
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an amine transaminase-lipase cascade for chiral amide synthesis under flow conditions†
Antía
Pintor
ab,
Ashley P.
Mattey
b,
Iván
Lavandera
 a,
Vicente
Gotor-Fernández
*a and
Alexey
Volkov
*b
a,
Vicente
Gotor-Fernández
*a and
Alexey
Volkov
*b
aOrganic and Inorganic Chemistry Department. University of Oviedo. Avenida Julián Clavería 8, 33006 Oviedo, Spain. E-mail: vicgotfer@uniovi.es
bEnginzyme AB. Tomtebodevägen 6, 17165 Stockholm, Sweden. E-mail: alexey@enginzyme.com
First published on 19th July 2023
Abstract
The use of multienzymatic systems has gained increasing attention as a method of choice for complex (asymmetric) syntheses. Incompatibilities between substrates, reagents and/or enzymes in one-pot batch conditions can hamper the applicability of a pursued cascade, so the use of flow systems provide useful synthetic solutions. The implementation of immobilised enzymes in continuous flow reactors allows the compartmentalisation and segregation of the enzymes in separate reactors, leading to otherwise disfavoured reaction cascades. Here, an amine transaminase and a lipase have been immobilised on polymer-coated controlled porosity glass carrier materials and studied for the first time together in the transamination of a prochiral ketone followed by acylation of the corresponding chiral amine in flow mode, two incompatible transformations under batch. Thus, the preparation of (R)-N-(1-phenoxypropan-2-yl)acetamide was accomplished after optimisation of the reaction conditions.
Introduction
Biocatalytic processes have gained maturity and are currently considered emerging alternatives to chemical synthesis of industrially relevant (chiral) complex molecules.1,2 Traditional stepwise chemical routes and successive batch-reactions are now often replaced by multienzymatic systems, thus avoiding the isolation of (unstable) intermediates, reducing work-up steps and usually leading to high-yielding overall processes.3,4 Advances in enzyme immobilisation provide several advantages compared to the use of enzymes in solution, especially related to the possibility to perform reactions in non-aqueous media, streamlined downstream processing and finally biocatalyst recovery and reusability.5,6 Nevertheless, there are still limitations to one-pot systems that require special consideration during cascade assembly such as: (i) enzyme inhibition by any of the reagents, products or catalysts; (ii) ideal substrate concentrations for the different enzyme classes; (iii) optimal pH ranges and temperatures for the individual enzymes; or (iv) reaction medium including the use of surfactants,7,8 or alternatively partial or full organic media9 to improve the substrate solubility.Some of these challenges can be circumvented with implementation of a flow setup, where different enzymes can be compartmentalised and segregated in separate reactors.10,11 The implementation of processes in continuous flow has several advantages compared to the work under batch conditions, such as improved scalability, higher volumetric productivity, good versatility in system design, energy saving, easier thermal control and handling of hazardous components.12,13 Moreover, multi-phase reactions can benefit from better mass transfer obtaining higher performances.14 Hence, the field of flow biocatalysis has seen increasing interest recently;15–19 the combination of enabling technologies that flow chemistry offers with the advantages of enzymatic catalysis has led to the development of a number of sustainable synthetic tools with expanded chemistries.
Combination of different enzyme classes has provided elegant access towards different families of chemical products. Based on the importance of nitrogenated compounds, and the possibilities that enzymes bring for their preparation in optically pure forms, we have focused on the efficient synthesis and selective modification of chiral amines. For that reason, the combination of ATAs20–28 and lipases29,30 was selected for investigation, especially since these two enzyme classes have been largely employed in single transformations with scarce examples of reported cascade reactions (Scheme 1). Kroutil and co-workers described the synthesis of enantiopure 3-substituted cyclohexylamine derivatives bearing two chiral centres through a three-step route.31 Starting from prochiral bicyclic diketones, the first chiral centre was introduced using a C–C hydrolase (6-oxocamphor hydrolase from Rhodococus ruber NCIMB 9784, OCH), leading to the corresponding keto acids that were subsequently esterified using Candida antarctica lipase B (CALB) and methanol. Final amination using stereocomplementary transaminases allowed the formation of the second chiral centre, furnishing the desired amino ester diastereoisomers (Scheme 1a). The first two steps were successfully developed in a one-pot cascade and mostly organic media, requiring the filtration of the hydrolases before performing the transamination reaction in the same medium.
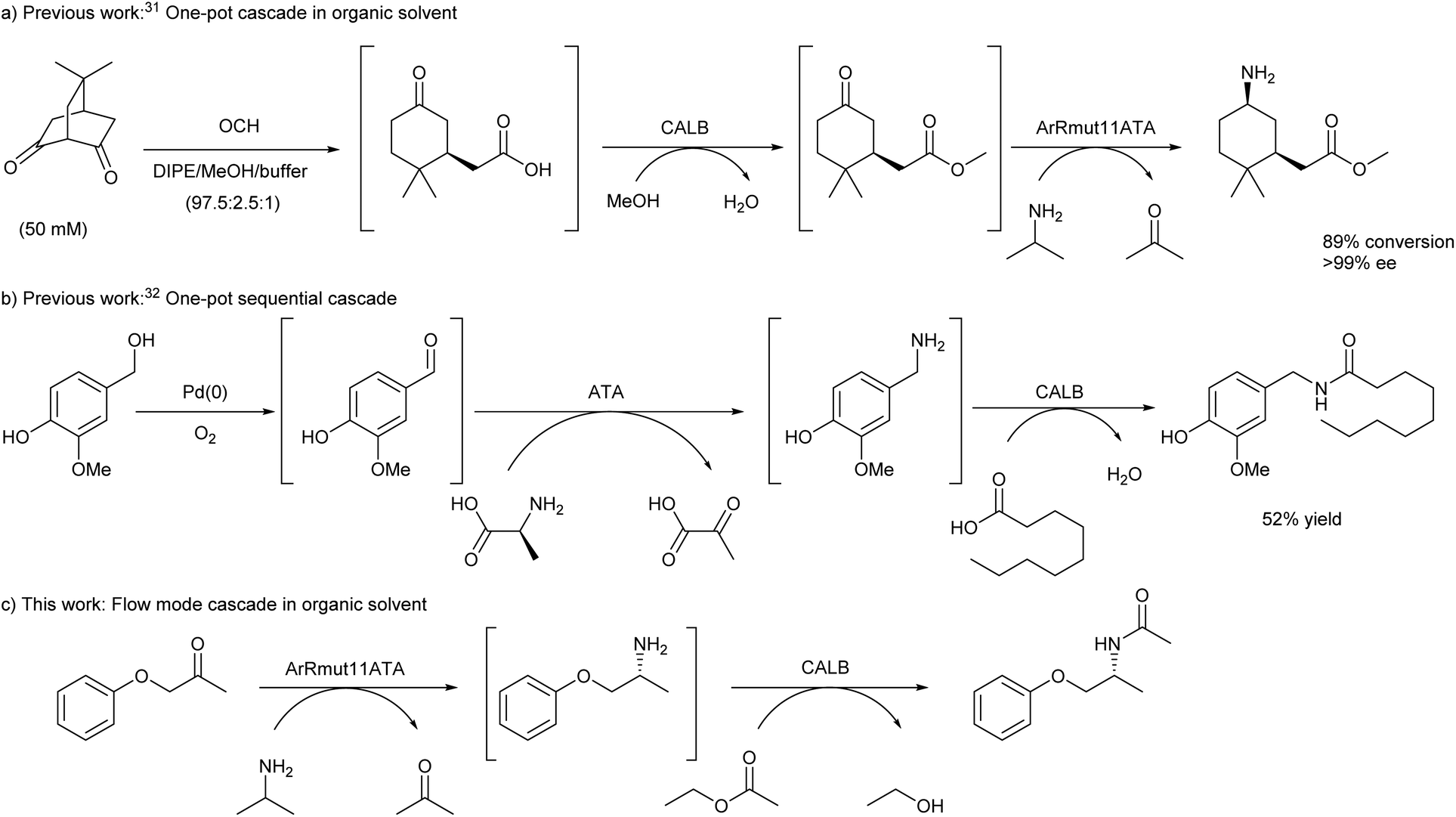 | ||
| Scheme 1 Enzymatic cascades combining ATAs and lipases. | ||
A year later, Berglund, Córdova and co-workers described the synthesis of capsaicinoids from vanillyl alcohol combining a palladium catalyst, an ATA, and a lipase (Scheme 1b).32 The cascade could be successfully performed in a sequential mode without any purification of intermediates, however the reaction medium had to be changed for each individual step. Herein, we sought to find suitable conditions for a lipase-ATA cascade for the formation of chiral nitrogenated compounds in organic solvent, exploiting the potential of flow chemistry to accomplish a stereoselective bienzymatic cascade to transform a ketone into a chiral amine in organic medium (Scheme 1c). With this purpose and selecting 1-phenoxypropan-2-one as model substrate, (R)-selective Arthrobacter sp. round 11 variant transaminase (ArRmut11ATA)33 and CALB, an efficient hydrolase for amide synthesis,34–36 were considered. The choice of both enzymes was based on the excellent selectivity displayed by ArRmut11ATA and CALB in the modification of structurally similar ketones,37 and racemic amines,38,39 respectively.
Results and discussion
Both enzymes were immobilised on EziG-Amber, a polymer coated glass with a semi-hydrophilic polymer surface. For each step, optimal conditions were investigated employing isopropylamine (2-PrNH2) and EtOAc as amine and acyl donors, respectively. The first reaction of the cascade, the transamination, has been previously studied with other ATAs on EziG carriers. It was shown that the amount of water in the system plays a key role in the ATA activity and is required to be optimised for each individual enzyme.40 Thus, one of the main objectives in the current investigation was to adopt a reaction engineering approach for ArRmut11ATA to facilitate a smooth transition between the two steps of the cascade, as well as to investigate the possibility of establishing a one-pot system. The motivation behind using EtOAc originated from the goal of employing it as both acyl donor and solvent in following steps of the cascade.17,41Enzyme immobilisation (ATA and lipase)
EziG, provided by EnginZyme AB (Sweden), is a material based on controlled pore glass, which is coated with organic polymer and chelated Fe(III) for His-tag binding. EziG is available in three different versions with varied surface properties. The first type of material, henceforth called EziG-Opal (LCAA CPG), is characterised with a hydrophilic surface, EziG-Coral (HybCPG VBC) has a hydrophobic surface polymer and EziG-Amber (HybCPG copo) is coated with a semi-hydrophilic polymer. The immobilisation of ArRmut11ATA was performed on the three types of EziG materials while CALB was immobilised on EziG-Amber as it was previously showed to be the most efficient support.42 The immobilisation of enzymes was performed by incubating the desired amount of cell free extract (CFE) in buffer supplied with EziG carrier material according to the protocol described in the Experimental section. The immobilisation progress was monitored by measuring the remaining enzymatic activity of the supernatant using a spectrophotometric assay for ArRmut11ATA and active-site titration assay for CALB.The immobilised ATA was used in aqueous media for the amination of 1-phenoxypropan-2-one with 2-PrNH2. Such model reaction was used to compare the activity of the immobilised enzyme on the three different supports. The model reaction for the immobilised CALB was the kinetic resolution of 1-phenylethan-1-ol by the transesterification with vinyl acetate in organic solvent. ArRmut11ATA was successfully immobilised on the three different EziG supports, from the CFE. Table 1 shows that the highest protein loading was achieved with EziG-Amber, which resulted in 5.7 w/w% loading and 43% recovered activity. Further improvement in protein loading up to 8.1 w/w% was possible when the enzyme was purified before immobilisation, as shown in Fig. S1 in ESI.† Although Opal provided higher recovered activity, the overnight reaction led to a higher conversion into the target amine when Amber was used. Therefore, support Amber was selected as the preferred option for this study. The immobilisation yield obtained for CALB was 67%, which corresponds to 7.5 w/w% protein loading, and the conversion towards the O-acetylated 1-phenylethan-1-ol was 15% after 0.5 h. Under the same conditions, Novozyme 435 (the most commercially applied enzyme preparation of CALB) gave 13% conversion.
| Enzyme | Support | Immobilisation yieldb [%] | Target protein loadingc [w/w%] | Recovered activityd [%] |
|---|---|---|---|---|
| a For the ArRmut11ATA: 7.5 mg of CFE containing 0.825 mg of target protein were used with 10 mg of support in all cases. For the CALB: 1 L of liquid CFE was used with 50 g of support. b Immobilisation yield = [(activity of the free enzyme − activity of the supernatant after immobilisation)/activity of the free enzyme] × 100. c Protein loading = (amount of target protein offered to the support × Immobilisation yield/10). d Recovered activity = (specific activity of immobilised enzyme/specific activity of the free enzyme) × 100. Activities for free and immobilised enzyme were obtained from the initial rates (Fig. S2 in ESI†). | ||||
| ArRmut11ATA | Amber | 69 ± 6 | 5.7 ± 0.5 | 43 ± 1.3 |
| ArRmut11ATA | Coral | 66 ± 6 | 5.4 ± 0.5 | 35 ± 1.4 |
| ArRmut11ATA | Opal | 57 ± 3 | 4.7 ± 0.2 | 66 ± 1.6 |
| CALB | Amber | 67 ± 2 | 7.5 ± 3.7 | — |
ATA-catalysed reaction in organic solvent
EziG-ATAs require a certain water activity in the reaction to remain active in an organic solvent. This preserves the functionality of the enzyme as water is required in the local environment around the protein, to ensure that 3-dimensional structure remains intact, and to prevent self-aggregation.43 Each ATA-catalysed reaction requires its own level of water activity in the reaction mixture for optimum functionality. Normally, less than a monolayer of water is required for an enzyme molecule to have activity in organic solvent. There are several ways to control and set this water activity with the most straightforward one being the addition of water to the reaction mixture.In this project the immobilised ATAs were used as wet formulations. Since the immobilisation was done in aqueous buffer, the immobilised enzyme could not be used directly in the reaction due to the excessive amount of water in the formulation. While such water excess could protect the enzyme from bulk organic solvent, it also hinders the uniform distribution of the catalyst within the reaction mixture and impedes efficient mass transfer of the target compound to the enzyme. Thus, an additional step was introduced to the immobilisation protocol aimed to reduce the amount of water in a controlled fashion for the final preparation, to later allow the remaining water to re-equilibrate with the reaction mixture. Such mild protocol is designed to maintain a certain amount of water within the immobilised enzyme preventing a decrease in enzymatic activity. Following this procedure, it was possible to set the water content in the immobilisation preparation through a series of washing steps: first with 2-propanol (2-PrOH) with set volume (v/v%) of deionised water and second with an organic solvent to remove 2-PrOH. Since the exact amount of water in the immobilised preparation was never quantified in this study, each water activity level will be characterised as “water content in 2-PrOH during ATA-EziG wash” referring to v/v% of water in 2-PrOH used during the washing steps after immobilisation. In future studies, Karl Fischer titration will be utilised to allow for better quantification of water content in the reaction media.
A set of reactions in EtOAc with 1-phenoxypropan-2-one (100 mM) and 2-PrNH2 (250 mM) as an amine donor was chosen to assess the activity of the ArRmut11ATA immobilised on the three different supports. The water amount was set by first washing with 10 v/v% water in 2-PrOH and second, running the reaction in EtOAc with 3 v/v% water. Based on experimental results, it was observed that additional amount of water in the reaction mixture was needed (apart from that present in the immobilised catalyst) leading to higher conversions (Fig. S3 in ESI†).
Under the target operational conditions, the yields obtained with the immobilised enzyme on Amber, Coral and Opal were 58%, 50% and 58%, respectively (Fig. S4 in ESI†). Apart from immobilised preparation, freeze-dried CFE was also explored in organic solvent reactions resulting in 26% conversion to the target amine. The amount of target protein used in the reactions depended on the catalyst formulation (CFE or immobilised) and on the immobilisation yield obtained for the enzyme on the different supports. It was 0.825 mg for the CFE and 0.621 mg, 0.588 mg and 0.495 mg for Amber, Coral and Opal, respectively. These results showed that although less amount of target protein was used in the reactions when using immobilised enzyme, higher activities were achieved due to potential stabilisation under non optimal conditions such as organic solvents as reaction media.
For all enzyme formulations when moving to organic solvent, a change in the stereoselectivity was observed, with an increase from 30% in buffer to 94% in organic solvent. The cause has not been investigated, but a similar behaviour was observed for the Halomonas elongata TA, where subtle changes in the reaction conditions affected the enzyme enantiopreference.44 Apart from the activity and selectivity, another important factor to be considered was the operational stability under process conditions. To assess the stability of the immobilised enzymes, batch recyclability studies were carried out for ArRmut11ATA and compared to the freeze dried CFE. The conditions were the same as those previously used and all the immobilised preparations and CFE were washed twice in between runs with 3 v/v% water in EtOAc to remove any residual substrate or product that could be adsorbed on the catalyst. It was observed that the activity decreased to the same extent for all the immobilised formulations and CFE. After one cycle all tested catalysts lost 20–25% of their initial activity (Fig. 1) and after four recycles 80% of initial activity was lost. The fact that there are no big differences between the three tested supports and the CFE indicates that the stability is an enzyme dependent characteristic and changing the support would not lead to any significant improvement. In order to streamline the experimental process, a decision regarding the support had to be made, leading to the selection of Amber. Although there were no significant differences observed in terms of activity towards the target reaction and recyclability among the various supports tested, Amber was chosen based on slightly higher protein loading. Additionally, selecting Amber was advantageous because it was the same support used for the immobilisation of the lipase enzyme. This decision aimed to maintain consistency and simplify the overall experimental setup.
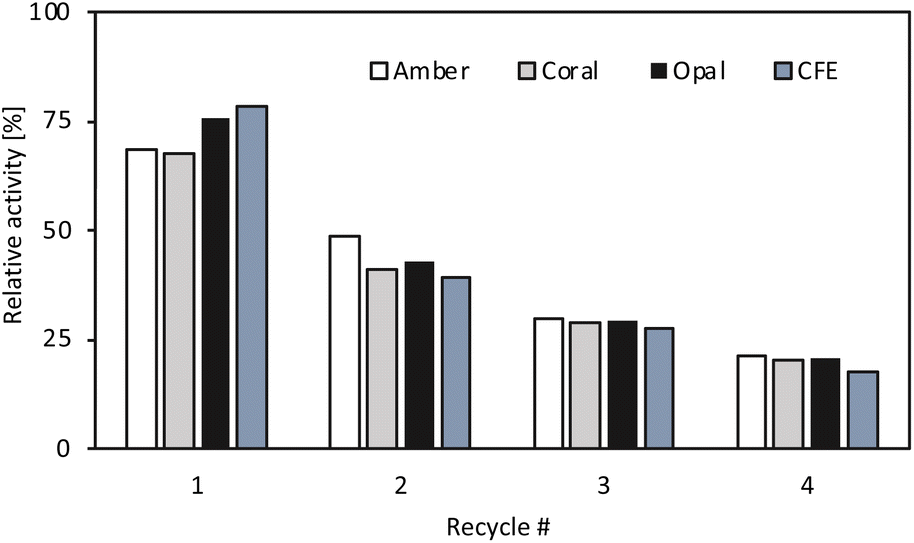 | ||
| Fig. 1 Recyclability studies of the CFE and immobilised ArRmut11ATA on the three different supports. Biotransformation conditions: 100 mM 1-phenoxypropan-2-one, 250 mM 2-PrNH2, 3 v/v% water in EtOAc, 10 mg of immobilised catalyst or 7.5 mg CFE in 1 mL total reaction volume, 18 h, 37 °C, 1200 rpm. Between runs the catalysts were washed twice with 1 mL of 3 v/v% water in EtOAc. Relative activity is the activity obtained in each recycle relative to that calculated in the first reaction cycle (set as 100%, Fig. S4 in ESI†). Reactions were performed in duplicate, and yields were determined using GC, after derivatisation of the samples with acetic anhydride. | ||
Stepwise ATA-lipase cascade in batch
In order to determine the operational window between transaminase and lipase reactions, it was necessary to explore the role of the reaction medium, particularly the water content in ethyl acetate. While the transaminase reaction benefits from higher water levels, excessive water could hinder the lipase-catalysed acylation step. To address this, both reactions were carried out using EtOAc with different water concentrations ranging from 0 to 3 v/v% (Fig. 2).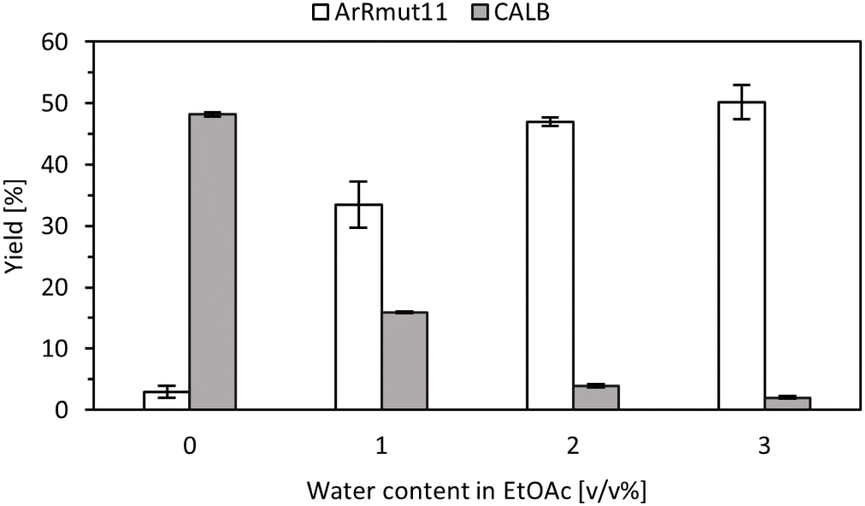 | ||
| Fig. 2 Yield data from the biotransformations of 1-phenoxypropan-2-one using ArRmut11ATA immobilised on Amber and for the kinetic resolution of the racemic 1-phenoxypropan-2-amine using CALB immobilised on the same support. ATA-catalysed reaction: 100 mM 1-phenoxypropan-2-one, 250 mM 2-PrNH2, different water contents in EtOAc, ArRmut11ATA-EziG (10 mg of EziG with 0.5 mL of 15 mg mL−1 CFE) in 1 mL total reaction volume, 18 h reaction time, 37 °C, 1200 rpm. Lipase reaction: 100 mM racemic 1-phenoxypropan-2-amine, 250 mM 2-PrNH2, different water contents in EtOAc, 1 mL total reaction volume, 24 h, 37 °C, 1200 rpm. Reactions were performed in duplicate, and yields were determined using GC. | ||
As previously described, ArRmut11ATA was immobilised on Amber and this preparation was washed with 10 v/v% water solution in 2-PrOH. The obtained results were consistent with previous observations. Hence, when the reactions were performed in neat EtOAc, target amine was not detected, however, with 1 v/v% water, conversions higher than 30% into the enantiopure amine were observed after 18 h. In parallel, the lipase-catalysed reactions were run in the presence of water to simultaneously confirm its influence in the amidation reaction. It was determined that the activity of the lipase with racemic 1-phenoxypropan-2-amine decreased even when 1 v/v% water was present in the reaction mixture (more than 50% activity decrease, Fig. 2), and higher amounts were incompatible with this step. The obtained results validated our hypothesis regarding the importance of water content in the reaction, enabling an operational window for both enzymes under the same reaction conditions. Specifically, it was determined that adding a 1 v/v% water in EtOAc was necessary for both enzymes to be sufficiently active.
Since the optimum water amount in the organic solvent was modified, as the next step it was necessary to re-evaluate the water content in the washes for immobilised ArRmut11ATA. Such study was aimed to ensure the compatibility between both steps. For this purpose, different immobilised ArRmut11ATA samples were prepared with variation in washing conditions (0–10 v/v% water in 2-PrOH). Later, catalysts were subjected to the previously identified conditions that were compatible with the lipase step (1 v/v% water in EtOAc as solvent). After 18 h, the supernatants were transferred to new tubes and the lipase was added, following a stepwise approach to run the cascade reaction in batch (Fig. 3).
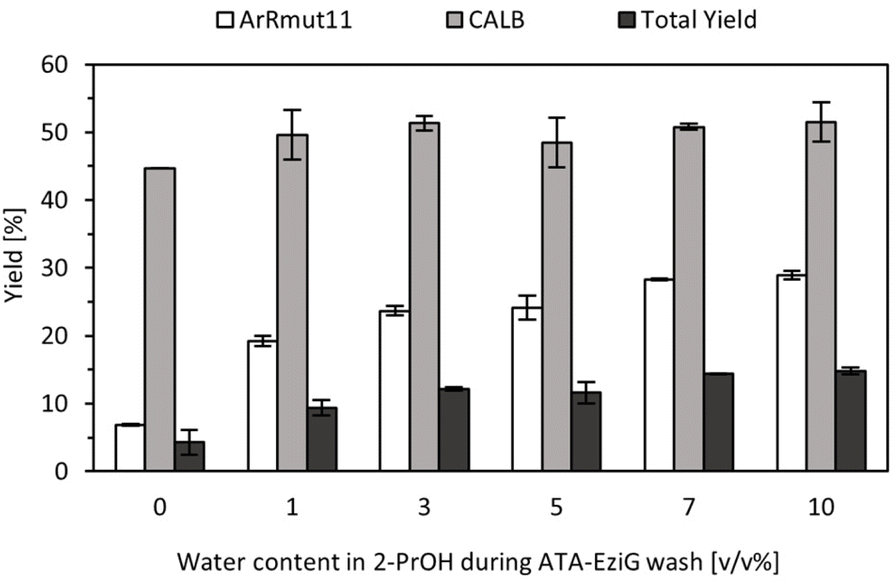 | ||
| Fig. 3 Yield data for the biocascades starting from 1-phenoxypropan-2-one using ArRmut11ATA immobilised on Amber containing varying levels of water and CALB immobilised on the same support. Biotransformation conditions: 100 mM 1-phenoxypropan-2-one, 250 mM 2-PrNH2, 1 v/v% water in EtOAc, ArRmut11ATA-EziG (10 mg) in 1 mL total reaction volume, 18 h, and then CALB-Amber (10 mg) was added, 20 h, 37 °C, 1200 rpm. Reactions were performed in duplicate, and yields were determined using GC. | ||
The yields obtained from the ATA-catalysed reaction were in agreement with previous results, approximately 30% was achieved when at least 10 v/v% water in 2-PrOH was used as washing solution. Washing conditions with higher water content were not included in this study since in previous experiments it was observed that between 10 to 20 v/v% water in 2-PrOH, the resulting activity for the enzymatic preparations were similar (Fig. S3 in ESI†). When ATA reaction mixture was used directly, lipase step proceeded as expected exhibiting similar activity as was observed previously at 1 v/v% water content in EtOAc with commercial racemic amine (Fig. 2). Despite incomplete conversions in ATA reactions and thus limited substrate availability in the second step of the cascade, it seemed that the lipase activity was not influenced by the water content in the washing solutions used for immobilised ArRmut11ATA (Fig. 3).
The total yield in the cascade was calculated based on the amide formation and initial substrate concentration, increasing from 4% to 15% at higher water percentage in the washing solutions (0–10 v/v%, Fig. 3). These results showed that the limiting step in this cascade is the ATA-catalysed reaction and that the water percentage during the washes had a limited effect on the lipase activity. For the following experiments, 10 v/v% water in 2-PrOH was chosen as the washing solution for the ATA to ensure the highest activity of immobilised catalyst in the first step of the cascade. The maximum yield to the amide under the best batch conditions was 15%, corresponding to a productivity value of 2.8 mg g−1 h−1. The full cascade was also run in batch in a one-pot approach with both enzymes present in the reaction vessel from the beginning. In this case, no conversion was observed to the intermediate or the final product (amine or amide).
A closer analysis of the composition of the reaction mixture revealed the formation of N-isopropylacetamide as by-product. Initially, it was speculated that the consumption of 2-PrNH2 by the acetylation reaction might deplete the offered amine donor for the transaminase reaction. Consequently, experiments were conducted with increasing concentrations of 2-PrNH2 to ensure sufficient equivalents for the transaminase. However, the results remained unchanged with no detected conversion to the intermediate amine. It is possible that the acetylated 2-PrNH2 or the co-product ethanol, formed during the acetylation reaction, may inhibit the transaminase enzyme. To investigate this hypothesis, the transaminase reaction was carried out in the presence of different concentrations of ethanol (1–10 mM, equivalent amounts of formed N-isopropylacetamide), however in all cases, conversion to the desired amine was observed.
Simultaneously, the potential inhibition of the transaminase by any component present in the lipase formulation was examined. The transaminase reaction was performed under the previously tested conditions, with the solvent (ethyl acetate with 1 v/v% water) preincubated with EziG-CALB. Interestingly, no conversion was observed under these reaction conditions. Furthermore, the addition of 2-PrNH2 to this preincubated solvent resulted in a liquid–liquid phase separation. Such behaviour could be attributed to salt-induced liquid–liquid phase separation caused by the buffer salts present in the immobilised CALB. Subjecting immobilised CALB to a washing procedure to remove impurities and buffer salts before the solvent incubation procedure did not eliminate phase separation. Although the cause behind this phenomenon remains unknown, it is evident that the addition of immobilised CALB alters the reaction mixture, causing phase separation and impairing the transaminase activity. These results highlight the need for compartmentalisation of both catalysts to make the cascade system feasible.
ATA-lipase cascade in continuous mode
The bienzymatic system was applied in a continuous flow setting. Since when the cascade was tested in batch one-pot mode no conversion was observed, physical catalyst separation was needed to achieve turnover of both catalysts. Therefore, the immobilised ArRmut11ATA and CALB were used as separate packed bed reactors (PBR), thus compartmentalising them. The reaction mixture composition used for the flow was identical to the best batch mode experiments: 100 mM 1-phenoxypropan-2-one, 250 mM 2-PrNH2 and 1 v/v% water in EtOAc as solvent.Two glass column reactors (15 mm i.d., 10 cm length) were filled with 1 gram of the corresponding immobilised catalyst. A slurry of immobilised ArRmut11ATA in 1 v/v% water in EtOAc was poured into the column, the bed was allowed to settle, and the solvent excess was drained. EziG-CALB catalyst was packed dry in a separate column.
Once the flow setup was assembled (Scheme 2 and Fig. S5 in ESI†), a washing step with 1 v/v% water in EtOAc was performed, to equilibrate the columns. Then, a substrate solution containing 1-phenoxypropan-2-one (100 mM) and 2-PrNH2 (250 mM) in EtOAc with 1 v/v% water was pumped through the reactors at 1.6 mL h−1 flow rate. The ArRmut11ATA PBR volume was calculated to 7.1 mL, corresponding to a residence time of 4.4 h. For the CALB PBR the volume was 5.3 mL, and the residence time was 3.3 h. The system was run for 11 days, which corresponds to 60.4 and 80.6 reactor volumes, for ArRmut11ATA and CALB, respectively.
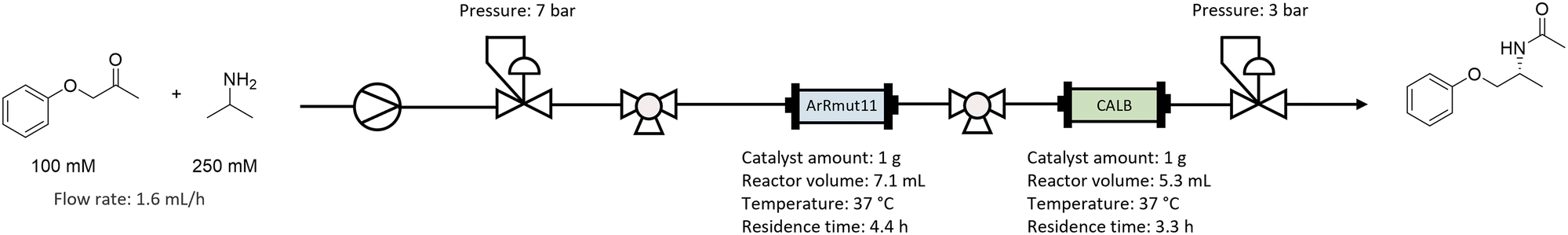 | ||
| Scheme 2 Continuous flow setup for the ArRmut11ATA and CALB cascade reaction for the production of (R)-N-(1-phenoxypropan-2-yl)acetamide. Residence time is defined here as the measured flow rate over the catalyst bed volume (calculated by using the bed height and internal diameter of the reactor). | ||
As depicted in Scheme 2, two additional 3-position valves were introduced in the system. The first one located after the pump and before the reactors, this allowed the analysis of the feed composition. The second was placed in between the ArRmut11ATA and CALB reactors, and was used to analyse the progress of the ArRmut11ATA-catalysed reaction to check the stability of this enzyme. Finally, the performance of the full cascade was measured at the outlet of the second reactor with CALB catalyst. The integration of consecutive sampling valves highlights a potential to adopt such an approach in an automated fashion and may allow for the design of closed loop self-optimising continuous biocatalytic systems.
To ensure that stable pressure and temperature throughout the experiment were obtained, the system was run for 25.5 h before the first samples which was considered as stabilisation time, corresponding to approximately three reactor volumes. Then, aliquots were taken and the composition of the feed, ArRmut11ATA outlet and CALB outlet were analysed. (R)-N-(1-phenoxypropan-2-yl)acetamide was obtained with 99% ee and an initial productivity of the cascade of 9.6 mg g−1 h−1. This means a 3.4-fold increase in productivity compared to the batch experiments. These initial values decreased to half after 11 days, due to the ATA activity loss (Fig. 4). The activity of the lipase catalyst appeared to be stable, however since the ATA activity was gradually decreasing, the substrate concentration for the lipase step also was diminishing throughout the runs. Thus, for the acylation CALB/amine ratio was continuously increasing, and for this reason activity losses for the lipase could not be detected.
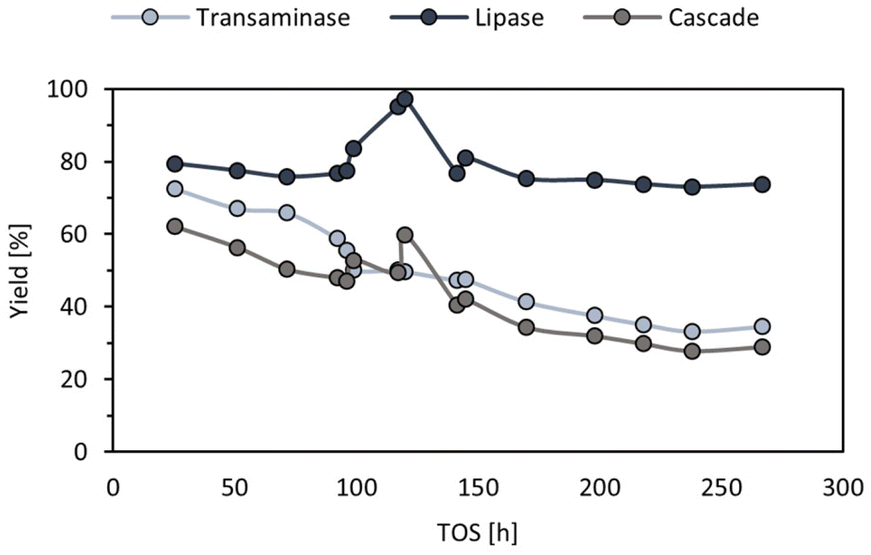 | ||
| Fig. 4 In flow stability test of the ArRmut11ATA and CALB cascade. For the ATA-catalysed amination the yield is calculated based on the initial 1-phenoxypropan-2-one concentration (100 mM) and (R)-1-phenoxypropan-2-amine measured at the outlet of the ATA reactor. For the lipase the yield is calculated based on the (R)-1-phenoxypropan-2-amine concentration measured at outlet of the ATA reactor and (R)-N-(1-phenoxypropan-2-yl)acetamide at the outlet of the lipase reactor. The cascade yield corresponds to the total conversion into (R)-N-(1-phenoxypropan-2-yl)acetamide and is based on the product formed and the initial ketone concentration (100 mM). The yields were determined using GC. TOS means time on stream. | ||
It is worth mentioning that one unknown peak was observed in all the outlets, i.e., feed, ATA, and lipase. The higher intensity of this peak was observed in the feed, and the relative area was stable throughout the flow run time (40%). It was not possible to isolate this compound, since after solvent evaporation (leaving the solvent to be evaporated at room temperature and atmospheric pressure) the area of the peak was decreasing to residual amount, which was not enough for characterisation. Instead, the reaction mixture was analysed by 1H-NMR (preparing the mixture in toluene-d8) and GC-MS (preparing the mixture in EtOAc). From both experiments, the peak was identified as the imine derived from the reaction of 1-phenoxypropan-2-one and 2-PrNH2, N-isopropyl-1-phenoxypropan-2-imine (Fig. S6–S9 in ESI†).
Transaminase stability
One important consideration for the application of a catalyst in industry is the long-term stability, and this can be improved by decreasing the operation temperature. Thus, in the next experiments the reactors were run under the same conditions but at room temperature (not controlled), to see whether mild conditions would lead to an improved stability of the immobilised enzymes.Aiming to obtain the same activity or conversion values as before, in order to compare both continuous mode experiments, the flow rate was decreased three-fold, to 0.54 mL h−1. The flow rate was adjusted according to the data obtained from batch reactions – at room temperature the activity was three times lower compared to 37 °C. The results showed a decrease in the initial productivity to 2.6 mg g−1 h−1. Moreover, the activity retention after 56 h on stream of the room temperature flow run appeared to be identical to the one at 37 °C, showing that in this case catalyst stability may not be temperature dependent (Fig. S10 in ESI†).
E factor calculation
The E factor was first introduced by Sheldon in 1992 and can be used to measure the sustainability of a process. This metric takes into account the product yield in addition to waste components such as solvent and material losses from (multi)step processes.45 To consider the environmental implications of our continuous biocatalytic process, we calculated E factor taking into consideration the waste generated due to the different components present. To determine the quantities wasted, the yield used was averaged over the whole flow run (39.6%), and total volume was calculated from the flow rate (Table 2). Obviously, the main component that contributed to E factor was EtOAc employed as both solvent and acylating agent, followed by 2-PrNH2 and water. The unreacted substrate was considered as material loss. The high E factor (117.5) in this case highlights the need to run continuous processes at high conversions with stable catalyst formulations.46 However, this number could largely diminish if a solvent recycling system would be implemented.| Component | Mass wasted for 3.23 g of amidea (g) | Contribution to E factor (kg kg−1) |
|---|---|---|
| a Taking an average yield value of 39.6%. | ||
| Ethyl acetate | 362.40 | 112.20 |
| Water | 4.22 | 1.31 |
| Isopropylamine | 5.29 | 1.64 |
| EziG-CALB | 1.00 | 0.31 |
| EziG-ArRmut11ATA | 1.00 | 0.31 |
| Acetone (co-product) | 0.97 | 0.30 |
| Ethanol (co-product) | 0.77 | 0.24 |
| Material loss | 3.83 | 1.19 |
| Total | 117.50 | |
Conclusions
A bienzymatic cascade for the stereoselective preparation of optically active (R)-N-(1-phenoxypropan-2-yl)acetamide from the corresponding prochiral 1-phenoxypropan-2-one was designed. As biocatalysts, (R)-selective Arthrobacter sp. round 11 variant transaminase and the Candida antarctica lipase type B were found to be active enzymes to catalyse the corresponding bioamination and acylation in a smooth manner. After immobilisation of the amine transaminase on different EziG supports, EziG-Amber resulted in the most active preparation for investigation of the bienzymatic cascade in EtOAc with different water contents. Under optimised conditions, the immobilised ATA showed higher activity compared to the CFE, which might be attributed to a potential stabilisation under non-native conditions such as the use of organic solvents as reaction media. To assemble the cascade, the amount of water in EtOAc was the most important parameter to be optimised. Enzymes under investigation required different water amounts in reaction mixture for optimum operation. In order for the cascade to produce the target compound, enzyme activities trade-off was unavoidable. Further screening revealed 1 v/v% water amount in the reaction mixture to be viable for target enzymes resulting in 30% activity retention for both of them, compared to the observed activities under optimal amount of water for the individual steps. Although low or no conversion were observed when the cascade was tested in batch one-pot mode, the system was successful in a continuous flow setting.Despite the exhaustive optimisation performed to identify the best reaction parameters and to understand the cascade performance dependencies, our approach remains as a proof of concept. Further development is necessary, which includes not only the identification and engineering of superior enzymes, but also the investigation of methods such as co-immobilisation or alternative compartmentalisation. These strategies could improve the bienzymatic approach outcome.7,8,47,48
To achieve good results, the immobilised ArRmut11ATA and CALB were packed in separate PBR, compartmentalising and segregating them. The implementation of the flow setup resulted in a great improvement of the metrics (up to 3.4-fold for productivity) towards the enantiopure (R)-amide when compared to the equivalent batch cascade in a stepwise approach. These results demonstrate the potential that modular PBR in flow setups can offer to overcome incompatibilities in cascade (bio)transformations.
Experimental
Materials
All chemicals were purchased from Sigma Aldrich unless stated otherwise. EziG supports were provided by EnginZyme AB.Enzyme expression and lysis
The enzymes were overexpressed in E. coli BL21(DE3), 50 μL of glycerol stock were inoculated to terrific broth autoinduction media (TB AIM 500 mL) with 100 μg mL−1 of kanamycin. The cultures were incubated at 200 rpm and 37 °C for 5 h and then at 200 rpm and 30 °C for 19 h. After 24 h the OD was measured, and the cells were harvested by centrifugation at 8000 rpm for 10 min at 4 °C. The collected cell pellets were resuspended in MOPS buffer and cell lysis of all the samples was performed using a Microfluidizer at 1200 bar over five passes. After lysis the OD was checked again to ensure the cells had been properly disrupted. The lysed cells were centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 1 h at 4 °C and the CALB was stored as liquid CFE in the fridge. For ArRmut11ATA the supernatant was recovered and PLP was added to a final concentration of 0.01 mM before freeze drying. Freeze drying of the CFE was performed using a freeze dryer and stored in the fridge.
000 rpm for 1 h at 4 °C and the CALB was stored as liquid CFE in the fridge. For ArRmut11ATA the supernatant was recovered and PLP was added to a final concentration of 0.01 mM before freeze drying. Freeze drying of the CFE was performed using a freeze dryer and stored in the fridge.
ArRmut11ATA protein purification
For the samples that were purified, the supernatant was collected and filtered (0.45 μm). The purification was performed on a Ni-IDA agarose column using standardised immobilised metal affinity chromatography (IMAC) protocol with ÄKTA explorer. The binding buffer was 50 mM MOPS, 150 mM NaCl, pH 7.6. The elution buffer was the same as the binding buffer but with 1 M imidazole. Fractions with containing target enzyme were merged and buffer exchanged to MOPS buffer (50 mM, pH 7.6) with 0.3 mM pyridoxal-5′-phosphate (PLP) using a PD10 desalting column. The concentration of pure proteins in solution was determined by measuring the absorbance of the solution at 280 nm.Determination of target enzyme content in CFE
To quantify the amount of target enzyme present in the CFE, a known quantity of the extract was subjected to purification, and the purified enzyme was quantified based on its absorbance at 280 nm. Specifically, 300 mg of CFE were purified, yielding 18 mL of purified enzyme solution with a concentration of 1.9 mg mL−1. Based on this measurement, the target enzyme constituted 11% of the CFE.Immobilisation procedure
The freeze-dried powder of ArRmut11ATA-His was rehydrated in 20 mM sodium phosphate buffer, pH 8 containing 0.3 mM pyridoxal phosphate to obtain a CFE concentration of 15 mg mL−1 (freeze-dried powder/buffer solution). The prepared CFE was resuspended on an end-over-end rotator for 1 h (20 rpm, rt) and then centrifuged for 5 min (7000 rpm, rt). After centrifugation, the CFE (500 μL) was transferred to a new tube containing the 10 mg of the support. Tubes were covered with foil and immobilisation was performed on an end-over-end rotator, 20 rpm, at room temperature for 2 h. Then, the supernatants were removed, and the immobilised supports were washed first with immobilisation buffer (1 × 1 mL, 30 s each), second with 2-PrOH washing solutions (2 × 1 mL, 30 s each) and third with the solvent used for the reaction (1 × 1 mL, 30 s), then the catalyst was used directly after removal of remaining solvent.When immobilised from the pure protein solution, the same procedure was done but 434 μL of pure enzyme solution were incubated with the support and PLP to 0.3 mM final concentration. In both cases the amount of target protein offered to the support (10 mg) was 0.825 mg.
For the immobilisation of CALB, the CFE was buffered with 20 mM MOPS pH 7.5 and EziG-Amber was then added. The ratio 1:20 EziG:CFE was chosen based on previous optimisations done at EnginZyme (data not shown). The immobilisation was performed with end-over-end mixing for 3 h before rinsing with the same buffer and vacuum drying for 16 h.
Immobilisation yield
For ArRmut11ATA, the CFE and supernatant from the immobilisations were diluted with 20 mM sodium phosphate buffer, pH 8. An aliquot of the diluted enzyme solution (100 μL) was mixed with a reaction mixture (100 μL) containing 10 mM 1-phenylethylamine, 20 mM sodium pyruvate in 20 mM sodium phosphate buffer, pH 8. The formation of acetophenone was measured by the absorbance (A245) at 245 nm every 49 s for 20 minutes using a plate reader. Reaction rates with each supernatant from immobilisations and fresh enzyme solution were extracted from linear regression of the data points (A245 min−1). The immobilised yield was calculated by determining the percentage of enzymatic activity left in the supernatant after immobilisation, relative to the enzymatic activity in the CFE.For CALB, the immobilised enzyme content was calculated by the tributyrin hydrolysis activity (TBU) assay, by comparing the activity of the starting CFE and the supernatant during immobilisation.
Recovered activity of immobilised catalysts
The recovered activity was obtained from the specific activity of immobilised enzyme compared to the specific activity of CFE in aqueous buffer as follows: recovered activity = (specific activity of immobilised enzyme/specific activity of the free enzyme) × 100. The specific activity was determined using initial reaction rates, considering the amount of enzyme used in the reaction. For the immobilised enzyme, the immobilisation yield was included in the calculations to consider only the amount of immobilised enzyme. The activity of ArRmut11ATA was determined by its ability to convert 1-phenoxypropan-2-one to 1-phenoxypropan-2-amine using isopropylamine as amine donor. A reaction mixture (1 mL) containing 50 mM 1-phenoxypropan-2-one, 250 mM isopropylamine and 5 v/v% DMSO in 20 mM sodium phosphate buffer, pH 8 was added to the immobilised catalyst, and the mixture was incubated for 5, 15, 30 and 45 min (1200 rpm, 37 °C). After that time, the reaction was quenched by adding 5 M NaOH. Then the reaction was extracted, and the yield of 1-phenoxypropan-2-amine was obtained by GC-FID analyses. Each reaction was performed in duplicate, and a single reaction was conducted for each time point. The activity of the immobilised CALB was tested in the kinetic resolution of racemic 1-phenylethan-1-ol with vinyl acetate as the acyl donor. The reaction mixture contained 1 M 1-phenylethan-1-ol, 600 mM vinyl acetate and 1 v/v% dodecane in tert-butyl methyl ether (MTBE).Continuous flow setup
Continuous flow reactions were performed using the following equipment: DIONEX dual piston HPLC pump (flow rate 1.6 mL h−1), PTFE/Steel tubing (1/16′′ ID), IDEX stainless steel BPR (back pressure regulator) holder fitted with a 7 bar cartridge, restek adjustable BPR with 5 μL dead volume, glass columns (15 mm ID), UNIQSIS heater block unit and OMNIFIT 3 way switching valves.GC-FID analysis
Gas chromatography coupled to flame ionisation detector (GC-FID) was used for analysis of the reactions and identification of unknown peaks (GC-MS). The ATA-catalysed reactions were analysed after derivatisation with acetic anhydride to facilitate the separation of both amine enantiomers. The lipase and cascade reactions in flow were analysed directly by sample dilution. Dodecane was used as internal standard. Method specifications on GC columns: CP-Chirasil Dex DB (25 m × 0.25 mm × 0.25 μm) or HP-5MS (30 m × 0.25 mm × 0.25 μm). 2 mL min−1 hydrogen, 47 kPa. Injection 1 μL with 20:1 split ratio. Injection temperature: 200 °C, Detector temperature: 250 °C, Detector type: FID Oven temperature: 100 °C, hold 2 min, 15 °C min−1 ramp to 195 °C, hold 2 min. Treatment of results: calibration curve with dodecane as an internal standard. Retention times (min) CP-Chirasil Dex DB: dodecane (4.5), 1-phenoxypropan-2-one (5.7), (R)-1-phenoxypropan-2-amine (5.9), N-isopropyl-1-phenoxypropan-2-imine (6.5) and (R)-N-(1-phenoxypropan-2-yl)acetamide (8.8). Retention times (min) HP-5MS: 1-phenoxypropan-2-one (4.4) and N-isopropyl-1-phenoxypropan-2-imine (6.1).
Author contributions
Conceptualisation: V. G.-F. and A. V.; investigation (enzyme immobilisation, flow continuous mode studies and development of analytical methods), A. P. and A. P. M.; experiment design and discussion: A. P. and A. P. M., I. L., V. G.-F. and A. V. Writing the original draft contribution, A. P., A. P. M. and A. V.; writing, review and editing, I. L., V. G.-F. and A. V. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
INTERfaces project has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 860414.References
- Biocatalysis for Practitioners. Techniques, Reactions and Applications, ed. G. de Gonzalo and I. Lavandera, Wiley-VCH, Weinheim, Germany, 2021 Search PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- J. M. Bolivar, J. M. Woodley and R. Fernandez-Lafuente, Chem. Soc. Rev., 2022, 51, 6251–6290 RSC.
- N. Borlinghaus, V. Wittmann and W. M. Braje, Curr. Opin. Green Sustainable Chem., 2022, 33, 100571 CrossRef CAS.
- H. Gröger, F. Gallou and B. H. Lipshutz, Chem. Rev., 2023, 123, 5262–5296 CrossRef PubMed.
- R. Kourist and J. González-Sabín, ChemCatChem, 2020, 12, 1903–1912 CrossRef CAS.
- A. P. Mattey, G. J. Ford, J. Citoler, C. Baldwin, J. R. Marshall, R. B. Palmer, M. Thompson, N. J. Turner, S. C. Cosgrove and S. L. Flitsch, Angew. Chem., Int. Ed., 2021, 60, 18660–18665 CrossRef CAS PubMed.
- S. C. Cosgrove and A. P. Mattey, Chem. – Eur. J., 2022, 28, e202103607 CAS.
- S. G. Newman and K. F. Jensen, Green Chem., 2013, 15, 1456–1472 RSC.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- G. Gambacorta, J. S. Sharley and I. R. Baxendale, Beilstein J. Org. Chem., 2021, 17, 1181–1312 CrossRef CAS PubMed.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 RSC.
- M. Santi, L. Sancineto, V. Nascimento, J. B. Azeredo, E. V. M. Orozco, L. H. Andrade, H. Gröger and C. Santi, Int. J. Mol. Sci., 2021, 22, 990 CrossRef CAS PubMed.
- S. Ötvös and C. O. Kappe, Green Chem., 2021, 23, 6117–6138 RSC.
- H. M. Arango, L. van den Biggelaar, P. Soumillion, P. Luis, T. Leyssens, F. Paradisi and D. P. Debecker, React. Chem. Eng., 2023, 8, 1505–1544 RSC.
- M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef.
- M. D. Truppo, J. D. Rozzell, J. C. Moore and N. J. Turner, Org. Biomol. Chem., 2009, 7, 395–398 RSC.
- J. Rudat, B. R. Brucher and C. Syldatk, AMB Express, 2012, 2, 11 CrossRef PubMed.
- S. Mathew and H. Yun, ACS Catal., 2012, 2, 993–1001 CrossRef CAS.
- M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965–6982 CrossRef CAS PubMed.
- R. Abu and J. M. Woodley, ChemCatChem, 2015, 7, 3094–3105 CrossRef CAS.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, Catalysts, 2018, 8, 254 CrossRef.
- P. Kelefiotis-Stratidakis, T. Tyrikos-Ergas and I. V. Pavlidis, Org. Biomol. Chem., 2019, 17, 1634–1642 RSC.
- F. Van Rantwijk and R. A. Sheldon, Tetrahedron, 2004, 60, 501–519 CrossRef CAS.
- V. Gotor-Fernández and V. Gotor, Curr. Org. Chem., 2006, 10, 1125–1143 CrossRef.
- E. Siirola, F. G. Mutti, B. Grischek, S. F. Hoefler, W. M. F. Fabian, G. Grogan and W. Kroutil, Adv. Synth. Catal., 2013, 355, 1703–1708 CrossRef CAS.
- M. Anderson, S. Afewerki, P. Berglund and A. Córdova, Adv. Synth. Catal., 2014, 356, 2113–2118 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- O. Kirk and M. W. Christensen, Org. Process Res. Dev., 2002, 6, 446–451 CrossRef CAS.
- V. Gotor-Fernández, E. Busto and V. Gotor, Adv. Synth. Catal., 2006, 348, 797–812 CrossRef.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- J. González-Sabín, V. Gotor and F. Rebolledo, Tetrahedron: Asymmetry, 2002, 13, 1315–1320 CrossRef.
- L. Muñoz, A. M. Rodriguez, G. Rosell, M. P. Bosch and A. Guerrero, Org. Biomol. Chem., 2011, 9, 8171–8177 RSC.
- W. Böhmer, A. Volkov, K. Engelmark Cassimjee and F. G. Mutti, Adv. Synth. Catal., 2020, 362, 1858–1867 CrossRef PubMed.
- V. Gotor-Fernández, R. Brieva and V. Gotor, J. Mol. Catal. B: Enzym., 2006, 40, 111–120 CrossRef.
- K. Engelmark Cassimjee, M. Kadow, Y. Wikmark, M. Svedendahl Humble, M. L. Rothstein, D. M. Rothstein and J.-E. Bäckvall, Chem. Commun., 2014, 50, 9134–9137 RSC.
- A. Zaks and A. M. Klibanov, J. Biol. Chem., 1988, 263, 8017–8021 CrossRef CAS PubMed.
- C. M. Heckmann, L. Robustini and F. Paradisi, ChemBioChem, 2022, 23, e202200335 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- Y. Liu, P. Liu, S. Gao, Z. Wang, P. Luan, J. González-Sabín and Y. Jiang, J. Chem. Eng., 2021, 420, 127659 CrossRef CAS.
- S. González-Granda, L. Escot, I. Lavandera and V. Gotor-Fernández, Angew. Chem., Int. Ed., 2023, 62, e202217713 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S10 including NMR, MS spectra and analytical data are included. See DOI: https://doi.org/10.1039/d3gc02426a |
| This journal is © The Royal Society of Chemistry 2023 |