
2D layered materials: structures, synthesis, and electrocatalytic applications
Lijia
Liu
a,
Wei
An
b,
Fengyun
Gu
c,
Lili
Cui
a,
Xingquan
He
 *a and
Meihong
Fan
*a
*a and
Meihong
Fan
*a
aSchool of Chemistry and Environmental Engineering, Changchun University of Science and Technology, 7089 Weixing Road, Changchun 130022, P. R. China. E-mail: hexingquan@hotmail.com
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China
cJilin Province Product Quality Supervision and Inspection Institute, 2699 Yiju Street, Changchun 130103, China
First published on 13th July 2023
Abstract
The discovery of graphene has opened a new world of 2D materials with abundant components and diverse bonding modes that have strong covalent bonds in a plane but weak interactions out of the plane. One of the many alluring features of two-dimensional materials is that they can be exfoliated in liquids. This peculiarity makes them able to be exfoliated into thin nanosheets with few layers or even monolayers. Exfoliation endows nanosheets with unique laterally extended topology, novel mechanical performance, and highly exposed surface, which are ideal for catalysis that mainly occurs on the material surface. In addition, the exfoliated nanosheets show unprecedented optical and electrical properties owing to the two-dimensional confinement of electrons. The unique electronic structure and surface structure of layered materials have attracted tremendous research interests in catalysis and energy conversion fields. This review first introduces the history of the development of layered materials and summarizes the structural chemistry based on connection modes of laminates and types of intercalation ions. Furthermore, rational methods are elaborated to achieve controllable exfoliation of different kinds of layered materials, with emphasis on the exfoliation mechanism and application sphere of each method. Furthermore, we discuss the latest research progress of layered materials as electrocatalysts and electrocatalyst support for application in energy conversion and highlight the relationship between catalytic performance and the layered structure based on theoretical and experimental results. Finally, we also explore the future prospects of 2D layered materials in the field of electrocatalysis, such as water splitting, CO2 reduction, and N2 fixation. At the same time, constructive suggestions on chemical synthesis, extended application, and improvement in structure stability are also proposed.
1. Introduction
Electrocatalysis, which can convert raw materials with low economic value into higher ones or harmful substances into harmless ones in an environmentally friendly manner, is widely involved in energy storage and conversion and plays an increasingly significant role in national economy and people's livelihood.1,2 More and more researchers turn attention to electrocatalysis for its cleanness and greenness, especially because it can couple with renewable energy powered generation.3,4 These features can meet the goal of carbon peaking and carbon neutralization perfectly. Hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), CO2 reduction reaction (CO2RR), and nitrogen reduction reaction (NRR) are the most common and basic specific electrochemical reaction processes in electrocatalysis.5 Reaction efficiency of these key reactions largely depends on the performance of electrocatalysts. Ideal catalysts should have high intrinsic activity and sufficient active site exposure.6,7 It has long been found that noble metal-based catalysts can catalyze these reactions efficiently.8 However, low reserves and high cost of precious metals severely hinder their large-scale investigation.9,10 Recently developed transition metal-based catalysts also show unsatisfactory performance, i.e. low activity, poor stability, and low selectivity.3,11 The goal of improving energy conversion efficiency puts forward new requirements for catalysts, which can provide ample active sites and access to identify the structure–activity relationship. Therefore, it is necessary to exploit new electrocatalysts with high activity, high stability, and low cost to push forward the scaling up of electrocatalysis in energy conversion.Two-dimensional (2D) structured materials receive tremendous research interest from various fields including nanotechnology, material science, and electrocatalysis. More than 70 years ago, scientists already knew the layered structure of some inorganic materials such as graphite, boron nitride, and metal chalcogenides.12,13 In the 1970s, hydrotalcite (also known as the layered double hydroxide, LDH) was confirmed to be layered structured metal hydroxide by X-ray diffraction method.14 Explorations on the electrocatalysis application of layered materials could be dated back to the 1980s, when MoS2 and LDHs, etc. were attempted to be used as electrodes in electrochemical water splitting.15 Layered materials have strong in-plane covalent bonds and weak inter-plane van der Waals interaction, suggesting their great potential to be exfoliated into nanosheets. The year 2004 marked the blossoming of ultrathin 2D nanomaterials when Novoselov et al. successfully exfoliated graphene using the Scotch tape method.16 This finding stimulated the thriving of 2D materials and the Nobel Prize was awarded. The discovery of MXene by Yury Gogotsi in 2011 further flourished the composition to transition metal carbides and nitrides.17 Nowadays, the family of 2D nanomaterials has developed and proliferated into a big family with several branches, including non-metallic layered materials (graphene, BN),18 layered double hydroxides,19 layered metal oxides (layered perovskite, P2-layered oxides), MXene,20 elemental 2D materials (silicene,21 germanene),22 and 2D transition metal compounds.23,24 In addition, new 2D materials are still emerging every year, and the time line of key milestones is shown in Fig. 1.
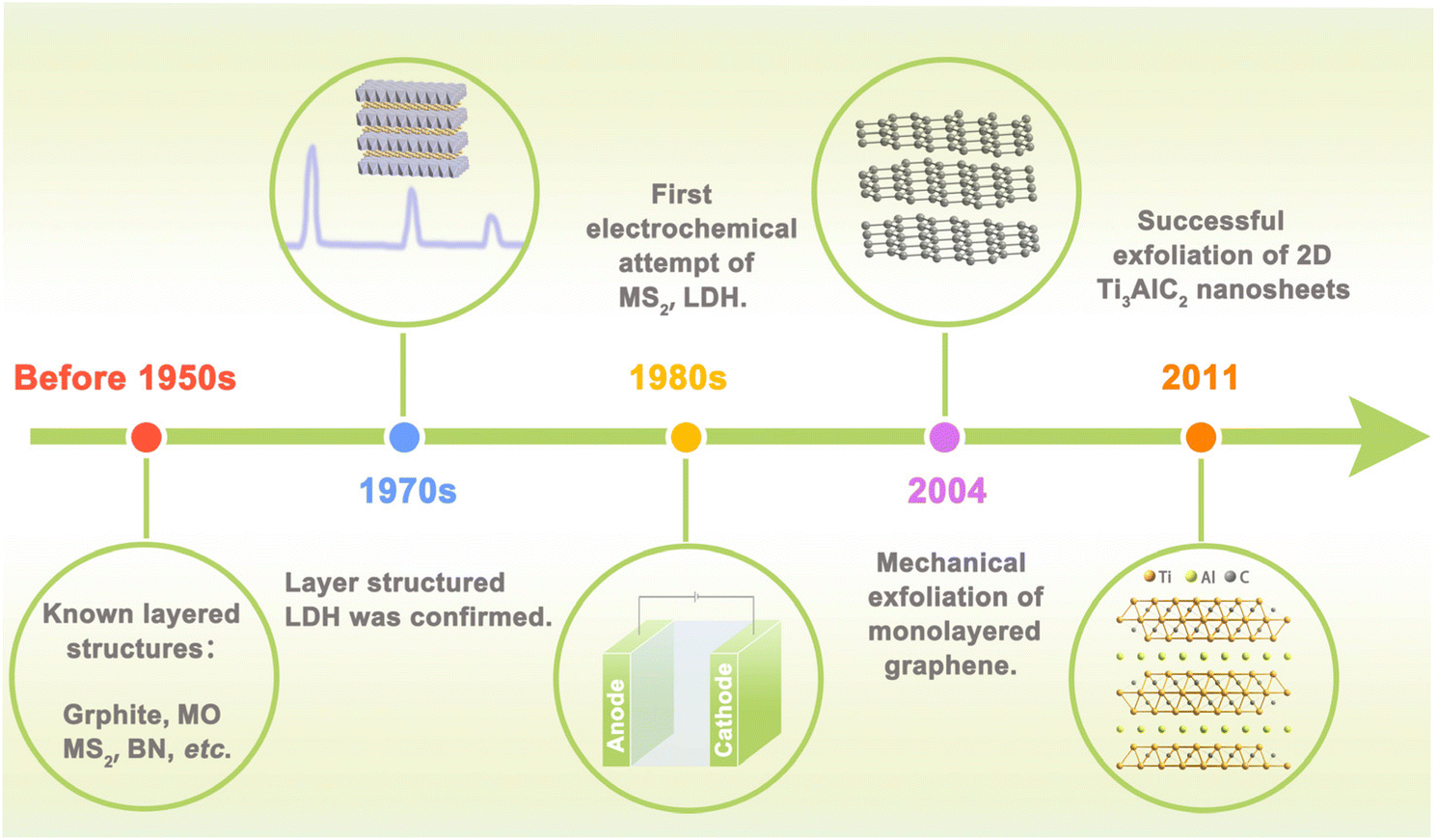 | ||
| Fig. 1 Time line of key milestones towards discovery and development of 2D materials. | ||
It is quite efficient to acquire highly active catalysts by converting the layered nanomaterials into nanosheets with distinctive physical, chemical, and electronic properties that are unattainable in bulk.25 (i) Ultrathin nanosheets with large lateral size and large specific surface area are favorable for exposing catalytic active sites and attractive for surface catalysis; (ii) highly exposed surface is helpful for the construction of uniform catalytic membrane electrode with low metal loading, which is beneficial for mass transfer. (iii) The confinement of electrons in the 2D ultrathin layer may give rise to compelling electronic properties, which are exceedingly attractive for catalytic reactions. (iv) 2D layer with defined components and crystal structure allows easy regulation of the electronic structures and properties by element doping, defect engineering, strain/phase engineering, making them an ideal platform for theoretical study. With the advancement of exfoliation methods such as mechanical exfoliation, liquid phase exfoliation, and chemical deposition, various types of inorganic layered nanomaterials have been fabricated and employed in electrocatalysis.26,27 With the combination of advanced characterization techniques and theoretical calculations, recognition and rational regulation of catalytic active sites has been greatly promoted.28 Further, with the rapid development of single-atom catalysts, 2D conductive materials are becoming suitable substrates for active-site anchoring so as to stabilize and regulate the catalysts. For example, graphene and MXene are widely used to support catalytic active phases due to their large surface/volume ratio and abundant functional groups, which are easy to incorporate with metal ions.29,30
Many reviews themed on 2D materials available now focus on one specific category of 2D material i.e. graphene, MXenes, transition metal chalcogenides or their derivatives, or center on the application of 2D materials in one field such as electrocatalysis, separation, and supercapacitor. Aiming at this situation, it is an impetus to present a comprehensive review containing various 2D materials, especially emphasizing the newly reported research progress. To this end, the review attempts to clarify the correlation between the layered structural characteristics and the catalytic performance of 2D materials. We first give a brief introduction about the structural chemistry of the 2D layered materials by dividing 2D materials into three categories, namely positively charged, negatively charged, and neutral laminates. Then, we summarize the preparation and exfoliation methods of layered structural materials. Following that, we discuss their application in electrocatalysis mainly including HER, OER, and ORR. Besides, 2D layered materials served as the support for electrocatalysts is also discussed in this paper. Finally, the review ends with personal opinions on the future potential opportunities for 2D materials in the catalytic field and the challenges existing in this promising field.
2. Structural chemistry of layered materials
If we want to classify these layered materials, the charge of the host layer may be a feasible criterion because it can intuitively display similarities and differences in different types of layered structures. Neutral, negatively charged, and positively charged layered compounds are three main streams of lamellated materials, and their representatives are illustrated in Fig. 2.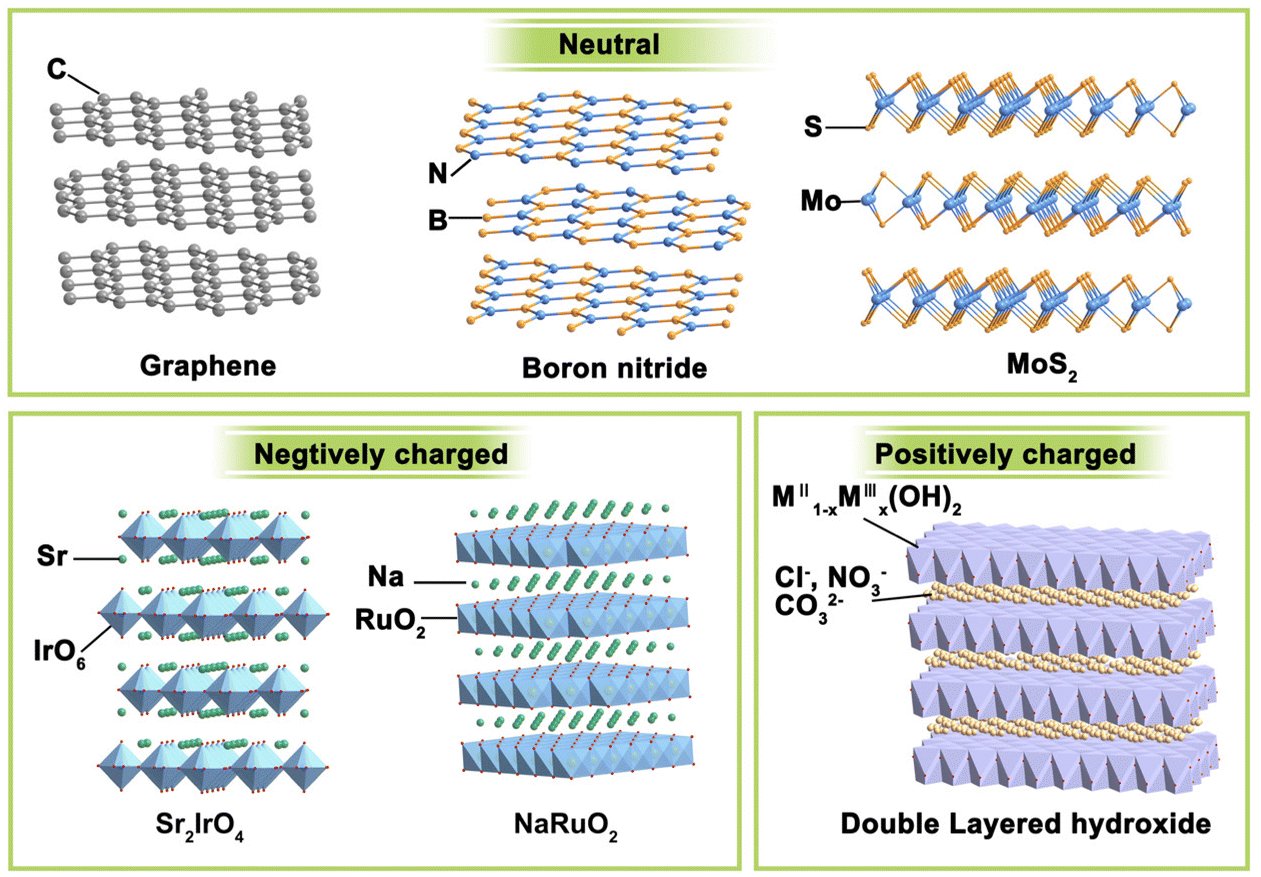 | ||
| Fig. 2 Classification of layered materials on the basis of the charge of the 2D layer. | ||
(i) Electrically neutral layered materials. Without ions intercalated between the host layers, neutral 2D materials are stacked by arranged laminate with relatively weak van der Waals or electrostatic attractive forces. Graphene, boron nitride, and MoS2 are typical representatives of this branch. For example, the most famous graphene consists of only sp2 hybrid C atoms interacting in a honeycomb arrangement with strong covalent bonding in the lateral plane.31 It can be warped into 0D fullerenes, rolled into 1D carbon nanotubes, or stacked into 3D graphite, making 2D graphene the most important basis of other graphite materials. Boron nitride has a similar structural lattice as graphene, in which equal numbers of boron and nitrogen atoms alternate in the six-membered ring to constitute the sp2-bonded layer.32 Besides, some metal compounds, mainly metal sulphides, and oxides, can also form a neutral laminate. Transition metal dichalcogenides (TMDs) are the most widely investigated and rapidly developed for their abundant compositions, structures, and wide range of applications. With a formula of MX2, the layered TMDs have sandwiched configurations and two crystal phases (i.e., 2H-MX2 and 1T-MX2) depending on the coordination and oxidation states of the metal atoms. In the 2H phase, the metals have a coordination of trigonal prismatic with a D3h point group symmetry, while that for the 1T phase is octahedral with a C3v symmetry. Their bandgap widths vary widely and layered TMDs can display semiconducting (e.g., MoS2, WS2) or metallic (e.g., Co9S8, NbS) properties. These layered TMDs have strong anisotropy and weak interlayer van der Waals interactions (∼40–70 meV), enabling the facile exfoliation of these layers and even mono-layer TMD.33 For instance, layered MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 and NbSe235 have been obtained through mechanical exfoliation as early as 1960s.
34 and NbSe235 have been obtained through mechanical exfoliation as early as 1960s.
