
A direct pathway for the coupling of arenes and alkylamines via a heterogeneous zeolite-based photocatalyst†
Vincent
Lemmens
a,
Kwinten
Janssens
 a,
Jorge
Gascon
b and
Dirk E.
De Vos
*a
a,
Jorge
Gascon
b and
Dirk E.
De Vos
*a
aCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
bKing Abdullah University of Science and Technology (KAUST), 23955 Thuwal, Saudi Arabia
First published on 14th June 2023
Abstract
The production of N,N-dialkyl arylamines is relevant for a range of high-value materials such as pharmaceuticals, agrochemicals and organic materials. In general, these molecules are assembled via multiple pre-functionalization steps of the aromatic compound and a subsequent cross-coupling with the arylamine under harsh conditions. In this manuscript, we develop a more sustainable and efficient method for the direct C–H amination of arenes and alkylamines using Ru(bipy)32+ entrapped in the supercages of a faujasite support (CBV-100). This creates a heterogeneous photocatalyst that can be used under visible light irradiation. Furthermore, the mesomeric and/or inductive effects of the substituent groups on the arene coupling partner are thoroughly investigated using Hammett plots, leading to a rationally guided solvent selection (HFIP or CH3CN). Using reduced reaction temperatures (<0 °C) drastically improves the product yield, while suppressing the generation of side products.
Introduction
The construction of carbon–nitrogen bonds is seen worldwide as an extremely important reaction for the manufacture of high value-compounds, such as bulk chemicals, polymers, organic materials, agrochemicals and pharmaceuticals.1–4 Moreover, the formation of (di)alkyl arylamines represents about 20–30% of all nitrogen compound manipulations in the current pharmaceutical industry and is therefore of utmost importance in global drug discovery programs.1,5,6Traditionally, the introduction of nitrogen functional groups on arene substrates is performed by multi-step syntheses, starting from nitration, followed by reduction and additional manipulation steps (Fig. 1).1 Unfortunately, nitration and subsequent manipulation steps are typically associated with harsh conditions (e.g. strongly acidic, high temperature/pressure) and low selectivity.1 Interest in the development of other approaches has therefore grown significantly. At the beginning of the twentieth century, Ullmann7,8 and Goldberg9 (Fig. 1) published their pioneering work on C–N coupling using stoichiometric or even catalytic amounts of copper at elevated temperatures (>200 °C).4,10–13 In this context, the Cu-catalyzed Chan–Lam14–16 and Pd-catalyzed Buchwald–Hartwig coupling17,18 (Fig. 1) were well adopted as new methodologies in industry and academia.13 Despite their versatility, all these approaches need pre-functionalized arenes (e.g. aryl-boron compounds or aryl halides), which requires additional functionalization steps. Hence, this approach is less suitable for late-stage applications.1,2
 | ||
| Fig. 1 Overview of the different approaches towards N-substituted arylamines. | ||
More recently, research has shifted towards greener C–H activation strategies, aiming at using unfunctionalized arenes.3 Generally, two different strategies are considered: (1) cross-dehydrogenative C–N couplings (CDC) and (2) electrophilic amination (EA).3,19 In the latter approach, the nitrogen coupling partner carries a leaving group on the nitrogen atom, which makes the nitrogen an appropriate electrophile for the direct C–H amination. With these new insights, Leonori and co-workers developed a photocatalytic method to synthesize arylamines, using Ru(bipy)3Cl2 as photocatalyst.1,2 In this approach, a specific leaving group is installed either ex situ2,20 or in situ1,5 on the nitrogen atom of the coupling partner, which is transformed into a highly electrophilic aminium radical after a single electron transfer (SET) with the appropriate photocatalyst (Fig. 1).1,2,5,20,21
Unfortunately, the homogeneous nature of these (photo)catalysts hampers their further adoption in large-scale processes due to the high cost and laborious post-synthetic removal of the catalyst from the reaction mixture. Therefore, heterogeneous catalysts are typically studied as promising and renewable alternatives, but they have rarely been considered for photocatalytic amination.22,23
Faujasite type zeolites, which are aluminosilicates made up of SiO4 and AlO4 tetrahedra, are an appealing group of microporous, crystalline materials with a three-dimensional framework structure connected by interlinked voids.24 These zeolite materials are currently applied in large-scale industrial processes (e.g. adsorption, FCC cracking,…) due to their low cost, high specific surface area (SSA), shape/size selectivity and high stability.25 Besides the catalytic activity inherent to zeolites, the ability for metal cation exchange and for entrapment of (photoactive) organometallic complexes in their supercages are other important features allowing design of single-site heterogeneous catalysts.25,26–33
In this work, a faujasite type zeolite Y material (CBV-100) is employed as host for the entrapment of Ru(bipy)32+ within its supercages via a “ship-in-a-bottle” technique (Fig. 1).26 The entrapped photoactive polypyridyl complex can be excited by visible light to ultimately form electrophilic aminium radicals as the nitrogen coupling partner in direct C–H aminations. In this way, a single-site heterogeneous catalyst is developed for the sustainable synthesis of arylamines, while avoiding harsh conditions, stoichiometric amounts of transition metal and pre-functionalized substrates (see ESI† for a detailed discussion on sustainability and economics).
Results and discussion
Synthesis and characterization of the catalyst
The faujasite CBV-100 ([Si2.55Al1O7.1Na1], with FAU topology and a silicon-to-aluminum ratio (SAR) of about 2.5 is commercially available; it is synthesized in aqueous media under hydrothermal conditions. This material is built up from sodalite cages, which are linked through double 6-membered rings (D6Rs).34 In this way, the sodalite cages of faujasite are arranged in such a way that interconnected supercages are formed. This topology makes the material excellent as a support material to encapsulate Ru(bipy)32+via a ship-in-a-bottle strategy.26,27 The zeolite material is first ion-exchanged with 1 wt% Ru using Ru(NH3)6Cl3 in ultrapure demineralized water and subsequently contacted with an excess of molten 2,2′-bipyridine (bipy) to obtain the cationic photoactive complex (12 Å) in the 13 Å wide supercages.27 The sample was excessively rinsed with acetone in order to remove excess ligand and complexes located on the outer surface. In order to determine the amount of entrapped photoactive molecules, the Ru-loaded zeolite was digested in aqua regia and HF; the Ru and the bipy were quantified by inductively coupled plasma atomic emission spectroscopy (ICP-OES) and 1H nuclear magnetic resonance (NMR) of the liquid phase using added standards. The Ru content was estimated at 0.91 wt% (Table S1†), while the molar amount of coordinated bipy ligands was about 3× higher (Fig. S2 and S3†). Consequently, 12.9% of the supercages contained the Ru complex, leading to the single-site heterogeneous catalyst CBV-100-Ru(bipy)3 [Si2.55Al1O7.1Na0.956(Ru(bipy)32+)0.022]. Additionally, N2 sorption analyses showed a significant decrease in micropore volume (45%) in comparison to the parent material (Fig. S4 and Table S2†).35The crystallinity and structure of the pristine and functionalized zeolite (CBV-100-Ru(bipy)3) were confirmed by powder X-ray diffraction (PXRD) (Fig. 2A) and SEM (Fig. S5†). Furthermore, the location of the extra-framework cations is studied by the relative intensities of the XRD peaks with Miller indices 220, 331 and 311 (Fig. 2A). The cations are assumed to be randomly distributed within the lattice if I331 > I220 > I311. However, upon impregnation of the zeolite with the photoactive complex, it is seen that I331 > I311 > I220. We therefore presume that this is an indication that the formation of Ru(bipy)32+ in the supercages is accompanied by the displacement of cations from their random positions in the supercages to locations inside the sodalite cages, at sites I′ and II.36–38
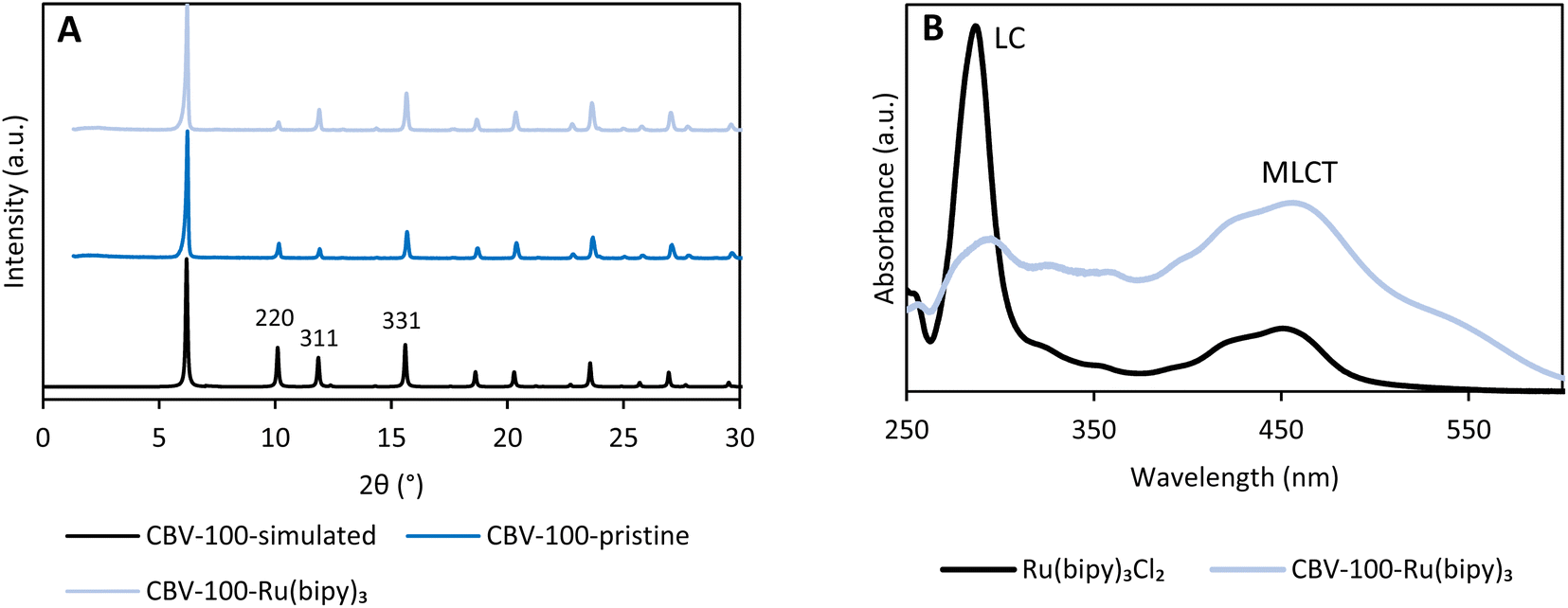 | ||
| Fig. 2 XRD diffractogram of simulated CBV-100 (black), pristine CBV-100 (dark blue) and Ru loaded CBV-100-Ru(bipy)3 (light blue) (A). UV-Vis spectrum of the homogeneous complex in acetonitrile (Ru(bipy)3Cl2, black) and a diffuse reflectance spectrum of CBV-100-Ru(bipy)3 (blue) (B). LC = ligand centered transition, MLCT = metal-to-ligand charge transfer. | ||
Additionally, spectroscopic techniques were employed in order to confirm the incorporation of the photoactive complex in the supercages of CBV-100. Fourier-transform infrared spectroscopy (FTIR) data confirm the presence of Ru(bipy)32+, since the characteristic bands in the region 1400 to 1500 cm−1 resembles the C–N, C–C stretching vibrations and/or the C–H in-plane bending vibrations of the bipyridyl ligand (Fig. S6†).35,39 Furthermore, a typical absorption spectrum of Ru(bipy)3Cl2 in acetonitrile was collected, showing: (i) an intense band at 285 nm attributed to a π–π* ligand-centered transition (LC), and (ii) a broad band from 380 nm to 500 nm in the visible region related to the metal-to-ligand charge transfer (MLCT) from the d-orbital of Ru to the π*-orbital of the ligand (Fig. 2B).40 A similar UV-Vis spectrum was measured for CBV-100-Ru(bipy)3 in diffuse reflectance mode, with a maximum at 456 nm, indicating that the photocatalyst can be excited in the visible-light region.
The oxidation state of the Ru center is of utmost importance, since only Ru(II) is susceptible to photoexcitation and can catalyze the amination reaction. X-ray absorption near-edge structure (XANES) of CBV-100-Ru(bipy)3 confirms that the pattern is very similar to that previously recorded for Ru(bipy)32+@EMC-1 (EMC-1 is a faujasite with lowered Al-content; SAR = 3.6) (Fig. 3A).27 Furthermore, the local environment of the encapsulated Ru center in the faujasite was studied by extended X-ray absorption fine structure (EXAFS) (Fig. 3B), which revealed that (i) the Ru-center is surrounded by six nitrogen atoms, (ii) no Ru–Cl interactions can be observed and (iii) the signals at higher radial distances (2–3 Å) can be assigned to the well-ordered carbon atoms of the 2,2′-bipyridine ligands. Moreover, the pattern and its intensity are similar to the pattern previously recorded for Ru(bipy)32+@EMC-1, which indicates that formation of the Ru-complex is successful. In addition, a good fit was obtained between the simulated spectrum of Ru(bipy)32+ and the measured spectrum of CBV-100-Ru(bipy)3 (Fig. S7†).
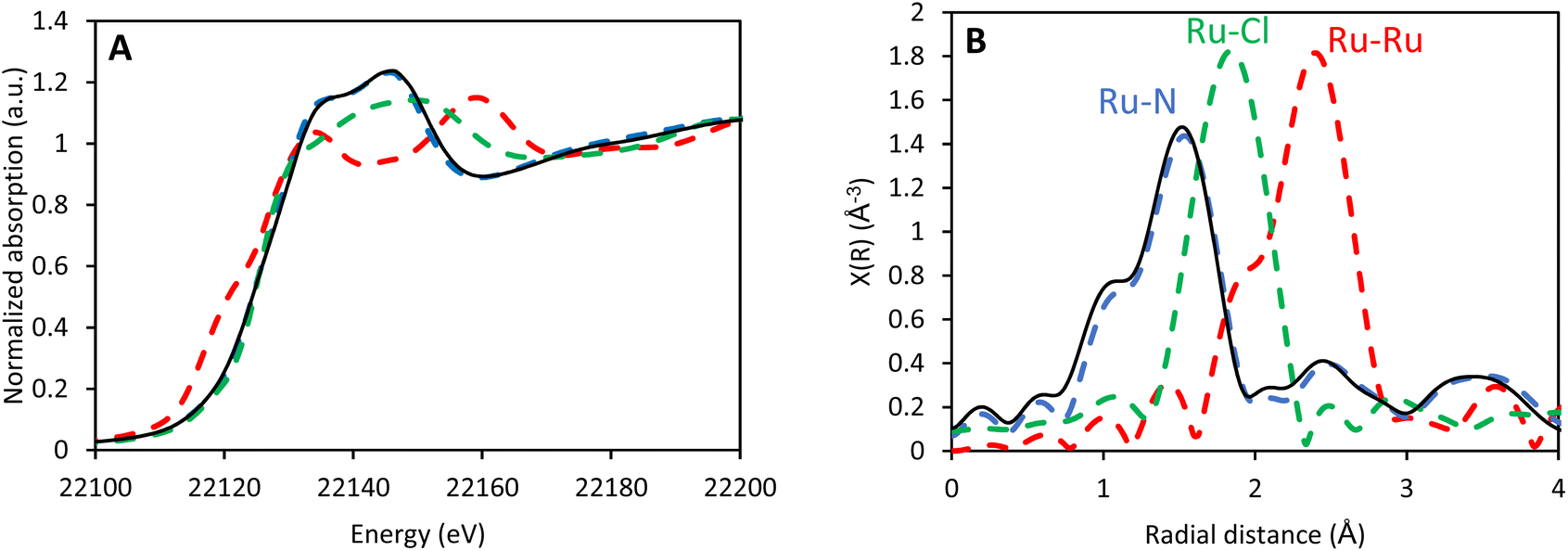 | ||
| Fig. 3 XANES (A) and EXAFS (B) data for CBV-100-Ru(bipy)3 (solid black) in comparison with reference samples (dashed lines): Ru(bipy)32+@EMC-1,27 blue; RuCl3, green; Ru-foil, red. | ||
Optimization of the reaction parameters
In order to check the performance of the heterogeneous single-site catalyst (i.e. CBV-100-Ru(bipy)3) for the formation of arylamines, piperidine and t-butylbenzene were employed as model substrates. N-Chlorosuccinimide (NCS) was first mixed together with the N-coupling partner in the solvent (HFIP). In this way, N-chloropiperidine was formed (Fig. S8†); next the solution was contacted with the strong acid (HClO4) and the catalyst under the optimized reaction conditions (Table 1, entry 1).|
|
||
|---|---|---|
| Entry | Conditions | Yield (Y3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b/Y4c) (%) b/Y4c) (%) |
| a 1 (0.1 mmol), NCS (1 equiv.), 1 mL HFIP were mixed together and stirred for 30 minutes, afterwards 2 (18 equiv.), CBV-100-Ru(bipy)3 (2 mol% Ru), HClO4 (4 equiv.) were added and exposed to blue light (455–470 nm, 40 W) for 3 h at room temperature. b Y3 is the yield of the arylamine 3. c Y4 is the summed yield of the chloro-tert-butylbenzenes. | ||
| 1 | Optimized conditions | 64/19 |
| 2 | Ru(bipy)3Cl2 as catalyst | 67/33 |
| 3 | No NCS | 0/0 |
| 4 | No light | 0/70 |
| 5 | No catalyst | 22/44 |
| 6 | 2 equiv. t-butylbenzene | 30/6 |
| 7 | 8 equiv. t-butylbenzene | 57/19 |
| 8 | 30 equiv. t-butylbenzene | 62/16 |
| 9 | 0 equiv. HClO4 | 0/0 |
| 10 | 2 equiv. HClO4 | 19/19 |
| 11 | 16 equiv. HClO4 | 61/24 |
| 12 | 1 mol% Ru | 49/15 |
| 13 | 5 mol% Ru | 61/15 |
| 14 | 0.5 mmol piperidine | 60/23 |
| 15 | N-Hydroxypiperidine | 0/0 |
| 16 | Acetoxy-N-piperidine | 0/0 |
| 17 | Trifluoroacetoxy-N-piperidine | 0/0 |

Control experiments in the absence of NCS, light and catalyst were performed to verify the role of each compound in the product formation (Table 1, entries 3–5). The NCS reagent is essential for the chlorine transfer onto piperidine; in its absence, no electrophilic aminium radical and subsequently aminated arene can be formed (Table 1, entry 3).1,21,41 In the absence of light, no aminated arene product is observed, while the yield of chlorinated arene (70%) was extremely high (Table 1, entry 4). This suggests that the N-chloropiperidine displays an enhanced electrophilic character on the chlorine atom, which makes it an excellent reagent for the acid-catalyzed electrophilic aromatic chlorination (Fig. S9†).1,42,43 Furthermore, in the absence of catalyst, a product yield of 22% is observed due to the homolytic cleavage of the N–Cl bond.21 Additionally, the yield of chlorinated product is also high in this scenario (44%), which indicates the importance of the photocatalyst to kinetically overcome this dark background reaction.
A clear optimum is reached as the concentration of t-butylbenzene is varied in the reaction mixture (Fig. S10†). The yield of aminated arene remains moderate (30%) at low arene concentrations (2 equiv., Table 1, entry 6), while a remarkable increase in yield is noticed at higher concentrations. However, at an excessively large amount of the arene substrate the product yield remains unchanged (Table 1, entry 8). Furthermore, (in)organic acids with a low pKa value (i.e. strong acids) are preferred, since protonation of N-chloropiperidine is more likely to occur (Fig. S11†).42–44 Nevertheless, a too strong acid (e.g. CF3SO3H, pKa = −14.7) excessively stimulates the acid-catalyzed chlorination reaction and a decrease in C–N products is noticed. By varying the amount of acid (Table 1, entries 9–11), a similar effect was observed; the yield increases until the acid concentration becomes so high that it promotes the unwanted chlorination reaction (Fig. S12†).42,43 Generally, an increase of product yield is expected with a higher catalyst loading. However, at higher catalyst loadings, the yield remains unchanged (Table 1, entries 12, 13 and Fig. S13†). In order to understand this observation, the corresponding homogeneously catalyzed reactions were performed, with Ru(bipy)3Cl2 as catalyst; these clearly showed an increase of initial reaction rate and a lower amount of side products at higher catalyst loading (Fig. S14†). Such trend is not observed for the heterogeneously catalyzed reactions. This could be due to light penetration problems: because of too much diffuse scattering, the incoming light will not travel deeply into the reaction suspension at higher catalyst loading (i.e. when the suspension contains more zeolite material); therefore the volume fraction of the suspension that remains ‘dark’, and in which the chlorination dominates, is increased. In this way, the addition of more catalyst at a certain level no longer has an additional effect, and the correct amount of catalyst needs to be carefully considered. A kinetic study revealed that the initial reaction rate is significantly slower than for the corresponding homogeneously catalyzed reaction, which is easily explained by the hampered penetration of light into the zeolite crystals and by the diffusion limitations of substrate and product molecules through the zeolite framework (Fig. S15†).
