
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Batch and flow electrochemical synthesis of allyl sulfones via sulfonation of allyl trifluoroborates: a robust, regioselective, and scalable approach†
Leonardo
Mollari‡
a,
Roberto
del Río-Rodríguez‡
 a,
Jose A.
Fernández-Salas
*ab and
José
Alemán
*abc
a,
Jose A.
Fernández-Salas
*ab and
José
Alemán
*abc
aDepartamento de Química Orgánica (módulo-1), Universidad Autónoma de Madrid, Cantoblanco, 28049-Madrid, Spain. E-mail: jose.aleman@uam.es; j.fernandez@uam.es
bInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049-Madrid, Spain
cCenter for Innovation in Advanced Chemistry (ORFEO-CINQA), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 5th October 2023
Abstract
This work describes the electrochemical synthesis of allyl sulfones via sulfonation of allyl trifluoroborates. The process involves a radical addition to the alkene followed by the elimination of the trifluoroborate moiety, giving access to different substituted allyl sulfones. The efficacy of this reaction is attributed to the β-σ(C–B) stabilization of the electrochemically-generated intermediate carbocation, which plays a pivotal role in enforcing regioselectivity during the process. The reaction system is robust and general, allowing for compatibility with aromatic, heteroaromatic, and aliphatic sulfinate and allylic systems. The protocol is considered an inexpensive, safe, and environmentally friendly method for synthesizing this recognizable organic motifs in both synthetic and medicinal chemistry in absence of mediators or even additional supportive electrolite, as only electrons applied through electrical current are required for the reaction to proceed. Moreover, the continuous flow conditions, which fits into green chemistry standards, have demonstrated the feasibility of electrochemically synthesizing allyl sulfones, resulting in highly favorable outcomes and enabling the potential scalability of the electrochemical process.
Introduction
Sulfones represent functional groups with a long history in pharmaceutical development as they are prevalent moieties in biologically active molecules.1 In particular, allyl sulfones could be identified as very relevant scaffolds due to their presence in pharmaceutically active derivatives such us2,3 protease inhibitors, fluorophores, the natural product ‘agelasidine’, or the antiinflammatory ‘resatorvid’ (Scheme 1A).1 In addition, they are versatile and interesting building blocks in organic synthesis.4–6 Consequently, the synthesis of allyl sulfones have attracted the attention of organic and medicinal chemists over the years, turning the development of straightforward methodologies for the preparation of allylic sulfones a focal point in organic chemistry research.7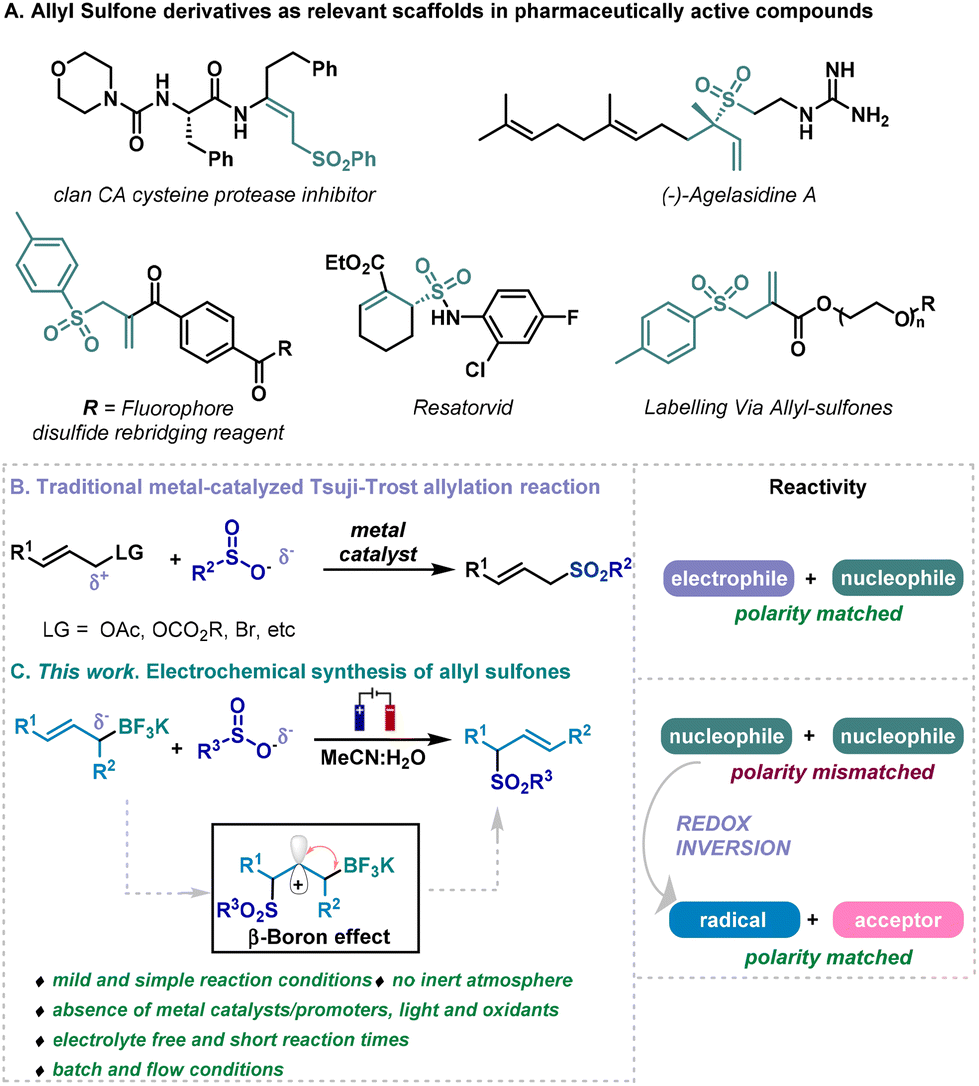 | ||
| Scheme 1 Biologically active allyl sulfones (A), previous work (B) and this work (C). | ||
In this context, different strategies have been studied in the literature over the last decades. Although some impressive advancements in the synthesis of allyl sulfones have been made, such as hydrosulfination, metal-catalyzed SO2 insertions, etc., tedious protocols, low atom economy and applicability may hamper the general application of this methodologies.8–20 Venerable Tsuji–Trost-type coupling can be clearly identified as the more prominent synthetic tool for the direct and rapid sulfonation of allyl derivatives.21–24 A plethora of metal-catalyzed systems have been described for the coupling of sulfonyl containing nucleophiles with various electrophilic allyl derivatives (acetates, carbonates, halides, alcohols, etc.) for the synthesis of allyl sulfones through allylic substitution reactions (Scheme 1B).25–30 These approaches involved a diverted allyl derivatives possessing electrophilic features, while the sulfonate anion would act as nucleophile, which engages a matched polarity (right, Scheme 1B). Aiming to discover novel approaches for synthesizing relevant allyl sulfones, we envisioned the use of sulfinates and allyl boron derivatives as allylating agent.31,32 We thought that a radical approach would circumvent the reactivity mismatched otherwise encountered when facing two nucleophilic species if a two electron process take place (Scheme 1B). Among the multiple strategies based on the single-electron activation of organic substrates, electrochemistry is quickly becoming one of the most popular avenues to access radical intermediates, as it utilizes “green” electrons as reagents through the application of an electric current.33–37 This technology enables mild conditions for redox reactions and enhances atom economy. Our hypothesis suggests an electrochemical protocol to alter the initial reactivity of sulfinates and allyl boronates by promoting a radical process through the anodic oxidation of sulfinates,38,39 allowing to include the borate counterpart to the portfolio of starting materials for allyl-sulfone synthesis. Similarly to well-known β-silicon effect (also known as σ(C–Si)-π-hyperconjugation),40 β-boron effect can stabilize carbocations through β-σ(C–B) interactions, imposing the regioselectivity of the process.41 The suitability of allyl trifluoroborate salts as allylating agents was then recognized,31,32 and this system would provide an alternative for synthesizing allyl sulfones by encountering two nucleophilic species. After the generation of a sulfur-based radical derivative from sulfinates, and its addition to the double bond of the allyl-boron derivative, a carbocation would be formed upon a second anodic oxidation.38 A trifluoroborate elimination process would then take place, generating a double bond in the vicinal carbon, involving an allylation process overall, resembling a formal Tsuji–Trost type allylation reaction (Scheme 1C). Contrary to what has been traditionally assumed in two electron processes, cross-coupling reactions where a sulfur-based nucleophile faced an electrophile bearing a leaving group, this system would confront two nucleophilic species as an alternative for the synthesis of allyl sulfones (Scheme 1C). This electrochemical approach would circumvent issues associated with previously described catalytic systems for the synthesis of allyl sulfones, as for example the use of metals as catalysts and the reaction conditions normally associated with their use, such as inert atmosphere or dry solvents, high temperatures or long reaction times21–24 Therefore, an electrochemical system for the synthesis of allyl sulfones would be highly desirable, pursuing and satisfying one of the main motives for the renaissance of electrochemistry in organic synthesis which has appeared as green alternative to conventional synthetic methods. This electrochemical approach that targets the synthesis of relevant organic molecules does offer both operational and practical advantages that make it a greener methodology (Scheme 1C). This new method is considered an inexpensive, safe, and environmentally friendly, that takes place under very mild and simple reaction conditions: (i) No need of stoichiometric oxidants, as only electrons, applied through electrical current, and the two substrates are needed. (ii) Avoids the use of metals as catalysts or promoters. (iii) Circumvents the necessity of using inert atmosphere or dry solvents, high temperatures or long reaction times traditionally required for the synthesis of allyl sulfones. (iv) The system eludes the use of supportive electrolyte, employs carbon-based electrodes and uses a reasonable benign mixture of solvents (H2O and CH3CN). In addition, this electrochemical synthesis of allyl sulfones has demonstrated its feasibility and potential for scalability under electrochemical continuous flow conditions, that fits into green chemistry standards as it allows the development of safer protocols and improves energy requirement of the system as shorter reaction times are needed.
Results
Optimization of the model reaction
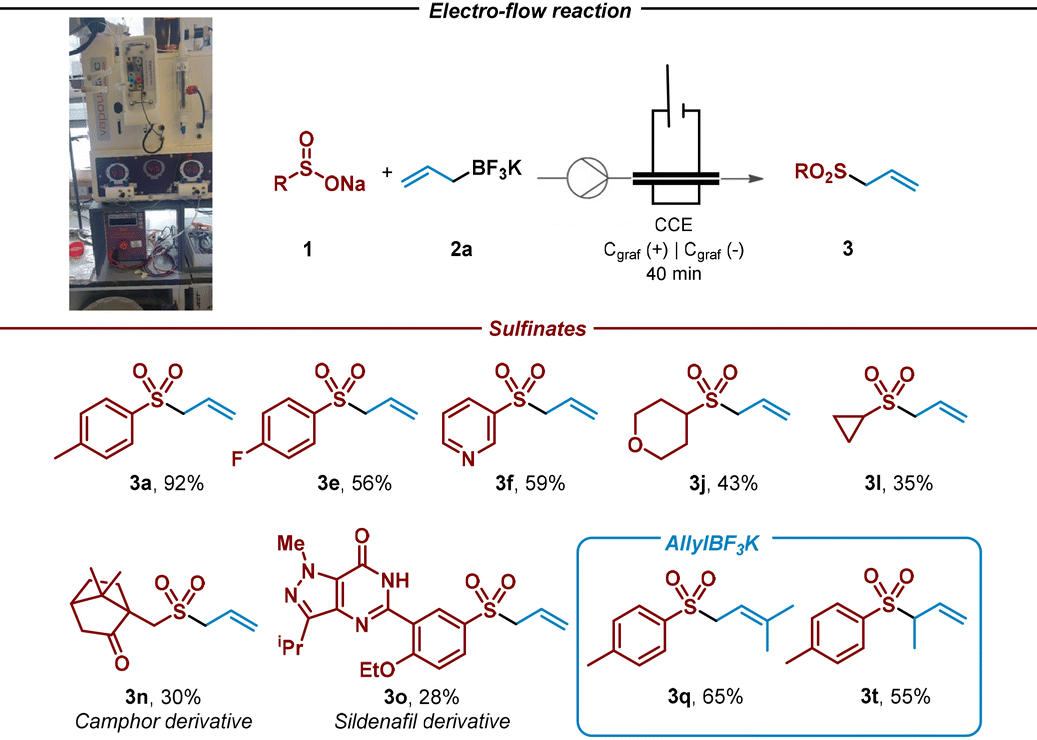
We began the study by choosing tosyl sulfinate (1a) as sulfonyl radical precursor and allyl trifluoroborate salt (2a) as allyl source (Table 1, entry 1). By using graphite electrodes in both anode and cathode under 2.5 mA constant current for 3 h in an undivided cell and absence of an additional electrolyte in a mixture 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of MeCN and H2O, the allyl sulfone 3a was isolated in very good yield (80%, entry 1). For the development of a synthetically useful electrochemical synthesis of allyl sulfones, various parameters such as solvent, electrolyte, electrode material and electrochemical parameters have been studied and summarized in Table 1.
:1 of MeCN and H2O, the allyl sulfone 3a was isolated in very good yield (80%, entry 1). For the development of a synthetically useful electrochemical synthesis of allyl sulfones, various parameters such as solvent, electrolyte, electrode material and electrochemical parameters have been studied and summarized in Table 1.
|
|
||
|---|---|---|
| Entrya | Deviation from optimized conditions | Yieldb (%) |
|
a Reaction conditions: 1a (0.1 mmol) and 2a (0.25 mmol) at constant current (2.5 mA) for 3 hours in MeCN:H2O (1:1, 3 mL) at r.t., under air, using Cgraf as anode and cathode.
b Isolated yields after purification by column chromatography.
|
||
| 1 | No deviation | 80 |
| 2 | TBAPF6 (0.5 equiv.) | 83 |
| 3 | TBAPF6 (1.0 equiv.) | 56 |
| 4 | 1 mA vs. 2.5 mA | 48 |
| 5 | 5 mA vs. 2.5 mA | 52 |
| 6 | MeCN/H2O 5:1 vs. 1:1 |
52 |
| 7 | H2O vs. MeCN/H2O 1:1 |
— |
| 8 | Ni foam (—) vs. Cgraf (—) | 50 |
| 9 | 2a: 1.5 equiv. vs. 2.5 equiv. | 46 |
| 10 | No current | 0 |
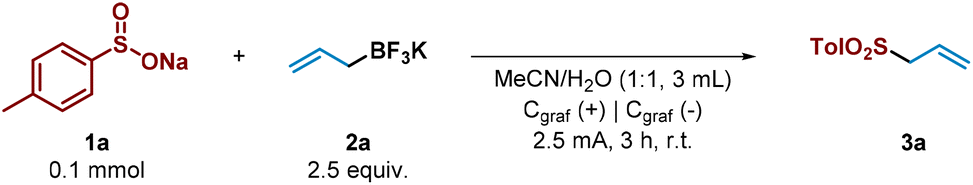
Firstly, the addition of an external electrolyte (entries 2 and 3) was studied. While the presence of 0.5 equiv. did not significantly improve the yield of the desired allyl sulfone 3a, 1 equiv. of the electrolyte greatly decreased the efficiency of the process. When the reaction was carried out at lower or higher current intensities (entries 4 and 5), allyl sulfone 3a was obtained in lower yield. Different solvent systems (entries 6 and 7) were then evaluated. Thus, when MeCN was employed in a major proportion (entry 6), the electrochemical system performed substantially worse. However, only H2O as sole solvent did not improve either, showing that MeCN as co-solvent was necessary to competently perform the alkylation (entry 7). The electrode material selection had an important impact on the reaction. Ni cathode led to the allyl sulfone in moderate yield (entry 8). We then tried to reduce the excess of the allyl trifluoroborate salt 2a (entry 9). However, in presence of 1.5 equiv. of it (2a), the reaction performance was notably diminished as it plays a double role (reagent and electrolyte). Finally, the reaction did not take place in absence of electrical current (entry 10).
Substrate scope
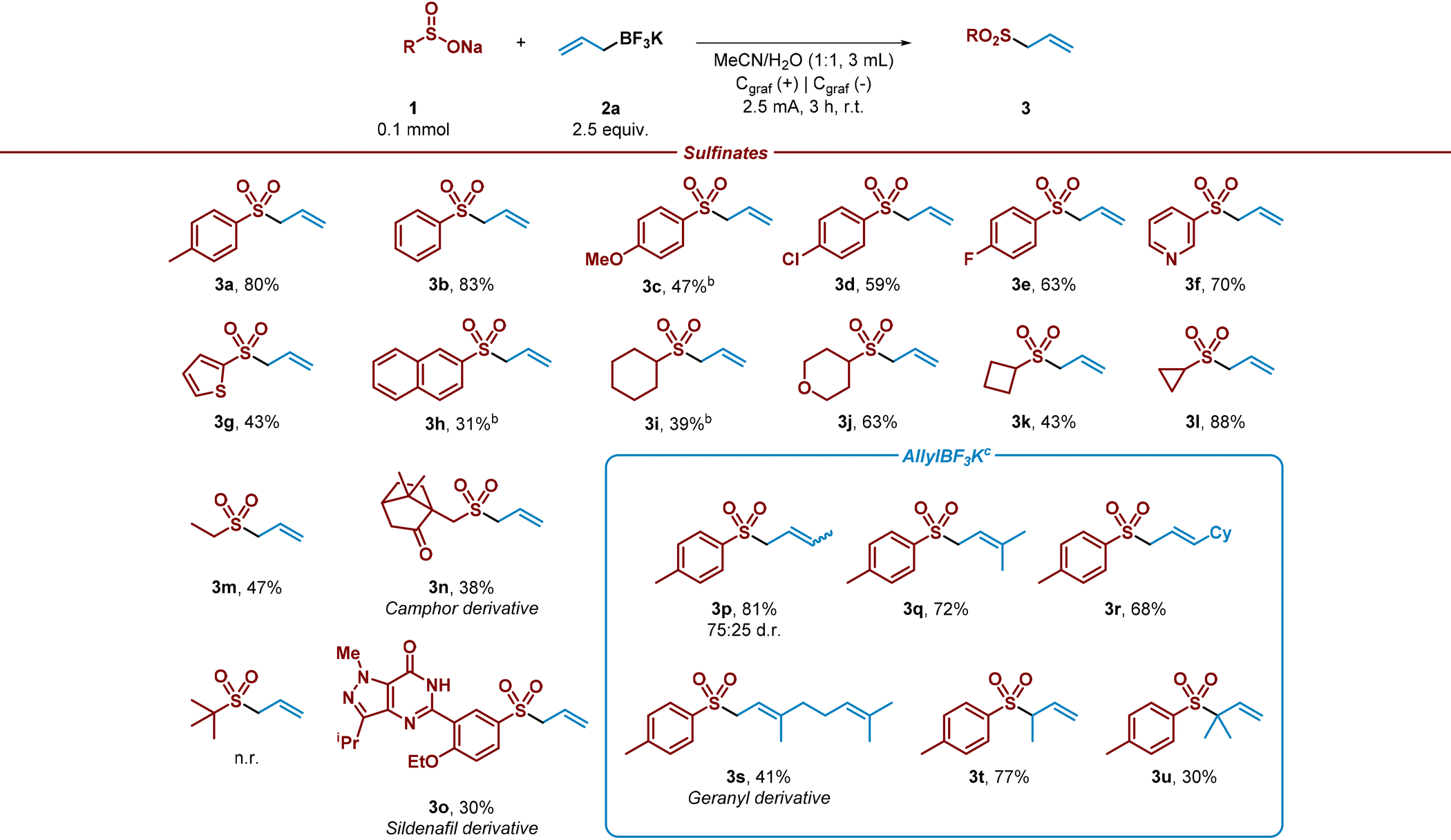
Once the reaction conditions had been optimized, a variety of aryl and alkyl sulfinates were tested under the electrochemical reaction conditions in an undivided cell with a readily available graphite anode/cathode, in absence of additional electrolyte and in a mixture of MeCN/H2O as solvent system (Table 2). This study demonstrated that both aryl and alkyl sulfinates precursors efficiently performed the sulfonation, leading to the desired allyl sulfones in good yields (3a–3o). Phenyl sulfinate and aryl sulfinates decorated with electron-donating groups were well tolerated (3a–3c), although a diminished performance was observed with methoxy-bearing aryl sulfinate (3c). In addition, halides containing aryl sulfinates accomplished the desired reaction, leading to the sulfonylated product in good yields (3d and 3e). Notably, heterocyclic sulfinates showed decent performance under the optimized reaction conditions, conducting to the allyl sulfones (3f and 3g).|