(ii) Negatively charged layered materials. In negatively charged layer compounds, the metal compound laminates are negatively charged, and interlayer cations balance the charge. Many transition metal-based layered metal salts are typical of this category, such as K0.8Ti1.73Li0.27O4,36 KCa2Nb3O10,37 Cs6W11O36,38 RbLaTa2O7,39 Na2Ti3O7,30 KNb3O8,40 K0.75Na0.25IrO2,41 and NaRuO2.42 The central metal coordinates with six surrounding oxygen to form MO6 octahedron and these octahedrons connect through corner-sharing, edge-sharing, or face-sharing manner, composing negatively charged laminates. Balancing metals are usually alkali metals or alkaline earth metals such as Na, K, and Sr. RP-type perovskites with the formula of An+1BnO3n+1 are a typical case. They are constituted by repetitious layers of ABO3 perovskite blocks with corner-shared BO6 octahedra and alternated AO rock salt layers along the c-axis. Due to the strong interaction between intercalation ions and laminates, the layered materials are difficult to exfoliate directly. It is often necessary to obtain protonated samples through proton exchange under the assistance of acid before further exfoliation.
Other representative materials are ternary layered transition carbides, nitrides, and carbonitrides discovered by Gogotsi and Barsoum in 2011.43 They can be formulated as Mn+1AXn, where M refers to early transition metals (Sc, Ti, V, Cr, Y, Zr, Nb and Mo), A refers to the IIIA or IVA group elements (Al and Si are often the cases), and X presents C and/or N elements. MXene, which is obtained from the MAX phase, has a graphene-like layered structure. M–X has strong bonding energy, and A is of high chemical activity in MAX and can be easily removed. Taking advantage of this feature, researchers have synthesized a family of 2D transition metal–carbon/nitride materials by extracting Mn+1Xn from the MAX phase via selectively etching the A site.
(iii) Positively charged layered materials. Correspondingly, the positively charged layer compounds have positively charged metal compound laminates, and the interlayers are filled with charge-balancing anions. The most representative example is layered double hydroxides (LDHs). LDHs are naturally occurring minerals that are composited with lamellar mixed metal hydroxides.44 LDHs have typical edge-sharing M–O octahedral units constructed with the main layers and anions in the hydrated interlayer regions, implying their potential for anion exchange and applications in catalysis, photochemistry, and electrochemistry. In the LDH structure, a fraction of the divalent cations are replaced by trivalent metal cations, resulting in the positively charged 2D host layer. Common LDHs have both divalent and trivalent metals and can be formulated as [MII1−xMIIIx(OH)2][An−]x/n·zH2O, where MII may be Mg2+, Fe2+, Cu2+, Zn2+, or Ni2+, MIII may be Al3+, Ga3+, Fe3+, Co3+, Cr3+, or Mn3+, the value of x is between 0.2 and 0.4, and An− represents inorganic/organic anions, i.e., NO3−, Cl−, CO32−, SO42− or CH3COO− to compensate charges but does not participate in constructing the layered framework.45 The interlayer anions have strong electrostatic interaction with the intralayer metal ions, making LDHs difficult to be exfoliated through conventional methods.
3. Preparation of layered structural materials
Layered materials are mainly prepared in two ways: top-down and bottom-up strategies. Exfoliation is a typical top-down method to prepare layered nanosheets with few layers or monolayers from a lamellar matrix by etching away the sandwich layer. Exfoliation significantly boosts materials’ surface area, which can greatly enhance the chemical and physical properties of catalytic materials. Related exfoliation methods mainly include mechanical exfoliation (i.e. conventional mechanical exfoliation and Au-assisted exfoliation) and liquid phase exfoliation (oxidation, ion intercalation, ion exchange). On the other hand, bottom-up strategies can achieve good control of the thickness and the number of defects on the laminate. For example, vapor phase deposition (chemical vapor deposition, physical vapor deposition) was used in the synthesis of α-Mo2C.46 We will discuss the areas of application, advantages, and disadvantages of each exfoliation method in this section.3.1 Mechanical exfoliation
Mechanical exfoliation was the first used strategy for the exfoliation of 2D materials in 2004 when Geim's team developed a new ribbon-based exfoliation method for producing single and few-layered graphene from graphite.47 It is a traditional method of manufacturing thin nanoflakes by peeling off large crystals in layers using a transparent tape. A common method involves attaching large crystals, such as graphite, to adhesive on the transparent tape and then peeling them into flakes using additional adhesive.48,49 This process can be repeated multiple times until the desired flake is obtained. After that, the freshly cleaved flake is stuck from the clear tape onto a clean and flat target surface and a tool such as plastic tweezers is used to cleave it further. Finally, by peeling off the transparent tape, a single or few-layered nanosheets can be obtained that remains on the substrate. With the development of 2D materials, this method is also employed to prepare other layered materials, such as TMD and h-BN. As shown in Fig. 3a, single or several layers of MoS2 can be efficiently manufactured by the transparent tape-assisted mechanical peeling method.48 This technique does not involve the use of chemicals or chemical reactions during manufacturing. Consequently, the exfoliated nanosheets, whether monolayer or few-layer ones, maintain their pristine crystal quality, with a very clean surface, and no chemicals are introduced. However, the conventional mechanical exfoliation method shows many demerits. Firstly, the size of the exfoliated 2D materials is relatively small, ranging from a few to several tens of microns. Secondly, the control of the size and thickness remains challenging, as the peeling process is performed by hand and lacks precision, controllability, and reproducibility are the main issues.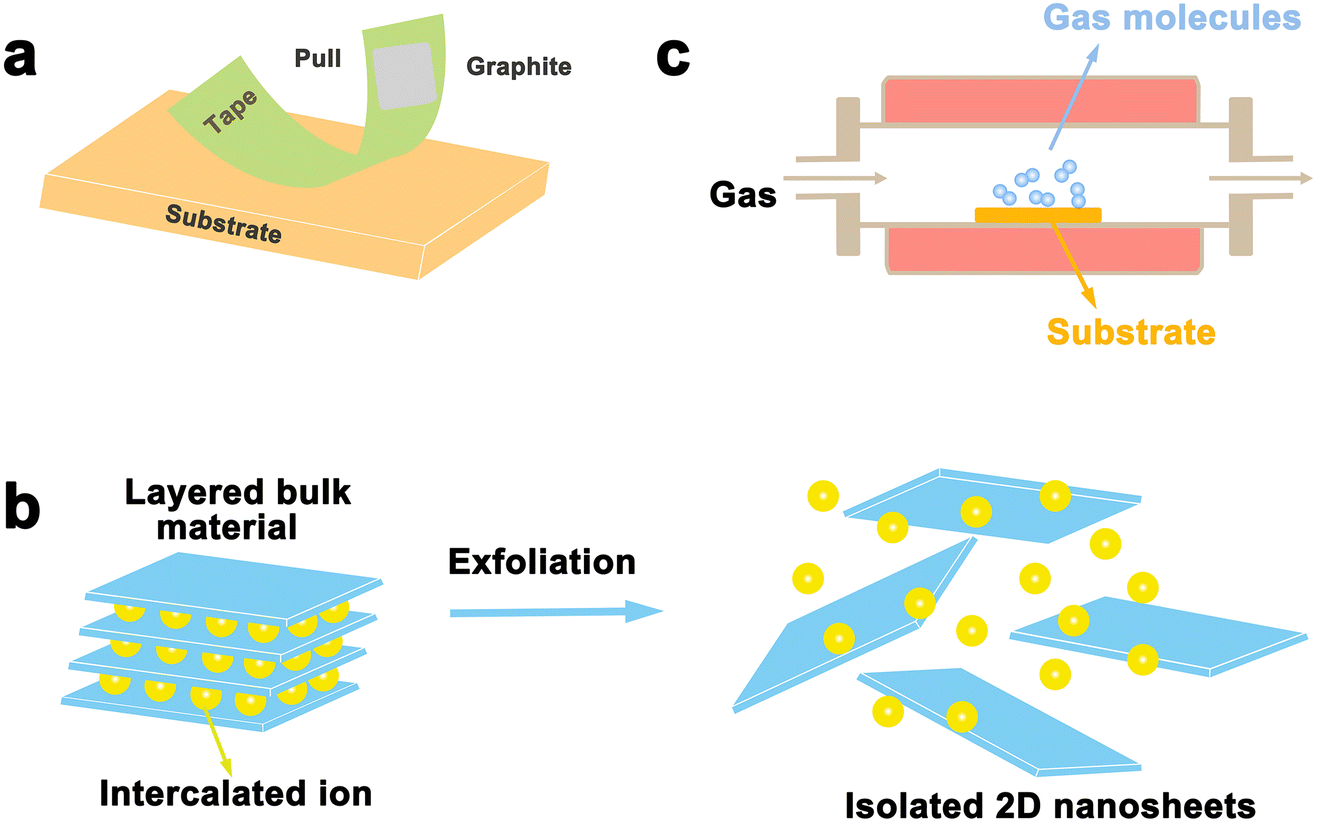 | ||
| Fig. 3 Schematic illustration of top-down and bottom-up synthesis methods. (a) Mechanical exfoliation method; (b) wet chemistry method; (c) chemical vapor deposition. | ||
To this end, in 2015, Sutter and his colleagues used a new oxygen plasma-enhanced exfoliation method that improved the exfoliation rate and area of Bi2Sr2CaCu2Ox nanosheets by making slight improvements to the traditional tape mechanical exfoliation technique.49 They noted that the oxide substrate had a surface layer of adsorbed molecules, which could be eliminated by treatment with oxygen plasma. This enhanced the interaction between the 2D material and the substrate. Briefly, the process begins with the treatment of the substrate with oxygen plasma to remove the surrounding adsorbed molecules. To achieve a more uniform interfacial contact between the substrate and the bulk crystal, a subsequent heat treatment is incorporated into the exfoliation process. This improved technique boosts productivity and increases the nanosheet area, and can be further used to produce large areas of monolayer or few-layer nanosheets and raise the production efficiency. This approach is effective in promoting graphite exfoliation but proves to be less efficient in exfoliating MoS2 and other various 2D materials lacking strong interactions with oxide substrates.
Javey et al. prepared single-layer TMDs nanosheets on different substrates such as SiO2/Si or quartz using a gold-assisted exfoliation method. The successful implementation of this method is attributed to the strong affinity between gold and sulfur, which can form a semi-covalent bond.50 In 2015, Magda and his team reported a gold-assisted exfoliation on mica with a larger atomically flat and clean Au (111) surface, unlike conventional SiO2/Si substrates. Later, the bulk MoS2 crystals were exfoliated on fresh Au substrates to produce monolayers of MoS2 with lateral dimensions up to several hundred microns.51 They also demonstrated that the method worked well for a variety of layered sulfur compounds, as well as selenides and tellurides. They successfully exfoliated bulk WSe2 and Bi2Te3 crystals to obtain monolayers on a lateral scale of several hundreds of micro-meters. Recently, a number of researchers have also exfoliated layered materials based on this method. For example, in 2020, Wu et al. prepared monolayered 2D MoS2 nanosheets.52 First, a thermal release tape (TRT) film of MoS2 crystals was pressed onto a flame-annealed 200 nm-thick Au (111) film on a mica substrate, and then lightly pressed on the back side to ensure good contact. Afterwards, the sample was transferred to a 90° etching plate to release the tape. At the same time, the MoS2 sheet was peeled off the edge of the sheet with fine tweezers. Finally, a millimeter monolayer of MoS2 was obtained on the Au surface.
3.2 Liquid phase stripping
Although mechanical exfoliation is commonly used for preparing high-quality 2D nanosheets, the yield of 2D nanosheets prepared by this method is low. As shown in Fig. 3b, liquid phase exfoliation allows for large quantities of dispersed nanosheets in a variety of organic, aqueous, or surfactant-containing solutions, which can be prepared in large quantities in industrial technology, and it enables the formation of thin sheets and composites with potential scalability. Such exfoliation results in materials with very large crystal surface areas of over 1000 m2,53 increasing surface activity, and extending their applications. There are three main techniques for liquid exfoliation of layered materials: oxidative sonication, ion intercalation, and ion exchange.Liquid exfoliation was first achieved through the oxidation of graphite, with the discovery of monolayer-thick graphene oxide (GO) flakes by Ruoff et al. in 2006.54 The oxidation method is mainly applied to the exfoliation of graphite, by using graphite powder as the raw material, that is Hummers’ method for preparation of graphene oxide.55 The strategy for preparing graphene oxide involves the use of strong oxidizing agents such as sulfuric acid and potassium permanganate to add hydroxyl and epoxide groups to the substrate, which can enhance the hydrophilicity of the substrate, enabling water intercalation and large-scale exfoliation under ultrasonic treatment to produce monolayer-thick graphene oxide flakes. The available solvent is, however, limited by the degree of surface tension matching between the layered material and the solvent, and the exfoliation of graphite requires a solvent with a surface tension close to 40 mJ m−2.56 Previous studies have shown that 2D nanosheets such as graphene, BN, TaSe2, MoS2, and MoSe2 can be successfully prepared by direct ultrasonic treatment of layered crystals in solvents such as dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF) and acetone.57 Recently, this method has also been used to strip other 2D materials, including PBi2.58 The amount and type of attached oxide can be controlled by oxidation, potentially enabling control of electrical conductivity. However, one of its drawbacks is that chemical groups and defective scattered electrons are inevitably introduced, giving relatively high resistivity.
Intercalation exfoliation of layered materials is a well-established and controlled method widely used for the exfoliation of layered materials, which has been greatly facilitated by the development of lithium-ion intercalation–exfoliation 2D TMDs since 2011.59 Since then, lithium-ion intercalation-based exfoliation is now commonly used to prepare 2D layered materials. For instance, in 2021, Tian et al. prepared several layers of WS2 nanosheets by a simple lithium-ion intercalation exfoliation method, which not only enabled the large-scale preparation of WS2 nanosheets, but also allowed the achievement of the obtained WS2 nanosheets with large lateral dimensions, excellent lattice structure, and no chemical impurity residues.60 With the increase in lithium costs and the susceptibility of lithium intercalation compounds to environmental factors and fire hazards, researchers have proposed many alternative cationic and anionic intercalators. For example, in 2018, Danae Gonzalez Ortiz and his team achieved exfoliation of BNs by embedding K+ and Zn2+ ions in h-BNs, and the obtained h-BNs typically showed a thickness of about a few nanometers (2 to 3).
The essence of the ion exchange method is the same as that of ion intercalation. The former utilizes exchangeable interlayers that contain cationic counter ions, specifically LDHs, clays, and certain metal oxides, to replace existing ions with larger radii present within the lamellar crystals.53 This process brings about interlayer expansion, which can be further enhanced through the application of ultrasound and shear to achieve the desired peeling effect. Adachi-Pagano et al., for the first time, successfully exfoliated LDH using dodecyl sulfate as an anionic surfactant and butanol as a dispersing agent.61 Wang et al. achieved gentle and sustainable exfoliation of bulk crystalline carbon nitride into ultrathin nanosheets in pure water through ion exchange in 2023. In summary, the ion-exchange liquid-phase exfoliation method can effectively exfoliate many layered massive crystals into ultrathin 2D nanosheets in solution. High-yield and high-volume production can be achieved by this method. However, it has certain requirements on the surface chemical activity and ion exchange reaction of the exfoliating materials, and it is difficult to achieve exfoliation for some materials that are difficult to perform surface modification or have low surface chemical activity, such as graphite and TMDs.
