
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural features of lignin–hemicellulose–pectin (LHP) orchestrate a tailored enzyme cocktail for potential applications in bark biorefineries†
Jinze
Dou
 *a,
Jincheng
Wang
b,
Sami
Hietala
c,
Dmitry V.
Evtuguin
d,
Tapani
Vuorinen
*a and
Jian
Zhao
b
*a,
Jincheng
Wang
b,
Sami
Hietala
c,
Dmitry V.
Evtuguin
d,
Tapani
Vuorinen
*a and
Jian
Zhao
b
aDepartment of Bioproducts and Biosystems, Aalto University, Espoo, Finland. E-mail: jinze.dou@aalto.fi; tapani.vuorinen@aalto.fi
bState Key Laboratory of Microbial Technology, Shandong University, Qingdao, China
cDepartment of Chemistry, University of Helsinki, Helsinki, Finland
dCICECO/Department of Chemistry, University of Aveiro, Aveiro, Portugal
First published on 16th June 2023
Abstract
Wood bark is a structurally complex by-product of the pulp and paper industry, which focuses primarily on the valorization of structurally more regular wood xylem components. The aim of this study was the elucidation of the less valorised willow wood counterparts (whole bark, inner bark, sclerenchyma bundles, and parenchymatous tissues) by NMR spectroscopic techniques. This allowed a better understanding of the structural features of macromolecular components of bark (i.e. pectin, hemicellulose, and lignin), thus providing a base for a more rational design of the customized biochemical processes prior to chemical processing of bark. This crucial knowledge contributed to the creation of a protocol/decision tool to select tailored enzymes (discarding the slightest substrate binding) for the biological pre-treatment of bark to a state suitable for chemical pulping. Such a protocol/decision-making tool would significantly improve the efficiency of enzyme selection by 60–70% due to the specific catalytic activity of the enzymes involved.
Introduction
Strips of bark or bast materials were first used to make paper in China around 105 AD. Today, industries set aside this resource and use debarked wood to produce pulp and paper because the chemistry of wood and its chemical processing are much simpler than those of bark.1 Wood bark (estimated 3.59 billion m3) – roughly 15–20% of the volume produced annually from a wood log2 – is used exclusively for energy at the mill, making it by far the greatest long-neglected biomass resource on earth. It is understandably challenging to valorize wood bark because bark has a rather heterogeneous composition both morphologically and chemically. It mainly consists of cellulose, hemicellulose, lignin, pectin, suberin, starch, and a variety of extractives (tannins, fatty acids, resin acids, terpenoids, etc.). Each of these ingredients reacts differently to chemicals for pulping and bleaching. Valorization of bark is underexploited because the focus has so long been on extractives of bark; here, we recommend turning the focus to the entire bark and re-examining how best to extract more natural resources from it.3 It is believed that the bark deserves equivalent attention as its counterpart wood.A major obstacle here is our lack of a holistic approach to understanding the structural features of the major constituents and their structural association (i.e. pectin, hemicellulose and lignin) in the cell walls of wood and bark. Wood cells of the lignocellulosic biomass is made of multiple layers of middle lamella, and primary and secondary cell walls. The cell wall usually comprises cellulose fibrils as reinforcing elements, which are embedded in the hemicellulose and lignin matrix, and non-structural components (extractives, starch and proteins). Pectin is also present in the primary wall. Structural proteins can become part of the cell wall, whereas starch is located elsewhere, like most extractives. In heartwood, some extractives can impregnate the cell walls and thus contribute to their properties. This supramolecular matrix architecture is bonded by complex carbohydrates and aromatics,4–6 providing cell walls with mechanical strength, rigidity, and inherent recalcitrance to (bio)chemical degradation.
Understanding the chemistry of pectin and hemicellulose is essential for designing a customized enzymatic cocktail as pretreatment to smartly implement chemical pulping for bark. Pectin consists mainly of linear, “smooth” segments of homogalacturonan (HG) and rhamnopyranosyl groups of rhamnogalacturonan I (RG-I) that are substituted at O-4 through the arabinan, galactan, and arabinogalactan side chains. The skeleton of RG-I is considered to be the “hairy region” of pectin, consisting of alternating 1,4-linked galacturonic acid (GalA) and 1,2-linked rhamnose units. Compared with strong mineral acids, extraction in the presence of citric acid is known for retaining pectin's structure to its maximum extent.7 Hemicelluloses, the second most abundant group of polysaccharides, have a biological function to strengthen the structural and material properties of cell walls.8 Glucuronoxylan, xyloglucan, galactomannan (GAMA, a (1–4)-β-mannopyranosidic main chain connected with one (1–4)-β-galactopyranosidic side chain), and glucomannan (GLMA, a (1–4)-β-glucopyranosidic main chain connected with one (1–4)-β-mannopyranosidic side chain) represent the prominent hemicellulose building units.8 Glucuronoxylan, as the primary hemicellulose in hardwood, contains xylose and glucuronic acid as its main constituents. It is characterized by a linear β-(1,4)-linked β-D-xylopyranosyl unit and is substituted by 4-O-methyl-D-glucuronic acid (–MG) and acetyl groups. Alkaline extraction,9 peracetic acid delignification followed by DMSO extraction,10 pressurized water extraction,11 and cellulolytic enzyme-aided extraction12 are the conventional methodologies (ESI Table 1†) to isolate hemicellulose from wood. However, hemicellulose extraction from tree bark has been rarely reported.
Lignin chemistry provides fundamental knowledge for designing chemical pulping. Lignins are cross-linked macromolecules consisting of three phenylpropanoid units: p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units. The dominating linkage types are β-O-4 (β-ether), β-5 (phenylcoumaran), β–β (resinol), 5–5 (biphenyl), and 5-O-4 (diaryl ether).13 Although much less research has been concentrated on the lignin structure of bark than that of wood, we do know from previous investigations that bark lignin contains more G-units than S-units from several species, including spruce, eucalyptus, blackwood acacia,14 and the willow hybrid Karin.15 A relative ratio of S-units/G-units plays an essential role in the durability and mechanical resistance of the bark tissue. The relative abundance of dominating linkage types and S/G ratio influence the pulping yield (or lignin depolymerization) as syringyl-type lignin is less reactive compared to guaiacyl-type lignin.16 Dioxane lignin17 and cellulolytic enzyme lignin (CEL) are currently the main protocols to prepare “native” lignin for characterization. Lignin features can be characterized by non-destructive 2D nuclear magnetic resonance (NMR) spectroscopy, for example.18
Herein, we follow conventional protocols to identify structural differences of pectin, hemicellulose, and dioxane lignin from willow wood to bark (whole bark; inner bark; fiber bundles; and parenchymatous tissues). We have developed new pretreatments to recover hemicellulose from bark. The distinct structural differences of the substrate (i.e., bark) can further orchestrate screening and selection strategies of the tailored enzymes for the prior recovery (or elimination) of these macromolecule components (e.g., pectin and hemicellulose). This strategy is considered an essential pretreatment for implementing chemical pulping for bark valorization, and it is also in line with the strategy of “tailor-made enzyme consortium based on the structural features of the substrate”.19,20 If all active components of wood bark can be utilized, the value of bark is likely to be comparable to that of wood.
Results
Staining and mass balance of biomass
Both microscopic and chromatographic techniques were applied to reveal the morphological distribution and chemical composition of the willow samples studied. Fig. 1a–c illustrate the morphological distribution of willow bark fiber bundles (WBFB) in both tangential and cross sections. Safranin-stained WBFB in red (Fig. 1a–c) shows that the WBFB section is heavily lignified and contains most of the lignin found in willow inner bark (WIB). In particular, safranin stained the middle lamella regions of WBFB deep red, demonstrating heavy lignification21 in the middle lamella (Fig. 1c). However, the characteristic red stain of lignin by safranin was not observed in parenchyma, indicating the absence of lignin in parenchymatous tissues. Moreover, toluidine stained the cambium layer and parenchyma cell walls deep blue (or purple), emphasizing their richness in acidic pectin,22 and a small concentration of pectin in WBFB (Fig. 1d) and willow wood (WW) were stained light blue. The richness of the blue color seems to be in proportion to the richness of pectin at the cell wall of wood and bark (Table 1).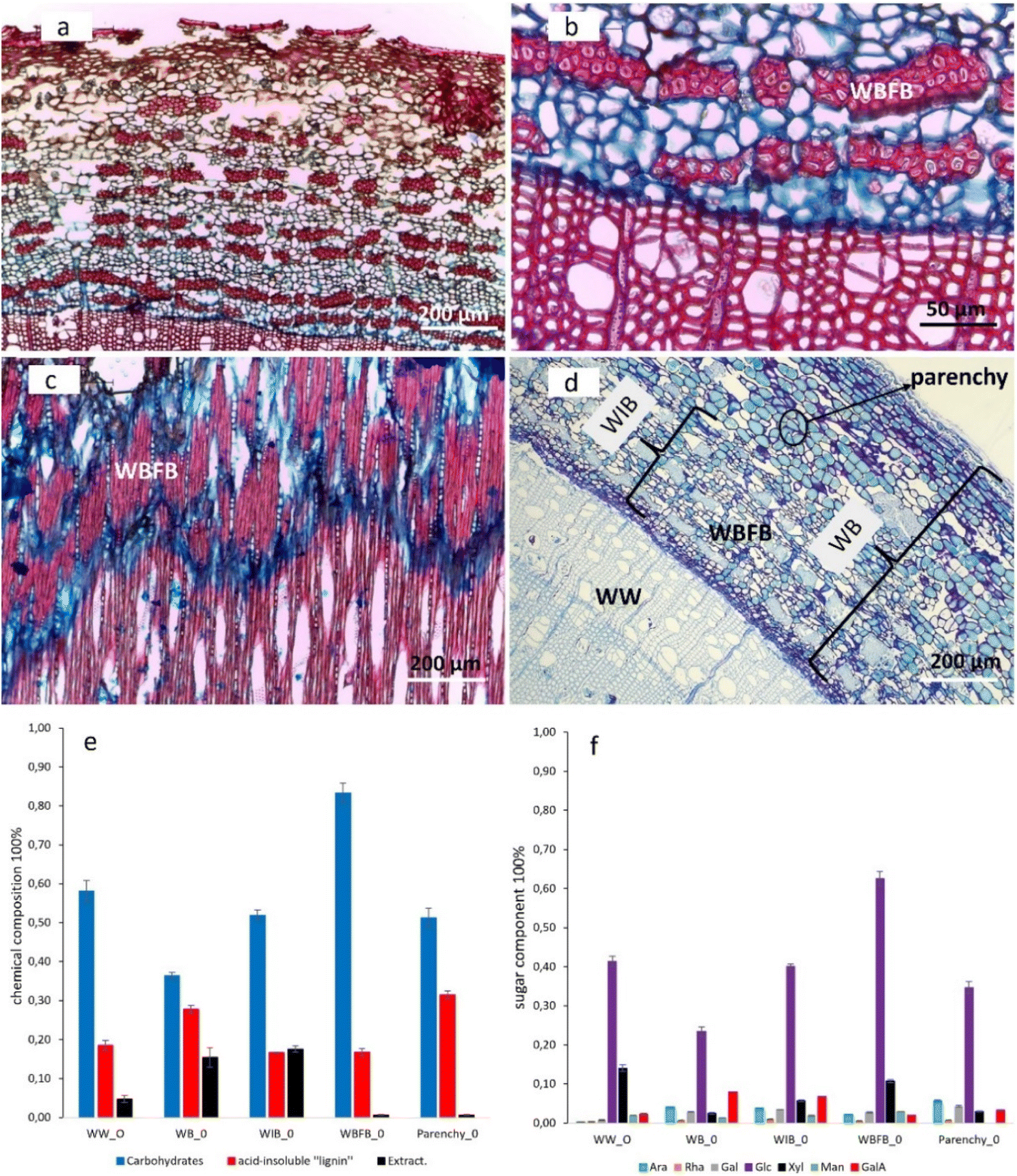 | ||
| Fig. 1 Optical microscopy staining images and the chemical composition from willow wood (WW); bark (WB); inner bark (WIB); fiber bundle (WBFB); and parenchyma (Parenchy) (ESI Fig. 1–6†). (a–c) Safranin and alcian blue stained transverse (a and b) and tangential (c) sections showing WBFB (red color) and phloem rays (vertically aligned cells with small lumina) from Salix caprea. (d) Toluidine blue O-stained transverse section showing the parenchyma with deep blue colored cell walls from Salix myrsinifolia. (e) Overall chemical composition (% of the dry mass). (f) Carbohydrate composition (% of sugars in the monosaccharide). Abbreviations: arabinose (Ara), rhamnose (Rha), galactose (Gal), glucose (Glc), xylose (Xyl), mannose (Man), galacturonic acid (GalA). Error bars are based on two independent measurements. | ||
| WW | WB | WIB | WBFB | Parenchyma | ||
|---|---|---|---|---|---|---|
| a Denotes the percentages of total volume of xylan (Xy-5a for xylan; Xy-MG-1 for Xy-MG; Xy-2-O-Ac-2 for Xy-2-O-Ac; Xy-3-O-Ac-3 for Xy-3-O-Ac; Xy-2,3-di-O-Ac-1 for Xy-2,3-di-O-Ac) and other hemicellulose (GAMA-2 for GAMA; GLMA-5 for GLMA) signals. b Denotes the estimated relative percentages of the total volume of inter-unit linkage signals of lignin (calculated from the α-C/H correlations). c Denotes the percentages of the total volume of G2, G′2, S2/6, and S'2/6 signals. Standard deviations are included in the parentheses. | ||||||
| Mass balance (% original) | Ash | 2.9 | 6.5 | 2.5 | 1.8 | 0 |
| Extracts | 4.5 (1.0) | 14.5 (2.5) | 10.8 (0.8) | 1.8 (0.1) | 0.7 (0.1) | |
| Pectin | 0.9 | 3.1 | 6.1 | 1.9 | 7.6 | |
| Cellulose | 43.2 | 24.2 | 28.5 | 52.2 | n.d. | |
| Hemicellulose | 12.1 | 3.8 | 5.5 | 7.2 | 3.5 | |
| Dioxane lignin | 8.9 (0.5) | 5.8 (0.03) | 1.7 (0.1) | 2.7 (0.03) | 0 | |
| Sum | 72.4 | 58.0 | 55.2 | 67.7 | 11.8 | |
| Structural characteristics of hemicellulose | ||||||
| Xylan | Xylan | 0.32 | 0.65 | 0.45 | 0.99 | 0.08 |
| Xy-MG | 0.06 | 0.002 | 0.01 | 0.01 | 0 | |
| Xy-2-O-Ac | 0.42 | 0.006 | 0.19 | 0 | 0 | |
| Xy-3-O-Ac | 0.20 | 0.12 | 0.26 | 0 | 0 | |
| Xy-2,3-di-O-Ac | 0 | 0 | 0 | 0 | 0.63 | |
| Other hemicellulose | GAMA | 0 | 0.02 | 0.05 | 0.01 | 0 |
| GLMA | 0 | 0.20 | 0.05 | 0 | 0.28 | |
| Structural characteristics of dioxane lignin | ||||||
| Inter-unit linkages (%)b | β-O-4′ aryl ethers | 89 | 91 | 80 | 85 | n.d. |
| Phenylcoumaran | 1 | 1 | 1 | 0 | n.d. | |
| Resinols | 10 | 8 | 19 | 14 | n.d. | |
| Aromatic unitsc | G (%) | 21 | 52 | 40 | 45 | n.d. |
| S (%) | 79 | 48 | 60 | 55 | n.d. | |
| S/G ratio | 3.8 | 0.9 | 1.5 | 1.2 | n.d. | |
Generally, differences in the chemical composition between wood and bark (WB, willow inner bark or WIB, WBFB, and parenchyma) are significant (Fig. 1e and f). Pectin characteristics (arabinose, rhamnose, galactose and GalA) are much more abundant in bark, although GalA was detected in wood. Furthermore, the sugar content of WW was roughly 20% higher than that of its counterpart WB, and this has an equal presence in WIB and parenchyma. Glucose was the main monosaccharide found in both WB and WW, whereas xylose and mannose were the dominant non-cellulosic sugars besides GalA. Comparison of xylose/mannose ratios indicated that xylan was the main hemicellulose component in WW, whereas the ratio drops in different sections of bark, suggesting that both xylan and GAMA or GLMA possibly has a relatively higher presence in WB than in WW. The acid-insoluble lignin content of WB was roughly 10% higher than that of WIB and WBFB, suggesting that the overestimation of acid-insoluble lignin was probably due to the heterogeneous chemicals that originated from parenchyma and storage cells of bark. Furthermore, it is clear that the acid-insoluble lignin of the parenchyma is not real lignin since there are no characteristic lignin peaks from FT-IR or CPMAS NMR (ESI Fig. 7†). Furthermore, the extractive content was much higher in WB than in WW, and the absence of extractives in WBFB indicates that extractives are stored mostly in the storage cells of WIB (Fig. 1d).23
The starting biomasses were successively treated to separate pectin, hemicellulose, and dioxane lignin from all parts of willow, with the exception that dioxane lignin cannot be recovered from parenchymatous tissues. The unidentified components from the mass balance (Table 1) can originate from tannin, suberin, proteins, and so forth. Diethyl ether-soluble HTS from the bark of willow hybrids included fatty acids (azelaic acid and hexadecanoic acid) and aromatics (4-hydroxybenzoic acid and protocatechuic acid) (ESI Fig. 8–10 and Table 2†). Catechol, as a thermal decomposition product of catechin and building block of polyflavonoid tannins,24 occurred in trace amounts in both hybrids. Interestingly, the detection of lactic acid may explain the degradation of hemicellulose (i.e., GLMA) under mild alkali treatment.25
Pectin characteristics
To liberate both pectic polysaccharides and metals from all samples, pH 2 citric acid was used as a chelating agent and the samples were correspondingly named CA-P.7,26 The pectin yield of WW is significantly smaller than the quantities purified from bark (Table 1). In particular, pectin has a more significant presence in parenchyma than the other tissues of bark. Dialysis removed almost half or two-thirds of the small Mw fractions from crude pectin. Although dialysis of CA-P had a minimal effect on the neutral monosaccharide composition (Table 2), the treatment resulted in a roughly two- to ten-fold increase of GalA in the pectin samples of WW, WB, and parenchyma, whereas a similar increase for WBFB was negligible. The ratio of Rha/GalA is roughly four times higher for pectin when recovered from bark (WBFB and parenchyma) than from WW, indicating that there are much fewer HG fragments in bark. This observation is also supported by the low presence of GalA in pectin from WB compared to that of WW (Table 2 and ESI Fig. 11†). Furthermore, a high ratio of (Gal + Ara)/Rha from WB indicates that RG-I domains are at least twice as likely to be branched compared to WW. The relative content of glucose in the parenchyma is much higher than that from the other fractions, which suggests that glucose may originate from starch, as reported for WB.20 The low DM and DA of pectin recovered from WBFB and parenchyma can be explained by the partial demethylation and deacetylation due to sodium bicarbonate treatment.27| WW-CA-P | WW-DCA-P | WB-CA-P | WB-DCA-P | WIB-CA-P | WIB-DCA-P | WBFB-CA-P | WBFB-DCA-P | Parenchyma-CA-P | Parenchyma-DCA-P | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Denotes that HG regions that also contain the branched GalA in the form of xylogalacturonan (XGA), particularly for pectin recovered from WW. | ||||||||||
| Pectin yield (% WB) | 0.9 (—) | 0.5 (—) | 3.1 (—) | 2.1 (—) | 6.1 (—) | 3.0 (—) | 1.9 (—) | 1.0 (—) | 7.6 (—) | 2.1 (—) |
| M w (kDa) | 318 | 389 | 288 | 91 | 147 | |||||
| M w/Mn | 4.5 | 6.3 | 5.4 | 4.1 | 2.5 | |||||
| Monosaccharides, mg g −1 | ||||||||||
| Ara | 43 (5.7) | 48 (0.1) | 84 (1.7) | 115 (0.3) | 79 (1.2) | 99 (0.1) | 66 (6.2) | 60 (0.3) | 65 (0.7) | 201 (0.2) |
| Rha | 24 (6.4) | 22 (5.4) | 23 (0.3) | 29 (0.01) | 25 (0.5) | 28 (0.2) | 35 (2.0) | 35 (0.2) | 14 (0.2) | 43 |
| Gal | 60 (4.0) | 71 (2.2) | 99 (0.7) | 107 (0.02) | 105 (1.4) | 122 (0.3) | 122 (0.2) | 181 (0.4) | 48 (0.4) | 155 (0.2) |
| Glc | 517 (61.9) | 307 (4.5) | 161 (0.5) | 202 (0.3) | 194 (2.2) | 116 (0.003) | 395 (39.4) | 391 (1.7) | 123 (0.3) | 437 (0.6) |
| GalA | 203 (0) | 544 (0) | 185 (0) | 446 (0) | 190 (0) | 89 (0) | 94 (12.4) | 112 (0) | 42 (0) | 399 (0) |
| Molar composition | ||||||||||
| Rha/GalA | 0.1 (0.04) | 0.05 (0.01) | 0.1 (0.002) | 0.1 (0) | 0.2 (0.003) | 0.4 (0.002) | 0.4 (0.1) | 0.4 (0.002) | 0.4 (0.004) | 0.1 (0.001) |
| (Gal + Ara)/Rha | 4.2 (0.7) | 5.6 (1.5) | 8.0 (0.001) | 7.7 (0.02) | 7.2 (0.003) | 7.7 (0.03) | 5.2 (0.5) | 6.5 (0.02) | 8.3 (0.01) | 8.4 (0.06) |
| HGa (mol %) | 16.6 (2.4) | 41.9 (0.5) | 25.8 (0.2) | 42.4 (0.004) | 24.5 (0.3) | 10.8 (0.06) | 6.7 (2.1) | 8.0 (0.06) | 7.8 (0.1) | 25.9 (0.004) |
| RG-I (mol %) | 16.7 (0.9) | 15.4 (0.8) | 44.2 (0.3) | 34.3 (0.04) | 42.0 (0.3) | 62.9 (0.03) | 38.1 (1.4) | 40.3 (0.001) | 51.5 (0.2) | 39.2 (0.003) |
| DM (%) | 21.2 (2.8) | 15.8 (n.d.) | 88.1 (14) | 67.2 (n.d.) | 81.4 (n.d.) | 77.1 (0.11) | 11.3 (0.1) | 6.6 (0.4) | 7.6 (2.0) | 1.0 (0.09) |
| DA (%) | 37.7 (0.2) | 21.3 (n.d.) | 22.0 (9.9) | 10.7 (n.d.) | 28.7 (n.d.) | 25.1 (1.55) | 8.5 (0.7) | 5.2 (0.3) | 10.0 (1.3) | 1.4 (0.08) |
A solution-state 2D HSQC NMR analysis (Fig. 2) revealed typical inter-unit linkages of pectin, and its spectra were assigned based on the literature data.20,28 Three clear signals at δC/δH of 56.0/3.80, 23.8/2.15, and 19.5/1.25 ppm indicate the presence of methyl (OMe) and acetyl (OAc) groups in 1,4-α-D-GalpA, and C6/H6 in rhamnose, respectively. Furthermore, the non-anomeric C/H atoms of GalA (δC/δH of 69.0/3.75, 69.6/3.8, 74.0/4.72, and 83.0/4.25 ppm) were also confirmed. Strong starch (1,4-α-D-Glcp)29 signals were also present in both wood and bark at δ 102.5/δ 5.41 (C1/H1), δ 74.5/δ 3.64 (C2/H2), δ 76.5/δ 3.98 (C3/H3), δ 79.7/δ 3.66 (C4/H4), δ 74.2/δ 3.84 (C5/H5), and δ 63.7/δ 3.83 (C6/H6). Specific non-anomeric and anomeric methine signals revealed the presence of arabinofuranosyl groups (terminal and 1,5-, 2,5-, and 2,3,5-linked) and galactopyranosyl groups from pectin that are recovered from bark. However, only terminal, 2,5- and 1,5-linked arabinofuranosyl groups were detected for WW. Interestingly, C1/H1–C5/H5 of terminal non-reducing xylopyranose residues linked at O-3 of the 1,4-GalpA backbone in XGA were identified for their respective characteristic signals at 101.5/5.08, 71.6/3.82, 79.4/3.84, 70.1/4.34, and 63.1/4.34 ppm (characteristic C1/H1 signal at 101.5/5.08 ppm, Fig. 2).30 The presence of XGA as part of a pectin complex has been previously reported in the flowering plant Arabidopsis thaliana.31
 | ||
| Fig. 2 2D HSQC NMR spectra (TSP-d4, δC/δH, 0/0 ppm) of dialyzed citric acid extracted pectin (DCA-P) of WW; WB; WIB; WBFB; and parenchyma. (a) Methyl (δC/δH, 0–59.38/0–4.02 ppm). (b) Non-anomeric methylene and methine (δC/δH, 58.93–95.48/3.16–4.59 ppm). (c) Anomeric methine regions (δC/δH, 95.92–113.89/4.21–5.66 ppm). For linkage feature differences, see ESI Table 3, and for chemical shift assignments, ESI Table 4.† | ||
The relatively high proportion of terminal arabinofuranosyl residues supported the highly branched arabinan side-chain structures at O-2 and O-3 branches from WW (ESI Table 3†). However, arabinofuranosyl was mostly 1,5-linked in bark pectin, indicating that the arabinan side chain from bark is much less branched compared to that from wood. Galactopyranosyl groups were mostly in the form of 1,4-linked with relatively few terminal groups, suggesting that side chains of galactan exist mostly in linear form in wood and bark. Overall, the characteristics of wood pectin are high acetylation, high proportion of HG domains, low proportion of less branched RG-I regions, and existence of XGA. The main feature of bark pectin is its heterogeneity from layer to layer. The pectin features of WB and WIB are a high DM (and DA) of HG domains and a high proportion of highly branched RG-I domains, and their arabinan and galactan side chains are abundant. However, for WBFB and parenchyma, the DM (and DA) of HG domains is relatively low. This knowledge is essential to select the optimized pectinase and other enzymes that target RG-I regions, because pectinases exhibit specific catalytic activity in degrading pectin depending on their structural features (Table 2).
Hemicellulose characteristics
Pretreatment using citric acid is important not only for pectin chelation, but also for removing metallic inorganic components,26 which is crucial to minimize the reactivity of peracetic acid (PAA).32 In WW, acid-insoluble lignin decreased progressively from original biomass (WW_O) to citric acid-treated biomass (WW_C), PAA-delignified solid residue (WW_P), and DMSO-extracted solid residue (WW_DMSO) (ESI Fig. 12†). The presence of high amounts of acid-insoluble lignin in WB_P and WB_DMSO indicated the complexity of bark since PAA is an electrophile with high selectivity in reactions, particularly with aromatic compounds.32 Meanwhile, the overall content of sugars and its monosaccharide glucose increased along with the treatment from “O” to “C”, “P”, “DMSO” and “H” (ESI Fig. 12†). As for hemicellulose, xylose was the main sugar constituent of all extracted hemicelluloses (Fig. 3), particularly at WW. Furthermore, the overall presence of galactose, glucose, and mannose is much higher in bark than in wood, indicating the presence of GAMA and GLMA in purified hemicellulose from bark (Fig. 3). In addition, the high glucose from parenchyma's hemicellulose indicates that DMSO is possibly capable of partially dissolving α-glucan starch33 in addition to hemicellulose. This is also consistent with the NMR results (Fig. 4) and iodine staining (ESI Fig. 5†).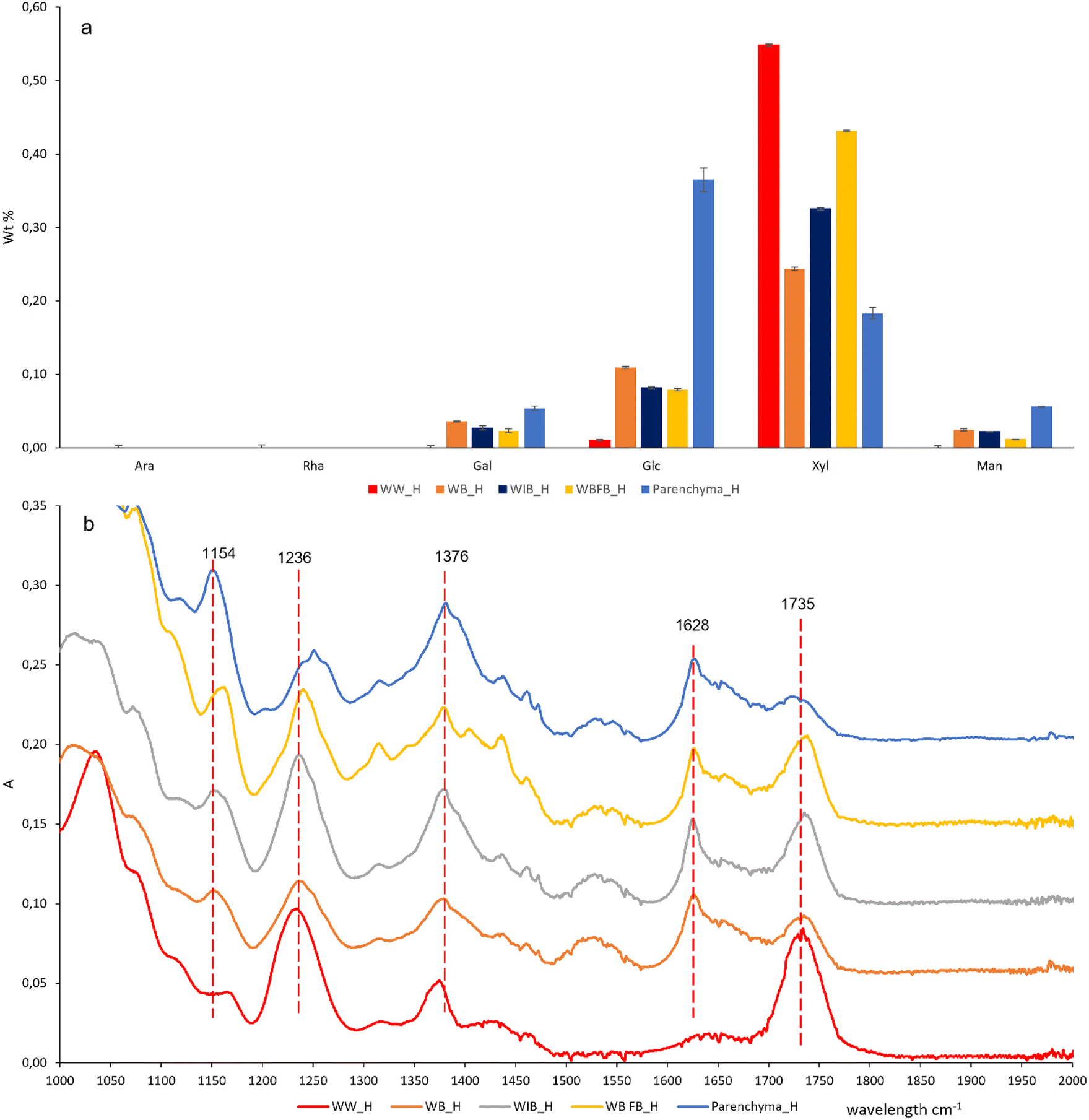 | ||
| Fig. 3 Chemical characteristics of purified hemicellulose from WW, WB, WIB, WBFB, and parenchyma. (a) Monosaccharide composition. (b) FT-IR spectrum (assignment in ESI Table 7†). | ||
 | ||
| Fig. 4 2D HSQC spectra (DMSO-d6/pyridine-d5, v/v 4/1) of the purified hemicellulose from WW, WB, WIB, WBFB, and parenchyma. (a) Methyl (δC/δH, 9.9–52.2/0.5–3.8 ppm). (b) Non-anomeric methylene and methine (δC/δH, 53.0–84.6/2.9–5.0 ppm). (c) Anomeric methine group (δC/δH, 95.6–106.0/3.6–5.5 ppm). See ESI Table 8† for the assignment. The chemical shifts between the branched D-mannose and the non-substituted D-mannose residues are very similar39 and therefore the same chemical label has been assigned. | ||
The absorbance bands (Fig. 3) at 1735 cm−1 and 1236 cm−1 were verified as the characteristic of hemicellulose.9,12,34 These two peaks have become more significant along with multiple stages (from “O”, “P”, “DMSO” to “H”) (ESI Fig. 14†). Moreover, there were no absorption bands of lignin at 1500 cm−1 and 1594 cm−1 in the PAA-treated samples (P) (ESI Fig. 14†) in comparison with raw wood (or bark) (O). The evidence of lignin removal is also justified from its color differences between raw sawdust (O) and its PAA-treated sawdust (P) (ESI Fig. 1–5†). The complete white color of the PAA-treated sawdust (P) is indicative of lignin removal for WW, WIB, and WBFB. However, the light-yellow color of the treated sawdust is indicative of some residual lignin chromophores, like quinones (1675 cm−1 at FT-IR),35 in WB (ESI Fig. 2†) and parenchyma (ESI Fig. 5†). Overall, PAA delignification is an essential step to break down the recalcitrant matrix and make hemicellulose become more accessible to DMSO. These multiple pretreatments eliminate most of the PAA-reactive compounds from bark. Most of the acetyl substituents are surprisingly stable (Fig. 3b and 4 and ESI Fig. 13†) after 0.1 M NaOH treatment, although alkaline extraction has been known for deacetylating acetyl groups from the chain,36 which is also supported by the absence of the acetyl group (1.5–1.8 ppm)37 (ESI Fig. 13†).
The structural features of hemicellulose were further comparatively studied by 1H and 13C NMR (ESI Fig. 13†). Xylan,9,12,34 GLMA,38 and GAMA39 were assigned according to a published database. Five clear strong signals of (1 → 4) linked β-D-Xylp residues were detected at 102.0 (C-1), 75.8 (C-4), 75.0 (C-3), 73.2 (C-2), and 63.6 (C-5) ppm, respectively. Weak signals at 169.3 (ESI Fig. 15†), 97.6, 82.0, 72.2, 72.1, 71.6, and 56.2 ppm (ESI Fig. 13†), corresponding to –COOH, C-1, C-4, C-3, C-5, C-2, and –OCH3 of the –MG group in hemicellulose were detected, respectively. Similar characteristics were also observed from the 1H NMR spectra. In particular, the major signals at 4.38 (H-1), 4.0 (H-5b), 3.58 (H-4), 3.42 (H-3), 3.27 (H-5a), and 3.20 (H-2) ppm originated from β-D-xylopyranosyl units, while the minor signals at 5.26 (H-1), 4.26 (H-5), 3.47 (H-2), and 3.33 (–OCH3) ppm could be assigned to the attached –MG units. Moreover, a signal of acetyl CH3 was observed at 2.01 and 20.85 ppm from 1H and 13C NMR, respectively. The signals at 169.3 and 172.0 ppm correspond to the –COOH and carbonyl groups of hemicellulose, respectively. These signals were present in all purified hemicelluloses. One significant signal at 165 ppm could be tentatively assigned to the non-protonated ester group in cutin40 that is present in the recovered hemicelluloses from bark. Furthermore, the aliphatic groups of suberin centering around 30.0 ppm40 appeared only at the hemicellulose of WB, indicating that suberin was possibly co-extracted with hemicellulose from DMSO and that suberin is mostly present in the outer bark of willow.41
2D HSQC NMR (Fig. 4) has been applied to elucidate the linkage features of hemicelluloses. WW's hemicellulose is a typical hardwood xylan containing the substituted –MG group. Specifically, C1/H1–C5/H5 of terminal xylose were identified for their characteristic peaks of 101.9/4.37, 72.4/3.16, 71.8/3.36, 75.5/3.68, and 63.0/3.29 (5a) and 3.98 (5b) ppm, respectively. The identified peak at δC/δH 20.5/1.99 ppm could be assigned to the acetyl group of xylans. The peaks at δC/δH 55.8/3.46 (Xy-MG-OCH3), 70.18/4.01 (Xy-MG-5), 81.3/3.23 (Xy-MG-4), 71.81/3.74 (Xy-MG-3), 71.72/3.57 (Xy-MG-2), and 97.2/5.31 ppm (Xy-MG-1) represent the C1/H1–C6/H6 correlations of the branched linkages between the 4-O-α-D-glucuronic acid (MeGlcA) and (1 → 4)-β-D-Xylp ((1 → 4)-β-D-Xylp-2-O-(4-OMe-D-GlcpA)), which was mostly identified at WW. Xy-2-O-Ac and Xy-3-O-Ac were assigned only at WW, WB, and WIB. To be specific, signals of 2-O-acetyl-β-D-Xylp-units (Xy-2-O-Ac) were detected at 99.6/4.57 (C1–H1) and 73.1/4.63 ppm (C2–H2), respectively. Similarly, signals of 3-O-acetyl-β-D-Xylp-units (Xy-3-O-Ac) were detected at 101.9/4.39 (C1–H1) and 74.66/4.92 ppm (C3–H3), respectively. Furthermore, a signal of 2.3-di-O-acetyl-β-D-Xylp-units (Xy-2,3-di-O-Ac) detected at 99.09/4.75 ppm (C1–H1) was present only in the parenchyma's hemicellulose.
The bark's hemicellulose contained much fewer –MG groups but included more of the substituted groups of GAMA and GLMA. Specifically, five dominant cross peaks at 98.9/4.76, 70.2/4.02, 71.73/3.77, 76.06/3.78, 74.02/3.65, and 66.6/4.12 ppm were assigned to C1/H1, C2/H2, C3/H3, C4/H4, C5/H5, and C6/H6 of GAMA units, respectively. In addition, the signals at 101.2/4.49, 71.82/3.59, 72.82/3.72, 76.97/3.76, 69.3/3.87, and 68.7/3.73 ppm were respectively assigned to C1/H1, C2/H2, C3/H3, C4/H4, C5/H5, and C6/H6 of GLMA units. Furthermore, signals from proteinaceous phenylalanine (δC/δH, 129/7.20 ppm) were identified in the bark samples (ESI Fig. 16b−e†), indicating that bark hemicellulose is possibly linked with protein. Table 1 summarizes the relative abundance of the main linkages of hemicellulose. WW hemicellulose was rich in acetyl substitutions at carbon 2 (42%) or 3 (20%), followed by xylan (32%) and the side branches of –MG (6%). In comparison with WW, hemicellulose from WB, WIB, and WBFB contained more unsubstituted xylan, GAMA, GLMA, and less Xy-MG. Parenchymal hemicellulose contained more Xy-2,3-di-O-Ac, GLMA and 1,4-α-D-Glcp starch.
All these data lead to the conclusion that wood hemicellulose is a typical polysaccharide (>40 kDa) (ESI Fig. 17 and Table 9†) made up of β-1,4-linked xylose residues with mainly side branches of –MG and minor acetyl substitutions at carbon positions of 2 or 3. The molar mass distribution is shifted to a shoulder of a lower molar mass peak (ESI Fig. 17†), which indicates that the shoulder peaks could be attributed to the unrecovered hemicelluloses.42 Overall, bark hemicellulose is chemically heterogeneous from WBFB to parenchymatous tissues. Hemicelluloses from WB (>38 kDa), WIB (>45 kDa), and WBFB (>27 kDa) were symbolized for their characteristic units of GAMA and GLMA in addition to the main xylan as the backbone. Interestingly, the hemicellulose from parenchyma (>14 kDa) featured more GLMA and starch in addition to the xylan and minor acetyl substitutions at both C2 and C3 (2.3-di-O-Ac-b-D-Xylp). Similar observations are reported in Table 1 and Fig. 4.
Dioxane lignin characteristics
The most well-known method for quantitatively determining lignin, Klason lignin, was originally designed for wood-based biomass. The presence of acid-insoluble lignin fractions, including protein, cutin/suberin, humins, and fats, is the main factor in the overestimation of lignin in bark. Similar observations have been reported by other researchers.43,44 Therefore, it is an essential step to achieve the maximal yield of dioxane lignin by prior removal of interfering substances (including extractives, proteins, pectins and tannins), which provides reliable acid-insoluble lignin quantitation for bark-derived samples. Briefly, the reported acid-insoluble “lignin” of bark (16 ± 3.3%) after all possible pretreatments was roughly 8–12% smaller than WB_original (28 ± 0.01%, no pretreatment) (ESI Table 10†) and those reported in the literature (24.7 ± 0.1%),15 which suggested that bark lignin has been frequently overestimated due to the undesired protein and HTS that are reported in Table 1. A similar trend was observed in WIB and WBFB. Moreover the removal efficiency of Klason lignin increased progressively from WB (22%), to WIB (42%), WBFB (52%), and WW (84%), which may show that there are much more complex compounds possibly present in the outer bark that interfere with dioxane lignin purification.Dioxane lignin purification is revealed by both compositional analysis (HPAEC-PAD) and spectroscopic characterization (FT-IR and CP-MAS 13C NMR) from all samples, including the original sample (O), 0.1 M NaOH-treated solid residue (N), solid residues after dioxane/water extraction (dioxane), and the recovered dioxane lignin (L). Glucose and xylose were the main monosaccharides from all samples (from “O” to “N”, and “dioxane”). The determined acid-insoluble “lignin” decreased progressively along with the treatment (from “O” to “N”, “dioxane”) except for parenchyma (ESI Fig. 18†). The most characteristic absorption signals of lignin45 were at ca. 1462, 1423, 1506, and 1594 cm−1, which were almost absent after the dioxane–water extraction (ESI Fig. 19†) and showed up in the dioxane lignin (Fig. 5) for all samples.
 | ||
| Fig. 5 Chemical characteristics of the dioxane lignin (L) from WW, WB, WIB, and WBFB. (a) FT-IR spectra. (b) CP-MAS 13C NMR spectra. (c) 2D HSQC NMR spectra (DMSO-d6/pyridine-d5, v/v 4/1) showing the aromatic (δC/δH, 96–150/6.0–8.2 ppm) and side-chain (δC/δH, 48–92/2.0–6.0 ppm) regions. See ESI Tables 11 and 12† for the assignments. | ||
Clear signal intensity differences of lignin were seen between 110 and 165 ppm46 throughout the treatment (ESI Fig. 20†) by CP-MAS 13C NMR spectroscopy. Specifically, all spectra showed characteristic signals of lignin at 154, 148, 135, and 53 ppm (ESI Table 11†). These were absent in the “dioxane” samples in comparison with the “O” and “N”, and all these characteristic peaks appeared clearly at the dioxane lignin (Fig. 5b). Cellulose and hemicellulose characteristics were shown at stages including “O”, “N”, and “dioxane” (ESI Fig. 20†) for all samples, indicating that the dioxane extraction succeeded in recovering dioxane lignin without significant degradation of holocellulose. It has been reported that dioxane lignin can be extracted with tannins and fatty acids from the bark of spruce or birch.47,48 In WW, the disappearance of the peak centering around 20 ppm in WW_N can be attributed to the C4–C8 of the inter-flavonoid linkages of tannins or fats, and these were completely removed after the 0.1 M NaOH treatment48 (Fig. 5). Well-resolved aliphatic carbon resonances (30 and 33 ppm)49 and –C(O)O– (174.8 ppm) of the recovered dioxane lignin were attributed to the suberin from WB and WIB. Other intense signals at δC 160–180 ppm (ester and –COOH groups) and δC 10–35 ppm (–CH3 and –CH2 of aliphatic) indicate that the dioxane lignin from bark was also contaminated by ferulic acid, indicating the difficulty of suberin and ferulic acid removal by extraction solely with 0.1 M NaOH. This was observed both in solid- (Fig. 5) and liquid-state 13C NMR analysis (ESI Fig. 21†).41
The structural features of the dioxane lignin were further investigated using HSQC NMR (Fig. 5c). For the inter-unit linkage characterization, the Cα/Hα, Cβ/Hβ, and Cγ/Hγ correlations of β-O-4 were reflected at δC/δH of 71.9/5.03, 84.0/4.45, and 58.9–59.8/3.52–3.76 ppm, respectively. Additionally, Cα/Hα, Cβ/Hβ, and Cγ/Hγ correlations of β-5 were identified at δC/δH of 87.2/5.59, 53.3/3.51 and 62.9/3.77 ppm, respectively, while the β–β bond showed the corresponding correlations at δC/δH of 85.1/4.72, 53.8/3.10 and 71.1–71.2/3.87–4.22 ppm, respectively. For the lignin monomers, the S-units showed correlations of C2,6/H2,6 at δC/δH of 104.2/6.80 and 106.5/7.40 ppm. C2/H2 correlation of G-units was shown at δC/δH of 110.8/6.95 ppm, and C5/H5 and C6/H6 correlations at δC/δH of 114.8/6.80 and 119.1/6.90 ppm, respectively. The characteristic resonances from ferulic acid (Fig. 5c)50 and suberin (ESI Fig. 22†)41,51 have been identified in the dioxane lignin of willow bark. Ferulic acid has been known to be responsible for the structural association with cell wall components through suberin.52 All these assignments (ESI Table 12†) were based on a database from the literature.15,19 The phenylalanine and polysaccharide peaks disappeared in the recovered dioxane lignin compared to the whole cell wall and CEL of the willow bark.15
The S/G ratios of dioxane lignin from WW, WB, WIB, and WBFB, determined by HSQC, were 3.9, 0.9, 1.5, and 1.2 (Table 1), respectively. As characteristic of most G/S lignins, WW-L and WB-L were rich in β-aryl ether structures (89% and 91%, respectively), followed by resinols (10% and 8%) and phenylcoumarans (1%). In comparison with WB-L, both WIB-L and WBFB-L contained less β-aryl ether (80–85%) and more resinols (14–19%). Overall, these structural features of dioxane lignin expressed as relative proportions of aforementioned lignin substructures were similar to those of CEL from willow bark.15 Furthermore, the dioxane lignin preparation appeared to contain much fewer impurities (e.g., protein and polysaccharides) compared to the CEL (Fig. 5c). Moreover, the molecular weight of dioxane lignin was shown to be similar to that of the CEL from WW and WIB (ESI Fig. 23†), although the molecular weight of dioxane lignin from WB was nearly three times higher than that of the CEL of WB (ESI Table 13†), indicating the possible presence of contaminating suberin macromolecules. Based on these results, we concluded that willow bark lignin has a significantly higher proportion of guaiacyl units than wood lignin, although β-O-4 linkages are dominant in both. The yield of dioxane lignin is significantly higher than CEL, which indicates that dioxane lignin could be more representative of its original lignin structure than CEL.15 However, there is still a high number of impurities, tentatively attributed to ferulic acid, suberin, or the tannin/lignin complex50,53 (Fig. 5b and c and ESI Fig. 22†), present in the dioxane lignin of bark.
Tailored enzyme cocktail according to structural features
Compared with WW, WB and WIB were more chemically heterogeneous because of the concomitant tissues of WBFB and parenchyma (Table 3). Therefore, the enzyme cocktail application (Table 3) needs to be customized based on the substrate structure and specific characteristics of the enzymes. Fig. 6 shows the process of screening and selection of the enzymatic cocktail based on the structural features of WIB. For pectin, the ratio and composition of the HG and RG-I regions affect its degradation by pectinases. Pectin polysaccharides of bark showed a high proportion of highly branched RG-I and were rich in non-branched arabinan and galactan side-chains, indicating the necessity of selecting galactanase and arabinase as part of the cocktail. In contrast, when the content of RG-I in pectin is low (e.g., WW), only the enzymes targeting the HG region need to be considered.21 The DM also influences the selection of HG-degrading enzymes. Pectate lyase and polygalacturonase show higher activity on low esterified pectin, while pectin lyase and polymethyl galacturonase prefer highly esterified pectin with a high degree of esterification. In addition, acetylated GalpA residues can be removed by pectin/rhamnogalacturonan acetyl esterase. In WW, the unique XGA units require xylogalacturonan hydrolase, because the activities of other pectinases can be inhibited by T-xylp residues. | ||
| Fig. 6 Step-by-step (in black) screening and selection process (in red) of the tailor-made enzyme cocktail (in dark blue) based on the structural features of the substrate. (a) Pectin. (b) Hemicellulose. See Table 4 and ESI Tables 14–17† for further information of the enzyme. | ||

|
In WW, acetylxylan esterase can assist xylanase in the degradation of xylans more effectively since xylans show a high degree of acetylation. Different acetyl xylan esterases remove the acetyl groups in O-2 or O-3 (Table 4). In bark, xylans show a lower degree of acetylation, but they contain galactan, mannan, galactomannan, and glucomannan, which require mannanase and galactosidase to hydrolyze the respective hemicelluloses. Although the concentration of different enzymes acting on substrates needs to be further confirmed by experiments and optimized by response surface methodology (RSM), characterizing the structure can significantly narrow down the selection range of enzymes, and this has been successfully applied in the separation of fiber bundles from willow bark using the tailored pectinase cocktail.20 Several enzyme cocktails (Table 3) were tailored for different sections of willow, and this will be systematically investigated in another study, but is not within the scope of this work. Selection and screening of enzymes can filter out approximately 60–70% of the candidate enzymes based on the structural features of the substrates (Table 4 and ESI Tables 14–17†). For future implementation of the bark biorefinery concept, a higher ratio of S/G unit samples (WIB or WBFB) can be more easily delignified since they have fewer lignin carbohydrate complexes, providing a much higher pulping efficiency in comparison with WB. By combining knowledge of staining images and their chemical profiles, our understanding about the morphological and structural linkage differences from wood and bark has been significantly enhanced, which provides fundamental knowledge to realize the strategy of a “tailor-made enzyme consortium based on the structural features of the substrate”.20 Overall, prior enzyme recovery or elimination of macromolecule components (e.g., pectin and hemicellulose) is considered an essential pretreatment for implementing chemical pulping to produce dissolving-grade pulps for bark valorization.

|

|
Discussion
In summary, we performed systematic analysis, recovery, and follow-up characterization of the major non-cellulosic components (pectin, hemicellulose and lignin) from willow wood and its bark. Structural feature differences of the pectin, hemicellulose, and dioxane lignin were elucidated. As for willow wood pectin, those with high acetylation, a high proportion of HG domains, a low proportion of the less branched RG-I region, and the presence of xylogalacturonan are the main structural features to consider. However, the main features of wood bark pectin are its high proportion of the RG-I domain compared to willow wood. The degree of methylation (acetylation) and the branching degree of the RG-I domain are highly heterogeneous between the inner bark, fiber bundles, and parenchymatous tissues. Hemicellulose recovery from bark is demonstrated here for the first time, highlighting the importance of prior removal of highly peracetic acid-reactive compounds. Willow wood contains typical hardwood xylan with one substituted –MG group. Bark generally contains much less –MG substituents and a higher amount of GAMA and GLMA substituted groups linked to proteins. Willow bark dioxane lignin has a significantly higher proportion of guaiacyl units in comparison with wood lignin, although β-aryl ether inter-unit linkages are dominant in dioxane lignin of both bark and wood, indicating that delignification of bark to individual fibers requires more severe pulping conditions than what is required for its counterpart wood. The disclosed lignin chemistry will be highly useful for tailoring delignification technologies that would be facilitated by biological pretreatment of wood bark. Moreover, parenchyma cells have been known as thin-walled cells, acting as storage cells containing hemicellulose, pectin, and starch. The screening efficiency of the enzyme cocktail has been substantially increased based on the substrate's structural features. Knowledge of the structural characteristics of the different components of processed wood and bark materials is essential to design a successful tailored bio(chemical) approach for valorizing not only willow bark but also other types of bark from different wood sources.Materials and methods
Materials and chemicals
Two-year-old willow Klara hybrid stems were harvested from Carbons Finland Oy (Kouvola, Finland) on 5 May 2019. The willow wood (WW), bark (WB), and inner bark (WIB) were manually peeled. Willow bark fiber bundles were recovered through a mild chemical treatment using sodium bicarbonate as described previously.27 Meanwhile, the parenchyma tissues were carefully recovered (ESI Fig. 5†). All the samples were Wiley-milled (<1 mm mesh) and stored at −20 °C before further use. Another four-year-old willow Karin was included in the analysis of hydrolysable tannin-like substances (HTS). Salix myrsinifolia and goat willow (Salix caprea) were included for the staining studies; a profile about their growth and harvest was reported previously.23 Acetone, arabinose, N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) containing 10% trimethylchlorosilane (TMCS), chloroform, citric acid, dichloromethane, diethyl ether, dioxane, dimethyl sulfoxide (DMSO), DMSO-d6, ethanol, formic acid, fructose, galactose, glucose, hydrochloric acid, hydrogen peroxide, mannose, methanol, pepsin, peracetic acid, pyridine-d5, rhamnose, sodium bicarbonate, sodium hydroxide, sodium sulphate, starch, tetracosane (C24), and xylose were supplied from Sigma-Aldrich, Finland.Experimental flow
The Wiley-milled (<1 mm mesh) samples (O) were extracted under the Soxhlet unit (ColeParmer Extractors, Lenz) with three different solvents (i.e., dichloromethane, acetone and water) for 2–3 h to remove both lipophilic and hydrophilic extracts (ESI Fig. 6†). The experimental flow was composed of four major steps: pectin recovery using citric acid treatment; multiple pretreatments and HTS purification; hemicellulose recovery; and dioxane lignin purification.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, w/v, pH 2) at 90 °C for 60 min for chelating pectic polysaccharides. The slurry was filtered through a membrane (a diameter size of 15–20 μm) and the citric acid-treated solid residue was preserved for further analyses. Ethanol was added to the liquid (final ethanol concentration: 75 v/v%) and the mixture was kept cold (+5 °C) for pectin precipitation. Centrifugation (8000 rpm, Eppendorf 5804R) and freeze drying were implemented to obtain freeze-dried crude citric acid pectin (CAP). The CAP was further purified using dialysis membranes (Spectra/Por, MWCO 6–8 kDa, 96 h). Finally, the collected pectin precipitates were further centrifuged and freeze dried to obtain dialyzed citric acid pectin (DCAP).
:1) at 37 °C for 16 h using an incubation shaker with a speed of 300 rpm (CERTOMAT, Sartorius Biotech Inc.). The protein-free solid residue (P) was then washed with hot water until the washing liquid was neutral. HTS was removed by a further treatment with 0.1 M NaOH (1/50, w/v) under nitrogen flow at 100 °C for 1 h (ESI Fig. 6†) and the solid residue (N) was washed with water. The concentrated diethyl ether-soluble portion was finally vacuum dried overnight before further GC-MS analysis. The yield of protein and HTS was calculated from their weight differences before and after the treatments. The alkali-extracted bark was prepared for both hemicellulose purification and dioxane lignin isolation.
:MeOH mixture (7/3, v/v), and the hemicellulose was recovered after centrifugation (8000 rpm, Eppendorf 5804R). Once the residual solvent was eliminated from the hemicellulose under the fume hood, freeze drying was implemented to obtain the dried hemicellulose (H). The solid residues after DMSO extraction were collected for further analysis.
:1, v/v) containing 0.2 M HCl under a reflux unit under a nitrogen atmosphere using a 2 L three-necked flask also equipped with a reflux condenser. 250 mL of dioxane/water with the sample were refluxed at 90–95 °C for a period of 30–40 min. A pore size of 3–4 crucible was employed to collect the purified fractions. The same extraction procedure was repeated three times. The fourth extraction was conducted without the addition of hydrochloric acid to the dioxane/water mixture. The combined extracts were finally concentrated using a rotavapor, and lignin was precipitated by introducing the concentrated dioxane solution (roughly 150 mL) into cold water (final volume roughly 1600 mL). The dioxane lignin was centrifuged (8000 rpm, Eppendorf 5804R) at 5 °C and freeze dried for further analysis.
:30, w/v, pH 2) at 90 °C for 60 min for chelating pectic polysaccharides. The slurry was filtered through a membrane (a diameter size of 15–20 μm) and the citric acid-treated solid residue was preserved for further analyses. Ethanol was added to the liquid (final ethanol concentration: 75 v/v%) and the mixture was kept cold (+5 °C) for pectin precipitation. Centrifugation (8000 rpm, Eppendorf 5804R) and freeze drying were implemented to obtain freeze-dried crude citric acid pectin (CAP). The CAP was further purified using dialysis membranes (Spectra/Por, MWCO 6–8 kDa, 96 h). Finally, the collected pectin precipitates were further centrifuged and freeze dried to obtain dialyzed citric acid pectin (DCAP).
:1) at 37 °C for 16 h using an incubation shaker with a speed of 300 rpm (CERTOMAT, Sartorius Biotech Inc.). The protein-free solid residue (P) was then washed with hot water until the washing liquid was neutral. HTS was removed by a further treatment with 0.1 M NaOH (1/50, w/v) under nitrogen flow at 100 °C for 1 h (ESI Fig. 6†) and the solid residue (N) was washed with water. The concentrated diethyl ether-soluble portion was finally vacuum dried overnight before further GC-MS analysis. The yield of protein and HTS was calculated from their weight differences before and after the treatments. The alkali-extracted bark was prepared for both hemicellulose purification and dioxane lignin isolation.
:MeOH mixture (7/3, v/v), and the hemicellulose was recovered after centrifugation (8000 rpm, Eppendorf 5804R). Once the residual solvent was eliminated from the hemicellulose under the fume hood, freeze drying was implemented to obtain the dried hemicellulose (H). The solid residues after DMSO extraction were collected for further analysis.
:1, v/v) containing 0.2 M HCl under a reflux unit under a nitrogen atmosphere using a 2 L three-necked flask also equipped with a reflux condenser. 250 mL of dioxane/water with the sample were refluxed at 90–95 °C for a period of 30–40 min. A pore size of 3–4 crucible was employed to collect the purified fractions. The same extraction procedure was repeated three times. The fourth extraction was conducted without the addition of hydrochloric acid to the dioxane/water mixture. The combined extracts were finally concentrated using a rotavapor, and lignin was precipitated by introducing the concentrated dioxane solution (roughly 150 mL) into cold water (final volume roughly 1600 mL). The dioxane lignin was centrifuged (8000 rpm, Eppendorf 5804R) at 5 °C and freeze dried for further analysis.
Analytics
Several chromatographic (HPAEC-PAD; GPC; HPLC and GC-MS) and spectroscopic (FT-IR and NMR) techniques were employed for the characterization (ESI Table 18†).Chromatographic techniques
The molar mass for dioxane lignin was determined. Samples were dissolved in an eluent (0.1 M NaOH) at the concentration of 2 mg ml−1. The HPLC system used was Agilent 1100, and the columns used were Polymer Standards Service MCX 300 × 8 mm (three columns with pore sizes of 100 Å, 500 Å and 1000 Å). The flow rate was 0.7 ml min−1, and the injection volume was 50 μl. The calibration curve was accomplished with polystyrene sulfonate standards (1000–64000 g mol−1), ascorbic acid (176 g mol−1), and NaCl (58 g mol−1; detection with a refractive index detector). Molar masses were determined based on the UV signal at 280 nm.
The cellulose samples were dissolved in an eluent (0.9% LiCl in DMAc) via a solvent exchange procedure (water/acetone/DMAc). The instrument consists of a Dionex Ultimate 3000 HPLC module, Shodex DRI (RI-101) detector, and Viscotek/Malvern SEC/MALS 20 multi-angle light-scattering (MALS) detector. The columns used were Agilent PLgel MIXED-A (×4), and the flow rate was 0.75 ml min−1. The injection volume was 100 μl. Detector constants (MALS and DRI) were determined using a narrow polystyrene sample (Mw = 96000 g mol−1, Đ = 1.04) dissolved in 0.9% LiCl in DMAc. A broad polystyrene sample (Mw = 248000 g mol−1, Đ = 1.73) was used to check the calibration of the detector. The ∂n/∂c value of 0.136 ml g−1 was used for cellulose in 0.9% LiCl in DMAc.
Spectroscopic techniques
Nuclear magnetic resonance (NMR) spectroscopy was applied for the analyses of the chemical structures of pectin, hemicellulose, and dioxane lignin, respectively. Spectra were processed using a Topspin 4.0 (Bruker). The detailed experimental parameters are summarized below.
Data availability
The authors declare that all the data that support the findings of this study are available within the article and its ESI files or from the corresponding author upon reasonable request.Author contributions
J. D. designed the concept, performed the experimental work, and wrote the manuscript draft. J. W. assisted with the designing of the enzyme cocktail based on the structural features of willow wood and bark under the supervision of J. Z., and S. H. performed the CPMAS NMR test. D. E. instructed the work for preparing hemicellulose and dioxane lignin. T. V. supervised the work and provided critical review of the manuscript. All authors proofread the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work made use of the RawMatTERS Finland infrastructure (RAMI) facilities based at Aalto University. This work was a part of the Academy of Finland's Flagship Programme under project no. 318890 and 318891 (Competence Center for Materials Bioeconomy, FinnCERES). Special thanks go to Dr Heidi Henrickson from Aalto University for the language check. GC-MS infrastructure from the University of Aveiro is also appreciated for analyzing the tannin-like substances. Dr Leena Pitkänen from Aalto University provided the assistance for the molecular weight analysis using gel permeation chromatography. The NMR premises from the University of Helsinki are also appreciated.References
- E. Sjöstrom, Wood Chemistry: Fundamentals and Applications, Academic Press, New York, 1981 Search PubMed.
- H. Chen, P. Chauhan and N. Yan, Green Chem., 2020, 22, 6874–6888 RSC.
- M. Jablonsky, J. Nosalova, A. Sladkova, A. Haz, F. Kreps, J. Valka, S. Miertus, V. Frecer, M. Ondrejovic, J. Sima and I. Surina, Biotechnol. Adv., 2017, 35, 726–750 CrossRef CAS PubMed.
- X. Kang, A. Kirui, M. C. D. Widanage, F. Mentink-Vigier, D. J. Cosgrove and T. Wang, Nat. Commun., 2019, 10, 347 CrossRef CAS PubMed.
- A. Kirui, W. Zhao, F. Deligey, H. Yang, X. Kang, F. Mentink-Vigier and T. Wang, Nat. Commun., 2022, 13, 538 CrossRef CAS PubMed.
- O. M. Terrett, J. J. Lyczakowski, L. Yu, D. Iuga, W. T. Franks, S. P. Brown, R. Dupree and P. Dupree, Nat. Commun., 2019, 10, 4978 CrossRef CAS PubMed.
- F. Dranca and M. Oroian, Food Res. Int., 2018, 113, 327–350 CrossRef CAS PubMed.
- J. Berglund, D. Mikkelsen, B. M. Flanagan, S. Dhital, S. Gaunitz, G. Henriksson, M. E. Lindström, G. E. Yakubov, M. J. Gidley and F. Vilaplana, Nat. Commun., 2020, 11, 4692 CrossRef CAS PubMed.
- S. Sun, X. Cao, H. Li, F. Xu and R. Sun, Int. J. Biol. Macromol., 2014, 69, 158–164 CrossRef CAS PubMed.
- D. V. Evtuguin, J. L. Tomás, A. M. S. Silva and C. P. Neto, Carbohydr. Res., 2003, 338, 597–604 CrossRef CAS PubMed.
- M. Borrega and H. Sixta, Cellulose, 2013, 20, 2803–2812 CrossRef CAS.
- J. Ding, C. G. Yoo, Y. Pu, X. Meng, S. Bhagia, C. Yu and A. J. Ragauskas, Green Chem., 2019, 21, 3902–3910 RSC.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- D. M. Neiva, J. Rencoret, G. Marques, A. Gutiérrez, J. Gominho, H. Pereira and J. C. del Río, ChemSusChem, 2020, 13, 4537–4547 CrossRef CAS PubMed.
- J. Dou, H. Kim, Y. Li, D. Padmakshan, F. Yue, J. Ralph and T. Vuorinen, J. Agric. Food Chem., 2018, 66, 7294–7300 CrossRef CAS PubMed.
- S. Shimizu, T. Yokoyama, T. Akiyama and Y. Matsumoto, J. Agric. Food Chem., 2012, 60, 6471–6476 CrossRef CAS PubMed.
- D. V. Evtuguin, C. P. Neto, A. M. S. Silva, P. M. Domingues, F. M. L. Amado, D. Robert and O. Faix, J. Agric. Food Chem., 2001, 49, 4252–4261 CrossRef CAS PubMed.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 RSC.
- J. Dou, M. Kögler, K. K. Kesari, L. Pitkänen and T. Vuorinen, Green Chem., 2023, 25, 1908–1909 RSC.
- J. Dou, J. Wang, J. Zhao and T. Vuorinen, Green Chem., 2022, 24, 2576–2587 RSC.
- A. Maceda and T. Terrazas, Polymers, 2022, 14, 961 CrossRef CAS PubMed.
- V. C. Ribeiro and C. A. E. Leitão, Protoplasma, 2020, 257, 993–1008 CrossRef CAS PubMed.
- J. Dou, L. Galvis, U. Holopainen-Mantila, M. Reza, T. Tamminen and T. Vuorinen, ACS Sustainable Chem. Eng., 2016, 4, 3871–3876 CrossRef CAS.
- A. Arbenz and L. Avérous, Green Chem., 2015, 17, 2626–2646 RSC.
- C. Alvarez-Vasco and X. Zhang, Bioresour. Technol., 2013, 150, 321–327 CrossRef CAS PubMed.
- D. Xu, J. Wan, D. Xu, T. Luo, Y. Zhong, X. Yang, L. Yang, Z. Zhang and X. Wang, Can. J. Chem. Eng., 2020, 98, 665–675 CrossRef CAS.
- J. Dou, M. Rissanen, P. Ilina, H. Mäkkylä, P. Tammela, S. Haslinger and T. Vuorinen, Ind. Crops Prod., 2021, 164, 113387 CrossRef CAS.
- E. N. Makarova and E. G. Shakhmatov, Carbohydr. Polym., 2020, 246, 116544 CrossRef CAS PubMed.
- H. N. Cheng and T. G. Neiss, Polym. Rev., 2012, 52, 81–114 CrossRef CAS.
- L. Shen, X. Chu, Z. Zhang and T. Wu, Int. J. Biol. Macromol., 2022, 194, 100–109 CrossRef CAS PubMed.
- J. K. Jensen, S. O. Sørensen, J. Harholt, N. Geshi, Y. Sakuragi, I. Møller, J. Zandleven, A. J. Bernal, N. B. Jensen, C. Sørensen, M. Pauly, G. Beldman, W. G. T. Willats and H. V. Scheller, Plant Cell, 2008, 20, 1289–1302 CrossRef CAS PubMed.
- J. Kim and C. H. Huang, ACS EST Water, 2021, 1, 15–33 CrossRef CAS.
- S. Santacruz, K. Koch, R. Andersson and P. Åman, J. Agric. Food Chem., 2004, 52, 1985–1989 CrossRef CAS PubMed.
- T. Yuan, F. Xu, J. He and R. Sun, Biotechnol. Adv., 2010, 28, 583–593 CrossRef CAS PubMed.
- U. P. Agarwal, J. Wood Chem. Technol., 1998, 18, 381–402 CrossRef CAS.
- F. Peng, P. Peng, F. Xu and R. Sun, Biotechnol. Adv., 2012, 30, 879–903 CrossRef CAS PubMed.
- A. Svärd, E. Brännvall and U. Edlund, Carbohydr. Polym., 2015, 133, 179–186 CrossRef PubMed.
- H. Nishimura, A. Kamiya, T. Nagata, M. Katahira and T. Watanabe, Sci. Rep., 2018, 8, 6538 CrossRef PubMed.
- K. A. Nobre, C. E. A. Soares, Í. G. P. Vieira, R. R. de Almeida, R. A. Moreira, T. G. Araújo, M. E. N. P. Ribeiro and N. M. P. S. Ricardo, Quim. Nova, 2018, 41, 607–612 CAS.
- C. P. Neto, J. Rocha, A. Gil, N. Cordeiro, A. P. Esculcas, S. Rocha, I. Delgadillo, J. D. P. D. Jesus and A. J. F. Correia, Solid State Nucl. Magn. Reson., 1995, 4, 143–151 CrossRef PubMed.
- J. Dou, D. V. Evtuguin and T. Vuorinen, J. Agric. Food Chem., 2021, 69, 10848–10855 CrossRef CAS PubMed.
- T. Schult, T. Hjerde, O. I. Optun, P. J. Kleppe and S. Moe, Cellulose, 2002, 9, 149–158 CrossRef CAS.
- H. Kim, D. Padmakshan, Y. Li, J. Rencoret, R. D. Hatfield and J. Ralph, Biomacromolecules, 2017, 18, 4184–4195 CrossRef CAS PubMed.
- F. Lu, C. Wang, M. Chen, F. Yue and J. Ralph, A facile spectroscopic method for measuring lignin content in lignocellulosic biomass, Green Chem., 2021, 23, 5106–5112 RSC.
- Z. Shi, L. Xiao, J. Deng and R. Sun, BioEnergy Res., 2013, 6, 1212–1222 CrossRef CAS.
- G. Gilardi, L. Abis and A. E. G. Cass, Enzyme Microb. Technol., 1995, 17, 268–275 CrossRef CAS.
- A. V. Faleva, I. I. Pikovskoi, S. A. Pokryshkin, D. G. Chukhchin and D. S. Kosyakov, Polymers, 2022, 14, 964 CrossRef CAS PubMed.
- A. V. Faleva, A. V. Belesov, A. Y. Kozhevnikov, D. I. Falev, D. G. Chukhchin and E. V. Novozhilov, Int. J. Biol. Macromol., 2021, 166, 913–922 CrossRef CAS PubMed.
- J. Dou, H. Bian, D. J. Yelle, M. Ago, K. Vajanto, T. Vuorinen and J. Zhu, Ind. Crops Prod., 2019, 129, 15–23 CrossRef CAS.
- D. G. Branco, C. Santiago, A. Lourenço, L. Cabrita and D. V. Evtuguin, J. Agric. Food Chem., 2021, 69, 8555–8564 CrossRef CAS PubMed.
- A. Bento, R. Escórcio, A. S. Tomé, M. Robertson, E. C. Gaugler, S. J. Malthus, L. G. Raymond, S. J. Hill and C. S. Pereira, Ind. Crops Prod., 2022, 185, 115172 CrossRef CAS.
- A. V. Marques, J. Rencoret, A. Gutiérrez, J. C. del Río and H. Pereira, Holzforschung, 2016, 70, 275–289 CrossRef CAS.
- M. Bunzel and J. Ralph, J. Agric. Food Chem., 2006, 54, 8352–8361 CrossRef CAS PubMed.
- N. Sharma, M. Rathore and M. Sharma, Rev. Environ. Sci. Biotechnol., 2013, 12, 45–60 CrossRef CAS.
- P. Wu, S. H. Yang, Z. C. Zhan and G. M. Zhang, Appl. Microbiol. Biotechnol., 2020, 104, 7247–7260 CrossRef CAS PubMed.
- J. Zandleven, G. Beldman, M. Bosveld, J. Benen and A. Voragen, Biochem. J., 2005, 387, 719–725 CrossRef CAS PubMed.
- J. Navarro-Fernández, I. Martínez-Martínez, S. Montoro-García, F. García-Carmona, H. Takami and Á. Sánchez-Ferrer, J. Bacteriol., 2008, 190, 1375–1382 CrossRef PubMed.
- M. Mutter, C. M. G. C. Renard, G. Beldman, H. A. Schols and A. G. J. Voragen, Carbohydr. Res., 1998, 311, 155–164 CrossRef CAS PubMed.
- I. R. Silva, C. Jers, A. S. Meyer and J. D. Mikkelsen, New Biotechnol., 2016, 33, 41–54 CrossRef CAS PubMed.
- E. G. S. Farro, A. E. T. Leite, I. A. Silva, J. G. Filgueiras, E. R. de Azevedo, I. Polikarpov and A. S. Nascimento, Int. J. Biol. Macromol., 2018, 117, 7–16 CrossRef CAS PubMed.
- E. Luonteri, C. Laine, S. Uusitalo, A. Teleman, M. Siika-aho and M. Tenkanen, Carbohydr. Polym., 2003, 53, 155–168 CrossRef CAS.
- A. Pollet, J. A. Delcour and C. M. Courtin, Crit. Rev. Biotechnol., 2010, 30, 176–191 CrossRef CAS PubMed.
- M. A. S. Correia, K. Mazumder, J. L. A. Brás, S. J. Firbank, Y. Zhu, R. J. Lewis, W. S. York, C. M. G. A. Fontes and H. J. Gilbert, J. Biol. Chem., 2011, 286, 22510–22520 CrossRef CAS PubMed.
- F. J. St John, J. M. González and E. Pozharski, FEBS Lett., 2010, 584, 4435–4441 CrossRef CAS PubMed.
- P. M. A. Pawar, S. Koutaniemi, M. Tenkanen and E. J. Mellerowicz, Front. Plant Sci., 2013, 4, 118 Search PubMed.
- H. R. Sørensen, C. T. Jørgensen, C. H. Hansen, C. I. Jørgensen, S. Pedersen and A. S. Meyer, Appl. Microbiol. Biotechnol., 2006, 73, 850–861 CrossRef PubMed.
- V. Poria, J. K. Saini, S. Singh, L. Nain and R. C. Kuhad, Bioresour. Technol., 2020, 304, 123019 CrossRef CAS PubMed.
- M. Yamabhai, S. Sak-Ubol, W. Srila and D. Haltrich, Crit. Rev. Biotechnol., 2016, 36, 32–42 CrossRef CAS PubMed.
- H. Wang, H. Luo, J. Li, Y. Bai, H. Huang, P. Shi, Y. Fan and B. Yao, Bioresour. Technol., 2010, 101, 8376–8382 CrossRef CAS PubMed.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5, 87187–87199 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00808h |
| This journal is © The Royal Society of Chemistry 2023 |