
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2-assisted hydrolytic hydrogenation of cellulose and cellulose-based waste into sorbitol over commercial Ru/C†
Daniele
Polidoro
 a,
Giancarmelo
Stamilla
a,
Matteo
Feltracco
bc,
Andrea
Gambaro
bc,
Alvise
Perosa
*a and
Maurizio
Selva
*a
a,
Giancarmelo
Stamilla
a,
Matteo
Feltracco
bc,
Andrea
Gambaro
bc,
Alvise
Perosa
*a and
Maurizio
Selva
*a
aDepartment of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice, Via Torino 155, 30175 – Venezia Mestre, Italy. E-mail: selva@unive.it
bInstitute of Polar Sciences-CNR, Via Torino 155, 30175 – Venezia Mestre, Italy
cDepartment of Environmental Sciences, Informatics and Statistics, Ca’ Foscari University of Venice, Via Torino 155, 30175 – Venezia Mestre, Italy
First published on 31st July 2023
Abstract
A single-step protocol was developed for the hydrolytic hydrogenation of microcrystalline cellulose into sorbitol over commercial carbon-supported Ru, in the presence of gaseous CO2 as an acid source and molecular hydrogen as a reductant. Under these conditions, cellulose was first hydrolysed to glucose by reversibly formed carbonic acid in water and then instantaneously hydrogenated on Ru/C. By tuning the reaction parameters, such as temperature, time and the relative pressure of CO2 and hydrogen gas, cellulose was fully converted at 220 °C in 18 h under 30 and 40 bar of H2 and CO2, respectively, with a sorbitol yield of 81%. Blank experiments revealed that without a catalyst and hydrogen, the reaction exhibited <5% conversion and glucose was the only detected product when the reaction was performed under CO2 pressure. XRD measurements on CO2-treated cellulose surprisingly revealed no noticeable changes in the crystallinity index (<10% with respect to microcrystalline cellulose), suggesting that hydrolytic hydrogenation took place on crystalline, not amorphous, cellulose. Furthermore, not only several cellulosic feedstocks, including filter paper, cotton wool, and cotton fiber, but also typical cellulose-based wastes such as a cardboard pizza box were also tested and under the optimized conditions sorbitol was obtained with yields ranging from 56% up to 72% in all cases. No less significant was the Ru/C catalyst stability, which could be recycled at least six times without any noticeable activity loss.
Introduction
The depletion of fossil feedstocks and the enormous environmental issues posed by their combustion and chemical manipulation are among the most pressing concerns of our society, to which the scientific/industrial community is responding through massive investments in the research of new strategies for a sustainable economy aimed at the conversion/valorisation of biomass into biofuels, renewable molecules and bio-based materials.1–4 In this context, given its abundance and its reduced impact on the food chain, lignocellulose (LC) is perhaps the best alternative to fossil resources,5–7 and even more specifically, cellulose, the major component of LC, is the most promising biopolymer for the synthesis of a variety of chemicals.8 Among them, sorbitol and its derivatives have widespread applications as dispersing agents, humectants in pharmaceuticals, low-calorie sweeteners, cosmetics and textiles.9,10–13 Sorbitol is also one of the ten building block chemicals obtainable from cellulosic resources listed as strategic by the US Department of Energy.14The synthesis of sorbitol from cellulose is generally performed via a two-step reaction that includes the acid-catalysed hydrolysis of cellulose to glucose followed by the hydrogenation of glucose to sorbitol over metal catalysts (Scheme 1).15
 | ||
| Scheme 1 Two-step catalytic cellulose conversion into sorbitol, via hydrolysis and hydrogenation. | ||
Even though the robust crystalline structure of cellulose makes its hydrolytic breakdown to glucose still a challenge, many reaction protocols have been developed over the years.8 Homogeneous catalysts such as H2SO4 and HCl have been extensively employed for this purpose. The use of strong liquid acids, however, is not sustainable from an environmental standpoint and suffers from serious drawbacks such as low selectivity, difficult product separation, corrosion and the need for acid recovery.16–18
As an alternative, several heterogeneous (acid) catalysts have been proposed for the direct conversion of cellulose into polyols.19–22 Due to the vast body of literature in this area, only a selection of representative works are commented on here. In a first seminal study, Fukuoka and Dhepe described the hydrolytic hydrogenation of cellulose using different metal-based catalysts of which Pt/γ-Al2O3 showed the best performance with a production of sorbitol and mannitol in 25% and 6% yields, respectively.23 Subsequent studies highlighted that Ru-based catalysts, even commercial ones, were probably the best option for biomass and biomass-derived compounds processing through reductive protocols,24–26 including the conversion of cellulose into sorbitol, not only because they displayed good activity and selectivity, but also due to their competitive costs since Ru was available at a far lower price (ca. ∼4%) compared to other metals such as Au and Pt of comparable activity.27 Luo et al. reported that at 245 °C and pH2 = 60 bar, in the presence of carbon-supported ruthenium (Ru/C), a high conversion of cellulose was achieved (86%) with a 30% sorbitol yield,28 though the high temperature caused both a partial degradation of glucose29 and the hydrogenolysis of sorbitol.30 Several Ru catalysts supported on acidic carriers such as sulfonated carbon,6 phosphate,9 and molecular sieves31 allowed significant improvements by making the reaction possible at lower temperatures and pressures (<200 °C, 30–50 bar H2) and with a higher sorbitol yield of up to 71%.6 Notwithstanding this, the process was slow due to the moderate acidity of the support that brought about a low hydrolysis rate. More performant catalysts were obtained using acid–Ru binary systems where the ratio of acidity to reduction activity could be adjusted: for example, heteropolyacids coupled with Ru/C could effectively improve cellulose conversion, giving a mixture of sorbitol and mannitol in a 68% yield in only 1 h, at 180 °C under 50 bar H2.29 The poor water solubility of heteropolyacids, however, made difficult the catalyst handling/recovery, thereby hampering any large-scale applications of the procedure. To improve the reusability of the solid acid, zirconium phosphate (ZrP) instead of heteropolyacids was considered, in combination with Ru/C. This system was apparently highly active, affording an 85% yield of C6 alcohol in 2.5 h, at 190 °C and 50 bar H2.32 However, it required an acidic pre-treatment of cellulose to reduce its crystallinity; otherwise, the rate-determining hydrolysis step was problematic.8,33,34 Deng et al. reported that the crystallinity of cellulose could be decreased by treating it with phosphoric acid. After this preliminary step, a 69% sorbitol yield was reached via hydrolytic hydrogenation catalysed by Ru/CNT (carbon nanotubes) at 185 °C and pH2 = 50 bar.35 Mechanochemical treatments were also evaluated to reduce the crystallinity index of cellulose, especially by ball-milling. A comparative analysis demonstrated that a catalytic mixture of Ru/C and H4SiW12O40 allowed an 85% and a 36% yield of sugar alcohols starting from ball-milled cellulose and pristine microcrystalline cellulose, respectively.29 In another work by Pereira et al., a conversion close to 90% with 80% selectivity to sorbitol was reported by ball-milling Ru/C and cellulose together, at 205 °C under 50 bar H2 in 1 h.36
Whichever the approach, the use of acids, both as solids and even more so as liquids, always implies concerns related to safety, corrosion (especially at high temperatures and pressures), and disposal. Therefore, the design of more sustainable and low-environmental-impact protocols for the direct conversion of cellulose into sorbitol remains a highly desirable target of a modern biorefinery.