a Reaction conditions: 1 (0.1 mmol) and 2 (0.25 mmol), CH3CN:H2O (1:1, 3 mL), r.t, under air, undivided cell (Cgraphite anode, Cgraphite cathode) at constant current (2.5 mA) for 3 h. Isolated yields.
b Reaction conditions: 1 (0.1 mmol), 2 (0.25 mmol), TBAPF6 (0.05 mmol), CH3CN:H2O (1:1, 3 mL), r.t, under air, undivided cell (Cgraphite anode, Cgraphite cathode) at constant current (2.5 mA) for 3 h.
c Reaction conditions: 1 (0.1 mmol), 2 (0.25 mmol), CH3CN:H2O (5:1, 3 mL), r.t, under air, undivided cell (Cgraphite anode, Ni foam cathode) at constant current (5 mA) for 2 h.
|
|---|
|
2-Naphthyl substituted sulfinate was also subjected to the allylation reaction and led to the desired product with moderate yield (3h). Aliphatic sulfinates were then evaluated. To our delight, secondary and primary sulfinates showed good performance, leading to the desired coupled product in good yields (3i–3m). Sterically encumbered tert-butyl sulfinate did not lead to the desired allylated product. In addition, as we wished to extend the applicability of the method, recognizable scaffolds decorated with a sulfinate moiety were also studied under the optimized electrochemical conditions. To our delight, camphor and sildenafil sulfinate derivatives provided the corresponding allylated products (3n and 3o). The potential of the electrochemical method was further evaluated by testing various allylic trifluoroborate salts. Secondary trifluoroborate derivatives α-substituted with methyl and sterically hindered cyclohexyl group were very well tolerated (3p and 3r). Remarkably, tertiary salts led efficiently to the corresponding allylic sulfones (3s and 3q). To our delight, β-substituted (3t) and β-disubstituted (3u) trifluoroborate salts conducted to the corresponding allylated products successfully. It should be noted, that iso-prenyl sulfone 3u has been utilized in the literature as ideal prenyl transfer reagent.42 This may help to overcome limitations in prenylation reactions, which is consider a challenging and essential reaction on which nature relies to modify properties of molecules and build terpenoids. In addition, and considering the electrons consumed and the isolated yields under the optimised reaction conditions, this process presents respectable faradaic efficiency (23–69%).43
Notably, the reaction between sulfinate 1a and allyl trifluoroborate salts 2a and 2c exhibited remarkable efficiency, even when scaled up by a factor of 10, resulting in the formation of desired allyl sulfones in good yield after 16 hours with slightly modified reaction conditions (Scheme 2).
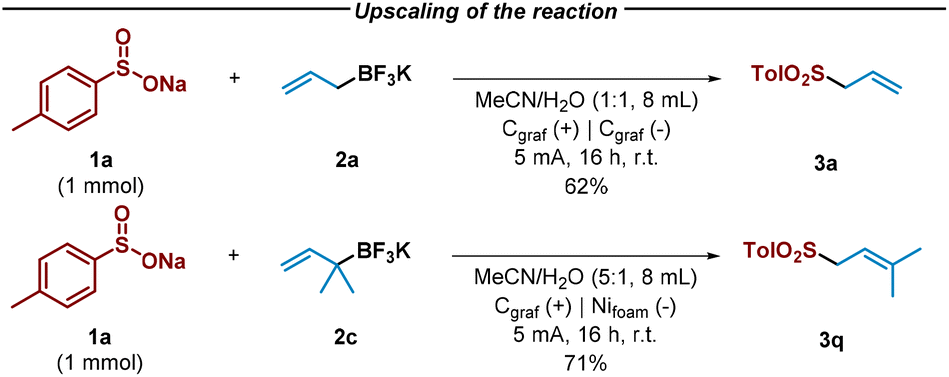 | ||
| Scheme 2 Upscaling of the reaction and synthesis of allyl sulfone 3a and 3q. | ||
Given the encouraging results achieved and the synthetic potential of the developed method, we decided to perform the electrochemical reaction under continuous flow conditions (Table 3, see ESI†). Flow chemistry fits into the current regimen that requires the development of new high safety, low waste generation, and energy efficiency processes. Gratifyingly, the reaction showed a remarkable enhancement in the efficiency of the process for allyl sulfone 3a that could be isolated in 92% yield after 40 minutes. In addition, and to test the robustness of the method, the flow reaction was tested with aromatic, heteroaromatic and aliphatic sulfinates, including more sophisticated camphor and sildenafil sulfinate derivatives, providing the corresponding allylated products with similar efficiency when compared with batch conditions (3a, 3e, 3f, 3j, 3l, 3n and 3o). In addition, tertiary and β-substituted trifluoroborate salts led efficiently to the corresponding allyl sulfones (3q and 3t) under continuous flow conditions. It should be noted that the space–time yield calculated for the preparation of 3a under flow reactions conditions is 44.7 g per (Lh), which leads to a more efficient process when compared with batch conditions (1.7 g per (Lh)).44,45
|
a Reaction conditions: 1 (0.1 mmol) and 2 (0.25 mmol), CH3CN:H2O (1:1, 4 mL), r.t, under air, undivided cell (Cgraphite anode, Cgraphite cathode) at constant current (2.5 mA). Isolated yields.
|
|---|
|
We then performed a reaction in presence of TEMPO (2,2,6,6-tetramethylpiperidinooxy) as control experiment in order to gain an insight into the reaction mechanism. As shown in Scheme 3, in the presence of this radical scavenger, the reaction was almost inhibited as only 8% of the allyl sulfone was observed by NMR. In addition, the corresponding TEMPO adduct (4) resulted from the trapping event of the sulfonyl radical species could be identified (see ESI†). This result strongly indicate that radical species are involved in the reaction pathway. Based on this experiment, the literature and the previously showed results, we propose a mechanism for the electrochemical reaction (Scheme 3). At first, sulfinate (1a) gets oxidized (Eox = −0.37 V vs. SCE, see ESI†)46 at the anode, generating the sulfonyl radical (I). Then, addition of the sulfur-based radical to the alkene (2) generates secondary alkyl radical II, which undergoes anodic oxidation to generate the cation intermediate III.38,47–49 Finally, elimination of BF3 as in a Sakurai-type allylation reaction50 would generate the desired allylsulfone 3. We believe that the reaction should follow an addition/oxidation sequence.
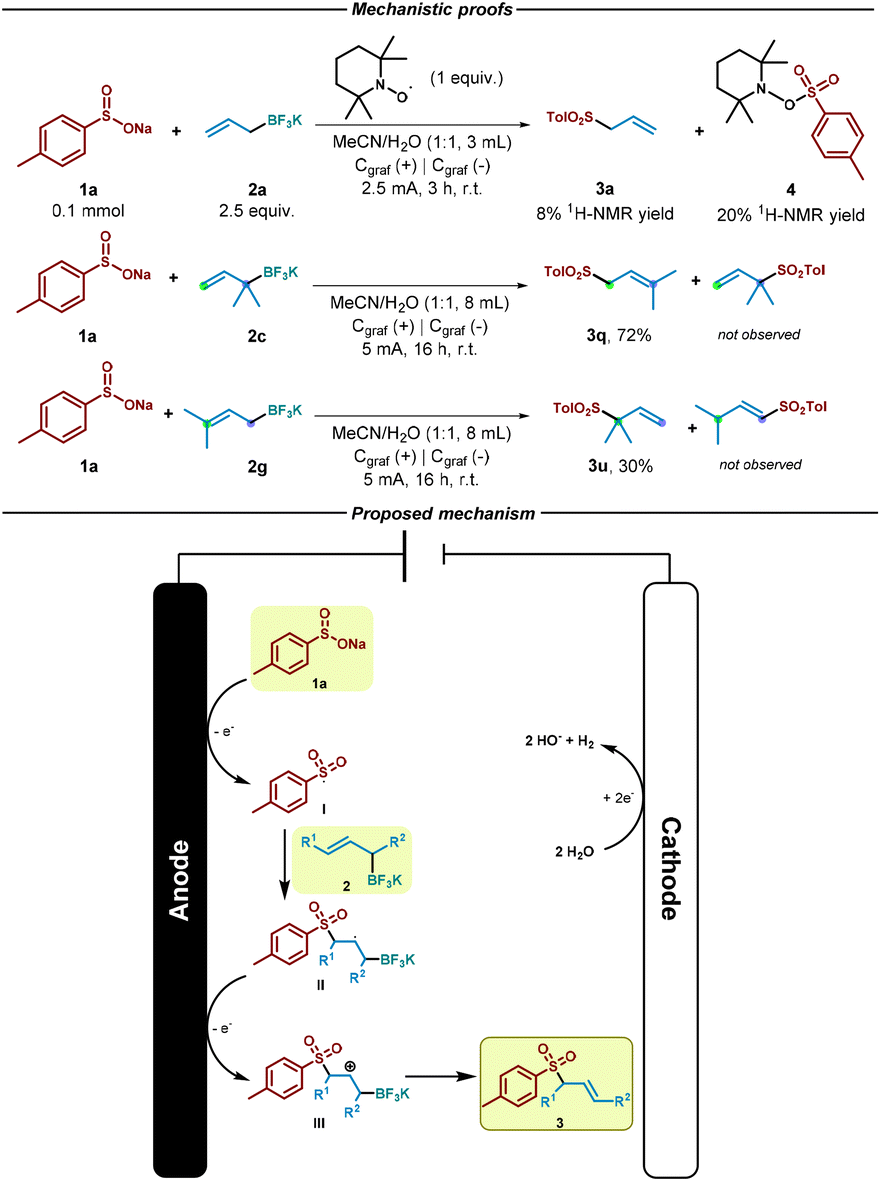 | ||
| Scheme 3 Control experiments and reaction mechanism. | ||
Conclusions
In summary, we have presented a versatile and straightforward electrochemical system for the synthesis of allyl sulfones using readily accessible sulfinates and allyl trifluoroborate salts. The reaction system has exhibited robustness and generality, accommodating aromatic, heteroaromatic, and aliphatic sulfinates. Considering these advantages, this protocol offers an economical, safe, and environmentally friendly approach to produce allyl sulfones, which serve as prominent organic motifs in both synthetic and medicinal chemistry. In addition, the synthesis of allyl sulfones has been efficiently implemented to an electrochemical continuous flow system.Author contributions
L.M., R.R.-R., J.A.F.-S. and J.A. designed the experiments and analyzed the data. L.M., and R.R.-R. performed the experiments. R.R.-R and J.A.F.-S. described the ESI.† J.A.F.-S. and J.A. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Spanish Government (PID2021-122299NB-I00, TED2021-130470B-I00, TED2021-129999B-C32), European Structural Funds (S2018/NMT-4367), Proyectos Sinérgicos I + D (Y2020/NMT6469) and ‘Comunidad Autónoma de Madrid’ (SI1/PJI/2019-00237). J. A. F.-S. thanks the Spanish Government for a Ramón y Cajal contract.References
- I. Ahmad and Shagufta, Int. J. Pharm. Pharm. Sci., 2015, 7, 19 CAS.
- A. El-Awa, M. N. NoShi, X. Mollat du Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315 CrossRef CAS PubMed.
- J. Corpas, S.-H. Kim-Lee, P. Mauleón, R. Gómez Arrayás and J. C. Carretero, Chem. Soc. Rev., 2022, 51, 6774 RSC.
- F. Reck, F. Zhou, M. Girardot, G. Kern, C. J. Eyermann, N. J. Hales, R. R. Ramsay and M. B. Gravestock, J. Med. Chem., 2005, 48, 499 CrossRef CAS PubMed.
- F. Velázquez, M. Sannigrahi, F. Bennett, R. G. Lovey, A. Arasappan, S. Bogen, L. Nair, S. Venkatraman, M. Blackman, S. Hendrata, Y. Huang, R. Huelgas, P. Pinto, K.-C. Cheng, X. Tong, A. T. McPhail and F. G. Njoroge, J. Med. Chem., 2010, 53, 3075 CrossRef PubMed.
- X. Chen, S. Hussain, S. Parveen, S. Zhang, Y. Yang and C. Zhu, Curr. Med. Chem., 2012, 19, 3578 CrossRef CAS PubMed.
- N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939 CrossRef CAS.
- For selected examples of hydrosulfination, see: V. Khakyzadeh, Y.-H. Wang and B. Breit, Chem. Commun., 2017, 53, 4966 RSC.
- A. B. Pritzius and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 15818 CrossRef CAS PubMed.
- A. B. Pritzius and B. Breit, Angew. Chem., Int. Ed., 2015, 54, 3121 CrossRef CAS PubMed.
- For selected examples of SO2 insertion, see: J. Zhang, K. Zhou, G. Qiu and J. Wu, Org. Chem. Front., 2019, 6, 36 RSC and the references cited therein.
- R. Mao, Z. Yuan, R. Zhang, Y. Ding, X. Fan and J. Wu, Org. Chem. Front., 2016, 3, 1498 RSC.
- J. Li, G. Qin, Y. Liu and H. Huang, Org. Chem. Front., 2016, 3, 259 RSC.
- P.-X. Zhou, Y.-Y. Ye, L.-B. Zhao, J.-Y. Hou, X. Kang, D. Q. Chen, Q. Tang, J.-Y. Zhang, Q.-X. Huang, L. Zheng, J.-W. Ma, P.-F. Xu and Y.-M. Liang, Chem. – Eur. J., 2014, 20, 16093 CrossRef CAS PubMed.
- X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC.
- K. Xu, V. Khakyzadeh, T. Bury and B. Breit, J. Am. Chem. Soc., 2014, 136, 16124 CrossRef CAS PubMed.
- L. Wang and L.-F. Zhang, Synlett, 2022, 33, 1929 CrossRef CAS.
- X. Li, X. Xu and C. Zhou, Chem. Commun., 2012, 48, 12240 RSC.
- X. Li, X. Xu and Y. Tang, Org. Biomol. Chem., 2013, 11, 1739 RSC.
- F.-L. Yang and S.-K. Tian, Tetrahedron Lett., 2017, 58, 487 CrossRef CAS.
- B. M. Trost and C. A. Kalnmals, Chem. – Eur. J., 2020, 26, 1906 CrossRef CAS PubMed.
- S. Parisotto and A. Deagostino, Synthesis, 2019, 51, 1892 CrossRef CAS.
- Y.-N. Wang, L.-Q. Lu and W.-J. Xiao, Chem. – Asian J., 2018, 13, 2174 CrossRef CAS PubMed.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed.
- K. Inomata, T. Yamamoto and H. Kotake, Chem. Lett., 1981, 10, 1357 CrossRef.
- E. Blart, J.-P. Genêt, M. Safi, M. Savignac and D. Sinou, Tetrahedron, 1994, 50, 505 CrossRef CAS.
- F.-X. Felpin and Y. Landais, J. Org. Chem., 2005, 70, 6441 CrossRef CAS PubMed.
- S. Chandrasekhar, V. Jagadeshwar, B. Saritha and C. Narsihmulu, J. Org. Chem., 2005, 70, 6506 CrossRef CAS PubMed.
- R. J. Reddy and A. H. Kumari, RSC Adv., 2021, 11, 9130 RSC.
- Y. Xu, M. Salman, S. Khan, J. Zhang and A. Khan, J. Org. Chem., 2020, 85, 11501 CrossRef CAS PubMed.
- D. Fernandez Reina, A. Ruffoni, Y. S. S. Al-Faiyz, J. J. Douglas, N. S. Sheikh and D. Leonori, ACS Catal., 2017, 7, 4126 CrossRef CAS.
- I. Suzuki, N. Esumi and M. Yasuda, Asian J. Org. Chem., 2016, 5, 179 CrossRef CAS.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef PubMed.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484 CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386 RSC.
- W. Liu, L. Hao, J. Zhang and T. Zhu, ChemSusChem, 2020, 15, e202102557 CrossRef PubMed.
- Y.-L. Lai, Y. Mo, S. Yan, S. Zhang, L. Zhu, J. Luo, H. Guo, J. Caia and J. Liao, RSC Adv., 2020, 10, 33155 RSC.
- J. B. Lambert, Y. Zhao, R. W. Emblidge, L. A. Salvador, X. Liu, J.-H. So and E. C. Chelius, Acc. Chem. Res., 1999, 32, 183 CrossRef CAS.
- Y. Li, W.-X. Fan, S. Luo, A. Trofimova, Y. Liu, J.-H. Xue, L. Yang, Q. Li, H. Wang and A. K. Yudin, J. Am. Chem. Soc., 2023, 143, 7548 CrossRef PubMed.
- M. D. Rathnayake and J. D. Weaver, Eur. J. Org. Chem., 2020, 1433 CrossRef CAS.
- A. Uwe Meyer, S. Jäger, D. P. Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050 CrossRef.
- P. A. Kempler and A. C. Nielander, Nat. Commun., 2023, 14, 1158 CrossRef CAS PubMed.
- C. A. Hone and C. O. Kappe, Chem.: Methods, 2021, 1, 454 CAS.
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502 CrossRef CAS PubMed.
- M.-J. Luo, B. Liu, Y. Li, M. Hu and J.-H. Li, Adv. Synth. Catal., 2019, 361, 1538 CrossRef CAS.
- H. Mei, J. Liu, Y. Guo and J. Han, ACS Omega, 2019, 4, 14353 CrossRef CAS PubMed.
- X.-Q. Mou, L.-C. Ren, M. Wang, H.-H. Zhang, A. Cai, K.-X. Wan, S.-M. Zhang, B.-D. Cui, Y. Zhang and Y.-Z. Chen, J. Org. Chem., 2023, 88, 3238 CrossRef CAS PubMed.
- F. Nowrouzi, A. N. Thadani and R. A. Batey, Org. Lett., 2009, 11, 2631 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc02408c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |