
The design of alternative anodic reactions paired with electrochemical CO2 reduction†
Honglei
Chen‡
,
Chenglong
Ding‡
,
Caitao
Kang‡
,
Jiahong
Zeng
,
Yao
Li
,
Yanming
Li
,
Yuanli
Li
,
Changli
Li
 * and
Jingfu
He
*
* and
Jingfu
He
*
School of Materials, Shenzhen Campus of Sun Yat-sen University, No. 66, Gongchang Road, Guangming District, Shenzhen, Guangdong 518107, P.R. China. E-mail: lichli5@mail.sysu.edu.cn; hejf27@mail.sysu.edu.cn
First published on 5th July 2023
Abstract
Electrochemical reduction of CO2 to produce value-added products is a promising way to utilize CO2 in the atmosphere. The anode side of the conventional carbon dioxide reduction reaction (CO2RR) system is not fully exploited, significantly limiting the overall energy conversion efficiency and economic benefits. In this review, we summarized and analyzed CO2RR-paired anodic reactions that can enhance the overall commercial profit in four broad categories, including wastewater treatment, organic synthesis, the chloride oxidation reaction and the convergent reaction. The existing scale, key factors for performance, energy efficiency and suitability to combine with CO2RR for each type of anodic reaction are all systematically compared. Finally, a detailed description of the challenges and future directions of the coupling systems are also analyzed.
 Honglei Chen | Honglei Chen is currently a PhD student at Sun Yat-sen University, China. She received her Master's degree from Guangxi University in 2022. Currently, her research is supervised by Prof. Jingfu He. Her research interests are HMF electrocatalytic chemical oxidation and design of electrocatalysts. |
 Chenglong Ding | Chenglong Ding received his Bachelor's degree from the Hefei University of Technology in 2021. He is currently a graduate student in Materials and Chemicals at Sun Yat-sen University. His research interests focus on solar water splitting and carbon dioxide reduction. |
 Caitao Kang | Caitao Kang received her bachelor's degree from Hunan University. She is currently a graduate student in Materials and Chemical Engineering at Sun Yat-sen University. Her research interests focus on the synthesis of electrocatalysts and their applications in energy conversion and storage. |
 Changli Li | Changli Li is an Associate Professor at the School of Materials, Sun Yat-sen University, China. He received his PhD degree in Mechanical Engineering from the University of Tokyo, Japan, in 2015. After his Post-Doctoral studies at Tsinghua University and the University of British Columbia, he began his independent career as a faculty member at Sun Yat-sen University. His research activities focus on the synthesis of nanostructured materials and semiconductor heterojunctions for electrochemical and photoelectrochemical energy conversion. |
 Jingfu He | Jingfu He is currently an associate professor in the School of Materials, Sun Yat-sen University. He received his Ph.D. in 2012 from the University of Science and Technology of China. He joined the Berlinguette group at the University of British Columbia as a postdoctoral associate in 2015 and conducted research on film synthesis and CO2 electrocatalysis. His current research interests include photoelectrochemistry and electrochemistry for CO2 reduction, water splitting and in situ synchrotron radiation spectroscopy. |
1. Introduction
Developing a renewable energy system is crucial to mitigate the dilemma of sufficient energy supply and high CO2 atmospheric concentration. To overcome the discontinuity of renewable energy, finding an efficient way to restore renewable energy and facilitate its storage and transportation is highly desired.1–8 The electrochemical reduction of CO2 driven by renewable electricity can convert CO2 and water into fuels and valuable chemicals like hydrocarbons and alcohol, which is a solid foundation of a recyclable energy system.9–15The selectivity of CO2 reduction is strongly related to the bonding strength of catalysts with CO2, CO and other intermediate products.16–21 To enhance the activity and selectivity of a specific product, researchers have tried different design strategies for catalysts and electrolyzers of CO2RR.22–27 Catalyst-design strategies such as nanostructuring, alloying and metal–organic coordination have been developed and exhibit promising potential in tuning the selectivity of CO2RR.16,23,28–44 To meet the needs of the reaction rate and selectivity for industrialization, standardized gas phase flow cell design is systematically studied, including the ion permeability membrane structure, gas flow, pH environment, cation concentration, and water management.45–54 In the electrolytic cell for CO2 reduction, 90% CO selectivity or 70% ethylene selectivity can be realized at a high current density of 1000 mA cm−2.55 However, the whole cell voltage to support 200 mA cm−2 is still higher than 3 V due to the high potential of the anode reaction, cathode reaction and mass transport. The overall energy conversion efficiencies of the unique systems nowadays are <40% at a current density of >200 mA cm−2. Moreover, most of the products of CO2 reduction can also be produced from fossil raw materials with mature technology and low prices.56 Therefore, the commercial application of CO2RR is greatly hindered due to the low overall energy efficiency and inconspicuous commercial products.
To realize the commercialization of electrochemical CO2RR, the most crucial challenge is to improve the overall commercial value of products per unit of electricity. At present, the traditional anode reaction of CO2 reduction is the oxygen evolution reaction (OER) from water oxidation, with a theoretical potential of 1.23 V vs. RHE and it yields a valueless product (O2). Notably, there is another type of water oxidation reaction that undergoes a two-electron process and generates high-value H2O2 with a higher theoretical potential of 1.76 V vs. RHE.57 However, this reaction requires a very fine catalyst design, and the selectivity is currently not ideal. Thus, a high voltage is required to drive the current density of the CO2RR system to meet the commercialization demand.58–63 To this end, it is necessary to seek an alternative anodic reaction that can replace OER and can be coupled with CO2RR to achieve considerable improvement in terms of energy consumption and commercial value.
Two approaches attract significant attention, one is to reduce the energy required for the anode reaction, and the other is to enhance the value of anode products. For instance, using wastewater treatment as an alternative anode reaction can greatly reduce the potential required by the reaction. On the other hand, synthesizing organic compounds, such as alcohol oxidation, and inorganic compounds, like chlorine gas production, can provide high-value products. It is worth noting that different anodic coupling reactions for cathodic HER have been heavily studied,64,65 and they can indeed provide a lot of reference for the design of the anodic reaction of CO2RR. However, due to the unique requirements of CO2RR on the electrolyte environment and the influence of CO2RR on the electrolyte composition, the design of alternative anodic reactions for CO2RR has particular features compared with that in the HER system. Due to the deficiency of research data and the great potential of this research strategy, it is very urgent to systematically analyze the difficulty and practicability of various alternative anodic reactions that are compatible with CO2 reduction (typical anode reactions are shown in Fig. 1). For instance, whether the industrial scale of the alternative anodic reaction can support the expected industrial scale of CO2RR in the future should be pointed out. The compatibility and theoretical energy efficiency of each anode reaction with the development of catalysts should also be analyzed.
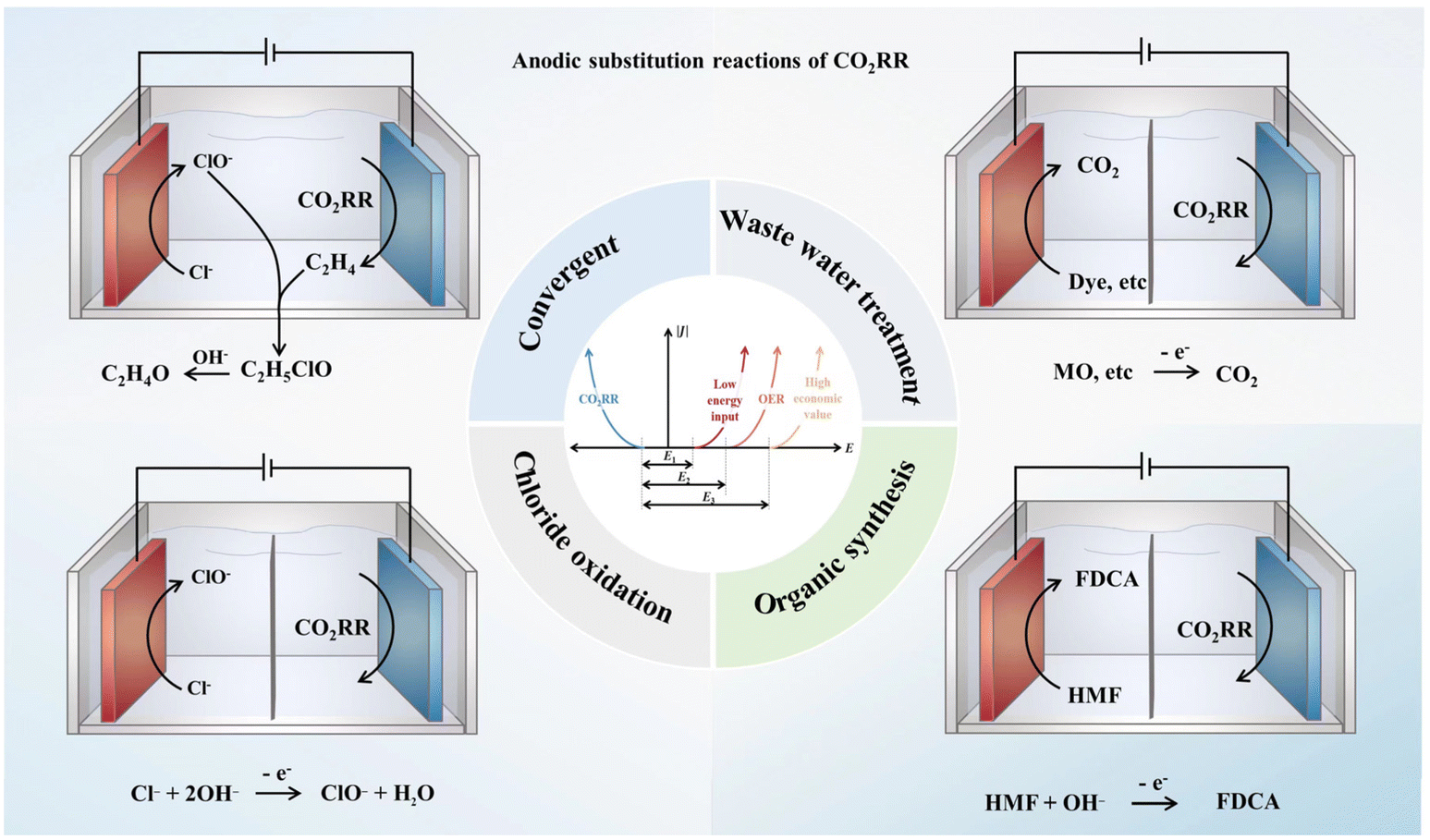 | ||
| Fig. 1 Four typical types of alternative anode reactions of the CO2RR coupled system. | ||
For this purpose, this article systematically analyzes how to construct an economically efficient electrochemical reduction system of an alternative anode reaction coupled with CO2RR. We especially sort out the potential required to oxidize different reactants, the market scale of different products, and the reaction environment compatibility of the anode reaction and CO2 reduction. In addition, we further collate and analyze the designs and possible development ideas of the convergent coupling reactions. Finally, several standardized considerations are provided for the design of paired anode reactions of electrochemical CO2 reduction for future research.
2. Reaction environment of the CO2RR
The characteristics of an electrolyte (aqueous phase/non-aqueous phase), such as pH, ionic species, buffer capacity and mass transfer, are all important factors that can affect the activity and selectivity of CO2 reduction (Fig. 2). Therefore, a summary and analysis of the design of the CO2RR are required to clarify the possible challenges in the coupling of different anode reactions.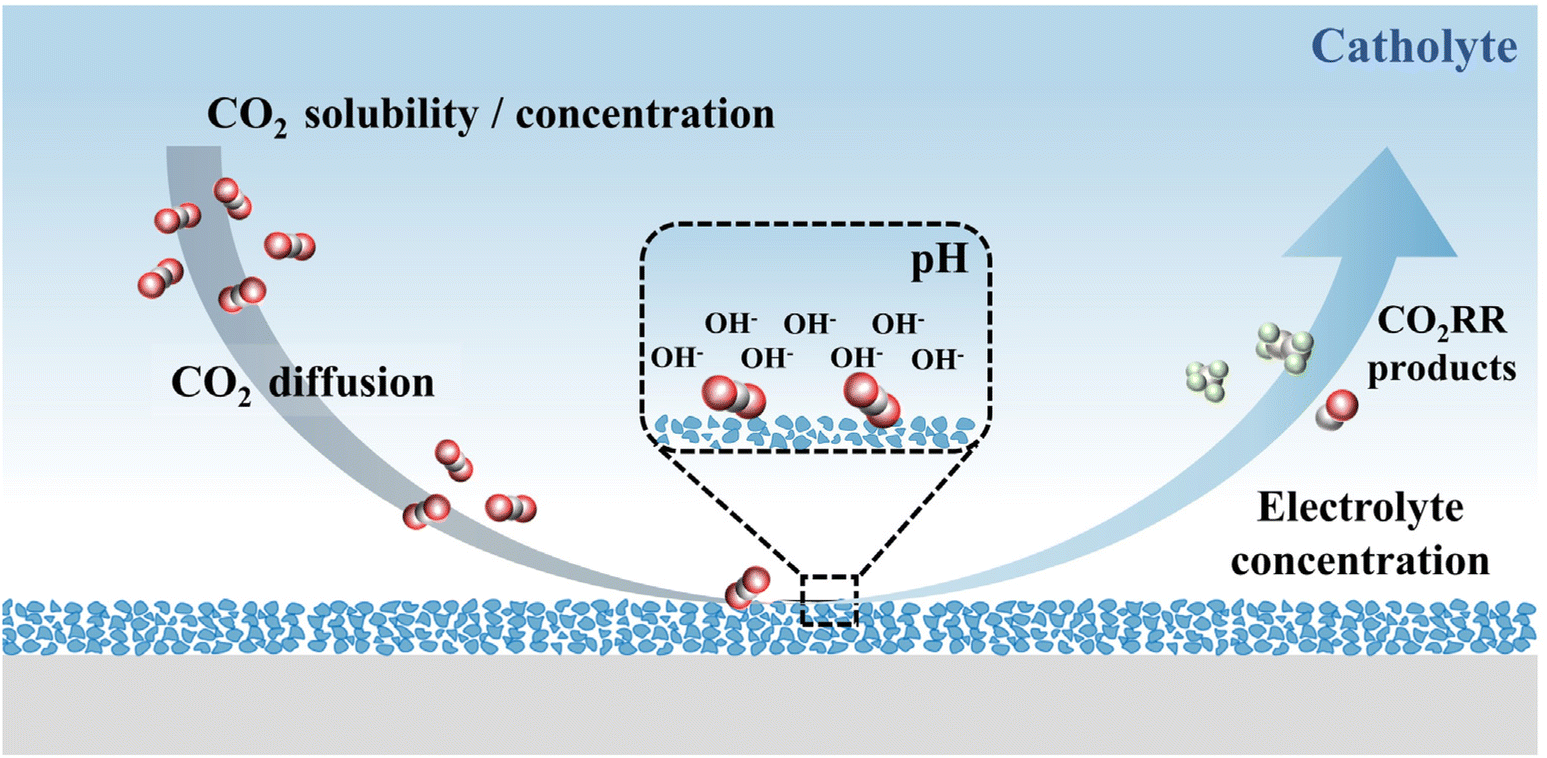 | ||
| Fig. 2 The influence factors of CO2RR performance, such as CO2 solubility, CO2 concentration, CO2 diffusion coefficient, pH values and electrolyte concentration. | ||
The concentration of CO2 can significantly change the activity and selectivity of the reaction.66 Thus, the solubility and diffusivity of CO2 should be carefully considered. Compared with CO2 dissolved in water, the diffusion coefficient is three orders of magnitude larger in the gas phase.67 As a result, the flow cell mode is used in the actual industrial design. Gas-phase CO2 is used for mass transfer and a three-phase interface could be maintained at the catalyst surface, which helps rapidly replenish the consumed protons through the electrolyte layer.53
In the design of the membrane electrolysis cell, the reactants and products of the cathode and anode will be effectively isolated and cannot be cross-mixed, but the membrane must consume additional electrical energy when carrying current through it. The alkaline environment of the CO2RR with an anion exchange membrane can take advantage of the high electrical mobility of OH− in the aqueous phase and inhibit H2 evolution,68–72 so the alkaline environment is currently considered to be the best operating environment for the CO2RR. However, the alkaline electrolyte may absorb CO2 and form bicarbonate, which can be sequentially transported across the AEM and released as CO2.73–75 Other operating conditions are urgently required to minimize energy consumption and maximize carbon efficiency, such as acidic environments. In this situation, the highest electrical mobility across the membrane can be provided by a proton exchange membrane and the CO2 dissolution in the electrolyte could be suppressed.76 Notably, the CO2RR performance in the acidic electrolyte is inferior to that in the alkaline electrolyte. For alkaline and acid electrolytes, cations of high concentration are proven beneficial for the CO2RR.77–79 The organic electrolyte may be required for some special requirements of the dissolution characteristics of reactants and products. This type of electrolyte has unique advantages in inhibiting hydrogen production and specific products such as oxalic acid formation.18
Although the current electrolyte environment of the CO2RR can be well compatible with the traditional OER, the compatibility of the electrolyte environment needs to be comprehensively considered when alternative reactions are used to couple the CO2RR.
3. Waste treatment
Water contamination resulting from industrial and domestic sewage is a serious sanitation problem (the global wastewater production is 380 billion m3 per year),80 which contains various aromatic compounds, nitrogen-containing compounds, fluorine-containing compounds, inorganic salts, dyes, etc. Due to the high toxicity, high concentration, and complexity of wastewater pollutants, it is difficult to remove them by conventional methods. Thus, removing pollutants by electrochemical oxidation is environmentally-friendly and is an effective strategy to mineralize non-biodegradable organics completely.81–83 Recently, the strategy of combining CO2 reduction and wastewater treatment in a separate electrolytic cell to reduce the overall input energy has been proposed. Notably, many organic pollutants contain reductive functional groups like hydroxyl and carbonyl, and many inorganic pollutants contain non-metals of nitrogen and sulfur. Thus, these pollutants are easy to electrochemical oxidize at a relatively low potential compared with the OER, which has significant advantages for the CO2 coupling reaction. Unlike conventional HER cathodes, the CO2RR requires a neutral or weakly alkaline solution environment and has a strong influence on the electrolysis environment due to the dissolution of CO2 and the production of liquid phase products. Furthermore, the degradation of organic pollutants has the potential to in situ provide raw materials for the CO2RR. As a result, unique designs for the overall reaction environment and catalysts are required.Kenis and coworkers proposed alternative anodic oxidation of glycerol, which has great economic benefits.84 Waste glycerol, as the byproduct of biodiesel production, usually contains about 50% by weight of glycerol and other organic matter.85 The anodic oxidation of glycerol to the product of lactic acid has a theoretical electrode potential of ca. 0.25 V vs. RHE, and the anodic oxidation of glycerol to glyceraldehyde has a theoretical electrode potential of ca. 0.4 V vs. RHE.84 In their experiment, 2 M KOH + 2 M glycerol is used as the anolyte, which successfully reduces the total reaction voltage from 1.6 V to 0.75 V and reduces the energy input by 53% compared to the combination of the OER and CO2RR (Fig. 3b).
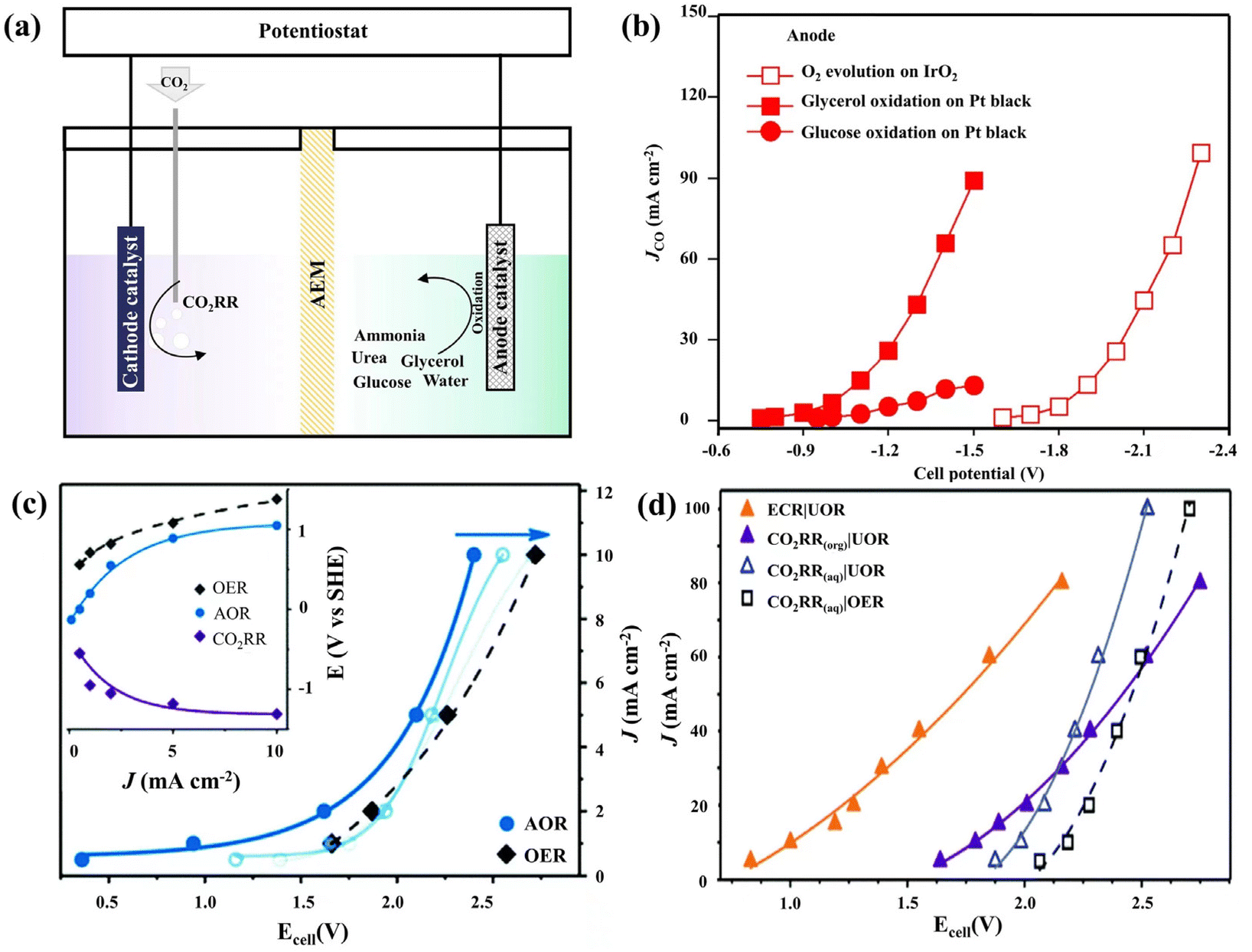 | ||
| Fig. 3 (a) The schematics and performance of the CO2RR coupled with the UOR, the AOR, the OER and glycerol/glucose oxidation. (b) The electrochemical reduction performance for CO2-to-CO on silver, coupled to O2 evolution, glycerol electro-oxidation or glucose electro-oxidation at the anode. (c) The total current density of the [CO2RR|AOR] electrolyzer as a function of the cell voltage is shown for the fresh catalyst (solid blue line) and after 1 h and 3 h of operation (fading blue lines). The shift is attributed to the catalyst poisoning effect. The reference plot for the [CO2RR|OER] electrolyzer (black dashed line) does not shift during the experiment. Inset: individual electrode potentials as a function of total current density (fresh catalyst). (d) The total current density as a function of the cell voltage for the cathodic CO2RR performed in both protic and aprotic environments coupled with the OER and UOR. The orange line presented that ECR (ECR represents electrochemical activation of R–Br followed by the reaction with CO2) decreased the cathodic potential requirement by up to an additional 0.8 V. (a and c, d) Adapted with permission from ref. 91 Copyright © 2020, the Royal Society of Chemistry. (b) Adapted with permission from ref. 84 Copyright © 2019, Springer Nature. | ||
As the main pollutant of wastewater and human body production,80,86,87 nitrogen-containing compounds have high-energy chemical bonds and are prone to produce other toxic byproducts.88,89 Therefore, replacing the OER with ammonia or urea oxidation reactions (the AOR and UOR, respectively) for electrochemical CO2 utilization results in significant energy savings.84,90 Klinkova and coworkers confirmed that using Pt/Pt-based catalysts for the AOR and Ni/Ni-based catalysts for the UOR could decrease about 0.2 V of the operating cell voltage compared with the anodic OER in a two-compartment H-cell divided by an AEM (Fig. 3). For AOR coupled with CO2RR, this voltage advantage can be maintained at a high current density after 3 hours (Fig. 3c). Notably, changing the electrolyte of the CO2RR/UOR system from an aqueous to an organic solution can not only improve the FECO from 90% to 93%, but also have about a 0.2 V improvement in cell voltage (Fig. 3d). Even more, if ECR (ECR represents electrochemical activation of R–Br followed by the reaction with CO2) is replaced by the normal CO2RR, the cathodic potential requirement would decrease up to an additional 0.8 V.91
Other kinds of substitutable anode reactants with significant market benefits are dyes.92–95 It is promising to degrade such considerable dyes (such as 4-nitrophenol, acid orange 7, crystal violet, etc.) by co-electrolysis with CO2. Using the above pollutants as reactants on the anode side can reduce the voltage by 0.2–1 V compared to the OER.96–101 In terms of traditional dyes, the degradation efficiency is usually close to 100%. For example, in a co-electrolysis system of CO2 and p-nitrophenol, the current efficiency of the formate yield is 84.63% ± 3.09% at an anode potential of 1.8 V.99 In addition, the decomposition of 4-nitrophenol (60 mL, 10 mg L−1) can be as high as 99%, when coupled with the CO2RR, whose CH3OH and C2H5OH production are 98.29 mmol L−1 and 40.95 mmol L−1 after 2 h.100 It can also be found that the contaminant removal rate can be effectively improved by cleverly designing the catalyst so that the electrode exposes more active sites. For instance, when increasing the amount of urea, the 3D gear-shaped Co3O4 changed the morphology to the filled hexagonal and nanoparticles decorated. The larger ECSA provides more active sites so that the superior performance of 4-NP degradation compared with other electrolytes and catalyst morphologies is shown (Fig. 4).100,101 For acid orange 7 degradation, the very high removal of both color (>99%) and TOC (& 92%) could be reached, although its degradation may come with a few main byproducts (oxalic and formic acids).102 Another common degradable dye is crystal violet (CV). After being coupled with the CO2RR, the dye removal efficiency of 71.5 and 60.9% can be obtained in the KHCO3 electrolyte at 2.8 V for 25 min.98 For the design of electrolytic systems for contaminant degradation, it is necessary to pay attention to the applied potential value and the sensitivity to different pH (Fig. 4h).
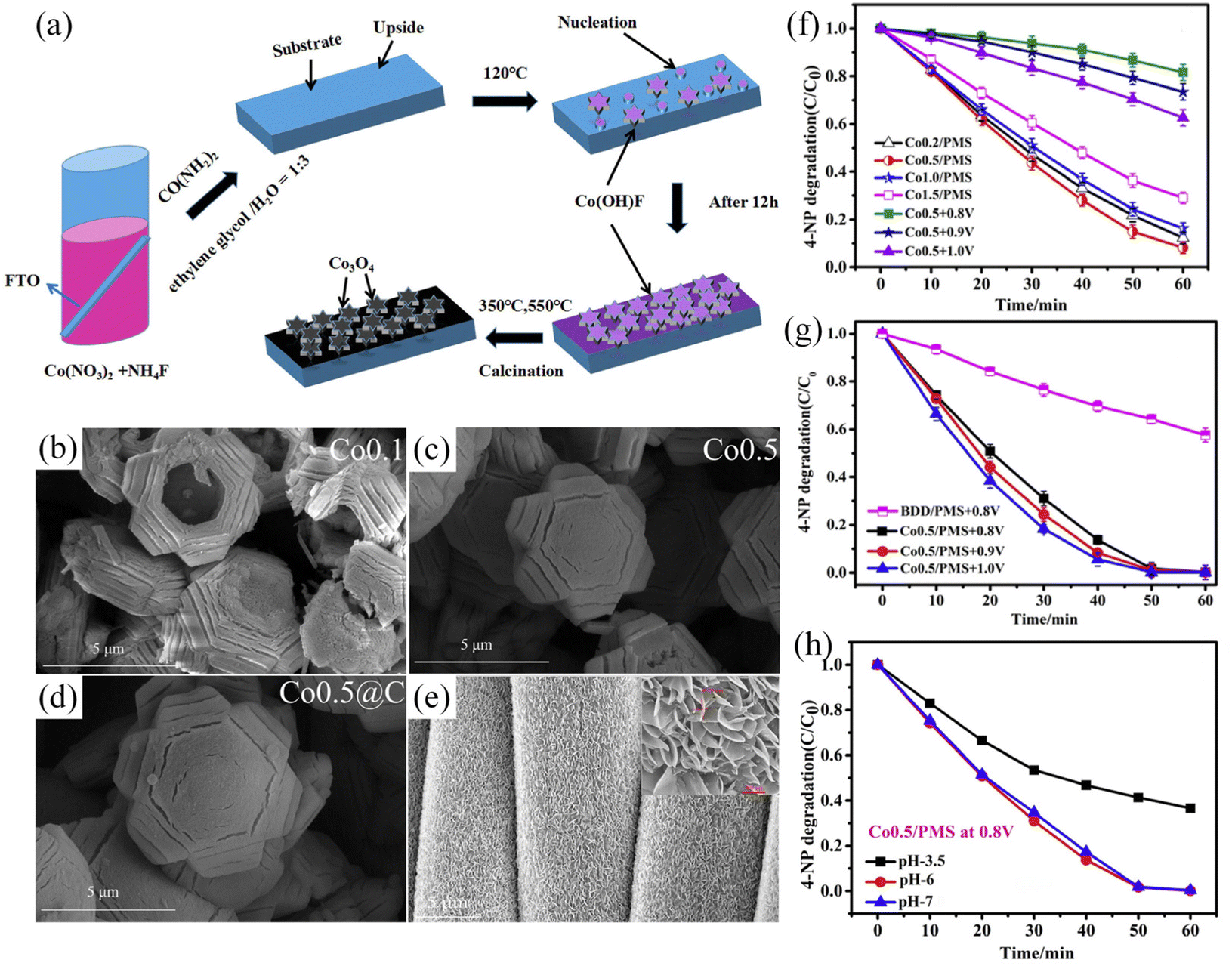 | ||
| Fig. 4 Influence of catalyst design, electrode morphology and electrolyte environment for the anodic reaction. (a) Formation mechanism of the 3D-hexagonal Co3O4 arrays. (b–d) different morphologies of cobalt oxide catalysts for the cathode. Upon increasing the amount of urea, the 3D gear-shaped Co3O4 changed the morphology to the filled hexagonal and nanoparticles decorated. The larger ECSA provides more active sites. (e) The anode SnO2/CC catalyst shows a uniform nanosheet structure, which is good for CO2RR activity. (f) 4-NP degradation during carbon dioxide reduction for the Co0.5/PMS system. (g) The Co0.5/PMS system at different potentials and the BDD/PMS system at 0.8 V vs. Ag/AgCl. (h) The removal efficiency of 4-NP at different pH values in the systems of Co0.5/PMS at 0.8 V vs. Ag/AgCl. (a and f–h) Adapted with permission from ref. 100 Copyright © 2019, Elsevier. (b–e) Adapted with permission from ref. 101 Copyright © 2020, Elsevier. | ||
Despite these results, the strategy of combining CO2 reduction and wastewater treatment still meets many challenges. A gap between combining CO2 reduction and wastewater treatment in a lab and the industrial system is the annual production and concentration of the waste. An ideal reactant should be a byproduct of a very large-scale chemical industry. Urea production is one of the key chemical industries, with an annual production of >100 million tons. It is known that 25 million tons of urea wastewater containing 0.05–0.07 wt% ammonia and 0.5–1 wt% urea will be produced yearly in China, where more than half of the world's urea is produced.103 Glycerol is a cheap byproduct of biodiesel and soap manufacturing which has an annual production of 45 billion L and >2 million tonnes, respectively.85,104,105 In general, producing 100 kg of biodiesel yields approximately 10 kg of waste glycerol, containing about 50% by weight of glycerol and other organic matters, including 1%–3% oil and grease, 15%–18% methyl ester and 12%–16% soaps.85 These two types of wastes have excellent potential for industrialized degradation, and the possible CO2 generation in the factory may also be treated simultaneously at the system's cathode. The degradation of dye-like molecules has also been extensively studied, and about 7 × 107 tons of synthetic dyes are produced worldwide.92 Inefficient textile dyeing processes may cause 15–50% of dyes to be released into generated wastewater with a concentration of 300 mg L−1.93,94 However, it is worth noting that >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different dye molecules are used in different commercial applications and not all of them have the potential to support the CO2RR.
000 different dye molecules are used in different commercial applications and not all of them have the potential to support the CO2RR.
The concentration of waste can greatly influence the selectivity of the oxidation reaction, and the exponential decay of waste observed in experiments indicates that the oxidation reaction rate is proportional to the waste concentration.100 In fact, the electrical energy required to reduce the concentration of pollutants by one magnitude is generally the same, and the OER is the main anodic reaction when the concentration of the pollutant is low. In the aforementioned studies of combining CO2 reduction and wastewater treatment, the concentration of the pollutant is always chosen to be 1 mM–1 M to support a pollutant degradation current density of >1 mA cm−2.102,106,107 The reality is that domestic and environmental sewage contains many degradable substances, usually ca. 1 mg L−1.86 Considering such a situation, waste emitted from industry with a high concentration rather than the low concentration waste in the environment should be a more suitable choice to couple with the CO2RR.
The oxidation reaction of a considerable number of pollutants can provide the same current density at a smaller overpotential than the OER (Fig. 5). However, considering the overpotential required to complete the reaction is still much higher than theoretical values, optimizing the anode side of the reaction cell is highly desired. The catalysts and electrolyte environment of wastewater oxidation are developed based on understanding the reaction mechanism. There are two reaction pathways for the electrochemical water pollution treatment: direct oxidation and indirect oxidation. In the direct oxidation reaction pathway, pollutants will be adsorbed on the electrode surface and sequentially react with the adsorbed OH radicals generated on the electrode surface.108 Indeed, Co-based catalysts that can produce abundant OH* on the catalyst surface are now commonly used in wastewater treatment. For the indirect oxidation pathway, hypochlorite or other free radicals are generated in the solution to drive the pollutants’ oxidation further. This mechanism could explain that when KCl or SO42− is used as an anolyte, the removal rate of MO, acid orange 7 and crystal violet pollutants can be improved.97,98,102 Importantly, the current research does not have a specific evaluation standard for the coupling of pollutant removal and CO2 reduction, which makes it challenging to clarify the individual contribution of different reaction conditions, pollutant concentration, catalyst electrode and other factors. Moreover, the time scale of the current related research is still relatively short, and the stability of the catalyst under long-term work also needs attention.
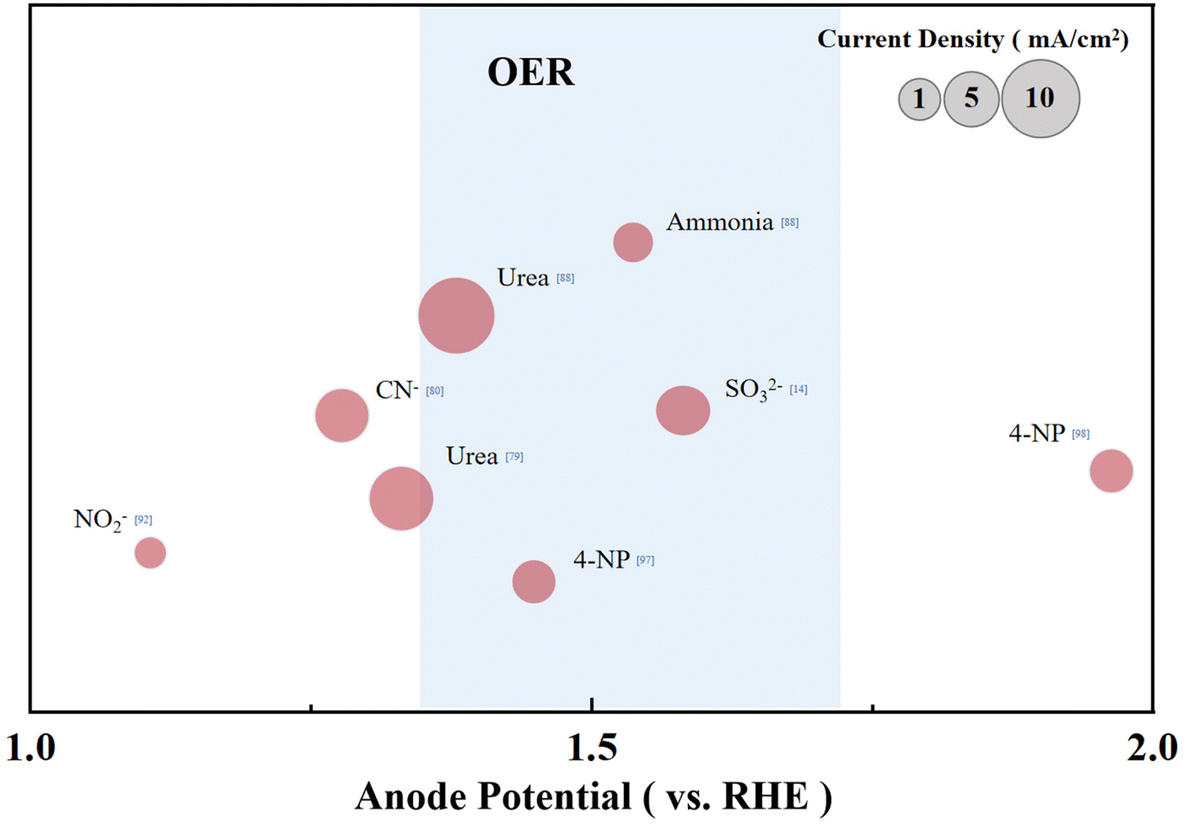 | ||
| Fig. 5 Standard organic degradation potential (the blue shade represents the potential range of the OER under the same conditions), and the anode current density is represented by a circle. | ||
4. Electrochemical organic synthesis
Electrochemical conversion of organic reactants is a pathway that has received much attention in the chemical industry. For instance, the electrolytic dimerization of acrylonitrile was once the most common industrial approach to producing adiponitrile.109 The chemical synthesis of C–H activation and the addition of functional groups are also realized by electrochemical methods.110 Compared with the degradation of pollutants, the control of the reaction conditions in organic chemical synthesis needs to be finer because it is necessary to ensure that the reaction products have considerable purity. At the same time, organic chemical synthesis also needs to avoid the complete oxidation of reactants to CO2. Hence, controlling the catalyst and catalytic environment differs from pollutant degradation.Although the current experimental oxidation potential of the alcohol oxidation still has inferior to the theoretical value, the alcohol oxidation reaction in an aqueous solution usually has very good selectivity compared to water oxidation and the FE is ca. 70–95% at the start of the reaction (Fig. 6).107,111,112 The oxidation of secondary alcohols yielded the respective ketones in exclusivity, whereas the oxidation of primary alcohols could produce aldehydes and/or carboxylic acids. Berlinguette and coworkers focused on converting the CO2RR at a copper-indium cathode and oxidative chemistry at a platinum anode with the redox mediator 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO). This study shows that the concomitant oxidation of alcohol at a platinum anode could reach >75% yield and the FE for the conversion of CO2 into CO is over 70%.107 Coupling the CO2RR with benzyl alcohol oxidation to benzaldehyde also shows excellent application potential. The formation of benzaldehyde could be realized with the simultaneous syngas generation with varying ratios of H2 and CO at the cathode, leading to the maximum energy efficiency for an electrochemical cell of 17.6% with ∼70% FE.112 Besides, electrolysis of syringaldehyde and o-phenylenediamine oxidative condensation to produce 2-(3,5-dimethoxy-4-hydroxyphenyl)benzimidazole can be coupled with the CO2RR.113 The CO2RR can exhibit a FECO of 100% after 2 h of bulk electrolysis, and the syringaldehyde oxidative condensation product was isolated and purified in 65% yield. Besides the organic product with a benzene ring, researchers have tried the electrochemical reduction of CO2 to CO paired with the electrochemical oxidation of 1,2-propanediol to lactic acid. This exploration can decrease the energy consumption by ∼35% and increase the production value of the system, resulting in a combined FE of up to 160% compared to the OER in the anode.111 Therefore, coupling the CO2RR and organic synthesis is a feasible paired electrochemical process that maximizes energy efficiency by utilizing the energy input to form valuable products on both sides of the electrochemical cell.
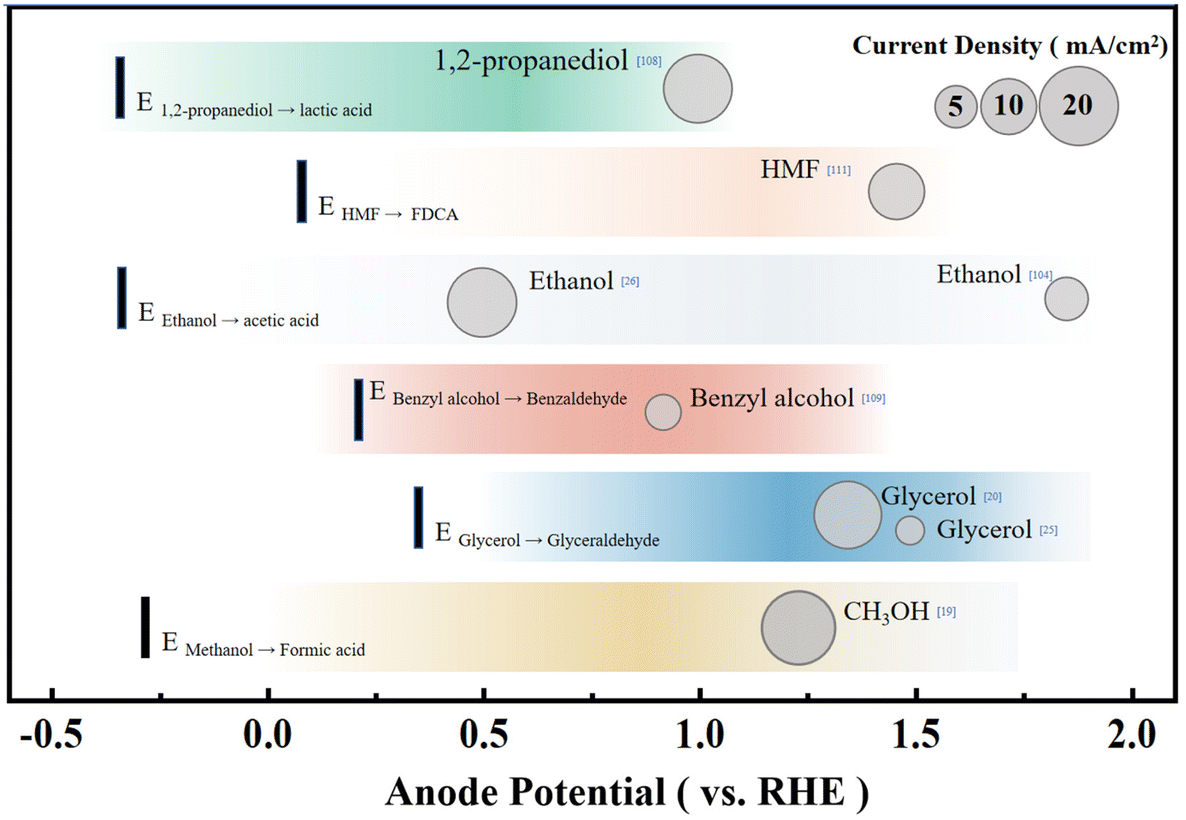 | ||
| Fig. 6 The oxidation potential and current densities for the oxidation reaction of typical organics. The theoretical electrode potential of the reaction is represented by a black vertical short line on the left, and the anode current density is represented by a circular size. | ||
From an economic point of view, the price factors of raw materials and synthetic substances also need to be carefully measured as well as the industrial demand for products, which are the factors that could affect the selection of the anode reaction. For many popular anode raw materials like alcohols, their derivatives are not significantly more expensive or even cheaper than themselves, which greatly limits their practical application value. This is supported by a systematic analysis of the commercial viability of a series of organic oxidation reactions,106 in which the calculation of the comprehensive cost, depreciation cost and operating cost (including labor, raw materials, etc.) is considered. The 5-hydroxymethylfurfural (HMF), as one of the essential intermediate biomass products, is believed to be able to provide valuable products.106,114,115 HMF can be converted into various high-value-added compounds such as 2,5-diformylfuran (DFF), dimethylfuran, levulinic acid, 2,5-bisaminomethylfuran, dimethylfuran, 2,5-furandicarboxylic acid (FDCA), etc. (Fig. 7).115 In particular, the economic value of FDCA is very high, whose market price is US$32–580 kg−1, with great industrial application potential. For instance, 2,5-furandicarboxylic acid (FDCA) is a feedstock to produce renewable polymers such as polyethylene 2,5-furandicarboxylate (PEF) as a promising alternative to polyethylene terephthalate (PET) that has an annual production of 50 million metric tons.116,117
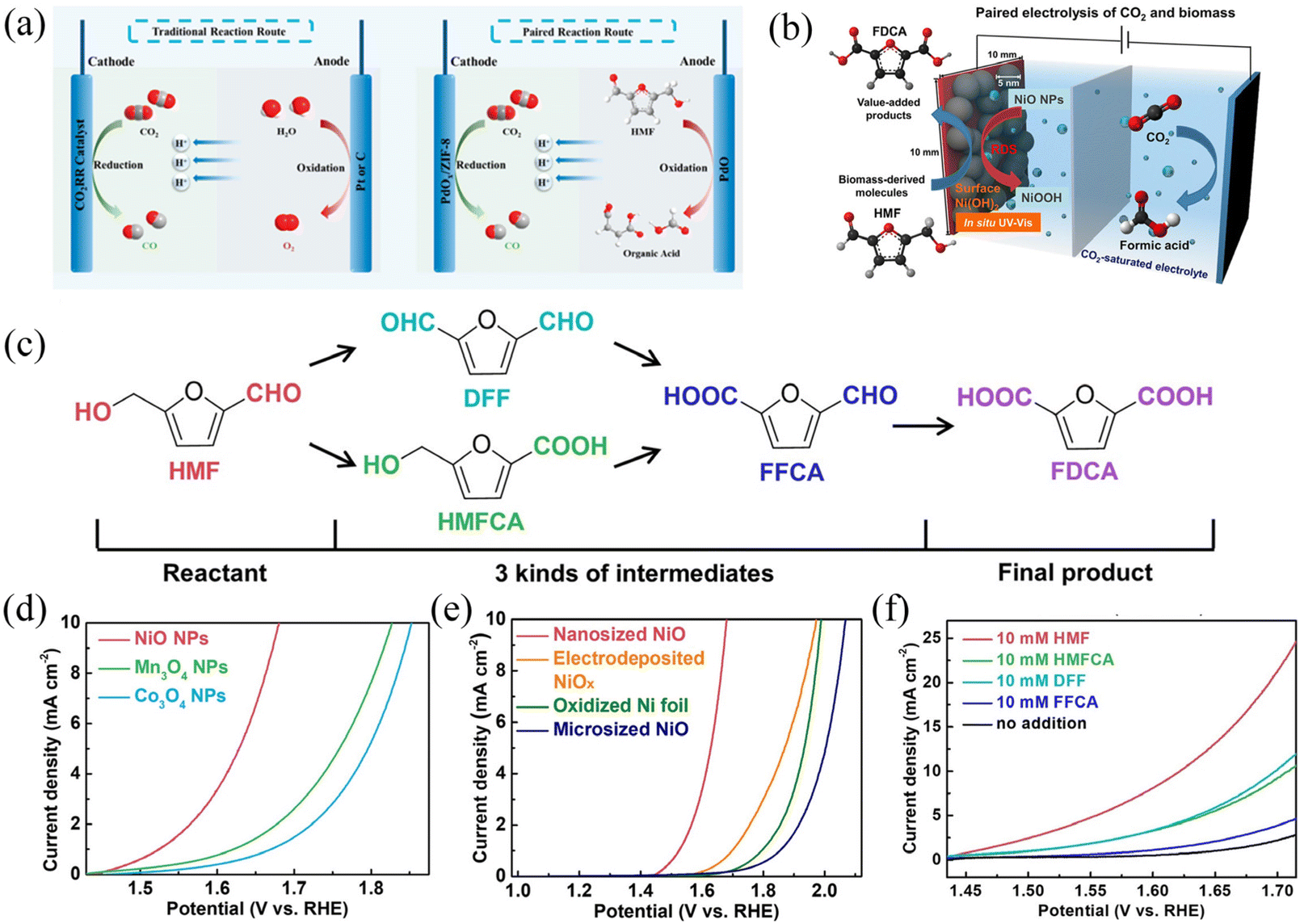 | ||
| Fig. 7 Mechanism and performance of HMF oxidation and the CO2RR coupling system. (a and b) HMF oxidation and the CO2RR system; (c) HMF oxidation path and mechanism. (d–f) Electrochemical performance of metal oxide nanoparticles (NPs) for HMF oxidation in a CO2-saturated 0.5 M potassium bicarbonate solution (pH 7.2) at a scan rate of 10 mV s−1. (d) LSV curves of three different transition metal oxide NPs (NiO, Mn3O4, and Co3O4) in an electrolyte containing 10 mM HMF. (e) LSV curves of NiO NPs in a CO2-saturated 0.5 M KHCO3 electrolyte with 10 mM HMF compared with various nickel oxides. (f) LSV curves of NiO NPs for oxidation of HMF and its intermediates (HMFCA, DFF, and FFCA). (a) Adapted with permission from ref. 115 Copyright © 2022, American Chemical Society. (b–f) Adapted with permission from ref. 114 Copyright © 2020, American Chemical Society. | ||
Besides the economic consideration, the development of electrochemical organic oxidation still meets several challenges in terms of selectivity, high potential and/or low limited current density due to the lack of understanding of the reaction mechanism. Theoretically, alcohol oxidation has 1–1.5 V more negative standard electrode potential than water oxidation.85 However, recent experiments usually exhibit an ca. 300–500 mV negative shift of anodic current of alcohol oxidation compared to that of water oxidation, which is much lower than the theoretical expectation of 1–1.5 V. Considering that many alcohols, such as benzyl alcohol, can be naturally oxidized to aldehydes in an air atmosphere, the performance of the catalyst still needs to be greatly improved. Therefore, optimization of the complex reaction conditions (such as catalysts and electrolytic environment) of organic oxidation is needed (Fig. 7). The most common precious metal catalysts for HMF oxidation include Pt, Au, Pd, and Ru, which can support the conversion of HMF to a mixture of DFF and FDCA at a relatively low overpotential of 0.1–0.2 V compared with the standard electrode potential of HMF oxidation of 0.3 V vs. RHE. However, precious metal catalysts can only support a saturation current below 10 mA cm−2, and the strong binding of carbonyl species quickly poisons them. Transition metals of Ni and Co, on the other hand, have very high overpotentials of ca. 1 V. However, the overall current is more significant than that of noble metals, and the product can be mainly FDCA with higher purity. Although the combination of HMF and the CO2RR has been tried, the overall current and energy conversion efficiency is relatively low.114 Further improvements in catalyst performance are fundamental to realizing the commercialization of HMF oxidation, which involves an in-depth mechanistic understanding of the direct-indirect oxidation pathway and the selectivity of the oxidation of alcohols and aldehydes.
In addition to the HMF oxidation for the anode reaction, the directional functionalization of some alcohols or aldehydes has also been widely studied, including valuable phosphorylated alcohols,118 the large-scale synthesis of tetrahydropyran glycine,119 synthesis of biologically active piperidine metabolites of clopidogrel,120etc. From this research, it is found that the oxidation pathway can be regulated by the branches or functional groups of organic compounds, which provide a reference for the oxidation path of HMF. Complex biomass-derived reagents such as syringaldehyde can also be applied as anodic reagents.113
To guarantee the successful combination of electrochemical conversion of organic matter and the CO2RR, it is necessary to pay great attention to the choice of the solution environment. Many electrochemical conversions require an organic solution environment, while the CO2RR generally uses an aqueous environment. This separation of organic/inorganic phases is very important, and the ion permeation membrane and the electrolytic cell configuration design will have additional requirements.
5. Chloride oxidation reaction
The Chlor-alkali industry is an industrialized system that can simultaneously produce hydrogen and other valuable chemicals, such as chlorine gas and NaOH. Chlorine gas is an important industrial raw material for pulp, pharmaceuticals, sewage treatment, etc., with global production of 88 million tons per year.121 In fact, the hydrogen produced in the Chlor-alkali industry accounts for 4% of hydrogen production globally and dominates the commercial electrochemical hydrogen evolution.In the most common design of the membrane electrode reaction cell, the input solution of the cathode is dilute salt water, which produces hydrogen and NaOH simultaneously. The input solution of the anode is saturated brine, and its chloride ions are oxidized to chlorine gas. The sodium ions could penetrate the membrane and enter the cathode side, which plays the role of charge transfer. Although the reaction of oxidizing chloride ions to chlorine gas requires a relatively positive potential (1.36 vs. SHE) compared with the OER, the high value of chlorine gas makes the entire reaction very profitable.
The combination of the CO2 reduction reaction and the Chlor-alkali reaction in a laboratory is not difficult, which only required to change the reaction environment of the cathode to a CO2-saturated environment. Anna and coworkers proved that in a coupled system, the NaHCO3 electrolyte or NaCl electrolyte saturated with CO2 could support a CO faradaic efficiency of >99% at a current density of <10 mA cm−2.91 Sun and coworkers confirmed in a flow cell with a catholyte of 2M KHCO3, the outstanding performance of 100 mA cm−2 and 99.3% faradaic efficiency can be realized at −1.10 V and 250 mA cm−2 with 98.5% efficiency at −1.19 V.122 In a mature coupling system of the Chlor-alkali reaction, Lu and coworkers designed mesoporous catalysts that can be used in both cathodes and anodes (Fig. 8a).123 Amazingly, this coupled electrolysis system showed a 97% FE of CO at a cell voltage of ca. 2.5 V and an 87% FE for ClO− with a current density of 20 mA cm−2. However, it should be noted that FECO is not completely positively correlated with the potential and KCl concentration (Fig. 8b and c). In their research, a reduction in cell voltage by 1.8 V and an increase in energy efficiency by 40% could be obtained compared to the conventional system composed of cathodic CO2 reduction and anodic O2 evolution (Fig. 8e). The CO and Cl2 produced by the system can be used to produce phosgene, an acylating agent that can support many downstream high-value chemicals. Dai and co-workers have constructed a membrane electrolysis cell with cathode CO2 reduction and anode HCl oxidation.124 This attempt allows the cathodic current density to stabilize at 15.70 mA cm−2 at −2.241 V (vs. SHE) with a FECO of 92.23%. On the anode, the detected current density reached 2.53 mA cm−2 at 1.36 V (vs. SHE), with the FE of Cl2 formation maintained at 82.5%.
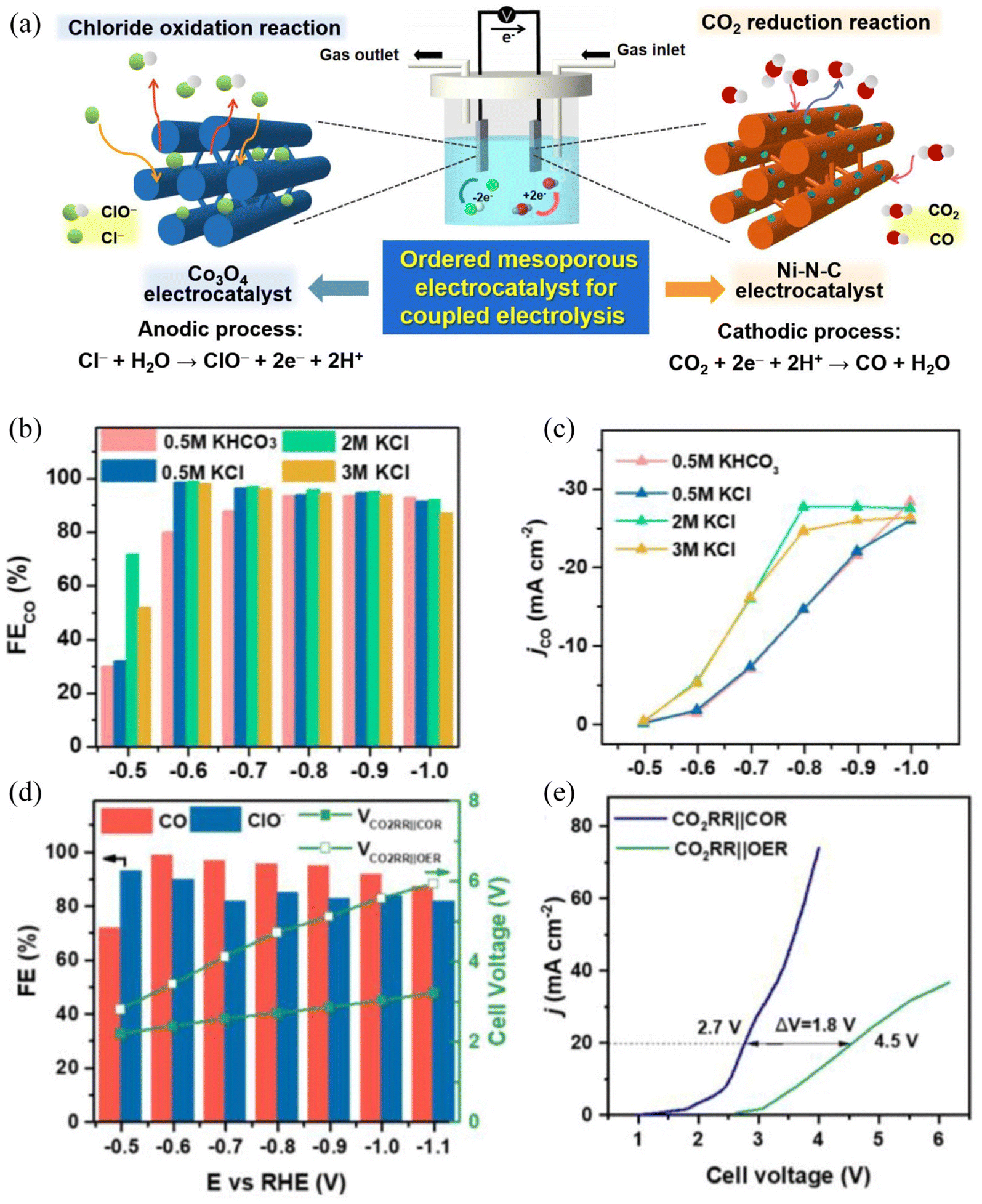 | ||
| Fig. 8 The catalyst preparation and coupled electrolysis of the CO2RR and anodic chloride oxidation reaction. (a) Hexagonally ordered mesoporous electrocatalysts for the coupled electrolysis composed of the cathodic CO2 reduction and the anodic chloride oxidation reaction; (b) the CO faradaic efficiency and (c) partial CO current density of OMP-Ni-N-C under different catholytes. (d) The performance of coupled electrolysis in a 2.0 M KCl electrolyte. Dependence of the FECO, FEClO− and cell voltage on the OMP-Ni-N-C||OMP-Co3O4 cathode in an H-type cell (CO2RR||OER: 0.5 M KHCO3||0.5 M KHCO3); (e) LSV curves of OMP-Ni-N-C||OMP-Co3O4 at a scan rate of 10 mV s−1 in an H-type cell. (CO2RR||OER: 0.5 M KHCO3||0.5 M KHCO3). Adapted with permission from ref. 123 Copyright © 2022, Elsevier. | ||
However, for the industrialization of this coupled system, energy efficiency and a long-term equilibrium state should be carefully considered. As mentioned above, the best environment for the CO2RR nowadays is neutral to alkaline electrolytes, and two different cell configurations can be schemed by the choice of the PEM or AEM (Fig. 9). For both cell configurations 1 (Fig. 9a) and 2 (Fig. 9b), the saturated anolyte of KCl is oxidized to chlorine gas, leaving the anolyte with excess K+ ions. In cell configuration 1 (Fig. 9a), these K+ ions sequentially penetrate the PEM and enter the cathode side to combine with the HCO3− formed by the CO2RR, which eventually leads to the precipitation of KHCO3 in the cathode cell. In cell configuration 2 (Fig. 9b), the HCO3− formed in the cathode cell is the carrier of charge to transport across the AEM, resulting in the precipitation of KHCO3 in the anode cell. Indeed, a precipitation rate of 3.5 mmol and 7.1 mmol were observed for the current density of 50 mA cm−2 and 100 mA cm−2 in a cell configuration similar to configuration 1.122
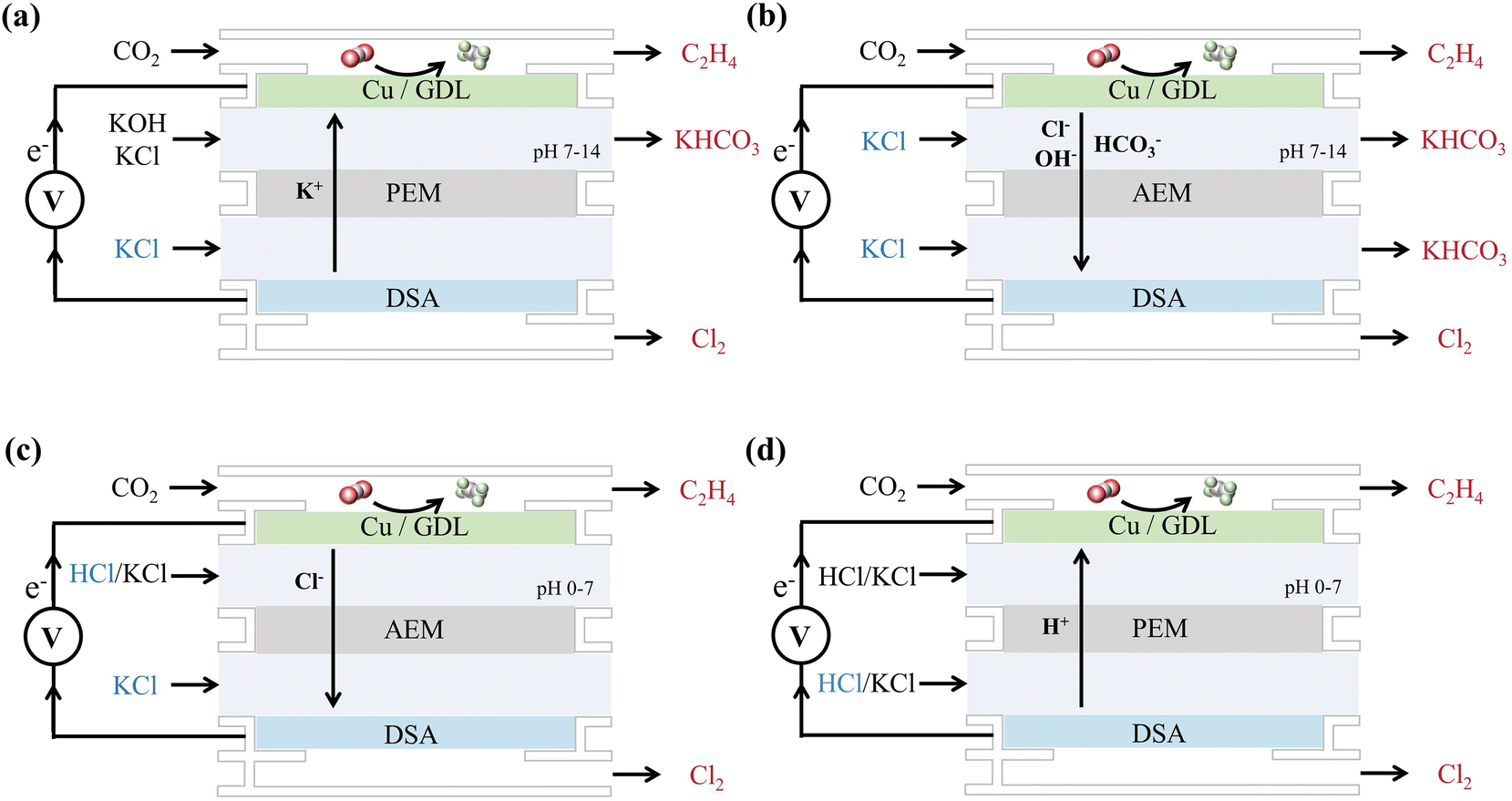 | ||
| Fig. 9 Four typical cell configurations of the CO2RR and Chlor-alkali reaction coupling system. The products are marked with a red color and the reactants that need to be supplemented are marked in blue color. (a) Cathodic alkaline environment with PEM; (b) Cathodic alkaline environment with AEM; (c) Cathodic acidic environment with AEM; (d) Cathodic acidic environment with PEM. | ||
Notably, the mobility of the cation carrier of K+ and the anion carrier of HCO3− is about 5 times lower than that of H+ and OH−, which leads to a high voltage of ca. 4 V across the membrane at a high current density of 100 mA cm−2.87,122
| 2Cl− → Cl2 + 2e− |
| CO2 + (x − 2y + 2)H2O + (x − 2y + 4)e− → CHxOy + (x − 2y + 4)OH− |
| (x − 2y + 4)OH− + (x − 2y + 4)CO2 → (x − 2y + 4)HCO3− |
To overcome the precipitation of bicarbonate salts and enhance the carbon utilization ratio, an acid electrolyte environment might be a better choice.76 In cell configuration 3 (Fig. 9c), we can keep replenishing the HCl electrolyte into the cathode cell, in which the CO2RR will consume the proton, and Cl− could be transported across the AEM and act as a reactant for Cl2 evolution. By this design, clean equilibrium can be realized with efficient carbon utilization. However, the cost of HCl might be a new consideration in this system. To further reduce the cell voltage, the system can be designed with a PEM and both the anode and the cathode operated in an acid environment, as cell configuration 4 (Fig. 9d). In this design, the HCl electrolyte is replenished in the anode cell, in which Cl− acts as the reactant for Cl2 evolution, and H+ will be transported across the PEM and consumed by the CO2RR.
| CO2 + (x − 2y + 4)H+ + (x − 2y + 4)e− → CHxOy + (2 − y)H2O |
Although possible cell configurations can be deduced from previous research and basic theory, combining the CO2 reduction reaction with the Chlor-alkali reaction still needs to be deeply investigated. Unlike the HER as a cathodic reaction, CO2RR-coupled chloride oxidation reactions accumulate HCO3− instead of OH− on the anode side when using the AEM configuration (configurations 3 (Fig. 9c)), which is the unavoidable result of the CO2RR at the cathode. Therefore, the specificity and environmental suitability of the cathodic reaction, as well as the value accounting of the overall reaction, need to be properly considered when designing the reaction configuration. Cu catalysts are very sensitive to the electrochemical environment, and Cl− might significantly influence the selectivity of the CO2RR. It is also crucial to improve the activity and stability of RuO2–TiO2 and Cu/Sn/Ag catalysts for the acidic Cl2 evolution and the CO2RR in configurations 3 and 4 (Fig. 9c–d). However, considering the large potential of the actual reaction and the pH of the electrolyte, these factors need to be considered when designing the catalyst.
6. Convergent reaction
The convergent reaction is very complex compared with the reaction types mentioned above. In this design, the intermediate products of the anode and cathode could react to form a more complex and valuable product, which is also the unique advantage of using the CO2RR for the cathode. We should keep in mind that the membrane structure in the electrolyzer is against the high energy conversion efficiency due to the large voltage bias across the membrane at a high current density. Introducing a membrane in the electrolyzer is necessary to avoid reverse/side reactions and simplify the purification process. For a convergent reaction, the system should be carefully designed to ensure that the side reactions are minimized to avoid the drawbacks of the membraneless configuration.A very interesting attempt in CO2 reduction research is the convergent reaction of halide oxidation and CO2 reduction (Fig. 10). The Cl2 and Br2 products from halide oxidation could further react with water and result in the formation of oxidants HClO or HBrO. HClO and HBrO can react with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, forming CH3–CH2ClO and CH3–CH2BrO. Classic single-chamber cells designed by Zhong's team allow the catalytic intermediate, which comes from the cathodic CO2RR, to be in situ brominated by Br2 generated from the anode to produce 2-bromoethanol.125 This attempt not only overcomes the limitations of CO2RR products (usually hydrocarbons and oxygenates), but also makes the functional chemicals (chemistry and drug synthesis) brought by this continuous reaction more economically attractive (BrCH2CH2OH of $21000 per ton, CO of $1300 per ton, HCOOH of $1300 per ton, and C2H4 of $800 per ton). The results show that the onset potential of producing 2-bromoethanol is around −0.72 V vs. RHE and the maximum FE of converting CO2 to 2-bromoethanol is 40% at −1.01 V vs. RHE with a partial current density of −19 mA cm−2. In particular, the study showed the testing of different kinds of electrolytes for the complex environmental impact of the membrane-free electrolytic cell and confirmed that the simultaneous presence of a bromine source and absence of a membrane is a prerequisite to ensure full contact between Br2 and the intermediate for subsequent reactions. The halogen ion oxidation at an anode cell can also oxidize catalysts for homogeneous catalysis. Nam and coworkers used halogen ion oxidation to further oxidize the Pd/C suspended particle catalyst (Fig. 10).126 It was observed that CO from the CO2RR and methanol in the solution can be assembled into dimethyl carbonate (DMC, $1207 million) with an FE of 57% at 12 mA cm−2 (Fig. 10c). Br− is the best halogen ion for this application considering both the requirement for oxidation ability and aqueous solubility of halogen molecules. When Br2 is in the environment where CO2 and CO coexist, the FEDMC efficiency is the highest (∼68%) (Fig. 10d) because the presence of CO2 has the effect of inhibiting Br2 loss and CO can be used as an additional reactant to react with MeO−. Therefore, when designing the convergent reaction of the single chamber electrolytic cell, the pH environment, gas composition, the valence state, fluidity, and halogen species in the electrolyte environment, especially for the anode side reaction, deserved special consideration.
C double bonds, forming CH3–CH2ClO and CH3–CH2BrO. Classic single-chamber cells designed by Zhong's team allow the catalytic intermediate, which comes from the cathodic CO2RR, to be in situ brominated by Br2 generated from the anode to produce 2-bromoethanol.125 This attempt not only overcomes the limitations of CO2RR products (usually hydrocarbons and oxygenates), but also makes the functional chemicals (chemistry and drug synthesis) brought by this continuous reaction more economically attractive (BrCH2CH2OH of $21000 per ton, CO of $1300 per ton, HCOOH of $1300 per ton, and C2H4 of $800 per ton). The results show that the onset potential of producing 2-bromoethanol is around −0.72 V vs. RHE and the maximum FE of converting CO2 to 2-bromoethanol is 40% at −1.01 V vs. RHE with a partial current density of −19 mA cm−2. In particular, the study showed the testing of different kinds of electrolytes for the complex environmental impact of the membrane-free electrolytic cell and confirmed that the simultaneous presence of a bromine source and absence of a membrane is a prerequisite to ensure full contact between Br2 and the intermediate for subsequent reactions. The halogen ion oxidation at an anode cell can also oxidize catalysts for homogeneous catalysis. Nam and coworkers used halogen ion oxidation to further oxidize the Pd/C suspended particle catalyst (Fig. 10).126 It was observed that CO from the CO2RR and methanol in the solution can be assembled into dimethyl carbonate (DMC, $1207 million) with an FE of 57% at 12 mA cm−2 (Fig. 10c). Br− is the best halogen ion for this application considering both the requirement for oxidation ability and aqueous solubility of halogen molecules. When Br2 is in the environment where CO2 and CO coexist, the FEDMC efficiency is the highest (∼68%) (Fig. 10d) because the presence of CO2 has the effect of inhibiting Br2 loss and CO can be used as an additional reactant to react with MeO−. Therefore, when designing the convergent reaction of the single chamber electrolytic cell, the pH environment, gas composition, the valence state, fluidity, and halogen species in the electrolyte environment, especially for the anode side reaction, deserved special consideration.
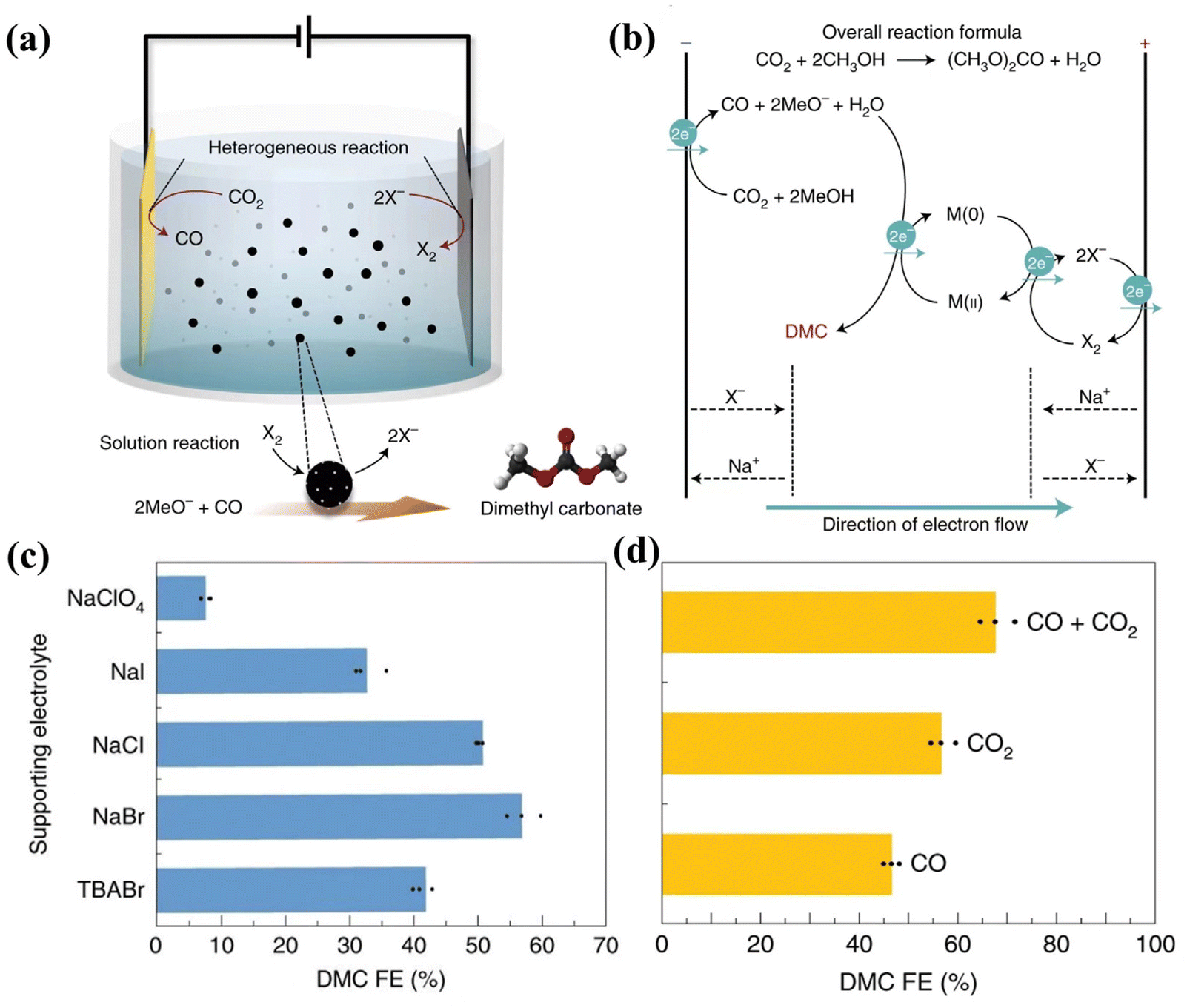 | ||
| Fig. 10 Schematic and performance of the coupled system of the convergent electrochemical DMC synthesis reaction and the CO2RR. (a) Schematic diagram of an all-in-one integrated electrolytic cell for the multistep coupling reaction to synthesize DMC. (b) Schematic of a multi-step reaction to produce DMC involving M(0)/M(II), X−/X2 redox pairs, and CO2 reduction to CO. (c) The FEDMC results obtained by using different supporting electrolytes in the H-cell electrolytic cell with Pd/C as the anode under 12 mA cm−2. NaClO4 was used to demonstrate electrolyte systems that did not produce extra oxidants, and TBABr was used to exclude cationic influences. (d) FEDMC obtained by the multi-step reaction after pressurizing methanol solution with different gases (CO2, CO2 + CO, CO). Adapted with permission from ref. 126 Copyright © 2021, Nature Publishing Group. | ||
There are similar experimental designs that have the potential to be further developed into a convergent coupling reaction. Sargent and coworkers reported that the reaction of the obtained ethylene chlorohydrin hydroxide with hydroxide could yield the desired ethylene oxide and regenerate Cl− (Fig. 11).127 First, ethylene produced by the membrane electrode of the CO2RR (anodic paired reaction is water oxidation) is collected by a gas diffusion device, and then ethylene is sent to an ethylene oxide flow cell for the synthesis reaction by finely controlling the flow rate. The reported FE of these reactions towards 2-bromoethanol and ethylene oxide are 40% and 45%, respectively, mainly limited by the ethylene selectivity of the CO2RR. Notably, the oxidation of Br− and Cl− can effectively stabilize final products and other CO2RR products, such as CO, against further oxidation. It is worth mentioning that this integrated coupling convergent reaction demanding secondary transport has quite high requirements for the selectivity and conversion of gas in the first step.
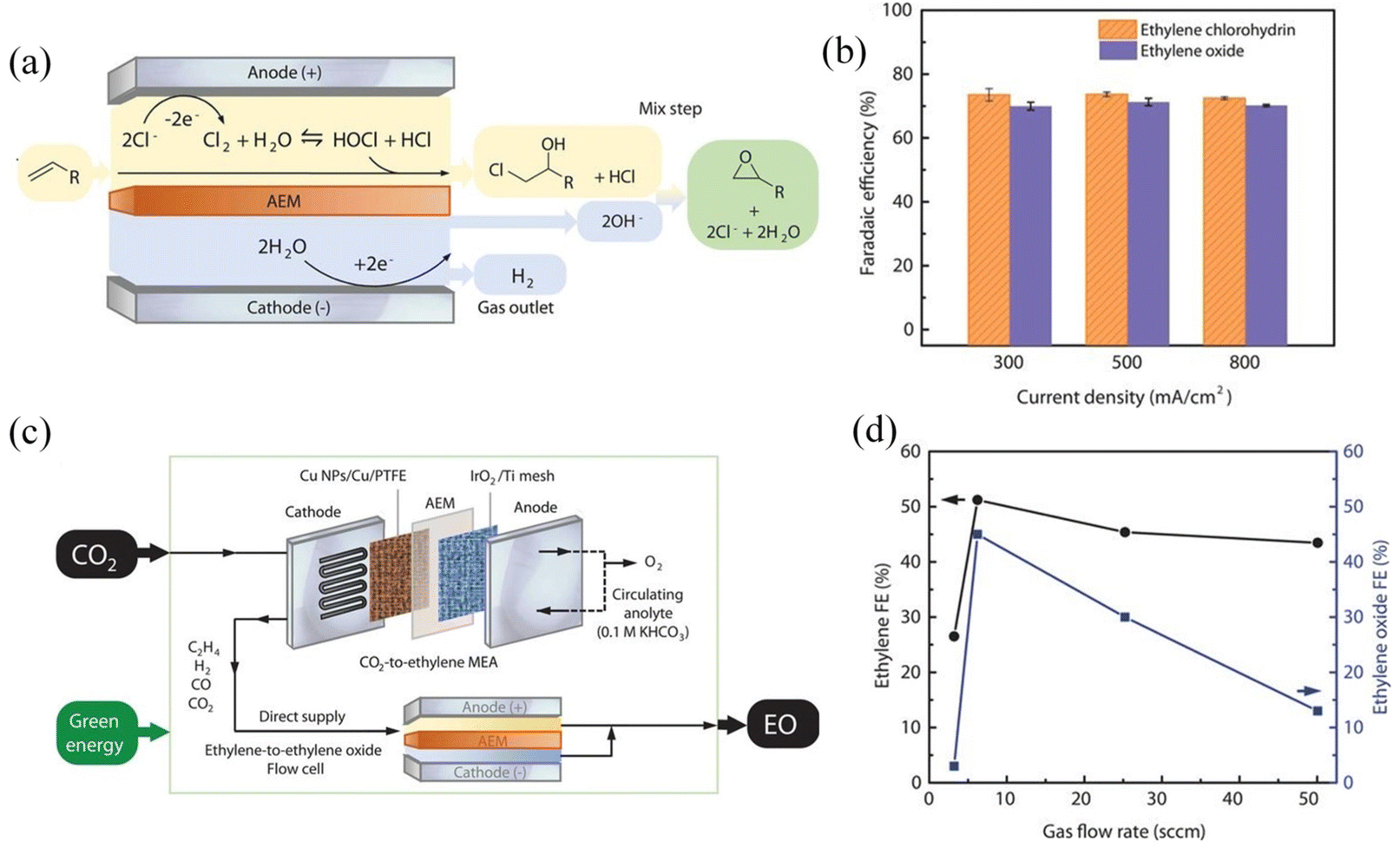 | ||
| Fig. 11 The ethylene-to-ethylene oxide and CO2RR coupling electrochemical system. (a) Schematic of the ethylene-to-ethylene oxide electrochemical system. (b) Faradaic efficiencies of ethylene oxide and ethylene chlorohydrin at different current densities. (c) Schematic of the CO2-to-ethylene oxide (EO) process in which the ethylene-to-EO cell was directly supplied with the gas output from a CO2-to-ethylene MEA. (d) Faradaic efficiencies of ethylene (in MEA) and ethylene oxide (in the flow cell) as a function of the gas flow rate. Adapted with permission from ref. 127 Copyright © 2020 Science Publishing Group. | ||
There are also other reduced or oxidized free radicals that can react with CO2 or the intermediates of the CO2RR. For instance, the reduction of organic halides,91 imines,128 and benzyl halides129 all can create free radicals that can directly bind CO2. R4NX, CH3CN or phenol can also be activated and react with CO2RR intermediates to form cyanoacetic acid or diphenyl carbonate.130,131 It is worth noting that the convergent is a very commonly used mode in electrochemical organic synthesis, and many other potential reagents can be activated to free radicals for future convergent reaction design.132–135 At present, the development of convergent reactions is still in a very early stage. The selectivity and yield of products need to be greatly improved, which requires great optimization of the catalytic environment. This kind of reaction is no longer aimed at primary products and has a stronger connection to the fine chemical industry, which makes it more applicable to the initial commercialization stage of the CO2RR.
7. Summary and perspective
Considering fuel and environmental sustainability, the effective electrochemical strategy for CO2 utilization is rapidly evolving. Although the products of carbon dioxide reduction have certain economic benefits, the overall energy efficiency and added value are hindered since the anodic OER has a high overpotential and produces no economic value product. Selecting a suitable anode reaction to improve the overall energy conversion efficiency or the commercial value of the overall product is a very promising solution. In this review, we have discussed and summarized the research status of several potential alternative anodic reactions, including wastewater treatment, organic synthesis, chlorine evolution and the convergent reaction. These strategies can all effectively increase the commercial output of CO2RR per unit of energy. Wastewater treatment as an anodic reaction can effectively reduce the input energy of the reaction without increasing the cost of reagents. Organic synthesis as the anodic reaction has the potential to simultaneously optimize the input energy and output product value. Chlorine production as the anodic reaction is a very mature industrial reaction with extremely high production value. The convergent reaction is very complex and challenging to control, but it has great potential to produce some special fine chemical products. Despite these achievements, the current overall research is still in a very rough stage, lacking guiding theories and systematic rules. For the rapid development of related fields, we believe that there are many aspects that need special attention and further optimization.(1) The market size of alternative reactions. In the early stage of CO2 reduction research and application, an anode replacement reaction can greatly reduce the energy requirements and commercial benefits of the reaction. When designing this reaction, it is necessary to consider whether the reaction can be commercially profitable and the commercial scale of the reaction. Industrial wastewater from some chemical plants can provide very cheap anode reactants that can greatly reduce the potential required for the reaction. For example, urea production, soap production, and dye textiles can all produce waste liquids or byproducts of about 10% of their annual production of 1000–1000000 tons. It would be even more advantageous if CO2 is produced simultaneously in the factory and can be treated with wastewater simultaneously. On the other hand, the concentration of the anodic reactant in domestic sewage is ca. 1 mg L−1, which is significantly lower than that in industrial wastewater (0.1–10 wt%). The low concentration of contamination suggests a much lower catalytic current. Therefore, it may not be suitable for domestic sewage treatment to couple with CO2 reduction. For the choice of organic chemical synthesis as alternative anodic reactions, the price difference between raw materials and products mainly needs to be considered because raw materials are generally not cheap. For example, alcohol oxidation, which is often studied, is unlikely to achieve real commercial value. Compared to conventional alcohol oxidation, the commercial value of biomass oxidation is much higher. In the search for alternative reactions, it should try to target the reactants with a larger scale, higher concentration, and higher commercial value so that the research results can be more specific to the future development of the field. Industrially mature electrochemical reactions are also worth considering. The scale of the Chlor-alkali reaction is 88 million tons, which is even larger than those of all the reactants mentioned above, and the commercial profit of chlorine is very high.
(2) The optimization of the anodic reaction. When we design alternative anodic reactions, there are requirements for the catalytic properties of the anode. For wastewater treatment, the pollutants should be completely oxidized, and for organic chemical synthesis, it is necessary to ensure that the reaction products have considerable purity. For all reactions, the energy conversion efficiency should be optimized as high as possible. It is worth noting that many reports use organic synthesis or wastewater treatment as an alternative reaction and the potential is indeed reduced by 0.3–0.4 V compared to the OER. However, theoretically, the potential of these reactions can often be 0.8–1 V lower than that of the OER. This suggests that the overpotentials of these reactions are even higher than that of the OER. Reactions such as HMF oxidation are still in a very early stage with the catalytic current on the order of mA and the selectivity not optimized. These deficiencies require intense research on the catalytic mechanism and catalyst design, including the respective roles of non-precious metals, noble metals, and catalytic electrolysis environments in the catalytic system.
(3) Reaction compatibility issues. A mature electrochemical synthesis system can form a stable ionic flow and a long-term stable equilibrium by simply replenishing the reactants. Unfortunately, most of the current research in the field of alternative reactions has only tested short-term reaction efficiencies and did not prove that these systems can operate in the long term. For many potential organic oxidation types of alternative reactions, utilizing an organic solvent environment may be necessary. This organic–inorganic hybrid system will complicate the anti-corrosion design of the entire system. Moreover, the ion-permeable membrane will be more prone to instability as an organic–inorganic interface. At the same time, there must be ion exchange between the anode and cathode in the electrochemical reaction, and the current ion-permeable membrane cannot prevent the penetration of small organic molecules, which may affect the long-term operational stability of the system. The current optimal alkaline reaction environment for CO2 reduction easily leads to the formation, crossover and precipitation of carbonate and bicarbonate, which directly affects the long-term ion balance of the system. In a system like the Chlor-alkali reaction, introducing the CO2 reduction reaction makes NaOH no longer a product, which greatly reduces the commercial output of the entire system. Thus, switching from HER to CO2RR in the Chlor-alkali system may even lower the total commercial profit and prevent people from making such an attempt. When designing relevant responses, we should pay close attention to compatibility issues to provide a reference for future industrialization development.
(4) Establishment of evaluation criteria. Different from the evaluation index of traditional electrolysis systems (the voltage when the current density reaches 10 mA cm−2), there is no unified evaluation standard for the anode of the carbon dioxide coupling system. For the anodic reaction of pollutant degradation, most of the work only focuses on the changing rate of pollutant concentration at a certain current, and completely ignores the overall energy conversion efficiency. Moreover, the final degradation product is not determined in many research works, which can have a great impact on the current efficiency. To better compare the catalytic system, we need to clarify the potential and current relationship of the anode reaction. For organic synthesis, it is necessary to clarify the overpotential and selectivity at standard currents such as 10 mA cm−2 and 100 mA cm−2 to determine its potential for coupling with the CO2RR.
(5) Other possible anodic reactions. In addition to the above four reaction categories, there are many anodic alternative reaction strategies that can be further developed. In addition to the treatment of pollutants in aqueous solution systems, gaseous pollutants such as H2S can also be used for anode reactants.136,137 Besides small organic molecule synthesis, anode reactions for polymeric growth can significantly reduce the potential required for reactions and provide high-value anode products.138 Apart from the valuable chemical of Cl2 from chloride oxidation, the high-value chemical H2O2 can also be produced from anodic water oxidation to improve the overall reaction product value.65 In general, all electrochemical anodic reaction systems that can reduce system operating costs or increase the product value have the potential for further analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22002191), the Natural Science Foundation of Guangdong Province (No. 2022A1515010928, 2023A1515030236 and 2022A1515012661), the Shenzhen Science and Technology Program (Grant No. JCYJ20220530150200002), and the Fundamental Research Funds for the Central Universities, Sun Yat-sen University (No. 22qntd0206).References
- Z. Z. Wu, F. Y. Gao and M. R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC
.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC
- L. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C. J. Yang and K. Caldeira, Science, 2018, 360, 9793 CrossRef PubMed
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 3506 CrossRef PubMed
- P. T. T. Phuong, D. V. N. Vo, N. P. H. Duy, H. Pearce, Z. M. Tsikriteas, E. Roake, C. Bowen and H. Khanbareh, Nano Energy, 2022, 95, 107032 CrossRef CAS
- Z. F. Liang, J. H. Wang, P. Y. Tang, W. Q. Tang, L. J. Liu, M. Shakouri, X. Wang, J. Llorca, S. L. Zhao, M. Heggen, R. E. D. Borkowski, A. Cabot, H. B. Wu and J. Arbiol, Appl. Catal., B, 2022, 314, 121451 CrossRef CAS
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Y. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- K. Ying, W. Lizhang, J. Hao, L. Fei, Z. Tingting, Z. Mengning, C. Qianwen, M. Mingxiao and X. Yuehua, J. Electroanal. Chem., 2019, 847, 113264 CrossRef
- W. Ying, G. Sergio, M. Ulaganatha Raja, L. Yanming, W. Degao, M. Alexander and M. Thomas, ACS Appl. Energy Mater., 2019, 2, 97–101 CrossRef
- J. F. He, C. H. Wu, Y. M. Li and C. L. Li, J. Mater. Chem. A, 2021, 9, 19508 RSC
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed
-
Y. Hori, Modern Aspects of Electrochemistry, Springer, 2008 Search PubMed
- X. Wei, Y. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 3148–3155 CrossRef CAS PubMed
- M. A. Bajada, S. Roy, J. Warnan, K. Abdiaziz, A. Wagner, M. M. Roessler and E. Reisner, Angew. Chem., Int. Ed., 2020, 59, 15633–15641 CrossRef CAS PubMed
- J. F. He, C. H. Wu, Y. M. Li and C. L. Li, J. Mater. Chem. A, 2021, 9, 19508–19533 RSC
- F. P. Pan and Y. Yang, Energy Environ. Sci., 2020, 13, 2275–2309 RSC
- V. K. Abdelkader-Fernández, D. M. Fernandes and C. Freire, J. CO2 Util., 2020, 42, 101350 CrossRef
- A. S. Varela, Curr. Opin. Green Sustainable Chem., 2020, 26, 100371 CrossRef
- H. Mohamed, S. Reza, N. Uzoma, R. Thibault, B. Gianluigi, K. Paul, B. Stève, C. Christophe and B. Elena, ACS Appl. Energy Mater., 2020, 3, 8725–8738 CrossRef
- B. Manuela, F. Jonathan, L. Alessandro, M. Andrea, M. Hamish, O. Werner, V. Erik and V. Francesco, Energy Technol., 2014, 2, 522–525 CrossRef
- V. N. Gopalakrishnan, J. Becerra, E. F. Pena, M. Sakar, F. Béland and T. Do, Green Chem., 2021, 23, 8332–8360 RSC
- J. F. He, Y. L. Li, A. X. Huang, Q. H. Liu and C. L. Li, Electrochem. Energy Rev., 2021, 4, 680–717 CrossRef
- L. Zeng, J. W. Chen, L. X. Zhong, W. H. Zhen, Y. T. Tay, S. Z. Li, Y. G. Wang, L. M. Huang and C. Xue, Appl. Catal., B, 2022, 307, 121154 CrossRef CAS
- X. Dong, Z. F. Xin, D. He, J. L. Zhang, Y. Q. Lan, Q. F. Zhang and Y. F. Chen, Chin. Chem. Lett., 2022, 3, 107459 Search PubMed
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed
- J. F. He, J. N. Johnson, A. X. Huang and C. P. Berlinguette, ChemSusChem, 2018, 11, 48–57 CrossRef CAS PubMed
- J. F. He, K. E. Dettelbach, A. X. Huang and C. P. Berlinguette, Angew. Chem., Int. Ed., 2017, 56, 16579–16582 CrossRef CAS PubMed
- J. F. He, K. E. Dettelbach, D. A. Salvatore, T. Li and C. P. Berlinguette, Angew. Chem., Int. Ed., 2017, 56, 6068–6072 CrossRef CAS PubMed
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed
- Y. H. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed
- M. Liu, Y. J. Pang, B. Zhang, P. D. Luna, O. Voznyy, J. X. Xu, X. L. Zheng, C. T. Dinh, F. J. Fan, C. H. Cao, G. Pelayo, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. D. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed
- R. Reske, H. Mistry, F. Behafarid, B. R. Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed
- A. Vasileff, C. C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem, 2018, 4, 1809–1831 CAS
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Y. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. T. Wang, Nat. Catal., 2018, 1, 111–119 CrossRef CAS
- Z. G. Geng, X. D. Kong, W. W. Chen, H. Y. Su, Y. Liu, F. Cai, G. X. Wang and J. Zeng, Angew. Chem., 2018, 130, 6162–6167 CrossRef
- Y. Z. Xu, C. L. Li, Y. Q. Xiao, C. H. Wu, Y. M. Li, Y. B. Li, J. G. Han, Q. H. Liu and J. F. He, ACS Appl. Mater. Interfaces, 2022, 14, 11567–11574 CrossRef CAS PubMed
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC
- D. A. Salvatore, D. M. Weekes, J. F. He, K. E. Dettelbach, T. G. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS
- D. G. Wheeler, B. W. Mowbray, A. Reyes, F. Habibzadeh, J. F. He and C. P. Berlinguette, Energy Environ. Sci., 2020, 13, 5126–5134 RSC
- C. T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. D. Arquer, A. Kiani, J. P. Edwards, P. D. Luna and O. Bushuyev, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. F. Jiao, Nat. Commun., 2014, 5, 1–6 Search PubMed
- F. P. G. D. Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani and X. X. Wang, Science, 2020, 367, 661–666 CrossRef PubMed
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. X. Huang and C. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed
- Z. C. Liu, H. Z. Yang, R. Kutz and R. R. Masel, J. Electrochem. Soc., 2018, 165(15), J3371 CrossRef CAS
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. T. Liang, E. Stavitski, H. N. Alshareef and H. T. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave and E. B. Duoss, Nat. Energy, 2022, 7, 130–143 CrossRef CAS
- C. T. Kang, Y. M. Li, Y. Z. Xu, C. L. Ding, H. L. Chen, J. H. Zeng, Y. Li, C. L. Li and J. F. He, J. Phys. Chem. Lett., 2023, 14, 2983–2989 CrossRef CAS PubMed
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS
- N. Wanninayake, Q. X. Ai, R. X. Zhou, M. A. Hoque, S. Herrell, M. I. Guzman, C. Risko and D. Y. Kim, Carbon, 2020, 157, 408–419 CrossRef CAS
- X. Hu, Z. Sun, G. Mei, X. Zhao, B. Y. Xia and B. You, Adv. Energy Mater., 2022, 12, 2201466 CrossRef CAS
- S. Ghosh and R. Basu, Nanoscale, 2018, 10, 11241–11280 RSC
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave and E. B. Duoss, Chem. Soc. Rev., 2022, 7, 130–143 CAS
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. Y. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik and Y. S. Zhu, Nat. Energy, 2020, 5, 222–230 CrossRef
- L. Wang, Y. Pan, D. Wu, X. K. Liu, L. L. Cao, W. Zhang, H. L. Chen, T. Liu, D. Liu, T. Chen, T. Ding, Y. Wang, C. L. Ding, C. T. Kang, C. L. Li, J. F. He and T. Yao, J. Mater. Chem. A, 2022, 10, 20011–20017 RSC
- L. Gouda, L. Sévery, T. Moehl, E. Mas-Marzá, P. Adams, F. Fabregat-Santiago and S. D. Tilley, Green Chem., 2021, 23, 8061–8068 RSC
- W. H. Lie, C. Deng, Y. W. Yang, C. Tsounis, K. H. Wu, H. Chandra, V. Maria, N. M. Bedford and D. W. Wang, Green Chem., 2021, 23, 4333–4337 RSC
- G.-F. Chen, Y. Luo, L.-X. Ding and H. Wang, ACS Catal., 2018, 8, 526–530 CrossRef CAS
- X. Hu, G. Mei, X. Chen, J. Liu, B. Y. Xia and B. You, Angew. Chem., Int. Ed., 2023, e202304050 Search PubMed
- M. Loloei, S. Kaliaguine and D. Rodrigue, Sep. Purif. Technol., 2022, 296, 121391 CrossRef CAS
- H. Ahmadi, M. Jamialahmadi, B. S. Soulgani, N. Dinarvand and M. S. Sharafi, Fluid Ph. Equilibria, 2020, 523, 112584 CrossRef CAS
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. Gabardo, F. P. G. D. Arquer, A. Kiani, J. Edwards, P. D. Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. J. Pang, D. Sinton and E. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- S. Lee, S. Hong and J. Lee, Catal. Today, 2017, 288, 11–17 CrossRef CAS
- B. Kim, S. Ma, H. M. Jhong and P. Kenis, Electrochim. Acta, 2015, 166, 271–276 CrossRef CAS
- A. S. Varela, M. Kroschel, T. Reier and P. Strasser, Catal. Today, 2016, 260, 8–13 CrossRef CAS
- K. J. P. Schouten, Z. S. Qin, E. Pérez Gallent and M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed
- P. Mardle, S. Cassegrain, F. Habibzadeh, Z. Q. Shi and S. Holdcroft, J. Phys. Chem. C, 2021, 125, 25446–25454 CrossRef CAS
- J. E. Huang, F. W. Li, A. Ozden, A. S. Rasouli, F. P. G. D. Arquer, S. J. Liu, S. Z. Zhang, M. C. Luo, X. Wang and Y. W. Lum, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed
- K. Xie, R. K. Miao, A. Ozden, S. J. Liu, Z. Chen, C. T. Dinh, J. E. Huang, Q. C. Xu, C. M. Gabardo and G. Lee, Nat. Commun., 2022, 13, 1–12 Search PubMed
- J. N. Huang, F. W. Li, A. Ozden, A. S. Rasouli, F. P. G. DeArquer, S. J. Liu, S. Z. Zhang, M. C. Luo, X. Wang and E. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed
- M. R. Singh, Y. Kwon, Y. W. Lum, J. W. Ager and A. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed
- V. J. Ovalle, Y. S. Hsu, N. Agrawal, M. J. Janik and M. Waegele, Nat. Catal., 2022, 5, 624–632 CrossRef CAS
- M. Qadir, P. Drechsel, B. J. Cisneros, Y. Kim, A. Pramanik, P. Mehta and O. Olaniyan, Nat. Resour. Forum, 2020, 44, 40–51 CrossRef
- D. Kanakaraju, B. D. Glass and M. Oelgemoller, J. Environ. Manage., 2018, 219, 189–207 CrossRef CAS PubMed
- S. Periyasamy, P. Subramanian, E. Levi, D. Aurbach, A. Gedanken and A. Schechter, ACS Appl. Mater. Interfaces, 2016, 8, 12176–12185 CrossRef CAS PubMed
- S. C. Cheng, M. Gattrell, T. Guena and B. MacDougall, Electrochim. Acta, 2002, 47, 3245–3256 CrossRef CAS
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS
- S. S. Yazdani and R. Gonzalez, Curr. Opin. Biotechnol., 2007, 18, 213–219 CrossRef CAS PubMed
- E. Urbańczyk, M. Sowa and W. Simka, J. Appl. Electrochem., 2016, 46, 1011–1029 CrossRef
- F. J. Quan, G. M. Zhan, H. Shang, Y. H. Huang, F. L. Jia, L. Z. Zhang and Z. H. Ai, Green Chem., 2019, 21, 3256–3262 RSC
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS
- A. Afif, N. Radenahmad, Q. Cheok, S. Shams, J. H. Kim and A. K. Azad, Renewable Sustainable Energy Rev., 2016, 60, 822–835 CrossRef CAS
- R. Hannah, P. Francois and T. H. Boyer, Environ. Sci.: Water Res. Technol., 2019, 5, 1993–2003 RSC
- X. V. Medvedeva, J. J. Medvedev, S. W. Tatarchuk, R. M. Choueiri and A. Klinkova, Green Chem., 2020, 22, 4456–4462 RSC
- V. Chandanshive, S. Kadam, N. Rane, B. H. Jeon, J. Jadhav and S. Govindwar, Chemosphere, 2020, 252, 126513 CrossRef CAS PubMed
- P. V. Nidheesh, M. H. Zhou and M. A. Oturan, Chemosphere, 2018, 197, 210–227 CrossRef CAS PubMed
-
K. Singha, P. Pandit, S. Maity and S. R. Sharma, in Green Chemistry for Sustainable Textiles, eds. N. Ibrahim and C. M. Hussain, Woodhead Publishing, 2021, ch. 11, pp. 153–164 Search PubMed
- L. Chiayu, A. Balamurugan, L. Yihsuan and H. Kuochuan, Talanta, 2010, 82, 1905–1911 CrossRef PubMed
- Q. N. Wang, X. Q. Wang, C. Wu, Y. Y. Cheng, Q. Y. Sun and H. B. Yu, J. CO2 Util., 2018, 26, 425–433 CrossRef CAS
- Q. N. Wang, C. Q. Zhu, C. Wu and H. B. Yu, Electrochim. Acta, 2019, 319, 138–147 CrossRef CAS
- V. S. K. Yadav and M. K. Purkait, Energy Fuels, 2016, 30, 3340–3346 CrossRef CAS
- Q. N. Wang, W. L. Wang, C. Q. Zhu, C. Wu and H. B. Yu, J. CO2 Util., 2021, 47, 101497 CrossRef CAS
- J. P. Zou, Y. Chen, S. S. Liu, Q. J. Xing, W. H. Dong, X. B. Luo, W. L. Dai, X. Xiao, J. M. Luo and J. Crittenden, Water Res., 2019, 150, 330–339 CrossRef CAS PubMed
- M. Zhu, L. S. Zhang, S. S. Liu, D. K. Wang, Y. C. Qin, Y. Chen, W. L. Dai, Y. H. Wang, Q. J. Xing and J. P. Zou, Chin. Chem. Lett., 2020, 31, 1961–1965 CrossRef CAS
- S. Sabatino, A. Galia, G. Saracco and O. Scialdone, ChemElectroChem, 2017, 4, 150–159 CrossRef CAS
- H. L. Hu, C. L. Deng, X. J. Wang, Z. G. Chen, Z. Zhong and R. X. Wang, Chemosphere, 2020, 258, 127228 CrossRef CAS PubMed
- M. Simoes, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef CAS PubMed
- IEA, Global biofuel production in 2019 and forecast to 2025, 2020 Search PubMed
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H. S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed
- T. F. Li, Y. Cao, J. F. He and C. Berlinguette, ACS Cent. Sci., 2017, 3, 778–783 CrossRef CAS PubMed
- J. Qiao and Y. Z. Xiong, J. Water Process. Eng., 2021, 44, 102308 CrossRef
- M. M. Baizer, J. Electrochem. Soc., 1964, 111, 215 CrossRef CAS
- M. Yan, K. Yu and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed
- E. Pérez-Gallent, S. Turk, R. Latsuzbaia, R. Bhardwaj, A. Anastasopol, F. Sastre-Calabuig, A. C. Garcia, E. Giling and E. Goetheer, Ind. Eng. Chem. Res., 2019, 58, 6195–6202 CrossRef
- Y. Wang, S. Gonell, U. R. Mathiyazhagan, Y. M. Liu, D. G. Wang, J. M. Alexander and T. J. Thomas, ACS Appl. Energy Mater., 2018, 2, 97–101 CrossRef
- M. Llorente, B. Nguyen, C. Kubiak and K. Moeller, J. Am. Chem. Soc., 2016, 138, 15110–15113 CrossRef CAS PubMed
- S. Choi, M. Balamurugan, K. Lee, K. Cho, S. Park, H. Seo and K. Nam, J. Phys. Chem. Lett., 2020, 11, 2941–2948 CrossRef CAS PubMed
- J. H. Bi, Q. G. Zhu, W. W. Guo, P. S. Li, S. Q. Jia, J. Y. Liu, J. Ma, J. L. Zhang, Z. M. Liu and B. X. Han, ACS Sustainable Chem. Eng., 2022, 10, 8043–8050 CrossRef CAS
- S. Rajendran, R. Raghunathan, I. Hevus, K. Retheesh, A. Ugrinov, M. P. Sibi, D. C. Webster and J. Sivaguru, Angew. Chem., Int. Ed., 2015, 54, 1159–1163 CrossRef CAS PubMed
- L. Dissanayake and L. Jayakody, Front. Bioeng. Biotechnol., 2021, 9, 656465 CrossRef PubMed
- M. Ociepa, K. W. Knouse, D. He, J. Vantourout, D. Flood, N. Padial, J. Chen, B. Sanchez, E. Sturgell, B. Zheng, S. J. Qiu, M. Schmidt, M. Eastgate and P. S. Baran, Org. Lett., 2021, 23, 9337–9342 CrossRef CAS PubMed
- A. Mathur, B. Wang, D. Smith, J. Li, J. Pawluczyk, J. H. Sun, M. K. Wong, S. Krishnananthan, D. R. Wu, D. Sun, P. Li, S. Yip, B. C. Chen, P. S. Baran, Q. Chen, O. D. Lopez, Z. Yong, J. A. Bender, V. N. Nguyen, J. L. Romine, D. R. S. Laurent, G. Wang, J. F. Kadow, N. A. Meanwell, M. Belema and R. Zhao, J. Org. Chem., 2017, 82, 10376–10387 CrossRef CAS PubMed
- S. A. Shaw, B. Balasubramanian, S. Bonacorsi, J. C. Cortes, K. Cao, B. C. Chen, J. Dai, C. Decicco, A. Goswami, Z. Guo, R. Hanson, W. G. Humphreys, P. Y. Lam, W. Li, A. Mathur, B. D. Maxwell, Q. Michaudel, L. Peng, A. Pudzianowski, F. Qiu, S. Su, D. Sun, A. A. Tymiak, B. P. Vokits, B. Wang, R. Wexler, D. R. Wu, Y. Zhang, R. Zhao and P. S. Baran, J. Org. Chem., 2015, 80, 7019–7032 CrossRef CAS PubMed
- Y. H. Wang, Y. Y. Liu, D. Wiley, S. L. Zhao and Z. Y. Tang, J. Mater. Chem. A, 2021, 9, 18974–18993 RSC
- J. H. Guo and W. Y. Sun, Appl. Catal., B, 2020, 275, 119154 CrossRef CAS
- R. Ge, L. Y. Dong, X. Hu, Y. T. Wu, L. He, G. P. Hao and A. H. Lu, Chem. Eng. J., 2022, 438, 135500 CrossRef CAS
- C. J. Li, J. Shi, J. X. Liu, Y. J. Duan, Y. X. Hua, S. Wu, J. Z. Zhang, X. G. Zhang, B. Yang and Y. N. Dai, Electrochim. Acta, 2021, 389, 138728 CrossRef CAS
- S. D. Zhong, Z. Cao, X. L. Yang, S. M. Kozlov, K. W. Huang, V. Tung, L. G. Cavallo, L. J. Li and Y. Han, ACS Energy Lett., 2019, 4, 600–605 CrossRef CAS
- K. M. Lee, J. H. Jang, M. Balamurugan, J. E. Kim, Y. I. Jo and K. T. Nam, Nat. Energy, 2021, 6, 733–741 CrossRef CAS
- W. Leow, Y. W. Lum, A. Ozden, Y. H. Wang, D. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. W. Li, D. Sinton and E. Sargent, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed
- C. H. Li, X. Z. Song, L. M. Tao, Q. G. Li, J. Q. Xie, M. N. Peng, L. Pan, C. Jiang, Z. Y. Peng and M. F. Xu, Tetrahedron, 2014, 70, 1855–1860 CrossRef CAS
- H. Senboku, K. Nagakura, T. Fukuhara and S. Hara, Tetrahedron, 2015, 71, 3850–3856 CrossRef CAS
- B. Batanero, F. Barba, C. M. Sánchez-Sánchez and A. Aldaz, J. Org. Chem., 2004, 69, 2423–2426 CrossRef CAS PubMed
- R. Kanega, T. Hayashi and I. Yamanaka, ACS Catal., 2013, 3, 389–392 CrossRef CAS
- W. Z. Zhang, N. M. Hong, L. Song and N. K. Fu, Chem. Rec., 2021, 21, 2574–2584 CrossRef CAS PubMed
- Y. Y. Ma, X. T. Yao, L. Zhang, P. F. Ni, R. H. Cheng and J. X. Ye, Angew. Chem., Int. Ed., 2019, 58, 16548–16552 CrossRef CAS PubMed
- F. Eisner, M. Azzouzi, Z. P. Fei, X. Y. Hou, T. Anthopoulos, J. Dennis, M. Heeney and J. Nelson, J. Am. Chem. Soc., 2019, 141, 6362–6374 CrossRef CAS PubMed
- D. Lehnherr, Y. H. Lam, M. Nicastri, J. C. Liu, J. Newman, E. Regalado, D. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 468–478 CrossRef CAS PubMed
- B. Zhang, J. Bai, Y. Zhang, C. Zhou, P. Wang, L. Zha, J. Li, A. Simchi and B. Zhou, Environ. Sci. Technol., 2021, 55, 14854–14862 CrossRef CAS PubMed
- W. Ma, H. Wang, W. Yu, X. Wang, Z. Xu, X. Zong and C. Li, Angew. Chem., Int. Ed., 2018, 57, 3473–3477 CrossRef CAS PubMed
- D.-D. Ma, S.-G. Han, C. Cao, W. Wei, X. Li, B. Chen, X.-T. Wu and Q.-L. Zhu, Energy Environ. Sci., 2021, 14, 1544–1552 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01215h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |