
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReengineering of the carbon-to-acetylene process featuring negative carbon emission†
Miao
Li
 ab,
Hong
Zhao
*a,
Siyuan
Chen
a,
Siyuan
liu
a,
Long
Yan
c,
Chen
Hou
a and
Biao
Jiang
*a
ab,
Hong
Zhao
*a,
Siyuan
Chen
a,
Siyuan
liu
a,
Long
Yan
c,
Chen
Hou
a and
Biao
Jiang
*a
aGreen Chemical Engineering Research Center, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201210, China. E-mail: zhaoh@sari.ac.cn; jiangb@sari.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cSchool of Chemistry and Chemical Engineering, Yulin University, Yulin 719000, China
First published on 16th August 2023
Abstract
Various sources of carbon can be converted into acetylene (C2H2) by using the key intermediate calcium carbide (CaC2). However, the production of CaC2 is a typical energy-intensive process, accompanied by considerable carbon dioxide (CO2) emissions and a large amount of industrial solid waste. In this study, a sustainable methodology for carbon-to-acetylene and carbon monoxide (CO) co-production as well as CO2 capture based on BaCO3−BaC2−Ba(OH)2−BaCO3 looping was first established, in which BaC2 replaced CaC2 as the key intermediate of the carbon-to-acetylene process to generate C2H2. The kinetic behavior investigation of the BaC2 formation indicated that the solid-phase synthesized BaC2 is a promising intermediate for the carbon-to-acetylene conversion owing to its faster kinetics, lower formation temperature, and no carbon dioxide release compared with those observed for the CaC2 production. Moreover, the lab-scale recovery of barium to carbide formation was conducted as the proof-of-concept to validate the coupling process of carbon-to-acetylene with CO2 capture based on Ba looping, resulting in less carbide slag waste and negative carbon emission. The facile co-production of carbon monoxide, environmentally friendly process, and convenience of large-scale production, as well as possible independent manufacturing of fossil resources, make barium carbide-based carbon-to-C2H2 -CO a promising key chemical platform for sustainable development. The proposed technology would provide new insights into the reengineering process of carbon to chemicals.
Introduction
While artificial synthetic chemicals are rapidly gaining importance as a cornerstone of modern society, they rely heavily on unsustainable fossil resources and pose a serious threat to the environment. Currently, one of the key global concerns in industrial chemistry is to design and invent green reactions and processes to sustainably prepare chemicals by using renewable resources, clean technologies, and revalorization of waste.1–4 With a thermodynamically unstable carbon–carbon triple bond, acetylene can undergo diverse coupling/addition reactions, serving as a versatile building block for the syntheses of organic compounds, including vinyl chloride, acrylonitrile, vinyl acetate, 1,4-butanediol (BDO), etc.5–8 C2H2 is also widely used in welding, brazing, cutting, hardening, texturing, and thermal spraying.9 Recently, C2H2 has been identified as a primary core for new drugs and biologically active molecules, with potential access to novel carbon nanomaterials and is promising for molecular 3D printing.10–13 In particular, with the increasing limitations on the fossil resource supply, C2H2, which can be conveniently produced from renewable biomass instead of fossil feedstocks, could usher in new opportunities to be a key chemical platform for sustainable development.14Since 1892, C2H2 has been industrially produced by the calcium carbide method using coal as a carbon source.15 In fact, inexpensive and available sources of carbon materials, including coke,16,17 anthracite,18,19 biochar,20,21 pyrolysis char,22etc., can react with lime (CaO) to produce calcium carbide (3C + CaO = CaC2 + CO), which is a key intermediate that reacts with water to produce acetylene (CaC2 + H2O = Ca(OH)2 + C2H2). In the coal chemical industry, the coal-to-acetylene process is shorter and lower equipment investment is required, compared with the coal-to-olefin process led by coal gasification. However, because of the highly endothermic nature of CaC2 formation, this seemingly simple chemical reaction has to be performed industrially in an electric arc furnace at 2000–2300 °C for approximately 2 h to overcome dynamic and thermodynamic restrictions (poor mass and heat transfer as well as a slow reaction rate).23–26 In addition, considerable carbon dioxide (CO2) emissions generated from the conversion of limestone (CaCO3) to CaO, as well as a large amount of solid waste of the by-product CaC2 slag (the main component is insoluble Ca(OH)2),27 originated from C2H2 generation, have exacerbated the calcium-based carbon-to-acetylene process.28,29 The disadvantages of high temperature, high energy consumption, and high waste emissions in calcium carbide-based C2H2 production have seriously restricted the whole acetylene chemical industry. In the past few decades, the plasma reactor,30,31 the rotary kiln process,32 the microwave heating method,33,34 the oxygen-thermal method,35,36 and catalytic synthesis37,38 have been tried to produce CaC2. However, these efforts, mainly focused on the dynamic condition optimization and the new reactor development for CaC2 synthesis, have made limited progress in achieving green manufacturing of calcium carbide-based C2H2.
As mentioned above, the drawbacks in calcium carbide-based C2H2 production are inherently related to the corresponding chemical reactions associated with the calcium cycling of CaCO3−CaO–CaC2−Ca(OH)2. That is, on the basis of calcium carbide, it should be very difficult to achieve a sustainable acetylene chemical industry, as shown in Fig. 1a.
