
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expanding lignin thermal property space by fractionation and covalent modification†
Luke A.
Riddell
a,
Floris J. P. A.
Enthoven
a,
Jean-Pierre B.
Lindner
b,
Florian
Meirer
c and
Pieter C. A.
Bruijnincx
 *a
*a
aUtrecht University, Organic Chemistry & Catalysis, Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl
bBASF SE, Group Research, Carl-Bosch-Str. 38, 67056 Ludwigshafen am Rhein, Germany
cUtrecht University, Inorganic Chemistry & Catalysis, Debye Institute for Nanomaterial Science and Institute for Sustainable and Circular Chemistry, Faculty of Science, Utrecht, The Netherlands
First published on 13th July 2023
Abstract
To fully exploit kraft lignin's potential in material applications, we need to achieve tight control over those key physicochemical lignin parameters that ultimately determine, and serve as proxy for, the properties of lignin-derived materials. Here, we show that fractionation combined with systematic (incremental) modification provides a powerful strategy to expand and controllably tailor lignin property space. In particular, the glass transition temperature (Tg) of a typical kraft lignin could be tuned over a remarkable and unprecedented 213 °C. Remarkably, for all fractions the Tg proved to be highly linearly correlated with the degree of derivatisation by allylation, offering such tight control over the Tg of the lignin and ultimately the ability to ‘dial-in’ this key property. Importantly, such control over this proxy parameter indeed translated well to lignin-based thiol–ene thermosetting films, whose Tgs thus covered a range from 2–124 °C. This proof of concept suggests this approach to be a powerful and generalisable one, allowing a biorefinery or downstream operation to consciously and reliably tailor lignins to predictable specifications which fit their desired application.
Introduction
Efficient and viable biorefining necessitates the valorisation of lignocellulose's most complex and variable component, lignin. Current industrial delignification methods focus on accessing high purity carbohydrate streams to the detriment of the structure of the resulting technical, i.e. isolated, lignin.1–5 Of the delignification technologies, the kraft process is the most dominant, and structural analyses show kraft lignins to be a complex and heterogeneous combination of (a few remaining) common lignin inter-unit linkages, as well as kraft-specific structures and functionalities.6–8 Moreover, the distribution of functionalities and structures within the lignin is a function of molecular weight (MW). The complexity (and recalcitrance) of kraft lignin's polymeric structure is evident and suggests that kraft lignin valorisation via lignin-to-materials strategies, as opposed to lignin-to-chemicals via depolymerisation,5,9 perhaps is a more fruitful endeavour. Whilst important advances have been made in the many efforts focussing on direct lignin-to-materials approaches, which include lignin application in thermoplastics,10,11 road bitumen,12,13 films14,15 and polymer composites/blends,16–18 the pervasive problem of lignin's intrinsic functional group, linkage and MW heterogeneity has generally hampered many valorisation efforts. Indeed, intra-sample variation often leads to ill-defined and undesirable properties of the as-is technical lignin, as each individual component of the lignin contributes to the average with property values that span over a broad range, making it challenging to fit the lignin to an application. As an example, in lignin-polymer blends, this lack of property homogeneity (e.g. in solubility19–21) between the lignin (macro)molecules within a sample can lead to incompatibility between the lignin and the material matrix.10,22 In particular, kraft lignins are often not well suited for direct use in materials applications as they give rise to brittleness and themselves are only partially soluble in many conventional solvents, instead requiring less desirable solvents (e.g. DMF, DMSO) for total dissolution.19 Clearly, methods that (1) reduce lignin heterogeneity and (2) allow for fine-tuning the properties of a homogenised lignin sample would provide a powerful tool for more effective lignin valorisation. One often-used method to reduce heterogeneity is by fractionation.23,24 In one example, Duval et al. efficiently obtained lower-dispersity fractions from LignoBoost kraft lignin by sequential solvent extraction.19 Fine-tuning the properties of a (typically non-fractionated) lignin in turn is typically done by modification of the functional groups on the lignin. For example, Koivu et al. demonstrated the impact of fatty acid chain length and loading upon a lignin's glass transition temperature (Tg).25 While both fractionation and modification are potentially powerful, the two methods are rarely combined in systematic fashion.The ability to ‘dial-in’ a property is particularly important, as the feedstock typically must meet product needs and not vice versa. Hence with a chosen performance parameter in mind, exploration of its property space is thus an important exercise in outlining the potential and flexibility of a feedstock material for application (Fig. 1a). Once the property space is mapped, correlating the relation between structural and material properties would provide better understanding and control over the latter, allowing for informed tailoring of the lignin to suit specific applications. In particular, the Tg is a key physicochemical characteristic as it determines the suitability of lignin use in many possible applications. As the Tg defines the temperature of a material's transition between glassy and brittle, to ductile and elastomeric, it can also be considered a proxy for other industrially relevant materials performance parameters (e.g., viscosity). Homogenisation of the lignin via fractionation combined with partial/full modification of the lignin could considerably extend Tg space depending on the nature or degree of modification.
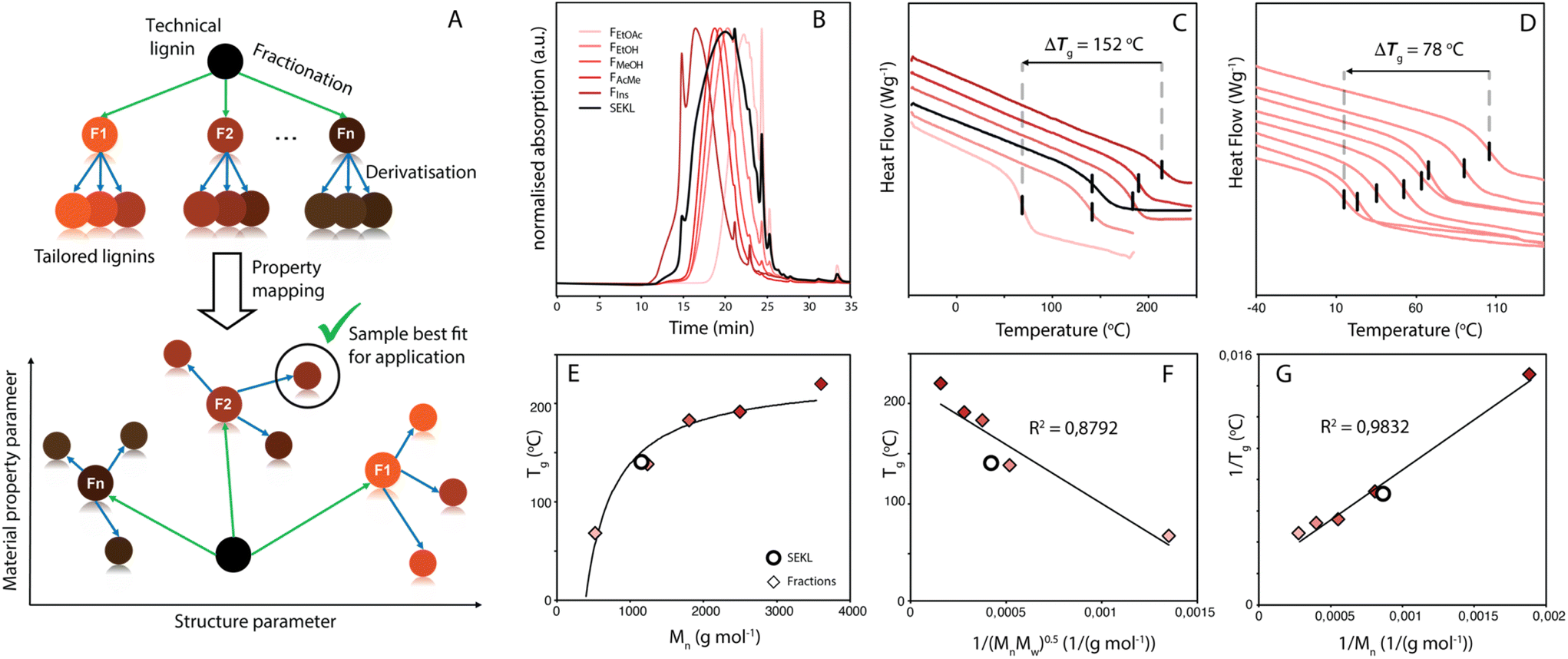 | ||
| Fig. 1 (a) Strategy for informed lignin selection by fractionation and derivatisation (b) normalised GPC traces of SEKL (black) and its subsequent fractions FEtOAc, FEtOH, FMeOH, FAcMe, FIns (red gradient) (c) DSC traces of SEKL (black) and its fractions (red gradient) (d) DSC traces of FEtOAc and its allylated derivatives FEtOAc − 0 to FEtOAc − 6. (e) Flory-Fox fit for SEKL and fractions (f) Flory-Fox-Ogawa fit for SEKL and fractions (g) Fox-Loshaek fit for SEKL and fractions. Note that for (c) and (d), traces were shifted vertically for clarity. | ||
In addition to property homogenisation and adjustment, insertion of reactive handles is typically also required for downstream applications. For this, the –OH functionalities are most often targeted using a wide array of modification reactions, which have the added effect of altering the physicochemical properties of the lignin as well.25–30 Some recent examples have shown how either lignin fractionation, modification, or in rare cases fractionation combined with a single (full) modification can greatly improve applicability for materials use.31–35 Here, we build on this approach and present a powerful, translational strategy that systematically combines fractionation and incremental modification to explore, expand and fully map the physicochemical parameter space of a chosen lignin (Fig. 1a). Using the well-established Williamson-type phenol etherification to introduce reactive allyl handles for application in thiol–ene based thermosets, we demonstrate that by combining these two relatively simple approaches, lignin property space can be systematically and predictably expanded upon and in fact, controlled by the appropriate degree of modification of the proper fraction.32,35 This opens up avenues for lignin utilisation in material applications for which it hasn't previously been considered. Rewardingly, the control over the lignin's properties is directly reflected in its derived materials, i.e., in the Tg of the thermosetting films. Such thermosets can be used in a wide range of applications, including in shape-memory materials,36 tissue scaffolds,37 coatings,38 and aerospace resins,39,40 thus providing many potential avenues for the valorisation of an underutilised bio-based feedstock.