3.3 Vapor deposition
Mechanical exfoliation and liquid phase exfoliation are both top-down preparation techniques, while vapor deposition is a bottom-up preparation methodology that can directly prepare the desired 2D materials, and the size of the prepared material depends on the size of the substrate, which is expected to achieve high-volume preparation (Fig. 3c). Vapor phase deposition is divided into two main categories: chemical vapor deposition (CVD) and physical vapor deposition (PVD).CVD techniques have been demonstrated to produce large-sized 2D crystals with few defects and controlled atomic layer thickness, with reasonably good quality and uniformity.62 To prepare 2D materials, a chosen substrate is placed into a furnace with gas or vapor precursors circulating inside. These precursors can then react or decompose on the substrate's surface. Under suitable experimental conditions, this process can yield ultrathin 2D nanosheets on the substrate.62 Since the successful growth of monolayer graphene on Cu foil by CVD,63 CVD has gradually become the main method for the preparation of various 2D materials, including graphene,64,65 h-BN,66 and metal dichalcogenides (MX2, M = Mo, W, Ta, Cr, Nb, Re, etc.; X = Se, S, Te).67,68 For example, Ren et al. successfully synthesized MoSe2 nanosheets on Au with different Mo contents by CVD, as shown in Fig. 4. The early precursors are mainly metals/metal oxides, and the high melting point and thermodynamic instability of such precursors, as well as the high production costs, making them difficult to be grown on substrates directly through CVD for many 2D crystals. For this reason, intensive research has been carried out to solve the issues that existed in traditional CVD technology. Among them, plasma technology has facilitated the development of CVD. Using the plasma CVD method, 2D materials can be grown on non-catalytic substrates such as SiO2/Si or sapphire at a heat-treatment temperature lower than that utilizing conventional CVD technology. This is achieved by decomposing precursors into highly reactive materials using plasma. For example, Wei et al. successfully synthesized monolayer graphene crystals on SiO2/Si substrates via methane/hydrogen plasma CVD, highlighting the significant role of plasma technology in facilitating the production of high-quality 2D materials via CVD.69,70 A CVD method using metal organics as precursors, named MOCVD, has also been developed. For example, in 2018, D. Andrzejewski and colleagues grew MoS2 on sapphire (0001) substrates using molybdenum hexacarbonyl and di-tert-butyl sulfide as precursor materials. Compared to conventional CVD, the pyrolysis temperature of the organic precursors is distinctly decreased via MOCVD technology.71
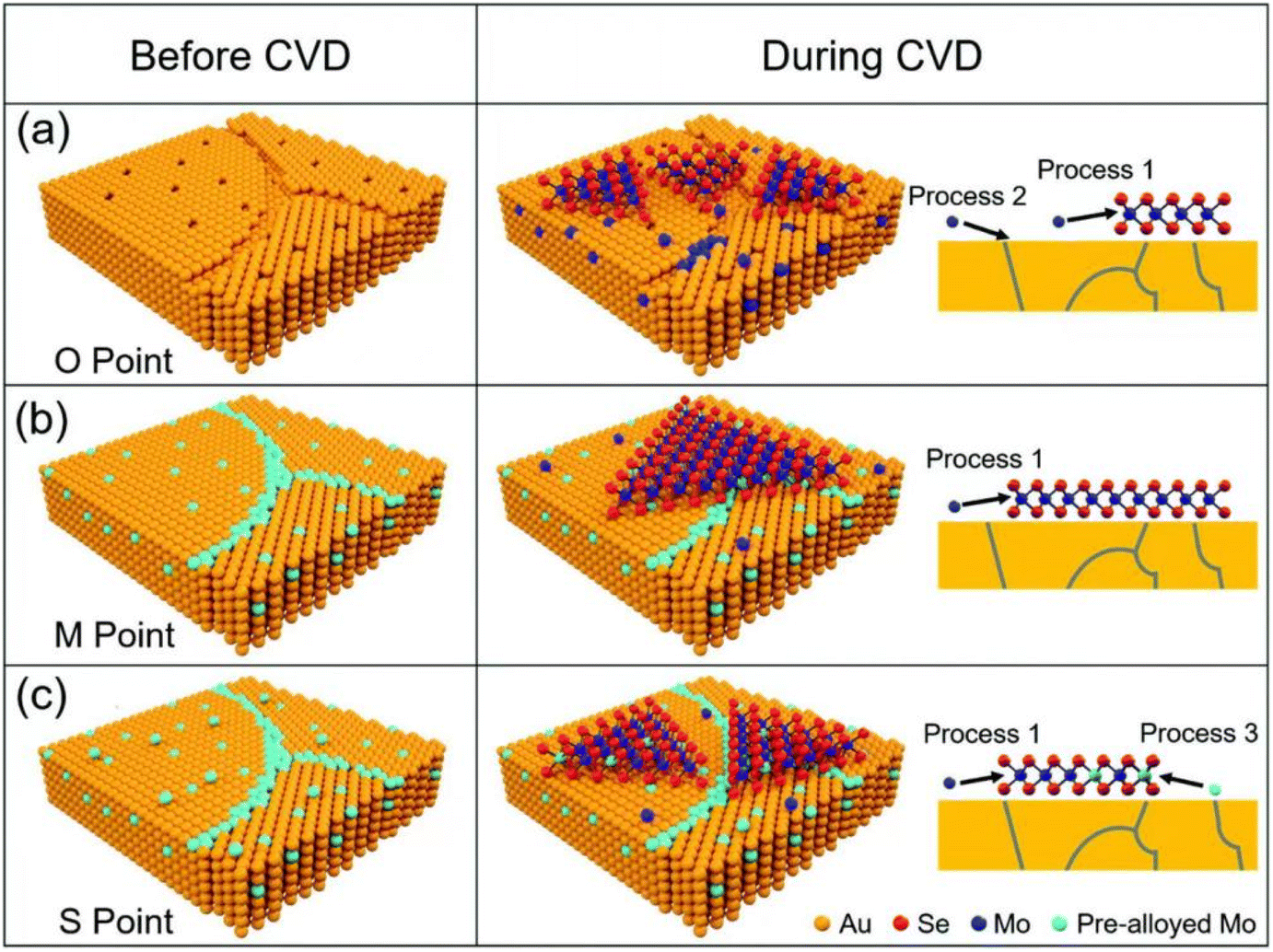 | ||
| Fig. 4 Schematic diagram of MoSe2 growth on pre-alloyed Au with different Mo content. (a) The original polycrystalline Au surface with many defects, including vacancies and grain boundaries (b) Mo content got saturated at 900 °C. (c) Mo was oversaturated at 900 °C (reproduced from ref. 68 with permission from the Royal Society of Chemistry, copyright 2022).68 | ||
Physical vapor deposition refers to the use of physical methods to vaporize the surface of a material into gaseous molecules, atoms, or ions under vacuum conditions, which are then passed through a low-pressure gas and deposited into thin films. The three main types of physical vapor deposition are vacuum vapor deposition, cathodic sputtering and ion plating.72 Of these, sputter coating and pulsed laser deposition are the most effective methods for large-area production of continuous two-dimensional (2D) materials.73 Sputtering enables control over the physical and chemical properties of thin films through adjusting process parameters. For instance, Zhang et al. utilized magnetron sputtering to produce three-dimensional (3D) graphene on nickel foam, demonstrating the technique's efficacy.74 The resulting graphene exhibited high conductivity and promising applications in electrochemistry. Pulsed laser deposition is another method for producing high-quality 2D materials. Zhu and his colleagues successfully produced high-quality MoS2 films on sapphire (0001), Si(001), and graphene using laser deposition technology, showcasing the effectiveness of pulsed laser deposition as a method for preparing 2D materials.75 While there have been notable strides in the fabrication of two-dimensional (2D) materials through physical vapor deposition (PVD), certain limitations persist that hinder the material's properties. Specifically, during sputtering, defects such as twinning and dislocations can be produced, which will lead to negative impact on the overall performance of the materials. In addition, it requires expensive equipments, professional operating techniques, and rich experiences. All in all, each preparation method has its own merits and deficiencies, and needs further development.
Though remarkable progress has been made in the preparation of 2D layered materials, still there are some issues to be addressed: (i) more than 1000 kinds of layered structures exist according to the inorganic crystal structure database (ICSD), while only a limited few dozen phases have been exfoliated.28 Exploring innovative preparation strategies applicable to desirable structures to obtain corresponding 2D nanosheets will be a hotspot. (ii) Most of the current well-developed methods depend on experimental trial-and-error approaches with low production yield and quality. It is necessary to develop rational design principles to guide the preparation and exfoliation of layered materials. (iii) Preparation of pure phase or monolayer 2D nanosheets is still challenging. (iv) Present preparation is faced with the dilemma of high cost due to complex preparation steps and harsh conditions. Thus, developing facile and environmentally friendly synthetic approaches to scale-up production of 2D layered materials is appealing for industrialization.
4. Layer structured electrocatalysts
Due to their characteristic two-dimensional structural features as well as fascinating physical and chemical properties, 2D materials have long been focused on by the research communities in various fields from biomedicine to the petrochemical industry.76 Maximized surface, good stability, and huge potential for modification make them especially appealing in electrocatalysis. Discussions on reactions of the 2D layered materials involved here are mainly hydrogen evolution reaction (HER), oxygen evolution reaction (OER), and oxygen reduction reaction (ORR).4.1 Layered transition metal oxides
Transition metal oxides (TMOs) are an important family of functional materials. They have the advantages of easy synthesis, environmental friendliness, great flexibility, diversity of composition, crystal structure, and functionality, and permit extensive potential in catalysis.77 While they are congenitally deficient in catalysis due to the semiconducting characteristics. Downsizing TMOs to 2D structure with an extended lateral dimension can bring about enhanced catalytic performance when compared to bulk counterparts.78Many transition metal-based layered oxides have been successfully prepared, including Ti, Nb, Ta, Mn, Mo, and W.79 These layered oxides have different structural frameworks, which typically include layered perovskite structure, layered α-NaFeO2-type structure, and honeycomb layered structure. Layered oxides comprise various TMO6 octahedral host layers (TM = transition metal elements), and large-radius alkaline or alkaline-earth cations in the interlayer channels.79 For example, in layered perovskite structure, the corner-sharing TMO6 octahedra form a perovskite layer, while in honeycomb layered structure, TMO6 octahedra are connected in an edge-sharing manner to form a regular honeycomb layer. Traditionally, the preparation of well-defined layered oxides depends on solid-state methods under high-temperature and high-pressure conditions. The resulting layered oxides are composed of micrometer-sized particles with low surface areas, which significantly limits the catalytic study of layered oxides. Until liquid-phase exfoliation methods are developed to generate novel 2D oxide nanomaterials, the layered metal oxides provoke wide interests in electrocatalytic applications.
Layered Mn-based oxides have shown great promise in OER application for their variable oxidation states, capricious bonding modes, thermodynamic stability, and low toxicity.80 For instance, manganese is an O2-evolution active element as first inspired by the biological systems that μ-oxo-bridged tetrameric Mn4Ca clusters almost exist in all photosynthetic organisms.81 This finding have triggered numerous studies to mimic the composition of biological enzymes, particularly manganese oxide with various crystallographic forms and morphological structures. δ-MnO2 has a 2D layered structure with MnO6 units orderly arranged in an edge-sharing pattern. Arno Bergmann synthesized δ-MnO2 and γ-MnO2 by symproportionation and impregnation methods, respectively. They found that the di-μ-oxo-bridged Mn ions in δ-MnO2 was related to the pronounced charge capacity behavior and efficient use of surface.82 Besides, the Spiccia group realized a circulation from layered Mn3+/4+ oxide to disordered Mn2+ ions by impregnating a synthetic tetranuclear-manganese cluster into a Nafion matrix and placing it under illumination. In situ X-ray absorption spectroscopy and transmission electron microscopy evidenced the transformation. These experiments suggested that the disordered Mn3+/4+ oxide catalyst functioned through a dissolution and reformation mechanism, and the cycle of manganese in different valence states was the origin of its catalysis.83 Modifications of the layered MnO2 lattice have proven to be efficient to enhance the water oxidation activity of the material. I. G. McKendry and coworkers introduced intralayer cobalt and interlayer iron to promote OER catalytic activity of MnO2. The enhanced activity was considered to be the synergistic effect of charge mobility caused by Co3+ doping into the sheets and band structure tuning caused by interlayer dopants.
Designing OER catalysts available for acidic media is important in the application of a proton-exchange membrane water electrolyzer. Most non-noble metal (e.g. Mn, Fe, Co, Ni) oxides-based catalysts work well in alkaline electrolytes but cannot resist corrosion in acid. Iridium oxide and ruthenium oxide are state-of-the-art electrocatalysts toward OER in an acidic environment for their optimum binding energy with the key intermediates. Traditionally, iridium oxides and ruthenium oxides do not exist in layered crystal phases.84 Recently, researchers have turned attention to layered iridates and ruthenates for acidic OER, including Sr2IrO4, K0.75Na0.25IrO2, and NaRuO2.85 The intercalation ions can be exchanged with acid to obtain protonated iridates/ruthenates and then layered iridium oxide and ruthenium oxide through further exfoliation. The monolayer or few-layered oxides often exhibit strikingly high performance for OER originating from the extended surface and fully exposed active sites. For example, D. Weber et al. obtained IrOOH nanosheets by acid treatment of K0.75Na0.25IrO2 in an aqueous TBAOH solution. After exfoliation, the protonated IrOOH nanosheets maintained the triangular arrangement of the edge-sharing Ir(OOH)6 octahedra. IrOOH nanosheets showed excellent catalytic performance for OER with an overpotential of 344 mV to deliver a current density of 10 mA cm−2 in 0.1 M HClO4, outperforming bulk rutile-IrO2 and bulk IrOOH.86 The Shao group directly prepared layered IrO2 of the 3R phase by a microwave-assisted mechano-thermal method in a strongly alkaline medium. The edge-sharing IrO6 octahedron constituted 2D layers and the layers packed in ABCABC were arranged in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, different from the rutile phase IrO2 (P42/mnm). 3R-IrO2 also showed ultrahigh activity toward OER with an overpotential of 188 mV to drive a current density of 10 mA cm−2 and turnover frequency (TOF) of 5.7 sUPD−1 at 1.50 V vs RHE.87 By modulating the thermal treatment temperature, recently, the group synthesized 1T-IrO2. 1T-IrO2 also had a layered structure composed of edge-sharing IrO6 octahedrons but ABAB stacking modes, different from that of the 3R phase. 1T-IrO2 also gave notable catalytic activity with a low overpotential of ca. 197 mV to deliver a current density of 10 mA cm−2 and TOF of 4.2 sUPD−1.88
m space group, different from the rutile phase IrO2 (P42/mnm). 3R-IrO2 also showed ultrahigh activity toward OER with an overpotential of 188 mV to drive a current density of 10 mA cm−2 and turnover frequency (TOF) of 5.7 sUPD−1 at 1.50 V vs RHE.87 By modulating the thermal treatment temperature, recently, the group synthesized 1T-IrO2. 1T-IrO2 also had a layered structure composed of edge-sharing IrO6 octahedrons but ABAB stacking modes, different from that of the 3R phase. 1T-IrO2 also gave notable catalytic activity with a low overpotential of ca. 197 mV to deliver a current density of 10 mA cm−2 and TOF of 4.2 sUPD−1.88
Layered perovskite oxides are derivatives of cubic perovskite ABO3, consisting of corner-sharing BO6 octahedra and interlayer A cations. The layered perovskite can be described as the Ruddlesden–Popper phase (e.g., A2BO4), Dion–Jacobson phase (e.g., A′A2B3O10), and Aurivillius phase (e.g., ABi2B2O9).89 A-site cations are commonly rare-earth or alkaline earth metals, B-site cations are commonly transition metals, and both sites can be easily substituted by other elements, leading to a wide variety of compositions and diversified properties. The most significant application of the perovskite is OER catalysis in acid since the discovery of SrIrO3 to catalyze OER efficiently by L. C. Seitz, in 2016.90 Subsequently, layered Ir-based perovskites have gained attention due to their ability to enhance OER performance. For example, the Grimaud group prepared Ruddlesden–Popper layered perovskite Sr2IrO4, which was then changed into protonated phase H3.6IrO4·3.7H2O through an Sr2+/H+ cation exchange method at room temperature. Compared to commercial IrO2 nanoparticles, H3.6IrO4·3.7H2O showed an enhanced specific catalytic activity and high mass activity.91 Huang et al. prepared ultrathin 2D Ir-based nanosheets with a thickness of about 1.3 nm by thermal treatment of ZnIr(OH)6 perovskite hydroxide (Fig. 5a). The nanosheet could catalyze OER efficiently in both acidic and alkaline environments that needed overpotentials of 278 mV and 252 mV to drive a current density of 10 mA cm−2, respectively.92
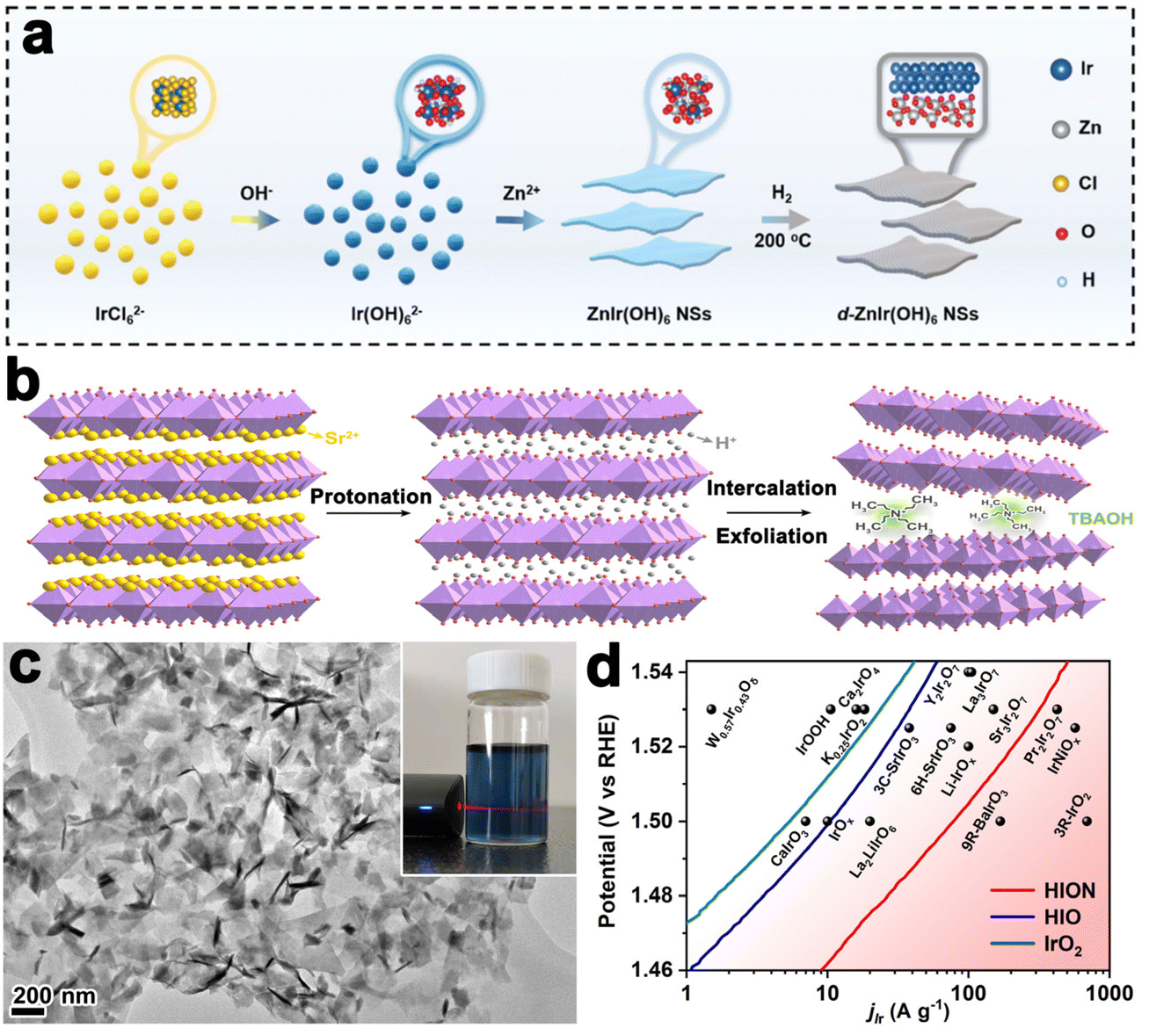 | ||
| Fig. 5 (a) Schematic illustration of the synthesis of ZnIr(OH)6 NSs and ZnIr(OH)6 NSs (reproduced from ref. 92 with permission from the Royal Society of Chemistry, copyright 2022).92 (b) Liquid-phase exfoliation of Sr2IrO4. (c) TEM image of HION and the inset showing the photograph of HION colloidal suspension with typical Tyndall light scattering under laser irradiation. (d) Comparison of Ir mass activity of HION with previously reported OER catalysts in acid media (reproduced from ref. 93 with permission from the American Chemical Society, copyright 2022).93 | ||
For bulk layered metal oxides, their catalytic efficiencies are usually medieval due to the low electrical conductivity, slow mass transfer kinetics, and inadequate active sites. Exfoliation of layered TMOs into monolayer or multilayer nanosheets with thickness in sub-micrometer or micrometer scale has been an effective way to improve the catalytic performance since the successful exfoliation of graphene. Their properties are usually different from the corresponding bulk materials due to the vanished interlayer chemical bond and strong surface polarization. Sourav Laha et al. fabricated highly active hexagonal ruthenium oxide nanosheets towards OER, reaching 10 mA cm−2 at an overpotential of only ≈255 mV. Characterization after OER indicated that the morphology and oxidation states of ruthenium nanosheets kept almost unchanged. Unlikely, in another report, the single-layered IrOOH nanosheets prepared by Daniel Weber and coworkers underwent protonation in the acidic OER process.86 Further, the Zou group realized the preparation of protonated iridate colloidal nanosheets exfoliated from Ruddlesden–Popper layered perovskite Sr2IrO4 for the first time (Fig. 5b). The TEM image in Fig. 5c revealed fine monodispersity of the fully protonated iridate nanosheets with good structural uniformity and dispersibility. The stable 2D morphological structure enabled a highly active catalyst with an ultralow-Ir-loading that showed 10 times higher activity than the IrO2 (Fig. 5d).93
4.2 Layered metal non-oxide compounds
Layered transition metal sulfides (LTMS), layered transition metal carbides (LTMC), layered transition metal nitrides (LTMN), and MXene are the main streams that constitute the family of layered metal non-oxide. In this section, the four transition-metal-based layer-structured materials will be discussed in sequence.Layered transition metal sulfides (LTMS) with a general formula of MS2, where M mainly represents Mo, W, Nb, Re, Ti, Ta, etc.,94 were first used in the petroleum and chemical industry in the early 1920s and later applied to hydrogenation of olefins, ketones and aromatics, hydrodesulfurization (HDS) as well.95 Molybdenum disulfide (MoS2) is a prototypical LTMS, containing two-dimensional S–Mo–S sheets stacked alternatively. In each monolayer unit, the Mo atom covalently bonds with three S atoms in the top layer and another three S atoms in the bottom. The adjacent S–Mo–S sheets are held together by weak van der Waals force, making laminate relatively amenable to slide and lubricant applications.96 The application of MoS2 as HER catalysts was realized decades ago for the wide band gap and semiconducting characteristics. In 2005, Jens K. Nørskov et al. first reported the Pt-like ΔGH* value of the edge sites at (1010) crystal facet of MoS2 and the catalytical activity of MoS2 toward HER using density functional calculations.97 Later Thomas F. Jaramillo's group evidenced the theory and proposed the linear relationship between activity and number of edges by preparing nanoparticulate MoS2 with different perimeters in the experiment.98
This breakthrough finding has raised tremendous research fanaticism to enhance the fraction of MoS2 exposed edge sites. To this end, two major treatment directions are formed: (i) activation of inert in-plane sites and (ii) exposure of edges as many as possible.94 Heteroatom doping is a common and effective method to convert in-plane sites into active ones. For instance, single Ag atoms doping was proved to be effective in regulating the electronic structures of the MoS2 basal plane. DFT calculations indicated that in-plane S sites neighbored to and between the Ag atoms were activated and a distance synergy of single Ag atoms in catalytic performance optimization was extracted. The authors also presented the minimum ΔGH* of 0.03 eV with optimum HER activity.99 Single fluorine atom is another element able to modulate the electronic structures of the MoS2 for its electronegativity.100 F-doped MoS2 electrode was fabricated through a plasma etching strategy as illustrated in Fig. 6a. F doping produced the activated basal plane of MoS2 and led to a fivefold activity enhancement compared to pristine edges. As revealed by the DFT calculations in Fig. 6b and c, the etched sites had more negative ΔGH* value and downshifted d-band center, which relieved the over-binding of H on F-doped Mo sites and was favorable for the desorption of hydrogen products. In another study of transition metal doping modification, the authors found that the incorporated Co ions bonded with edge S toms, forming a hexagonal morphology and hence changed both the morphology and the intrinsic activity of MoS2.101 In addition, coupling with Ru nanoparticles on the inert basal plane is another plausible way to activate interfacial S sites. As revealed by the investigation of Hou et al., thanks to Ru nanoparticles hybridizing, the electronic structure of interfacial S was modulated and the ΔGH* was optimized, giving an enhanced activity toward HER.102
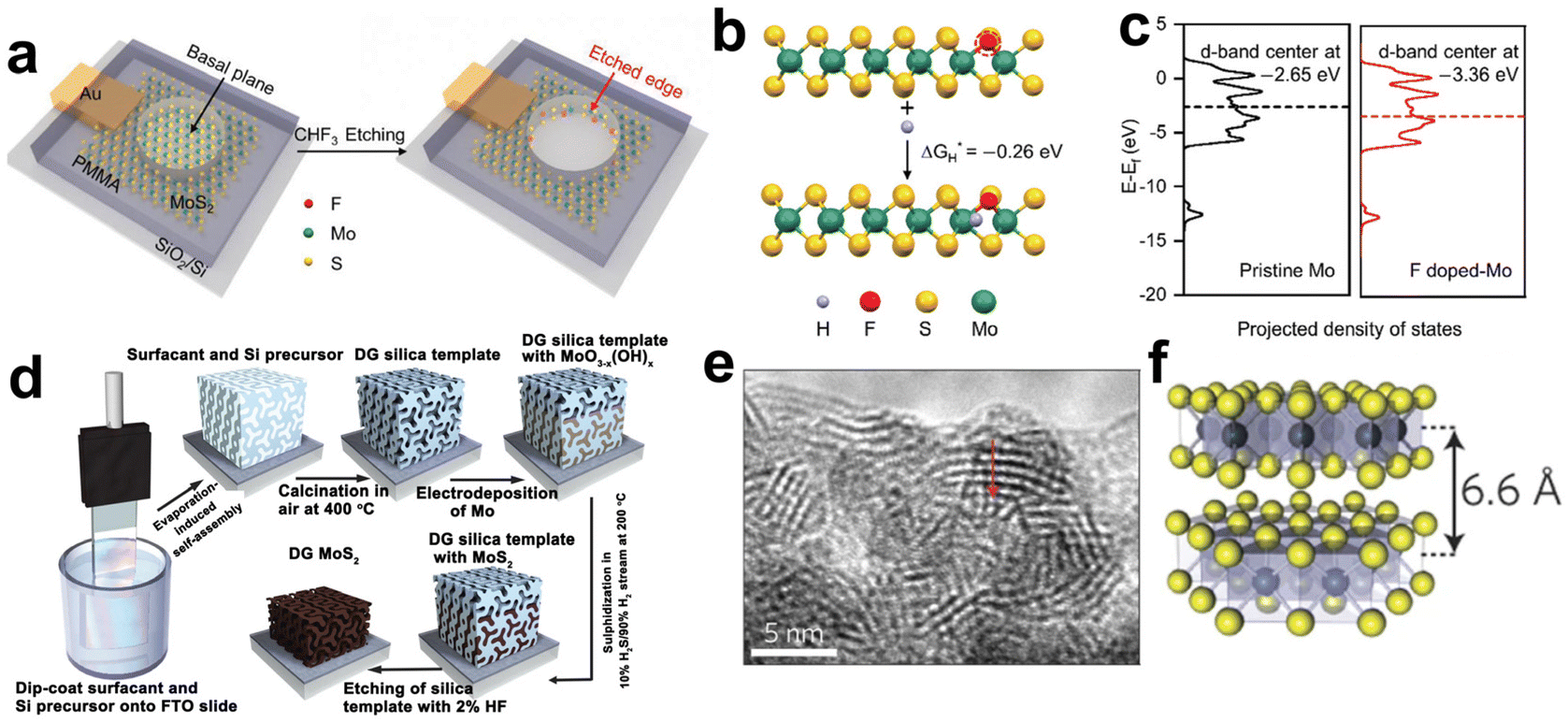 | ||
| Fig. 6 (a) Schematic illustration of in situ creating and doping of etched edges on MoS2 by CHF3 plasma. (b) The calculation of ΔGH* based on an S-terminated edge structure with one sulfur atom substituted by fluorine. (c) The projected electronic density of states of the d-bands for Mo atoms on pristine and F-doped MoS2 (reproduced from ref. 100 with permission from Wiley, copyright 2018).100 (d) Synthesis procedure and structural model for mesoporous MoS2 with a double-gyroid (DG) morphology. (e) TEM image where the S–Mo–S layers are resolved. (f) Model of MoS2 along the [110] projection (reproduced from ref. 106 with permission from Nature Publishing Group, copyright 2012).106 | ||
To expose as many edges as possible, one can maximize the edges by nanosizing, pore-creating and exfoliating into monolayers.103–105 For example, the Jaramillo group engineered the surface structure of MoS2 to preferentially expose edge sites by synthesizing contiguous large-area highly-ordered double-gyroid MoS2 thin films through a template-assisted electrodeposition method (Fig. 6d).106 TEM characterization magnified the mesoporous double-gyroid and the TEM image in Fig. 6e revealed that the S–Mo–S layers had a layer-to-layer spacing of 6.6 nm, in accordance with that indicated in the model (Fig. 6f). A large fraction of edge sites together with its high surface area led to excellent activity towards HER. Yang's group grew MoS2 thin films on a substrate by a CVD method with controlled layers and induced the correlation between catalytic activity and a number of MoS2 layers. Different from previous views that addressed the number of edge sites, the authors attributed layer-dependent electrocatalysis to the hopping of electrons in the vertical direction.107
From bulk MoS2, new perspectives on its structure, catalytic active site, and performance improvement have significantly proceeded. These advances make it a promising noble-metal-free electrocatalyst and provide insight into the investigation of other materials.
4.3 Layered multicomponent layered materials
In addition to single metal compounds, some niche multicomponent metal-based compounds such as transition metal phosphorous sulfides, transition metal phosphorous selenides, and transition metal phosphorous nitrides, also have typical layered structures. Here we take thio(seleno)phosphates as an example to illustrate their status in electrocatalysis.Layered transition metal phosphorous sulfides, with a general formula MPSx, have variable metal components ranging from Cr, Ga, Sn, Cd to Mn, Fe, Co, Ni.108 Their crystal structure belongs to monoclinic space group C2/m with D3h symmetry. Usually, the metal is divalent and ionically bonded with P2S6 clusters, and the chemical formula can be written as M22+(P2S6)4−. They have an S–M–S sandwiched structure constituting the layer, similar to that of MoS2. The interlayers interact by the van der Waals force. Due to the wide band gap and unique ferromagnetic properties, they are widely used in magnetism and spintronic devices, while their application in electrocatalysis is rare.
Until now, only NiPS3, CoPS3, and BiPS4 have been reported to be active towards HER, OER, and ORR, respectively.108 The catalytic activity of such materials remains unsatisfactory but can be modulated by proper modification. The Zhang team electrochemically activated inert layered Pd3P2S8 through a lithiation method. Lithiation resulted in the formation of amorphous lithium-incorporated palladium phosphosulfide nanodots with abundant vacancies. The structural evolution contributed to its remarkable catalytic activity, achieving an onset potential of only −52 mV vs RHE and outstanding long-term stability for HER.109 Similarly, Wang et al. activated the basal planes of NiPS3 by carbon doping that moderated the filled state of the valence band of NiPS3 and hence the hydrogen adsorption strength of surface sites.110 These reports enlighten the regulation of surface and electronic structures of 2D nanomaterials towards catalytic performance optimization. Layered transition metal phosphorous sulfides provide a broad platform for material functionalization. With regard to the metal phosphorous sulfides, their application in catalysis is not widespread. To be really practical to replace noble metal Pt, in addition to achieving the performance of Pt-like activity, scale-up preparation of catalysts is also a huge challenge.
These research advances demonstrate that the layered materials are ideal models to identify active sites and establish structure–activity relationship. Structural stability of the layered electrocatalysts, however, is a major concern. On one hand, the 2D layers with large lateral size have high surface energy, and therefore tend to agglomerate or deactivate, which will lead to diminishment of the peculiar 2D structural features. On the other hand, 2D materials are prone to undergo structural evolution during catalysis. Hence, more attention is expected to focus on the structural stability improvement of 2D nanomaterials. Moreover, electrochemical applications of 2D materials mainly concentrate on HER, OER, and ORR at present. Extending their applications to CO2RR and NRR seems to have great potential if the rational design and construction of ultrathin 2D nanosheets can be achieved.
5. Layer-structured electrocatalyst support
Anchoring catalytically active species onto 2D layered materials has been confirmed to be an efficient strategy to enhance catalytical performance.111,112 Benefiting from the extended lateral size, supporting 2D materials permits a maximized exposure of active sites, reduction of noble metal usage, and unique electronic interaction with the supported metal.113 Employing 2D materials as supports has received widespread attention and several reviews have referred to 2D layered materials as advanced support for electrocatalysts and application in energy conversion.114–118 It is important to offer an updated overview of the latest progress in a systematic summary of 2D material-supported catalysts, understanding of metal–support electronic interaction, and applicable scenarios.5.1 Graphene support
The discovery of graphene has stimulated significant interest in the scientific community due to its alluring intrinsic properties such as large surface area, high electrical conductivity, and exceptional electrochemical stability. These properties have led to the identification of numerous potential applications of graphene in the field of electrocatalysis. As a result, graphene has been extensively studied as an ideal supporting material for catalysts, with a particular focus on its suitability as a conducting support.119,120 Catalysts supported on graphene can be classified into three kinds based on the degree of aggregation of the loading material, namely nanoparticles, metal clusters, and single atoms. Typically, nanoparticles with sizes larger than 1 nm, while those with sizes smaller than or equal to 1 nm are classified as metal clusters or sub-nanoclusters. Single-atom catalysts are those in which the atoms are individually distributed.121Graphene shows good corrosion resistance and favorable electronic metal–support interaction and considerable stability. These desirable properties make it an attractive option for supporting metal/metal oxide/metal sulfide nanoparticles such as palladium,122,123 platinum,124–126 ruthenium,127 cobalt,128 nickel, nickel oxide,129 zinc oxide,130 manganese oxide129 and cobalt sulfide.131,132 Recently, Zhao et al. successfully synthesized ultra-fine platinum–cobalt nanocatalysts protected by graphene nano-pockets (Fig. 7a) with low metal loading and high stability. The graphene shell effectively limited catalyst aggregation and ensured excellent catalytic stability, while also restrained nanoparticle sintering and aggregation, thereby preventing oxidative dissociation and diffusion. Furthermore, the graphene pocket largely retained dissolved Pt atoms, which could be re-deposited onto the platinum–cobalt nanoparticles, helping to maintain their sizes (Fig. 7b and c).133
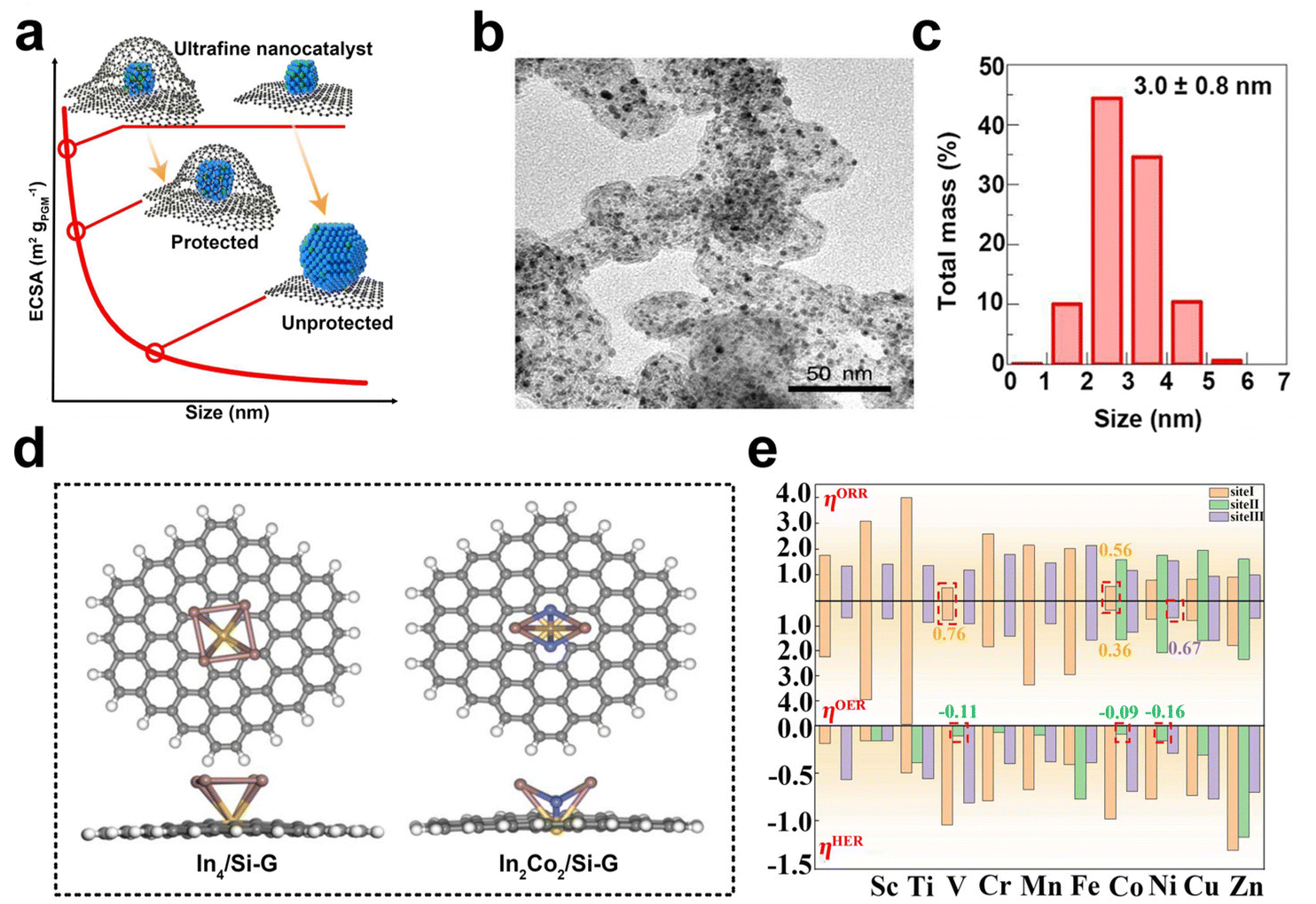 | ||
| Fig. 7 (a) Schematic illustration of ultrafine nanocatalysts encaged in graphene pockets and their impact on ECSA retention after an ADT. (b and c) Characterization of the PtCo@Gnp before the catalysis test: (b) TEM image, (c) size distribution (reproduced from ref. 133 with permission from Nature Publishing Group, copyright 2022).133 (d) Top and side views of optimized In4/Si-G and In2Co2/Si-G. The brown, blue, yellow, gray, and white atoms represent the In, Co, Si, C, and H atoms, respectively. (e) ORR, OER, and HER overpotentials of In2M2/Si-G. The red dotted boxes represent sites of catalysts with better multifunctional catalytic activity (reproduced from ref. 139 with permission from Elsevier, copyright 2023).139 | ||
The catalytic properties of metal clusters and single atoms differ significantly from those of metal nanoparticles with larger particle sizes. Metal clusters possess a molecule-like electronic structure and offer numerous surface-active sites that can effectively adsorb, activate, and transform reactant molecules due to their distinctive geometric and electronic structural properties.134 Currently, the reported graphene-supported metal cluster catalysts mainly include monometallic and multi-metallic catalysts. Palladium,135 platinum,136 iron, cobalt, and nickel nanoclusters are typical examples of the former. The latter includes bimetallic cobalt/tungsten clusters-loaded graphene,137 platinum/nickel clusters-anchored graphene, etc.138 Chen et al. systematically investigated the catalytic performance of silica-doped graphene-supported indium-based bimetallic clusters (In2M2/Si-G, M = Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn) (Fig. 7d) as efficient multifunctional electrocatalysts for ORR, OER, and HER by using molecular simulation techniques. The results showed that all these bimetallic clusters could be loaded onto Si-G substrates with good activity (Fig. 7e).139 Recently, significant advances have been made in the area of graphene-supported metal-cluster catalysts. Tian and his colleagues successfully developed an innovative technique referred to as the graphene-confined ultrafast radiant heating (GCURH) method. This approach has been shown to be highly effective in synthesizing metal cluster catalysts with exceptional loading capacity in a fraction of a microsecond. The GCURH method leverages the unique properties of graphene, including ultra-low permeability, flexibility, and high-temperature resistance. They also utilized this method to synthesize sub-nano Co cluster catalysts with a metal loading up to 27.1 wt% through the pyrolysis of Co-based metal–organic frameworks (MOFs). This catalyst displayed remarkable activity in oxygen evolution reactions and offered great convenience in catalyst recovery and refinement due to its single-metal composition. This breakthrough technology is poised to deal with the complex and environmentally sustainable difficulties faced by metal cluster catalysts.140
SACs have gained much attention in the field of electrocatalysis because of their maximum atom utilization, uniform active center distribution, and diverse metal–support interactions.141 To enhance their electrochemical energy conversion performance, suitable support is necessary to prevent the aggregation of metal atoms and ensure their ultrafine dispersion. The graphene-single atom catalyst (G-SAC) nano-platform, which utilizes graphene-supported single atoms as a catalyst, provides a way to study how metal–support interactions affect electrocatalytic performance on an atomic level. Two types of G-SACs are prevalent: one is loaded with noble metals, including Pd,142,143 Pt,144–146 Au, and Ru.147 The other is single atoms of non-precious metals, such as alkaline earth metals148 and transition metals, including Fe,149 Co,150 Ni,151,152 Cu,153 and W154 among others. Qiu et al. used a template-assisted approach to anchor iron single atoms onto highly stable hollow graphene nanospheres, resulting in catalysts with exceptional ORR performance due to the combination of atomically dispersed Fe active centers and the stable substrate.149 Similarly, in 2023, Wu and coworkers148 prepared alkaline earth metal single-atom catalysts (AE-SACs) supported on graphene and explored the feasibility of AE metals as multiphase catalytic active centers for electrocatalytic nitrate (NO3−) reduction reactions (ENO3RR). AE metal active centers can strongly adsorb and activate NO3−, while AE metal elements contribute to charge transfer between catalyst supports and NO3−.
In addition to pure graphene,127,129,131,155 heteroatoms doped graphene125,128 and graphene with different micro-structures,126,132,156 have been explored as material supports. Besides, Au,157 polyethylene, calcium oxide,122etc. have also been reported for the modification of graphene. These modified graphene materials show enhanced catalytic activity and stability to some extent. For example, in 2013, Xiong et al.158 deposited Pt nanoparticles on pure and nitrogen-doped graphene and compared the structure and catalytic performance of two catalysts. They found that Pt nanoparticles grown on an N-G substrate had smaller particle sizes, narrower size distributions, and better dispersions, and therefore displayed excellent catalytic performance of methanol oxidation reaction (MOR)-the onset potential of nitrogen-doped samples for MOR was approximately 0.4 V vs Ag/AgCl (3.5 M KCl), whereas this value of undoped nitrogen samples was about 0.5 V vs Ag/AgCl (3.5 M KCl). This suggested that nitrogen doping could be a viable strategy to enhance the MOR activity of graphene-based catalysts.
Graphene has been demonstrated to be a promising substrate for application in electrochemical energy conversion. However, there is still some debate surrounding the potential impact of defects in graphene-based catalysts on metal active sites. Firstly, existing studies have shown that vacancies in graphene can provide active sites for metal catalysis, but excessive vacancies may lead to the dilution of the catalytic sites. Future research should investigate the optimal amount of vacancies in graphene-based catalysts to deliver the best catalytic performance.159 Secondly, there are controversies related to surface oxidation groups. Several studies have shown that the oxygen-containing functional groups on graphene surfaces can provide active sites and enhance the catalytic activity of metal catalytic sites. Alternative research finding has suggested that the oxygen-containing functional groups may act as a competitor to the metal catalytic sites concerning the adsorption of active species, which may result in reduced catalytic activity.160 Therefore, the control of surface modifications or surface oxygen-containing functional groups can be used to tailor catalytic activity. Thirdly, the introduction of additional electronic states and defect sites via heteroatom doping can also enhance the activity of metal catalytic sites. Nonetheless, the impact of heteroatom type and concentration on catalytic properties is intricate, leading to the ongoing debate. Current understanding of the effect of heteroatom doping on catalyst activity remains limited, despite extensive research efforts in this area.161 Therefore, given the complexity of the material at hand, a comprehensive analysis is necessary to optimize the design of catalysts with graphene-based defects. In conclusion, a more comprehensive approach is needed to fully understand the effect of heteroatom doping on catalyst activity, especially for graphene-based defects.
In addition, graphene as a support currently faces several issues: (1) active site problem. Graphene's surface contains a variety of functional groups, such as carboxyl and hydroxyl groups, but these functional groups are not ideal catalytic active sites. Additional active sites need to be introduced through surface modification and other methods. (2) Reduction rate. A certain amount of oxygen-containing functional groups are often left in the preparation process of graphene, which requires reduction treatment. However, if the reduction rate is not high enough, it can affect its electrocatalytic performance. (3) Cyclic stability. Graphene may be deactivated during long-term cycling use, so it is necessary to improve the cyclic stability by optimizing the preparation method and structural design of catalysts. All in all, employing graphene as a supporting material in catalysts has demonstrated notable advances in electrocatalysis. Nevertheless, the questions referred to above still remained and should be addressed in future investigations.
5.2 BN support
Hexagonal boron nitride (h-BN), namely white graphene, is composed of alternating B and N atoms with equal numbers bonded in a honeycomb arrangement and stretched into two-dimensional layers.162 The h-BN has exceptional chemical stability, superb mechanical strength, and ultrahigh thermal conductivity, making them widely investigated as thermal conducting materials, dielectric layers, and lubricants.163 Due to interlayer insulation and electrochemical inertness originating from the wide band gap, investigation of BN in the field of catalysis is still rare.Catalysis mainly proceeds on the surface of materials and inert supports can be converted into electrocatalysts through surface and/or interface engineering strategies. By the merits of abundant sites on the 2D substrate surface, the support can regulate the electronic structure and hence catalytic performance of the active sites through the electronic metal–support interaction.164 Catalytic ability of a given material depends on the electronic configuration and microstructure of active sites, which directly determines the adsorption energy, reaction energy barrier, and the number of exposed active sites. In this point of view, regulating the electronic structure of the active centers is the key to optimizing the intrinsic properties.165
Some physical or chemical modification approaches including hybridizing with conductive additives and compounding have been adopted to activate h-BN,166,167 robust BN is deemed as dominant support for anchoring metal sites when thinking of its high N content and high stability. Taketsugu et al. predicted that Ni-modified h-BN can serve as an efficient ORR catalyst from the theoretical calculation in 2013.168 Not alone, Chen's group also put forward a highly stable NRR catalyst by theoretically constructing a single Mo atom catalyst model onto a defective BN layer.169
On the basis of preliminary work and experience, researchers have devoted themselves to activating BN through three experimental methods, mainly including defect, band structure, and interface engineering.169,172
(i) Defect engineering. It has been evidenced that creating unsaturated N atoms originating from B vacancies in the h-BN sheet is responsible for the anchoring of metal atoms and the formation of M–Nx sites as active sites. For example, Zhao et al. introduced Mo single atoms onto the surface of the h-BN monolayer with boron monovacancy. The defect-rich hybrid material exhibited high catalytic activity for N2 fixation at room temperature owing to selective stabilization of N2H* species, or destabilizing NH2* species (Fig. 8a).169,173 The introduction of C into the BN lattice is also effective to create C–N and/or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, which is beneficial to the fixation and dispersion of metal sites. Du et al. fabricated an unusual boron nitride with C, O dopants (L-BN) under the assistance of pulsed laser ablation (PLA) as illustrated in Fig. 8b and c. Based on enhanced electrical conductivity stemming from the interlayer B–B dipolar interaction, L-BN seemed to be an attractive support for oxygen evolution reaction. When IrOx was loaded on L-BN, the IrOx/L-BN exhibited inspiringly high activity that needed an overpotential of 259 mV to drive 10 mA cm−2 of current density due to strong interaction between N–CN in L-BN and IrOx. As revealed from DFT calculations of N–CN bonds in Fig. 8d, after PLA, the C atom in N–CN bonds of L-BN lost 0.403 electrons and the unsaturated electronic structure showed strong electron affinity.170
N bonds, which is beneficial to the fixation and dispersion of metal sites. Du et al. fabricated an unusual boron nitride with C, O dopants (L-BN) under the assistance of pulsed laser ablation (PLA) as illustrated in Fig. 8b and c. Based on enhanced electrical conductivity stemming from the interlayer B–B dipolar interaction, L-BN seemed to be an attractive support for oxygen evolution reaction. When IrOx was loaded on L-BN, the IrOx/L-BN exhibited inspiringly high activity that needed an overpotential of 259 mV to drive 10 mA cm−2 of current density due to strong interaction between N–CN in L-BN and IrOx. As revealed from DFT calculations of N–CN bonds in Fig. 8d, after PLA, the C atom in N–CN bonds of L-BN lost 0.403 electrons and the unsaturated electronic structure showed strong electron affinity.170
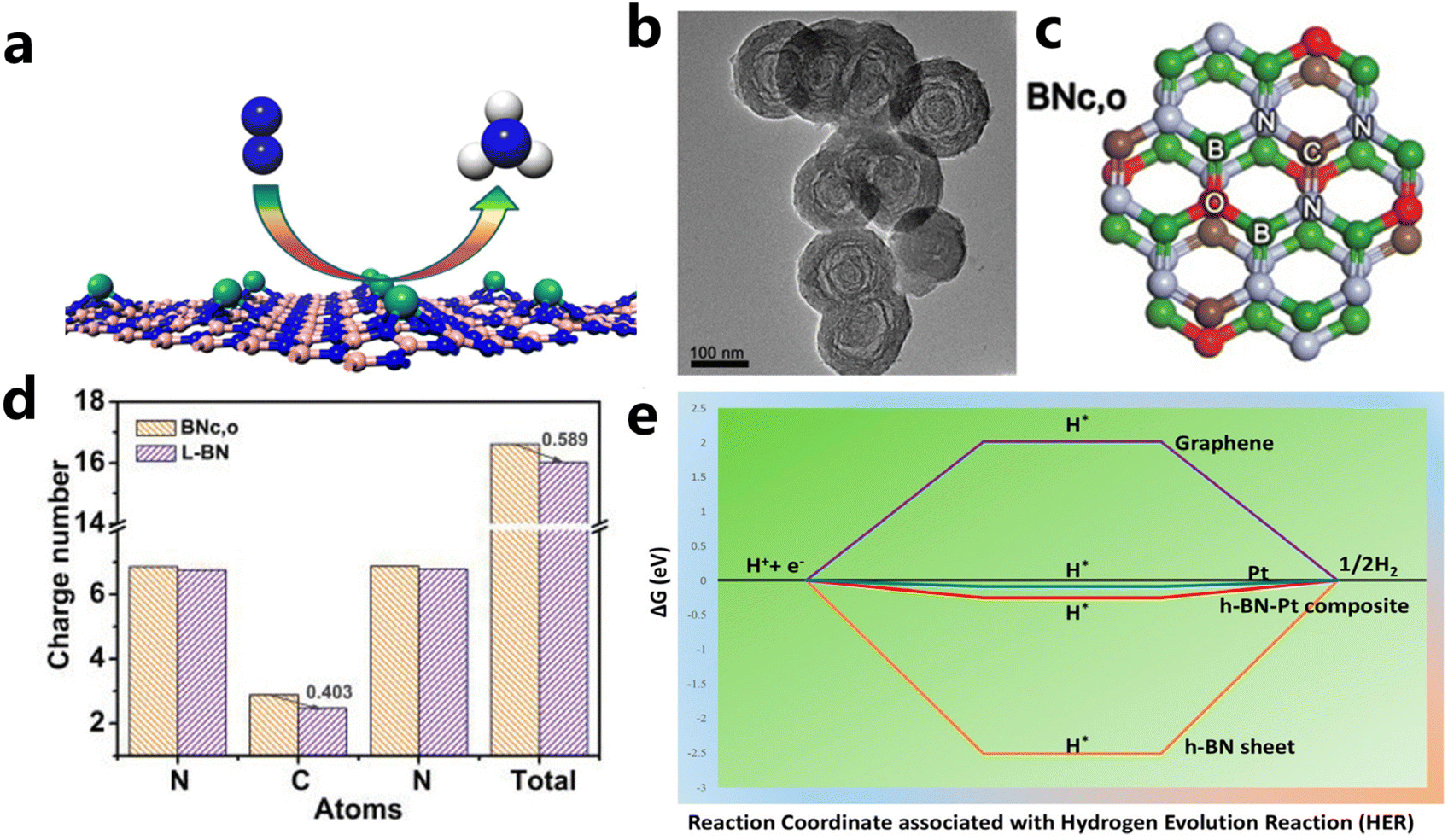 | ||
| Fig. 8 (a) Schematic illustration of single Mo atom supported on defective boron nitride monolayer (reproduced from ref. 169 with permission from the American Chemical Society, copyright 2017).169 (b and c) TEM and structural models of BNc,o (d) the number of charges per atom in the N–CN bond (reproduced from ref. 170 with permission from wiley, copyright 2020).170 (e) Free energy diagram for HER on Pt-modified h-BN, graphene, Pt, and Pt/h-BN (reproduced from ref. 171 with permission from the American Chemical Society, copyright 2018).171 | ||
(ii) Band structure engineering. The potential catalytic application is limited by the wide conduction band structure, and electronic regulation of the h-BN surface is essential for their catalytic performance.168,172 Creating B- and N-vacancies and defects through heteroatom doping has been evidenced to be effective to narrow its band structure.174 Besides, the introduction of metal elements can also modulate the band structure of BN. By loading the wide band gap BN nanosheet onto the surface of Au, Uosaki et al. observed slight protrusion of the unoccupied BN states toward the Fermi level and reduced the band gap of BN. The BN–Au hybrid was also theoretically suggested and experimentally proved to be an electrocatalyst for ORR.175 Pt decorating on BN sheets could also enhance its catalytic property for HER by band gap modulation. As illustrated in the free energy diagram in Fig. 8e, compared to BN, Pt loading had a significantly optimized ΔG value near that of pristine Pt and enhanced the charge transfer kinetics due to synergistic interaction between h-BN and Pt.171
(iii) Interface engineering. Catalytic reactions mainly proceed on the surface of a given material, and hence microstructures and local electronic environments play a crucial role in the catalytic behavior. Sun et al. reported hetero-structured h-BN/Pd hybrid material with well-defined interfaces that was used as a stable ORR catalyst. Both theoretical calculation and experimental results showed the strong interaction between the interface of BN support and Pd nanoparticles, which lowered the d-band center of Pd and optimized the adsorption of reaction intermediates.176 Similarly, Tang et al. achieved well dispersion of a single Mo atom on h-BN by constructing a hetero-interface with graphene (BCN). Compared with the one supported on bare h-BN, the Mo single-atom catalyst on hybrid BCN exhibited a metallic property due to the strong interaction between the two. The interfacial interaction facilitated electron transfer and the NRR process.177 A similar effect was also observed for the gold clusters supported on the h-BN/Au(111) electrodes towards HER.178
Compared with carbon-based supports, the BN layer shows strong corrosion resistance at high potential, which is crucial for the structural stability and catalytic stability of materials. This also implies the potential application of BN in industrial catalysis, which often run at elevated temperature and voltage.
5.3 MXene support
Layered transition metal carbides and nitrides can be collected into a large class of layered ternary compounds, Mn+1AXn with hexagonal symmetry and space group P63/mmc. As revealed by the chemical formula, “M” is an early transition metal, “A” is a main group element (usually Al, Si, Sn, and Ga), “X” is C and/or N, and n can be 1, 2 and 3.179 The MAX structure is quite stable for the existence of metallic, covalent and ionic bonding in the structure. While the A layer has relatively weak bonds and is more reactive to be etched away by HF to produce MXene. After the Al layer is etched away, MXene presents clearly separated layers with accordion-like morphology (Fig. 9a and b). Due to good electrical and thermally conductivity, excellent ductility and hydrophilicity, and inherent low capacities of the layered structure, the MXene family has been growing rapidly since the first inception in 2011.181 The MAX phases have generated more than 70 members of ternary, layered, machinable transition metal carbides, nitrides, and carbonitrides. The component elements cover Ti, Al, C, Ta, Nb, V, Cr, Zr, Hf, etc. Among them, Ti3AlC2 is the most widely studied and becomes one of the most promising members in the energy conversion field.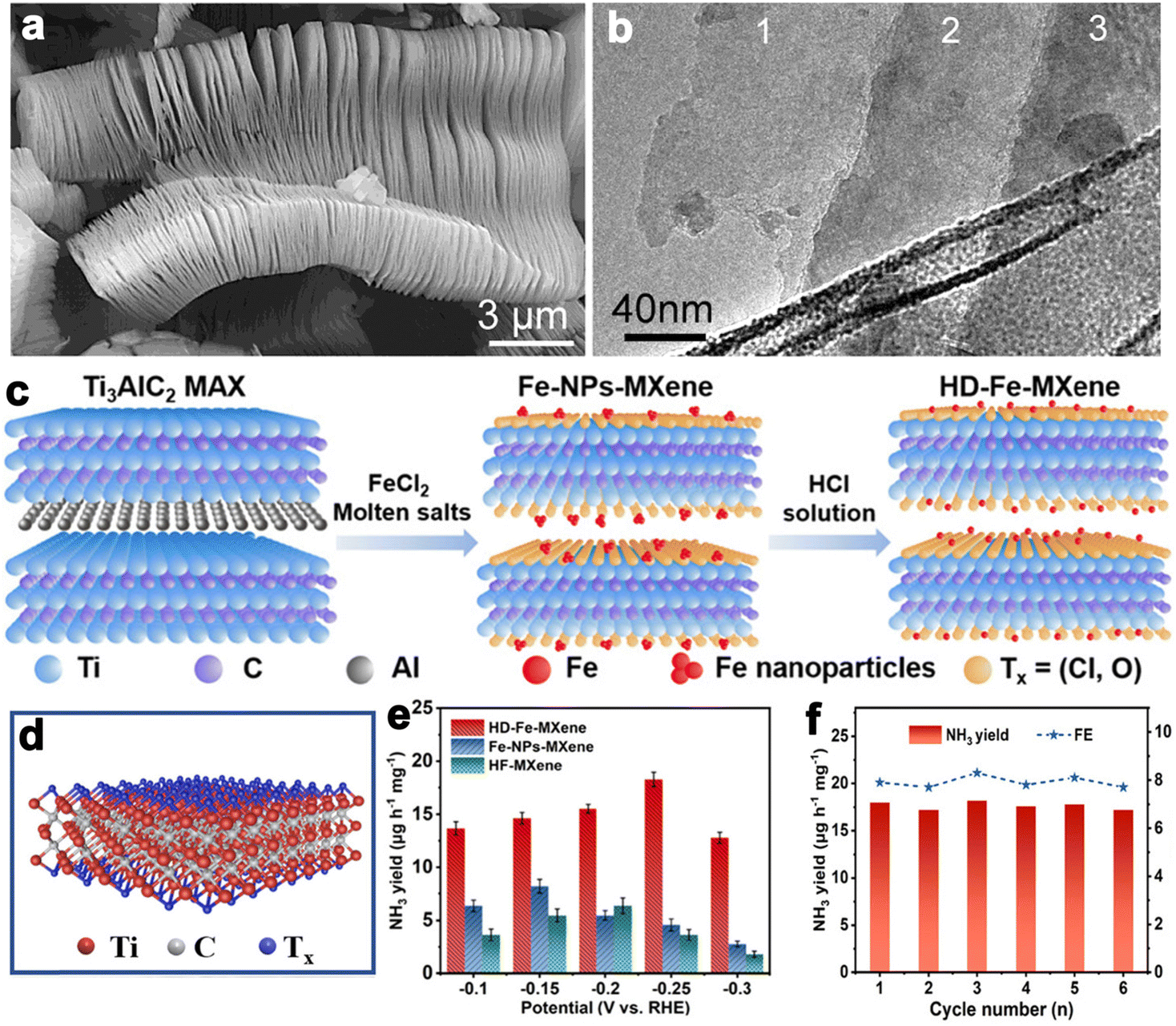 | ||
| Fig. 9 (a and b) SEM and TEM images of Ti3AlC2 layers (reproduced from ref. 179 with permission from the American Chemical Society, copyright 2012).179 (c) Schematic synthesis process of HD-Fe-MXene. (d) Crystal structure of Ti3C2Tx MXene. (e) NH3 yields for the samples at different potentials. (f) NH3 yields of HD-Fe-MXene and FEs during six cycles at −0.25 V versus RHE in 0.1 M Na2SO4 (reproduced from ref. 180 with permission from Wiley, copyright 2022).180 | ||
After the removal of the A layer, MXene displays a unique 2D layered structure with abundant functional groups on the surface such as –O, –F, –Cl, etc. This permits flexibility in further enriching their functionalities, making them remarkable catalytic electrodes. It is found that the material properties are closely related to the following factors, such as surface compositions, microstructures, and chemistry states of MXene.182 Strategies to optimize material properties are proposed accordingly (i) surface functional groups tailoring; (ii) surface/interface engineering; (iii) heterostructure construction.
MXene is generally prepared by etching a layer in a liquid phase reaction, leaving abundant functional groups (–Cl, –F, –O, –OH, –H) on the surface and endowing them with unique surface hydrophilicity and adsorptive property. Surface chemical properties, especially terminal functional groups play a significant role in activity modeling. By regulating the type of surface terminations on MXene, the catalytic performance of specific reactions can be selectively improved. For instance, –O terminations are deemed to facilitate HER, while –F terminations deteriorate HER but are favorable to NRR.183 Wang et al. found that introducing TiOF2 nanospheres on the surface of Ti3C2Ox–MXene could effectively stabilize the oxygen-containing terminals on the surface of the substrate and showed excellent oxidation resistance and HER catalytic activity.184 In another report, Gao et al. indicated that O-terminals could act as the HER active sites as revealed by the near zero Gibbs free energy (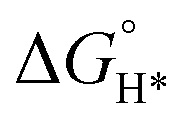 ) of the terminal O atom site.183 While some researchers claimed that surface F terminals were also detrimental to NRR because they covered active sites. The Zhi group prepared a highly active NRR catalyst by ironing out inactive F*/OH* terminals and introducing Fe to greatly reduce the surface work function.185 Similarly, Ma and co-workers also dispersed Fe in the fluorine-free Ti3C2Tx by immobilizing Fe nanoclusters from aggregation and exposing abundant active sites as illustrated in Fig. 9c. After exfoliation, Ti3C2Tx MXene inherited the hexagonal close-packed structure (Fig. 9d). In addition, the HD-Fe-MXene delivered outstanding NRR activity with an FE of 21.8% and good stability for 6 cycles (Fig. 9e and f).180
) of the terminal O atom site.183 While some researchers claimed that surface F terminals were also detrimental to NRR because they covered active sites. The Zhi group prepared a highly active NRR catalyst by ironing out inactive F*/OH* terminals and introducing Fe to greatly reduce the surface work function.185 Similarly, Ma and co-workers also dispersed Fe in the fluorine-free Ti3C2Tx by immobilizing Fe nanoclusters from aggregation and exposing abundant active sites as illustrated in Fig. 9c. After exfoliation, Ti3C2Tx MXene inherited the hexagonal close-packed structure (Fig. 9d). In addition, the HD-Fe-MXene delivered outstanding NRR activity with an FE of 21.8% and good stability for 6 cycles (Fig. 9e and f).180
Microstructural control is effective in exposing active sites and modulating the electronic structure and hence the adsorption ability. Qiao et al. constructed a hybrid film composed of overlapped g-C3N4 and Ti3C2 nanosheets through homogeneous assembly. The free-standing flexible films with porous architecture and high surface area delivered high OER catalytic activity comparable to precious metals benefiting from the coupling of the components through Ti–Nx interaction.186 Zhang et al. reported highly efficient cobalt-tipped carbon nanotube/Ti3C2 nanosheet composites (Co–CNT/Ti3C2) for ORR through in situ growth of ZIF-67 particles on Ti3C2 nanosheets. The incorporation of Co–CNT brought about abundant active Co–N/C sites, a high degree of graphitization, and a large surface area of the Co–CNT/Ti3C2 samples, making it an efficient electrocatalyst.187
From the above discussion, modeling MXene with appropriate termination and interfacial interaction is crucial to predict and optimize their chemistry and activity. More trials are needed in this direction.
Developing efficient techniques to support the catalytic species on 2D nanomaterials is a promising way to obtain well-dispersed nanoparticles with high surface exposure and low usage. Some classical supporting strategies such as impregnation, alcohol reduction, and liquid phase reduction can be extended to support catalysts on 2D nanomaterials.188,189 In addition, the stability and utilization of catalysts can be regulated by modulating the crystallinity, size, and dispersibility of catalytic active phases, as well as electronic metal–support interaction. The investigation of the electronic interactions between active species and supports needs to be stressed. Further, the catalytic mechanism and evolution of active sites can be revealed with the help of in situ spectroscopy methods and isotope tracing techniques with a combination of first-principles calculations.190,191
6. Outlook and conclusion
2D materials, showing fascinating physical and chemical properties that are distinctly different from their 3D counterparts, have received tremendous research enthusiasm since their discovery. The emergence and advance of 2D materials have not only enriched our understanding of nanoscience, speeded up investigations in the fields of nanomaterials and catalysis, but also given birth to many new interdisciplinary disciplines and promoted the development of many emerging disciplines. Especially in recent years, with the development of nanoscience and theoretical computing technology, two-dimensional materials play an increasingly important role in the field of catalysis. To further prosper the design, synthesis, and application of these 2D materials, the following issues still need to be addressed.(i) Developing highly controllable synthetic methods to realize high-yield and massive production of ultrathin 2D nanomaterials. At present, almost all 2D materials are still prepared at laboratory level, while one of the basic requirements for the practical application of materials is large-scale production at low cost, especially for those with metastable phases. For example, the studies on layered IrO2 (e.g., 1T-IrO2, 3R-IrO2) have revealed that the meta-stable phases have high intrinsic activity toward oxygen evolution when compared to common rutile IrO2.88,192 However, production of ultrathin 2D layered IrO2 is still far from the criteria that are required for industry. Developing scalable and controllable preparation methodology is still an arduous task. In addition, the current well-developed exfoliation of layered materials is mainly achieved in the liquid phase, which is often low-yield and high-cost. Experimental methods for 2D materials preparation with precise control of high quality and predefined numbers of layers need to be developed. Nanosheets with definite composition and structure offer an ideal platform for theoretical investigation, which will deepen our understanding of basic physics and intrigue unexpected phenomena and applications.
(ii) Extending the electrochemical application of 2D layered materials. More than 1000 kinds of layered materials have been discovered from the crystal structure database, but only a small portion has been explored for electrocatalytic applications. Many newly emerging conductive 2D materials, such as siloxane and germanene, with graphene-like bonding mode and stacking mode, have broadened the range of the 2D family. Lately, several reports indicated the potential of siloxane to act as excellent support of noble metal catalysts for hydrogen and oxygen evolution through topotactic transformation of layered CaSi2.193 Exploiting exfoliation techniques to convert silicide, germanide into corresponding siloxane and germanene efficiently is a cue for more potential electrochemical applications. Further, research on the application of layered iridium oxides (e.g., layered perovskite structure and layered honeycomb structure)194 for oxygen evolution reaction in an acidic environment has implied that 2D layered iridium oxides have unique synergistic advantages in balancing high activity and high structural stability. These advances will trigger extensive exploration interest for more opportunities in the field of electrocatalysis.
(iii) Stabilizing the exfoliated 2D layered nanosheets. The exfoliated ultrathin 2D layered nanosheets have a tendency to irreversible aggregation that makes them hard to store and thus lose the merits originating from 2D structure, which is challengeable to catalysis. Besides, most layered nanosheets are prone to oxidization in ambient conditions, leading to structural damage.195 When involved in catalysis, as substrates or active phases, 2D materials may go through structural evolution, especially under oxidative environments. The exploration of simple but reliable methods to stabilize these ultrathin 2D nanomaterials to dramatically prolong their stability is of vital importance.
(iv) Strengthening mechanism study of layered catalysts. Understanding the electrocatalytic mechanism of 2D nanomaterials is significantly important, but it is not easy. Fortunately, many advanced in situ characterization techniques such as in situ TEM, in situ XPS, in situ Raman spectroscopy, and in situ electrochemical mass spectrometry have been employed to track the electrocatalytic reaction processes.196,197 Moreover, the structure variations of some layered catalysts are a complex process involving many chemical reactions. For example, layered metal non-oxides are easily oxidized to corresponding metal oxides/(oxy)hydroxides, giving birth to new structures to act as real active OER catalysts.198 It is urgent to uncover the mechanism behind the phenomenon and explore factors that influence catalytic activity and stability. Structural stability evaluation and structural stability–activity relationships can be established in future investigations. Further, taking advantage of these characteristics to guide the design of novel layered catalysts with desired properties is feasible.
Conflicts of interest
There is no conflicts to declare.Acknowledgements
X. He thanks the Natural Science Foundation of Jilin Province, China (Grant No. 20210101120JC) for the financial support. M. Fan thanks the Science and Technology Research Program of the Education Department of Jilin Province, China (No. JJKH20230828KJ) for the financial support.Notes and references
- J. Lee, B. Jeong and J. D. Ocon, Curr. Appl. Phys., 2013, 13, 309–321 CrossRef.
- M. Fan, L. Cui, X. He and X. Zou, Small Methods, 2022, 6, 2200855 CrossRef CAS PubMed.
- Y. Wang, J. Li and Z. Wei, J. Mater. Chem. A, 2018, 6, 8194–8209 RSC.
- Y. Miao, L. Ouyang, S. Zhou, L. Xu, Z. Yang, M. Xiao and R. Ouyang, Biosens. Bioelectron., 2014, 53, 428–439 CrossRef CAS PubMed.
- H. Chen, Y. Liu, B. Zhang and X. Zou, Pure Appl. Chem., 2021, 93, 1411–1421 CrossRef CAS.
- F. Yu, L. Yu, I. K. Mishra, Y. Yu, Z. F. Ren and H. Q. Zhou, Mater. Today Phys., 2018, 7, 121–138 CrossRef.
- M. Fang, W. Gao, G. Dong, Z. Xia, S. Yip, Y. Qin, Y. Qu and J. C. Ho, Nano Energy, 2016, 27, 247–254 CrossRef CAS.
- J. Guo, C. Lin, C. Jiang and P. Zhang, Appl. Surf. Sci., 2019, 475, 237–255 CrossRef CAS.
- B. Wang, X. Cui, J. Huang, R. Cao and Q. Zhang, Chin. Chem. Lett., 2018, 29, 1757–1767 CrossRef CAS.
- C. Zhao, D. Li and Y. Feng, J. Mater. Chem. A, 2013, 1, 5741–5746 RSC.
- X. Long, W. Qiu, Z. Wang, Y. Wang and S. Yang, Mater. Today Chem., 2019, 11, 16–28 CrossRef CAS.
- F. H. Horn, Nature, 1952, 170, 581–581 CrossRef.
- F. W. Fenning and F. R. Holt, Nature, 1950, 165, 722–723 CrossRef CAS PubMed.
- B. R. Shaw, Y. Deng, F. E. Strillacci, K. A. Carrado and M. G. Fessehaie, J. Electrochem. Soc., 1990, 137, 3136–3143 CrossRef CAS.
- M. Jaksic, Int. J. Hydrogen Energy, 1987, 12, 727–752 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- C.-H. Lin, H.-C. Fu, B. Cheng, M.-L. Tsai, W. Luo, L. Zhou, S.-H. Jang, L. Hu and J.-H. He, npj 2D Mater. Appl., 2018, 2, 23 CrossRef.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- M. Naguib, J. Come, B. Dyatkin, V. Presser, P.-L. Taberna, P. Simon, M. W. Barsoum and Y. Gogotsi, Electrochem. Commun., 2012, 16, 61–64 CrossRef CAS.
- H. Oughaddou, H. Enriquez, M. R. Tchalala, H. Yildirim, A. J. Mayne, A. Bendounan, G. Dujardin, M. A. Ali and A. Kara, Prog. Surf. Sci., 2015, 90, 46–83 CrossRef CAS.
- S. Suzuki, T. Iwasaki, K. K. H. D. Silva, S. Suehara, K. Watanabe, T. Taniguchi, S. Moriyama, M. Yoshimura, T. Aizawa and T. Nakayama, Adv. Funct. Mater., 2021, 31, 2007038 CrossRef CAS.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 17033 CrossRef CAS.
- Y. Li, Z. Li, C. Chi, H. Shan, L. Zheng and Z. Fang, Adv. Sci., 2017, 4, 1600430 CrossRef PubMed.
- C. Tan, X. Cao, X. J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G. H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- P. Chen, Y. Tong, C. Wu and Y. Xie, Acc. Chem. Res., 2018, 51, 2857–2866 CrossRef CAS PubMed.
- X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909–921 CrossRef CAS.
- N. Karmodak and O. Andreussi, ACS Energy Lett., 2020, 5, 885–891 CrossRef CAS.
- W. Peng, M. Luo, X. Xu, K. Jiang, M. Peng, D. Chen, T. S. Chan and Y. Tan, Adv. Energy Mater., 2020, 10, 2001364 CrossRef CAS.
- X. Qiu, X. Zhang and L.-Z. Fan, J. Mater. Chem. A, 2018, 6, 16186–16195 RSC.
- H. Lee, K. Paeng and I. S. Kim, Synth. Met., 2018, 244, 36–47 CrossRef CAS.
- A. Pakdel, C. Zhi, Y. Bando and D. Golberg, Mater. Today, 2012, 15, 256–265 CrossRef CAS.
- Z. Zhao, H. Zhang, H. Yuan, S. Wang, Y. Lin, Q. Zeng, G. Xu, Z. Liu, G. K. Solanki, K. D. Patel, Y. Cui, H. Y. Hwang and W. L. Mao, Nat. Commun., 2015, 6, 7312 CrossRef CAS PubMed.
- R. F. Frindt, J. Appl. Phys., 1996, 37, 1928–1929 CrossRef.
- R. F. Frindt, Phys. Rev. Lett., 1972, 28, 299–301 CrossRef CAS.
- K.-y. Chen, W.-x. Zhang, Y. Liu, H.-p. Zhu, J. Duan, X.-h. Xiang, L.-h. Xue and Y.-h. Huang, Chem. Commun., 2015, 51, 1608–1611 RSC.
- Y.-S. Han, I. Park and J.-H. Choy, J. Mater. Chem., 2001, 11, 1277–1282 RSC.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 12, 41–58 RSC.
- D. Arney and P. A. Maggard, ACS Catal., 2012, 2, 1711–1717 CrossRef CAS.
- R. Li, L. Liu, B. Ming, Y. Ji and R. Wang, Appl. Surf. Sci., 2018, 439, 983–990 CrossRef CAS.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- Z. Lin, Y. Liu, U. Halim, M. Ding, Y. Liu, Y. Wang, C. Jia, P. Chen, X. Duan, C. Wang, F. Song, M. Li, C. Wan, Y. Huang and X. Duan, Nat. Biotechnol., 2018, 562, 254–258 CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- K.-H. Goh, T.-T. Lim and Z. Dong, Water Res., 2008, 42, 1343–1368 CrossRef CAS PubMed.
- M. F. P. Duarte, I. M. Rocha, J. L. Figueiredo, C. Freire and M. F. R. Pereira, Catal. Today, 2018, 301, 17–24 CrossRef CAS.
- C. Xu, L. Wang, Z. Liu, L. Chen, J. Guo, N. Kang, X. L. Ma, H. M. Cheng and W. Ren, Nat. Mater., 2015, 14, 1135–1141 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- M. Yi and Z. Shen, J. Mater. Chem. A, 2015, 3, 11700–11715 RSC.
- Y. Huang, E. Sutter, N. N. Shi, J. Zheng, T. Yang, D. Englund, H. J. Gao and P. Sutter, ACS Nano, 2015, 9, 10612–10620 CrossRef CAS PubMed.
- S. B. Desai, S. R. Madhvapathy, M. Amani, D. Kiriya, M. Hettick, M. Tosun, Y. Zhou, M. Dubey, J. W. Ager, 3rd, D. Chrzan and A. Javey, Adv. Mater., 2016, 28, 4053–4058 CrossRef CAS PubMed.
- P. Süle, M. Szendrő, G. Z. Magda, C. Hwang and L. Tapasztó, Nano Lett., 2015, 15, 8295–8299 CrossRef PubMed.
- F. Wu, Z. Liu, N. Hawthorne, M. Chandross, Q. Moore, N. Argibay, J. F. Curry and J. D. Batteas, ACS Nano, 2020, 14, 16939–16950 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 2002, 80, 1339–1339 CrossRef.
- H.-g. Jee, J.-H. Han, H.-N. Hwang, B. Kim, H.-s. Kim, Y. D. Kim and C.-C. Hwang, Appl. Phys. Lett., 2009, 95, 093107 CrossRef.
- D. Hanlon, C. Backes, E. Doherty, C. S. Cucinotta, N. C. Berner, C. Boland, K. Lee, A. Harvey, P. Lynch, Z. Gholamvand, S. Zhang, K. Wang, G. Moynihan, A. Pokle, Q. M. Ramasse, N. McEvoy, W. J. Blau, J. Wang, G. Abellan, F. Hauke, A. Hirsch, S. Sanvito, D. D. O'Regan, G. S. Duesberg, V. Nicolosi and J. N. Coleman, Nat. Commun., 2015, 6, 8563 CrossRef CAS PubMed.
- S. Sinha, T. Zhu, A. France-Lanord, Y. Sheng, J. C. Grossman, K. Porfyrakis and J. H. Warner, Nat. Commun., 2020, 11, 823 CrossRef CAS PubMed.
- Y. Jung, Y. Zhou and J. J. Cha, Inorg. Chem. Front., 2016, 3, 452 RSC.
- L. Tian, H. Qiao, Z. Huang and X. Qi, Cryst. Res. Technol., 2021, 56, 2000165 CrossRef CAS.
- M. Adachi-Pagano, C. Forano and J.-P. Besse, Chem. Commun., 2000, 7, 91–92 RSC.
- J. Yu, J. Li, W. Zhang and H. Chang, Chem. Sci., 2015, 6, 6705–6716 RSC.
- H. Zheng, R. K. Smith, Y. W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS PubMed.
- W. H. Park, I. Jo, B. H. Hong and H. Cheong, Nanoscale, 2016, 8, 9822–9827 RSC.
- S. G. Ramaraj, M. Muruganathan, O. G. Agbonlahor, H. Maki, Y. Onda, M. Hattori and H. Mizuta, Carbon, 2022, 190, 359–365 CrossRef CAS.
- S. Jia, W. Chen, J. Zhang, C. Y. Lin, H. Guo, G. Lu, K. Li, T. Zhai, Q. Ai and J. Lou, Mater. Today Nano, 2021, 16, 100135 CrossRef CAS.
- Y. Zhang, Y. Yao, M. G. Sendeku, L. Yin, X. Zhan, F. Wang, Z. Wang and J. He, Adv. Mater., 2019, 31, e1901694 CrossRef PubMed.
- X. Xin, J. Chen, Y. Zhang, M. L. Chen, Y. Bao, W. Liu, Y. Liu, H. Xu and W. Ren, Nanoscale Horiz., 2022, 7, 743–751 RSC.
- D. Wei, Y. Lu, C. Han, T. Niu, W. Chen and A. T. Wee, Angew. Chem., Int. Ed., 2013, 52, 14121–14126 CrossRef CAS PubMed.
- D. Wei, L. Peng, M. Li, H. Mao, T. Niu, C. Han, W. Chen and A. T. Wee, ACS Nano, 2015, 9, 164–171 CrossRef CAS PubMed.
- D. Andrzejewski, M. Marx, A. Grundmann, O. Pfingsten, H. Kalisch, A. Vescan, M. Heuken, T. Kümmell and G. Bacher, Nanotechnology, 2018, 29, 295704 CrossRef CAS PubMed.
- M. J. Molaei, M. Younas and M. Rezakazemi, ACS Appl. Electron. Mater., 2021, 3, 5165–5187 CrossRef CAS.
- C. Muratore, A. A. Voevodin and N. R. Glavin, Thin Solid Films, 2019, 688, 137500 CrossRef CAS.
- H. Zhang, Y. Zhou, Y. Ma, J. Yao, X. Li, Y. Sun, Z. Xiong and D. Li, J. Alloys Compd., 2018, 740, 174–179 CrossRef CAS.
- K. Jagannadham, J. Cui and Y. Zhu, J. Electron. Mater., 2017, 46, 1010–1021 CrossRef CAS.
- K. Huang, Z. Li, J. Lin, G. Han and P. Huang, Chem. Soc. Rev., 2018, 47, 5109–5124 RSC.
- B. Raveau, J. Eur. Ceram. Soc., 2005, 25, 1965–1969 CrossRef CAS.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 12, 41–58 RSC.
- R. Ma and T. Sasaki, Acc. Chem. Res., 2015, 48, 136–143 CrossRef CAS PubMed.
- Z. Ma, H. Xu, Y. Liu, Q. Zhang, M. Wang, Y. Lin, Z. Li, X. He, J. Sun, R. Jiang, Z. Lei, Q. Li, L. Yang and Z.-h. Liu, J. Mater. Chem. A, 2022, 10, 24216–24225 RSC.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Energy Environ. Sci., 2013, 6, 2745 RSC.
- R. K. Hocking, R. Brimblecombe, L. Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Nat. Chem., 2011, 3, 461–466 CrossRef CAS PubMed.
- J. Gao, Y. Liu, B. Liu and K. W. Huang, ACS Nano, 2022, 16, 17761–17777 CrossRef CAS PubMed.
- Q. Zhang, H. Chen, L. Yang, X. Liang, L. Shi, Q. Feng, Y. Zou, G.-D. Li and X. Zou, Chin. J. Catal., 2022, 43, 885–893 CrossRef CAS.
- D. Weber, L. M. Schoop, D. Wurmbrand, S. Laha, F. Podjaski, V. Duppel, K. Müller, U. Starke and B. V. Lotsch, J. Mater. Chem. A, 2018, 6, 21558–21566 RSC.
- A. K. Stephan, Joule, 2021, 5, 1–2 CrossRef.
- Q. Dang, H. Lin, Z. Fan, L. Ma, Q. Shao, Y. Ji, F. Zheng, S. Geng, S. Z. Yang, N. Kong, W. Zhu, Y. Li, F. Liao, X. Huang and M. Shao, Nat. Commun., 2021, 12, 6007 CrossRef CAS PubMed.
- A. Kumar, A. Kumar and V. Krishnan, ACS Catal., 2020, 10, 10253–10315 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- R. Zhang, P. E. Pearce, V. Pimenta, J. Cabana, H. Li, D. Alves Dalla Corte, A. M. Abakumov, G. Rousse, D. Giaume, M. Deschamps and A. Grimaud, Chem. Mater., 2020, 32, 3499–3509 CrossRef CAS.
- S. Liu, Y. Zhang, X. Mao, L. Li, Y. Zhang, L. Li, Y. Pan, X. Li, L. Wang, Q. Shao, Y. Xu and X. Huang, Energy Environ. Sci., 2022, 15, 1672–1681 RSC.
- H. Chen, L. Shi, K. Sun, K. Zhang, Q. Liu, J. Ge, X. Liang, B. Tian, Y. Huang, Z. Shi, Z. Wang, W. Zhang, M. Liu and X. Zou, ACS Catal., 2022, 12, 8658–8666 CrossRef CAS.
- A. Kuc, N. Zibouche and T. Heine, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 245213 CrossRef.
- R. R. Chianelli, A. F. Ruppert, M. José-Yacamán and A. Vázquez Zavala, Catal. Today, 1995, 23, 269–281 CrossRef CAS.
- H. Bao, Y. Huang, F. Ma, Z. Yang, Y. Miao, K. Xu and P. K. Chu, J. Mater. Sci., 2016, 51, 6850–6859 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jorgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- X. Gao, Y. Zhou, Z. Cheng, Y. Tan, T. Yuan and Z. Shen, Appl. Surf. Sci., 2021, 547, 149113 CrossRef CAS.
- R. Zhang, M. Zhang, H. Yang, G. Li, S. Xing, M. Li, Y. Xu, Q. Zhang, S. Hu, H. Liao and Y. Cao, Small Methods, 2021, 5, e2100612 CrossRef PubMed.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515 RSC.
- S. Geng, F. Tian, M. Li, Y. Liu, J. Sheng, W. Yang, Y. Yu and Y. Hou, Nano Res., 2021, 15, 1809–1816 CrossRef.
- L. Zhao, C. Hong, L. Lin, H. Wu, Y. Su, X. Zhang and A. Liu, Carbon, 2017, 116, 223–231 CrossRef CAS.
- S. Y. Cho, S. J. Kim, Y. Lee, J. S. Kim, W. B. Jung, H. W. Yoo, J. Kim and H. T. Jung, ACS Nano, 2015, 9, 9314–9321 CrossRef PubMed.
- Y. Wan, Z. Zhang, X. Xu, Z. Zhang, P. Li, X. Fang, K. Zhang, K. Yuan, K. Liu, G. Ran, Y. Li, Y. Ye and L. Dai, Nano Energy, 2018, 51, 786–792 CrossRef CAS.
- J. Kibsgaard, Z. Chen, B. N. Reinecke and T. F. Jaramillo, Nat. Mater., 2012, 11, 963–969 CrossRef CAS PubMed.
- Y. Yu, S. Y. Huang, Y. Li, S. N. Steinmann, W. Yang and L. Cao, Nano Lett., 2014, 14, 553–558 CrossRef CAS PubMed.
- C. C. Mayorga-Martinez, Z. Sofer, D. Sedmidubsky, S. Huber, A. Y. Eng and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 12563–12573 CrossRef CAS PubMed.
- X. Zhang, Z. Luo, P. Yu, Y. Cai, Y. Du, D. Wu, S. Gao, C. Tan, Z. Li, M. Ren, T. Osipowicz, S. Chen, Z. Jiang, J. Li, Y. Huang, J. Yang, Y. Chen, C. Y. Ang, Y. Zhao, P. Wang, L. Song, X. Wu, Z. Liu, A. Borgna and H. Zhang, Nat. Catal., 2018, 1, 460–468 CrossRef CAS.
- J. Wang, X. Li, B. Wei, R. Sun, W. Yu, H. Y. Hoh, H. Xu, J. Li, X. Ge, Z. Chen, C. Su and Z. Wang, Adv. Funct. Mater., 2020, 30, 1908708 CrossRef CAS.
- S. Li, C. Cheng, H. W. Liang, X. Feng and A. Thomas, Adv. Mater., 2017, 29, 1700707 CrossRef PubMed.
- Y. Yao, Z. Jin, Y. Chen, Z. Gao, J. Yan, H. Liu, J. Wang, Y. Li and S. Liu, Carbon, 2018, 129, 228–235 CrossRef CAS.
- G. Shi, C. Yu, Z. Fan, J. Li and M. Yuan, ACS Appl. Mater. Interfaces, 2019, 11, 2662–2669 CrossRef CAS PubMed.
- Y. Wang, D. Yan, S. El Hankari, Y. Zou and S. Wang, Adv. Sci., 2018, 5, 1800064 CrossRef PubMed.
- J. Xiong, J. Di and H. Li, Adv. Sci., 2018, 5, 1800244 CrossRef PubMed.
- X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909–921 CrossRef CAS.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- J. Di, C. Yan, A. D. Handoko, Z. W. Seh, H. Li and Z. Liu, Mater. Today, 2018, 21, 749–770 CrossRef CAS.
- J. Liu, Q. Ma, Z. Huang, G. Liu and H. Zhang, Adv. Mater., 2019, 31, e1800696 CrossRef PubMed.
- C. Wang and D. Astruc, Prog. Mater. Sci., 2018, 94, 306–383 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed.
- U. Shamraiz, Z. Ahmad, B. Raza, A. Badshah, S. Ullah and M. A. Nadeem, ACS Appl. Mater. Interfaces, 2020, 12, 4396–4404 CrossRef CAS PubMed.
- X. Song, Q. Shi, H. Wang, S. Liu, C. Tai and Z. Bian, Appl. Catal., B, 2017, 203, 442–451 CrossRef CAS.
- C. V. Rao, A. L. M. Reddy, Y. Ishikawa and P. M. Ajayan, Carbon, 2011, 49, 931–936 CrossRef CAS.
- H. Xu, B. Yan, S. Li, J. Wang, C. Wang, J. Guo and Y. Du, Chem. Eng. J., 2018, 334, 2638–2646 CrossRef CAS.
- H. Huang, J. Zhu, D. Li, C. Shen, M. Li, X. Zhang, Q. Jiang, J. Zhang and Y. Wu, J. Mater. Chem. A, 2017, 5, 4560–4567 RSC.
- J. Huang, S. B. Scott, I. Chorkendorff and Z. Wen, ACS Catal., 2021, 11, 12745–12753 CrossRef CAS.
- M. Li, C. Yan, R. Ramachandran, Y. Lan, H. Dai, H. Shan, X. Meng, D. Cui, F. Wang and Z.-X. Xu, Chem. Eng. J., 2022, 430, 133050 CrossRef CAS.
- M. Prabu, P. Ramakrishnan, H. Nara, T. Momma, T. Osaka and S. Shanmugam, ACS Appl. Mater. Interfaces, 2014, 6, 16545–16555 CrossRef CAS PubMed.
- Y. Liu, H. Jiang, J. Hao, Y. Liu, H. Shen, W. Li and J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 31841–31852 CrossRef CAS PubMed.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 RSC.
- X. Hu, T. Huang, Y. Tang, G. Fu and J. M. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 4028–4036 CrossRef CAS PubMed.
- Z. Zhao, Z. Liu, A. Zhang, X. Yan, W. Xue, B. Peng, H. L. Xin, X. Pan, X. Duan and Y. Huang, Nat. Nanotechnol., 2022, 17, 968–975 CrossRef CAS PubMed.
- Q. Liu, Q. Zhang, W. Shi, H. Hu, J. Zhuang and X. Wang, Nat. Chem., 2022, 14, 433–440 CrossRef CAS PubMed.
- D. Franz, U. Schroder, R. Shayduk, B. Arndt, H. Noei, V. Vonk, T. Michely and A. Stierle, ACS Nano, 2021, 15, 15771–15780 CrossRef CAS PubMed.
- Z. Jia, M. Peng, X. Cai, Y. Chen, X. Chen, F. Huang, L. Zhao, J. Diao, N. Wang, D. Xiao, X. Wen, Z. Jiang, H. Liu and D. Ma, ACS Catal., 2022, 12, 9602–9610 CrossRef CAS.
- J. Lei, X. X. Fan, T. Liu, P. Xu, Q. Hou, K. Li, R. M. Yuan, M. S. Zheng, Q. F. Dong and J. J. Chen, Nat. Commun., 2022, 13, 202 CrossRef CAS PubMed.
- L. Yang, G. Li, R. Ma, S. Hou, J. Chang, M. Ruan, W. Cai, Z. Jin, W. Xu, G. Wang, J. Ge, C. Liu and W. Xing, Nano Res., 2021, 14, 2853–2860 CrossRef CAS.
- X. Chen, H. Zhu, J. Zhu and H. Zhang, Chem. Eng. J., 2023, 451, 138998 CrossRef CAS.
- Y.-C. Han, J. Yi, B. Pang, N. Wang, X.-C. Li, T. Yao, K. S. Novoselov and Z.-Q. Tian, Natl. Sci. Rev., 2023 DOI:10.1093/nsr/nwad081.
- X. Wei, Y. Liu, X. Zhu, S. Bo, L. Xiao, C. Chen, T. T. T. Nga, Y. He, M. Qiu, C. Xie, D. Wang, Q. Liu, F. Dong, C. L. Dong, X. Z. Fu and S. Wang, Adv. Mater., 2023, 35, e2300020 CrossRef PubMed.
- D. Huang, D. J. Kim, K. Rigby, X. Zhou, X. Wu, A. Meese, J. Niu, E. Stavitski and J. H. Kim, Environ. Sci. Technol., 2021, 55, 13306–13316 CAS.
- S. Zhou, L. Shang, Y. Zhao, R. Shi, G. I. N. Waterhouse, Y. C. Huang, L. Zheng and T. Zhang, Adv. Mater., 2019, 31, e1900509 CrossRef PubMed.
- Z. Sun, Y. Yang, C. Fang, Y. Yao, F. Qin, H. Gu, Q. Liu, W. Xu, H. Tang, Z. Jiang, B. Ge, W. Chen and Z. Chen, Small, 2022, 18, e2203422 CrossRef PubMed.
- S. Liu and S. Huang, Carbon, 2017, 115, 11–17 CrossRef CAS.
- X. Song, N. Li, H. Zhang, H. Wang, L. Wang and Z. Bian, J. Power Sources, 2019, 435, 226771 CrossRef CAS.
- D. Yang, J.-H. Yang, Y.-P. Yang and Z.-Y. Liu, Appl. Catal., B, 2023, 326, 122402 CrossRef CAS.
- D. Wu, P. Lv, J. Wu, B. He, X. Li, K. Chu, Y. Jia and D. Ma, J. Mater. Chem. A, 2023, 11, 1817–1828 RSC.
- X. Qiu, X. Yan, H. Pang, J. Wang, D. Sun, S. Wei, L. Xu and Y. Tang, Adv. Sci., 2019, 6, 1801103 CrossRef PubMed.
- P. Kumar, K. Kannimuthu, A. S. Zeraati, S. Roy, X. Wang, X. Wang, S. Samanta, K. A. Miller, M. Molina, D. Trivedi, J. Abed, M. A. Campos Mata, H. Al-Mahayni, J. Baltrusaitis, G. Shimizu, Y. A. Wu, A. Seifitokaldani, E. H. Sargent, P. M. Ajayan, J. Hu and M. G. Kibria, J. Am. Chem. Soc., 2023, 145, 8052–8063 CrossRef CAS PubMed.
- X. Song, N. Li, H. Zhang, L. Wang, Y. Yan, H. Wang, L. Wang and Z. Bian, ACS Appl. Mater. Interfaces, 2020, 12, 17519–17527 CrossRef CAS PubMed.
- X. Zhao, X. Liu, B. Huang, P. Wang and Y. Pei, J. Mater. Chem. A, 2019, 7, 24583–24593 RSC.
- G. Han, Y. Zheng, X. Zhang, Z. Wang, Y. Gong, C. Du, M. N. Banis, Y.-M. Yiu, T.-K. Sham, L. Gu, Y. Sun, Y. Wang, J. Wang, Y. Gao, G. Yin and X. Sun, Nano Energy, 2019, 66, 104088 CrossRef CAS.
- C. Chen, W. Luo, H. Li, T. Hu, Y. Zhao, Z. Zhao, X. Sun, H. Zai, Y. Qi, M. Wu, Y. Dong, J. Dong, W. Chen, X. Ke, M. Sui, L. Zhang, Q. Chen, Z. Wang, E. Zhu, Y. Li and Y. Huang, Chem. Mater., 2021, 33, 3639–3649 CrossRef.
- S. S. Li, J. J. Lv, L. N. Teng, A. J. Wang, J. R. Chen and J. J. Feng, ACS Appl. Mater. Interfaces, 2014, 6, 10549–10555 CrossRef CAS PubMed.
- X. Zhang, F. Meng, S. Mao, Q. Ding, M. J. Shearer, M. S. Faber, J. Chen, R. J. Hamers and S. Jin, Energy Environ. Sci., 2015, 8, 862–868 RSC.
- P. Gnanaprakasam, S. E. Jeena and T. Selvaraju, J. Mater. Chem. A, 2015, 3, 18010–18018 RSC.
- B. Xiong, Y. K. Zhou, Y. Y. Zhao, J. Wang, X. Chen, R. O. Hayre and Z. P. Shao, Carbon, 2013, 52, 181–192 CrossRef CAS.
- G. Zhang, G. Wang, Y. Liu, H. Liu, J. Qu and J. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef CAS PubMed.
- T. H. Tan, J. Scott, Y. H. Ng, R. A. Taylor, K.-F. Aguey-Zinsou and R. Amal, ACS Catal., 2016, 6, 8021–8029 CrossRef CAS.
- Z. Zhao and Z. Xia, ACS Catal., 2016, 6, 1553–1558 CrossRef CAS.
- A. Yamanaka and S. Okada, Sci. Rep., 2016, 6, 30653 CrossRef CAS PubMed.
- X. Y. Wang, F. D. Zhuang, R. B. Wang, X. C. Wang, X. Y. Cao, J. Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764–3767 CrossRef CAS PubMed.
- Z. Li, Y. Chen, T. Ma, Y. Jiang, J. Chen, H. Pan and W. Sun, Adv. Energy Mater., 2021, 11, 2101202 CrossRef CAS.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- Ş. G. İrim, A. A. Wis, M. A. Keskin, O. Baykara, G. Ozkoc, A. Avcı, M. Doğru and M. Karakoç, Radiat. Phys. Chem., 2018, 144, 434–443 CrossRef.
- D. Pan, S. Luo, Y. Feng, X. Zhang, F. Su, H. Liu, C. Liu, X. Mai, N. Naik and Z. Guo, Composites, Part B, 2021, 222, 109039 CrossRef CAS.
- A. Lyalin, A. Nakayama, K. Uosaki and T. Taketsugu, J. Phys. Chem. C, 2013, 117, 21359–21370 CrossRef CAS.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- H. Liu, X. H. Zhang, Y. X. Li, X. Li, C. K. Dong, D. Y. Wu, C. C. Tang, S. L. Chou, F. Fang and X. W. Du, Adv. Energy Mater., 2020, 10, 1902521 CrossRef CAS.
- A. Guha, T. Veettil Vineesh, A. Sekar, S. Narayanaru, M. Sahoo, S. Nayak, S. Chakraborty and T. N. Narayanan, ACS Catal., 2018, 8, 6636–6644 CrossRef CAS.
- A. Lyalin, A. Nakayama, K. Uosaki and T. Taketsugu, Phys. Chem. Chem. Phys., 2013, 15, 2809–2820 RSC.
- C. Deng, R. He, W. Shen and M. Li, Phys. Chem. Chem. Phys., 2019, 21, 18589–18594 RSC.
- S. Azevedo, J. R. Kaschny, C. M. C. de Castilho and F. de Brito Mota, Eur. Phys. J. B, 2009, 67, 507–512 CrossRef CAS.
- K. Uosaki, G. Elumalai, H. Noguchi, T. Masuda, A. Lyalin, A. Nakayama and T. Taketsugu, J. Am. Chem. Soc., 2014, 136, 6542–6545 CrossRef CAS PubMed.
- Y. Chen, J. Cai, P. Li, G. Zhao, G. Wang, Y. Jiang, J. Chen, S. X. Dou, H. Pan and W. Sun, Nano Lett., 2020, 20, 6807–6814 CrossRef CAS PubMed.
- Y. Huang, T. Yang, L. Yang, R. Liu, G. Zhang, J. Jiang, Y. Luo, P. Lian and S. Tang, J. Mater. Chem. A, 2019, 7, 15173–15180 RSC.
- M. Gao, M. Nakahara, A. Lyalin and T. Taketsugu, J. Phys. Chem. C, 2021, 125, 1334–1344 CrossRef CAS.
- M. Naguib, O. Mashtalir, J. Carle, V. Presser, J. Lu, L. Hultman, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2012, 6, 1322–1331 CrossRef CAS PubMed.
- Y. Wang, Y. Sun, H. Li, W. Zhang, S. Wu, C. Liu, Y. Gao, B. Jia, J. Qiu and T. Ma, Carbon Neutralization, 2022, 1, 117–125 CrossRef.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Zhang, C. Lai, B. Li, S. Liu, D. Huang, F. Xu, X. Liu, L. Qin, Y. Fu, L. Li, H. Yi and L. Chen, Small, 2021, 17, e2007113 CrossRef PubMed.
- G. Gao, A. P. O'Mullane and A. Du, ACS Catal., 2016, 7, 494–500 CrossRef.
- Z. Wang, K. Yu, Y. Feng, R. Qi, J. Ren and Z. Zhu, Appl. Surf. Sci., 2019, 496, 143729 CrossRef CAS.
- Y. Guo, T. Wang, Q. Yang, X. Li, H. Li, Y. Wang, T. Jiao, Z. Huang, B. Dong, W. Zhang, J. Fan and C. Zhi, ACS Nano, 2020, 14, 9089–9097 CrossRef CAS PubMed.
- T. Y. Ma, J. L. Cao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 1138–1142 CrossRef CAS PubMed.
- J. Chen, X. Yuan, F. Lyu, Q. Zhong, H. Hu, Q. Pan and Q. Zhang, J. Mater. Chem. A, 2019, 7, 1281–1286 RSC.
- T. Zhao, S. Boullosa-Eiras, Y. Yu, D. Chen, A. Holmen and M. Ronning, Top. Catal., 2011, 54, 1163–1174 CrossRef CAS.
- Y. Ukisu, React. Kinet., Mech. Catal., 2014, 114, 385–394 CrossRef.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2018, 4, 60–67 CrossRef.
- M. Deng, M. Xia, Y. Wang, X. Ren and S. Li, J. Mater. Chem. A, 2022, 10, 13066–13073 RSC.
- Z. Fan, Y. Ji, Q. Shao, S. Geng, W. Zhu, Y. Liu, F. Liao, Z. Hu, Y.-C. Chang, C.-W. Pao, Y. Li, Z. Kang and M. Shao, Joule, 2021, 5, 3221–3234 CrossRef CAS.
- C. Chen, H. Tian, Z. Fu, X. Cui, F. Kong, G. Meng, Y. Chen, F. Qi, Z. Chang, L. Zhu, H. Huang, B. Y. Xia and J. Shi, Appl. Catal., B, 2022, 304, 121008 CrossRef CAS.
- L. Wang, L. Shi, Q. Liu, Y. Huang, W. Yan, X. Liang, X. Zhao, H. Chen and X. Zou, ACS Catal., 2023, 13, 7322–7330 CrossRef CAS.
- Y. Guo, Y. Wei, H. Li and T. Zhai, Small, 2017, 13, 1701649 CrossRef PubMed.
- Y. Zhu, J. Wang, H. Chu, Y.-C. Chu and H. M. Chen, ACS Energy Lett., 2020, 5, 1281–1291 CrossRef CAS.
- T. Xia, Y. Yang, Q. Song, M. Luo, M. Xue, K. K. Ostrikov, Y. Zhao and F. Li, Nanoscale Horiz., 2023, 8, 146–157 RSC.
- O. Neumann, A. D. Neumann, S. Tian, C. Thibodeaux, S. Shubhankar, J. Müller, E. Silva, A. Alabastri, S. W. Bishnoi, P. Nordlander and N. J. Halas, ACS Energy Lett., 2016, 2, 8–13 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |