
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A phosphine free, inorganic base free, one-pot tandem Mizoroki–Heck olefination/direct arylation/hydrogenation sequence, to give multicyclic alkylated heteroarenes†
Roberta A.
Kehoe
ab,
Mark E.
Light
c,
David J.
Jones
 d and
Gerard P.
McGlacken
*ab
d and
Gerard P.
McGlacken
*ab
aSchool of Chemistry, Analytical and Biological Chemistry Research Facility (ABCRF), University College Cork, T12 YN60, Ireland. E-mail: g.mcglacken@ucc.ie
bSynthesis and Solid State Pharmaceutical Centre, University College Cork, T12 YN60, Ireland
cDepartment of Chemistry, University of Southampton, SO17 1BJ, UK
dEaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK
First published on 29th June 2023
Abstract
One-pot processes which facilitate a number of tandem reactions, represent an environmentally friendly approach to building molecular complexity. In addition, there are significant cost and time saving benefits. However, unearthing multiple reactions that display substrate, reagent and solvent compatibility across two or more reactions, is not straightforward. This is even more pronounced when using catalysts, which by their nature, are present in small amounts and thus susceptible to poisoning. Herein we describe a phosphine-free, inorganic base free, multi-step, one-pot reaction sequence which enables the rapid synthesis of complex, medicinally relevant heterocycles in excellent yields. This Pd-catalysed approach combines Heck olefination, C–H activation, and hydrogenation, and the same pre-catalyst is involved in the three mechanistically distinct processes. Quinolines as substrates are used in the main, but we also provide a neat extension to pyridines and simple aryl substrates. Deuterium (D2) incorporation can be facilitated through use of the COware apparatus in the reduction step. Favourable mass productivity (MP), environmental impact factor (EF), solvent intensity (SI) and process mass intensity (PMI) values are reported. While the motivation to avoid problematic additives (inorganic base and P-ligands) and problematic solvent (DMA and NMP) was driven by a green agenda, the removal of reagents and additives in the context of one-pot strategies might also serve to reduce the bad actors and improve yields in one-pot processes.
Introduction
The design of one-pot, multi-step reaction sequences is highly desirable in the context of green organic synthesis, as molecular complexity is rapidly achieved, whilst avoiding wasteful isolation and purification of reaction intermediates.1 Achieving substrate, reagent and solvent compatibility across two or more reactions is not straightforward and, thus, unearthing multiple compatible reactions, which work together in a single medium, is very challenging. This problem is even more pronounced when considering transition metal-catalysed reactions, as catalytic performance can be dramatically influenced by the various reaction inputs and outputs. This presents a barrier to the optimisation of multi-step, one-pot processes as the structure of the initial catalyst is not static, and changes over the course of one or more steps due to interactions with intermediates and by-products. Consequently, individual reaction optimisation is less applicable and due regard must be given to the potential role of other upstream or downstream reaction components. Auto-tandem catalysis is defined as a domino reaction process in which one catalyst promotes two or more fundamentally distinct chemical transformations in a single reactor.2 There has been tremendous progress in the development of Pd-catalysed auto-tandem processes, in a bid to develop greener synthetic methods. A key feature of this approach is that Pd acts as the catalyst in two sequential, one-pot reactions.3 Interestingly, extending this concept to the development of processes with three or more mechanistically distinct Pd-catalysed reactions has proven challenging and examples are sparse in the literature.4Fagnou and co-workers previously reported a rare example of a three-reaction process, wherein a series of benzochromenes were prepared in good yields (Scheme 1). While this exquisite example demonstrates the power of auto-tandem catalysis, there is clear potential for further green chemistry improvement and expansion.
 | ||
| Scheme 1 Comparison to previous work from a green perspective. | ||
Quinolines, pyridines and furans represent important molecule classes in medicinal and natural product chemistry.5 Core quinoline, pyridine and furan motifs are the 22nd, 2nd and 25th most abundant of the 351 ring systems present in FDA-approved drugs respectively.6 Direct arylation via C—H activation of these moieties has received significant attention in recent years, as the protocol avoids the introduction of activating groups, and the associated waste. Direct arylation via C—H activation also facilitates the introduction of new aryl groups which could be difficult using traditional (Het)Ar–(Het)Ar bond forming processes such as the Suzuki–Miyaura and Stille reactions.7 This is especially the case with intramolecular reactions, where the installation of halides and boronic acids (or stannanes) onto a single substrate is often difficult.
We posited that intramolecular direct arylation of quinolines could be coupled with a Mizoroki–Heck alkenylation and a hydrogenation in a one-pot sequence to give decorated tri- and tetra-cyclic quinolines with a built-in benzofuran. However, we were also aware that the quinoline nitrogen atom can ligate Pd8a and thus effect a synergistic or deleterious effect on any of the three Pd-mediated process.8 In any case, we were keen to apply a ‘bottom-up’ green chemistry approach to develop tricyclic alkylated heterocycles, bearing key functionalities relevant to medicinal chemistry.
We set ourselves the specific challenges of (1) minimising catalyst loading (2) eliminating the need for a P-ligand (3) avoiding the use of toxic solvents such as DMA and NMP and (4) carrying out the reactions in air. Herein we describe the successful execution of these goals (Scheme 1). We expand the substrate scope from quinolinyl to pyridinyl and simple aryl ethers. Also, for the first time in these types of processes, we introduce deuterium via reduction of the Heck products using the COware apparatus (which circumvents the difficulty in accessing D2 gas). Finally, some initial investigation of the catalytic species required for each step is presented.
Results and discussion
Optimisation study
As a starting point, we revisited our previously reported one-pot, two-step methodology involving a Mizoroki–Heck/direct arylation sequence with a view toward obviating the need for N-methylpyrrolidone (NMP) as solvent.8a,9,10 We began our optimisation efforts with a number of more desirable solvents (Table 1 and ESI† for further information).10 Initially, we found that 1,4-dioxane gave rise to moderate conversion (Table 1, entry 1), which was improved slightly through the addition of 18-crown-6 as an additive (Table 1, entry 2). Jeffery and co-workers have previously shown that the use of quaternary ammonium salts as additives in Pd-catalysed olefination reactions led to superior reaction performance.11,12|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive | Base | Conversiona |
| a Conversion to product 3a determined by 1H NMR analysis, isolated yields following chromatographic purification in parentheses. b 3.00 eq. TBAOAc used. | ||||
| 1 | 1,4-Dioxane | — | K2CO3 | 65% |
| 2 | 1,4-Dioxane | 18-Crown-6 | K2CO3 | 73% |
| 3 | 1,4-Dioxane | TBACl | K2CO3 | 100% (80%) |
| 4 | 1,4-Dioxane | TBACl | — | 15% |
| 5 | 1,4-Dioxane | TBACl | NaOAc | 77% |
| 6 | 1,4-Dioxane | TBAOAc | K2CO3 | 98% (79%) |
| 7 | 1,4-Dioxane | TBAOAc | — | 84% |
| 8b | 1,4-Dioxane | TBAOAc | — | 98% (77%) |
| 9b | EtOAc | TBAOAc | — | 100% |
| 10b | CPME | TBAOAc | — | 98% (78%) |
| 11b | Cyrene | TBAOAc | — | 98% |
| 12b | H2O | TBAOAc | — | 88% |

Gratifyingly, the use of tetrabutylammonium chloride (TBACl) in 2.00 equivalents in place of 18-crown-6 resulted in near quantitative formation of olefin 2a (Table 1, entry 3). Conducting the reaction in the absence of base or using NaOAc in place of K2CO3 gave lower conversion (Table 1, entries 4 and 5). We also explored the use of tetrabutylammonium acetate (TBAOAc).13 Exchanging TBAOAc for TBACl resulted in similarly excellent results (Table 1, entry 6 cf.Table 1, entry 3). Encouraged by this initial result, we wondered whether TBAOAc could also act as the base for the reaction and replace K2CO3. We knew the acetate ion could act as base from previously tried NaOAc, which gave 77% conversion to the target compound (Table 1, entry 5). Using TBAOAc in the absence of K2CO3, these conditions gave rise to 2a in good conversion (Table 1, entry 7), which was the rendered quantitative on increasing the equivalents of TBAOAc from 2.00 to 3.00 (Table 1, entry 8). Although preferable to NMP, 1,4-dioxane is still not an ideal solvent for this transformation from a green chemistry perspective. To our delight, the use of TBAOAc enabled this chemistry to proceed in several more desirable solvents, including EtOAc, CPME, Cyrene and water with 3.00 equivalents (Table 1, entries 9–12 and ESI† for further information).
With optimised conditions for the first two steps in hand, we next turned our attention to finding compatible conditions for the subsequent reduction step (Table 2). Alkene reduction with molecular hydrogen is ideal from a green chemistry perspective.14
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temperature | Conversion to 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Conversion to 3aa |
| a Conversion to products 2a and 3a determined by 1H NMR analysis from starting material 1a. b n.a. = not applicable: EtOAc did not support the hydrogenation step. c 5.00 eq. TBAOAc used as reaction medium; n.d. = not determined. | ||||
| 1 | CPME | 90 °C | 28% | n.d. |
| 2 | CPME | 110 °C | 98% | 100% |
| 3 | Cyrene | 100 °C | 74% | n.d. |
| 4 | Cyrene | 135 °C | 98% | 100% |
| 5 | EtOAc | 60 °C | n.d. | n.d. |
| 6b | EtOAc | 90 °C | 100% | n.a. |
| 7 | 2-MeTHF | 70 °C | 14% | n.d. |
| 8 | 2-MeTHF | 90 °C | 100% | 50% |
| 9c | — | 135 °C | 100% | 100% |
| 10c | — | 100 °C | 100% | 100% |

Although conducting the reaction in CPME, Cyrene, EtOAc and 2-MeTHF typically led to excellent conversions to olefin 2a, elevated temperatures were generally required to achieve quantitative hydrogenation (Table 2, entries 1–8). Furthermore, Cyrene appeared to polymerise during the course of the reaction, thus proving problematic.15 As TBAOAc melts at approximately 100 °C, we wondered whether molten TBAOAc could act as both base for the first two reactions and a reaction medium for the hydrogenation step.
Indeed, quantitative formation of reduced product 3a was recorded using TBAOAc at both 135 °C and 100 °C (Table 2, entries 9 and 10). Finally we attempted a number of transfer hydrogenation agents (in place of H2) but with less success. See ESI for details.†
Substrate scope
We then extended the scope of our optimised conditions to include several phenoxyquinoline substrates (Scheme 2). The products, prepared through our three-step, one-pot process were isolated in excellent yields following purification by column chromatography. The scope was established by varying the precursors for the Heck reaction. The reaction worked well irrespective of whether the aryl bromide Heck partner was on the pendant phenoxy-ring or the quinoline core. Both electron-rich and electron-poor olefin coupling partners were also well-tolerated. | ||
| Scheme 2 Scope of phenoxyquinoline substrates. | ||
Pyridines are often difficult substrates in direct arylation reactions.9 However, their prevalence in biologically active compounds6 prompted us to submit a series of phenoxypyridines to our optimal conditions (Scheme 3). We were pleased that both electron-rich and electron poor olefins were compatible with the Heck-hydrogenation sequence, giving novel benzofuropyridines 6a–6c in good yields. Finally, we were interested in exploring the synthesis of benzofurans via this approach (Scheme 4). Benzofuran derivatives are ubiquitous in bioactive molecules and have well-established uses as antimicrobial and anticancer chemotherapeutics.16 This class of substrates also constitutes an interesting extension as they do not possess a coordinating nitrogen atom. The reaction worked well with diaryl ether substrates (9a–9e).17
 | ||
| Scheme 3 Scope of phenoxypyridine substrates. | ||
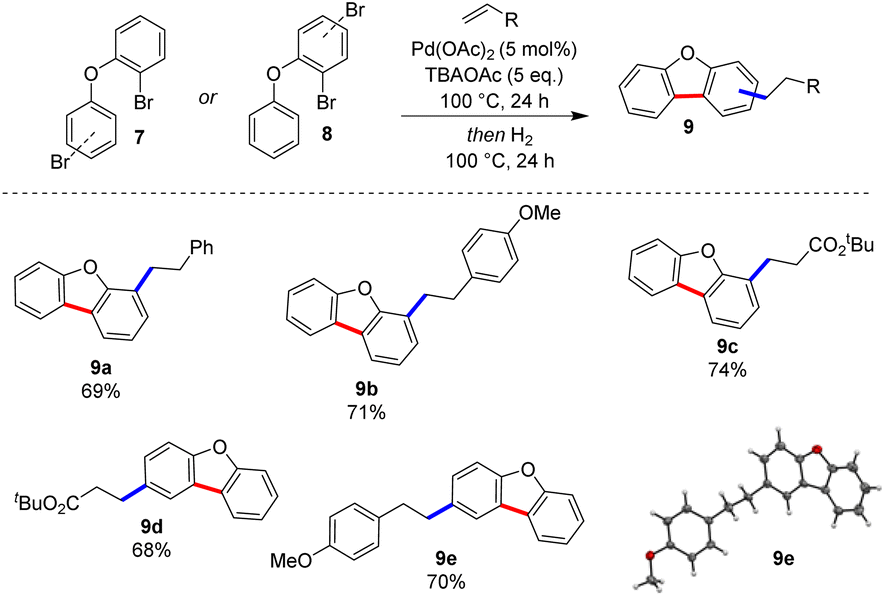 | ||
| Scheme 4 Scope of diaryl ether substrates. | ||
Deuterium incorporation and mechanistic studies
In a bid to better understand the underlying mechanistic processes which were operative in this complex transformation, we undertook a series of mechanistic studies.Deuterium incorporation
Incorporating deuterium into target molecules has many important applications in the context of mechanistic studies (e.g., kinetic isotope effects), but also by enhancing metabolic stability and pharmacokinetics, aiding ADME studies, bettering toxicity profiles in drugs and creating new chemical entities of commercial importance.18 To facilitate easy access to D2 for this study, we used the ‘COware’ apparatus developed by Skrydstrup (Scheme 5).19 This apparatus connects two chambers by a glass bridge which facilitates ex situ gas generation. Here D2 was generated (by addition of DCl to zinc) and successfully incorporated into the final structure 4h-d4 by addition across the double bond. The ability to isotopically label these densely functionalised molecules at a late stage in their preparation demonstrates the medicinal chemistry potential of this approach. Interestingly, some incorporation of D was observed at other locations on the quinoline, which might provide a rich avenue of further research. | ||
| Scheme 5 Deuterium incorporation using COware apparatus. | ||
Sequence of the direct arylation and olefination reactions
Initially, we were curious whether the direct arylation and Heck reactions took place concurrently or sequentially. 1H NMR analysis of the reaction between dibromoquinoline 1b and styrene, under the optimised conditions, revealed that the Heck reaction was significantly faster than the direct arylation reaction. This result suggests that oxidative addition to the less hindered quinoline Ar–Br bond is favoured over oxidative addition at the phenoxy Ar–Br. Indeed, we have previously observed a similar trend in reactivity when studying tandem Suzuki–Miyaura/C–H activation reactions on phenoxyquinoline substrates.8aProbing the catalytic species in the hydrogenation reaction
During the Heck olefination/C–H activation process, we observed the formation of palladium black, and we were interested to determine the speciation of Pd during the hydrogenation step. We considered it most likely that heterogeneous or soluble nanoparticulate Pd, formed in situ, was catalysing the hydrogenation reaction.3l,4c To discern the impact of soluble Pd sources, we undertook a filtration test. Initially, we performed the first two reaction steps (Heck and C–H activation) using a dibromoquinoline and styrene as per our established reaction conditions (see ESI† for more information). However, before inserting the hydrogen balloon, we filtered the reaction mixture through a 2 μm PTFE syringe filter and placed the filtrate back into a clean reaction vial. The reaction mixture was then purged with hydrogen and allowed to stir for a further 24 hours at 100 °C. As the reaction mixture was rid of visible aggregates of palladium, it appeared as a brown homogenous solution. Complete conversion to the hydrogenated product was observed by 1H NMR spectroscopy. Thus, interestingly, it appears that soluble Pd nanoparticles are active catalysts in the hydrogenation reaction.4cAnalysis of green chemistry metrics
Finally, as we set ourselves the challenge to design a ‘bottom-up’ green chemistry approach to the polycyclic alkylated heterocycles in this study, we turned to calculating green metrics to evaluate the greenness of our reaction versus the current state-of-the-art reported by Fagnou (Scheme 1).4d The metrics were determined as per previously reported calculations by Ma et al.20 Analysis was conducted on atom economy (AE), atom efficiency (AEf), carbon efficiency (CE), reaction mass efficiency (RME), optimum efficiency (OE), process mass intensity (PMI), E-factor (EF), solvent intensity (SI) and water intensity (WI) (Fig. 1 and see ESI† for the definition of the metrics and methodology). | ||
| Fig. 1 Quantitative comparison of green chemistry metrics for this work and previous work by Fagnou.4d For illustration purposes, EF, SI and PMI values were inverted and multiplied by 100 (calculation can be found in ESI†). | ||
As shown in Fig. 1, our process constitutes a significant green chemistry improvement under these metrics due to our system not requiring an ancillary phosphine ligand, added base and excess solvent. For comparison, in green metric calculations, we input our quinoline substrate with Fagnou's conditions and input Fagnou's chromene substrate into our new conditions. Both metrics show the more favourable greenness of our conditions (see ESI†).
The MP of the average one-pot values of 12% is six times higher than that of Fagnou's process, which has a value of just 2%. MP is an important metric that indicates the level of resource consumption as it includes all benign and non-benign reagents such as catalysts, base and solvent.
Conclusions
Achieving substrate, ligand and solvent compatibility across three mechanistically distinct catalytic reactions is challenging. In the methodology described here, no P-ligand or inorganic base is added, with TBAOAc acting as both ionic liquid and base. Decorated tri- and tetra-cyclic alkylated heteroarenes are conveniently and rapidly formed. Our new methodology greatly improves on the current state-of-the-art and demonstrates superior green chemistry values across many of the key metrics.Experimental
General information
All reagents and solvents were purchased from commercial suppliers and were used without further purification. Column chromatography was carried out using 60 Å (35–70 μm) silica. 1H NMR (600 MHz), 1H NMR (500 MHz), 1H NMR (400 MHz), and 1H NMR (300 MHz) spectra were recorded on Bruker Avance III 600, Bruker Avance 500, Bruker Avance 400, and Bruker Avance III 300 NMR spectrometers respectively. 13C NMR (150.9 MHz), 13C NMR (125 MHz), 13C NMR (100 MHz), and 13C NMR (75.5 MHz) spectra were recorded on Bruker Avance III 600, Bruker Avance 500, Bruker Avance 400, and Bruker Avance III 300 NMR spectrometers respectively. Chemical shifts are reported in parts per million (ppm) downfield from TMS internal standard. Additional information on the COware apparatus and setup is available in the ESI.†General procedure for the synthesis of phenoxyquinoline starting materials
A mixture of the 4-chloroquinoline substrate S1 (1 eq.), the phenol (3.5 eq.) and NaOH (1.5 eq., crushed pellets) was stirred at 120 °C until TLC analysis indicated the reaction was complete (2–24 hours). The reaction mixture was cooled to room temperature and 10% aqueous NaOH (10 mL) was added and stirred for 30 min. The aqueous layer was extracted with DCM (3 × 15 mL). The organic layers were combined and washed with 6 M NaOH (3 × 15 mL), water (10 mL) and brine (10 mL), dried over MgSO4, filtered, and concentrated in vacuo. Impure products were purified by column chromatography over silica gel using a gradient cyclohexane:EtOAc (70:30).
General procedure for the one-pot Mizoroki–Heck/direct arylation/hydrogenation reaction
To a screw capped vial was added the quinoline/pyridine/diaryl ether substrate (1 eq.), Heck coupling partner (1.1 eq.), palladium diacetate (5 mol%) and tetrabutylammonium acetate (5 eq.) which was stirred at 100 °C for 24 hours. The reaction mixture was then purged with hydrogen gas and stirred under a hydrogen atmosphere for a further 24 hours at 100 °C. The crude reaction mixture was loaded directly onto silica gel for purification by column chromatography. The benzofuroquinoline and benzofuropyridine products eluted in a gradient cyclohexane:EtOAc (70:30). The dibenzofuran products eluted in a gradient cyclohexane:EtOAc (90:10).
General procedure for the synthesis of deuterated benzofuroquinoline (4h-d4)
To chamber ‘A’ of the 20 mL COware19a apparatus was added the benzofuroquinoline substrate (1 eq.), Heck coupling partner (1.1 eq.), palladium diacetate (5 mol%) and tetrabutylammonium acetate (5 eq.). To the second chamber ‘B’ was added zinc powder (3.65 eq.). Chamber ‘A’ was submerged into an oil bath and was stirred at 100 °C for 24 hours. DCl (4 M in D2O, 7.3 eq.) was added via syringe to chamber ‘B’ which initiated the D2 formation [CAUTION] and reaction was allowed to stir at 100 °C for a further 24 hours. The crude product was purified loaded directly onto silica gel for purification by column chromatography, eluting with a gradient cyclohexane:EtOAc (70:30).
Crystallographic data
X-ray crystallographic data for compounds 3a [CCDC 2143462], 3b [CCDC 2143446], 3c [CCDC 2204946], 4d [CCDC 2211708], 4h [CCDC 2204910], 4j [CCDC 2201481], 6a [CCDC 2204956], 9a [CCDC 2204818], 9e [CCDC 2204795] (see ESI for details).†Author contributions
RK, DJ and GM conceived the ideas and experiments. RK carried out the experiments. ML solved the single crystal structures. All contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Denis Lynch of University College Cork for providing NMR services, the Science Foundation Ireland (grant number SFI/12/IP/1315) and the Synthesis and Solid State Pharmaceutical Centre (SSPC) (SFI/12/RC/2275 and SFI/12/RC2275_P2). We would like to thank Thermo Fisher Scientific in Cork for helping with the scale-up reactions.References
- Y. Hayashi, Chem. Sci., 2016, 7, 866 RSC.
- (a) N. Shindoh, Y. Takemoto and K. Takasu, Chem. – Eur. J., 2009, 15, 12168 CrossRef CAS PubMed; (b) F.-X. Felpin and E. Fouquet, ChemSusChem, 2008, 1, 718 CrossRef CAS PubMed; (c) J. E. Camp, Eur. J. Org. Chem., 2017, 425 CrossRef CAS.
- (a) K. Hu, Q. Zhen, J. Gong, T. Cheng, L. Qi, Y. Shao and J. Chen, Org. Lett., 2018, 20, 3083 CrossRef CAS PubMed; (b) A. A. Nechaev, P. R. Jagtap, E. Bažíková, J. Neumannová, I. Císařová and E. Matoušová, Org. Biomol. Chem., 2021, 19, 3434 RSC; (c) W. Hagui, N. Besbes, E. Srasra, T. Roisnel, J.-F. Soulé and H. Doucet, Org. Lett., 2016, 18, 4182 CrossRef CAS PubMed; (d) J. Koubachi, S. El Kazzouli, S. Berteina-Raboin, A. Mouaddib and G. Guillaumet, J. Org. Chem., 2007, 72, 7650 CrossRef CAS PubMed; (e) K. Geoghegan, S. Kelleher and P. Evans, J. Org. Chem., 2011, 76, 2187 CrossRef CAS PubMed; (f) S. P. Mulcahy and J. G. Varelas, Tetrahedron Lett., 2013, 54, 6599 CrossRef CAS PubMed; (g) H. Jong, S. T. C. Eey, Y. H. Lim, S. Pandey, N. A. B. Iqbal, F. F. Yong, E. G. Robins and C. W. Johannes, Adv. Synth. Catal., 2017, 359, 616 CrossRef CAS; (h) L. Mayer, R. Kohlbecher and T. J. J. Müller, Chem. – Eur. J., 2020, 26, 15130 CrossRef CAS PubMed; (i) G. A. Molander, S. L. J. Trice and S. M. Kennedy, J. Org. Chem., 2012, 77, 8678 CrossRef CAS PubMed; (j) R. Barroso, M. Paraja, M.-P. Cabal and C. Valdés, Org. Lett., 2017, 19, 4086 CrossRef CAS PubMed; (k) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 4720 CrossRef CAS PubMed; (l) F.-X. Felpin, O. Ibarguren, L. Nassar-Hardy and E. Fouquet, J. Org. Chem., 2009, 74, 1349 CrossRef CAS PubMed; (m) L. Nassar-Hardy, S. Fabre, A. M. Amer, E. Fouquet and F.-X. Felpin, Tetrahedron Lett., 2012, 53, 338 CrossRef CAS; (n) F.-X. Felpin, J. Coste, C. Zakri and E. Fouquet, Chem. – Eur. J., 2009, 15, 7238 CrossRef CAS PubMed; (o) J. Barluenga, A. Jiménez-Aquino, M. A. Fernández, F. Aznar and C. Valdés, Tetrahedron, 2008, 64, 778 CrossRef CAS; (p) J. Laudien, E. Fouquet, C. Zakri and F.-X. Felpin, Synlett, 2010, 1539 CAS; (q) B. Gabriele, P. Plastina, G. Salerno, R. Mancuso and M. Costa, Org. Lett., 2007, 9, 3319 CrossRef CAS PubMed; (r) R. Devlin, D. J. Jones and G. P. McGlacken, Org. Lett., 2020, 22, 5223 CrossRef CAS PubMed.
- (a) M. Szlosek-Pinaud, P. Diaz, J. Martinez and F. Lamaty, Tetrahedron Lett., 2003, 44, 8657 CrossRef CAS; (b) A. Pinto, L. Neuville and J. Zhu, Tetrahedron Lett., 2009, 50, 3602 CrossRef CAS; (c) J. Hitce and O. Baudoin, Adv. Synth. Catal., 2007, 349, 2054 CrossRef CAS; (d) J.-P. Leclerc, M. André and K. Fagnou, J. Org. Chem., 2006, 71, 1711 CrossRef CAS PubMed.
- (a) B. S. Matada, R. Pattanashettar and N. G. Yernale, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS PubMed; (b) E. Khan, ChemistrySelect, 2021, 6, 3041 CrossRef CAS; (c) M. Napiórkowska, M. Cieślak, J. Kaźmierczak-Barańska, K. Królewska-Golińska and B. Nawrot, Molecules, 2019, 24, 1529 CrossRef PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef; (c) D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636 CrossRef CAS; (d) T.-Y. Luh, M.-K. Leung and K.-T. Wong, Chem. Rev., 2000, 100, 3187 CrossRef CAS PubMed; (e) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed; (f) Y. Nishihara, Applied Cross-Coupling Reactions, Springer Science & Business Media, 2012, vol. 80 Search PubMed; (g) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC; (h) R. Cano, J. M. Pérez, D. J. Ramón and G. P. McGlacken, Tetrahedron, 2016, 72, 1043 CrossRef CAS.
- (a) R. M. Shanahan, A. Hickey, L. M. Bateman, M. E. Light and G. P. McGlacken, J. Org. Chem., 2020, 85, 2585 CrossRef CAS PubMed; (b) M. Wasa, K. S. L. Chan, X.-G. Zhang, J. He, M. Miura and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570 CrossRef CAS PubMed; (c) Y. He, Z. Wu, C. Ma, X. Zhou, X. Liu, X. Wang and G. Huang, Adv. Synth. Catal., 2016, 358, 375 CrossRef CAS; (d) K. Mackey, D. J. Jones, L. M. Pardo and G. P. McGlacken, Eur. J. Org. Chem., 2021, 495 CrossRef CAS.
- R. M. Shanahan, A. Hickey, F. J. Reen, F. O'Gara and G. P. McGlacken, Eur. J. Org. Chem., 2018, 6140 CrossRef CAS.
- (a) P. Gandeepan, N. Kaplaneris, S. Santoro, L. Vaccaro and L. Ackermann, ACS Sustainable Chem. Eng., 2019, 7, 8023 CrossRef CAS; (b) H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739 CrossRef CAS; (c) J. Sherwood, H. L. Parker, K. Moonen, T. J. Farmer and A. J. Hunt, Green Chem., 2016, 18, 3990 RSC; (d) A. Jordan, C. G. J. Hall, L. R. Thorp and H. F. Sneddon, Chem. Rev., 2022, 122, 6749 CrossRef CAS PubMed; (e) J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650 RSC.
- F. Chen, T. Dong, T. Xu, X. Li and C. Hu, Green Chem., 2011, 13, 2518 RSC.
- (a) T. Jeffery, Tetrahedron Lett., 1999, 40, 1673 CrossRef CAS; (b) T. Jeffery, Tetrahedron, 1996, 52, 10113 CrossRef CAS; (c) T. Jeffery and M. David, Tetrahedron Lett., 1998, 39, 5751 CrossRef CAS.
- (a) V. Caló, A. Nacci, A. Monopoli, A. Detomaso and P. Iliade, Organometallics, 2003, 22, 4193 CrossRef; (b) V. Calò, A. Nacci, A. Monopoli, A. Damascelli, E. Ieva and N. Cioffi, J. Organomet. Chem., 2007, 692, 4397 CrossRef; (c) B. C. Ranu, S. S. Dey and A. Hajra, Tetrahedron, 2003, 59, 2417 CrossRef CAS; (d) V. Calò, A. Nacci, L. Lopez and N. Mannarini, Tetrahedron Lett., 2000, 41, 8973 CrossRef; (e) V. Calò, A. Nacci, A. Monopoli, A. Fornaro, L. Sabbatini, N. Cioffi and N. Ditaranto, Organometallics, 2004, 23, 5154 CrossRef; (f) G. Battistuzzi, S. Cacchi and G. Fabrizi, Synlett, 2002, 0439 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- P. Ray, T. Hughes, C. Smith, M. Hibbert, K. Saito and G. P. Simon, Polym. Chem., 2019, 10, 3334 RSC.
- H. Khanam and Shamsuzzaman, Eur. J. Med. Chem., 2015, 97, 483 CrossRef CAS PubMed.
- For attempts at scale up see ESI.†.
- (a) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276 CrossRef CAS PubMed; (b) S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634 CrossRef CAS PubMed; (c) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022 CrossRef CAS PubMed; (d) A. Mullard, Nat. Rev. Drug Discovery, 2017, 16, 305 Search PubMed.
- (a) S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594 CrossRef CAS PubMed; (b) A. Modvig, T. L. Andersen, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Org. Chem., 2014, 79, 5861 CrossRef CAS PubMed.
- X. Ma, X. Zhang, J. M. Awad, G. Xie, W. Qiu and W. Zhang, Green Chem., 2019, 21, 4489 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterisation data on each isolated compound, further optimisation details and crystallographic data for nine novel compounds. CCDC 2143462, 2143446, 2204946, 2211708, 2204910, 2201481, 2204956, 2204818 and 2204795. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc01403g |
| This journal is © The Royal Society of Chemistry 2023 |