The use of CO2 may represent a further attractive choice to generate weakly acidic aqueous solutions. This has been proposed and used in several strategies for biomass conversion including pre-treatments of cellulose,37 rice straw,38 corn stover,39 and agroindustrial residues.40 In particular, it has been demonstrated that the yield of sugars can be significantly improved by introducing CO2 during lignocellulose hydrolysis in hot water,17 thereby confirming the potential of CO2-assisted hydrolysis as a green technology for biomass upgrading.
In this work, the combination of wet CO2 as an acid source and molecular hydrogen as a reductant was investigated for the hydrolytic/hydrogenation of microcrystalline cellulose into sorbitol, in the presence of commercial 5% Ru/C. By tuning the reaction parameters, such as temperature, time and pressure, cellulose was fully converted at 220 °C in 18 h under 30 and 40 bar of H2 and CO2, respectively, allowing sorbitol formation in 81% yield. To extend the scope, available and cheap cellulose feedstocks, such as filter paper, cotton wool, cotton fiber and a cardboard pizza box were also evaluated as starting materials and a sorbitol yield ranging from 56% to 72% was achieved. Remarkably, the catalytic activity of Ru/C was not altered by the reaction environment during the recycling tests for at least six consecutive runs.
Results and discussion
Catalyst and acidity
As mentioned above, supported Ru-based systems are among the preferred systems for the hydrogenation of sugars. In this work, with the aim of designing a protocol as easily accessible as possible, commercial 5% Ru/C (lot #MKBW5890 V) was exclusively used. This catalyst has been characterized for its structural, morphological, and acidic properties in recent papers by our group.13,41–43 Moreover, to cope with the need for an acidic environment that is crucial for the hydrolysis of cellulose to glucose, the use of pressurised CO2 able to generate weakly acidic aqueous solutions by the formation of carbonic acid was considered to avoid any issues related to more conventional acids, either liquids or solids.44 It was known that at 25 °C, the pH of the aqueous solution decreased to about 2.83 when CO2 was added (70 bar),45 but more importantly, the acidity was reversible since carbonic acid was (almost) completely removed by venting CO2 from the reactor. This simple operation did not involve solvent discharge or additional treatments, and (weak) H2CO3 strongly limited any corrosion issues.17,46,47 Last but not least, CO2 is nontoxic, and as a by-product of biorefinery processes for fuels and chemicals, it was available at almost no cost.46CO2-assisted hydrolysis of D-maltose into glucose
Exploratory tests were performed to investigate the use of wet CO2 by employing D-(+)-maltose as a model compound. The CO2-assisted hydrolysis of maltose to glucose was studied (Scheme 2).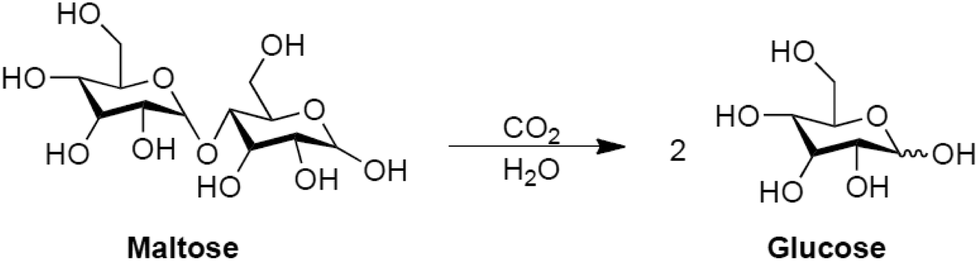 | ||
| Scheme 2 CO2-assisted hydrolysis of D-maltose. | ||
Experiments were carried out in a stainless-steel autoclave in which an aqueous solution of maltose (maltose: 100 mg; H2O: 5 mL) was allowed to react in the presence of CO2. The effects of temperature (T), time (h) and CO2 pressure (p) were investigated through three series of tests by varying the following parameters: (i) T in the range 25–150 °C at constant CO2 pressure (40 bar) and time t = 2 h; (ii) time in the range 2–15 h at constant pressure (40 bar) and temperature (150 °C); and (iii) p in the range 5–40 bar, at constant temperature (150 °C) and time (12 h). Maltose conversion and glucose selectivity were determined by HPAEC-MS. All the reported reactions were run in duplicate to ensure reproducibility: unless otherwise specified, conversions and selectivity differed by less than 5% from one test to another. The results are shown in Fig. 1.
 | ||
| Fig. 1 Effects of (a) temperature; (b) time and (c) CO2 pressure on the hydrolysis of D-maltose. Reaction conditions: maltose (100 mg) and H2O (5 mL). Conversion and selectivity were determined by HPAEC-MS. | ||
No maltose conversion was observed at pCO2 = 40 bar in 2 h in the temperature range 25–100 °C; however, increasing T from 120 to 150 °C prompted a gradual increase in the conversion of maltose from 7% to 35%, respectively (Fig. 1a). No products other than glucose, which was obtained in >99% selectivity, were observed under these conditions. Having set 150 °C as the operative temperature to continue this investigation, the effect of time was explored (Fig. 1b). An increase in the reaction time caused an increase in maltose conversion, which became quantitative after 12 h. A small, but not negligible, amount of fructose (1–2%) due to the isomerization of glucose was also detected.48 No further increase in conversion and selectivity was observed by extending the reaction time to 15 h. This allowed us to set T = 150 °C and t = 12 h as the conditions to study the effect of CO2 pressure (Fig. 1c). Notably, high maltose conversion (ca. 68–70%) was achieved even at the lowest investigated CO2 pressure (5 bar), but the reaction became quantitative with excellent glucose selectivity (98–99%) only at 40 bar. A blank experiment was performed at 150 °C for 12 h in the absence of CO2. The reaction reached only 27% maltose conversion into glucose, thereby confirming the crucial role of the acidity provided by carbonic acid. The limited extent of the hydrolysis process observed without CO2 was due to the thermal instability of the β-O-4 glycosidic bond.49,50
In summary, parametric analysis showed that the CO2-assisted hydrolysis of maltose was strictly dependent on the reaction conditions: however, 98–99% glucose selectivity was obtained with quantitative maltose conversion, at 150 °C, under 40 bar CO2 for 12 h.
CO2-assisted hydrolysis/hydrogenation of D-maltose into sorbitol
An initial experiment aimed at exploring the direct conversion of maltose into sorbitol was designed under the best conditions observed for maltose hydrolysis (maltose: 100 mg, H2O: 5 mL, 150 °C, 40 bar CO2, 12 h) with the addition of Ru/C (50 mg) as a hydrogenation catalyst and hydrogen (30 bar). The final pressure was given by pH2 + pCO2 at room temperature, i.e. 70 bar. The reaction allowed a quantitative conversion of maltose into a mixture of maltitol (24%) and sorbitol (76%), respectively. Given this promising result, further tests were carried out by varying both the reaction time and the temperature. The results are shown in Fig. 2. | ||
| Fig. 2 Effects of (a) time and (b) temperature on the CO2-assisted hydrogenation of maltose into sorbitol. Reaction conditions: maltose (100 mg), H2O (5 mL), 40 bar CO2, 30 bar H2, and Ru/C (50 mg). Conversion and selectivity were determined by HPAEC-MS. | ||
At 150 °C, the prolongation of the reaction from 12, to 15, 18 and 24 h induced a gradual increase in sorbitol selectivity, up to 87%, at the expense of maltitol (Fig. 2a). The desired process was further favoured by slightly increasing the reaction temperature, from 150 to 170 °C: at quantitative conversion, sorbitol was obtained with an excellent 96% selectivity (Fig. 2b). Last but not least, the formation of C4–C5 polyols (derived from hydrogenolysis reactions) during maltose hydrolysis/hydrogenation was never detected by HPAEC-MS.
In principle, different reaction pathways could be hypothesized for the conversion of maltose into sorbitol (Scheme 3). Maltose could be first hydrolysed to glucose, which could be further hydrogenated into sorbitol (pathway A, top); alternatively, the direct hydrogenation of maltose could provide maltitol (pathway B, bottom), followed by its hydrolysis to yield sorbitol and glucose in equimolar amounts. Finally, hydrolysis/hydrogenation of maltose could take place simultaneously to provide sorbitol (pathway C, centre). The results of Fig. 2 clearly highlighted that the catalytic hydrogenation of maltose to maltitol was faster than the hydrolysis reaction, confirming that the conversion of maltose into sorbitol proceeded via pathway B.
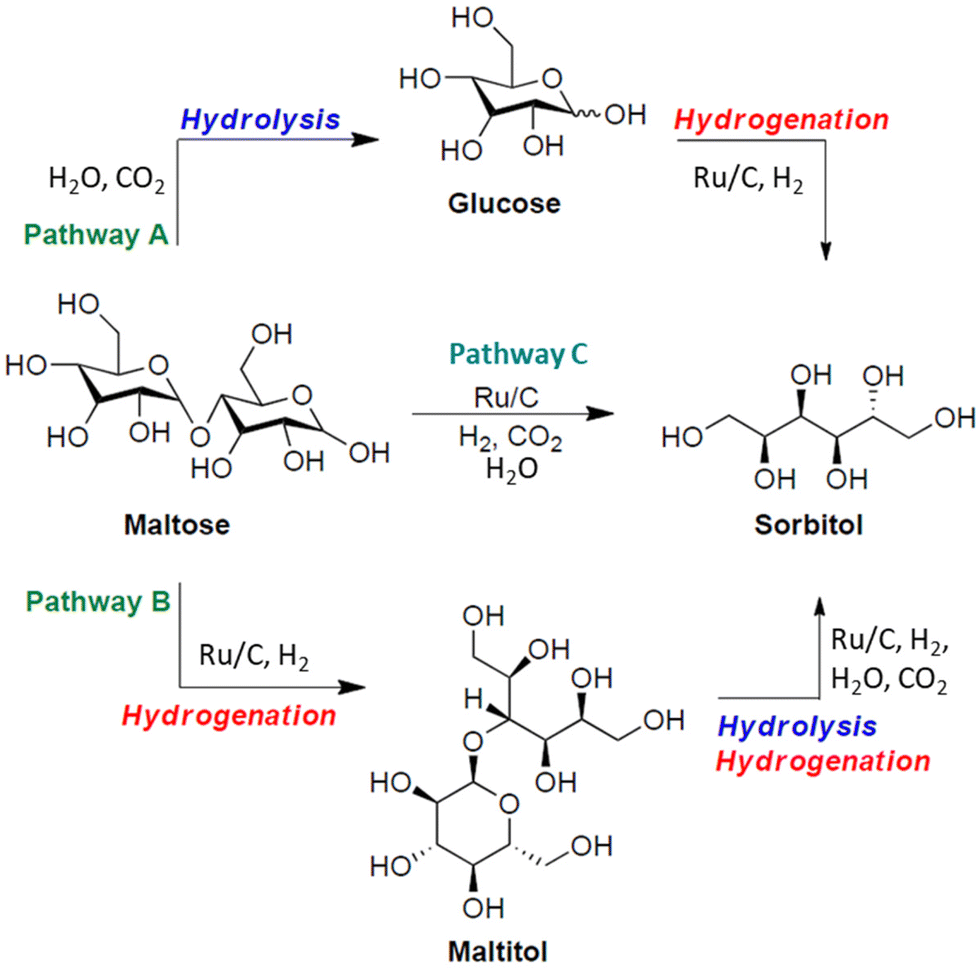 | ||
| Scheme 3 Reaction pathways A–C for the CO2-assisted hydrogenation of D-maltose into sorbitol. | ||
Moreover, while complete maltose hydrolysis to glucose took place under the standard conditions of 150 °C, 40 bar CO2, 12 h (Fig. 1), complete maltitol hydrolysis was slower and required harsher conditions (170 °C) and a longer time (24 h) with 40 bar CO2 and 30 bar H2. To better understand the reasons for the different behaviour, we first assumed that the presence of 30 bar H2 played a role in the hydrolysis kinetics of maltitol. In fact, when maltitol was set to react without H2 under the standard conditions observed for maltose hydrolysis (150 °C, 40 bar CO2, 12 h), glucose and sorbitol were achieved much faster with almost equal selectivity (48% and 52%, respectively) at 97% maltitol conversion, seemingly indicating that the presence of H2 from the preceding reduction step slowed the reaction. An experiment with He in place of H2 under the standard maltitol hydrolysis conditions (150 °C, 40 bar CO2, 12 h and 30 bar He) also showed a lower maltitol conversion (83%) with the glucose/sorbitol ratio remaining unaltered. Both experiments seemed to indicate that the CO2-assisted hydrolysis was slower in the presence of additional pressure (H2 or He). However, since the addition of H2 or He does not affect the partial pressure of CO2, and the pH of the solution presumably remains constant as it depends on CO2 concentration and thus its partial pressure, the reason for a slower hydrolysis rate does not seem ascribable to a change in pH. The explanation for this behaviour is still unclear and is currently under investigation in our lab.
CO2-assisted hydrolysis/hydrogenation of cellulose into sorbitol
Based on the results obtained for the CO2-assisted hydrolytic hydrogenation of maltose, the next step was to apply the reaction to more complex substrates starting from cellulose. A suspension of microcrystalline cellulose (100 mg) in 5 mL of H2O was set to react under 40 bar CO2 and 30 bar H2 for 24 h, in the presence of Ru/C (50 mg). Different temperatures in the range 150–250 °C were explored. Cellulose conversion was determined gravimetrically by the difference in weight of cellulose employed in the reaction and the solid recovered after the reaction, taking into consideration also the weight of the solid catalyst. The yield of water-soluble polyols such as sorbitol, mannitol and C4–C5 sugar alcohols was determined by HPAEC-MS (a detailed HPAEC-MS quantification protocol is reported in the ESI section†). The results are reported in Table 1.|
|
|||||
|---|---|---|---|---|---|
| Entry | Temperature (°C) | Cellulose conversion (%) | Yield (%) | ||
| Sorbitol | Mannitol | C4–C5 polyols | |||
| Reaction conditions: cellulose (100 mg), Ru/C (50 mg), H2O (5 mL), 40 bar CO2, 30 bar H2, and 24 h. Yield (mass%) was determined by HPAEC-MS. | |||||
| 1 | 150 | 32 | 7 | 2 | <1 |
| 2 | 180 | 50 | 41 | 2 | 2 |
| 3 | 200 | >99 | 67 | 4 | 5 |
| 4 | 220 | >99 | 81 | 4 | 7 |
| 5 | 250 | >99 | 54 | 3 | 26 |

At the lower temperature of 150 °C, the conversion of cellulose reached 32%, but the observed products, which included sorbitol, mannitol (as an isomerization product) and a mixture of C4–C5 polyols (hydrogenolysis products such as erythritol, xylitol and arabitol), were obtained in poor, if not negligible, yields of 7%, 2% and <1%, respectively (entry 1). This result was ascribed to the formation of water-soluble oligomers which could not be detected by HPAEC-MS. An increase in the reaction temperature from 150 to 180 and 200 °C improved the conversion to >99%, and considerably favoured the formation of sorbitol which was achieved in up to 67% yield (entries 2 and 3). The concurrent formation of small amounts of mannitol (4%) and C4–C5 polyols (5%) was also observed.
Finally, the yield of sorbitol was further increased to 81% at 220 °C (entry 4), one of the best results reported so far for this reaction. Raising the T to 250 °C, however, brought about an increase in hydrolysis products (C4–C5 sugar alcohols) which were obtained in a 26% yield at the expense of sorbitol (54%, entry 5).
In summary, the hydrolytic hydrogenation protocol proved highly efficient in the conversion of cellulose into sorbitol, using CO2 as an acid precursor and molecular hydrogen as a reductant. Sorbitol was obtained with the highest yield of 81% at 220 °C, under 40 bar CO2 and 30 bar H2 for 24 h. The formation of hydrogenolysis products such as erythritol, xylitol and arabitol (not exceeding 7% yield) was also observed.
As mentioned in the Introduction section, acidic pre-treatments are often employed to reduce the crystallinity of cellulose and increase its reactivity.35 To shed light on this aspect under the conditions explored in this work, the effect of the CO2 acidity on the crystalline structure of cellulose was investigated. Experiments were carried out in 5 mL H2O, at 220 °C and for 18 h, in the absence of Ru/C, under a variety of conditions by treating microcrystalline cellulose: (i) as such, without CO2; (ii) under 40 bar CO2; (iii) under 30 bar H2 and (iv) under 40 bar CO2 and 30 bar H2 (conditions of the CO2-assisted hydrolytic hydrogenation of cellulose). The crystallinity index of the solid recovered at the end of each test was then measured by XRD. The results are reported in Fig. 3.
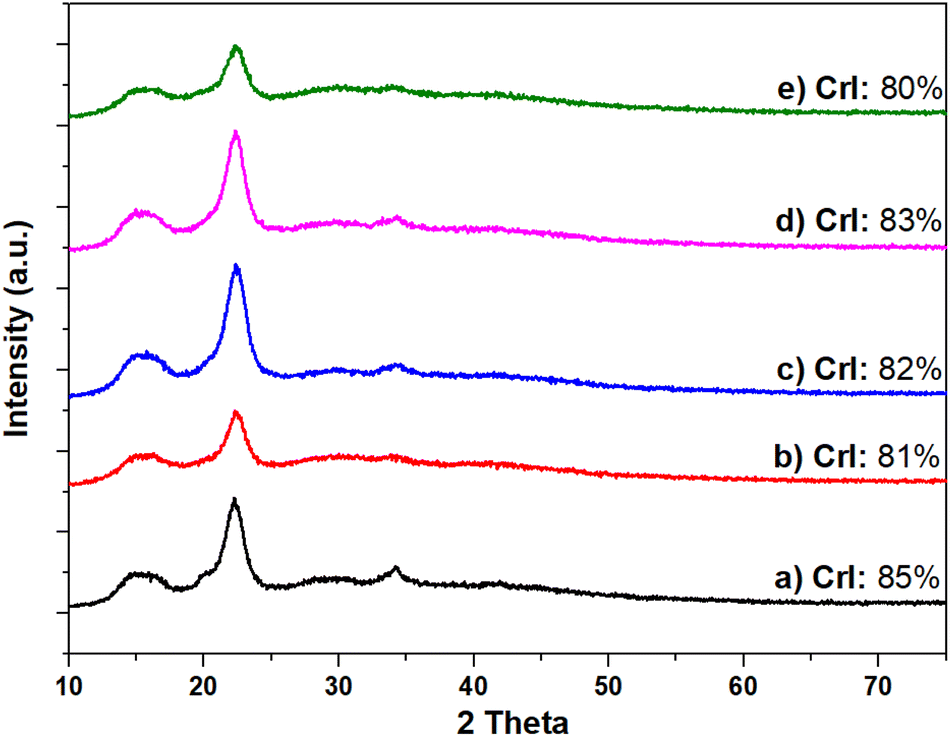 | ||
| Fig. 3 XRD profiles of (a) pristine microcrystalline cellulose, (b) microcrystalline cellulose after 18 h at 200 °C, (c) microcrystalline cellulose after 18 h at 200 °C under 40 bar CO2, (d) microcrystalline cellulose after 18 h at 200 °C under 30 bar H2 and (e) microcrystalline cellulose after 18 h at 200 °C under 40 bar CO2 and 30 bar H2. | ||
Quite unexpectedly, XRD profiles differed by less than 10% from one sample to another and compared to pristine microcrystalline cellulose (crystallinity index of ca. 85%). Indeed, in all profiles of Fig. 4(a–e), the strongest peak at 2θ = 22.6°, which originated from the cellulose crystalline plane (002), indicated that the degree of crystallinity of microcrystalline cellulose was substantially preserved regardless of the presence of CO2. Additionally, no cellulose conversion (<5%) was observed when reactions were carried out in the absence of CO2 (profiles b and c), while in contrast, in the presence of CO2, with or without additional hydrogen (d and e), measurements revealed ca. 67–70% cellulose conversion and glucose was the only detected product from HPAEC-MS. In other words, CO2 favoured the hydrolytic breakdown of the biopolymer, but in the unreacted cellulose, CO2 was apparently unable to modify the domains where the polymer chains were aligned with each other, against the hypothesis that CO2 acidity worked by reducing the crystallinity.
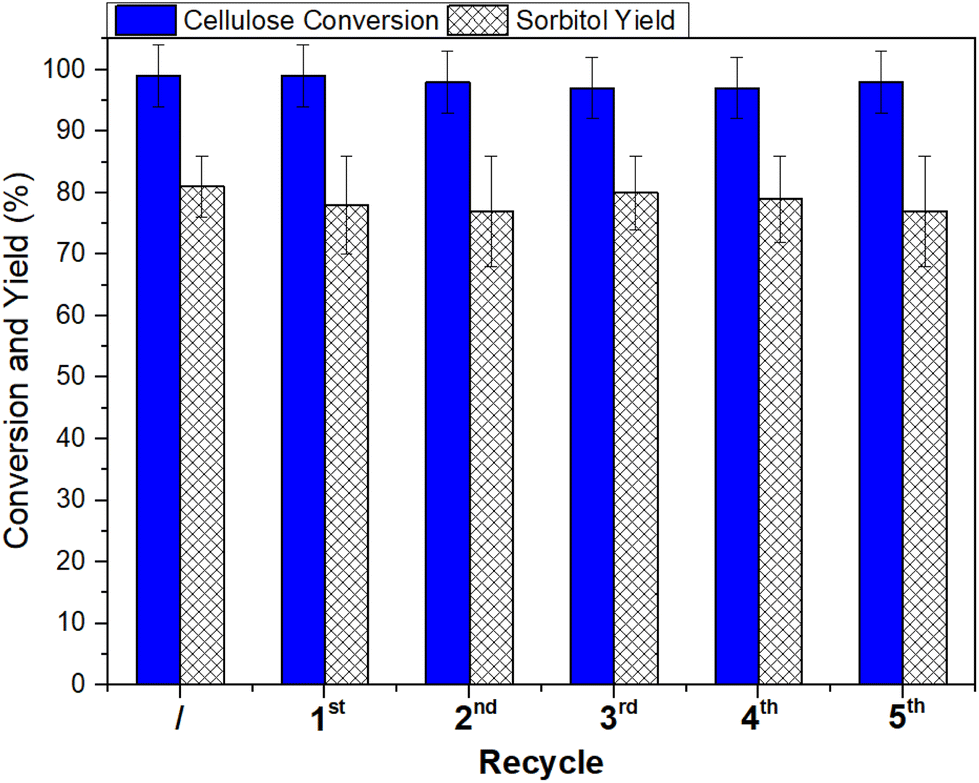 | ||
| Fig. 4 Ru/C recycling in the CO2-assisted hydrogenation of cellulose into sorbitol. Reaction conditions: cellulose (100 mg), Ru/C (50 mg), H2O (5 mL), 220 °C, 40 bar CO2, 30 bar H2, and 24 h. Yield was determined by HPAEC-MS. | ||
Catalyst recycling
The cost of the catalyst in a liquid-phase reaction may represent up to a third of the total cost of the process, implying that its loss by leaching or other reasons is critical, and its recovery and reuse are crucial.52 The stability and reusability of Ru/C were therefore investigated by designing recycling experiments under the conditions described in Table 1, entry 4 (cellulose: 100 mg, Ru/C: 50 mg, H2O: 5 mL, 220 °C, 40 bar CO2, 30 bar H2, 24 h). Once the first reaction was completed, the catalyst was filtered, washed with distilled water (30 mL) and dried overnight. During the recycling tests, the catalyst amount differed by less than 5% from one test to another and fresh Ru/C was added during the recycling tests to maintain the original catalyst/substrate ratio. The recovered catalyst was added to fresh microcrystalline cellulose (100 mg), water (5 mL) and the appropriate amount of Ru/C and a new reaction was started. The recycling procedure was repeated six times, and the whole set of reactions was run twice to ensure reproducibility. The results are illustrated in Fig. 4. Both the cellulose conversion and the sorbitol yield remained steady at 97–99% and 77–81%, respectively, during the six runs, thereby demonstrating that the overall performance of Ru/C was not altered over time by the reaction environment during the recycling tests and by the washing/restoring procedures.CO2-assisted hydrolysis/hydrogenation of cellulose feedstock/waste into sorbitol
Other real-world cellulose sources were explored to investigate the applicability and scalability of the CO2-assisted hydrolysis/hydrogenation protocol. In this scope, cheap and readily available cellulosic feedstocks, including filter paper, cotton wool, and cotton fiber, and common cellulose-based wastes such as a pizza carton made of cardboard were selected. A mixture of finely ground cellulose feedstock (100 mg, size <1 mm), Ru/C (50 mg) and H2O (5 mL) was set to react at 220 °C for 24 h, under 40 bar CO2 and 30 bar H2. The conversion and yield were determined by the same method described previously. The results are summarized in Table 2.| Entry | Cellulose feedstock | Conversion (%) | Yield (%) | ||
|---|---|---|---|---|---|
| Sorbitol | Mannitol | C4–C5 polyols | |||
| Reaction conditions: cellulose feedstock (100 mg), Ru/C (50 mg), H2O (5 mL), 40 bar CO2, 30 bar H2, and 24 h. Yield (mass%) was determined by HPAEC-MS. | |||||
| 1 | Filter paper | >99 | 62 | 3 | 21 |
| 2 | Cotton wool | >99 | 71 | 5 | 7 |
| 3 | Cotton fiber | >99 | 72 | 4 | 6 |
| 4 | Pizza carton | >99 | 56 | 6 | 13 |
First, the commonly employed laboratory filter paper was tested. Such a starting material was fully converted, allowing sorbitol formation in 62% yield with the concurrent formation of mannitol (3%) and, if compared with microcrystalline cellulose, a higher amount of C4–C5 polyols (21%). A remarkable improvement in sorbitol yield was observed when cotton wool and cotton fibers were tested. In these cases, sorbitol was obtained in 71% and 72% yields, respectively, with an almost equal amount of mannitol (5% and 4% respectively) and C4–C5 polyols (7% and 6%, respectively), while the conversion remained stable and quantitative in both cases. The pizza carton made of cardboard was also completely converted, with a sorbitol yield of 56%, while mannitol and C4–C5 polyols were observed in 6% and 13% yields, respectively. Overall, these results not only confirmed that the investigated reductive protocol was effective for the direct conversion of cellulose into sorbitol, but they also proved that the process was successfully applied to a wide range of cellulose feedstocks.
Comparison of the CO2-assisted hydrolytic hydrogenation of cellulose to literature results
A comparative assessment of the reported CO2-assisted protocol for the conversion of cellulose into sorbitol against other methods was carried out by selecting some of the most representative works from the vast body of literature in this area. To make the analysis as consistent as possible, batch liquid phase methods that used Ru/C catalysts and pristine microcrystalline cellulose were considered. Table 3 shows the results.| Entry | Catalyst | Acid | Experimental conditions (T, p H2, t) | Cellulose conversion (%) | Sorbitol yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a ZrP: zirconium phosphate. b Yield referred to C6-alditols. c 40 bar CO2. | ||||||
| 1 | Ru/C | — | 245, 60, 0.5 | 86 | 35 | 28 |
| 2 | Ru/C | H4SiW12O40 | 180, 50, 1 | 99 | 68 | 29 |
| 3 | Ru/C | ZrPa | 190, 50, 3 | 99 | 64b | 32 |
| 4 | Ru/C | CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
220, 30, 24 | >99 | 81 | This work |
An efficient cellulose conversion into polyols by combination of hydrolysis using H+ ions, reversibly formed in situ in hot water, with instantaneous hydrogenation over Ru/C was reported by Liu28 (entry 1). Upon considering the absence of any additional acid source and the reduced reaction time (0.5 h) as sustainable and environmentally friendly aspects, the higher temperature of 245 °C promoted the formation of a mixture of C1–C5 products and the final sorbitol yield was low (ca. 34–35%). However, the addition of heteropolyacids (entry 2) not only allowed us to reduce the reaction temperature to 180 °C but also helped us to increase the sorbitol yield to 68% after 1 h.29 However, as mentioned in Introduction, the low solubility of heteropolyacids in aqueous solutions made their handling/recovery difficult. To solve this problem, zirconium phosphate (ZrP) instead of heteropolyacids was employed as a solid acid source32 (entry 3). With this system, by tuning the reaction parameters, 64% yield of a mixture of C6-alditols was achieved after 3 h at 190 °C under 50 bar H2. On the other hand, employing CO2 as an acid precursor, sorbitol was obtained in 81% yield with quantitative cellulose conversion. To the best of our knowledge, this is the best result reported for this transformation. Among the green advantages of the reported procedure, it is worth noting that (i) carbonic acid is sufficiently acidic for the complete glycolysis of cellulose, and even more importantly, its formation is reversible by simply venting the reactor, ensuring a lack of product contamination, limiting corrosion and improving safety compared to using conventional liquid or solid acids; (ii) the catalyst (Ru/C) can be recycled at least six times without significant activity loss; and (iii) the protocol proved effective not only for microcrystalline cellulose but also for other cellulosic feedstocks, including filter paper, cotton wool, and cotton fiber as well as typical cellulose wastes such as a pizza carton. Based on this evidence, the proposed protocol paves the way for the design of innovative and simple methods for the recovery and valorisation of any cellulose-based feedstocks, including waste, to produce sorbitol in what is a typical circular economy approach.
Experimental section
Materials and equipment
D-(+)-Maltose (>99%), microcrystalline cellulose, and 5% Ru/C (lot #MKBW5890V) were commercially available compounds sourced from Sigma-Aldrich. If not otherwise specified, reagents were employed without further purification. Water was of Milli-Q grade. H2 gas was purchased from SIAD, Italy. Quantitative analyses were performed by high-pressure anion exchange chromatography and ion chromatography (Thermo Scientific™ Dionex™ ICS-5000) coupled to a single quadrupole mass spectrometer (Thermo Scientific™ MSQ Plus™) (HPAEC-MS). The crystallinity index of cellulose samples was investigated by X-ray diffraction (XRD) using a D8 Advance Bruker® AXS diffractometer, with Cu Kα radiation as the X-ray source, coupled to a Lynxeye detector, and monitoring the 2θ within 10–80° at a rate of 0.08° min−1. All reactions were performed in duplicate to verify reproducibility.Typical CO2-assisted hydrogenation experiments
Experiments were performed in a 25 mL tubular reactor made from borosilicate glass (Pyrex), which was charged with microcrystalline cellulose (100 mg), 5% Ru/C (50 mg) and water (5 mL). The vessel was placed in a jacketed stainless-steel autoclave equipped with a manometer and two needle valves, and pressurised with hydrogen to 30 bar and CO2 to 40 bar (the final pressure was given by pH2 + pCO2 at room temperature). The autoclave was then heated by oil circulation at T = 150–250 °C, and the mixture was kept under magnetic stirring at a rate of 1500 rpm. After the desired reaction time (24 h), the autoclave was cooled to room temperature and gently purged. The catalyst (Ru/C) was filtered on PTFE (0.2 μm) and the product solutions were analysed by HPAEC-MS.Catalyst recycling
The recycling/reuse of the catalyst was investigated after the CO2-assisted hydrogenation of cellulose under the following reaction conditions: cellulose (100 mg), Ru/C (50 mg), H2O (5 mL), 220 °C, 40 bar CO2, 30 bar H2, for 24 h. Once the first reaction was complete, the catalyst was filtered on PTFE, washed with H2O (30 mL) and dried overnight. The recovered catalyst was added with fresh cellulose (100 mg) and H2O (5 mL) and a new reaction was started. The overall sequence was repeated for six subsequent runs (five recycles), and each reaction was run twice to ensure reproducibility.Product analysis
Cellulose conversion (Ccellulose, %) was determined based on the weight of cellulose utilized in the reaction (mcellulose,0) and the solid recovered after the reaction, taking into consideration the fraction of the solid catalyst in the remainder with mcellulose = mrecovered solid − mcatalyst (eqn (1)).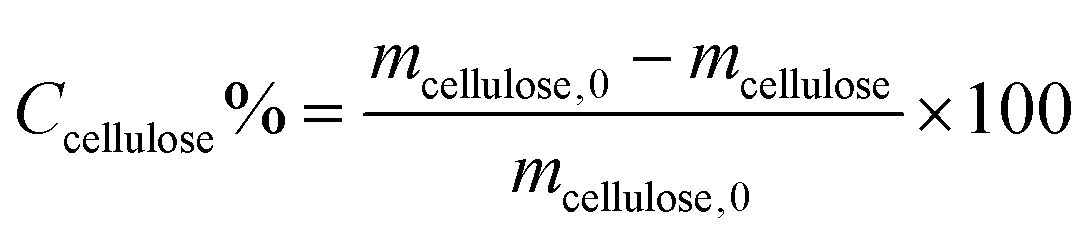 | (1) |
Product analysis and quantification were performed according to a validated method by Barbaro et al.53 A detailed protocol is reported in the ESI section.†
Crystallinity index determination
The crystallinity index (CrI) was calculated using the XRD peak height method,51 according to the following equation:where I002 is the maximum intensity of the (002) lattice diffraction (2θ = 22.6°) and IAM is the intensity diffraction at 2θ: 18°. I002 represents both crystalline and amorphous cellulose, whereas IAM represents amorphous cellulose only.

Conclusions
The herein reported study demonstrates the efficiency and robustness of a one-step CO2-assisted hydrolytic hydrogenation protocol for the conversion of cellulose into sorbitol. The use of maltose as a model substrate has been tested in the first part of the investigation to prove the role of CO2 in the hydrolytic breakdown of the glycosidic bond: at 150–170 °C, in an aqueous solution pressurised with CO2 (30 bar), the weak acidity due to carbonic acid is enough to make the glycolysis proceed to completion with the formation of glucose. Importantly, the reversible formation of carbonic acid, which is removed by venting the reactor, improves the safety and environmental impact of the procedure, and strongly limits any corrosion issues. Next, when Ru/C and hydrogen are added to the reaction system, a selectivity of up to 96% is achieved towards the product, sorbitol, derived from a tandem sequence: the hydrolysis of maltose followed by the hydrogenation of glucose. By tuning the reaction parameters, the protocol is also equally effective in the conversion of microcrystalline cellulose into sorbitol. At 220 °C, 40 bar CO2 and 30 bar H2 for 24 h, cellulose is fully converted, with a sorbitol yield of 81%, one of the best results reported to date for this transformation. Six consecutive recycling tests in the CO2-assisted hydrolysis/hydrogenation of cellulose prove the excellent stability of Ru/C that can be easily recovered by filtration from the reaction medium. Finally, the scope of the reaction can be extended to other largely available cellulose sources, including filter paper, cotton wool, cotton fiber and pizza cartons made from cardboard: sorbitol is obtained in good-to-excellent yields ranging from 56% to 72% in all cases. Overall, this study not only proves the concept of combining two selective processes of hydrolysis and hydrogenation to achieve an added-value polyol such as sorbitol straight from cellulose, through a single-batch process, but it also paves the way for the design of innovative and simple methods for the recovery and valorisation of any cellulose-based feedstocks including waste.Author contributions
D.P.: conceptualization, investigation, methodology, and writing – original draft preparation; G.S.: investigation and analytical methodology; M.F.: investigation and analytical methodology; M.F.: investigation and analytical methodology; A.G.: supervision and funding acquisition; A.P.: conceptualization, supervision, writing – reviewing and funding acquisition; M.S.: conceptualization, supervision, writing – reviewing and editing, and funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Ca’ Foscari Università di Venezia is gratefully acknowledged for its valuable support during the development of this work.References
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- Q. Di, Y. Wang, A. Zanobetti, Y. Wang, P. Koutrakis, C. Choirat, F. Dominici and J. D. Schwartz, N. Engl. J. Med., 2017, 376, 2513–2522 CrossRef CAS PubMed.
- D. R. Gentner, S. H. Jathar, T. D. Gordon, R. Bahreini, D. A. Day, I. El Haddad, P. L. Hayes, S. M. Pieber, S. M. Platt, J. De Gouw, A. H. Goldstein, R. A. Harley, J. L. Jimenez, A. S. H. Prévôt and A. L. Robinson, Environ. Sci. Technol., 2017, 51, 1074–1093 CrossRef CAS PubMed.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- J. W. Han and H. Lee, Catal. Commun., 2012, 19, 115–118 CrossRef CAS.
- Y. Zhou, G. Chen, Z. Long and J. Wang, RSC Adv., 2014, 4, 42092–42113 RSC.
- M. Yabushita, H. Kobayashi and A. Fukuoka, Appl. Catal., B, 2014, 145, 1–9 CrossRef CAS.
- J. Xi, Y. Zhang, Q. Xia, X. Liu, J. Ren, G. Lu and Y. Wang, Appl. Catal., A, 2013, 459, 52–58 CrossRef CAS.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J. C. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef PubMed.
- F. Brandi and M. Al-Naji, ChemSusChem, 2022, 15, e202102525 CrossRef CAS PubMed.
- J. Zhang, J. B. Li, S. B. Wu and Y. Liu, Ind. Eng. Chem. Res., 2013, 52, 11799–11815 CrossRef CAS.
- D. Polidoro, A. Perosa, E. Barbaro, M. Feltracco, E. Argiriadis and M. Selva, ACS Sustainable Chem. Eng., 2022, 10, 2844–2858 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–555 RSC.
- L. S. Ribeiro, J. J. Delgado, J. J. M. Órfão and M. F. R. Pereira, Appl. Catal., B, 2017, 217, 265–274 CrossRef CAS.
- X. Wang, L. Meng, F. Wu, Y. Jiang, L. Wang and X. Mu, Green Chem., 2012, 14, 758–765 RSC.
- J. Liang, X. Chen, L. Wang, X. Wei, H. Wang, S. Lu and Y. Li, Bioresour. Technol., 2017, 228, 147–155 CrossRef CAS PubMed.
- K. Wu, Y. Wu, Y. Chen, H. Chen, J. Wang and M. Yang, ChemSusChem, 2016, 9, 1355–1385 CrossRef CAS PubMed.
- A. Negoi, K. Triantafyllidis, V. I. Parvulescu and S. M. Coman, Catal. Today, 2014, 223, 122–128 CrossRef CAS.
- H. Kobayashi, Y. Ito, T. Komanoya, Y. Hosaka, P. L. Dhepe, K. Kasai, K. Hara and A. Fukuoka, Green Chem., 2011, 13, 326–333 RSC.
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS PubMed.
- M. Käldström, N. Kumar and D. Y. Murzin, Catal. Today, 2011, 167, 91–95 CrossRef.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- S. Gyergyek, D. Lisjak, M. Beković, M. Grilc, B. Likozar, M. Nečemer and D. Makovec, Nanomaterials, 2020, 10, 1–9 CrossRef PubMed.
- R. Šivec, M. Huš, B. Likozar and M. Grilc, Chem. Eng. J., 2022, 436, 32–40 CrossRef.
- R. Šivec, M. Grilc, M. Huš and B. Likozar, Ind. Eng. Chem. Res., 2019, 58, 16018–16032 CrossRef.
- D. Zhao, T. Su, Y. Wang, R. S. Varma and C. Len, Mol. Catal., 2020, 495, 111133 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS PubMed.
- J. Geboers, S. Van De Vyver, K. Carpentier, K. De Blochouse, P. Jacobs and B. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- G. Liang, L. He, H. Cheng, W. Li, X. Li, C. Zhang, Y. Yu and F. Zhao, J. Catal., 2014, 309, 468–476 CrossRef CAS.
- J. Zhang, L. Lin, J. Zhang and J. Shi, Carbohydr. Res., 2011, 346, 1327–1332 CrossRef CAS PubMed.
- Y. Liao, Q. Liu, T. Wang, J. Long, L. Ma and Q. Zhang, Green Chem., 2014, 16, 3305–3312 RSC.
- V. B. Agbor, N. Cicek, R. Sparling, A. Berlin and D. B. Levin, Biotechnol. Adv., 2011, 29, 675–685 CrossRef CAS PubMed.
- P. Yang, H. Kobayashi and A. Fukuoka, Chin. J. Catal., 2011, 32, 716–722 CrossRef CAS.
- W. Deng, X. Tan, W. Fang, Q. Zhang and Y. Wang, Catal. Lett., 2009, 133, 167–174 CrossRef CAS.
- L. S. Ribeiro, J. J. M. Órfão and M. F. R. Pereira, Green Chem., 2015, 17, 2973–2980 RSC.
- M. Muin, A. Adachi, M. Inoue, T. Yoshimura and K. Tsunoda, J. Wood Sci., 2003, 49, 65–72 CrossRef CAS.
- M. Gao, F. Xu, S. Li, X. Ji, S. Chen and D. Zhang, Biosyst. Eng., 2010, 106, 470–475 CrossRef.
- N. Narayanaswamy, A. Faik, D. J. Goetz and T. Gu, Bioresour. Technol., 2011, 102, 6995–7000 CrossRef CAS PubMed.
- A. R. C. Morais, A. C. Mata and R. Bogel-Lukasik, Green Chem., 2014, 16, 4312–4322 RSC.
- A. Bellè, T. Tabanelli, G. Fiorani, A. Perosa, F. Cavani and M. Selva, ChemSusChem, 2019, 12, 3343–3354 CrossRef PubMed.
- D. Polidoro, A. Perosa and M. Selva, ChemSusChem, 2022, 15, e202201059 CAS.
- A. Bellè, K. Kusada, H. Kitagawa, A. Perosa, L. Castoldi, D. Polidoro and M. Selva, Catal. Sci. Technol., 2022, 12, 259–272 RSC.
- C. Schacht, C. Zetzl and G. Brunner, J. Supercrit. Fluids, 2008, 46, 299–321 CrossRef CAS.
- K. L. Toews, R. M. Shroll, C. M. Wai and N. G. Smart, Anal. Chem., 1995, 67, 4040–4043 CrossRef CAS.
- L. V. A. Gurgel, M. T. B. Pimenta and A. A. da S. Curvelo, Ind. Crops Prod., 2014, 57, 141–149 CrossRef CAS.
- H. Lv, L. Yan, M. Zhang, Z. Geng, M. Ren and Y. Sun, Chem. Eng. Technol., 2013, 36, 1899–1906 CrossRef CAS.
- A. A. Marianou, C. M. Michailof, A. Pineda, E. F. Iliopoulou, K. S. Triantafyllidis and A. A. Lappas, ChemCatChem, 2016, 8, 1100–1110 CrossRef CAS.
- W. Yin, Z. Tang, R. H. Venderbosch, Z. Zhang, C. Cannilla, G. Bonura, F. Frusteri and H. J. Heeres, ACS Catal., 2016, 6, 4411–4422 CrossRef CAS.
- S. Yamaguchi, S. Fujita, K. Nakajima, S. Yamazoe, J. Yamasaki, T. Mizugaki and T. Mitsudome, ACS Sustainable Chem. Eng., 2021, 9, 6347–6354 CrossRef CAS.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 1–10 CrossRef PubMed.
- I. Sádaba, M. L. Granados, A. Riisager and E. Taarning, Green Chem., 2015, 17, 4133–4145 RSC.
- E. Barbaro, T. Kirchgeorg, R. Zangrando, M. Vecchiato, R. Piazza, C. Barbante and A. Gambaro, Atmos. Environ., 2015, 118, 135–144 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPAEC-MS quantification protocol and product analysis. See DOI: https://doi.org/10.1039/d3gc01813j |
| This journal is © The Royal Society of Chemistry 2023 |