Results and discussion
Fractionation, allylation and NMR analysis
The Lineo Classic lignin (SEKL) by Stora Enso is a commercially available softwood kraft lignin produced on an industrial scale and is thereby a relevant material for study. SEKL was fractionated by sequential extraction with ethyl acetate (EtOAc), ethanol (EtOH), methanol (MeOH) and finally acetone (AcMe), yielding four soluble fractions (FEtOAc, FEtOH, FMeOH, FAcMe) and a final insoluble residue (FIns) of improved Đ (Fig. 1b, Table S3†). These were then fully structurally characterised, see ESI† for details. Note that this sequence of organic solvents has been shown to be effective for fractionation of un-modified softwood kraft lignin.19,32,35 Use of a solvent series may result in a relatively high E-factor, but this can be avoided by distillation recovery to regenerate the fractionating solvents, overall closing the solvent loop. Without solvent recovery, however, fractionation with a solvent series is less green than, e.g., sequential precipitation methods,41 or ultrafiltration methodologies.42 Sequential precipitation methods, which have the disadvantage of generating contaminated water waste, have generally been explored for fractionation of modified lignins.41 Ultrafiltration methods are promising but suffer from filtrate flux decay due to membrane fouling, requiring its regeneration through cleaning. Overall, each fractionation method clearly has certain benefits and drawbacks which must be considered prior to selection of the method.The fractions were then each allylated to different degrees (samples denoted as Fsolvent − X, with higher X denoting higher degree of allylation). Post-allylation, the lignin samples were obtained as coloured powders, with a general brightening of the colour from brown to yellow with increasing %allyl; only FEtOAc−6 was obtained as a highly viscous liquid (Fig. S2†). The %allyl is defined by the difference in the total phenolic hydroxyl group content prior/post-allylation, as the derivatisation method is almost entirely selective for phenolic OH allylation (Fig. 2a). The phenolic OH content was quantified as a function of allylation degree by 31P NMR after phosphitylation of the (remaining) –OH groups, taking into account the additional mass from the new pendant groups, as described elsewhere.25 HSQC NMR analysis confirmed successful selective allylation of the phenolic hydroxyl functionalities, with few other changes to the structure being observed. Notably, two distinct resonances differing in both 1H and 13C shift were observed for the allyl α(C–H) correlation, corresponding to G-units and 5-substituted G-units respectively, as identified by overlay of the HSQC spectra of allylated model compounds and of SEKL (Fig. 2c), confirming a previous suggestion that this arises due to condensation modes of the G-rings by Jawerth et al.32
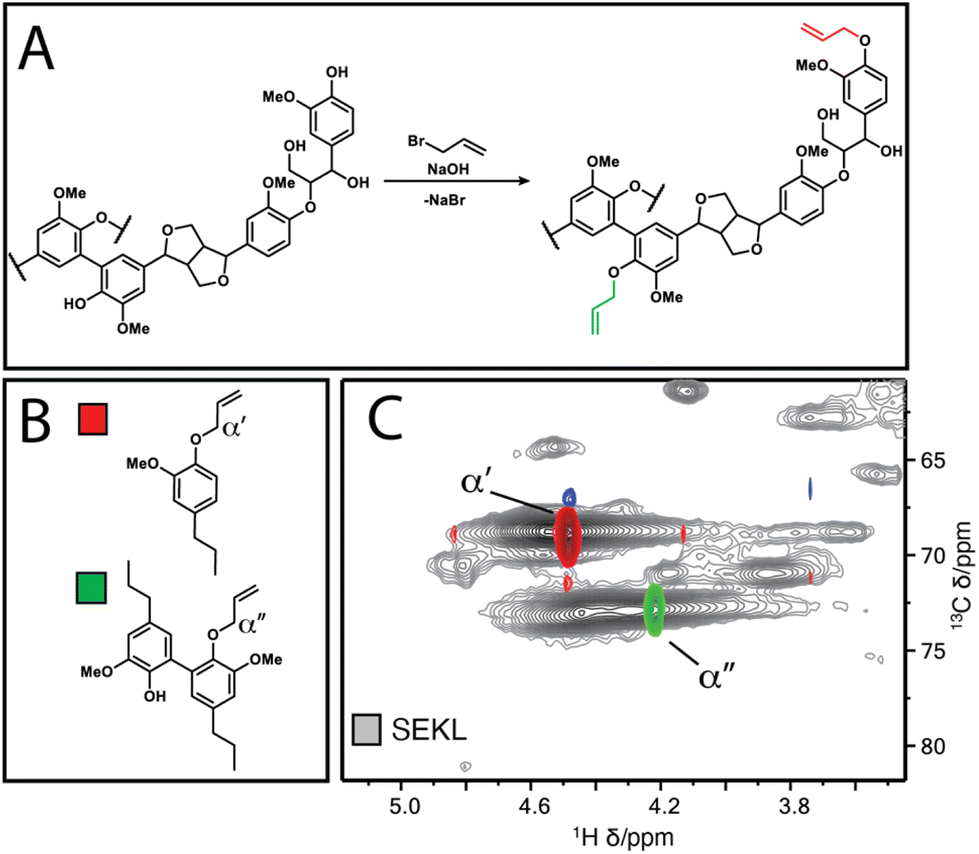 | ||
| Fig. 2 (a) Williamson allylation of lignin (b) Model compounds 1 & 2 (c) overlay of HSQC spectra of compounds 1 (red-blue) & 2 (green-orange) with SEKL (grey), respectively. | ||
Thermal properties
Kraft lignins typically bestow rigidity and brittleness to materials, owing to their intricate hydrogen bonding network,43,44 and polyaromatic and highly condensed structure,7,45 resulting in high Tg values, much above room temperature. As anticipated, the Tg values obtained by modulated differential scanning calorimetry (MDSC) proved to be strongly dependent on both MW and %allyl. The Tg values of fractions and SEKL are well described by the Flory-Fox relationship46 (Fig. 1d), previously shown to be a remarkably good model for both organosolv and kraft lignins.42,47,48 Alternative relationships that take dispersity (Đ) (Flory-Fox-Ogawa,49Fig. 1f) and crosslinking (Fox-Loshaek,50Fig. 1g) into account have been applied to lignin as well;47 in particular the latter relationship gave an excellent fit for our sample set, lending further credence to the crosslinked nature of SEKL as a typical kraft lignin. Fractionation alone thus already extended Tg property space by 152 °C with FEtOAc and FIns showing the lowest and highest measured Tgs of 68 °C and 219 °C, respectively. The Tg of as-is SEKL is 141 °C, an intermediate value between the EtOH and MeOH fractions but given that SEKL has a lower Mn than FEtOH, the larger Tg of SEKL is due to its Đ.Systematic allylation allowed for further coverage and extension of property space. Notably, in addition to allowing application-tailored Tg values, the sample set also revealed strong structure–property relationships between %allyl and Tg. Overall, the lowest Tg value was found to be that of FEtOAc−6, the most highly derivatised FEtOAc sample with Tg of 12 °C, a reduction of 56 °C when compared to the non-allylated EtOAc fraction (FEtOAc). The highest value was initially found to be from FIns, yielding an expanded ΔTg of 202 °C. The spread of values shown in Fig. 3a is quite remarkable when compared to literature values for fractionation (of an organosolv lignin) alone (∼150 °C),48 or for partial derivatisation of the same softwood kraft lignin with fatty acids (∼100 °C).25 Correlation plots of Tgvs. %allyl for each fraction's allyl derivatives, (Fig. 3c), show a remarkably strong linear relationship for all fraction series. The drop in Tg upon increasing %allyl is considered a consequence of the loss in hydrogen bonding capacity of lignin, which has been identified as a key factor in determining the Tg of a lignin.25,26,47 While strongly linear relationships between the Tg and %allyl were observed for all series, the non-derivatised samples (e.g. FEtOAc), as obtained from fractionation, did not (initially) fit the trends: all showed a lower Tg than anticipated from the fit through the derivatised samples. Inclusion of these samples would heavily skew the fit towards a curved, downward trajectory, similar to that reported by Duval and Avérous.51 The observed deviation from linearity could however be readily rationalised and attributed to incomplete precipitation of all lignin during recovery (i.e. a minor “fraction” of small components was not recovered). Control precipitation-recovery experiments conducted under reaction conditions without addition of allyl bromide yielded control samples (with suffix “−0”). These samples showed increased Tg values which fit excellently with the linear trends established with the derivatised samples (Fig. 3b & c; Table S5†). As expected, the difference with the control is larger for the lower Mw fractions, as with these samples, more small lignin fragments are present to begin with. Inclusion of the control samples further expanded the property space, with FIns−0's Tg = 225 °C, yielding an overall ΔTg = 213 °C. The maximum value afforded by this sample can indeed be considered as an “upper-limit” value to the property space of the Stora Enso lignin when treated under the fractionation and derivatisation conditions herein. Fig. 3c also shows a striking and unexpected co-linearity between each series, suggesting that the influence of incremental allylation appears to be uniform for each fraction, i.e. regardless of MW. This is tentatively attributed to the influence of aromatic-derived hydrogen bonding, a particularly dominant force in determining lignin's Tg, but this requires further investigation. Regardless, the strong structure–property correlation signifies a level of control over the physical parameters of the lignin and hence predictability of properties when generating new samples.
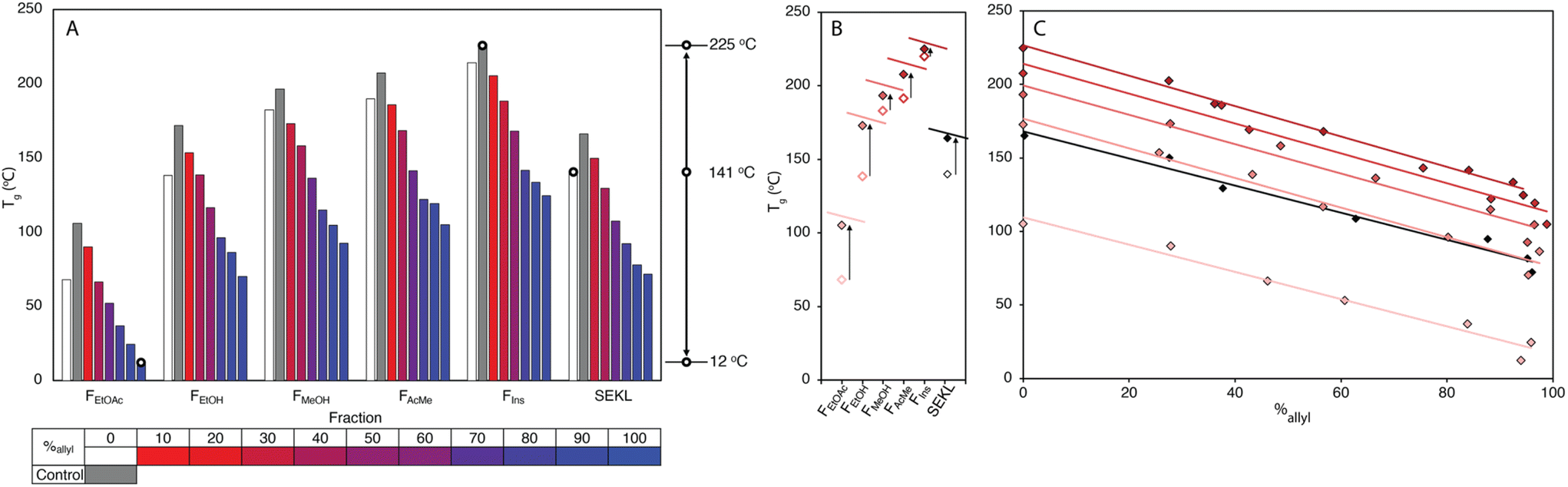 | ||
| Fig. 3 (a) Spread of Tg values for SEKL and its fractions through allylation, including control precipitation experiments for each fraction in grey (b) impact of control precipitation on SEKL and fractions (c) mapping of Tg values for SEKL and its fractions as a function of %allyl. | ||
To show that control over lignin Tg translates to application, a thiol–ene click reaction with was used to produce films with the FX − 6 series of samples, according to the protocol of Lawoko and co-workers with trimethylolpropane tris(3-mercaptopropionate) (TPTM) as the crosslinking agent (Fig. 4).32,33,35 The resulting films were orange-brown in colour (Fig. S2†) and varied in their opacity and tactility, from the FEtOAc − 6 film (Tg = 2 °C) which was a transparent and highly flexible solid, to the FIns − 6 film (Tg = 124 °C) which was transparent and somewhat brittle. The observed physical behaviours are reflected well in the Tg of the films and a range in ΔTg of 122 °C was observed across the samples. There was also a difference in how effectively the modified lignins could disperse within the reaction mixture. In particular, the FIns − 6 was only partially soluble in EtOAc, leading to poor dispersion and aggregate formation within the resulting film, hence the poor optical features of this sample. Notably, the film's Tg was found to clearly depend on the precursor (modified) lignin Tg (Fig. 5a), showing that the fractionation methodology, and therefore the lignin MW serves well to modify the resulting film Tg. Additionally, A blank reaction was performed on sample SEKL-1 under the above conditions, with the quantity of TPTM appropriate for sample SEKL-6; this material indeed showed poor solubility and did not form a film as expected, given lignin's relatively low abundance of unsaturated bonds (Fig. S4†). Finally, thiol–ene films were synthesised with selected samples from the FEtOAc and FMeOH series (see Table S6†). All but 1 lignin sample fully dissolved and formed homogeneous materials, with FMeOH − 1 being the exception. Film formation was unsuccessful with this fraction and the material omitted from the series. Mapping the film Tg values of these thermosets as a function of their lignin's %allyl (Fig. 5b) again showed a linear correlation, highlighting the strong influence of %allyl on the resulting thermosetting films’ properties.
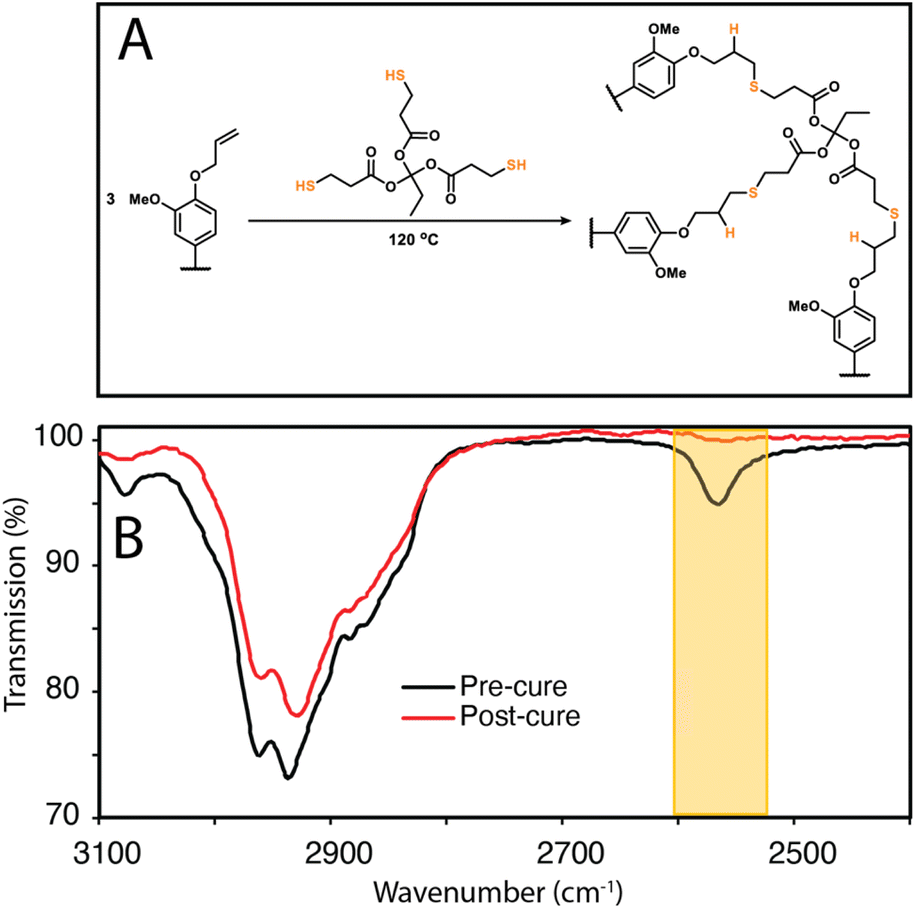 | ||
| Fig. 4 (a) Thiol–ene crosslinking reaction between lignin and TPTM (b) ATR-FTIR comparison of mixture of SEKL with TPTM before and after curing at 120 °C for 20 h. | ||
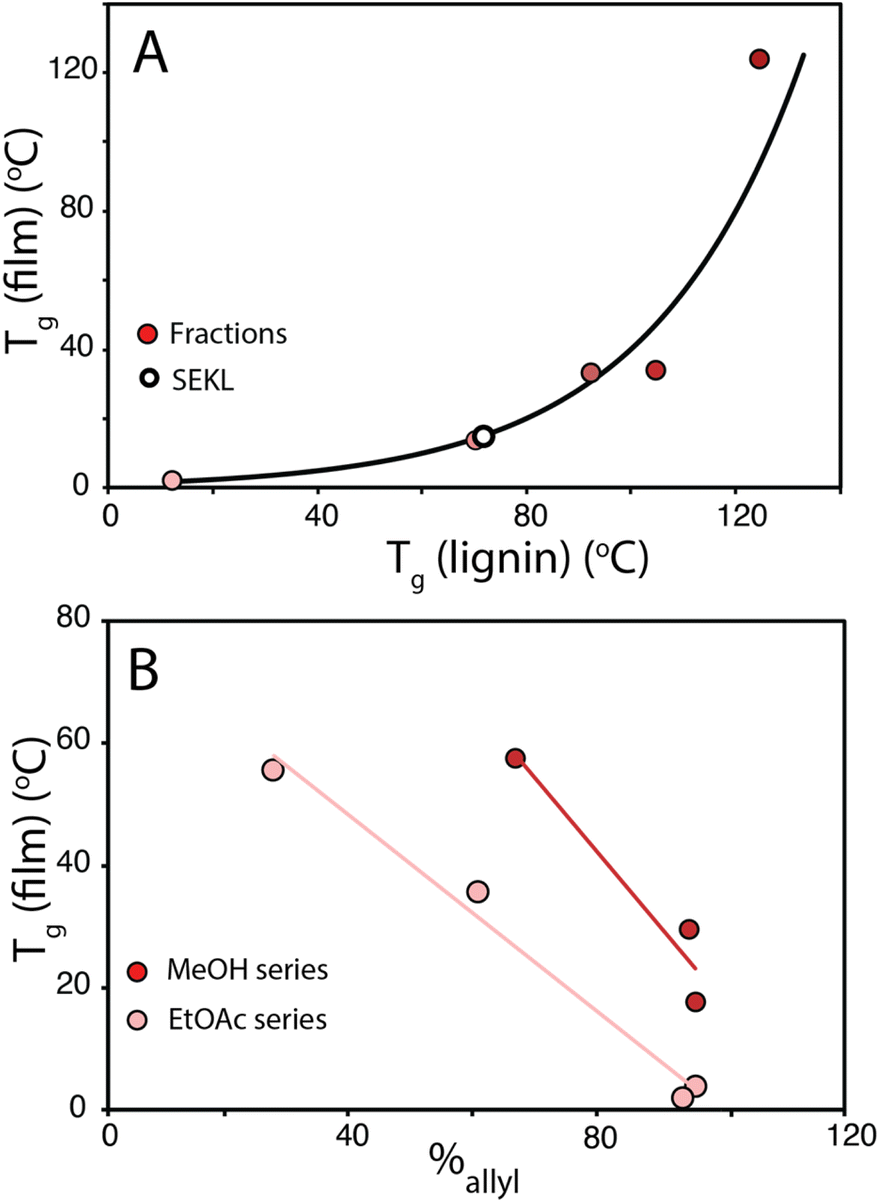 | ||
| Fig. 5 (a) Tg of films plotted as a function of their precursor sample (Fx − 6) Tg value (b) Tg of FEtOAc and FMeOH derived films as a function of %allyl. | ||
Conclusions
The combined fractionation–modification approach demonstrates how well the properties of a lignin can be controlled. The strong correlation of Tg with Mn for each fraction showed that the lignins well fit the Flory-Fox and Fox-Loshaek models, with the latter providing an indication of crosslinking within the lignin structure. A total Tg property space of 213 °C across a range of 12–225 °C was achieved, considerably larger than seen before elsewhere in the literature for a single lignin. Furthermore, remarkably strong linear relationships between increasing %allyl and decreasing Tg are reported for each of the fraction series, with notable collinearity between each series, suggesting that phenolic H-bonding is dominant in its influence over lignin Tg. Finally, thiol–ene thermosetting resins were produced to validate the utilisation of a lignin's property as a proxy parameter for resultant materials’ properties. This concept was shown to be translated well to the example thermosets, with ΔTg = 122 °C, covering a range from 2–124 °C. Much like with the lignins themselves, correlations were found between increasing %allyl of the lignin sample with decreasing film Tg, displaying excellent tuneability of the resulting material's properties. The systematic nature of the methodology and the strong structure–property relationships uncovered here reveal the possible levels of control over the lignin's properties. Fundamentally, this study (as a proof of concept) shows that within a property space range, lignins can be consciously tailored towards a desired material specification, thus providing a powerful toolbox to guide (industrial) end users towards fitting their lignin to a range of potential applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Netherlands Organisation for Scientific Research (NWO LIFT grant ENPPS.LIFT.019.17), as well as BASF SE. Stora Enso SE is acknowledged for providing the kraft lignin.References
- S. Bertella and J. S. Luterbacher, Trends Chem., 2020, 2, 440–453 CrossRef CAS.
- M. Lawoko and J. S. M. Samec, Curr. Opin. Green Sustainable Chem., 2023, 40, 100738 CrossRef CAS.
- C. S. Lancefield, S. Constant, P. de Peinder and P. C. A. Bruijnincx, ChemSusChem, 2019, 12, 1139–1146 CrossRef CAS PubMed.
- N. Shukry, S. M. Fadel, F. A. Agblevor and S. F. El-Kalyoubi, J. Appl. Polym. Sci., 2008, 109, 434–444 CrossRef CAS.
- Q. Meng, J. Yan, R. Wu, H. Liu, Y. Sun, N. N. Wu, J. Xiang, L. Zheng, J. Zhang and B. Han, Nat. Commun., 2021, 12, 1–12 CrossRef PubMed.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- N. Giummarella, P. A. Lindén, D. Areskogh and M. Lawoko, ACS Sustainable Chem. Eng., 2020, 8, 1112–1120 CrossRef CAS.
- C. S. Lancefield, H. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- X. Shen, C. Zhang, B. Han and F. Wang, Chem. Soc. Rev., 2022, 51, 1608–1628 RSC.
- M. Parit and Z. Jiang, Int. J. Biol. Macromol., 2020, 165, 3180–3197 CrossRef CAS PubMed.
- C. Wang, S. S. Kelley and R. A. Venditti, ChemSusChem, 2016, 9, 770–783 CrossRef CAS PubMed.
- A. Balaguera, G. I. Carvajal, J. Albertí and P. Fullana-i-Palmer, Resour., Conserv. Recycl., 2018, 132, 37–48 CrossRef.
- C. Moretti, B. Corona, R. Hoefnagels, I. Vural-Gürsel, R. Gosselink and M. Junginger, Sci. Total Environ., 2021, 770, 144656 CrossRef CAS PubMed.
- M. Farooq, Z. Tao, J. J. Valle-Delgado, M. H. Sipponen, M. Morits and M. Österberg, Langmuir, 2020, 36, 15592–15602 CrossRef CAS PubMed.
- E. M. Zadeh, S. F. O'Keefe and Y. T. Kim, ACS Omega, 2018, 3, 7388–7398 CrossRef CAS PubMed.
- S. Luo, J. Cao and A. G. McDonald, Ind. Crops Prod., 2017, 97, 281–291 CrossRef CAS.
- I. Spiridon, K. Leluk, A. M. Resmerita and R. N. Darie, Composites, Part B, 2015, 69, 342–349 CrossRef CAS.
- M. Cho, F. K. Ko and S. Renneckar, Biomacromolecules, 2019, 4485–4493 CrossRef CAS PubMed.
- A. Duval, F. Vilaplana, C. Crestini and M. Lawoko, Holzforschung, 2016, 70, 11–20 CrossRef CAS.
- C. M. Hansen, Prog. Org. Coatings, 2004, 51, 77–84 CrossRef CAS.
- J. Howell, M. Roesing and D. Boucher, J. Phys. Chem. B, 2017, 121, 4191–4201 CrossRef CAS PubMed.
- C. Pouteau, S. Baumberger, B. Cathala and P. Dole, C. R. Biol., 2004, 327, 935–943 CrossRef CAS PubMed.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- C. Cui, R. Sun and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2014, 2, 959–968 CrossRef CAS.
- K. A. Y. Koivu, H. Sadeghifar, P. A. Nousiainen, D. S. Argyropoulos and J. Sipilä, ACS Sustainable Chem. Eng., 2016, 4, 5238–5247 CrossRef CAS.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 1077–1087 RSC.
- S. Sen, H. Sadeghifar and D. S. Argyropoulos, Biomacromolecules, 2013, 14, 3399–3408 CrossRef CAS PubMed.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- A. Moreno and M. H. Sipponen, Mater. Horizons, 2020, 7, 2237–2257 RSC.
- A. Duval and M. Lawoko, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- M. Jawerth, M. Johansson, S. Lundmark, C. Gioia and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5, 10918–10925 CrossRef CAS.
- I. Ribca, M. E. Jawerth, C. J. Brett, M. Lawoko, M. Schwartzkopf, A. Chumakov, S. V. Roth and M. Johansson, ACS Sustainable Chem. Eng., 2021, 9, 1692–1702 CrossRef CAS.
- Y. Cui, M. Lawoko and A. J. Svagan, ACS Sustainable Chem. Eng., 2020, 8, 13282–13291 CrossRef CAS.
- M. E. Jawerth, C. J. Brett, C. Terrier, P. T. Larsson, M. Lawoko, S. V. Roth, S. Lundmark and M. Johansson, ACS Appl. Polym. Mater., 2020, 2, 668–676 CrossRef CAS.
- D. P. Nair, N. B. Cramer, T. F. Scott, C. N. Bowman and R. Shandas, Polymer, 2010, 51, 4383–4389 CrossRef CAS PubMed.
- Y. Chen, P. Han, L. J. Vandi, A. Dehghan-Manshadi, J. Humphry, D. Kent, I. Stefani, P. Lee, M. Heitzmann, J. Cooper-White and M. Dargusch, Mater. Sci. Eng. C, 2019, 95, 160–165 CrossRef CAS PubMed.
- M. Barletta, L. Lusvarghi, F. P. Mantini and G. Rubino, Surf. Coatings Technol., 2007, 201, 7479–7504 CrossRef CAS.
- S. Parker, R. Reit, H. Abitz, G. Ellson, K. Yang, B. Lund and W. E. Voit, Macromol. Rapid Commun., 2016, 37, 1027–1032 CrossRef CAS PubMed.
- I. Hamerton and J. Kratz, The Use of Thermosets in Modern Aerospace Applications, Elsevier Ltd., 2018 Search PubMed.
- L. Y. Liu, S. Chen, L. Ji, S. K. Jang and S. Renneckar, Green Chem., 2021, 23, 4567–4579 RSC.
- O. Sevastyanova, M. Helander, S. Chowdhury, H. Lange, H. Wedin, L. Zhang, M. Ek, J. F. Kadla, C. Crestini and M. E. Lindström, J. Appl. Polym. Sci., 2014, 131, 9505–9515 CrossRef.
- S. Kubo and J. F. Kadla, Biomacromolecules, 2005, 6, 2815–2821 CrossRef CAS PubMed.
- S. C. Qi, J. I. Hayashi, S. Kudo and L. Zhang, Green Chem., 2017, 19, 2636–2645 RSC.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5, 87187–87199 RSC.
- T. G. Fox and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef CAS.
- R. Ebrahimi Majdar, A. Ghasemian, H. Resalati, A. Saraeian, C. Crestini and H. Lange, ACS Sustainable Chem. Eng., 2020, 8, 16803–16813 CrossRef CAS.
- A. Duval, G. Layrac, A. van Zomeren, A. T. Smit, E. Pollet and L. Avérous, ChemSusChem, 2021, 14, 387–397 CrossRef CAS PubMed.
- T. Ogawa, J. Appl. Polym. Sci., 1992, 44, 1869–1871 CrossRef CAS.
- T. G. Fox and S. Loshaek, J. Polym. Sci., 1955, 15, 371–390 CrossRef CAS.
- A. Duval and L. Avérous, Green Chem., 2020, 22, 1671–1680 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01055d |
| This journal is © The Royal Society of Chemistry 2023 |