
Catalytic depolymerization of polyester plastics toward closed-loop recycling and upcycling
Yujing
Weng†
 ab,
Cheng-Bin
Hong†
a,
Yulong
Zhang
*b and
Haichao
Liu
*a
ab,
Cheng-Bin
Hong†
a,
Yulong
Zhang
*b and
Haichao
Liu
*a
aBeijing National Laboratory for Molecular Science, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, PR China. E-mail: haicliu@pku.edu.cn
bHenan Key Laboratory of Coal Green Conversion, College of Chemistry and Chemical Engineering, Henan Polytechnic University, Jiaozuo, Henan 454000, PR China. E-mail: zhangyulong@hpu.edu.cn
First published on 22nd November 2023
Abstract
Plastic waste is globally ubiquitous and ecologically harmful, but it can be recycled as an abundant carbon source to alleviate worldwide heavy dependence on fossil resources and reduce CO2 emissions. Therefore, research into the chemical recycling of plastic waste has become a critical and pressing area. Compared with polyolefins, polyesters, as represented by PET and PLA, can easily achieve selective depolymerization to their corresponding monomers due to the presence of weaker ester bonds, thus favoring their closed-loop recycling and upcycling. However, comprehensive reviews on this important topic remain scarce, especially from the standpoint of re/upcycling. In this review, we present significant progress in the catalytic depolymerization of different polyesters, including biodegradable polyesters and nonbiodegradable polyesters, and discuss the key factors that limit the efficacies of the different methods and formidable challenges towards closed-loop recycling and upcycling. Such insightful discussion may benefit the further development of advanced strategies to address the problems with the increasing polyester plastic wastes and stimulate their efficient recycling to value-added chemicals and materials.
1. Introduction
Today, plastics are used in almost all products. Consequently, plastic waste has become a globally ubiquitous and ecological threat.1 In 2020, global annual plastic waste production was about 367 million metric tons (Mt), and the total plastic waste is predicted to reach 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mt by 2050.2,3 Currently, less than 10% of plastic waste is recycled, with about 10% incinerated and the remaining 80% sent to landfill or randomly discarded in the environment.4 But landfill and incineration do not solve the problem of unsustainable plastics and ultimately contribute to environmental pollution by releasing substances such as greenhouse gases. Moreover, accumulating and poorly biodegradable plastic pollution is a serious global environmental problem, especially the bioaccumulation of micro- and nano-plastics in living organisms throughout the food web.5–7 Current consumption patterns of plastics have led to a large amount of plastic waste (e.g., packaging materials), which is becoming a major contributor to environmental pollution in solid waste streams. Clearly, plastic recycling is an urgent global issue concerning carbon neutrality and environmental remediation.8,9
000 Mt by 2050.2,3 Currently, less than 10% of plastic waste is recycled, with about 10% incinerated and the remaining 80% sent to landfill or randomly discarded in the environment.4 But landfill and incineration do not solve the problem of unsustainable plastics and ultimately contribute to environmental pollution by releasing substances such as greenhouse gases. Moreover, accumulating and poorly biodegradable plastic pollution is a serious global environmental problem, especially the bioaccumulation of micro- and nano-plastics in living organisms throughout the food web.5–7 Current consumption patterns of plastics have led to a large amount of plastic waste (e.g., packaging materials), which is becoming a major contributor to environmental pollution in solid waste streams. Clearly, plastic recycling is an urgent global issue concerning carbon neutrality and environmental remediation.8,9
Plastics are currently recycled through primary, secondary, tertiary, and energy recycling methods (Fig. 1).10–12 Primary recycling refers to the reprocessing of plastic, to produce a product that serves the purpose identical to the original plastic.10 For example, new polyethylene terephthalate (PET) bottles can be manufactured from recycled PET bottles. Secondary recycling refers to the physical processing of plastic waste and reuse, also known as mechanical recycling, but the value of recycled plastic products typically declines, with different use cases.13,14 For example, recycled low-molecular weight PET is used in fibre production. Mechanical processes such as sorting, grinding, cleaning, and extrusion are involved in both primary and secondary recycling methods, but mechanical reprocessing results in thermal and mechanical degradation of the polymers to varying degrees, therefore, the number of cycles of primary and secondary recycling is limited by polymer degradation.10,13 The incineration of plastic waste, also known as energy recycling, can partially recover energy in the form of heat. Although this method does not require recycling, CO2 and other harmful gases are released during combustion, with significantly less energy generated by the burning of plastics compared with the energy saved by recycling plastics.15 Therefore, instead of benefitting the economy, the incineration of plastic waste will exacerbate environmental pollution and resource consumption. Chemical recycling, or tertiary recycling, is the process of breaking down plastic waste into chemicals through chemical reactions, which can be either used to produce the same plastics with virgin-like material properties (i.e., closed-loop recycling) or other useful materials (i.e., upcycling).
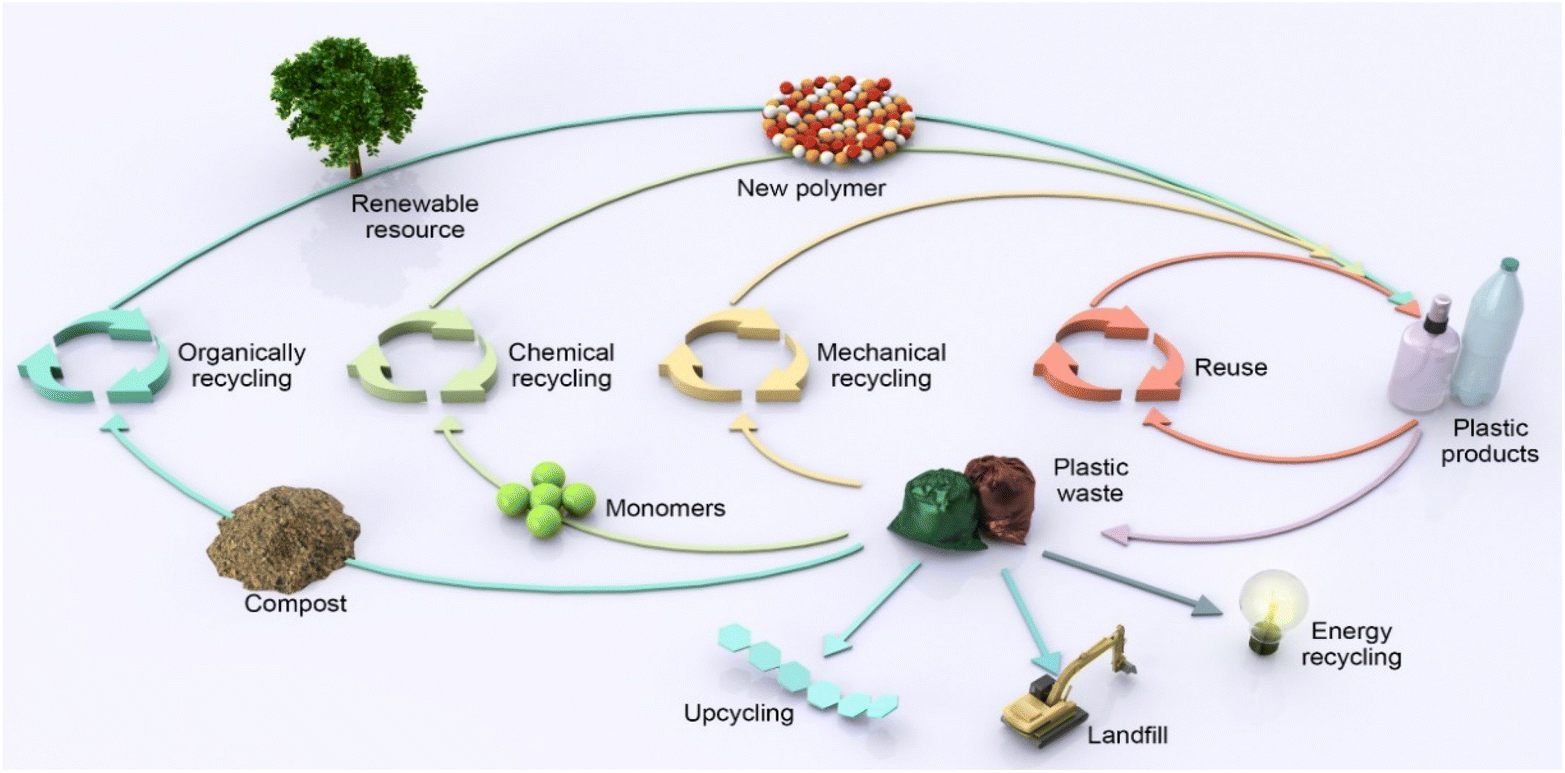 | ||
| Fig. 1 Recycling routes of polyester plastics. | ||
It is important to position plastic wastes as low-cost, abundant raw chemical materials and consider them as the beginning rather than the end of value chain.16 To this end, high-purity monomers or chemicals obtained from chemical depolymerization of plastic wastes can be recombined into new plastics via closed-loop recycling or other value-added products via upcycling.11 Primarily, it is difficult to adapt the design of traditional polyolefins into closed-loop recycling. Polyolefins (Fig. 2), such as polyethylene (PE), polypropylene (PP), and polystyrene (PS), are the most widely used petroleum-based polymer materials, with a global market size of USD 240 billion in 2020. Generally, polyolefin monomers are joined by C–C bonds, with the monomer skeleton structure also composed of C–C bonds. Thus, the selective recovery of monomers by polyolefin depolymerization is challenging under moderate reaction conditions, as the bond energies of the different C–C bonds are basically the same.11,17 Although some recent advances have been made in polyolefin depolymerization, with the capture of fuel additives such as alkanes and aromatics through the catalytic pyrolysis of waste polyolefins, this cannot solve the recycling issue.17,18 Therefore, there is still a need to produce new plastic from non-renewable resources including petroleum and coal.19 Moreover, if plastic waste is converted to fuel, the carbon atoms in the plastic will still end up in the atmosphere, which is not conducive to carbon neutrality in terms of carbon footprint.
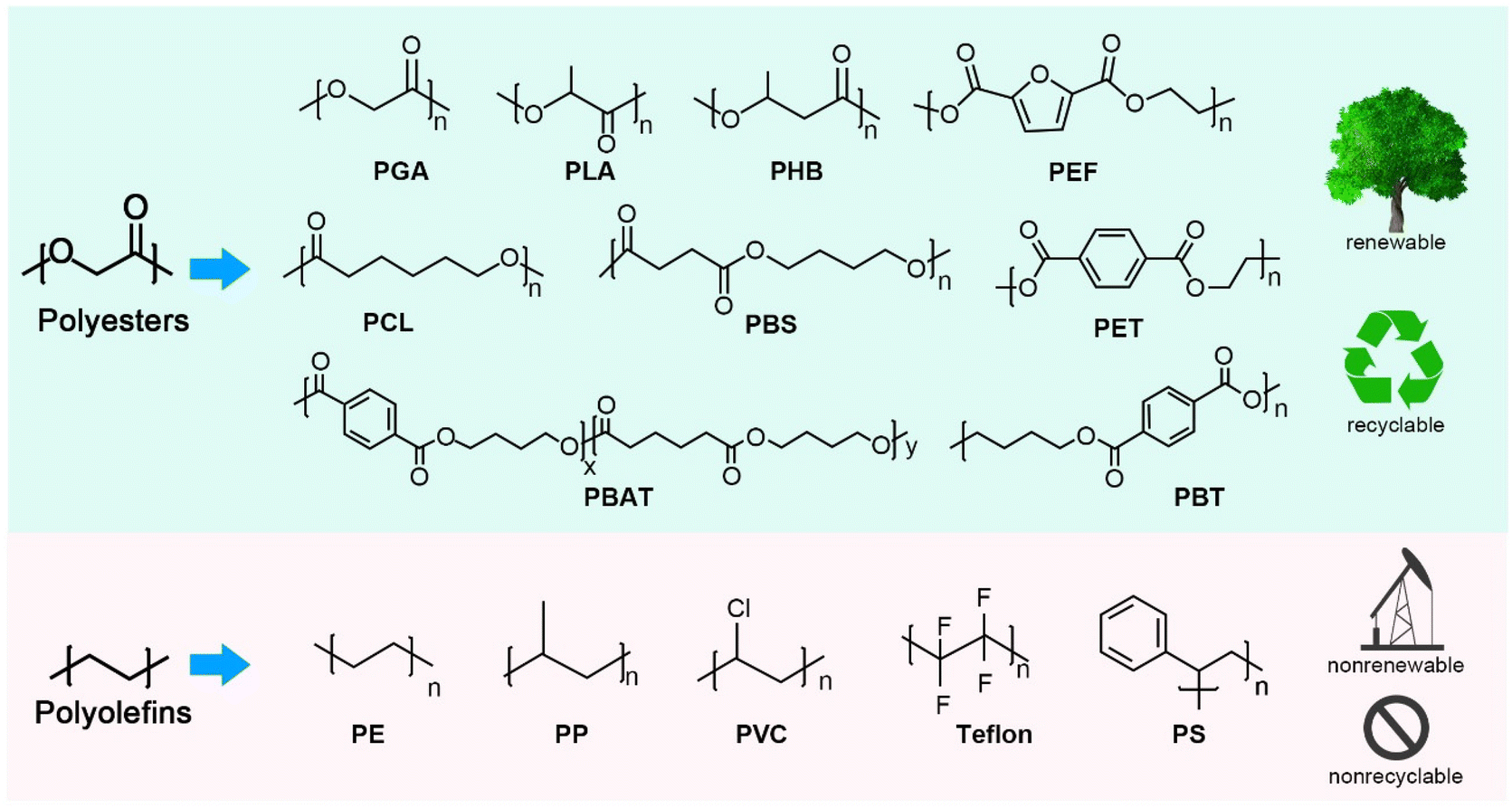 | ||
| Fig. 2 Comparison between polyesters and polyolefins. | ||
Compared with polyolefins, polyesters can be more easily depolymerized to their monomers via highly selective breakage of the ester bonds.20–23 More importantly, most polyesters, such as poly(lactic acid) (PLA), poly(3-hydroxybutyrate) (PHB), polycaprolactone (PCL), poly(ethylene-2,5-furandicarboxylate) (PEF), and PET, can be produced from a variety of sources, including non-renewable and renewable resources, and have become more attractive due to their good thermal and mechanical properties, processing versatility, and degradability (Fig. 2).24–27 Among these, PET is the most important and abundant one, and is widely used as thermoplastic polymers with excellent properties, including thermal stability, transparency, high strength, low density, chemical resistance and low cost.28 PBT and PEF are structurally and functionally similar to PET, and possess unique advantages in physicochemical properties.29,30 For example, PBT can be a viable substitute for the crystalline thermoplastics (e.g., PE and nylon) because of its lower melting point, lower strength and stiffness, lower glass transition temperature, and better impact resistance. More significantly, aliphatic-based polyesters are known as biodegradable plastics and prone to be degraded by natural microorganisms to water and dioxide. Among the biodegradable plastics, PLA is a promising industrial thermoplastic with the highest market share.29 Nowadays, polymer blending techniques have been used for PLA modification to improve its biodegradation rate and toughness for different applications.31 Polyhydroxyalkanoates (PHAs) are naturally synthesized by biological methods, and have attracted widespread interest in biomedical applications because of their excellent biodegradability and biocompatibility.32,33 PHB, as a target molecule from plant cells, is the simplest of PHAs. Aliphatic co-polyesters such as PBA, PBSA, PBAT, and PCL are also biodegradable and available from non-renewable and renewable resources, and are commercial thermoplastic polyesters with excellent manufacturing performance, flexibility, and toughness.34,35
Taken together, polyesters, as represented by PET and PLA, are important polymer materials, and their market size is rapidly growing with promising potential to replace polyolefins and use for preparing electronics, food and beverages, healthcare, and consumer goods.36–39 Therefore, it is important to study the catalytic depolymerization of waste polyester plastics toward closed-loop recycling and upcycling. Recently, a number of comprehensive reviews on plastic recycling have appeared.10,11,24,28,40,41 However, most previous reviews have mainly focused on summarizing the depolymerization methods of polyesters with less attention to the correlation between their depolymerization and re/upcycling. Meanwhile, the corresponding challenges still need to be clearly discussed, especially from the standpoint of closed-loop recycling and upcycling. In this review, we will summarize the significant progress in the catalytic depolymerization of various polyesters, and discuss the key factors limiting the efficacies of these methods and the formidable challenges in closed-loop recycling and upcycling. Such insightful discussion may benefit the further development of advanced strategies to address the problems of increasing amounts of waste polyester plastics. In addition, this review can also be used as a helpful guide for the recycling and upcycling of polycarbonate, polyurethane, polyamide, polyether, and other polyester-like polymers with easily decomposable chemical bonds.
2. Closed-loop recycling
Various technologies have been reported for the degradation of polyester, including gasification, pyrolysis, hydrogenolysis, enzymolysis, hydrolysis, and alcoholysis.16,42 While gasification, hydrogenolysis and pyrolysis can efficiently decompose plastics into chemicals or fuel additives, their products are difficult to use directly to synthesize new polyesters, making it difficult to achieve closed-loop recycling.43–45 Although chemical reagents are not required in pyrolysis, most pyrolysis processes require harsh reaction temperatures, which can easily lead to side reactions such as monomer dehydration and racemization.46,47 Hydrogenolysis uses hydrogen to depolymerize polyesters and produce saturated alcohols and alkanes in the presence of costly noble metal catalysts, which also rely on harsh reaction conditions and toxic reagents, and are not suitable for closed-loop recycling.48 Therefore, in view of closed-loop recycling, alcoholysis, hydrolysis, and enzymolysis will be mainly discussed below due to their capability to selectively cleave ester bonds to the corresponding monomers.2.1 Alcoholysis of polyesters
For polyester alcoholysis, methanolysis, i.e., using methanol as the solvent, compared with ethanolysis and glycolysis, exhibits the higher reactivity, due to the smaller steric hindrance and stronger nucleophilic ability of methanol, and thus it is widely reported in the depolymerization of polymers.49–55Collinson et al. reported the methanolysis and ethanolysis of PLA over the Zn(OAc)2 catalyst at the boiling point of the solvent, showing 70% methyl lactate and 21% ethyl lactate yields, respectively, under reflux conditions for 15 h.56 Zn–N-heterocyclic carbene alkoxide complexes ([(S, CNHC)ZnCl(OBn)]2 and [(O, CNHC)ZnCl(OBn)]2) can also efficiently depolymerize PLA to methyl lactate and oligomers in methanol at room temperature through extensive transesterification reactions.57 Magnesium and calcium alkoxides were also found to be efficient for the depolymerization of PLA, and 89% conversion of PLA and 86% yield of ethyl lactate (2.05 L) were achieved under the optimized reaction condition.51
Basic organic molecules, typically nitrogenous bases, have also been used in methanolysis. By employing triazabicyclodecene (TBD) as an organocatalyst, Leibfarth et al. reported PLA depolymerization in a methanol and methylene chloride mixture via rapid and quantitative transesterification, obtaining an optimal 95% yield of methyl lactate in only 2 min.58 Zinck et al. also used TBD and anhydrous toluene to depolymerize PLA to form oligomers with a tunable microstructure at 105 °C.59 Moreover, commercial-grade PGA resin obtained complete PGA depolymerization in only 30 min at 120 °C.58 In addition to TBD, Alberti et al. found that organic bases such as 4-dimethylaminopyridine (DMAP), triethylenediamine (DABCO), and diazacyclodecene (DBU) could serve as effective catalysts for the methanolysis of PLA to methyl lactate.60 Besides, they also used methanol as the only solvent, obtaining efficient PLA depolymerization at a relatively high temperature of 180 °C with a monomer yield of above 99% in 10 min. Brønsted acidic ionic liquids (ILs) of 1-butyl-3-methylimidazolium hydrogen sulfate ([BMim]HSO4) and 1-methyl-3-(3-sulfopropyl)-imidazolium hydrogen sulfate ([MimPS]HSO4) were reported in PLA depolymerization, and obtained 87.9% and 88.7% yields of methyl lactate at 115 °C, respectively.41,63 [MimPS]HSO4 was found to effectively catalyze PHB methanolysis with an 83% yield of methyl 3-hydroxybutyrate at 140 °C.64 In addition, the Lewis acid of ferric chloride (FeCl3) showed efficient catalytic performance in the methanolysis of PLA waste (Table 1, 87.2%).65 Moreover, Fe-containing magnetic IL (1-butyl-3-methylimidazolium tetrachloroferrate, [BMim]FeCl4) was found to exhibit high reactivity, catalyzing PLA methanolysis at 120 °C with 94.6% yield of methyl lactate, and PHB methanolysis at 140 °C achieved 94.6% yield of methyl 3-hydroxybutyrate.66,67 Furthermore, the Brønsted–Lewis acidic IL of 1-(3-sulfonic acid)-propyl-3-methylimidazole ferric chloride ([MIMPS]FeCl4) was found to be effective for the methanolysis of PHB with a yield of 87.4% for methyl 3-hydroxybutyrate under optimal conditions.68 In addition, the combination of acidic ILs and alkaline ILs was reported in the conversion. For example, 1-butyl-3-methylimidazolium acetate-promoted zinc acetate (2[BMim][OAc]-Zn(OAc)2) effectively co-catalyzed the methanolysis of PLA with an optimal methyl lactate yield of 92%.69 In addition to imidazole-cation-derived ILs, imidazole-anion-derived ILs have been synthesized and used in the alcoholysis of polyester. For example, 2-methylimidazolium-anions and DBU cations ([HDBU][2-MeIm]) showed an efficient catalytic performance in the depolymerization of PLA, PHB, PC, and PET at relatively low temperatures, with optimal yields of 87, 33, 96, and 75%, respectively, for methyl lactate, methyl 3-hydroxybutyrate, bisphenol A, and dimethyl terephthalate.50 Moreover, replacing the anion with acetate and propionate ([HDBU][AA] and [HDBU][PA]) further enhanced the catalytic performance in PLA depolymerization.70
| Catalyst | Solvent | Polymer | T (°C) | t (h) | Conv.a (%)a | Yieldma (%) |
Ref. |
|---|---|---|---|---|---|---|---|
| a T: reaction temperature, t: reaction time, conv.: conversion, yieldm: monomer yield. | |||||||
| Zn(OAc)2 | MeOH | PLA | 64.7 | 15 | — | 70 | 56 |
| Hf(OiPr)2 | MeOH/CH2Cl2 | PLA | 25 | 24 | — | 75 | 61 |
| Mg(OEt)2 | EtOH | PLA | 200 | 1 | 89 | 86 | 51 |
| Zn(HMDS)2 | MeOH | PLA | 25 | 2 | — | 99 | 62 |
| Zn(HMDS)2 | MeOH | PHB | 25 | 24 | — | 93 | 62 |
| TBD | EtOH/CH2Cl2 | PLA | — | 0.03 | 100 | >95 | 58 |
| DMAP | MeOH | PLA | 180 | 0.16 | — | 97 | 60 |
| [Bmim]HSO4 | MeOH | PLA | 115 | 3 | 90.2 | 87.9 | 41 |
| [MimPS]HSO4 | MeOH | PLA | 115 | 3.5 | 97.5 | 88.9 | 63 |
| [MimPS]HSO4 | MeOH | PHB | 140 | 3 | 91.2 | 83.7 | 64 |
| FeCl3 | MeOH | PLA | 130 | 4 | 96.0 | 87.2 | 65 |
| [Bmim]FeCl4 | MeOH | PLA | 120 | 3 | 99.3 | 94.6 | 66 |
| [Bmim]FeCl4 | MeOH | PHB | 140 | 3 | 94.1 | 85.0 | 67 |
| [MimPS]FeCl4 | MeOH | PHB | 140 | 3 | 98.5 | 87.4 | 68 |
| [Bmim]OAc | MeOH | PLA | 115 | 3 | 97.2 | 92.5 | 41 |
| [Bmim]OAc/Zn(OAc)2 | MeOH | PLA | 110 | 2 | 97 | 92 | 69 |
| [HDBU][2-MeIm] | MeOH | PLA | 70 | 1 | 100 | 87 | 50 |
| [HDBU][AA] | MeOH | PLA | 100 | 5 | 100 | 91 | 70 |
| Zn(OAc)2 | EG | PET | 196 | 3 | — | 85.6 | 71 |
| K6SiW11ZnO39 | EG | PET | 185 | 0.5 | 100 | 84 | 72 |
| [Bmim]OH | EG | PET | 190 | 2 | 100 | 71.2 | 73 |
| [Bmim]2[CoCl4] | EG | PET | 175 | 1.5 | 100 | 81.1 | 74 |
| [Bmim]ZnCl3 | EG | PET | 190 | 2 | 100 | 83.3 | 75 |
| [Ch][OAc] | EG | PET | 180 | 4 | 98.2 | 85.2 | 76 |
| 1,3-DMU/Zn(OAc)2 | EG | PET | 190 | 0.33 | 100 | 82.0 | 77 |
| Urea/ZnCl2 DES | EG | PET | 170 | 0.5 | 100 | 82.8 | 78 |
| Mn3O4@SiO2 | EG | PET | 300 | 1.3 | — | 90 | 79 |
| ZnMn2O4 | EG | PET | 260 | 1.3 | 100 | 92.2 | 80 |
| Fe3O4 | EG | PET | 300 | 1 | 100 | 90 | 81 |
| MnO2/HGO | EG | PET | 200 | 0.16 | 100 | 100 | 82 |
| Fe3O4@SiO2@[mim][FeCl4] | EG | PET | 180 | 24 | 100 | 100 | 83 |
| CHTs | EG | PET | 196 | 0.83 | — | 81.3 | 84 |
| CeO2 NPs | EG | PET | 196 | 0.25 | 98.6 | 90.3 | 85 |
For PET depolymerization, methanolysis is also an efficient method, in which methanol attacks the ester bond (180–280 °C, 2–4 MPa) in PET and undergoes transesterification to produce dimethyl terephthalate (DMT) and ethylene glycol (EG).28 Kumazawa et al. reported this reaction in supercritical methanol with a final DMT yield of 97% at 300 °C.86 Zhang et al. also reported on the methanolysis of PET in a high-performance manner under sub-supercritical conditions (200 °C, 3.5 h) without catalysts.87 Furthermore, Kurokawa et al. reported that aluminium triisopropoxide (AIP) may facilitate the methanolysis of PET, but the yields of DMT and EG were strongly dependent on the solubility of PET, with 88.5% DMT and 87.2% EG produced in 20 vol% toluene/methanol mixture at 200 °C.88 Mckeown et al. used a simple organocatalyst (tetramethylammonium methyl carbonate) for the transesterification of polyesters, such as PLA, PCL, PET and PEF.89 In addition, a nano-dispersed ZnO was prepared as a pseudo-homogeneous catalyst and employed for the methanolysis of PET, resulting in 97% DMT after only 15 min at 170 °C.90 However, although methanolysis is effective and promising, it is still not considered to be the best solution for the closed-loop recycling of PET, most likely related to its currently industrial production that is based on terephthalic acid (TPA) and bis(2-hydroxyethyl) terephthalate (BHET). The recycled DMT from PET methanolysis requires an additional transesterification or hydrolysis process to form BHET or TPA for the next PET reproduction, reducing the economic benefits of methanolysis.
Glycolysis is a simple method for commercial PET recycling worldwide, involving the transesterification of PET with an abundance of glycol at temperatures between 100 and 300 °C. The glycolysis agents include ethylene glycol, propylene glycol, diethylene glycol, and 1,4-butanediol.67 Among them, EG, due to its low toxicity and low vapor pressure, as well as the ability of the recovered BHET to repolymerize into new polyesters, represents the most attractive and commonly used agent for PET glycolysis.71,91 In the catalytic mechanism, the initial diffusion of EG into the polymer causes it to swell. Subsequently, a nucleophilic attack by free electrons on the oxygen of EG with the carbonyl carbon of the PET ester results in the formation of a new C–O bond with EG and breakage of the ester C–O bond.90 Next, the degradation of PET will be gradual, beginning with PET oligomers, then transitioning to BHET dimers, and finally to BHET monomers.28,92
Acid and base catalysts have also been found with high activity in PET glycolysis (Table 2), which can be classified as homogeneous and heterogeneous catalysts depending on whether they are soluble in EG. Metal acetates are the most commonly used homogeneous catalysts for PET glycolysis, such as Zn(OAc)2, Mn(OAc)2, Co(OAc)2, and Pb(OAc)2. For example, Zn(OAc)2 can reach the equilibrium state of PET glycolysis in 3–5 h with an optimal BHET yield of 66.9%.71,93 Chen et al. found that the combined use of Zn(OAc)2 and microwave irradiation shortened the equilibration time to 35 min at the same reaction temperature, with the BHET yield of 78% at equilibration.94 In addition, metal chlorides, metal carbonates, metal bicarbonates, and metal sulfates have been used to catalyze PET glycolysis, though they were less catalytic than Zn(OAc)2.91,95 Organic bases such as TBD are also efficient in catalyzing the glycolysis of PET. The corresponding TBD calculation and experimental results show that the hydrogen bonds between TBD and the carbonyl oxygen in PET can cause PET to become activated, thus facilitating PET depolymerization.96 Sardon et al. reported on a protic ionic salt (TBD:MSA (1:1)) formed by an equimolar quantity of TBD and methanesulfonic acid (MSA), which could completely depolymerize PET in less than 2 h with a 91% yield of BHET.97 The protic ionic complex could be recycled at least 5 times and remained stable up to >400 °C. Under mild conditions, the transition-metal-substituted polyoxometalates (POMs) of K6SiW11MO39(H2O) (M = Zn2+, Mn2+, Co2+, Cu2+, and Ni2+) also showed excellent catalytic activities in PET glycolysis.72,98 Zhang et al. conducted PET glycolysis over a K6SiW11ZnO39 catalyst at a low catalyst/PET molar ratio (0.13%) and a high PET/EG weight ratio (1:4), and the yield of BHET was found to be more than 84% under atmospheric pressure at 185 °C for 0.5 h.72 Moreover, the same research team reported on multiple transition metal Zn-substituted polyoxometalates (Na12[WZn3(H2O)2(ZnW9O34)2]) with more active sites, which could obtain complete PET conversion and 84.5% BHET yield at 190 °C for 40 min with a lower catalyst/PET molar ratio (0.018%) and high PET/EG weight ratio (1:4).98 Among the imidazolium-based ionic liquids, metal-containing ionic liquids such as [BMim]2[CoCl4], [BMim]2[ZnCl4], and [BMim]2[ZnCl3] were found with better thermal stability and higher performance than traditional metal catalysts and metal-free ionic liquids such as [BMim]Cl, [BMim]Br, [BMim]HCO3, [BMim]H2PO4, and [BMim]HSO4. This could be attributed to the strong interactions between the ester bond and the metal ions, causing the ester bonds to break more easily.73–75,99,100 In addition, a series of choline-based ionic liquids without metals have been developed for the glycolysis of PET, and under the optimum conditions (180 °C, 4 h), choline acetate ([Ch][OAc]) performed better in the glycolysis of PET, with a BHET yield of 85.2%.76 The promotion may be ascribed to the formation of hydrogen bonds between EG and [Ch][OAc]. Furthermore, deep eutectic solvents (DESs), such as urea/ZnCl2 and 1,3-dimethylurea/Zn(OAc)2 have been developed with good glycolysis performance under mild reaction conditions with corresponding BHET yields up to 83 and 82%, respectively.77,78
| Catalyst | Polymer | T (°C) | t (h) | Conv.a (%) | Yieldma (%) |
Ref. |
|---|---|---|---|---|---|---|
| a T: reaction temperature, t: reaction time, conv.: conversion, yieldm: monomer yield. | ||||||
| — | PHB | 200 | 6 | 100 | 84 | 101 |
| — | PET | 265 | 2 | 100 | ∼100 | 102 |
| — | PLLA | 250 | 0.25 | 100 | 90 | 103 |
| H2SO4 | PHB | 70 | 0.5 | 100 | 50 | 104 |
| H2SO4 | PHB | 200 | 2 | 100 | 73.9 | 43 |
| Zn(OAc)2 | PET | 265 | — | 100 | 100 | 105 |
| H2SO4 | PET | 135 | 5 | — | 90 | 106 |
| H2SO4/H3PO4 | PET | 140 | 2.3 | 100 | 97.8 | 107 |
| HNO3 | PET | 98 | 2.3 | 87.4 | 87.3 | 108 |
| HZSM-5 | PET | 230 | 0.67 | 100 | 100 | 109 |
| NaOH | PET | 99 | 2.5 | — | 85 | 110 |
| NaOH/Na2SO4 | PET | 150 | 1.5 | 98.5 | 98.5 | 111 |
| KOH | PET | 200 | 1 | — | 98 | 112 |
| KOH | PET | 160 | 0.5 | 92.2 | 90.9 | 113 |
| KOH/TOMAB | PET | 95 | 1 | — | 84 | 114 |
| KOH/TBAI | PET | 90 | 0.67 | 100 | 100 | 115 |
| NaOH/PTC | PET | 90 | 1 | — | 99 | 116 |
| NaOH/ethanol | PET | 80 | 0.33 | — | 95 | 117 |
| NaOH | PLLA | 180 | 0.33 | 100 | 100 | 118 |
Compared with the homogeneous catalysts, the heterogeneous catalysts can be readily extracted from the reaction system, thus they have attracted increasing attention in PET glycolysis. Imran et al. investigated PET glycolysis over supported metal oxides, in particular Mn3O4@SiO2 nanocomposites, and obtained a high BHET yield of over 90%.79 Imran et al. further compared the catalytic performance of ZnO, metal oxide spinels, and mixed metal oxide spinels, and found that ZnMn2O4 with a larger surface area and higher acidity afforded a BHET yield of 92.2% at 260 °C.80 They also reported that superparamagnetic γ-Fe3O4 nanoparticles, as a catalyst that can be readily restored, were efficient for PET glycolysis to BHET in more than 90% yield at 300 °C in 1 h.81 However, most of these oxide catalysts required high reaction temperatures (>250 °C) to achieve the complete depolymerization of PET. To address this problem, efforts have been made in developing efficient oxide catalysts for PET glycolysis at lower temperatures. Nanoporous MnO2/HGO nanosheets with a substantial surface area were prepared by the oxidation etching method and demonstrated a complete BHET yield of 100% within a brief 10 min at 200 °C, but required a substantial weight ratio of EG:PET (18.5:1) to drive the depolymerization reaction.82 Paramagnetic ionic liquid-coated SiO2@Fe3O4 nanoparticles (Fe3O4@SiO2@[mim][FeCl4]) were also used as catalyst for PET glycolysis with nearly 100% BHET yield over 12 consecutive reaction cycles at 180 °C for 24 h. Interestingly, the catalyst was readily recovered magnetically without tedious separation or purification processes.83 Chen et al. investigated the Mg–Al hydrotalcite catalyst which, upon calcination at 500 °C, with a Mg/Al molar ratio of 3, showed 81.3% yield of BHET at 196 °C in 50 min, although it tended to deactivate in the recycling tests.84 Recently, Wang et al. reported that ultrafine CeO2 nanoparticles with rich oxygen defects afforded 90.3% BHET yield in 15 min at 196 °C, but their activity also decreased after 3 cycles.85
The polyester alcoholysis by the above catalysts follows a similar reaction mechanism (Fig. 3). Typically, polyesters are first dissolved or swelled in alcohol. Then, the catalyst's cation interacts with –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in the polyester ester, enhancing the carbonyl carbon's electro-positivity. The anion reacts with the H atom in the –OH group of alcohol at the same time, forming the transition state of the six-membered ring. As a result, the O in the –OH group of alcohol will be more electronegative, making it easy to attack the carbonyl carbon in the ester. Thus, the central carbon's hybridization has shifted from sp2 to sp3, creating a tetrahedral middle. Next, the original ester bond (–C–O–) is broken as the hydrogen atom leaves alcohol and electrons on the oxygen atom transfer to form CO again.68
O in the polyester ester, enhancing the carbonyl carbon's electro-positivity. The anion reacts with the H atom in the –OH group of alcohol at the same time, forming the transition state of the six-membered ring. As a result, the O in the –OH group of alcohol will be more electronegative, making it easy to attack the carbonyl carbon in the ester. Thus, the central carbon's hybridization has shifted from sp2 to sp3, creating a tetrahedral middle. Next, the original ester bond (–C–O–) is broken as the hydrogen atom leaves alcohol and electrons on the oxygen atom transfer to form CO again.68
 | ||
| Fig. 3 Proposed catalytic mechanism for the depolymerization of polyester in alcoholysis.68 | ||
Overall, methanolysis is very efficient for the depolymerization of polyesters such as PLA, and the obtained products can be used directly for the synthesis of lactone or lactide which could be used for the ring-opening polymerization (ROP) reaction to produce new polyesters. Glycolysis is the most attractive method for PET recycling, and the corresponding monomeric products can be reused to produce plastics. Catalysts reported for alcoholysis consist of organometallic complexes, organic bases, ionic liquids, and heterogeneous oxide catalysts. However, many challenges still limit their future development and application. Firstly, the homogeneous catalysts are difficult to separate and recover from the alcoholysis system after reaction, and most heterogeneous catalysts exhibit relatively low activity in polyester depolymerization. Clearly, more efficient and green catalysts need to be developed, for example, based on the Earth-abundant and nonnoble metals or oxides. Secondly, alcohols are susceptible to forming ethers under the catalysis of acids. Although methanol is better suited to polyester depolymerization, its biotoxicity and high vapor pressure can cause potential environmental pollution and safety problems. For PET glycolysis, the BHET product is highly soluble in EG solution, leading to the notorious separation problem after the reaction (Fig. 3).
2.2 Hydrolysis of polyesters
Hydrolysis of polyesters refers to the depolymerization of polyesters into monomers in acidic, alkaline, or neutral aqueous solutions (Table 2). However, the hydrolysis of ester bonds in water generally has a higher activation energy than alcoholysis, leading to higher depolymerization temperatures.1 In addition, most polyesters are insoluble in water due to the high degree of polymerization and crystallinity, as well as strong hydrophobicity. Therefore, using solid acids to catalyze polyester hydrolysis is relatively difficult because the catalyst cannot effectively contact with polyester.The simplest polyester hydrolysis process is carried out in neutral aqueous solutions, which, however, require a high reaction temperature (200–400 °C) due to the strong hydrophobicity and rigid structure of the polymer. Saeki et al. found that PHB could undergo autocatalytic hydrolysis under sub-supercritical conditions without any external catalysts, in which carboxylic acid from hydrolysis also further catalyzes the hydrolysis of PHB.101 The results showed that the yield of 3-hydroxybutyric acid (3HB) decreased from 84% at 200 °C to 30% at 220 °C, possibly due to the influence of side reactions such as racemization and decomposition at high reaction temperatures. Campanelli et al. tested the breakdown of liquefied PET in an abundance of water at temperatures exceeding 250 °C and found that PET was fully depolymerized to TPA in 2 h.102 Moreover, the neutral hydrolysis of PLA in sub-supercritical water was investigated, and 90% lactic acid yield was obtained in 20 min at above 250 °C.103 Neutral hydrolysis can avoid the use of acid/base reagents. However, the high-temperature and high-vacuum reaction conditions are harsh, which will cause side reactions and reduce the selectivity of the monomers. As a result, current polyester hydrolysis reactions are mainly under acid or alkali catalysis.
Yu et al. investigated the hydrolysis of PHB by sulfuric acid (H2SO4) and found that neither 3HB nor crotonic acid (CA) was detected in acid solutions (0.1–4 N H+) at 70 °C, while PHB could be completely decomposed in concentrated H2SO4 solutions (80–98 wt%) with CA and 3HB yields of 90% and 2%, respectively.104 Therefore, 3HB was an intermediate with a volcano yield curve with the reaction time, and the optimal yield was about 50%. Bonartsev et al. investigated the hydrolysis reaction kinetics of PLA, PHB, and their derivatives in phosphate buffer at 37 °C and 70 °C.119 Interestingly, the total molecular weight of PHB remained unchanged at the initial stage of hydrolysis, but the atomic force microscopy (AFM) characterization showed macromolecule splits. Due to the large size and hydrophobicity of the PHB fragments, diffusion from the polyester matrix to the aqueous media remained difficult at the initial stage. During the second stage, the PHB molecular weight decreased gradually, and at a critical mass, the PHB fragments dissolved in the aqueous medium. Li et al. studied the conversion of PHB to 3HB and CA monomers under hydrothermal conditions, and found that 3HB and CA further dehydrated and decarboxylated to propylene and CO2 at 200 °C in H2SO4 solution (0.5 mol L−1) for 2 h (Fig. 4a).43
 | ||
| Fig. 4 (a) Proposed reaction network for PHB-to-propylene conversion,43 (b) proposed shrinking-core model for the hydrolysis of PET over Brønsted acids,120 (c) hydrolysis mechanism over alkaline catalysts and surfactants,115 and (d) the hydrolysis mechanism in acid and alkaline solutions.43 | ||
Zn(OAc)2 and other metallic salts with Lewis acidity were also frequently used for PET hydrolysis.105 However, these Lewis acids could undergo significant hydrolysis under hydrothermal conditions. For example, Zn(OAc)2 could be partially hydrolyzed to form acetic acid and zinc hydroxide. Currently, the acid catalysts used in PET hydrolysis mainly consist of strong Brønsted acids, such as sulfuric acid (H2SO4), nitric acid (HNO3), and phosphoric acid (H3PO4). Mancini et al. investigated the acid hydrolysis of post-consumer PET by using a H2SO4 solution (7.5 M), reaching 90% TPA conversion in 5 h at 135 °C.106 However, with increasing H2SO4 concentration, the EG yield decreased, as the strong dehydration effect of H2SO4 could lead to the carbonization of EG.120 To reduce the carbonization degree, H3PO4 was used to replace a part of H2SO4, thereby reducing the sulfuric acid concentration and increasing the EG yield.107 Using HNO3 instead of H2SO4 as an acid catalyst for hydrolysis offered the characteristics of oxidation side reactions. For example, during PET hydrolysis, HNO3 could oxidize the resulting EG into a value-added oxalic acid, which was detrimental to the closed-loop recycling of PET.108 Interestingly, it was reported that introducing Na2SO4 into the reaction solution could minimize the oxidation of nitric acid, leading to a higher EG yield through the ionic exchange reactions which protected EG during its recovery.28,108,111 To understand the Brønsted acid-catalyzed hydrolysis mechanism of PET, Yoshioka et al. proposed a widely accepted shrinking-core model (Fig. 4b).120 The kinetic studies revealed that the hydrolysis reaction took place on the PET powder's surface, influenced by the formation and expansion of pores and cracks, thus indicating that the apparent reaction rate was directly proportional to the concentration of esters and sulfuric acid.
PET materials have strongly hydrophobic and rigid structures, resulting in insufficient contact with solid acids; therefore, PET hydrolysis has been rarely conducted using solid acids. Recently, Cha et al. reported a microwave-assisted hydrolysis method to depolymerize PET into TPA by using ZSM-5-based zeolites as simply recoverable and facilely regenerable catalysts.109 100% TPA yield could be obtained after only 40 min by using the H+@ZSM-5-25 catalyst, and above 90% TPA yield was still maintained after 6 consecutive cycles. In particular, spent catalysts could be simply regenerated into new catalysts after annealing at 823 K for 6 h.
Alkaline hydrolysis with inorganic base catalysts is another commonly used method to hydrolyze polyesters. Yagihashi et al. reported the recovery of L-lactic acid from poly(L-lactic acid) (PLLA) in a dilute aqueous NaOH solution (0.6 mol L−1) at 160 °C for 60 min, and showed that PLLA almost completely converted to L-lactic acid without the formation D-lactic acid.118 Accordingly, the components solubilized from PLLA mainly consisted of L-lactic acid, suggesting that the degradation reaction was controlled by the dissolution of products on the polyester surface rather than by a chemical reaction. Tsuji et al. investigated the hydrolysis of PLLA films in 0.01 M NaOH at 37 °C and found that hydrolysis mainly occurred in the amorphous region via the surface erosion mechanism.121 Bonartsev et al. investigated the effects of molecular weight and morphology on the hydrolysis of PLA, PHB, the PHB–PLA blend, and the copolymer PHBV (20% of 3-hydroxyvalerate) at 37 and 70 °C.119 The results showed that compared with high molecular weight polymers, the low molecular weight polymers had a higher degradation rate, and the hydrolysis activity of polyester decreased with the increasing hydrophobicity. Therefore, the degradation was enhanced according to PHBV < PHB < PHB–PLA blend < PLA. Yu et al. investigated the proportion of PHB precipitate that was broken down into soluble monomeric substances in a sodium hydroxide solution at 70 °C.104 The results indicated that alkaline concentration was an important factor affecting PHB alkaline hydrolysis. The overall PHB degradation rate was found to be less than 5% at low concentrations of NaOH (0.1–0.4 mol L−1), reaching 70% when the NaOH concentration increased to 4 mol L−1. Moreover, a mechanism study suggested that the formation of crotonate did not occur through 3HB dehydration following the hydrolysis or dehydration of 3-hydroxyl groups prior to hydrolysis, but rather through the transient arrangement of a 6-membered ring comprising two adjacent 3HB units. Wan et al. carried out the depolymerization of PET flakes in a KOH solution at 120–160 °C and the ester linkage on the solid PET surface reacted with KOH in the solution, forming EG and terephthalic potassium salt.114,122 Interestingly, a number of surfactants as phase-transfer-catalysts have been found to significantly promote the alkaline hydrolysis of PET, such as trioctylmethyl ammonium bromide (TOMAB) and tetrabutyl ammonium iodide (TBAI).114–116 Hasan et al. reported on the alkaline hydrolysis of PET under microwave irradiation in the presence of quaternary ammonium salt and TBAI, with 99% of TPA obtained under the optimal conditions (10% NaOH, 60 min, 200 W power, and 3% wt./wt. TBAI and PET).115 In this reaction, there exists a solid organic phase and an aqueous phase, so the phase transfer catalyst might work following an interfacial mechanism.115,116 As shown in Fig. 4c, at the interface of the organic and aqueous phases, metal carbanion was created, and then surfactants were used to separate the species from the interface into the organic phase, creating reactive intermediates. As a result, the ester linkage in the PET macromolecule could be more easily attacked by the OH− ion to accelerate the depolymerization. The formed terephthalate anion then returned to the aqueous phase in the form of disodium salt.114 However, the addition of surfactants generally made the separation and recovery process more difficult. In addition to surfactants, Meester et al. found that the addition of ethanol to water could also promote PET alkaline hydrolysis, achieving approximately 95% TPA yield under the optimal condition (60:40 vol% EtOH:H2O, 5 wt% NaOH, 80 °C, 20 min).117 Recently, Wang et al. also reported a two-step alcoholysis and hydrolysis strategy for the efficient and selective depolymerization of PET-like polymers by a catalyst system composed of K3PO4/ethanol with a high degradation efficiency (>95%) under mild degradation conditions (110–130 °C).48
Although polyesters can be hydrolyzed into soluble products in both acids and bases, their tolerance to protons and hydroxyl anions was found to be significantly different (Fig. 4d). Taking PHB as an example, monomeric hydrolysis is rare in acid solutions of 0.1–4 mol L−1. Conversely, hydroxyl anions have the ability to effectively target the PHB backbone at alkali concentrations between 0.1 and 4 mol L−1.104 These hydroxyl anions have the ability to diminish the energy barrier of ester bond breakage, resulting in similar activation energies for the saponification and biodegradation of PHB.109 When an ester bond is broken down into carboxylic acid and alcohol in an alkaline solution, the hydroxyl anions will draw out protons from the acid and create carboxylate ions with a negative charge, which will be thermodynamically advantageous for nucleophilic substitution or re-esterification of the resulting alcohol and acid. Because hydroxyl anions can serve as a reagent rather than a catalyst, in this case, a high alkali concentration will promote the hydrolysis of polyester.1,104 However, in acidic solutions, protons will act as catalysts for hydrolysis and esterification.43,101 Thus, the polyester hydrolysis process yields a combination of carboxylic acid and alcohol, which can be further esterified with protons, and the hydrolysis step is preferred in concentrated acid solutions.
In summary, water is a green and inexpensive solvent for the hydrolysis of polyesters to monomers, and some challenges in this hydrolysis method need to be addressed for its application. Firstly, inorganic acids and bases are generally homogeneous and dissolve in water during the reaction, making product separation difficult and potentially causing serious environmental pollution. Secondly, owing to the weak nucleophilic ability of water molecules compared with alcohol molecules, the hydrolysis activity of polyesters is lower, and thus requires higher temperatures to achieve efficient depolymerization. Meanwhile, some polyester monomers are susceptible to side reactions (e.g., dehydration, decomposition, and racemization) at high temperatures, further lowering their selectivity. Thirdly, although inorganic bases (e.g., NaOH and KOH) can facilitate polyester hydrolysis, they will neutralize the carboxyl groups in the products to generate corresponding carboxylic salts during hydrolysis. As a result, alkali hydrolysis will not only deactivate the catalyst, but also requires the neutralization of the products, which will increase the operational cost of product separation and purification as well as waste discharges.
2.3 Enzymolysis of polyesters
Enzymolysis uses the microbial enzyme system to degrade the polyester polymers, has mild reaction conditions, low energy consumption, good selectivity and environmental friendliness. Thus, many efforts have been recently reported in the discovery of hydrolytic enzymes that can efficiently degrade polyesters, and make it become a viable recycling strategy.123Biodegradable aliphatic polyesters could be degraded and mineralized through natural microorganisms, and eventually convert to carbon dioxide and water. However, there are many factors that still influence the biodegradation process, including the properties of polyester (e.g., molecular weight, crystallinity and impurity), microbial factors (e.g., strain type, number of bacteria and enzymatic properties), and environmental factors (e.g., temperature, humidity, pH and oxygen concentration).124 Even widely used PLA, as an example, takes a long time to fully degrade in ambient conditions. Currently, PLA biodegrades by anoxic composting, which typically requires a composting temperature of about 60 °C and humidity of about 50%.7,125,126 In addition, most biodegradable polyesters are expensive; if they are biodegraded to carbon dioxide and water after one single use, this will waste significant resources and energy, and cause potential harm to environment. Thus, more attention should be paid to enzymatic strategies for the purpose of closed-loop recycling, but few are being reported. Yakunin et al. reported that two microbial carboxyl esterases (ABO2449 and RPA1511) were discovered to be capable of effectively breaking down PLA into lactic acid monomers, dimers, and larger oligomers, instead of CO2, as the end products.127 Myburgh et al. reported that the yeast Saccharomyces cerevisiae was equipped with a fungal cutinase-like enzyme (CLE1) which effectively catalyzed the enzymolysis of various PLA materials, resulting in the liberation of 9.44 g L−1 lactic acid from 10 g L−1 PLA films.128
For aromatic polyesters, such as PET, although they are naturally nonbiodegradable, recent studies showed that the PET enzymolysis is feasible toward closed-loop recycling. Müller et al. demonstrated PET can be depolymerized by a hydrolase (TfH) from the actinomycete Thermobifida fusca.129 Yoshida et al. reported a novel bacterium, Ideonella sakaiensis 201-F6, with an activity up to 120 fold of TfH (Fig. 5a).130 Tournier et al. used computer-aided enzyme engineering to produce a thermostable leaf-branch compost cutinase (LCC), showing a 90% PET conversion in less than 10 hours at 65 °C (Fig. 5b).131 Later, Alper et al. used a structure-based machine learning algorithm to engineer a robust and active FAST-PETase that can completely hydrolyse the untreated postconsumer-PET in 1 week, with a TPA yield of 94.9% at 30–50 °C.132 Similarly, Bell et al. obtained an evolved thermostable HotPETase (Tm = 82.5 °C) with the highest Tm among the active IsPETase derivatives.133 PET is known to be a semi-crystalline compound, and its amorphous region is more flexible at its glass transition temperatures (Tg, 65–71 °C), making it easier for enzymes to contact and react.134
 | ||
| Fig. 5 (a) Unrooted phylogenetic tree of known PET hydrolytic enzymes,130 (b) WTPETase protein structure rendered by the output of MutCompute,131 and (c) possible catalytic mechanism of PET enzymolysis.136 | ||
As to the enzymatic mechanism, Guo et al. proposed a PETase catalytic pathway based on microbe Ideonella sakaiensis (Fig. 5c).135,136 PETase adopts the canonical α/β hydrolase fold, strictly conserved catalytic triad S131-h208-D177, involving nucleophilic groups (e.g., serine), catalytic groups (e.g., aspartic acid) and alkaline groups (e.g., histidine).137 The enzyme's apo-form creates a slight opening for the binding of the substrate to the protein surface, exhibiting different conformations of the W156 side chain. When the enzyme binds PET, the carbonyl group of the 1st benzene ring is oriented towards the core of the substrate binding cleft through a nucleophilic assault, while the oxyanion hole aligns the ester linkage to stabilize the intermediate. The conventional method of cutinase entails the formation of an intermediate acyl enzyme, which is then subjected to a subsequent nucleophilic attack by a water molecule. Once the ester bond is severed, the residual benzoic acid group of the product forms a broad, level surface that can be conveniently piled up in front of the W156 side chain. Afterwards, the product is rotated and moved away from its original stacked T-position before it is released.136
Enzymolysis is superior in terms of its high selectivity, involving a large number of weak non-covalent interactions between substrate and enzyme that essentially impart stability and flexibility to the large molecules.138 Thus, when a polyester interacts weakly with a flexible enzyme, this may facilitate the mass transfer and depolymerization of the polyester chain.139–141 In addition, the short covalent connection between the enzyme and the substrate can trigger the substrate to perform the induced fit, thus promoting the catalytic depolymerization.131,139,142,143 However, challenges such as unbalanced enzyme–substrate interactions, low thermostability, low efficiency at high temperatures, and inhibiting intermediates, still hamper its further applications.134
3. Upcycling of polyesters
Different from closed-loop recycling, upcycling aims to chemically recycle or modify plastic waste to produce new products with higher market values, including small-molecule chemicals and polymer materials. For making small-molecule chemicals, like closed-loop recycling, upcycling also requires the complete depolymerization step. However, the target product is not the original monomer, but new high-value-added chemicals. Upcycling to materials is derived from incomplete depolymerization of polyesters, and involves the controlled degradation and post-functionalization of polyesters to obtain new materials with enhanced properties.16 Unlike polyolefins, polyesters, such as epoxy resins, polyurethanes, and unsaturated polyesters, have active functional groups for targeted deconstruction and subsequent reconstruction into new materials with improved properties.144 Thereby, the upcycling of plastic waste is a multidisciplinary approach that involves various depolymerization methods and techniques.1453.1 Hydrolysis/alcoholysis/enzymolysis of polyesters
Li et al. reported that the process of upcycling poly (3-hydroxybutyrate) (P3HB) into value-added polymerizable monomers, followed by polymerization towards degradable and recyclable polymers through four steps, including the efficient conversion of P3HB into ethyl 3-hydroxybutyrate (HBEt), selective ring-opening with CHO, cyclization to form the new bicyclic monomer 4-methyloctahydro-2H-benzo[b][1,4]dioxepin-2-one (4-MOHB), and its ROP, is made easier (Fig. 6a).146 Wang et al. developed an alternative cyclization-depolymerization strategy for integrating upcycling and closed-loop recycling of aliphatic polyester poly (p-dioxanone) (PPDO) by using an acidic ionic liquid as a catalyst/solvent bifunctional agent at a relatively low reaction temperature of 120 °C (Fig. 6b).147 In addition, they used an “amino-alcoholysis” strategy to upcycle the poly(bisphenol A carbonate) (BPA-PC) into BPA monomers and chiral 2-oxazolidinone chemicals via ZnX2-catalyzed depolymerization by chiral amino alcohols under mild conditions (25 °C, 3 h).148 Wang et al. found that the “polymer to polymer” approach enabled the recycling of PLA plastic waste from its end-of-life to new high-quality PLA materials (Fig. 6c). During depolymerization, organometallic complexes and organic bases were employed for selective methanolysis, while new lactide monomers were added to the repolymerization process to obtain new polyester with a higher molecular weight.149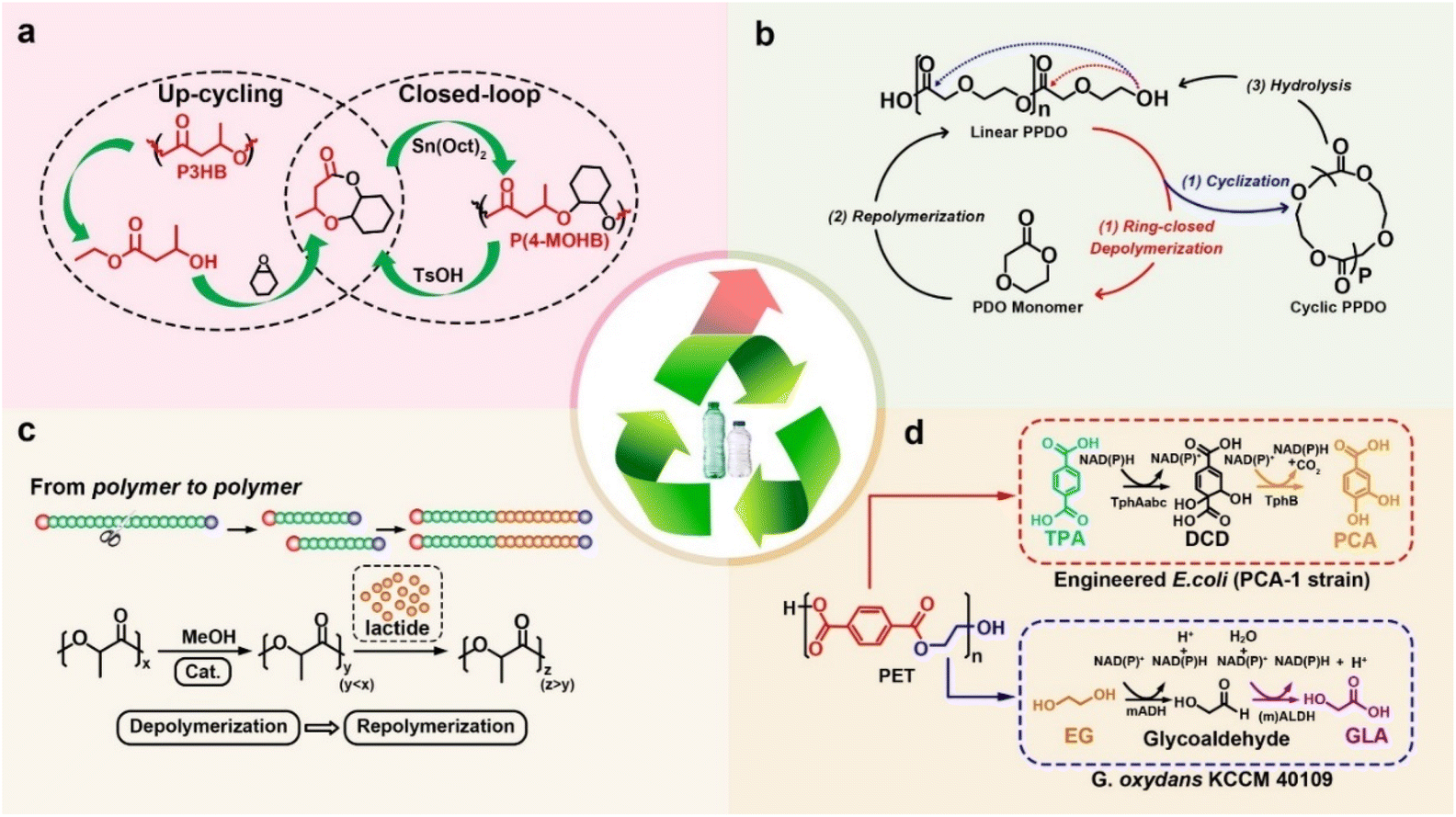 | ||
| Fig. 6 (a) The upcycling of P3HB based on alcoholysis method,146 (b) the upcycling of PPDO based on hydrolysis method,147 (c) the upcycling of PLA based on methanolysis method,149 and (d) the upcycling of PLA based on enzymolysis methods.152 | ||
The combination of hydrolysis/alcoholysis and bio-mediated transformation is also a promising strategy for upcycling polyester waste into new chemicals and polymers. O'Connor et al. reported three strains capable of accumulating medium chain length PHAs from PET hydrolysate, namely, GO16 (Pseudomonas putida), GO19 (Pseudomonas putida), and GO23 (Pseudomonas frederiksbergensis).150 Narancic et al. used GO16, a metabolically versatile Pseudomonas umsongensis, for the upcycling of PET hydrolysate into biodegradable polyester PHAs via microbial cultivation.151 Kim et al. investigated a one-pot chemobioprocess of PET depolymerization, first by depolymerizing PET into a glycolysis slurry on a biocompatible betaine catalyst, then by using IsPETase and IsMHET for enzymatic hydrolysis to produce TPA and EG, and finally through bioconversion to protocatechuic acid and glycolic acid (Fig. 6d).152,153 Wallace et al. upcycled the PET-derived monomer terephthalic acid into value-added small-molecule vanillin using a novel engineered Escherichia coli.154 Recently, Kunjapur et al. used an ω-transaminase from Chromobacterium violaceum (cvTA) to efficiently catalyze amine transfer to potential PET-derived aldehydes, such as terephthaladehyde from TPA and 4-formylbenzoic acid from MHET, to form monoamine para-(aminomethyl) benzoic acid (pAMBA, 70 ± 8% yield) or diamine para-xylylenediamine (pXYL, 69 ± 1% yield), both of which are valuable building blocks for polymeric materials and pharmaceuticals.155
Value-added materials can also be obtained from targeted deconstruction and subsequent reconstruction of PET upcycling. Szilagyi et al. produced desirable metal–organic frameworks (MOFs) with a Uio-66 topology by first depolymerizing PET into terephthalic acid, followed by subsequent purification and MOF synthesis.156 Beckham et al. demonstrated that PET could be upcycled into long-lifetime fibre-reinforced plastics via combination with renewably sourceable monomers.157 In the work, glycolyzed PET was obtained from controlled diol deconstruction by using titanium butoxide, which was reacted with renewably sourceable monomers, such as olefinic diacids and olefinic monoacids, to produce a series of unsaturated polyesters or diacrylic polymers. Similarly, Abdelaal et al. documented the transformation of PET waste into unsaturated polyester through the utilization of PET controlled glycolysis and subsequent interaction with maleic anhydride.158 Pitet et al. employed PET wastes in combination with a bioderived dimer fatty acid to fabricate engineering-grade segmented thermoplastic copolyesters through solvent-free melt polycondensation.159 Clearly, these examples of progress show that upcycling based on closed-loop recycling methods is efficient and promising. This strategy can be further improved by further improving the catalyst activity and stability and product separation efficiency as well as preventing the side reactions of solvents.
3.2 Ammonolysis/aminolysis of polyesters
Upcycling of polyester can also be performed by aminolysis or ammonolysis, involving the electrophilic attack of polyesters by amines or ammonia, more thermodynamically favorable than hydrolysis and alcoholysis.Harad et al. used ethanolamine (EA) with a 1:6 ratio of PET:EA under reflux conditions (180 °C) and yielded bis(2-hydroxyethylene) terephthalamide (BHETA).160 Fukushima, et al. used TBD as the organic catalyst and various amines as the depolymerization media for the aminolysis of wastes PET (Fig. 7a), yielding a versatile library of functional terephthalamides with great potential as building blocks for high-performance materials.37 Specifically, bis-amine-functionalized terephthalamides are desired monomers for highly thermostable polymers such as polyamides, polyimides, polyurethanes, and polyureas.161 Zhang, et al. used 2-aminoethanol for aminolysis of PLA to N-lactoyl ethanolamine, which was then isolated and purified to react with dimethylaminopyridine (DMAP) to synthesize dimethacrylate ester (DME), a new 3D printing material with a tensile strength of 58.6 MPa and a Young's modulus of 2.8 GPa (Fig. 7b).162 Recently, Ma et al. treated PLA with a simple ammonia solution over Ru/TiO2 and achieved a 94% selectivity to alanine at 140 °C, transforming PLA into lactamide and then further into ammonium lactate at higher temperatures.163 The final product alanine is one of the most important amino acids, and is widely used in food, forage, and pharmaceutical applications.
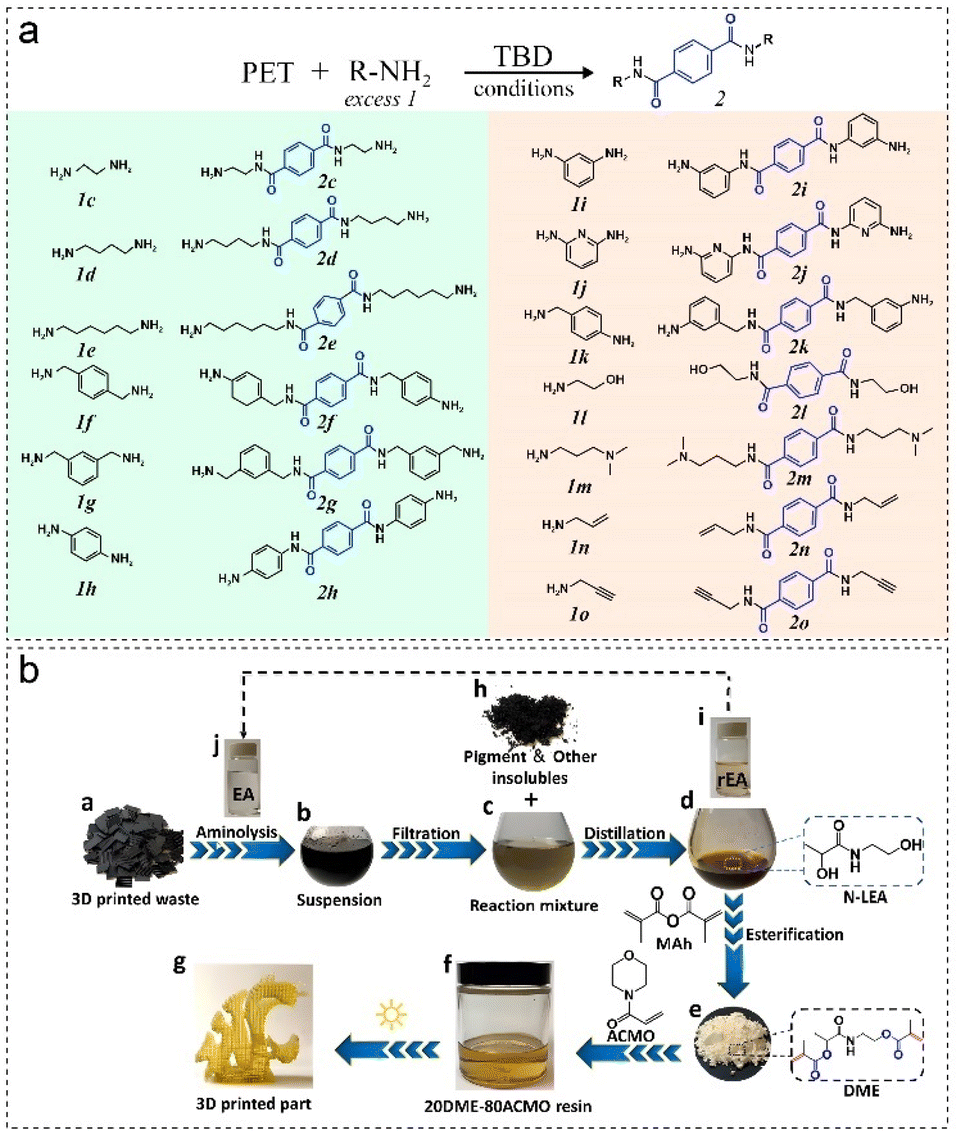 | ||
| Fig. 7 (a) Terephthalamides are produced through the organocatalytic aminolysis of PET,37 and (b) schematic upcycling process from FDM 3D printed PLA waste to MSLA 3D printed photocurable resins.162 | ||
The ammonolysis method facilitates the homogeneous distributions of functional groups on polyester fabrics. Lorusso et al. synthesized zwitterionic polymer brushes through the chemical modification of PET in an attempt to modify its hydrophobicity by the controlled insertion of amide moieties.164 In the work, the PET fabric was prefunctionalized by surface ammonolysis with diethyl amine, and then immobilized in the bromoisobutyryl bromide and TEA mixture to produce zwitterionic brushes by atomic transfer radical polymerization. Karpati et al. carried out ammonolysis of PET with an epoxy hardener isophoron-diamine as the solvent, and obtained 90% yield of the terephthalamide-diamine product over zinc acetate catalyst.165 Moreover, the raw aminolysis product could be further used as a cross-linking agent for epoxy resins without purification.
In summary, on the basis of ammonolysis/aminolysis, polyester can be upgraded to numerous nitrogen-containing chemicals and materials, a process worthy of further development. However, compared with water and alcohol as solvents, amines and ammonia are toxic and have high vapor pressures, which cause potential pollution and corrosion concerns.
3.3 Reductive and oxidative upcycling
Reductive and oxidative upcycling of polyesters generally involves their depolymerization and subsequent reduction and oxidation reactions.Lactic acid can be converted to a series of small molecules with high industrial values by hydrogenation and oxidation reactions, as shown in Fig. 8a, including dehydrating to acrylic acid, condensing/dehydrating to 2,3-pentanedione, decarbonylating/dehydrating to acetaldehyde, reducing to 1,2-propanediol, oxidizing to pyruvic acid, hydrodeoxygenating to methyl propionate, and ammonizing to alanine.161 Therefore, PLA depolymerization and lactic acid conversion can be efficiently coupled, and PLA waste can be upcycled to produce many value-added chemicals. Ma et al. recently reported the valorisation of waste PLA for the production of methyl methacrylate (Fig. 8b). The PLA is initially transformed into methyl propionate using an α-MoC catalyst in a methanol solution, and then the resulting methyl propionate is combined with formaldehyde to create methyl methacrylate (81% conversion, 90% selectivity), an essential monomer of polymethyl methacrylate that is used in paints, coatings, and adhesives.166 Furthermore, under the same reaction conditions, PGA and PCL were successfully converted into methyl acetate with a 94% yield and methyl hexanoate with a 79% yield, respectively.166
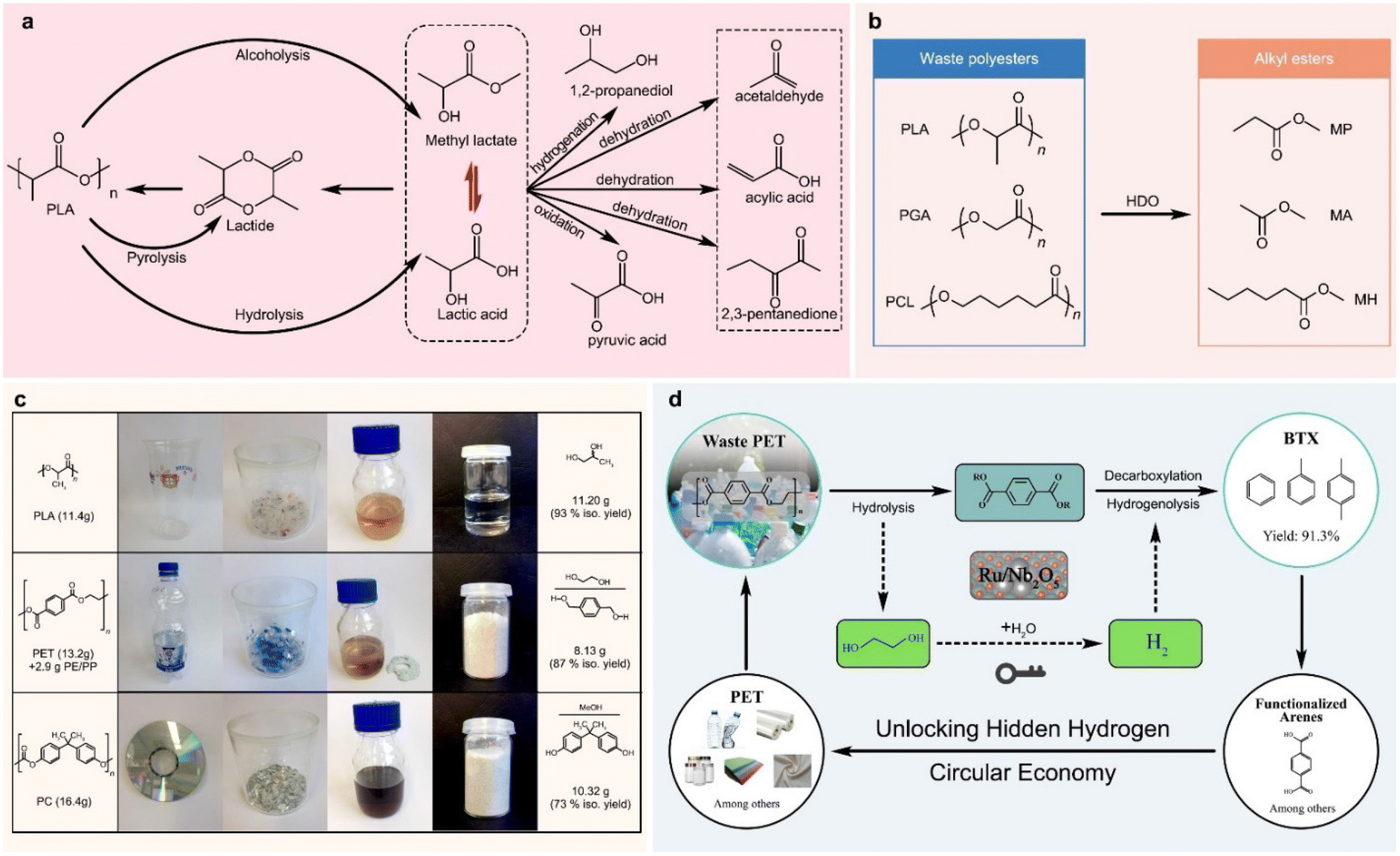 | ||
| Fig. 8 (a) Lactic acid as a platform molecule for the synthesis of chemical intermediates,161 (b) upcycling of polyesters via hydrodeoxygenation (HDO),166 and (c and d) upcycling waste PET to 1,4-phenylenedimethanol169 and aromatic products.173 | ||
PET composed of aromatic monomers is an ideal resource for the production of aromatic compounds.167 1,4-Benzenedimethanol is an essential component in the manufacture of pesticides, perfumes, and dyes, and can be obtained directly from PET hydrogenolysis. Clarke et al. tested a series of ruthenium(II)-catalysts bearing tridentate aminophosphine ligands for PET hydrogenolysis, and at 110 °C, 73% 1,4-benzenedimethanol was yielded using the ethylenediamine variant of the ruthenium(II)-sulfoxide complex in a 50/50% mixture of THF and anisole.168 By using two ruthenium(II)-complexes bearing tridentate phosphine ligands for PET hydrogenolysis in the presence of HNTf2 (1 mol%), Klankermayer et al. obtained high PET conversion (>99%) and 1,4-benzenedimethanol selectivity (>86%) under optimal conditions (140 °C, 100 bar H2 for 16 h) for a variety of commercial PET sources, such as bottles, yoghurt pots, and sports jersey (Fig. 8c).169 Feghali et al. reported two efficient organocatalysts, B(C6F5)3, and [Ph3C+, B(C6F5)4−], for the reductive depolymerization of PET in an Et3SiH and CH2Cl2 mixture, and obtained 1,4-phenylenedimethanol via hydrogenolysis and hydrosilyation cascade reactions.170 However, the use of toxic, costly, and homogeneous chemicals in these methods made them difficult to separate and recycle, and potentially causes serious environmental pollution. To overcome the limits of homogeneous catalysts, heterogeneous catalysts have also been recently reported for the selective reductive depolymerization of PET. By using CuFeCr catalysts derived from layered double hydroxides (LDHs), Ma et al. performed the co-transformation of PET and CO2, involving hydrogenation of CO2 to methanol, PET methanolysis, and subsequent dimethyl terephthalate hydrogenation.144,171 Yan et al. achieved a 95.2% overall yield of arenes and cyclic hydrocarbons on a Ru/Nb2O5 catalyst by transforming aromatic plastic wastes with C–O and C–C linkages into arenes in hydrogen.172 Ru/Nb2O5 was also applied to H2-free conversion of PET into toluene and p-xylene, instead of decarboxylation into benzene (Fig. 8d),173 where no external H2 was added, and the EG unit in PET acted as a hydrogen resource.167
3.4 Photolysis and electrolysis of polyesters
Photolysis refers to the breakdown of polymer chains through the breaking of their ester bonds with the combination of UV radiation and oxygen. At present, photosensitizers or photosensitive functional groups have been designed for introduction in the polyester materials, which will help the waste absorb energy from UV radiation to break polymer bonds, reduce molecular weight, and undergo enzymatic hydrolysis by microorganisms.174–177 Loyo et al. studied the photolysis of PLA materials, and found that calcium oxide additives contributed to the photolysis process of PLA, where it was hypothesized that the surface of calcium oxide nanoparticles may take up active hydroxyl groups, which contribute greatly to the free-radical reaction and accelerate the photolysis of PLA.174 However, this is rarely reported in the re/upcycling of polyester. Chen et al. recently reported that via efficient solar-thermal catalysis for recycling, various polyesters were recycled to form high-value-added monomers, and the BHET yields dramatically increased from 27 to 82% when the illumination time was prolonged from 1 to 4 h.178Coupling alkaline hydrolysis and electrocatalysis has also been performed for upcycling PET waste into terephthalate and formate in an H-type cell, where different electrocatalysts were examined, such as CuCo2O4/Ni foam,179 CuO nanowire,180 and CoNi0.25P/NF.181
3.5 Other upcycling methods
PET can also be used to prepare certain special carbon-based materials via high-temperature treatment. Topuz et al. reported the valorization of PET wastes to produce nanofibrous adsorptive membranes for application in oil removal, in which PET wastes were dissolved first in a trifluoroacetic acid solvent and then electrospun into nanofibrous membranes.182 Zhang et al. reported the mechanochemical extrusion of PET waste into porous carbon materials with a surface area of up to 1001 m2 g−1.183 PET and the pore-directing additives were first mixed and cyclically extruded in a twin-screw extruder, then carbonized under an inert atmosphere, and subsequently treated with hydrofluoric acid to remove additives, resulting in a porous carbon material. Yuan et al. pretreated PET wastes using physical and chemical activation methods to convert the PET waste into value-added porous carbons for CO2 capture.184 Nunes et al. used PET waste to produce a high-performance porous membrane using the no-solvent-induced phase separation method.185 Furthermore, Lee et al. demonstrated that the incorporation of carbon fibers into PET could practically upgrade the mechanical properties of the mixed plastics.186Ladewig et al. showed that PLA waste could be utilized for the synthesis of lactate-containing metal organic frameworks (MOFs) of ZnBLD, which maintained the chiral separation ability and exhibit great potential for application.187 Yang et al. investigated a two-step extrusion process for the re/upcycling of PHB to plasticized PLA.188 In the process, the initial step involved subjecting PHB to thermal degradation in an extruder at a temperature of 220 °C, resulting in the formation of PHB oligomers (1600 Da) equipped with functional end-groups that could be subsequently covalently attached to the PLA chains, thereby introducing flexibility into the system.
4. Insights into challenges and strategies
Clearly, the closed-loop recycling and upcycling of polyesters proceeds by their selective depolymerization, which requires efficient breaking of their crystalline structures and ester bonds to form oligomers and monomers. To address this issue, we next try to analyse the key challenges and tentatively propose the corresponding strategies.4.1 Catalysts
As discussed above, for polyester depolymerization, catalysts play essential roles in facilitating the selective breaking of ester bonds under mild conditions.At present, organic, inorganic and enzymatic catalysts are used for polyester depolymerization. Among them, inorganic acids and bases as homogeneous catalysts are effective in polyester depolymerization, but they encounter problems associated with their separation and recycling, and also serious environment pollution. Organic acids and bases are not so active and the post separation is also challenging. Organic/inorganic metal salts work well in catalyst–substrate interaction and catalysis, but their stability and separation are also problematic. Ionic liquids could play the roles of catalysts and solvents, thereby providing high activities. However, they are frequently synthesized by sophisticated methods with relatively high costs and difficulty in separation from homogeneous systems. Hydrolases are generally biosynthesized by microorganisms. They have unique flexible biological structures that induce substrates for adsorption and catalysis. In particular, enzymatic catalysts have high specificity and can obtain high selectivity to monomers at low depolymerization temperatures. However, most enzymes are expensive, thermally unstable, and easily inhibited by products. In addition, the efficiency of enzymolysis is still low with unsatisfactory monomer concentrations in the products. Heterogeneous catalysts can be readily separated from the reaction solutions, but have low efficiency in their contact with polyester substrates. Thereby compared with homogeneous catalysts, they require higher reaction temperatures, and are susceptible to deactivation by blockage or poisoning of active sites, leaching or sintering of catalytic species, and so on.
Accordingly, the design of catalysts should consider the accessibility and efficiency of the active sites and their stability along with ease of separation from the reaction solutions. For homogeneous catalysts, it may be useful to solve the separation issue by changing their solubility under different conditions, such as temperature and pH.189 While it is difficult to make the heterogeneous catalysts dissolvable, some process enhancers could be applied to improve the mass transfer efficiency, such as quaternary ammonium salts.115
In fact, enzymatic degradation is itself a natural behaviour, inspiring the design of chemical catalytic systems. For instance, the enzymolysis of cellulosic biomass is being engineered. During the process, the cellulose is generally degraded into oligosaccharides and disaccharides by the synergistic effect of endoglucanases and exoglucanases, and then hydrolysed to glucose by β-glucosidase (Fig. 9a).190,191 Mimicking this enzymatic system, we may design depolymerization catalysts with multiple sites to work synergistically, considering the different stages of processing polymers from macromolecules to small molecules. At present, polyester enzymolysis is usually carried out at the glass transition temperature of polyesters, at which the amorphous regions of polyesters are more flexible for contacting catalysts. Therefore, the design of chemical catalytic systems should consider the reactivity and selectivity at around the glass transition temperatures of polyesters. In addition, enzyme activity and specificity not only come from the active sites, but also from the physicochemical structures and surface groups of the enzymes. For cellulase, its molecular structure consists primarily of two regions: the first region is the carbohydrate-binding module that can absorb and bind with polysaccharide substrates, and the other one is the catalytic domain with an open cleft complementary to the substrate form (Fig. 9b). Therefore, coupling the adsorption and catalytic regions can also be adopted when designing chemical catalysts. Recently, Pickford et al. employed a thermostable form of leaf compost cutinase (LLC) to fabricate synthetic fusion constructs of LCC with five type-A carbohydrate-binding modules (CBMs), which facilitated the enzyme–substrate interaction and ultimately achieved 97% PET conversion (Fig. 9c).192 Perras et al. reported an ordered, mesoporous shell/active site/core catalyst architecture with benefits like processive enzymes, which can improve the polymer–surface interactions and the translocation of the macromolecules (Fig. 9d).18
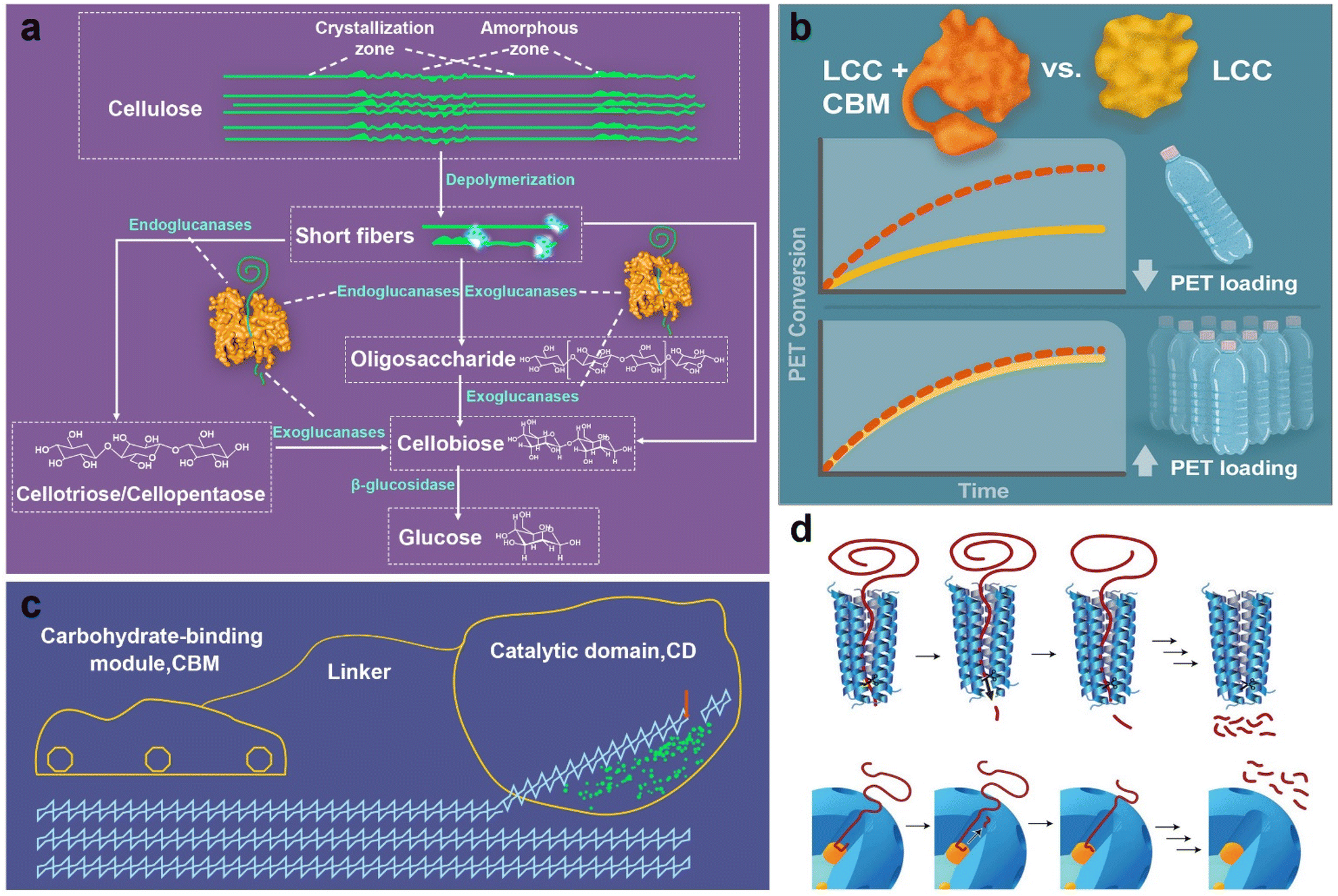 | ||
| Fig. 9 (a) Enzymatic hydrolysis of cellulose, (b) cellulase structure, (c) binding modules in enzymatic PET hydrolysis,192 and (d) catalytic upcycling of PE processive mechanism.18 | ||
4.2 Solvents
Solvents play a significant role in polyester depolymerization and re/upcycling, not only for dissolving the polymers, but also for promoting the reactions.Whatever methods are used to degrade the polyesters, the primary step is to break their crystalline structures and the protective polymer layers, making them accessible to catalysts. The solvents can be typically divided into good and poor solvents. In a so-called poor solvent, the attraction between the polymer chains is stronger than that between the chains and the solvent, the polymer tends to contract or phase separation occurs, and consequently does not easily mix with the solvent. Differently, in a good solvent, the attraction between the polymer chains is weaker, so the polymer chains tend to stay away from each other, leading to polymer swelling and dissolution. As shown in the phase diagram of a polymer solution (Fig. 10), at the temperature of θ, the phase diagram is divided into two parts: the lower part is the poor solvent region and the upper part is the good solvent region. The solid line in the diagram reveals that the phase separation of polymer solution occurs, and the dotted line shows the cryogenic boundary of a semi-thin good solvent. Clearly, polymer depolymerization requires the selection of a good solvent and appropriate reaction conditions to cause the polymer to swell and dissolve. High activity of polyester alcoholysis at room temperature was reported in the presence of alcohol and CH2Cl2, which can dissolve the polyester first, transforming a heterogeneous reaction into a homogeneous reaction.61,193 Notwithstanding the toxic and environmental concerns over CH2Cl2, this confirms the strategy, which can be advanced actually by choosing greener solvents for the polyester depolymerization.
 | ||
| Fig. 10 Solvent role in for dissolving the polymers and nucleophilic substitutions to the acyl group of polyesters. | ||
On the other hand, solvents can also promote the polyester depolymerization. Breaking the ester bonds involves essentially nucleophilic substitution. Therefore, the selection of nucleophilic reagents is important in polyester depolymerization and re/upcycling. Fig. 10 also shows some nucleophilic reagents classified by different nucleophilic elements. O-based reagents include water, alcohols, glycols, and carboxylic acids, which form new C–O bonds when the nucleophiles attack carbonyl carbon with the O-atom.194 However, these processes are generally reversible, and there is the need to tune reaction conditions to shift the reaction equilibrium. N-based nucleophilic reagents, including ammonia and amines, can efficiently form C–N bonds through N-atom nucleophilic attack on the carbonyl carbon. C-based nucleophilic reagents, such as hydrogen cyanide, alkynes, and Grignard reagents, can easily form C–C bonds. S-based nucleophilic reagents, such as hydrogen sulfide, thiol and sodium bisulfite, can form S–C bonds, which are also reversible due to a small energy difference between S–C and C–O bonds. Based on the properties of these reagents, they can be chosen for the closed-loop recycling or upcycling of polyesters.
4.3 Closed-loop recycling and upcycling
To study the closed-loop recycling of plastic wastes, their sources and industrial synthetic processes need to be preferentially considered, which provide the directions for choosing and designing their depolymerization methods. As shown in Fig. 11, two reaction routes are available to produce PET industrially. The first route involves the melt polycondensation of bis(2-hydroxyethyl) terephthalate (BHET) with ethylene glycol (EG) as a byproduct, and the second one involves the melt polycondensation of a mixture of terephthalic acid (TPA) and EG with water as a byproduct. At present, most biodegradable polyesters are synthesized by the ring-opening polymerization (ROP) of lactones or lactides, and corresponding acid and ester monomers cannot be directly repolymerized to targeted high-molecular polymers (Fig. 11). Taking PLA as an example, it is difficult to obtain high-molecular PLA by the direct polycondensation of lactic acid recycled from PLA depolymerization. Commercially, PLA is prepared by the ROP of lactide, which can maintain the properties of the functional groups of the monomers without releasing small molecules (i.e., H2O and CH3OH) in milder polymerization conditions and achieve the higher molecular weights. Similarly, the chemical recycling of PCL, PGA, PHB, and other polyesters with ROP requirement has not been well performed in a closed loop.40 Therefore, if the plastic waste can be selectively depolymerized to lactone or lactide intermediates, the closed-loop recycling will be more technologically and economically viable. | ||
| Fig. 11 Proposed closing-loop recycling routes of representative polyesters. | ||
Upcycling is an open-loop method with production of value-added chemicals and materials from plastic wastes. Thus, in order for it to be highly efficient, a combination of innovative depolymerization chemistry, breakthroughs in catalyst science, new bio/chemotechnologies and analytical characterization capabilities, novel approaches to separation science and waste management, and thorough economic and life-cycle assessments are necessary.195 In comparison with the aforementioned closed-loop recycling, upcycling has been rarely studied, and the resulting chemicals and materials are still limited, relative to the large volume of plastic wastes.16 Meanwhile, upcycling may involve heteroatoms to build new molecular structures, but this often requires the use of toxic chemicals. Consequently, the expenses, energy consumption, and ecological effects linked to the upcycling methods should not exceed those of manufacturing the same items from fresh materials obtained from the closed-loop recycling process (Fig. 12).
 | ||
| Fig. 12 Potential future development of upcycling. | ||
4.4 Chemical recycling and upcycling of mixed plastics
Polymers are often blended together, combined with small-molecule additives, or physically and chemically bound to other plastics to improve their performances; however, this creates difficulties and challenges when it comes to recycling. To some degree, the recycling issues are closely related to their classification and separation. Therefore, polymer products should be easily identified so that they can be initially classified, sorted, and processed by later consumers and recyclers. Therefore, it is now necessary to separate plastics from waste mixture streams using a variety of techniques, including manual sorting, sieving, gravity sorting, magnetic sorting, photoelectric sorting, and rebound sorting, based on differences in the size, density, magnetism and photoelectric properties of plastic particles. In addition, the chemical recycling of mixed plastic wastes is comprehensive, and has to be addressed by different approaches (Fig. 13). | ||
| Fig. 13 Proposed strategies for chemical recycling and upcycling of mixed plastic wastes. | ||
Currently, the chemical depolymerization and monomer recovery of mixed plastics are challenging.48 To our knowledge, only a few literature reports are available on the chemical recycling of mixed plastics. For hybrid of PLA and PET plastics, Collinson et al. developed a stepwise depolymerization strategy by taking advantage of the different reactivities to alcoholysis of the two plastics over zinc acetate. Hence, PLA could successfully undergo alcoholysis into lactate esters, while PET showed no reactivity under the same conditions due to its lower degradation, which facilitated post-separation.56 Sardon et al. also found that the protic ionic salt TBD: MSA catalyst could catalyze the selective and sequential depolymerization of PET and bisphenol A-based polycarbonate (BPA-PC), due to the energetic differences between BPA-PC and PET during glycolysis.196 These methods provide a stepwise depolymerization strategy suitable for mixed polyesters with significant differences in intrinsic depolymerization activity.197 Therefore, it is necessary to develop integrated methods compatible with different plastics degradation characteristics.
In cases where monomers are difficult to recycle from the mixtures of blended plastics, they can be considered for conversion, although not in an energy- or atom-economical way, into fuel additives (Fig. 13). One such method is gasification, and the plastic is partially oxidized at high temperatures to yield syngas (CO and H2) as the main product. Secondly there is pyrolysis, which is performed at high temperatures to thermally decompose mixed plastics to pyrolysis oil and gas products in the absence of oxygen. Thirdly, hydrocracking of plastic mixtures with hydrogen via catalytic hydrogenolysis at elevated temperatures forms saturated alcohols and hydrocarbons. In brief, these processes do not have specific requirements for the compositions of the plastic wastes and allow for recycling of a wide range of feedstocks.
5. Conclusions
Polyesters are very important polymers with excellent properties and large market shares, and thus the development of chemical recycling methods provides a viable solution to the efficient utilization of polyester wastes towards the global goal of carbon neutrality. This review has provided a comprehensive overview of the progress made in the depolymerization of polyesters and their recycling applications, demonstrating their practicality. However, there still need to be large improvements in terms of efficiency, versatility and cost. To address these problems, in this review, the key limiting factors and relevant challenges have been discussed, and then some strategies have been tentatively proposed for the closed-loop recycling and upcycling of polyester wastes.Currently, the main chemical depolymerization methods include hydrolysis, alcoholysis, enzymolysis, and ammonolysis. Hydrolysis uses green water solvent, but the hydrolysis activity is lower and thus requires higher reaction temperatures. Differently, methanolysis and glycolysis show superior depolymerization activities, but they are prone to side etherification reactions. Homogeneous catalysts have been largely used with high depolymerization efficiency, but their separation and recycling after reaction are difficult. Solid catalysts encounter low activity and stability, although they can be readily separated from the reaction solutions. Enzymolysis is highly selective under green and mild reaction conditions, but with low efficiency and the use of costly enzymatic agents. Aminolysis is of an excellent choice for upcycling, but the use of toxic and expensive chemicals should be considered.
These problems are closely related to the crystalline structures of polyesters and their accessibility to catalytic sites. These are the key factors that limit the depolymerization of polyesters to the corresponding oligomers and monomers. As such, the following suggestions are worth considering. Firstly, it is very critical to choose green solvents not only for efficiently dissolving or swelling polymers, but also for improving the activity and selectivity in polyester depolymerization. Secondly, the development of efficient catalysts is still the central issue for the depolymerization and recycling of polyesters, particularly based on cheaper and nontoxic metals, instead of toxic metals (e.g., Zn, Bi, Sn, and Cr). Meanwhile, it is worth designing enzyme-mimetic catalysts for higher efficiency and selectivity. Thirdly, the chemical recycling and upcycling of mixed plastic wastes is a comprehensive problem and should be addressed by multidisciplinary approaches. More process enhancers can be introduced to the depolymerization processes to improve the efficiency of the depolymerization and subsequent reactions of polyesters, such as photochemical, electrochemical, microwave-assisted and mechanochemical approaches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 22032001, 22208085, 21821004, and 21832001), the National Key Research and Development Program of China (Grant 2021YFA1501104), Henan Province Natural Science Foundation (GZS2020012), and the Beijing National Laboratory for Molecular Sciences (Grant BNLMS-CXXM-201905).References
- E. Feghali, L. Tauk, P. Ortiz, K. Vanbroekhoven and W. Eevers, Polym. Degrad. Stab., 2020, 179, 109241 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef.
- A. Stubbins, K. L. Law, S. E. Muñoz, T. S. Bianchi and L. Zhu, Science, 2021, 373, 51 CrossRef CAS PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. D. Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515 CrossRef CAS.
- R. G. Santos, G. E. Machovsky-Capuska and R. Andrades, Science, 2021, 373, 56 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61 CrossRef CAS.
- H. Y. Sintim, A. I. Bary, D. G. Hayes, M. E. English, S. M. Schaeffer, C. A. Miles, A. Zelenyuk, K. Suski and M. Flury, Sci. Total Environ., 2019, 675, 686 CrossRef CAS PubMed.
- P. K. Samantaray, A. Little, D. M. Haddleton, T. McNally, B. Tan, Z. Sun, W. Huang, Y. Ji and C. Wan, Green Chem., 2020, 22, 4055 RSC.
- W. W. Y. Lau, Y. Shiran, R. M. Bailey, E. Cook, M. R. Stuchtey, J. Koskella, C. A. Velis, L. Godfrey, J. Boucher, M. B. Murphy, R. C. Thompson, E. Jankowska, A. C. Castillo, T. D. Pilditch, B. Dixon, L. Koerselman, E. Kosior, E. Favoino, J. Gutberlet, S. Baulch, M. E. Atreya, D. Fischer, K. K. He, M. M. Petit, U. R. Sumaila, E. Neil, M. V. Bernhofen, K. Lawrence and J. E. Palardy, Science, 2020, 369, 1455 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501 CrossRef CAS.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347 CrossRef CAS.
- K. Hamad, M. Kaseem and F. Deri, Polym. Degrad. Stab., 2013, 98, 2801 CrossRef CAS.
- I. A. Ignatyev, W. Thielemans and B. V. Beke, ChemSusChem, 2014, 7, 1579 CrossRef CAS.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Waste Manage., 2009, 29, 2625 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. D. Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803 CrossRef CAS PubMed.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437 CrossRef CAS PubMed.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Huang, A. D. Sadow and F. A. Perras, Nat. Catal., 2020, 3, 893 CrossRef CAS.
- B. Kunwar, H. N. Cheng, S. R. Chandrashekaran and B. K. Sharma, Renewable Sustainable Energy Rev., 2016, 54, 421 CrossRef CAS.
- N. Malik, P. Kumar, S. Shrivastava and S. B. Ghosh, Int. J. Plast. Technol., 2017, 21, 1 CrossRef CAS.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185 CrossRef CAS.
- B. Geyer, G. Lorenz and A. Kandelbauer, eXPRESS Polym. Lett., 2016, 10, 559 CrossRef CAS.
- Y. Tokiwa and B. P. Calabia, Biotechnol. Lett., 2004, 26, 1181 CrossRef CAS PubMed.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041 CrossRef CAS.
- K. Min, J. D. Cuiffi and R. T. Mathers, Nat. Commun., 2020, 11, 727 CrossRef CAS.
- C. Li, C. Guo, V. Fitzpatrick, A. Ibrahim, M. J. Zwierstra, P. Hanna, A. Lechtig, A. Nazarian, S. J. Lin and D. L. Kaplan, Nat. Rev. Mater., 2020, 5, 61 CrossRef.
- T. Iwata, Angew. Chem., Int. Ed., 2015, 54, 3210 CrossRef CAS PubMed.
- E. Barnard, J. J. R. Arias and W. Thielemans, Green Chem., 2021, 23, 3765 RSC.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961 RSC.
- K. Hamad, M. Kaseem, M. Ayyoob, J. Joo and F. Deri, Prog. Polym. Sci., 2018, 85, 83–127 CrossRef CAS.
- Z. Li and X. J. Loh, Chem. Soc. Rev., 2015, 44, 2865 RSC; W. Zhang, IOP Conf. Ser.: Earth Environ. Sci., 2021, 647, 012156 CrossRef CAS.
- S. Y. Choi, M. N. Rhie, H. T. Kim, J. C. Joo, I. J. Cho, J. Son, S. Y. Jo, Y. J. Sohn, K. A. Baritugo, J. Pyo, Y. Lee, S. Y. Lee and S. J. Park, Metab. Eng., 2020, 58, 47 CrossRef CAS PubMed.
- X. Tang and E. Y.-X. Chen, Chem, 2019, 5, 284 CAS.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484 RSC.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610 RSC.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610 RSC.
- N. E. Kamber, Y. Tsujii, K. Keets, R. M. Waymouth, R. C. Pratt, G. W. Nyce and J. L. Hedrick, J. Chem. Educ., 2010, 87, 519 CrossRef CAS.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839 CrossRef CAS PubMed.
- G. Xu and Q. Wang, Green Chem., 2022, 24, 2321 RSC.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760 CrossRef CAS.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66 CrossRef CAS.
- Y. Li and T. J. Strathmann, Green Chem., 2019, 21, 5586 RSC.
- C. Alberti, N. Damps, R. R. R. Meißner, M. Hofmann, D. Rijono and S. Enthaler, Adv. Sustainable Syst., 2019, 4, 1900081 CrossRef.
- K. Hu, Y. Yang, Y. Wang, X. Duan and S. Wang, Chem. Catal., 2022, 2, 724 CrossRef CAS.
- S. D. A. Sharuddin, F. Abnisa, W. M. A. W. Daud and M. K. Aroua, Energy Convers. Manage., 2016, 115, 308 CrossRef.
- P. Dobrzynski, D. Fabbri, C. Torri, J. Kasperczyk, B. Kaczmarczyk and M. Pastusiak, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 247 CrossRef CAS.
- X. Zhao, X. Liu, K. Feng, W.-L. An, F. Tian, R. Du, S. Xu, L. Chen, G. Wu and Y.-Z. Wang, ChemSusChem, 2021, 15, e202101607 CrossRef PubMed.
- X. Zhou, C. Wang, C. Fang, R. Yu, Y. Li and W. Lei, Waste Manage., 2019, 85, 164 CrossRef CAS PubMed.
- M. Liu, J. Guo, Y. Gu, J. Gao and F. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15127 CrossRef CAS.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao, F. Liu and S. Yu, ACS Sustainable Chem. Eng., 2018, 6, 13114 CrossRef CAS.
- A. Kamimura, K. Ikeda, S. Suzuki, K. Kato, Y. Akinari, T. Sugimoto, K. Kashiwagi, K. Kaiso, H. Matsumoto and M. Yoshimoto, ChemSusChem, 2014, 7, 2473 CrossRef CAS.
- A. Kamimura, K. Kaiso, S. Suzuki, Y. Oishi, Y. Ohara, T. Sugimoto, K. Kashiwagi and M. Yoshimoto, Green Chem., 2011, 13, 2055 RSC.
- S. Kobayashi, H. Uyama and T. Takamoto, Biomacromolecules, 2000, 1, 3 CrossRef CAS PubMed.
- A. C. Sánchez and S. R. Collinson, Eur. Polym. J., 2011, 47, 1970 CrossRef.
- C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357 CrossRef CAS.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814 CrossRef CAS.
- J. Meimoun, A. Favrelle-Huret, M. Bria, N. Merle, G. Stoclet, J. De Winter, R. Mincheva, J.-M. Raquez and P. Zinck, Polym. Degrad. Stab., 2020, 181, 109188 CrossRef CAS.
- C. Alberti, N. Damps, R. R. R. Meißner and S. Enthaler, ChemistrySelect, 2019, 4, 6845 CrossRef CAS.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004 RSC.
- R. Yang, G. Xu, C. Lv, B. Dong, L. Zhou and Q. Wang, ACS Sustainable Chem. Eng., 2020, 8, 18347 CrossRef CAS.
- X. Song, H. Wang, X. Zheng, F. Liu and S. Yu, J. Appl. Polym. Sci., 2014, 131, 40817 CrossRef.
- X. Song, H. Wang, F. Liu and S. Yu, Polym. Degrad. Stab., 2016, 130, 22 CrossRef CAS.
- H. Liu, X. Song, F. Liu, S. Liu and S. Yu, J. Polym. Res., 2015, 22, 135 CrossRef.
- H. Liu, R. Zhao, X. Song, F. Liu, S. Yu, S. Liu and X. Ge, Catal. Lett., 2017, 147, 2298 CrossRef CAS.
- X. Song, H. Wang, C. Wang, F. Liu, S. Yu, S. Liu and Z. Song, J. Polym. Environ., 2019, 27, 862 CrossRef CAS.
- X. Song, F. Liu, H. Wang, C. Wang, S. Yu and S. Liu, Polym. Degrad. Stab., 2018, 147, 215 CrossRef CAS.
- X. Song, Z. Bian, Y. Hui, H. Wang, F. Liu and S. Yu, Polym. Degrad. Stab., 2019, 168, 108937 CrossRef CAS.
- F. Liu, J. Guo, P. Zhao, Y. Gu, J. Gao and M. Liu, Polym. Degrad. Stab., 2019, 167, 124 CrossRef CAS.
- G. Xi, M. Lu and C. Sun, Polym. Degrad. Stab., 2005, 87, 117 CrossRef CAS.
- Y. Geng, T. Dong, P. Fang, Q. Zhou, X. Lu and S. Zhang, Polym. Degrad. Stab., 2015, 117, 30 CrossRef CAS.
- Q. F. Yue, C. X. Wang, L. N. Zhang, Y. Ni and Y. X. Jin, Polym. Degrad. Stab., 2011, 96, 399 CrossRef CAS.
- Q. Wang, Y. Geng, X. Lu and S. Zhang, ACS Sustainable Chem. Eng., 2015, 3, 340 CrossRef CAS.
- Q. Yue, L. Xiao, M. Zhang and X. Bai, Polymers, 2013, 5, 1258 CrossRef.
- Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122 RSC.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2018, 7, 3292 CrossRef.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. Zhang, Green Chem., 2015, 17, 2473 RSC.
- M. Imran, K. G. Lee, Q. Imtiaz, B. K. Kim, M. Han, B. G. Cho and D. H. Kim, J. Nanosci. Nanotechnol., 2011, 11, 824 CrossRef CAS.
- M. Imran, D. H. Kim, W. A. Al-Masry, A. Mahmood, A. Hassan, S. Haider and S. M. Ramay, Polym. Degrad. Stab., 2013, 98, 904 CrossRef CAS.
- L. Bartolome, M. Imran, K. G. Lee, A. Sangalang, J. K. Ahn and D. H. Kim, Green Chem., 2014, 16, 279 RSC.
- S. B. Jin, J.-M. Jeong, S. G. Son, S. H. Park, K. G. Lee and B. G. Choi, Mater. Today Commun., 2021, 26, 101857 CrossRef CAS.
- I. Cano, C. Martin, J. A. Fernandes, R. W. Lodge, J. Dupont, F. A. Casado-Carmona, R. Lucena, S. Cardenas, V. Sans and I. de Pedro, Appl. Catal., B, 2020, 260, 118110 CrossRef CAS.
- F. Chen, G. Wang, W. Li and F. Yang, Ind. Eng. Chem. Res., 2013, 52, 565 CrossRef CAS.
- L.-X. Yun, H. Wu, Z.-G. Shen, J.-W. Fu and J.-X. Wang, ACS Sustainable Chem. Eng., 2022, 10, 5278 CrossRef CAS.
- B.-K. Kim, G.-C. Hwang, S.-Y. Bae, S.-C. Yi and H. Kumazawa, J. Appl. Polym. Sci., 2001, 81, 2102 CrossRef CAS.
- H. Tang, N. Li, G. Li, A. Wang, Y. Cong, G. Xu, X. Wang and T. Zhang, Green Chem., 2019, 21, 2709 RSC.
- H. Kurokawa, M. Ohshima, K. Sugiyama and H. Miura, Polym. Degrad. Stab., 2003, 79, 529 CrossRef CAS.
- P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721 RSC.
- J.-T. Du, Q. Sun, X.-F. Zeng, D. Wang, J.-X. Wang and J.-F. Chen, Chem. Eng. Sci., 2020, 220, 115642 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312 CrossRef.
- A. M. Al-Sabagh, F. Z. Yehia, A.-M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443 CrossRef CAS.
- S. Baliga and W. T. Wong, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071 CrossRef CAS.
- F. Chen, G. Wang, C. Shi, Y. Zhang, L. Zhang, W. Li and F. Yang, J. Appl. Polym. Sci., 2013, 127, 2809 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022 CrossRef.
- H. W. Horn, G. O. Jones, D. S. Wei, K. Fukushima, J. M. Lecuyer, D. J. Coady, J. L. Hedrick and J. E. Rice, J. Phys. Chem. A, 2012, 116, 12389 CrossRef CAS.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205 RSC.
- P. Fang, B. Liu, J. Xu, Q. Zhou, S. Zhang, J. Ma and X. Lu, Polym. Degrad. Stab., 2018, 156, 22 CrossRef CAS.
- H. Wang, Y. Liu, Z. Li, X. Zhang, S. Zhang and Y. Zhang, Eur. Polym. J., 2009, 45, 1535 CrossRef CAS.
- F. Scé, I. Cano, C. Martin, G. Beobide, Ó. Castillo and I. de Pedro, New J. Chem., 2019, 43, 3476 RSC.
- T. Saeki, T. Tsukegi, H. Tsuji, H. Daimon and K. Fujie, Polymer, 2005, 46, 2157 CrossRef CAS.
- J. R. Campanelli, M. R. Kamal and D. G. Cooper, J. Appl. Polym. Sci., 1993, 48, 443 CrossRef CAS.
- H. D. H. Tsuji and K. Fujie, Biomacromolecules, 2003, 4, 835 CrossRef CAS PubMed.
- J. Yu, D. Plackett and L. X. L. Chen, Polym. Degrad. Stab., 2005, 89, 289 CrossRef CAS.
- J. R. Campanelli, D. G. Cooper and M. R. Kamal, J. Appl. Polym. Sci., 1994, 48, 985 CrossRef.
- S. D. Mancini and M. Zanin, Polym.-Plast. Technol. Eng., 2007, 46, 135 CrossRef CAS.
- S. Mishra, A. S. Goje and V. S. Zope, Polym.-Plast. Technol. Eng., 2007, 42, 581 CrossRef.
- S. Mishra, A. S. Goje and V. S. Zope, Polym. React. Eng., 2003, 11, 79 CrossRef CAS.
- M. J. Kang, H. J. Yu, J. Jegal, H. S. Kim and H. G. Cha, Chem. Eng. J., 2020, 398, 125655 CrossRef CAS.
- S. Mishra, V. S. Zope and A. S. Goje, Polym. Int., 2002, 51, 1310 CrossRef CAS.
- S. Mishra and A. S. Goje, Polym. React. Eng., 2003, 11, 963 CrossRef CAS.
- G. P. Karayannidis, A. P. Chatziavgoustis and D. S. Achilias, Adv. Polym. Technol., 2002, 21, 250 CrossRef CAS.
- B.-Z. Wan, C.-Y. Kao and W.-H. Cheng, Ind. Eng. Chem. Res., 2001, 40, 509 CrossRef CAS.
- D. S. A. Vassilis, A. Kosmidis and G. P. Karayannidis, Macromol. Mater. Eng., 2001, 286, 640 CrossRef.
- N. R. Paliwal and A. K. Mungray, Polym. Degrad. Stab., 2013, 98, 2094 CrossRef CAS.
- H. I. Khalaf and O. A. Hasan, Chem. Eng. J., 2012, 192, 45 CrossRef CAS.
- S. Ügdüler, K. M. V. Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. D. Meester, Green Chem., 2020, 22, 5376 RSC.
- M. Yagihashi and T. Funazukuri, Ind. Eng. Chem. Res., 2010, 49, 1247 CrossRef CAS.
- A. P. Bonartsev, A. P. Boskhomodgiev, A. L. Iordanskii, G. A. Bonartseva, A. V. Rebrov, T. K. Makhina, V. L. Myshkina, S. A. Yakovlev, E. A. Filatova, E. A. Ivanov, D. V. Bagrov and G. E. Zaikov, Mol. Cryst. Liq. Cryst., 2012, 556, 288 CrossRef CAS.
- T. Yoshioka, T. Motoki and A. Okuwaki, Ind. Eng. Chem. Res., 2001, 40, 75 CrossRef CAS.
- H. Tsuji and Y. Ikada, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 59 CrossRef CAS.
- C.-C. Chen, X. Han, X. Li, P. Jiang, D. Niu, L. Ma, W. Liu, S. Li, Y. Qu, H. Hu, J. Min, Y. Yang, L. Zhang, W. Zeng, J.-W. Huang, L. Dai and R.-T. Guo, Nat. Catal., 2021, 4, 425 CrossRef CAS.
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. T. Avelino, P. Lamers, A. Bhatt, Y. Zhang, G. Avery, L. Tao, A. R. Pickford, A. C. Carpenter, J. E. McGeehan and G. T. Beckham, Joule, 2021, 5, 2479 CrossRef CAS.
- Y. Oda, A. Yonetsu, T. Urakami and K. Tonomura, J. Polym. Environ., 2000, 8, 29 CrossRef.
- H. Serrano-Ruiz, L. Martin-Closas and A. M. Pelacho, Sci. Total Environ., 2021, 750, 141228 CrossRef CAS PubMed.
- L. Manfra, V. Marengo, G. Libralato, M. Costantini, F. D. Falco and M. Cocca, J. Hazard. Mater., 2021, 416, 125763 CrossRef CAS PubMed.
- M. Hajighasemi, B. P. Nocek, A. Tchigvintsev, G. Brown, R. Flick, X. Xu, H. Cui, T. Hai, A. Joachimiak, P. N. Golyshin, A. Savchenko, E. A. Edwards and A. F. Yakunin, Biomacromolecules, 2016, 17, 2027 CrossRef CAS PubMed.
- M. W. Myburgh, L. Favaro, W. H. van Zyl and M. Viljoen-Bloom, Bioresour. Technol., 2023, 378, 129008 CrossRef CAS.
- R.-J. Müller, H. Schrader, J. Profe, K. Dresler and W.-D. Deckwer, Macromol. Rapid Commun., 2005, 26, 1400 CrossRef.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196 CrossRef CAS.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guemard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. Andre, S. Duquesne and A. Marty, Nature, 2020, 580, 216 CrossRef CAS PubMed.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662 CrossRef CAS.
- E. L. Bell, R. Smithson, S. Kilbride, J. Foster, F. J. Hardy, S. Ramachandran, A. A. Tedstone, S. J. Haigh, A. A. Garforth, P. J. R. Day, C. Levy, M. P. Shaver and A. P. Green, Nat. Catal., 2022, 5, 673 CrossRef CAS.
- R. Wei, G. von Haugwitz, L. Pfaff, J. Mican, C. P. S. Badenhorst, W. Liu, G. Weber, H. P. Austin, D. Bednar, J. Damborsky and U. T. Bornscheuer, ACS Catal., 2022, 12, 3382 CrossRef CAS.
- J. Lai, H. Huang, M. Lin, Y. Xu, X. Li and B. Sun, Front. Microbiol., 2022, 13, 1113705 CrossRef.
- X. Han, W. Liu, J.-W. Huang, J. Ma, Y. Zheng, T.-P. Ko, L. Xu, Y.-S. Cheng, C. C. Chen and R.-T. Guo, Nat. Commun., 2017, 8, 2106 CrossRef PubMed.
- H. F. Son, I. J. Cho, S. Joo, H. Seo, H.-Y. Sagong, S. Y. Choi, S. Y. Lee and K.-J. Kim, ACS Catal., 2019, 9, 3519 CrossRef CAS.
- Y. Li, J. Rodrigues and H. Tomas, Chem. Soc. Rev., 2012, 41, 2193 RSC.
- F. Kawai, ChemSusChem, 2021, 14, 4115 CrossRef CAS.
- C.-C. Chen, L. Dai, L. Ma and R.-T. Guo, Nat. Rev. Chem., 2020, 4, 114 CrossRef.
- S. Joo, I. J. Cho, H. Seo, H. F. Son, H.-Y. Sagong, T. J. Shin, S. Y. Choi, S. Y. Lee and K.-J. Kim, Nat. Commun., 2018, 9, 382 CrossRef.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50 CrossRef CAS PubMed.
- F. Kawai, T. Kawabata and M. Oda, Appl. Microbiol. Biotechnol., 2019, 103, 4253 CrossRef CAS PubMed.
- M.-Q. Zhang, M. Wang, B. Sun, C. Hu, D. Xiao and D. Ma, Chem, 2022, 8, 2912 CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Roman-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539 CrossRef CAS.
- Z. Li, Y. Shen and Z. Li, ACS Sustainable Chem. Eng., 2022, 10, 8228 CrossRef CAS.
- G.-Q. Tian, Z.-H. Yang, W. Zhang, S.-C. Chen, L. Chen, G. Wu and Y.-Z. Wang, Green Chem., 2022, 24, 4490 RSC.
- Z. Wang, R. Yang, G. Xu, T. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 4529 CrossRef CAS.
- R. Yang, G. Xu, B. Dong, H. Hou and Q. Wang, Macromolecules, 2022, 55, 1726 CrossRef CAS.
- S. T. Kenny, J. N. Runic, W. Kaminsky, T. Woods, R. P. Babu, C. M. Keely, W. Blau and K. E. O'Connor, Environ. Sci. Technol., 2008, 42, 7696 CrossRef CAS PubMed.
- T. Narancic, M. Salvador, G. M. Hughes, N. Beagan, U. Abdulmutalib, S. T. Kenny, H. Wu, M. Saccomanno, J. Um, K. E. O'Connor and J. I. Jimenez, Microb. Biotechnol., 2021, 14, 2463 CrossRef CAS PubMed.
- D. H. Kim, D. O. Han, K. I. Shim, J. K. Kim, J. G. Pelton, M. H. Ryu, J. C. Joo, J. W. Han, H. T. Kim and K. H. Kim, ACS Catal., 2021, 11, 3996 CrossRef CAS.
- H. T. Kim, J. K. Kim, H. G. Cha, M. J. Kang, H. S. Lee, T. U. Khang, E. J. Yun, D.-H. Lee, B. K. Song, S. J. Park, J. C. Joo and K. H. Kim, ACS Sustainable Chem. Eng., 2019, 7, 19396 CrossRef CAS.
- J. C. Sadler and S. Wallace, Green Chem., 2021, 23, 4665 RSC.
- D. H. Kim, D. O. Han, K. I. Shim, J. K. Kim, J. G. Pelton, M. H. Ryu, J. C. Joo, J. W. Han, H. T. Kim and K. H. Kim, ACS Catal., 2021, 11, 3996 CrossRef CAS.
- M. C. Ribadeneyra, J. King, M. M. Titirici and P. A. Szilagyi, Chem. Commun., 2022, 58, 1330 RSC.
- N. A. Rorrer, S. Nicholson, A. Carpenter, M. J. Biddy, N. J. Grundl and G. T. Beckham, Joule, 2019, 3, 1006 CrossRef CAS.
- M. Y. Abdelaal, T. R. Sobahi and M. S. I. Makki, Constr. Build. Mater., 2011, 25, 3267 CrossRef.
- A. A. Karanastasis, V. Safin and L. M. Pitet, Macromolecules, 2022, 55, 1042 CrossRef CAS.
- S. R. Shukla and A. M. Harad, Polym. Degrad. Stab., 2006, 91, 1850 CrossRef CAS.
- M. Dusselier, P. V. Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415 RSC.
- L. Shao, Y.-C. Chang, C. Hao, M.-e. Fei, B. Zhao, B. J. Bliss and J. Zhang, Green Chem., 2022, 24, 8716 RSC.
- S. Tian, Y. Jiao, Z. Gao, Y. Xu, L. Fu, H. Fu, W. Zhou, C. Hu, G. Liu, M. Wang and D. Ma, J. Am. Chem. Soc., 2021, 143, 16358 CrossRef CAS PubMed.
- E. Lorusso, W. Ali, M. Leniart, B. Gebert, M. Oberthur and J. S. Gutmann, Polymers, 2019, 12, 6 CrossRef.
- L. Karpati, M. Fejer, D. Kalocsai, J. Molnar and V. Vargha, eXPRESS Polym. Lett., 2019, 13, 618 CrossRef CAS.
- B. Sun, J. Zhang, M. Wang, S. Yu, Y. Xu, S. Tian, Z. Gao, D. Xiao, G. Liu, W. Zhou, M. Wang and D. Ma, Nat. Sustain., 2023, 6, 712 CrossRef.
- T. Tan, W. Wang, K. Zhang, Z. Zhan, W. Deng, Q. Zhang and Y. Wang, ChemSusChem, 2022, 15, e202200522 CrossRef CAS.
- J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Slawin and M. L. Clarke, Chem. – Eur. J., 2015, 21, 10851 CrossRef CAS.
- S. Westhues, J. Isel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS.
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980 CrossRef CAS PubMed.
- Y. Li, M. Wang, X. Liu, C. Hu, D. Xiao and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202117205 CrossRef CAS PubMed.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hulsey, H. Wang, Y. Guo, X. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527 CrossRef CAS PubMed.
- S. Lu, Y. Jing, B. Feng, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2021, 14, 4242 CrossRef CAS PubMed.
- C. Loyo, V. Moreno-Serna, J. Fuentes, N. Amigo, F. A. Sepúlveda, J. A. Ortiz, L. M. Rivas, M. T. Ulloa, R. Benavente and P. A. Zapata, Polym. Degrad. Stab., 2022, 197, 109865 CrossRef CAS.
- Y. Luo, Y. Cao and G. Guo, J. Appl. Polym. Sci., 2018, 135, 46509 CrossRef.
- J. Salač, J. Šerá, M. Jurča, V. Verney, A. A. Marek and M. Koutný, Materials, 2019, 12, 481 CrossRef.
- X. Zhang, M. Xia, X. Su, P. Yuan, X. Li, C. Zhou, Z. Wan and W. Zou, J. Hazard. Mater., 2021, 413, 125321 CrossRef CAS.
- Y. Liu, Q. Zhong, P. Xu, H. Huang, F. Yang, M. Cao, L. He, Q. Zhang and J. Chen, Matter, 2022, 5, 1305 CrossRef CAS.
- F. Liu, X. Gao, R. Shi, E. C. M. Tse and Y. Chen, Green Chem., 2022, 24, 6571 RSC.
- J. Wang, X. Li, T. Zhang, Y. Chen, T. Wang and Y. Zhao, J. Phys. Chem. Lett., 2022, 13, 622 CrossRef CAS.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS.
- F. Topuz, D. G. Oldal and G. Szekely, Ind. Eng. Chem. Res., 2022, 61, 9077 CrossRef CAS.
- J. Xu, X. Duan, P. Zhang, Q. Niu and S. Dai, ChemSusChem, 2022, 15, e202201576 CrossRef CAS PubMed.
- X. Yuan, N. M. Kumar, B. Brigljevic, S. Li, S. Deng, M. Byun, B. Lee, C. S. K. Lin, D. C. W. Tsang, K. B. Lee, S. S. Chopra, H. Lim and Y. S. Ok, Green Chem., 2022, 24, 1494 RSC.
- B. A. Pulido, O. S. Habboub, S. L. Aristizabal, G. Szekely and S. P. Nunes, ACS Appl. Polym. Mater., 2019, 1, 2379 CrossRef CAS.
- A. N. Gaduan, K. Singkronart, C. Bell, E. Tierney, C. Burgstaller and K.-Y. Lee, ACS Appl. Polym. Mater., 2022, 4, 3294 CrossRef CAS PubMed.
- B. Slater, S.-O. Wong, A. Duckworth, A. J. P. White, M. R. Hill and B. P. Ladewig, Chem. Commun., 2019, 55, 7319 RSC.
- X. Yang, J. Clénet, H. Xu, K. Odelius and M. Hakkarainen, Macromolecules, 2015, 48, 2509 CrossRef CAS.
- X. Zhang, D. Zhang, Z. Sun, L. Xue, X. Wang and Z. Jiang, Appl. Catal., B, 2016, 196, 50 CrossRef CAS.
- Y. Weng, X. Wang and Y. Zhang, Trends Chem., 2022, 4, 374 CrossRef CAS.
- L. Shuai and X. Pan, Energy Environ. Sci., 2012, 5, 6889 RSC.
- R. Graham, E. Erickson, R. K. Brizendine, D. Salvachúa, W. E. Michener, Y. Li, Z. Tan, G. T. Beckham, J. E. McGeehan and A. R. Pickford, Chem. Catal., 2022, 2, 2644 CrossRef CAS.
- L. A. Román-Ramírez, P. McKeown, M. D. Jones and J. Wood, ACS Catal., 2018, 9, 409 CrossRef.
- Y. Peng, J. Yang, C. Deng, J. Deng, L. Shen and Y. Fu, Nat. Commun., 2023, 14, 3249 CrossRef CAS PubMed.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390 CrossRef CAS PubMed.
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruiperez, J. L. Hedrick, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2021, 60, 6710 CrossRef CAS PubMed.
- J. Veskova, F. Sbordone and H. Frisch, Macromol. Chem. Phys., 2022, 223, 2100472 CrossRef CAS.
Footnote |
| † These authors contributed equally and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2024 |