Different leaving groups were installed on the piperidine molecule (viz. HO−, CH3C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O− and CF3C(O)O−) via a well-established pre-synthesis procedure.45,46 All these piperidine coupling partners were tested, but unfortunately barely any yield is observed (Table 1, entries 15–17 and Fig. S16–S18†).
O)O− and CF3C(O)O−) via a well-established pre-synthesis procedure.45,46 All these piperidine coupling partners were tested, but unfortunately barely any yield is observed (Table 1, entries 15–17 and Fig. S16–S18†).
The support that was used for the synthesis of the heterogeneous single-site catalyst was the faujasite-type zeolite CBV-100. This zeolitic support is excellently suitable due to appropriate cage dimensions, which prevent the photoactive complex from leaching (Table S3†). By increasing the Si/Al ratio (SAR), a remarkable decline in product yield (Fig. 4) is observed due to a lower Ru-content in the zeolite framework after catalyst synthesis (Table S3†). Typically, these high SAR faujasites are produced from NaY via steaming processes, during which water steam is responsible for gradually etching away the alumininum from the framework, resulting in defects and mesopores.47,48 Therefore, some photocatalyst molecules are located in these mesopores with less pore size restriction, which results in Ru-leaching during the washing steps of the synthesis. On the other hand, high product yields were obtained with zeolite MCM-22 (MWW topology), even if its Ru-content is rather low (Table S3†). However, a large portion (∼90%) of the Ru is leached during the reaction. This could be due to the fact that Ru here is located in the large half-cages located at the outer surface of the material (Table S4†). Even if a higher product yield of ∼75% is obtained, the high activity is in fact due to the homogeneous nature of the catalyst during the reaction.
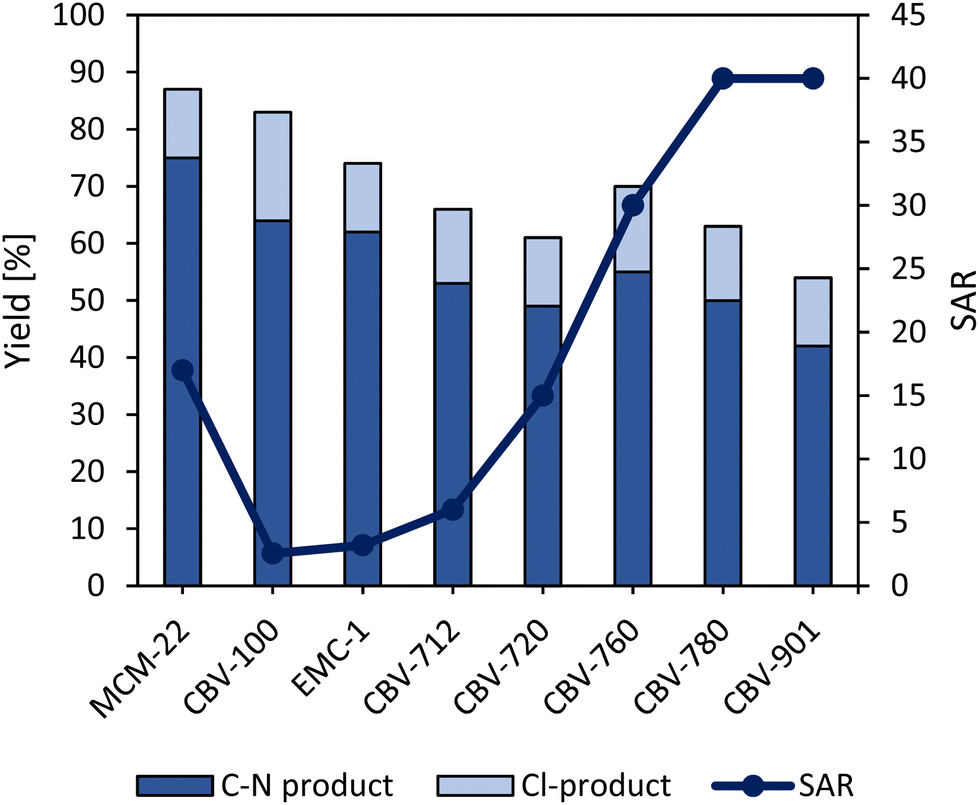 | ||
| Fig. 4 Product yields (dark blue line) for Ru(bpy)32+ immobilized on zeolite supports with different SAR ratios (MCM-22 (SAR = 17), EMC-1 (SAR = 3.6), CBV-100 (SAR = 2.55), CBV-712 (SAR = 6), CBV-720 (SAR = 15), CBV-760 (SAR = 30), CBV-780 (SAR = 40) and CBV-901 (SAR = 40)). Reaction conditions: 0.1 mmol piperidine, 1 equiv. NCS, 18 equiv. t-butylbenzene, 4 equiv. HClO4, 1 mL HFIP, 2 mol% Ru as zeolite-Ru (theoretically), blue LEDs 3 h, R.T. | ||
Strategies to overcome electrophilic chlorination: role of the solvent
Even if reaction conditions were thoroughly scrutinized, a significant amount of unwanted chlorinated products is still observed. To understand this better from a mechanistic point-of-view, we studied the homogeneous chlorination/amination of different arene substrates with N-chloropiperidine in more detail, using aromatics with weakly and increasingly strongly electron-donating substituents. The homogeneous photocatalyst Ru(bipy)3Cl2 was used in either HFIP or acetonitrile (CH3CN) (Fig. 5). Generally, a stronger electron donating effect of the substituent group results in a higher amount of chlorinated product, which is fully in line with the concept of electrophilic aromatic chlorination (Fig. 5A).49,50 While the trend is similar for both solvents, a slightly more negative ρ-value was noticed for HFIP, resulting in larger amounts of chlorinated arene as the substituent becomes more electron donating. The ability of HFIP to form H-bonds with the nitrogen atom of N-chloropiperidine may destabilize the N–Cl bond, which leads to the production of more chlorinated products if electron-rich arenes are available.51,52 Furthermore, the acidity of HFIP compared to that of CH3CN may as well boost the acid catalyzed chlorination reaction.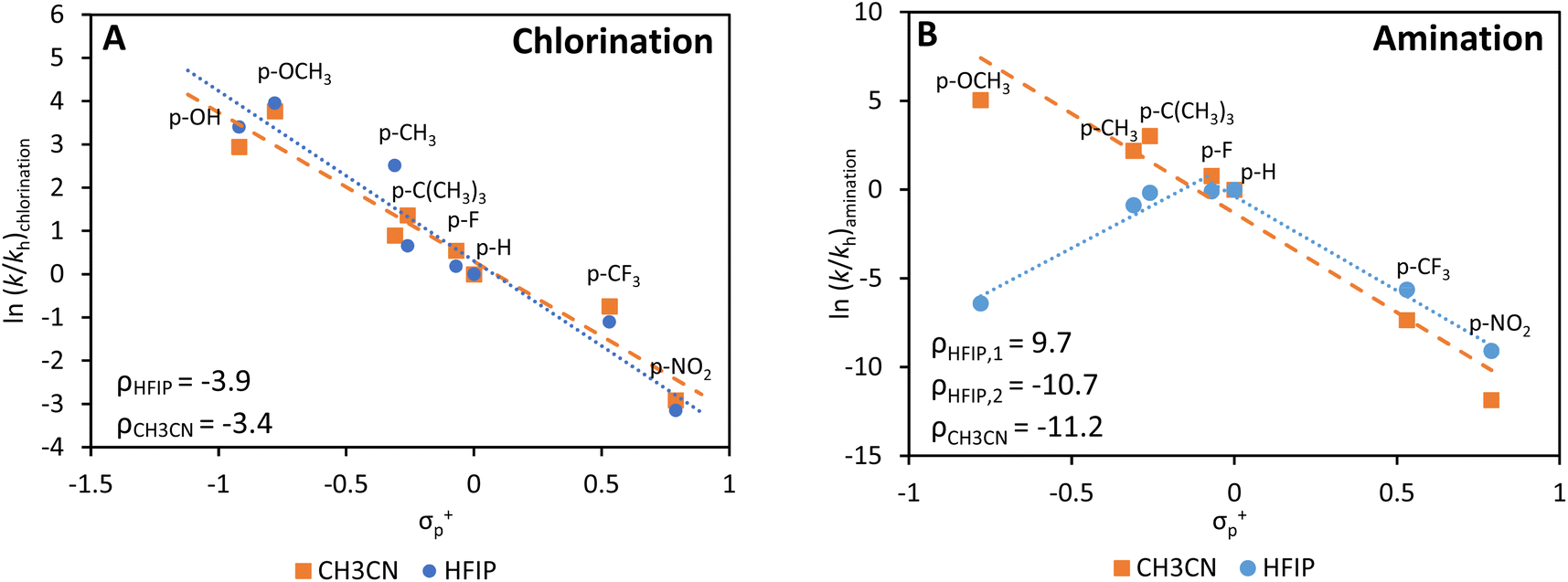 | ||
| Fig. 5 Hammett plots of the homogeneous chlorination (A) and amination (B) reaction in HFIP and CH3CN. Only the product with the Cl− or amino-substituent on the para position is considered in these plots. Reaction conditions: 0.1 mmol piperidine, 1 equiv. NCS, 18 equiv. arene, 4 equiv. HClO4, 1 mL HFIP or CH3CN, 2 mol% Ru(bipy)3Cl2, 15 min, R.T. | ||
On the other hand, a completely different trend is observed for the production of arylamines in HFIP and CH3CN (Fig. 5B). The electrophilic nature of the aminium radical is readily confirmed by the monotonously linear plot in CH3CN (ρ = −11.2); however, the Hammett plot obtained in HFIP displays a maximum, with lower rates for strongly electron-donating and electron-withdrawing groups. Surprisingly, by submitting the most electron-rich arenes to the reaction in HFIP, chlorination is preferred over amination due to the acidity and H-bond formation of this solvent (Fig. S19†).1,51,52 Overall, these experiments indicate that the preferred choice of the solvent is highly dictated by the electron density of the arene coupling partner: HFIP is suitable for electron-poor arenes, while CH3CN is better used for electron-rich arenes.1
Strategies to overcome electrophilic aromatic chlorination: role of temperature
The previous paragraph already revealed that the choice of a proper solvent for each arene molecule is important. However, the amount of chlorinated product is still considerable (Fig. 6) and therefore we considered the effect of temperature on the heterogeneous reaction yield. As the temperature of the reaction mixture rises, the content of Cl-product increases and therefore, it seems more of interest to cool the reaction (Fig. S20†). tert-Butylbenzene, toluene and anisole are employed as coupling substrates in HFIP; the zeolite-based photocatalyst is used in conditions that are slightly adapted to minimize the amount of chlorination. By changing the solvent to CH3CN for toluene and anisole, an increase in product yield was already observed, as explained in the previous paragraph (Fig. 6). By placing the samples in either an ice bath (0 °C) or an o-xylene/dry ice bath (−20 °C), the yield increases even further and the amount of chlorinated products becomes negligible (Fig. 6). Furthermore, upon cooling, the excited state of the photocatalyst is preserved longer, which could again favor the amination reaction.53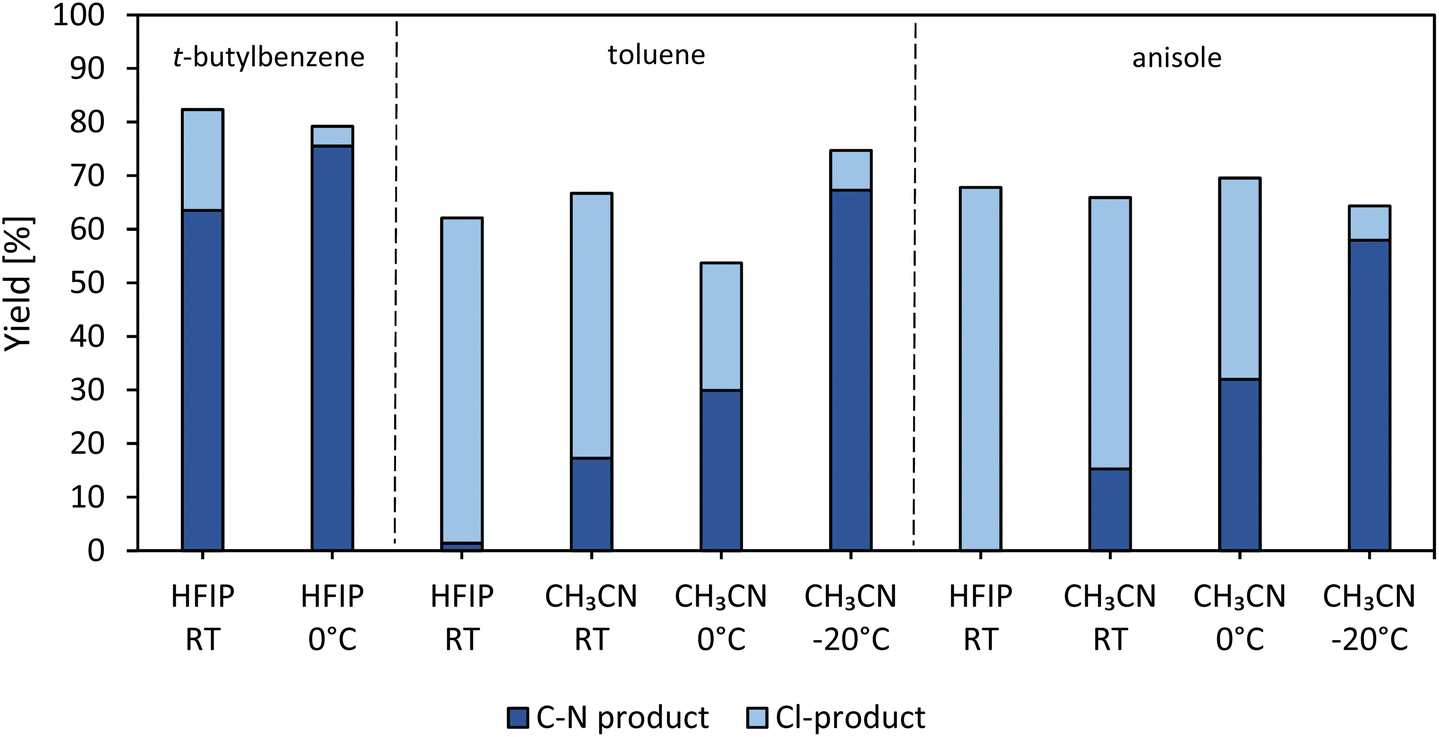 | ||
| Fig. 6 Yields of heterogeneously catalyzed reactions with varying solvent and/or temperature (RT = room temperature, 0 °C and −20 °C). Reaction conditions: 0.1 mmol piperidine, 1 equiv. NCS, 18 equiv. arene, 4 equiv. HClO4, 1 mL HFIP or CH3CN, 2 mol% Ru as CBV-100-Ru(bipy)3, blue LEDs, 3 h, R.T. Mixtures containing HFIP cannot be cooled until −20 °C. | ||
Substrate scope
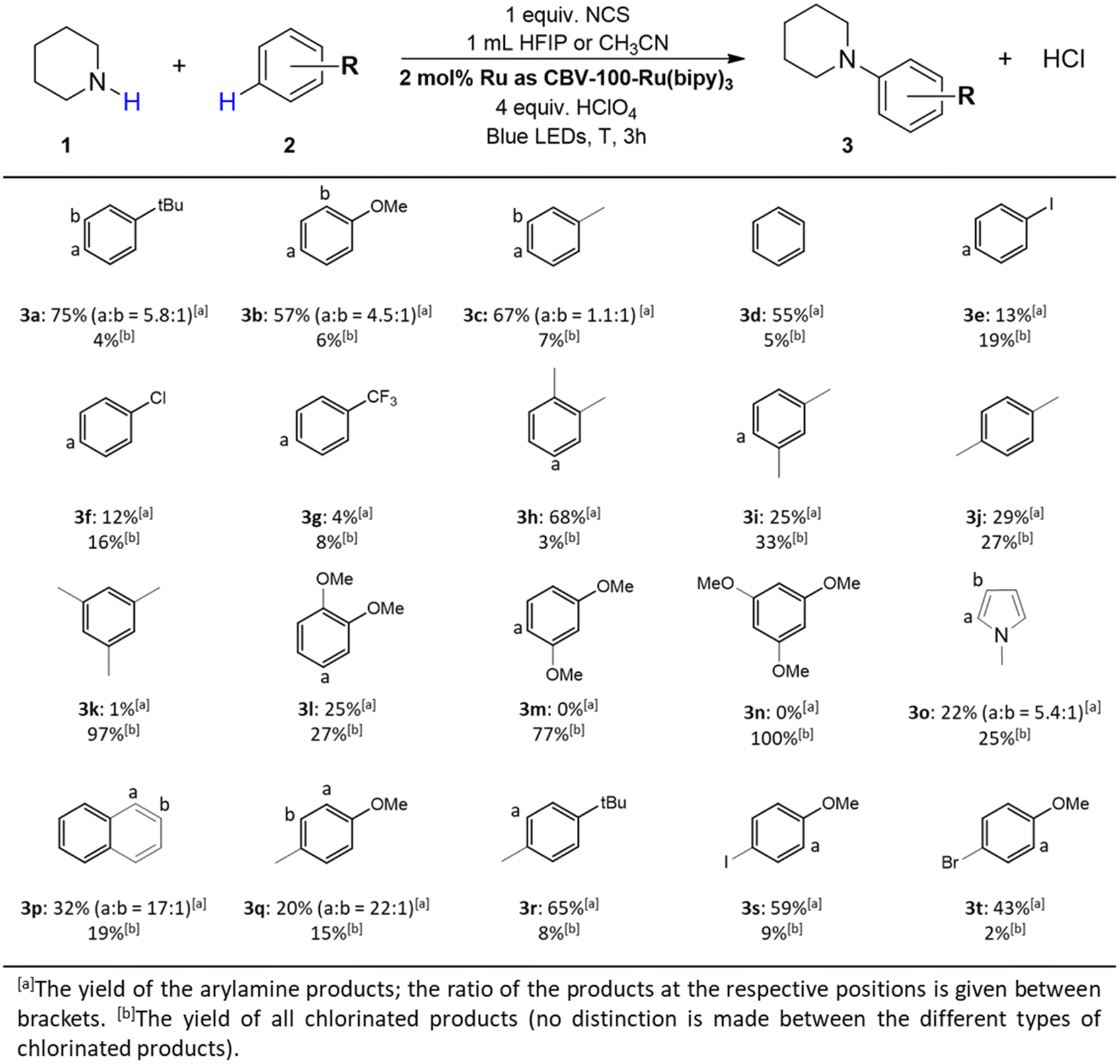 | ||
| Fig. 7 Substrate scope of different arene substrates. Reaction conditions: 1 (0.1 mmol), NCS (1 equiv.), 1 mL HFIP or CH3CN were mixed together and stirred for 30 minutes, afterwards 2 (18 equiv.), CBV-100-Ru(bipy)3 (2 mol% Ru), HClO4 (4 equiv.) were added and the suspension was exposed to blue lights (455–470 nm, 40 W), for 3 h at 0 °C, −20 °C or −40 °C. | ||
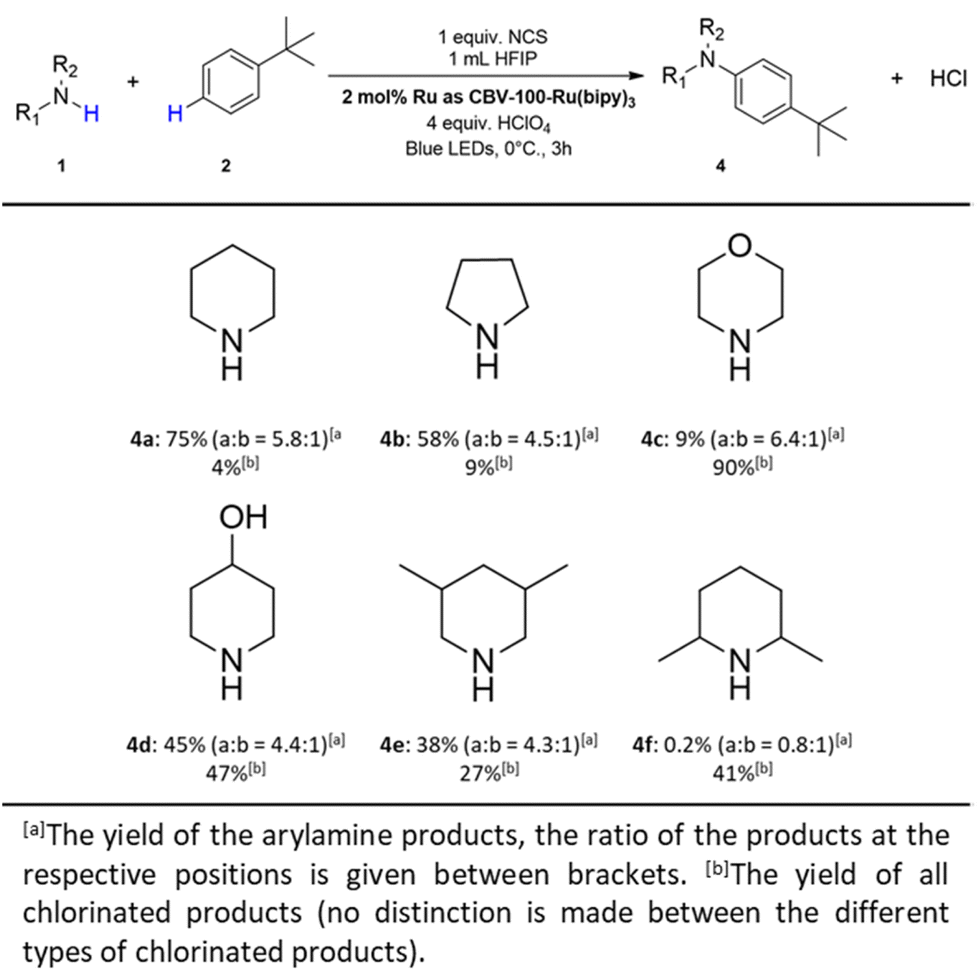 | ||
| Fig. 8 Substrate scope of different N-coupling partners. Reaction conditions: 1 (0.1 mmol), NCS (1 equiv.), 1 mL HFIP were mixed together and stirred for 30 minutes, afterwards 2 (18 equiv.), CBV-100-Ru(bipy)3 (2 mol% Ru), HClO4 (4 equiv.) were added and the suspension was placed under blue lights (455–470 nm, 40 W), for 3 h at 0 °C. | ||
Heterogeneity and recyclability of the catalyst
A recyclability test is executed to verify the heterogeneous nature of the catalyst. This test demonstrates a small decline in product yield after four recycling runs (Fig. 9A). ICP-OES analysis demonstrates that over 99% of the catalyst's activity is retained since the total amount of Ru-leaching in the reaction solvent is estimated at <1%, which is probably attributed to the fine fraction of the zeolite powder (Table S5†). Furthermore, the PXRD diffractogram at the end of the four consecutive runs shows an identical pattern as the pristine CBV-100 material; and no significant loss in long-range order is observed (Fig. S21†). Moreover, the filtrate collected after 30 min during a so-called filtration test (Fig. 9B) shows only a small increase in product yield after 3 h due to the homolytic, light induced cleavage of the N–Cl bond (Table 1, entry 4). | ||
| Fig. 9 Recyclability test for four consecutive runs. A small reduction in yield was observed after four runs (A). After the reaction, the mixture was centrifuged, the reaction solution was removed and the zeolite was washed with ultrapure water and dried. Subsequently, fresh reaction mixture was added to the zeolite and a second/third/fourth run was executed. Filtration test was performed and filtrate was collected after 30 minutes to check the heterogeneity of the reaction (B). | ||
Conclusion
In this work, we reported a direct coupling of amine compounds and aromatics under photoredox conditions. The photoactive complex Ru(bipy)32+ is synthesized within the supercages of a faujasite type zeolite (i.e. CBV-100) and its entrapment was successfully verified by PXRD, N2-physisorption, FTIR, UV-Vis and XAS. CBV-100 was selected as the ideal support, since leaching during catalyst synthesis and/or amination reaction was not observed. Furthermore, the direct C–H amination reaction was studied in detail using Hammett plots, and the proper reaction conditions for each arene substrate were selected, with acetonitrile being preferred for electron-rich arenes and HFIP for electron-poor arenes. Knowing these optimal conditions, the arene and amine substrate scope were explored to indicate the suitability of the heterogeneous catalyst in this coupling reaction. Finally, the heterogeneity of the catalyst was confirmed via recycling/filtration tests and metal analysis of the reaction mixture indicated that Ru-leaching was very limited. These results provide new insight into the development of heterogeneous zeolite-based photocatalysts, since Ru(bipy)32+ is commonly used in photocatalytic applications.Conflicts of interest
The Authors declare no conflict of interest.Acknowledgements
The research leading to these results has received funding from the KAUST-CRG Program (OSR-2018-CRG7-3741.2). K. J. and D. E. D. V. thank FWO for project funding (grant no. G0F2320N). We acknowledge the SUPERXAS beamline at Swiss Light sourc (SLS, Switzerland) for providing the beamtime and Dr. Aram Bugaev for the professional support during the measurements. V. L. is thanking Carlos Marquez for the ICP-OES measurements.References
- A. Ruffoni, F. Julia, T. D. Svejstrup, A. J. McMillan, J. J. Douglas and D. Leonori, Nat. Chem., 2019, 11(5), 426–433 CrossRef CAS PubMed.
- T. D. Svejstrup, A. Ruffoni, F. Julia, V. M. Aubert and D. A. Leonori, Angew. Chem., Int. Ed., 2017, 56(47), 14948–14952 CrossRef CAS.
- L. Van Emelen, M. Henrion, R. Lemmens and D. De Vos, Catal. Sci. Technol., 2022, 12(2), 360–389 RSC.
- P. Patnaik, A comprehensive guide to the hazardous properties of chemical substances, 2007, pp. 251–268 Search PubMed.
- S. C. Cosgrove, J. M. C. Plane and S. P. Marsden, Chem. Sci., 2018, 9(32), 6647–6652 RSC.
- S. L. Rössler, B. J. Jelier and P. F. Tripet, Angew. Chem., Int. Ed., 2019, 58(2), 526–531 CrossRef PubMed.
- F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36(2), 2382–2384 CrossRef.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43(10), 3525–3550 RSC.
- I. Goldberg, Ber. Dtsch. Chem. Ges., 1906, 39, 5–6 CrossRef.
- F. Benaskar, V. Engels, E. V. Rebrov, N. G. Patil, J. Meuldijk, P. C. Thüne, P. C. M. M. Magusin, B. Mezari, V. Hessel, L. A. Hulshof, E. J. M. Hensen, A. E. H. Wheatley and J. C. Schouten, Chem. – Eur. J., 2012, 18, 1800–1810 CrossRef CAS.
- K. Mullick, S. Biswas, C. Kim, R. Ramprasad, A. M. Angeles-Boza and S. L. Suib, Inorg. Chem., 2017, 56, 10290–10297 CrossRef CAS.
- W. Long, W. Qiu, C. Guo, C. Li, L. Song, G. Bai, G. Zhang and H. He, Molecules, 2015, 20, 21178–21192 CrossRef CAS PubMed.
- D. Berthold, A. M. Haydl, J. C. Leung, U. Scholz, Q. Xiao and Z. Zhu, Methodol. Amine Synth., 2021, 10, 377–444 Search PubMed.
- D. M. T. Chan, K. L. Monaco, R. P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39(19), 2933–2936 CrossRef CAS.
- P. Y. S. Lam, C. G. Clark and S. Saubern, Tetrahedron Lett., 1998, 39(19), 2941–2944 CrossRef CAS.
- M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119(24), 12491–12523 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47(34), 6338–6361 CrossRef CAS.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41(11), 1534–1544 CrossRef CAS.
- H. Kim and S. Chang, ACS Catal., 2016, 6(4), 2341–2351 CrossRef CAS.
- L. J. Allen, P. J. Carbrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136(15), 5607–5610 CrossRef CAS PubMed.
- J. M. Ganley, P. R. D. Murray and R. R. Knowles, ACS Catal., 2022, 10(20), 11712–11738 CrossRef PubMed.
- M. Xu, Y. Hua, X. Fu and J. Liu, Chem. – Eur. J., 2022, 28(15), e202104394 CrossRef CAS.
- Y. Y. Liu, D. Liang, L. Q. Lu and W. J. Xiao, Chem. Commun., 2019, 55(33), 4853–4856 RSC.
- R. Broach, D. Y. Jan and D. Lesch, Ullmann’s Encyclopedia of Industrial Chemistry, 2012 Search PubMed.
- A. Corma and H. Garcia, Chem. Commun., 2004, 1443–1459 RSC.
- W. DeWilde, G. Peeters and J. H. Lunsford, J. Phys. Chem., 1980, 2306–2310 CrossRef CAS.
- V. Lemmens, C. Vos, A. L. Bugaev and D. De Vos, ACS Appl. Mater. Interfaces, 2022, 14(1), 971–977 CrossRef CAS.
- N. Taira, M. Saitoh, S. Hashimoto, H. R. Moon and K. B. Yoon, Photochem. Photobiol. Sci., 2006, 5(9), 822–827 CrossRef CAS PubMed.
- Y. S. Park, E. J. Lee, Y. S. Chun, Y. D. Yoon and K. B. Yoon, J. Am. Chem. Soc., 2002, 124(24), 7123–7135 CrossRef CAS PubMed.
- F. Binder, G. Calzaferri and N. Gfeller, Sol. Energy Mater. Sol. Cells, 1995, 38(1–4), 175–186 CrossRef CAS.
- M. Sykora, K. Maruszewski, S. M. Treffert-Ziemelis and J. R. Kincaid, J. Am. Chem. Soc., 1998, 120(14), 3490–3498 CrossRef CAS.
- A. Moisette, M. Hureau, P. Col and H. Venzin, Microporous Mesoporous Mater., 2017, 254, 128–135 CrossRef.
- H. Garcia and H. D. Roth, Chem. Rev., 2002, 102(11), 3947–4007 CrossRef CAS.
- D. Martinez and J. Perez-Pariente, Zeolites and Ordered Porous Solids: Fundamentals and Applications, 2011 Search PubMed.
- S. Komaty, H. Özçelik, M. Zaarour, A. Ferre, S. Valable and S. Mintova, J. Colloid Interface Sci., 2021, 581, 919–927 CrossRef CAS PubMed.
- W. H. Quayle and J. H. Lunsford, Inorg. Chem., 1982, 21(1), 97–103 CrossRef CAS.
- M. Nakayama, J. Yano, K. Nakoaka and K. Ogura, Synth. Met., 2003, 138(3), 419–422 CrossRef CAS.
- A. Kozlov, K. Asakura and Y. Iwasawa, Microporous Mesoporous Mater., 1998, 21(4–6), 571–579 CrossRef CAS.
- M. U. Munshi, J. Martens, G. Berden and J. Oomens, J. Phys. Chem. A, 2020, 124(12), 2449–1459 CrossRef CAS PubMed.
- K. Mori, M. Kawashima, K. Kagohara and H. Yamashita, J. Phys. Chem. C, 2008, 112(49), 19449–19455 CrossRef CAS.
- C. Pastoriza, J. M. Antelo, J. Crugeiras and A. Pena-Gallego, J. Phys. Org. Chem., 2014, 27(5), 407–418 CrossRef CAS.
- J. R. L. Smith, L. C. McKeer and J. M. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 1529–1536 RSC.
- S. J. Lee, M. S. Terraza, D. J. Pippel and P. Beak, J. Am. Chem. Soc., 2003, 125, 7307–7312 CrossRef CAS PubMed.
- F. Minisci, Synthesis, 1973, 1–24 CAS.
- Z. Zhang, Y. Yu and L. S. Liebeskind, Org. Lett., 2007, 10(14), 3005–3008 CrossRef.
- J. K. Kerkovius and F. A. Menard, Synthesis, 2016, 48(11), 1622–1629 CrossRef CAS.
- W. Lutz, C. H. Rüscher and T. M. Gesing, Stud. Surf. Sci. Catal., 2004, 154, 1411–1417 CrossRef.
- R. A. Beyerlein, C. Choi-feng, J. B. Hall, B. J. Huggins and G. J. Ray, Top. Catal., 1997, 4(1–2), 27–42 CrossRef CAS.
- B. Ellis and R. Smith, Polymers: A Property Database, 2nd edn, 2000 Search PubMed.
- H. C. Brown and Y. Okamoto, J. Am. Chem. Soc., 1957, 79(8), 1913–1917 CrossRef CAS.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1(11), 0088 CrossRef CAS.
- R. J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83(2), 930–938 CrossRef CAS PubMed.
- M. K. Brennaman, J. H. Alstrum-Acevedo, C. N. Fleming, P. Jang, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2002, 124(50), 15094–15098 CrossRef CAS PubMed.
- R. J. Tang, P. Retailleau, T. Milcent and B. Crousse, ACS Omega, 2019, 4(5), 8690–8966 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, optimization of the reaction conditions, kinetic experiments, additional characterization data and a thorough discussion on sustainability and economics. See DOI: https://doi.org/10.1039/d3gc01225e |
| This journal is © The Royal Society of Chemistry 2023 |