
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-step oxidative carboxylation of olefins with carbon dioxide by combining electrochemical and 3D-printed flow reactors†
Diego
Iglesias
a,
Cristopher
Tinajero
a,
Simone
Marchetti
ab,
Ignazio
Roppolo
 b,
Marcileia
Zanatta
*a and
Victor
Sans
*a
b,
Marcileia
Zanatta
*a and
Victor
Sans
*a
aInstitute of Advanced Materials (INAM), Universitat Jaume I, Avda Sos Baynat s/n, 12071, Castellón, Spain. E-mail: zanatta@uji.es; sans@uji.es
bDipartimento di Scienza Applicata e Tecnologia, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy
First published on 8th November 2023
Abstract
The selective oxidation of alkenes to form epoxides followed by the cycloaddition of CO2 is a sustainable and cost-efficient method to generate functional cyclic carbonates. The use of a continuous-flow process allows seamless integration of both reactions sequentially under tailored and optimised conditions for each of the transformations to produce the cyclic carbonates. Here, we successfully demonstrate olefin electrooxidation, followed by the cycloaddition of CO2 to produce cyclic carbonates employing 3D printed (3DP) reactors in continuous flow and without the need for intermediate purification steps. This approach is highly convenient since the electrolyte (ammonium salt) from the electrochemical reaction acts also as a catalyst in the cycloaddition reaction. Different parameters in the electrochemical oxidation were evaluated (e.g. solvent, electrode, electrolyte, concentrations and current intensity). Complete conversion and high selectivity (>80%) towards the formation of epoxide were observed. The electrolyte served as a catalyst for the cycloaddition reaction. The digital design of the 3DP reactor played a crucial role in efficient performance of the cycloaddition reaction, showing increased productivity (a space-time yield of 4.38 gprod h−1 L−1) compared to that of a coil and a packed bed reactor. Consecutive CO2 cycloaddition reactions were also evaluated and a global yield of 83% of cyclic carbonates was observed for styrene. The system exhibited stability and stable activity for at least 20 h.
Introduction
The use of CO2 as a building block for the synthesis of high-value heterocyclic compounds has been receiving more interest in the framework of sustainable and green chemistry.1,2 The reaction between carbon dioxide and epoxides is 100% atom efficient and one of the few industrially feasible processes for producing cyclic carbonates. Organic carbonates are functional molecules that can be used as starting materials for the synthesis of polycarbonate and polyurethane, green solvents, fuel additives, and fine chemical intermediates. Recently, one-pot “oxidative carboxylation,” or the direct synthesis of cyclic carbonates from alkenes and CO2, has drawn a lot of attention since alkenes are more readily available and less expensive than the related epoxides. Such a procedure would eliminate the necessity for isolating and purifying epoxides, which are frequently unstable and extremely reactive. In this field, the synthetic approach can be divided into two main groups: (i) direct oxidation and carboxylation in only one step and3,4 (ii) one-pot.4,5 Despite the CO2 cycloaddition to epoxide being well-known in the literature,6–8 catalytic methods for generating organic carbonates from olefins are considerably scarcer.9 Additionally, designing a multifunctional catalyst that is efficient and selective for both the reactions still remains a big challenge.5,10An oxidant is essential to carry out the epoxidation reaction, but it is incompatible with the carboxylation catalytic system which usually employs a Lewis base. The use of organic oxidants, like tert-butyl hydroperoxide (TBHP), is common for epoxidation reactions but has several disadvantages due to their hazardous nature, flammability, toxicity, poor atom-economy, and the generation of waste. A greener alternative is hydrogen peroxide, as it produces water as the only by-product; however, this leads to a biphasic epoxidation reaction (as most olefins are hydrophobic) and hence requires long reaction times as mass transfer across the aqueous–organic interface controls the rate of the reaction.11 In this context, electrochemical oxidation appears as a green and non-toxic alternative for this reaction. Recently, Cantillo et al. (2021)12 presented the development of a selective approach for the synthesis of epoxides, diols, and aldehydes from a single set of electrochemical reaction components. The modularity permits the selection of the desired product under batch conditions. Alternatively, the use of continuous flow processes emerges as a solution for reducing heat and mass transfer limitations, something particularly important in multiphasic reaction systems.13–15 Even though the CO2 cycloaddition to epoxides under continuous flow has been explored,16–21 the direct oxidative carboxylation of olefins to yield cyclic carbonates using it is still an almost unexplored field.11,14,22 In 2021, Perosa et al.9 highlighted the necessity for the development of continuous-flow direct oxidative carboxylation processes, since only two studies have been reported to date. In 2014, Jamison et al.22 demonstrated a multi-step flow system to perform the oxidative carboxylation of olefins using N-bromosuccinimide (NBS) and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), resulting in 43–89% yields of cyclic carbonates. In 2017, Rioux and co-workers11 described a flow reactor based on a rhenium catalyzed epoxidation of olefins, followed by trapping of the epoxide by CO2 in the presence of an aluminum catalyst and iodide salt, resulting in yields from 48 to 98%.
Motivated by these research studies, the main focus of the present study will be on implementing a continuous flow setup to enhance the production capacity of olefin carboxylation.12 The epoxidation of olefins will be performed using electrochemical oxidation, avoiding the use of reagents incompatible with the next step. The cycloaddition step will be carried out in a bespoke reactor produced using 3D printing. Additive manufacturing (AM) techniques, also known as three-dimensional printing (3DP), have been recently receiving attention for designing and fabricating objects with specific shapes.23 3DP allows the production of reactor geometries that improve mass transfer during flow reactions,24 and the development of tailored formulations for 3DP to use in specific applications, such as catalysis or materials, is a growing and attractive area.25–31 In this regard, recently, we have demonstrated the application of a 3D-printed catalytic reactor in a flow system to convert CO2 into a cyclic carbonate using an epoxide as the starting point.28
In this work, we successfully demonstrate the oxidative carboxylation of olefins with CO2 to produce cyclic carbonates under continuous flow. The system performs the transformation in two steps under compatible conditions, avoiding the need for intermediate purification. Furthermore, the electrolyte employed in the first reaction serves as a catalyst for the CO2 cycloaddition, reducing the atom economy of the process. The geometry of a tailored designed 3DP reactor played a crucial role in the improvement of the contact between the phases, thus allowing selective production of the cyclic carbonates under mild conditions of pressure (P < 8 bar). Overall, a global yield of the transformation from olefin to cyclic carbonate of 83% was observed.
Results and discussion
Electrochemical epoxidation
A Vapourtec electrochemical ion cell with a parallel electrode configuration was employed to explore the electrochemical oxidation of styrene as the model substrate. The effects of the electrode nature, electrolyte, solvent and current on the reaction were extensively examined. Hydrogen was generated in the cathode, which was observed in the outlet, where the flow was multiphasic. An initial screening of the solvent and electrolyte was performed in order to select the best parameters to proceed with the reaction (Table S1†). A mixture of MeCN/H2O (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v:v) showed promising selectivity towards the products that can be used as reagents for the cycloaddition reaction (styrene oxide and halohydrin, Fig. 1A), while other solvents (DMSO, THF, and acetone) showed more selectivity towards the aldehyde, which is undesirable in this case. The use of TBA·Br resulted in higher selectivity towards the epoxide, compared to other ammonium salts evaluated (TBA·Cl, TBA·Br and TEA·BF4, Tables S1 and S2†). Conveniently, TBA·Br is an efficient catalyst for the subsequent cycloaddition reaction.32 The effect of the working electrode was also evaluated (flexible graphite and glassy carbon, Table S2†). Glassy carbon was selected due to its higher long-term stability and better reproducibility.
:20, v:v) showed promising selectivity towards the products that can be used as reagents for the cycloaddition reaction (styrene oxide and halohydrin, Fig. 1A), while other solvents (DMSO, THF, and acetone) showed more selectivity towards the aldehyde, which is undesirable in this case. The use of TBA·Br resulted in higher selectivity towards the epoxide, compared to other ammonium salts evaluated (TBA·Cl, TBA·Br and TEA·BF4, Tables S1 and S2†). Conveniently, TBA·Br is an efficient catalyst for the subsequent cycloaddition reaction.32 The effect of the working electrode was also evaluated (flexible graphite and glassy carbon, Table S2†). Glassy carbon was selected due to its higher long-term stability and better reproducibility.
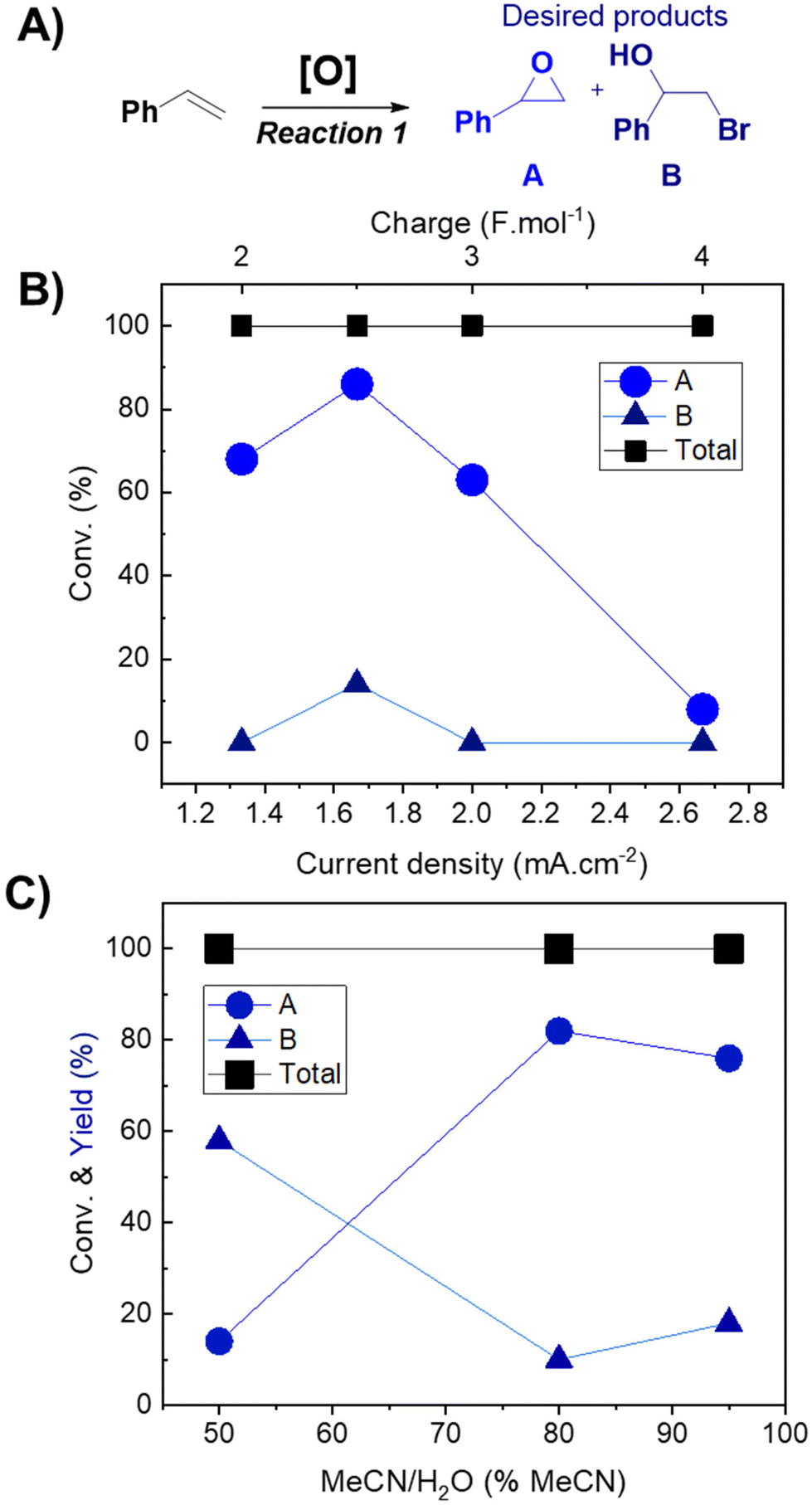 | ||
| Fig. 1 Optimization of the epoxidation flow reaction using styrene. (A) General reaction. Reaction conditions: 1 mmol of styrene, 2 mmol of electrolyte, 20 mL of solvent, and 0.05 mL min−1 flow rate, resulting in 12 min of residence time. Glassy carbon is used as the working electrode and stainless steel as the counter electrode. (B) Current density effect. (C) Solvent effect. | ||
The effects of current density (Fig. 1B) and solvent proportion (Fig. 1C) were then studied, using previous reaction parameters (solvent, electrode and electrolyte) where only products A and B were formed. Both products (A and B) were of interest since it is possible to further convert them into cyclic carbonates. The current density range to be studied was selected based on previously reported studies in the literature,12 with the charge varying from 2 F mol−1 to 4 F mol−1. Calculation of the respective current densities at the electrode surface was performed employing eqn (1) and (2) (see the Experimental section for more details). A current density of 1.66 mA cm−2 clearly presented better results (Fig. 1B), whereas other values of current density facilitated the formation of byproducts, such as aldehydes and bromo alkenes. Notably, the success of this reaction depended on the presence of a small amount of water, as observed in the solvent effect (Fig. 1C). On changing the amount of water in the MeCN solution, the major product also changed, as well as the conversion (Fig. 1C). The best result was observed with 80% MeCN, resulting in 86% selectivity towards epoxide, using a current of 20 mA (a charge of 2.5 F mol−1) with a residence time of 12 minutes, and this mixture was used for further reactions. Finally, control tests under different pressure conditions were performed; however, the reaction was not successful, resulting in the formation of solid products that began to accumulate on the electrodes. For this reason, atmospheric pressure was selected for further oxidation reactions. Overall, the results obtained for the reaction performed in flow highlighted its potential over batch experiments. In 2021, Cantillo et al.12 reported a selectivity of 53% using a current of 20 mA, with a charge of 3.5 F mol−1. Meanwhile, Qian et al. (2022)33 reported a selectivity of 97% towards epoxide using a current of 30 mA for 4 hours.
CO2 cycloaddition with a 3DP reactor
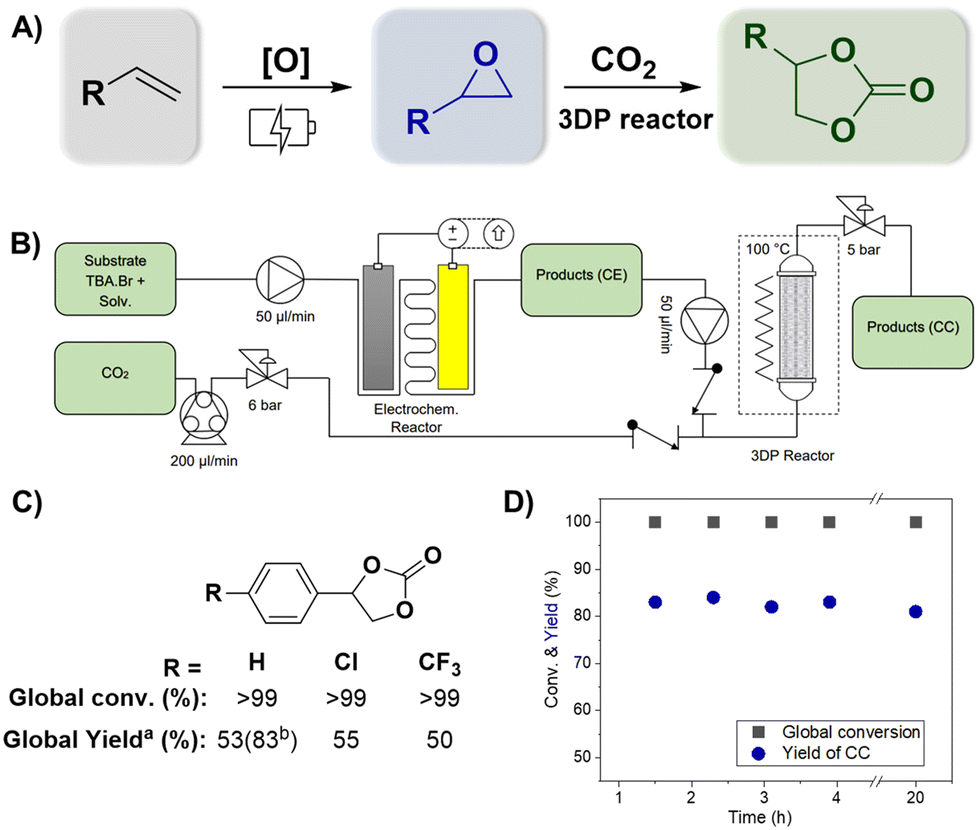
The evaluation of CO2 cycloaddition to an epoxide to produce cyclic carbonates was also conducted. Building upon our prior research, a variety of 3DP reactors were specifically designed to investigate their impact on the reaction.28 A new formulation was employed to improve the printing resolution (see the ESI† for more details). Two reactor designs with a helicoidal shape, namely R1 and R2 (as shown in Fig. 2A), were successfully printed using masked stereolithography (mSLA). These structures were designed to fit into a commercially available Omnifit™ column (L: 15 cm, ∅: 1.0 cm) enabling their use under flow conditions (Fig. 2A). Both designs are based on a helicoidal structure with four built-in columns to improve their mechanical resistance and flow distribution by representing obstacles to the pathway. Each geometry featured a different helix pitch: R1 had a distance of 4 mm between each complete spin, while R2 had a distance of 2.5 mm. STL files of the geometries can be found in the ESI.† | ||
| Fig. 2 Optimization of the CO2 cycloaddition reaction in flow using styrene oxide. Reaction conditions: 1 mmol of styrene oxide, 2 mmol of TBAB, and 20 mL of solvent (MeCN/H2O) (80:20) (v:v). Standard flow conditions: 0.05 mL min−1 liquid flow rate, 41 μL min−1 gas flow rate, and 100 °C. (A) Top left: CO2 cycloaddition reaction; top right: computer-aided design (SolidWorks or CAD) of structured reactors; bottom left: image of R2 3D printed; bottom right: image of R2 in an Omnifit™ column. (B) Evaluation of different flow reactors; productivity normalized according to residence time (see the Experimental section): R1 = 47 min; R2 = 51 min; PBR = 55 min; and coil = 60 min. (C) Temperature effect. (D) Liquid flow evaluation. (E) CO2 flow evaluation. | ||
In order to optimize the parameters of the cycloaddition reaction (Fig. 2A), a mixture of styrene oxide and TBA·Br in MeCN:H2O (80:20) (v:v) was used. The variation of the reactor structure demonstrated its strong influence over the selectivity and reproducibility of the reaction (Fig. 2B). Notably, the employment of 3DP reactors led to a substantial improvement in both conversion and selectivity (R1 = 93 ± 3% conversion and 82 ± 2% selectivity), as compared to the use of a conventional packed bed reactor (PBR) (97 ± 2% conversion and 50 ± 12% selectivity) containing beads of a commercial polymeric resin (Purolite ECR8209M, with an average diameter of 500 μm) of a similar formulation to our 3DP polymers, or a coiled tubular reactor (73 ± 5% conversion and 70 ± 6% selectivity). This result was more evident when comparing the space-time yield (STY) of the different systems; the values of R1 (4.38 gprod h−1 L−1) and R2 (4.11 gprod h−1 L−1) significantly decreased to 2.22 gprod h−1 L−1 for the PBR and 2.51 gprod h−1 L−1 for the coil. This result suggested a more uniform flow distribution in the 3DP reactor compared to that of the PBR. Furthermore, conventional reactors (PBR and coil) were affected by several issues, such as challenges regarding pressure control and difficulty in achieving a steady state. This was evidenced by the difficulty in collecting fractions of product solution with equal volumes at even time distributions, and also the low reproducibility of the results, as denoted by the high error bars (Fig. 2B). Alternatively, the use of 3DP structures improved the homogeneity of the fluid flow, sample collection volume and internal pressure of the column; overall it led to more stable flows and higher productivities. The spiral configuration of the reactor, complemented with four internal columns, apparently enhanced the interfacial area between the liquid and gas phases,34 thus improving the mass transfer, contact between reactants and catalyst and overall reaction rates (Fig. S1†). Moreover, the internal columns improved the stability of the structure: without them, some deformation of the helicoidal structure was observed due to swelling (Fig. S2†). The long-term stability of the 3DP reactor with internal columns was further demonstrated and will be discussed in more detail in Fig. 3D. Although the results obtained from R1 and R2 were similar, R1 presented slightly superior productivity, evidenced by the higher value of STY. Additionally, R1 showed superior durability and longer potential usage (see ESI Fig. S2†); for this reason, it was chosen for the subsequent phase of the experiment. In addition, noteworthy achievements are evident when comparing our system with previous PBRs documented in the literature for CO2 cycloaddition to epoxide. In fact, Valverde et al. (2021)35 reported 53% conversion under the operating conditions of 140 bar and 150 °C. Likewise, Yin et al. (2021)36 reported >76% conversion while operating at 20 bar and 90 °C.
 | ||
| Fig. 3 (A) Overview of the consecutive reactions for direct oxidative carboxylation of olefins (Fig. S12†). (B) Schematic representation of the continuous-flow system. (C) Substrates used for the scope (a47 min of residence time and b94 min of residence time). (D) System stability using styrene as the substrate in combined reactions. | ||
The increase in the temperature to 120 °C demonstrated a decrease in the selectivity favoring the formation of diols (Fig. 2C). For this reason, 100 °C was selected for further reactions. The flow of liquid was optimized, and as observed in Fig. 2D, the best results were obtained with smaller flow to represent higher residence time (47 min for 0.05 mL min−1; 30 min for 0.10 mL min−1; and 22 min for 0.15 mL min−1, respectively).
The flow of CO2 is also directly related to selectivity. While less concentration favored the hydrolysis yielding diols (Fig. 2E), higher concentration of CO2 resulted in greater formation of cyclic carbonates until a maximum point (40 μL min−1). From this point, the increase in the CO2 flow did not cause any significant change in the reaction. Another interesting point to consider is that passing from 15 μL min−1 to 40 μL min−1, the system transitioned from 1 phase (liquid) to 2 phases (gas–liquid). This discovery stems from the constrained solubility of CO2 within our solvent under our working pressure conditions (6 bar). At lower flow rates, we observed complete dissolution of CO2 in the liquid phase. In contrast, as the flow rate increased, we witnessed partial solubility, resulting in a two-phase system. This alteration in phase behavior may indeed impact the reaction mechanism and kinetics within the reactor, which can be responsible for the different conversions observed at this point.37 It has been studied how a two-phase flow promotes the apparition of vortexes inside the different phases.38 These generated vortexes enhance the mixing and mass transfer, and thus they can also be linked to the observed results.
Combined reactions
After optimizing the individual reactions, we combined the reactor sequence to perform direct oxidative carboxylation (Fig. 3A and Fig. S12†). The epoxidation and carboxylation reactors were identical to those described above. The solution containing the olefin and TBA·Br was pushed employing an HPLC pump towards the inlet of the ionic electrochemical reactor. The resulting product (a mixture of liquid and H2 gas) is collected in an intermediate collection vessel, allowing gas separation. The liquid phase was subsequently pumped with a second HPLC pump into a T-mixer and mixed with CO2 driven by a peristaltic pump. A back-pressure regulator (BPR) of 6.5 bar was added between the peristaltic pump and the T-mixer to ensure control over the gas flow rate. This, in addition to the use of two check valves at the respective flow inlets, allowed for a uniform mixing between the two phases. The multiphasic reaction mixture entered the 3DP reactor at a temperature of 100 °C, where the cycloaddition takes place. A BPR regulator controlled the pressure of the reactor at 6 bar. The products were collected employing an automated fraction collector in triplicate in a vial after reaching the steady state. The complete system can be observed in Fig. 3B.Using this system, different olefins were tested, and the production of the corresponding cyclic carbonate can be seen in Fig. 3C. Complete global conversion was observed for all reactions, and no residual olefins were detected in the 1H NMR spectrum (refer to Fig. S9–S11†). However, a slight decrease in the reaction rate was observed for this combined reaction. After the first hour, the yields of the cyclic carbonate were 53% for styrene, 55% for 4-(chloro)styrene, and 50% for 4-(trifluoromethyl)styrene. However, this was easily fixed by increasing the residence time, and styrene increased the global yield from 53% to 83%. Additional tests using electron-donor substituents (–OMe) were conducted; however, these experiments did not yield the desired product in the oxidation step. This outcome highlights the significance of the substituent's role in activating the epoxide. It is likely that substituents with stronger electron-withdrawing properties create a more electron-deficient carbon, which makes it more susceptible to nucleophilic attack of the bromide of TBA·Br.39,40 Finally, the stability of the system can be observed in Fig. 3D, where, after reaching the steady state, the yield of the desired product (CC) remained constant for at least 4 hours.
Conclusions
In this study, we have effectively showcased the continuous flow synthesis of cyclic organic carbonates from olefins and carbon dioxide. Our system accomplishes this transformation in two sequential steps, maintaining compatible conditions throughout the process, thus eliminating the requirement for intermediate purification steps. An integrated system for synthesizing a combination of electrochemical and chemical steps with TBA·Br as the electrolyte and catalyst reduces the use of expensive catalysts and waste generating oxidizing agents. The utilization of 3DP reactors presents significant advancements in terms of conversion and selectivity when compared to conventional PBRs or coiled tubular reactors. The improvement is more evident when comparing productivity, calculated using STY, where approximately half of the STY was observed for the commercial reactor compared to the 3DP reactor (R1 = 4.38 gprod h−1 L−1, R2 = 4.11 gprod h−1 L−1, PBR = 2.22 gprod h−1 L−1, and coil = 2.51 gprod h−1 L−1). Our methodology employs cost-effective catalysts, enables the execution of reactions at relatively low temperatures and pressures, and achieves rapid reaction times. These findings highlight the promising potential of 3D-printed reactors in enhancing the efficiency and sustainability of chemical reactions in various industries. While scaling up is not the primary focus of this paper, these factors combined with the continuous flow are relevant in terms of scalability and may represent a promising alternative for the future. Future research can further optimize this approach and explore its applicability to other catalytic systems.Experimental section
Reaction conditions
The current density (mA cm−2) and faradaic efficiency were calculated as follows:
 | (1) |
 | (2) |
The residence time was calculated using the eqn (3):
 | (3) |
The productivity was calculated considering the space-time yield (STY) (eqn (4)):
 | (4) |
Author contributions
Conceptualization: all the authors; methodology: MZ, VS and IR; software programming: CT; investigation: DI, CT and SM; writing – original draft: MZ and DI; writing – review and editing: all authors; supervision: MZ, VS and IR; project administration: VS; and funding acquisition: MZ, VS and IR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MICIN/AEI/10.13039/501100011033 (PID2020-119628RB-C33) and by the EU NextGenerationEU/PRTR (TED2021-130288B-I00). Generalitat Valenciana is gratefully acknowledged for funding for infrastructure (IDIFEDER/2021/029), GenT (CIDEGENT 2018/036) and Santiago Grisolía Programme (CIGRIS/2021/075). This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101026335.References
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- S. Dabral and T. Schaub, Adv. Synth. Catal., 2019, 361, 223–246 CrossRef CAS.
- Q. Han, B. Qi, W. Ren, C. He, J. Niu and C. Duan, Nat. Commun., 2015, 6, 10007 CrossRef PubMed.
- N. Podrojková, A. Oriňak, E. Garcia-Verdugo, V. Sans and M. Zanatta, Catal. Today, 2023, 418, 114128 CrossRef.
- L. Wang, S. Que, Z. Ding and E. Vessally, RSC Adv., 2020, 10, 9103–9115 RSC.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- T. K. Pal, D. De and P. K. Bharadwaj, Coord. Chem. Rev., 2020, 408, 213173 CrossRef CAS.
- R. Calmanti, M. Selva and A. Perosa, Green Chem., 2021, 23, 1921–1941 RSC.
- X. Yang, J. Wu, X. Mao, T. F. Jamison and T. A. Hatton, Chem. Commun., 2014, 50, 3245–3248 RSC.
- A. A. Sathe, A. M. K. Nambiar and R. M. Rioux, Catal. Sci. Technol., 2017, 7, 84–89 RSC.
- W. Jud, C. O. Kappe and D. Cantillo, Electrochem. Sci. Adv., 2021, 1, e2100002 CrossRef CAS.
- H. Seo, L. V. Nguyen and T. F. Jamison, Adv. Synth. Catal., 2019, 361, 247–264 CrossRef CAS.
- H. Seo, L. V. Nguyen and T. F. Jamison, Adv. Synth. Catal., 2019, 361, 247–264 CrossRef CAS.
- H. Luo, J. Ren, Y. Sun, Y. Liu, F. Zhou, G. Shi and J. Zhou, Chin. Chem. Lett., 2023, 34, 107782 CrossRef CAS.
- H. Yasuda, L.-N. He, T. Takahashi and T. Sakakura, Appl. Catal., A, 2006, 298, 177–180 CrossRef CAS.
- M. North, P. Villuendas and C. Young, Chem. – Eur. J., 2009, 15, 11454–11457 CrossRef CAS PubMed.
- X. Wu, M. Wang, Y. Xie, C. Chen, K. Li, M. Yuan, X. Zhao and Z. Hou, Appl. Catal., A, 2016, 519, 146–154 CrossRef CAS.
- A. Sainz Martinez, C. Hauzenberger, A. R. Sahoo, Z. Csendes, H. Hoffmann and K. Bica, ACS Sustainable Chem. Eng., 2018, 6, 13131–13139 CrossRef CAS.
- J. A. Kozak, J. Wu, X. Su, F. Simeon, T. A. Hatton and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 18497–18501 CrossRef CAS PubMed.
- X.-B. Lu, J.-H. Xiu, R. He, K. Jin, L.-M. Luo and X.-J. Feng, Appl. Catal., A, 2004, 275, 73–78 CrossRef CAS.
- J. Wu, J. A. Kozak, F. Simeon, T. A. Hatton and T. F. Jamison, Chem. Sci., 2014, 5, 1227–1231 RSC.
- S. Miralles-Comins, M. Zanatta and V. Sans, Polymers, 2022, 14, 5121 CrossRef CAS PubMed.
- C. Parra-Cabrera, C. Achille, S. Kuhn and R. Ameloot, Chem. Soc. Rev., 2018, 47, 209–230 RSC.
- M. R. Penny and S. T. Hilton, React. Chem. Eng., 2020, 5, 853–858 RSC.
- C. R. Tubío, J. Azuaje, L. Escalante, A. Coelho, F. Guitián, E. Sotelo and A. Gil, J. Catal., 2016, 334, 110–115 CrossRef.
- J. Wu, Y. Yan, L. Zhang, Z. Qin and S. Tao, Adv. Mater. Technol., 2019, 4, 1800515 CrossRef.
- D. Valverde, R. Porcar, M. Zanatta, S. Alcalde, B. Altava, V. Sans and E. García-Verdugo, Green Chem., 2022, 24, 3300–3308 RSC.
- S. Rossi, A. Puglisi and M. Benaglia, ChemCatChem, 2018, 10, 1512–1525 CrossRef CAS.
- V. Saggiomo, Catalyst Immobilization, 2020, pp. 369–408 Search PubMed.
- M. R. Penny, Z. X. Rao, R. Thavarajah, A. Ishaq, B. J. Bowles and S. T. Hilton, React. Chem. Eng., 2023, 8, 752–757 RSC.
- Y. Chen, P. Xu, M. Arai and J. Sun, Adv. Synth. Catal., 2019, 361, 335–344 CrossRef CAS.
- Y. Zhang, A. Iqbal, J. Zai, S.-Y. Zhang, H. Guo, X. Liu, I. ul Islam, H. Fazal and X. Qian, Org. Chem. Front., 2022, 9, 436–444 RSC.
- M. Berton, J. M. de Souza, I. Abdiaj, D. T. McQuade and D. R. Snead, J. Flow Chem., 2020, 10, 73–92 CrossRef.
- D. Valverde, R. Porcar, P. Lozano, E. García-Verdugo and S. V. Luis, ACS Sustainable Chem. Eng., 2021, 9, 2309–2318 CrossRef CAS.
- J. Sun, Z. Li and J. Yin, J. CO2 Util., 2021, 53, 101723 CrossRef CAS.
- J. Lefebvre, S. Bajohr and T. Kolb, Fuel, 2019, 239, 896–904 CrossRef CAS.
- C. Yao, Y. Zhao, H. Ma, Y. Liu, Q. Zhao and G. Chen, Chem. Eng. Sci., 2021, 229, 116017 CrossRef CAS.
- N. Panza, R. Soave, F. Cargnoni, M. I. Trioni and A. Caselli, J. CO2 Util., 2022, 62, 102062 CrossRef CAS.
- S. Marmitt and P. F. B. Gonçalves, J. Comput. Chem., 2015, 36, 1322–1333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03360k |
| This journal is © The Royal Society of Chemistry 2023 |