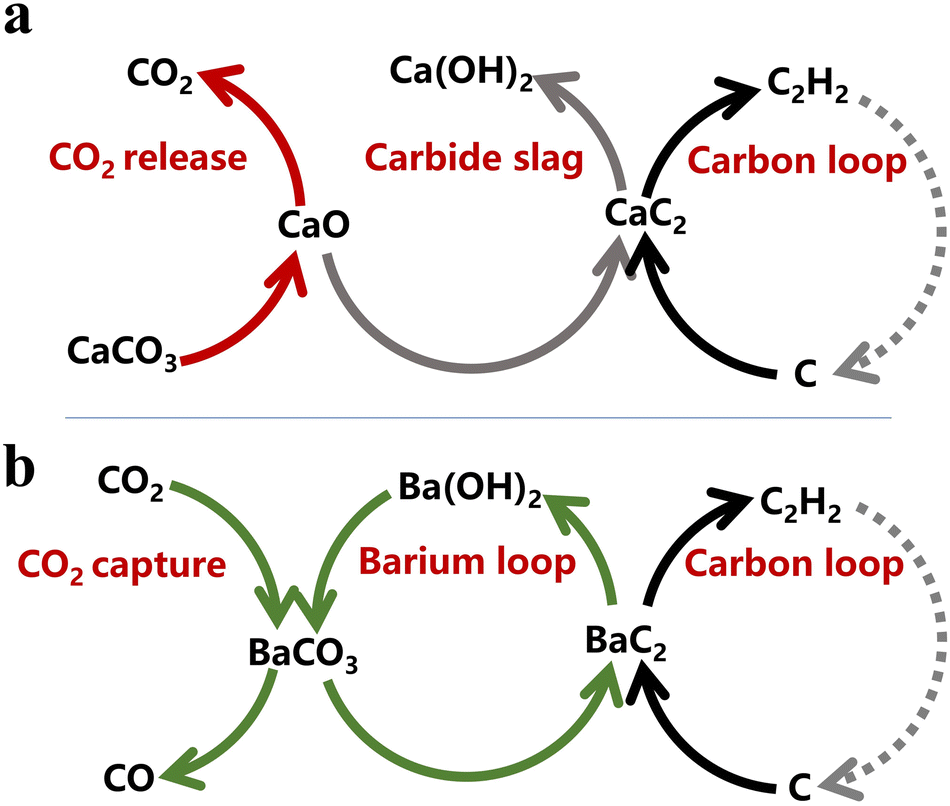 | ||
| Fig. 1 Schematic of the carbon-to-acetylene process. (a) Calcium-based carbon-to-acetylene process. (b) Coupling process of carbon-to-acetylene with CO2 capture based on Ba looping. | ||
Bearing the same C22− anion as CaC2, barium carbide can react with water to release C2H2,39,40 indicating its potential as the key intermediate in carbon-to-acetylene production. As early as 1892, a product containing approximately 38 wt% BaC2 was first synthesized by heating a mixture of barium carbonate, carbon, and magnesium at 700–800 °C (BaCO3 + 3Mg + C = BaC2 + 3MgO + CO).41 Subsequently, Thomson confirmed the formation of BaC2 by the reaction of BaO with carbon black in a vacuum at 1400 °C (BaO + 3C = BaC2 + CO).42 Later, Napier reported that barium carbide could be produced at approximately 1650 °C under normal pressure.43 The above studies indicated a more moderate synthesis temperature and less energy consumption for barium carbide. Unfortunately, owing to the global prevailing petrochemical processes based on the cost-effectiveness of ethylene and propene, the research on the coal-to-acetylene process has been interrupted considerably, and barium carbide has not received sufficient attention, not to mention its consideration as a commercial intermediate for carbon-to-acetylene production. Recently, with the expanding market demand for degradable plastics, polyvinylidene fluoride, etc., the importance of carbon-to-acetylene chemistry has been reassessed, especially in areas containing more coal and less oil. For example, more than 42 million metric tons of CaC2 were produced in 2021 in China. Hence, the carbon-to-acetylene process based on barium carbide must be re-evaluated.
In this study, a proof-of-concept of the negative carbon emission technology for the carbon-to-acetylene process and carbon dioxide utilization based on the barium cycle was first proposed (Fig. 1b). Pellets of a BaCO3 and coconut shell carbon mixture were used as the reactants to demonstrate the novel methodology of C2H2 production based on the solid-state synthesis of barium carbide. Emphasis was placed on the dynamic behaviours during the solid-phase formation of carbide, which was investigated using a thermogravimetric (TG) analyser and a high-temperature fixed-bed reactor equipped with an online gas chromatography (GC) instrument, respectively. In addition, the barium recovery and reuse for the carbide production were explored to demonstrate the coupling process of carbon-to-acetylene with CO2 capture based on practical, not conceptual, barium looping, resulting in less carbide slag waste. This work indicates that the barium-based carbon-to-acetylene process was more economical, more environmentally friendly than that based on calcium carbide, and would be executable with bright application prospects.
