkage and detachment of collagen hydrogel from PDMS channels allowing capillary morphogenesis.43,46 Treatment with NH2 ended chemicals such as poly(ethyleneimine) (PEI), pol-y(allylamine hydrochloride) (PAH), hexamethylenediamine (HMDA) and 1,3-diaminopropane (DAP) has been reported in PMMA69 and porous silicon substrates70 for antibody immobilisation. Recently, substrate amination with PLL to immobilise ECM protein has been used in PDMS devices to promote better bonding in cell culture applications.47,71 To the authors' knowledge, this is the first time that amination with PDL is used for collagen immobilisation on COP surfaces for 3D culture applications. In this work, heterogeneous results were observed in combined physisorption and covalent bond treatment with PDL. Followed by GA activation, PDL amino groups covalently crosslink with collagen amino groups. Thus, PDL treatment delayed the hydrogel contraction, maintaining the cell area occupied over 90% up to 3 days with 1.2 mg mL−1 collagen hydrogels, which was significantly prolonged with 4 mg mL−1 collagen hydrogels. Hence, our results point to PDL treatment as the second-best immobilisation technique after PAA-PG.
Grafting is a widely used technique for modifying the surface chemistry with polymer brushes for further modification in both PDMS and COC/COP. It allows a stable hydrophilic surface, can provide stable biomolecule attachment and provides biocompatibility with the possibility of patterning enclosed microchannels.27 Covalent attachment of graft chains onto a polymer surface offers stable surfaces in contrast to the physical coatings which can be delaminated.41 In microfluidics, the applications for grafted substrates are principally: reducing protein adsorption and platelet adhesion, providing anti-fouling properties, and immobilising antibodies and biomolecules.72 Wang et al.,31 successfully photografted PAA onto PDMS substrates and subsequently immobilised a mixture of ethylenediamine, chitosan, PLL and RGD peptides to promote cell attachment and growth in the modified regions. Similarly, in COP/COC substrates photografting has been predominantly used to generate microfluidic sensors by bioanalytical probes immobilisation,42,73 the reduction of nonspecific adsorption of plasma proteins or BSA74 and cell adhesion.40 Remarkably, photografting has not been reported to immobilise collagen either to avoid hydrogel collapse or to improve coating efficiency in COP-MD. In this work, we demonstrate that COP-MD previously treated by PAA-PG reached the longest fibroblast 3D culture, preserving hydrogel structure without collapsing at either 1.2 or 4 mg mL−1 collagen concentration after 8 days. Comparing the effect of PAA-PG and PDL immobilisation treatments, PDL-treated chips did not preserve the 3D structure as long as UV-photografting, possibly due to the chemical strength differences between both treatments. In fact, while PAA-PG presents double covalent bonding between COP-PAA and PAA–collagen, PDL is physisorbed onto the COP surface by electrostatic interaction and covalently bonded to collagen through the amino groups exposed by GA linked to PDL.
We also studied immobilisation by silanization with APTES, since this technique is widely applied for antibody immobilisation for immunoassays diagnosis in materials such as COP or COC.36–38 However, although ECM proteins immobilisation with APTES in PDMS microfluidic devices is successfully reported in cell sheet cultures (enhancing cell adhesion66,75 under shear stress conditions64 or for cell stretching applications76), there are no studies in 3D culture within microfluidic devices. Interestingly, although PAA-PG and APTES treatments are supposed to establish stable covalent linkages between proteins and chemically modified COP surface, only PAA-PG showed significant outcomes. APTES was shown as a non-appropriate immobilisation technique for collagen I-based hydrogels in COP-MD under the conditions applied in this work. Further investigation exploring extended surface activation times applied to 3D culture must be done.
Increasing collagen hydrogel concentration induced a delay in contraction behaviour. As reported, higher collagen concentration gels exhibit minor contraction than less concentrated ones.77 Consequently, the contraction tendency between 1.2 mg mL−1 and 4 mg mL−1 collagen hydrogels was very similar, so the efficacy of the treatments was almost equal. Thus, collagen hydrogel concentration directly affects the time required for cell contraction, delaying the phenomenon, but not avoiding it. Nevertheless, increasing collagen hydrogel concentration modifies mechanical properties such as elastic modulus, pore radius or permeability78 inducing changes in cell behaviour.77 Therefore, this strategy is not always close to physiological microenvironment stiffness.
Cancer cell spheroids (also known as multicellular tumour spheroids MCTS) are a successful in vitro model to mimic cell-to-cell interactions and tumour/ischaemia models. The core of solid tumours is highly hypoxic due to poor blood circulation. Moreover, hypoxia is considered to contribute to drug resistance,79 being one of the major hallmarks of solid tumours and a requirement for 3D tumour culture models. Due to the limited diffusion within spheroids, biochemical gradients are created (oxygen, nutrients, growth factors, signalling molecules and molecular waste).80 Nevertheless, microscopic imaging of large spheroids (>150 μm) and organoids is extremely challenging or requires expensive equipment due to poor light and label penetration, and attenuation of the fluorescent signal by light scattering.81,82 On the other hand, scaffolds based on hydrogels provide extracellular support, mimicking the biochemical and mechanical properties of ECM, allowing for cell–ECM interactions,10 cell growth and migration. Their tuneable porosity allows the diffusion of oxygen, nutrients and drugs to reach the cells while facilitating the waste removal. Combined with COP-MD, hydrogels permit the generation of ischaemia and necrotic core models due to the gas impermeability of COP, with the generation of nutrients and gas gradients inside the central chamber.52,54 However, the power of this type of ischaemia-on-chip model lies not only in the capacity to control the gas concentration inside the device (material properties) but also in the preservation of the three-dimensionality of the culture within the chamber. When including cells with contractile capacity within the hydrogel, its structure may be compromised and, therefore, the preservation of three-dimensionality and gradient generation. This phenomenon encouraged us to study PAA-PG surface functionalisation to validate an ischaemia-on-chip model with contractile cells. This treatment demonstrated long-term 3D culture, preserving collagen hydrogel structure. We were able to generate oxygen and nutrient gradients that led to the formation of necrotic cores. In fact, different necrotic core sizes were developed depending on initial HCF density seeding. PAA-PG also permitted a glioblastoma necrotic formation with U87-MG expressing high contractility capacity. The dimensions of the necrotic cores generated in this work are conditioned by the nature of the extracellular matrix (type of protein, concentration, porosity), the density and cell type (cell requirements and metabolism) and the characteristics of the device (design and dimensions). The latter can affect the diffusion of the culture medium. Relying on the size of the medium channels, the volume of culture medium available will vary. Another relevant feature is the presence of microchannels connecting the central chamber and the medium channels. These structures limit the area of contact and diffusion. Likewise, the culture medium flow rate constitutes an important factor to control the medium diffusion speed through the hydrogel.
Not only fibroblast-like cell types do manifest contractility events, but also endothelial cells retract hydrogels away from the walls of microfluidic devices in long-term experiments for in vitro angiogenesis.46 Hence, our approach could open the door to the application of the described surface treatments to other models. Moreover, adapting spheroids assays to hydrogel-embedded culture would allow easy real-time monitoring of ischaemia events and other applications such as drug testing and immunotherapy. This constitutes a basis for the development of closer patho-physiological and relevant co-culture models of angiogenesis, wound healing, tumour microenvironment and ischaemia within COP-MD.
Conclusions
We have characterised different protein immobilisation methods for 3D cell culture in COP microfluidic devices showing that collagen hydrogel contraction can be delayed when cultured with highly contractile cells such as fibroblasts. A combination of physisorption and covalent bonding methods for collagen immobilisation using PDL can be applied for long-term cultures in 3D hydrogels. However, their effectiveness on 3D collagen structure maintenance must be determined in advance for each condition (hydrogel concentration, experiment time) or cell type. In this work, we present PAA-PG as the most effective immobilisation method that allows long-term culture at high cell densities of contractile cell types such as fibroblasts. Moreover, PAA-PG allows the generation of gradients and necrotic core within COP-MD cultured with a high fibroblast density. The implementation of PAA-PG treatment to COP-MD opens new possibilities for in vitro model generation where collagen hydrogel structure preservation is required, such as ischaemia or tumour microenvironment.Author contributions
SGL: conceptualization, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing. TR: formal analysis, investigation, methodology, visualization, writing – review & editing. JC: formal analysis, supervision, writing – original draft, writing – review & editing. MLV: investigation, methodology. RM: conceptualization, funding acquisition, project administration, supervision, writing – review & editing. CSS: conceptualization, funding acquisition, project administration, supervision, writing – review & editing. IO: conceptualization, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
IO and RM are promoters for BeOnChip S.L. (Zaragoza, Spain). SGL is developing her PhD in collaboration with BeOnChip S.L. BeOnChip S.L. does not benefit or take part in any economic decisions of this work. TR, JC, MLV and CSS have no conflicts to declare.Acknowledgements
SGL was funded by a Spanish MINECO fellowship (DI-17-09585). TR studentship was supported by Instituto de Salud Carlos III (i – PFIS IFI16/00050). This work was funded by CDTI (SNEO-20181019), Spanish Government “Ministerio de Ciencia, Innovación y Universidades (MCIU)” through AEI/FEDER(UE) project PID2020-118485RB-I00; “Ministerio de Asuntos Económicos y Transformación Digital – Agencia Estatal de Investigación” through PID2020-118485RB-I00 and PID2021-126051OB-C41 projects. Government of Aragon project LMP221_21, FEDER (EU); through the “Fondo Social Europeo” (DGA T62_20R, E15_20R and E47_20R). This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 829010 (PRIME) and from H2020-MSCA-RISE-2017 programme under grant agreement No. 778354 (CISTEM); and also from the Key Digital Technologies Joint Undertaking with 876190. Authors would like to acknowledge the use of Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza. The authors want to especially thank Arantxa González for the donation of human cardiac fibroblasts and Jacobo Ayensa for statistical advisory.Notes and references
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- W. Tan and T. A. Desai, Biomed. Microdevices, 2003, 5, 235–244 CrossRef CAS.
- J. Padron, Crit. Rev. Oncol. Hematol., 2000, 36, 141–157 CrossRef CAS PubMed.
- R. M. Sutherland, Science, 1988, 240, 177–240 CrossRef CAS PubMed.
- A. Berglund, B. Glimelius, J. Bergh, O. Brodin, M.-L. Fjällskog, H. Hagberg, A. von Heideman, R. Larsson, B. Tholander, M. de la Torre, G. Aström, K. Oberg, G. Parö and P. Nygren, Med. Oncol., 2002, 19, 151–159 CrossRef CAS PubMed.
- Y. Fang and R. M. Eglen, SLAS Discovery, 2017, 22, 456–472 CrossRef CAS PubMed.
- S. Breslin and L. O'Driscoll, Drug Discovery Today, 2013, 18, 240–249 CrossRef CAS PubMed.
- K. E. Sung, G. Su, C. Pehlke, S. M. Trier, K. W. Eliceiri, P. J. Keely, A. Friedl and D. J. Beebe, Biomaterials, 2009, 30, 4833–4841 CrossRef CAS PubMed.
- S. R. Caliari and J. A. Burdick, Nat. Methods, 2016, 13, 405–414 CrossRef CAS PubMed.
- V. van Duinen, S. J. Trietsch, J. Joore, P. Vulto and T. Hankemeier, Curr. Opin. Biotechnol., 2015, 35, 118–126 CrossRef CAS PubMed.
- W. J. Polacheck, R. Li, S. G. M. Uzel and R. D. Kamm, Lab Chip, 2013, 13, 2252–2267 RSC.
- A. Boussommier-Calleja, R. Li, M. B. Chen, S. C. Wong and R. D. Kamm, Trends Cancer, 2016, 2, 6–19 CrossRef PubMed.
- A. M. Malek and S. Izumo, J. Cell Sci., 1996, 109, 713–726 CrossRef CAS PubMed.
- N. E. Ajami, S. Gupta, M. R. Maurya, P. Nguyen, J. Y.-S. Li, J. Y.-J. Shyy, Z. Chen, S. Chien and S. Subramaniam, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 201707517 CrossRef PubMed.
- B. M. Baker, B. Trappmann, S. C. Stapleton, E. Toro and C. S. Chen, Lab Chip, 2013, 13, 3246–3252 RSC.
- P. Fernandez and A. R. Bausch, Integr. Biol., 2009, 1, 252–259 CrossRef CAS PubMed.
- J. A. Hubbell, Nat. Mater., 2008, 7, 1–2 CrossRef PubMed.
- M. Ahearne, Interface Focus, 2014, 4, 20130038 CrossRef PubMed.
- B. M. Gillette, J. A. Jensen, B. Tang, G. J. Yang, A. Bazargan-Lari, M. Zhong and S. K. Sia, Nat. Mater., 2008, 7, 636–640 CrossRef CAS PubMed.
- F. Grinnell, Trends Cell Biol., 2000, 10, 362–365 CrossRef CAS PubMed.
- M. E. Smithmyer, L. A. Sawicki and A. M. Kloxin, Biomater. Sci., 2014, 2, 634–650 RSC.
- E. Bell, B. Ivarsson and C. Merrill, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1274–1278 CrossRef CAS PubMed.
- M. Yamato, E. Adachi, K. Yamamoto and T. Hayashi, J. Biochem., 1995, 117, 940–946 CrossRef CAS.
- S. I. Montanez-Sauri, K. E. Sung, J. P. Puccinelli, C. Pehlke and D. J. Beebe, J. Lab. Autom., 2011, 16, 171–185 CrossRef CAS PubMed.
- D. Kim and A. E. Herr, Biomicrofluidics, 2013, 7, 1–47 CrossRef PubMed.
- A. Shakeri, N. A. Jarad, S. Khan and T. F. Didar, Anal. Chim. Acta, 2022, 1209, 339283 CrossRef CAS PubMed.
- C. Cha, E. Antoniadou, M. Lee, J. H. Jeong, W. W. Ahmed, T. A. Saif, S. A. Boppart and H. Kong, Angew. Chem., Int. Ed., 2013, 52, 6949–6952 CrossRef CAS PubMed.
- Z. Yue, X. Liu, P. J. Molino and G. G. Wallace, Biomaterials, 2011, 32, 4714–4724 CrossRef CAS PubMed.
- A. Gokaltun, M. L. Yarmush, A. Asatekin and O. B. Usta, Technology, 2017, 05, 1–12 CrossRef PubMed.
- Y. Wang, H. H. Lai, M. Bachman, C. E. Sims, G. P. Li and N. L. Allbritton, Anal. Chem., 2005, 77, 7539–7546 CrossRef CAS PubMed.
- W. J. Polacheck, L. Kutys, J. Yang, J. Eyckmans, Y. Wu, H. Vasavada, K. K. Hirschi and C. S. Chen, Nature, 2017, 552, 258–262 CrossRef CAS PubMed.
- W. J. Polacheck, M. L. Kutys, J. B. Tefft and C. S. Chen, Nat. Protoc., 2019, 14, 1425–1454 CrossRef CAS PubMed.
- W. S. R. Lago, C. Aymes-Chodur, A. P. Ahoussou and N. Yagoubi, J. Mater. Sci., 2017, 52, 6879–6904 CrossRef CAS.
- P. S. Nunes, P. D. Ohlsson, O. Ordeig and J. P. Kutter, Microfluid. Nanofluid., 2010, 9, 145–161 CrossRef CAS.
- J. Raj, G. Herzog, M. Manning, C. Volcke, B. D. MacCraith, S. Ballantyne, M. Thompson and D. W. M. Arrigan, Biosens. Bioelectron., 2009, 24, 2654–2658 CrossRef CAS PubMed.
- C. Jönsson, M. Aronsson, G. Rundström, C. Pettersson, I. Mendel-Hartvig, J. Bakker, E. Martinsson, B. Liedberg, B. MacCraith, O. Öhman and J. Melin, Lab Chip, 2008, 8, 1191–1197 RSC.
- D. Sung, D. H. Shin and S. Jon, Biosens. Bioelectron., 2011, 26, 3967–3972 CrossRef CAS.
- Y. Qi, Y. Wang, C. Chen, C. Zhao, Y. Ma and W. Yang, ACS Appl. Bio Mater., 2020, 3, 3203–3209 CrossRef CAS PubMed.
- R. K. Jena and C. Y. Yue, Biomicrofluidics, 2012, 6, 1–12 CrossRef PubMed.
- S. Roy, C. Y. Yue, S. S. Venkatraman and L. L. Ma, Sens. Actuators, B, 2013, 178, 86–95 CrossRef CAS.
- F. Luna-Vera, J. D. Ferguson and J. C. Alvarez, Anal. Chem., 2011, 83, 2012–2019 CrossRef CAS PubMed.
- Y. Shin, S. Han, J. S. Jeon, K. Yamamoto, I. K. Zervantonakis, R. Sudo, R. D. Kamm and S. Chung, Nat. Protoc., 2012, 7, 1247–1259 CrossRef CAS PubMed.
- L. Wang, B. Sun, K. S. Ziemer, G. A. Barabino and R. L. Carrier, J. Biomed. Mater. Res., Part A, 2010, 93, 1260–1271 Search PubMed.
- Y. Shin, H. Kim, S. Han, J. Won, H. E. Jeong, E. S. Lee, R. D. Kamm, J. H. Kim and S. Chung, Adv. Healthcare Mater., 2013, 2, 790–794 CrossRef CAS.
- S. Chung, R. Sudo, I. K. Zervantonakis, T. Rimchala and R. D. Kamm, Adv. Mater., 2009, 21, 4863–4867 CrossRef CAS PubMed.
- A. Chhabra, H. H. G. Song, K. A. Grzelak, W. J. Polacheck, H. E. Fleming, C. S. Chen and S. N. Bhatia, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 1–11 CrossRef.
- Y. J. Chuah, Y. T. Koh, K. Lim, N. V. Menon, Y. Wu and Y. Kang, Sci. Rep., 2015, 5, 1–12 Search PubMed.
- T. B. Stachowiak, F. Svec and J. M. J. Fréchet, Chem. Mater., 2006, 18, 5950–5957 CrossRef CAS.
- T. Rohr, D. F. Ogletree, F. Svec and J. M. J. Frøchet, Adv. Funct. Mater., 2003, 13, 264–270 CrossRef CAS.
- G. T. Hermanson, Bioconjugate techniques, 2013 Search PubMed.
- J. M. Ayuso, M. Virumbrales-Muñoz, A. Lacueva, P. M. Lanuza, E. Checa-Chavarria, P. Botella, E. Fernández, M. Doblare, S. J. Allison, R. M. Phillips, J. Pardo, L. J. Fernandez and I. Ochoa, Sci. Rep., 2016, 6, 1–16 CrossRef.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- M. Virumbrales-Muñoz, J. M. Ayuso, A. Lacueva, T. Randelovic, M. K. Livingston, D. J. Beebe, S. Oliván, D. Pereboom, M. Doblare, L. Fernández and I. Ochoa, Sci. Rep., 2019, 9, 1–14 CrossRef PubMed.
- P. Pakshir and B. Hinz, Matrix Biol., 2018, 68–69, 81–93 CrossRef CAS PubMed.
- I. Beaulieu, M. Geissler and J. Mauzeroll, Langmuir, 2009, 25, 7169–7176 CrossRef CAS PubMed.
- P. Ganser, C. Baum, D. Chargin, A. F. Sauer-Budge and A. Sharon, Biomed. Microdevices, 2018, 20, 24 CrossRef.
- C. E. O'Neil, S. Taylor, K. Ratnayake, S. Pullagurla, V. Singh and S. A. Soper, Analyst, 2016, 141, 6521–6532 RSC.
- A. U. Haq, A. Boyd, J. Acheson, J. McLaughlin and B. J. Meenan, Surf. Coat. Technol., 2019, 362, 185–190 CrossRef.
- Å. Ö. B. Johansson, A. Larsson and A. Ocklind, J. Appl. Polym. Sci., 2002, 86, 2618–2625 CrossRef.
- A. J. S. Ribeiro, A. K. Denisin, R. E. Wilson and B. L. Pruitt, Methods, 2016, 94, 51–64 CrossRef CAS PubMed.
- A. Oláh, H. Hillborg and G. J. Vancso, Appl. Surf. Sci., 2005, 239, 410–423 CrossRef.
- S. J. Hwang, M. C. Tseng, J. R. Shu and H. Her Yu, Surf. Coat. Technol., 2008, 202, 3669–3674 CrossRef CAS.
- A. Siddique, T. Meckel, R. W. Stark and S. Narayan, Colloids Surf., B, 2017, 150, 456–464 CrossRef CAS PubMed.
- Y. J. Chuah, S. Kuddannaya, M. H. A. Lee, Y. Zhang and Y. Kang, Biomater. Sci., 2015, 3, 383–390 RSC.
- Z. Qian, D. Ross, W. Jia, Q. Xing and F. Zhao, Bioact. Mater., 2018, 3, 167–173 CrossRef PubMed.
- Y. H. Kim, N. S. Baek, Y. H. Han, M. A. Chung and S. D. Jung, J. Neurosci. Methods, 2011, 202, 38–44 CrossRef CAS PubMed.
- W. Liu, K. Han, M. Sun and J. Wang, Lab Chip, 2019, 19, 3162–3167 RSC.
- Y. Bai, C. G. Koh, M. Boreman, Y. J. Juang, I. C. Tang, L. J. Lee and S. T. Yang, Langmuir, 2006, 22, 9458–9467 CrossRef CAS PubMed.
- J. Yakovleva, R. Davidsson, A. Lobanova, M. Bengtsson, S. Eremin, T. Laurell and J. Emnéus, Anal. Chem., 2002, 74, 2994–3004 CrossRef CAS PubMed.
- S. L. Das, B. P. Sutherland, E. Lejeune, J. Eyckmans and C. S. Chen, Am. J. Physiol., 2022, 323, H738–H748 CAS.
- A. Shakeri, S. Khan and T. F. Didar, Lab Chip, 2021, 21, 3053–3075 RSC.
- Y. Qi, Y. Wang, C. Chen, C. Zhao, Y. Ma and W. Yang, ACS Appl. Bio Mater., 2020, 2020, 3203–3209 CrossRef PubMed.
- Q. Pu, O. Oyesanya, B. Thompson, S. Liu and J. C. Alvarez, Langmuir, 2007, 23, 1577–1583 CrossRef CAS PubMed.
- S. Kuddannaya, Y. J. Chuah, M. H. A. Lee, N. V. Menon, Y. Kang and Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 9777–9784 CrossRef CAS PubMed.
- P. J. Wipff, H. Majd, C. Acharya, L. Buscemi, J. J. Meister and B. Hinz, Biomaterials, 2009, 30, 1781–1789 CrossRef CAS PubMed.
- R. B. Vernon and E. Helene Sage, J. Cell. Biochem., 1996, 60, 185–197 CrossRef CAS PubMed.
- V. L. Cross, Y. Zheng, N. Won Choi, S. S. Verbridge, B. A. Sutermaster, L. J. Bonassar, C. Fischbach and A. D. Stroock, Biomaterials, 2010, 31, 8596–8607 CrossRef CAS PubMed.
- S. Raz, D. Sheban, N. Gonen, M. Stark, B. Berman and Y. G. Assaraf, Cell Death Dis., 2014, 5, 1–9 Search PubMed.
- T. Stanković, T. Ranđelović, M. Dragoj, S. Stojković Burić, L. Fernández, I. Ochoa, V. M. Pérez-García and M. Pešić, Drug Resistance Updates, 2021, 55, 100753 CrossRef PubMed.
- S. J. Edwards, V. Carannante, K. Kuhnigk, H. Ring, T. Tararuk, F. Hallböök, H. Blom, B. Önfelt and H. Brismar, Front. Mol. Biosci., 2020, 7, 1–10 CrossRef PubMed.
- S. Nath and G. R. Devi, Pharmacol. Ther., 2016, 163, 94–108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lc00075c |
| This journal is © The Royal Society of Chemistry 2023 |