Results and discussion
Reaction behaviours of BaCO3 and C in the thermogravimetric analyser
A milligram-scale reaction was conducted in the simultaneous thermal analyser to explore the reaction behaviours of C and BaCO3 and the result is shown in Fig. 2a and Table S1.† A two-stage process was clearly observed for the mass loss at temperatures ranging from room temperature to 1300 °C, implying that two reactions occurred during the heating process. The first stage started at approximately 580 °C and ended at 950 °C, where a maximum mass loss rate was observed at approximately 800 °C. In this stage, the mass loss was 22.6 wt%, which was very close to the theoretical mass loss of 22.8 wt% in the thermodynamically advantageous reaction of barium carbonate with carbon to form barium oxide and carbon monoxide (BaCO3 + C = BaO + 2CO), as predicted by thermodynamic analysis.44 The result indicated that there should be less CO2 emissions caused by direct BaCO3 decomposition (BaCO3 = BaO + CO2), benefiting from the noticeable distinction in thermodynamic reaction temperatures between 1047 °C (BaCO3 = BaO + CO2) and 700 °C (CO2 + C = CO).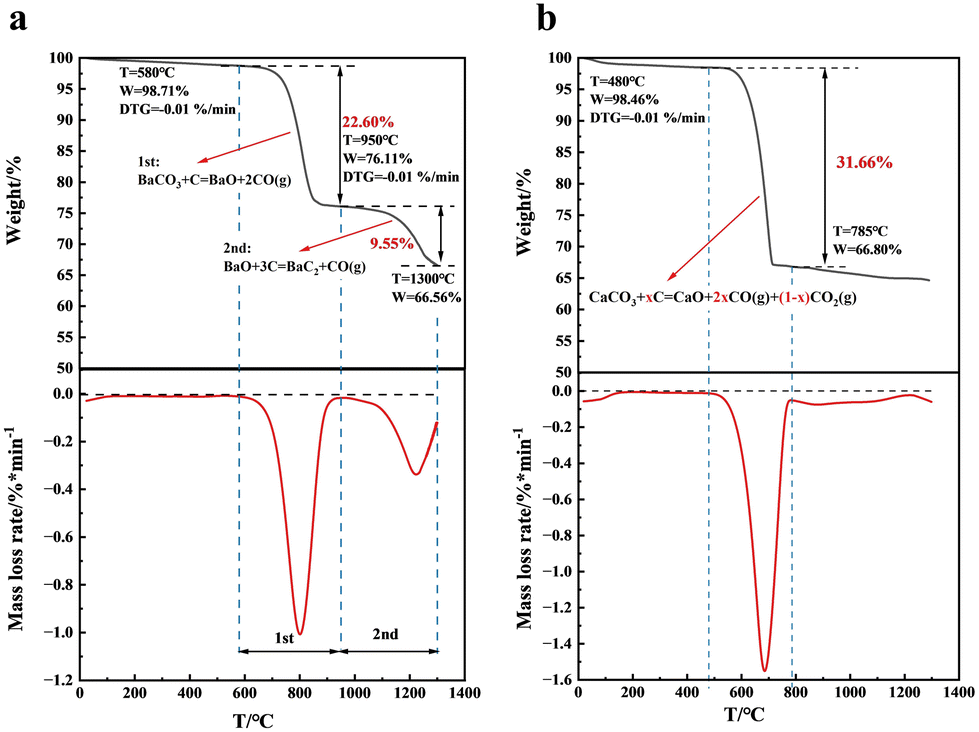 | ||
| Fig. 2 Reaction behaviours of BaCO3 and C. (a) TG/DTG curves of a mixture of C/BaCO3. (b) TG/DTG curves of a mixture of C/CaCO3. | ||
When the heating temperature was greater than 950 °C, the first mass loss process ended, while the second mass loss process started slowly, due to the formation of BaC2 from the prepared BaO and the remaining C. In this stage, the DTG curve displayed a comparable trend, but with lower values in terms of the loss rate than those in the first stage. The result suggested a lower gas release amount and a slower reaction rate of BaC2 formation than those of BaCO3 decomposition. With an increase in the heating temperature, the mass loss rate constantly increased and then peaked at approximately 1065 °C, where the maximum formation rate of BaC2 was observed. Subsequently, the mass loss rate progressively decreased because of the constant consumption of BaO and C. At a temperature of 1300 °C, the mass loss was 9.6 wt% in the second stage of the reaction. According to the theoretical mass loss of 14.8 wt% for the BaC2 formation reaction (BaO + 3C = BaC2 + CO), 64.9% of the BaO obtained in the first stage should be converted into BaC2 during the second process.
Different from that observed in the barium system, the mass loss of the CaCO3 and C mixture with a C/Ca molar ratio of 4 occurred mainly in the low temperature range, shown in Fig. 2b. The DTG curve clearly exhibited only one major mass loss peak in the whole investigated temperature range, which started at 480 °C and peaked at 685 °C with a mass loss of 31.4 wt%, and no clear DTG peak was observed at higher temperatures. The result implied that the main mass loss was attributed to the decomposition of CaCO3, and that CaC2 was hardly formed during the heating process. As shown in Table S1,† if CaCO3 was completely decomposed into CO2 and CaO (CaCO3 = CaO + CO2), the weight loss should be 29.7 wt%, while if CaCO3 was decomposed to CO and CaO (CaCO3 = CaO + CO2, CO2 + C = 2CO), the weight loss should be 37.8 wt%. The experimental mass loss related to the decomposition of CaCO3 was 31.7 wt%, which was between 29.7 and 37.8 wt%, signifying that approximately 60.9% CO and 39.1% CO2 were produced during CaCO3 decomposition. The result should be due to the insufficient distinction in the thermodynamic reaction temperature between CaCO3 decomposition (886 °C) and the Boudouard reaction (700 °C), showing that a large amount of produced CO2 from the CaCO3 decomposition would release directly from the reaction system before reacting with C to convert to CO. The above comparative TG investigation results indicated that BaC2, as a key intermediate in the carbon acetylene process, has advantages over CaC2 due to its lower formation temperature and no CO2 emissions.
Reaction behaviours of BaCO3 and C in the fixed-bed reactor
To confirm the inference obtained by TG analysis, BaC2 and CaC2 were synthesized by using their corresponding pellet reactants in the high-temperature fixed-bed reactor, and the gas-phase products obtained during the heating processes were monitored by online GC. Fig. 3a and b show the temperature dependence of the gas by-products by time-on-stream in barium and calcium systems, respectively. Not surprisingly, Fig. 3a clearly shows two separate CO peaks centred at 1025 °C and 1500 °C, respectively. Their integral area ratio is 1.96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, close to the theoretical value of 2:1 (BaCO3 + C = BaO + 2CO and BaO + 3C = BaC2 + CO), conforming the two-step reaction route for the formation of BaC2 from BaCO3 and C. CO2 was not detected in the outlet gas of the barium system during the entire reaction, confirming the absence of CO2 emissions in the barium system. Compared with the TG analysis result, the two CO peaks appear at higher temperatures. This should be due to the negative effects of mass and heat transfer in a large dosage reaction system, where pellet reactants replaced the powder materials used in TG analysis. Moreover, the solid products obtained at the ends of the above two reaction stages were separately collected and characterized by XRD analysis, and their XRD spectra are shown in Fig. 4a. The XRD pattern of the solids obtained at the end of the first reaction stage (heating for 250 min) revealed characteristic peaks of BaO (JCPDS 65-2923) with the Fm
:1, close to the theoretical value of 2:1 (BaCO3 + C = BaO + 2CO and BaO + 3C = BaC2 + CO), conforming the two-step reaction route for the formation of BaC2 from BaCO3 and C. CO2 was not detected in the outlet gas of the barium system during the entire reaction, confirming the absence of CO2 emissions in the barium system. Compared with the TG analysis result, the two CO peaks appear at higher temperatures. This should be due to the negative effects of mass and heat transfer in a large dosage reaction system, where pellet reactants replaced the powder materials used in TG analysis. Moreover, the solid products obtained at the ends of the above two reaction stages were separately collected and characterized by XRD analysis, and their XRD spectra are shown in Fig. 4a. The XRD pattern of the solids obtained at the end of the first reaction stage (heating for 250 min) revealed characteristic peaks of BaO (JCPDS 65-2923) with the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, and no characteristic peaks of BaCO3 were detected. The results indicated that BaCO3 was completely transformed into BaO when the reactants were heated to approximately 1150 °C. In the XRD pattern of the solids obtained after heating for 600 min, i.e., the end of the second reaction stage, the characteristic diffraction peaks of BaO disappeared; meanwhile, new diffraction peaks were observed at 23.8°, 25.2°, 28.6°, 38.5°, 40.9°, 43.5°, and 47.9°, which are indexed to BaC2 (JCPDS 65-2661) with the I4/mmm space group. The BaC2 content in it was 97.5% by measuring the corresponding C2H2 amount from the hydrolysis of the solid products (BaC2 + H2O = C2H2 + Ba(OH)2).
m space group, and no characteristic peaks of BaCO3 were detected. The results indicated that BaCO3 was completely transformed into BaO when the reactants were heated to approximately 1150 °C. In the XRD pattern of the solids obtained after heating for 600 min, i.e., the end of the second reaction stage, the characteristic diffraction peaks of BaO disappeared; meanwhile, new diffraction peaks were observed at 23.8°, 25.2°, 28.6°, 38.5°, 40.9°, 43.5°, and 47.9°, which are indexed to BaC2 (JCPDS 65-2661) with the I4/mmm space group. The BaC2 content in it was 97.5% by measuring the corresponding C2H2 amount from the hydrolysis of the solid products (BaC2 + H2O = C2H2 + Ba(OH)2).
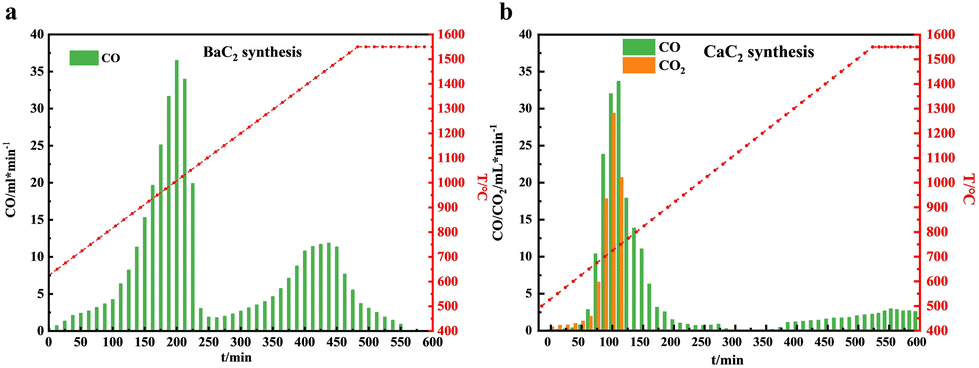 | ||
| Fig. 3 Gas phase product analysis of the Ca system and Ba system. Temperature dependence of the gas products by time-on-stream in the (a) barium system and (b) calcium system. | ||
 | ||
| Fig. 4 XRD patterns of reactants and their corresponding products. (a) Ba system. (b) Ca system. (1) reactants and (2) heating products at 250 min (Ba system) and 325 min (Ca system), and (3) heating products at 600 min. | ||
Fig. 3b shows the temperature dependence of the gas by-products by time-on-stream in the calcium system. It is obvious that in the lower temperature region, the CO peak overlapped significantly with the CO2 peak, corresponding to CaCO3 decomposition. The integral area ratio of CO to CO2 was 2.5:1, indicating 71.4% of CO and 28.6% of CO2 in the outlet gas. In the high temperature range, with a continuous increase in the temperature to 1550 °C, the CO concentration increased, but still remained at a low level, and no clear CO peak was observed in the outlet gas. Fig. 4b shows the XRD pattern of the solid products obtained by heating for 325 min and 600 min. Both the solid products only exhibited obvious characteristic peaks of CaO (JCPDS 37-1497), and the CaC2 characteristic diffraction peaks were not clearly detected, indicating that although a small amount of CO was observed when the temperature was beyond 1150 °C, the amount of CaC2 formed was very small. The results are in good agreement with those obtained from TG analysis, confirming that BaC2 was a promising candidate to replace CaC2 as the key intermediate of carbon-to-acetylene for the lower manufacturing temperature without CO2 emission.
To explore the effects of kinetic conditions on the formation of BaC2, a series of experiments were conducted at 1400–1550 °C with different temperatures and holding times in a high-temperature fixed-bed reactor. Fig. 5a shows the BaC2 yields while Fig. 5b shows the corresponding average reaction rates. As shown in Fig. 5a, the BaC2 yield exhibited a strong dependence on the temperature and reaction time. Notably, the reaction proceeded rather rapidly in the first 30 min, then gradually slowed down, and finally reached a steady state after approximately 1 h of preservation. Moreover, with an increase in the reaction temperature, a shorter heating time was needed to reach the same BaC2 yield. For example, a 75.2% BaC2 yield was obtained after heating for 60 min at 1400 °C, and a 75.8% BaC2 yield was obtained after heating for 30 min at 1500 °C. At 1550 °C, an 81.6% BaC2 yield was achieved after heating for 20 min, and up to a 97.5% yield after heating at 60 min. Pillai et al.33 reported that only a 14.1% CaC2 yield was obtained after heating for 30 min at 1700 °C when 10 g of pressed pellets of the graphite and CaO fine powder mixtures were used as the reactant. These results confirmed that the reaction conditions for the synthesis of barium carbide were considerably milder than those utilized for the synthesis of CaC2.
 | ||
| Fig. 5 The effects of kinetic conditions on the formation of BaC2.(a) Dependence of the BaC2 yield and Ba mass balance on the reaction temperature and time; (b) BaC2 reaction rate dependence on the reaction temperature and time. | ||
Considering the fact that calcium would be lost as calcium vapor during the production of CaC2,45 the mass balance of barium before and after barium carbide formation was investigated further. In our experimental ranges, elemental Ba was not detected in the outlet gas, and the loss of Ba, defined as the relative difference between the total amount of barium in the reactants and solid products, was less than 0.3% (Table S2†); this value was possibly related to a systematic error in the experimental operation. The results were in good agreement with the thermodynamic prediction that barium carbide cannot react further with barium oxide to form barium vapor and carbon monoxide (BaC2 + 2BaO = 3Ba + 2CO) at temperatures less than 2175 °C,44 indicating the good stability of BaC2 at high temperatures.
Morphology of the synthesized solid BaC2
Fig. 6 shows the SEM images and Fig. S1† shows the corresponding EDS mapping images of the reactants and the heated solid products obtained under different reaction conditions. As shown in Fig. 6a and a1, the rod-shaped BaCO3 particles and the irregularly shaped coconut shell carbon particles were physically mixed together before the reaction. These particles exhibited clear sharp edges, and the particle size of the majority of BaCO3 was about 5–10 μm and the carbon was about 5–30 μm. After heating at 1150 °C, the rod-shaped BaCO3 particles disappeared (Fig. 6b and b1), and some new aggregated fine particles were observed and easily distinguished from the larger carbon particles with no obvious morphological changes. The result was in good agreement with the TG and XRD analysis results that BaCO3 was decomposed into BaO first. Fig. 6c and c1 show the SEM images of the heated solid products with a BaC2 content of 74.1 wt% obtained at 1400 °C for 60 min. The aggregated fine BaO particles, mainly observed in the solids heated at 1150 °C, seemed to have migrated to carbon particles and developed into new larger ones with an irregular shape. Moreover, the less isolated carbon particles were observed. The particle surface with several small pores was flatter compared with the BaO particles. It can be speculated that a partial liquid-phase formed from a microscopic perspective, during the BaC2 formation process, and the pores could be due to the release of CO from the reactive interfaces. The results provided evidence to propose the diffusion of barium into the carbon materials and the formation of BaC2 on the internal and external surfaces of carbon particles, which was similar to the mass-transfer behaviours of CaO and C during CaC2 formation.17Fig. 6d and d1 show the SEM images of the BaC2 product with a 91.4 wt% content obtained by heating at 1550 °C for 30 min. The isolated carbon particles that were clearly in the other samples disappeared, which was in good agreement with the high BaC2 content. The particles developed into new larger ones rather than that formed at 1400 °C and all particles in these samples exhibited clear shapes and edges. As shown in Fig. S2,† all of the heated pellets rather retained their original shape and did not melt or stick together, confirming the solid conversion mechanism of BaC2, from a macroscopic perspective. Differing from the liquid-state synthesis of the industrial production of CaC2, the solid conversion mechanism of BaC2 was confirmed facilitating continuous operation during future large-scale industrial production. Moreover, Fig. 6c1 and d1 clearly show that the obtained barium carbide products are very porous, and would be very easy to react with water to generate C2H2, and would greatly facilitate the industrial operation in the carbon-to-C2H2 process. We know that in the calcium-based carbon-to-acetylene process, the liquid-phase formed CaC2 products are microstructure-densified and must be broken into small pieces of several millimetres before reacting with water to generate C2H2. The CaC2 dust generated in the crushing process is so hazardous that this process is typically carried out underground. | ||
| Fig. 6 SEM images of the reactant and products: (a and a1) reactant and (b and b1) heating products at 1150 °C, (c and c1) at 1400 °C for 60 min, and (d and d1) at 1550 °C for 30 min. | ||
Ba recovery to carbide synthesis
In the industrial calcium-based C2H2 generation process, each ton of industrial CaC2 (about 80% pure) would produce 1.2 tons of dry CaC2 slag, composed primarily of Ca(OH)2 and other water-insoluble inorganic salts originating from the ashes of carbon raw materials. In practice, CaC2 slag barely can be recycled for the production of carbide. One reason is the lower reactivity of the recovered CaO from the slag than that of the fresh one.46 The other important reason is no economic method to remove the inorganic impurities from the recovered CaO, which would accumulate in the arc furnace and lead to increased energy consumption and operational risks, as well as a lower quality of the carbide product.Fortunately, the carbon-to-C2H2 process based on barium looping is another matter. Different from Ca(OH)2, the soluble concentration of Ba(OH)2 in water was strongly dependent on the temperature (Fig. S4†). In cold water, the solubility of barium hydroxide was almost as low as that of Ca(OH)2, but in hot water of 80 °C, the solubility of Ba(OH)2 reached 100 g/100 g water. Hence, in the barium system, water-insoluble inorganic impurities can be easily separated by filtration from the hot Ba(OH)2 solution, which was obtained from the exothermic reaction of the generation of C2H2 gas. This excellent water solubility of Ba(OH)2 suggested that less solid waste would be produced in the barium-based carbon-to-acetylene process.
Considering that Ba(OH)2 can easily absorb CO2 to form BaCO3, the recovered Ba(OH)2 can be used directly as a CO2 adsorbent to regenerate BaCO3. Here, BaCO3 was regenerated from its carbide product by simple exposure to water-saturated CO2 gas, because there were little impurities in the reactants. As can be observed from Fig. S5a,† the regenerated BaCO3 samples exhibited a smaller particle size than that of the fresh one. When the regenerated BaCO3 was used as the reactant under the same conditions, the BaC2 yield reached 97.0%, similar to that of the fresh one (97.2%), indicative of the good activity of the recovered BaCO3.
On this basis, the carbon-to-C2H2 process based on barium looping is proposed and shown in Fig. 7. The entire system of the new process can be divided into three units: the barium carbide production unit, the C2H2 production unit, and the CO2 capture and barium carbonate recovery unit. In the barium carbide production unit, the powder mixture of barium carbonate and carbon-rich materials (biochar, coke, pyrolytic char, etc.) would be granulated into small pellets using a pellet mill. After drying, the dried pellets would be put into a high-temperature reactor to produce barium carbide. In this unit, the off-gas mainly containing CO and dust would be delivered into a separator and a heat exchanger to obtain dust-free cooled CO gas, which would be transferred to the next process, while the recovered energy would be used to preheat the reactants. The hot barium carbide solids would be cooled by a heat exchanger either directly or indirectly; the as-obtained cooled products would be transported to storage bins. In the C2H2 production unit, the carbide from the storage bins would be delivered into the C2H2 generator, where crude C2H2 and the hot barium hydroxide aqueous solution with some insoluble impurities would be formed. In this unit, the obtained barium carbide is porous and can react quickly with water to generate acetylene, so the industrial acetylene generator for the calcium-based process could be fit for the new process after small modifications. The crude C2H2 would be transferred to the refining process to obtain pure C2H2, while the hot slurry comprising barium hydroxide and the insoluble salts would be fed into a hot solid–liquid separator to obtain a hot barium hydroxide solution. In the CO2 capture process, CO2-rich gas can be bubbled into the hot barium hydroxide solution to accelerate the conversion of barium hydroxide to barium carbonate, which would be fed to the barium carbide production unit and used as a raw material. Notably, an appropriate amount of fresh barium carbonate should be supplied to the reaction system to compensate for the loss of barium, caused by the side-reaction of barium oxide with acidic impurities, mainly silicon dioxide, and is generally present in various carbon sources, to produce insoluble barium salts, which are important raw materials for producing anti-electromagnetic radiation concrete.
 | ||
| Fig. 7 Schematic diagram of the carbon-to-acetylene process with the co-production of carbon monoxide using BaC2 as an intermediate. | ||
We know well that gasification is one of the most proven approaches to large-scalely transform carbonaceous fuels into syngas, which mainly consists of a mixture of H2 and CO, and then into a wide range of chemicals via the F–T (Fischer–Tropsch) process, the MTO (Methanol to Olefin) process and so on. In the coal chemical industry, syngas production by coal gasification is regarded as the leading process among all and has been used on a large scale for over 150 years. Shell Coal gasification technology, one of the world-renowned gasification technology, can process petroleum coke, anthracite, bituminous coal, lignite and biomass into syngas at 4.0 MPa operating pressure and 1400–1700 °C gasification temperature.
Compared with the gasification technology, the barium-looping based new process, shown in Fig. 8, has the ability to directly convert a large variety of carbonaceous materials into CO and the advanced acetylene but not H2 under even mild reaction conditions. The novel method for carbon-to-chemical conversion based on barium-looping is more competitive with the shorter and simpler process flow, which would provide new insights for the coal chemical industry and organic synthesis industry.
 | ||
| Fig. 8 Schematic of the barium-based carbon-to-acetylene process with the co-production of carbon monoxide. | ||
Conclusions
The carbide-based carbon-to-acetylene process proposes a pathway for converting various sources of carbon into acetylene directly, which has an irreplaceable place in the acetylene chemical industry. However, the traditional calcium carbide production is a typical energy-intensive and heavy waste discharge process, which greatly limited its development. In this study, a novel sustainable coupling technology based on barium looping for carbon-to-C2H2 conversion and CO2 capture is designed and implemented for the first time to realize the simultaneous production of C2H2 and CO with a lower energy consumption and lower waste discharge. The research results demonstrate that this method is technically simple, feasible, and environmentally friendly, which is expected to achieve the sustainable reconstruction of the carbon-to-C2H2 process. Moreover, because of the high efficiency of co-production of acetylene and carbon monoxide, this technology is expected to have the prospect of partially replacing coal gasification, which is usually the first process in the modern coal chemical industry to produce CO and H2 as platform chemicals. Our work would provide new insights into the conversion of carbon into chemicals by a simpler, greener, and more efficient method.Finally, it should be noted that, unlike the denser, more transportable calcium carbide produced by electric arc furnaces, solid-state synthesized granular barium carbide is porous and easily reacts with water to form acetylene. As a result, it is unstable in air, and the use of barium carbide far from the production site may lead to unsafe transportation over long distances. Thus, it is better to produce acetylene on site.
Experimental
Preparation of reaction powder and pellets
As a carbon resource, coconut shell carbon (99.8% fixed carbon, Ningxia Guanghua Activated Carbon Co., Ltd) was ground and sieved to obtain a carbon powder with a particle size of 150–180 μm. Barium carbonate (BaCO3, >99.0%, Shanghai Titan Scientific Co., Ltd) with a particle size of 5–10 μm was used as the barium resource. A mixture of the sieved coconut shell carbon powder and barium carbonate powder in a C/Ba molar ratio of 4 was completely mixed and then dried at 120 °C for 12 h to obtain the reactant powder.BaCO3-C pellets prepared by wet granulation were used as the reactants for the synthesis of BaC2 in a fixed-bed reactor (Fig. 9a). First, a mixture of the sieved coconut shell carbon powder and barium carbonate powder in a C/Ba molar ratio of 4 was completely mixed, followed by the addition of 20 wt% deionized water and continuous stirring to obtain a reactant slurry. After the slurry was granulated in a disc granulator, wet reactant pellets with a diameter of 5 mm were obtained. Next, the wet pellets were dried at 120 °C for 12 h to obtain the reactant pellets.
 | ||
| Fig. 9 Schematic of the (a) fixed-bed reactor and (b) lab-made acetylene gas generator equipment. | ||
Reaction behaviour investigation of BaCO3-C
The reaction behaviours of the BaCO3-C reactants were first observed using a simultaneous thermal analyser SDT Q600 (TA, USA). First, 10 mg of the BaCO3–C reactant mixture with a C/Ba molar ratio of 4:1 was loaded in a TG crucible, and the temperature was increased from room temperature to 1300 °C at a rate of 5 °C min−1 under an Ar flow of 120 mL min−1. Blank experiments (without a sample) were conducted under the same conditions to correct the TG data by eliminating the buoyancy effect caused by the ramp in the temperature.
The reaction behaviours of the BaCO3-C reactants were further explored in a fixed-bed reactor (Fig. 9a). The fixed-bed reactor used for barium carbide synthesis was adapted from a high-temperature furnace reactor (Y-Feng Shanghai Co., Ltd), and it was equipped with an on-line GC instrument. The length and internal diameter of the reaction tube were 1200 mm and 50 mm, respectively, which was fitted with a corundum tube lined with graphite. Molybdenum silicide was used as the heating element, and the reaction temperature was regulated using an automatic temperature controller. Ar was used as the carrier gas, which was precisely regulated using a mass-flow meter (Beijing Seven Star Co., Ltd). In this section, 10 g of the reactant pellets placed in a uniform temperature zone were heated to the target temperature (1550 °C) at a rate of 2 °C min−1. After the reaction, the heated solid samples were cooled to a temperature of less than 100 °C and rapidly transferred to a glove compartment filled with N2 for subsequent analysis and characterization.
Barium carbide synthesis
Typically, 200 g of the reactant pellets placed in a uniform temperature zone were heated to the target temperature (1400–1550 °C) at a rate of 2 °C min−1, which were maintained at the target temperature for different times (20–60 min). After the reaction, the heated solid samples were cooled to a temperature less than 100 °C and rapidly transferred to a glove compartment filled with N2 for subsequent analysis and characterization.Product analysis
The crystalline phase structures of the heated solid samples were detected by X-ray diffraction (XRD, Mini Flex 600) with Cu Kα radiation (40 kV, 15 mA) at a scan rate of 3° min−1. The powder sample was sealed in an airtight sample bin to avoid possible air contact during the XRD tests. The morphology and microstructure of the synthesized solid samples were monitored using a field-emission scanning electron microscope (FE-SEM, Gemini SEM 300). The barium balance before and after the reaction was determined by inductively coupled plasma spectrometry (ICP, Thermo iCAP7600 USA). The yields of barium carbonate to carbide were calculated by measuring the corresponding C2H2 amounts from the hydrolysis of the synthesized solid samples in lab-made C2H2 gas generator equipment (Fig. 9b), which was qualified with standard CaC2 samples.The gas by-products obtained during BaC2 synthesis were analysed online on a SHIMADZU 2014C GC system equipped with a thermal conductivity detector (TCD) and a Porapak Q column (2 m × 4 mm).
Author contributions
Conceptualization: Biao Jiang and Hong Zhao; data curation: Hong Zhao, Miao Li, Siyuan Chen, and Chen Hou; formal analysis: Hong Zhao and Miao Li; funding acquisition: Hong Zhao and Long Yan; investigation: Hong Zhao and Miao Li; methodology: Hong Zhao and Miao Li; project administration: Hong Zhao; resources: Hong Zhao; software: Miao Li, Siyuan Chen, and Siyuan Liu; supervision: Hong Zhao; validation: Hong Zhao and Miao Li; visualization: Hong Zhao and Miao Li; writing – original draft: Miao Li; writing – review & editing: Hong Zhao.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Grant YLU-DNL Fund (2021013, 2021014) and the National Natural Science Foundation of China (22278417). The authors acknowledge the user experiment assistance system of SSRF.References
- G. Naray-Szabo and L. T. Mika, Green Chem., 2018, 20, 2171–2191 RSC.
- I. T. Horvath, Chem. Rev., 2018, 118, 369–371 CrossRef CAS PubMed.
- K. Kummerer, Angew. Chem., Int. Ed., 2017, 56, 16420–16421 CrossRef PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- S. P. Bedenko, K. I. Dement'ev and A. L. Maximov, Pet. Chem., 2022, 62, 989–1026 CrossRef CAS.
- I. T. Trotus, T. Zimmermann and F. Schuth, Chem. Rev., 2014, 114, 1761–1782 CrossRef CAS PubMed.
- H. Schobert, Chem. Rev., 2014, 114, 1743–1760 CrossRef CAS PubMed.
- J. Wu, Y. Ma, Y. Wang, C. Wang, H. Luo, D. Li and J. Yang, Green Chem., 2023, 25, 3425–3430 RSC.
- P. Pässler, W. Hefner, K. Buckl, H. Meinass, H. Wernicke, G. Ebersberg, R. Müller, J. Bässler, H. Behringer and D. Mayer, Ullmann's Encycl. Ind. Chem., 2011, 1, 277–326 Search PubMed.
- K. I. Galkin and V. P. Ananikov, Russ. Chem. Rev., 2016, 85, 226–247 CrossRef CAS.
- V. V. Voronin, M. S. Ledovskaya, A. S. Bogachenkov, K. S. Rodygin and V. P. Ananikov, Molecules, 2018, 23, 1–84 CrossRef CAS PubMed.
- G. P. Gakis, S. Termine, A. F. A. Trompeta, I. G. Aviziotis and C. A. Charitidis, Chem. Eng. J., 2022, 445, 136807 CrossRef CAS.
- A. Ramirez, C. Royo, N. Latorre, R. Mallada, R. M. Tiggelaar and A. Monzon, Mater. Res. Express, 2014, 1, 045604 CrossRef CAS.
- Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Green Chem., 2019, 21, 3499–3535 RSC.
- J. Morehead and G. d. Chalmot, J. Am. Chem. Soc., 1896, 18, 311–331 CrossRef.
- T. Xubo, M. Caixia, L. Qingya, L. Guodong and L. Zhenyu, J. Fuel Chem. Technol., 2010, 38, 539–543 Search PubMed.
- G. D. Li, Q. Y. Liu and Z. Y. Liu, Ind. Eng. Chem. Res., 2012, 51, 10742–10747 CrossRef CAS.
- Elion Resources Group, China Pat, CN103121685A; CN103121685B, 2013 Search PubMed.
- W. Xu and S. Zhang, China Pat, CN104355312A; CN104355312B, 2015 Search PubMed.
- Z. K. Li, Z. Y. Liu, R. X. Wang, X. J. Guo and Q. Y. Liu, Chem. Eng. Sci., 2018, 192, 516–525 CrossRef CAS.
- G. D. Li, Q. Y. Liu, Z. Y. Liu, Z. C. Zhang, C. Y. Li and W. Z. Wu, Angew. Chem., Int. Ed., 2010, 49, 8480–8483 CrossRef CAS PubMed.
- K. S. Rodygin, K. A. Lotsman, D. E. Samoylenko, V. M. Kuznetsov and V. P. Ananikov, Int. J. Mol. Sci., 2022, 23, 11828 CrossRef CAS PubMed.
- X. K. Zhang, Z. X. Tong, Y. L. He and X. Hu, Int. J. Heat Mass Transfer, 2021, 164, 120593 CrossRef CAS.
- R. Li, S. Ma, H. Ma, S. Liu and H. Wang, Appl. Therm. Eng., 2020, 181, 115877 CrossRef CAS.
- Q. Xu, Y. Li, S. Deng, Y.-L. He, L. Li and H. Yu, ACS Sustainable Chem. Eng., 2019, 7, 12510–12519 CAS.
- Y. Xiaomin, W. Jingsong, S. Xuefeng, J. Zeyi and X. Qingguo, Chem. Eng. Res. Des., 2022, 187, 516–528 CrossRef.
- K. A. A. Lotsman and K. S. S. Rodygin, Green Chem., 2023, 25, 3524–3532 RSC.
- X. Y. Liu, B. Zhu, W. J. Zhou, S. Y. Hu, D. J. Chen and C. Griffy-Brown, Int. J. Greenhouse Gas Control, 2011, 5, 1240–1249 CrossRef CAS.
- H. Yang, J. W. Cao, Z. Wang, H. H. Chen and X. Z. Gong, Int. J. Miner. Process., 2014, 130, 66–73 CrossRef CAS.
- C. W. Zhu, G. Y. Zhao and V. Hlavacek, J. Mater. Sci., 1995, 30, 2412–2419 CrossRef CAS.
- M. H. El-Naas, R. J. Munz and F. Ajersch, Plasma Chem. Plasma Process., 1998, 18, 409–427 CrossRef CAS.
- J. J. Mu and R. A. Hard, Ind. Eng. Chem. Res., 1987, 26, 2063–2069 CrossRef CAS.
- R. C. Pillai, E. M. Sabolsky, S. L. Rowan, I. B. Celik and S. Morrow, Ind. Eng. Chem. Res., 2015, 54, 11001–11010 CrossRef CAS.
- M. Li, S. Chen, H. Dai, H. Zhao and B. Jiang, Molecules, 2021, 26, 2568 CrossRef CAS PubMed.
- J. Guo, D. X. Zheng, X. H. Chen, Y. Mi and Z. Y. Liu, Ind. Eng. Chem. Res., 2013, 52, 17773–17780 CrossRef CAS.
- J. Guo and D. X. Zheng, Ind. Eng. Chem. Res., 2012, 51, 13414–13422 CrossRef CAS.
- Z. Chen, P. Cai, J. Cai, S. Hao, Y. Chen, W. Yu, F. Xu, K. You and H. a. Luo, J. Solid State Chem., 2022, 309, 123012 CrossRef CAS.
- S. H. I. Dejun, Q. Ke and Y. A. N. Zifeng, Front. Chem. Sci. Eng., 2011, 5, 372–375 CrossRef.
- U. Ruschewitz, Coord. Chem. Rev., 2003, 244, 115–136 CrossRef CAS.
- J. Feng, X. Bing and C. JingChao, Sci. China, Ser. B: Chem., 2008, 51, 545–550 CrossRef CAS.
- S. Barry, H. Fowler, R. Wallace, C. V. Boys, H. E. Jones, J. Aspinal, T. Parker, F. G. Worth, A. Kennedy and W. H. Hunter, Minutes of the Proceedings, 1898, vol. 134, pp. 34–60.
- M. deKay Thompson, Trans. Am. Electrochem. Soc., 1928, 54, 1–15 Search PubMed.
- D. H. Napier, Br. Coal Util. Res. Assoc., Mon. Bull., 1954, XVIII, 517 Search PubMed.
- L. Miao, Z. Hong, J. Biao, C. Siyuan and Y. Long, CIESC J., 2022, 73, 1908–1919 Search PubMed.
- L. M. Ji, Q. Y. Liu and Z. Y. Liu, Ind. Eng. Chem. Res., 2014, 53, 2537–2543 CrossRef CAS.
- X. Z. Gong, T. Zhang, J. Q. Zhang, Z. Wang, J. H. Liu, J. W. Cao and C. Wang, Renewable Sustainable Energy Rev., 2022, 159, 112133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01775c |
| This journal is © The Royal Society of Chemistry 2023 |