
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkylation of amines with allylic alcohols and deep eutectic solvents as metal-free and green promoters†
Stephany
Zárate-Roldán
 ab,
M. Concepción
Gimeno
*b and
Raquel P.
Herrera
*a
ab,
M. Concepción
Gimeno
*b and
Raquel P.
Herrera
*a
aLaboratorio de Organocatálisis Asimétrica, Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, C/Pedro Cerbuna 12, E-50009 Zaragoza, Spain. E-mail: raquelph@unizar.es
bDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, C/Pedro Cerbuna 12, E-50009 Zaragoza, Spain. E-mail: gimeno@unizar.es
First published on 1st June 2023
Abstract
A novel approach for the allylic alkylation of anilines, hydrazides, indole derivatives and additional interesting nucleophiles is described, which involves the direct use of a variety of allylic alcohols under very mild conditions, such as room temperature, and the use of sustainable deep eutectic solvents (DESs). The search for the optimum DES to be used in the reaction revealed that a simple mixture of choline chloride (ChCl) and lactic acid provides excellent results for a wide range of substrates with high isolated yields. This methodology represents a significant improvement compared to other procedures described in the literature, for which high temperatures, stronger reaction conditions or metal catalysts are usually required. In some cases, this protocol provides the first examples of trapping allylic carbocations with indole derivatives. In addition, challenging nucleophiles such as amides, carbamates, azides or sulfonamides, among others, have also been successfully used. All these features render this procedure an appealing and green alternative compared to other examples reported in the literature on the alkylation of amines by allylic alcohols. Preliminary mechanistic studies using unsymmetrically substituted alcohols support that the reaction could proceed via an SN1 pathway.
Introduction
Nitrogen-containing compounds, particularly anilines, indoles, or hydrazides, are important intermediates and building blocks for pharmaceuticals, agrochemicals, polymers, bioactive compounds, and dyes, among others.1 In addition to their reactivity, the alkylation of amines is a widely used organic reaction that involves an amine and an alkylating reagent with a good leaving group. Hence, some pivotal alkylated amines are also used for drug design in the pharmaceutical industry.2Although this approach is quite versatile because of the broad scope of alkylating agents, the traditional protocol is not very sustainable, due to the generation of stoichiometric amounts of leaving groups. In contrast, the N-alkylation of amines via nucleophilic substitution using alcohols is of paramount importance because the only waste generated in the reaction would be a molecule of water. Depending on the catalytic system, the generation of water could also be a problem, but the low price and ease of handling of alcohols used in the alkylation of amines make this approach widely popular in the industry.3 A broad variety of metal catalysts have been extensively explored since the first example of a catalysed alkylation of aniline using an alcohol via the so-called “hydrogen borrowing”.4 However, this metal-catalysed N-alkylation of amines usually requires high temperatures. Consequently, using lower temperatures and developing greener catalytic systems are considered challenges from both an economic and an environmental point of view.5
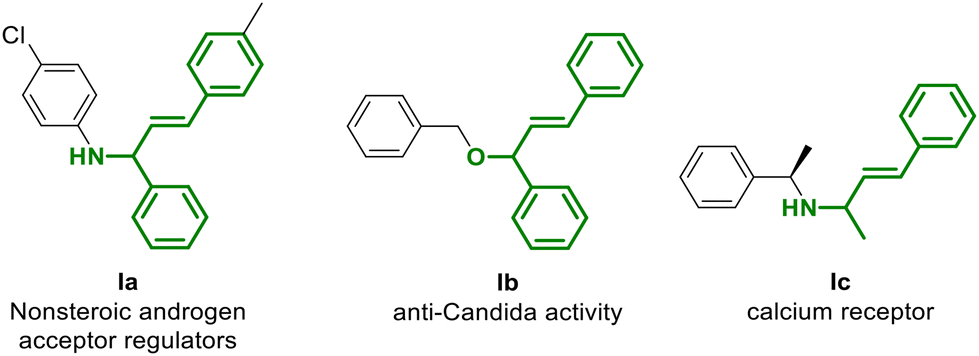
Moreover, the use of allylic alcohols in this procedure is of foremost importance,6 since the resulting allyl compounds are also fundamental building blocks and intermediates for the synthesis of some products such as those reported in Fig. 1.
Nowadays, compliance with the principles of green chemistry has become extremely important.10 Hence, achieving more sustainable processes that are cheaper, produce less waste, and use renewable sources is of great interest to our society. Among the improved processes that employ green solvents or more sustainable conditions, a special mention should be made of the use of ionic liquids (ILs),11 water12 or solvent-free conditions.13 Although these approaches avoid the use of organic solvents, they still require metals or high temperatures.
More recently, deep eutectic solvents (DESs), which are considered a class of ILs, have been proposed as greener alternatives to ILs.14 DESs are eutectic mixtures of Lewis or Brønsted acids and bases that contain anionic and cationic species.15 In spite of being considered a new type of IL, sharing many physical properties with them, the composition and application areas of DESs differ significantly. DESs offer several advantages over ILs, including lower cost, biodegradability, ease of preparation, and high atom economy.16 In addition, DESs have gained tremendous popularity due to their applications in two main research areas as synthetic and catalytic media.17 These green solvents are obtained from complexation with a hydrogen-bond acceptor (HBA), such as a quaternary salt, and a hydrogen-bond donor (HBD). One of the earliest works in the literature using DESs in the context of enzymatic catalysis was reported by Gill and co-workers in 1994. There, they showed that DESs could provide better reaction media than the conventional organic solvents.18
To the best of our knowledge, DESs have not been used previously for the N-alkylation of amines or their derivatives using allylic alcohols. While only a few examples of allylic amination of alcohols with anilines in the absence of metal systems have been reported,19 the closest approach for this process is available in the seminal works by Pastor's group.20 In 2018, the authors demonstrated the possibility of achieving a free-metal synthesis of N-allylanilines using 1,3-bis(carboxymethyl)imidazolium chloride (bcmim-Cl), an organic salt, as a catalyst. Although this ionic organic solid (IOS) is apparently similar to ionic liquids and despite the advantages of using IOSs in the N-alkylation of amines by allylic alcohols, temperatures between 80 °C and 100 °C are required for the success of this process. Hence, in search of an alternative for IOSs, the use of lower temperatures is still interesting from a green and energetic perspective. A pivotal example that also deserves a special mention is the work reported by Alemán's group using photocatalysis to obtain similar compounds.21,22
Results and discussion
Motivated by the necessity of looking for greener conditions to obtain allylamines, in this work, we envisioned the use of an environmentally friendly DES as the medium and as the promoter of the reaction as one of the simplest, greener, and straightforward approaches.23 First, different DESs were tested as media and as catalysts in the reaction model between (E)-1,3-diphenyl-2-propen-1-ol (1a) and aniline 2a in the absence of any additional solvent at room temperature (Scheme 1). Since one of the most frequently used HBA for DESs is (2-hydroxyethyl)trimethylammonium chloride, or choline chloride (ChCl), a member of the vitamin B family, we prepared our medium as a mixture of this component and different hydrogen-bond donors.24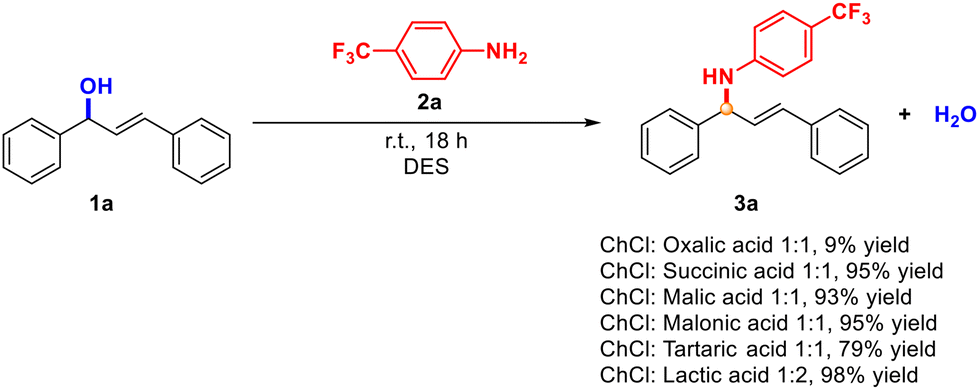 | ||
| Scheme 1 Screening of DESs in the model reaction between alcohol 1a and aniline 2a. | ||
The variety of DESs tested in this reaction were prepared at 60 °C as described in the literature, but then, the medium was cooled down. After the addition of both reagents, the reaction was performed at room temperature for 18 h, to compare the results of the process. We selected these DESs as model media because we hypothesised that the Brønsted acid used, as the HBD component, was also able to promote the formation of the carbocation necessary for the subsequent attack of the aniline. Moreover, we analysed these HBDs to evaluate the possible effect of the high viscosity at room temperature of all these media. This is a crucial effect that must still be considered since the resulting mixture can be difficult to handle when employed.25
Remarkably, in almost all cases, we obtained excellent and almost quantitative yields. It is worth noting that the aniline used is expected to be less nucleophilic than other anilines with electron-donating groups. Even though similar results were achieved in some cases, we proceeded to the next step with the DES formed by ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :lactic acid, since the crude product of the reaction was cleaner, and it was easier to work with this medium (for more information about the formation of the DES, see the experimental part). Finally, we also tested the impact of temperature, concluding that room temperature was more appropriate from a sustainable point of view.
:lactic acid, since the crude product of the reaction was cleaner, and it was easier to work with this medium (for more information about the formation of the DES, see the experimental part). Finally, we also tested the impact of temperature, concluding that room temperature was more appropriate from a sustainable point of view.
To the best of our knowledge, this is the first example where this reaction has been performed using a DES as both medium and promoter, at room temperature in the absence of another additional catalyst or solvent. With these conditions in hand, the scope and limitations of the substituents and functional group tolerance were evaluated using representative examples of different anilines, allylic alcohols and hydrazides (Schemes 2–4).
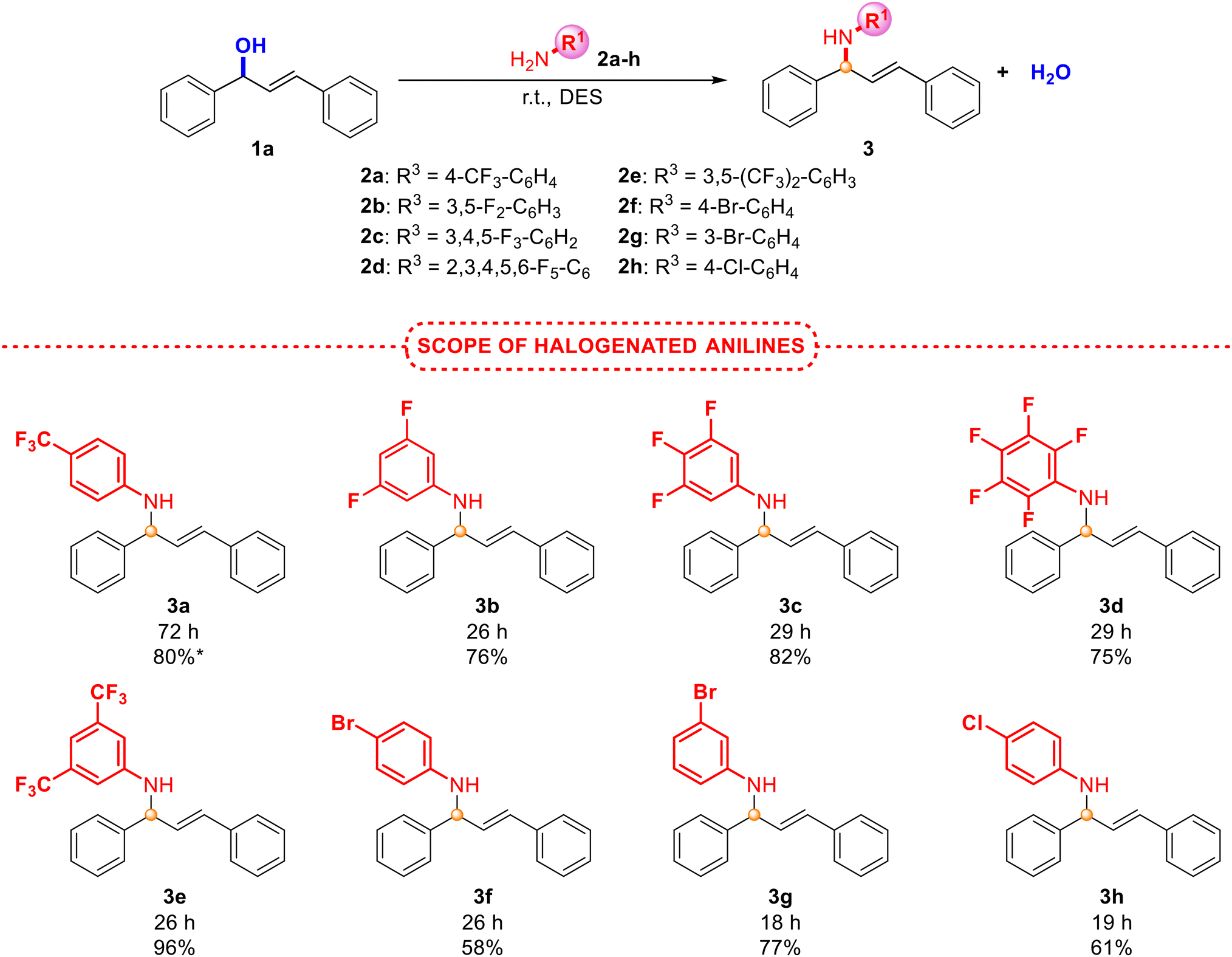 | ||
| Scheme 2 Scope of the allylic amination employing alcohol 1a and anilines 2a–h under the optimised reaction conditions. *Yield for 1 mmol scale. | ||
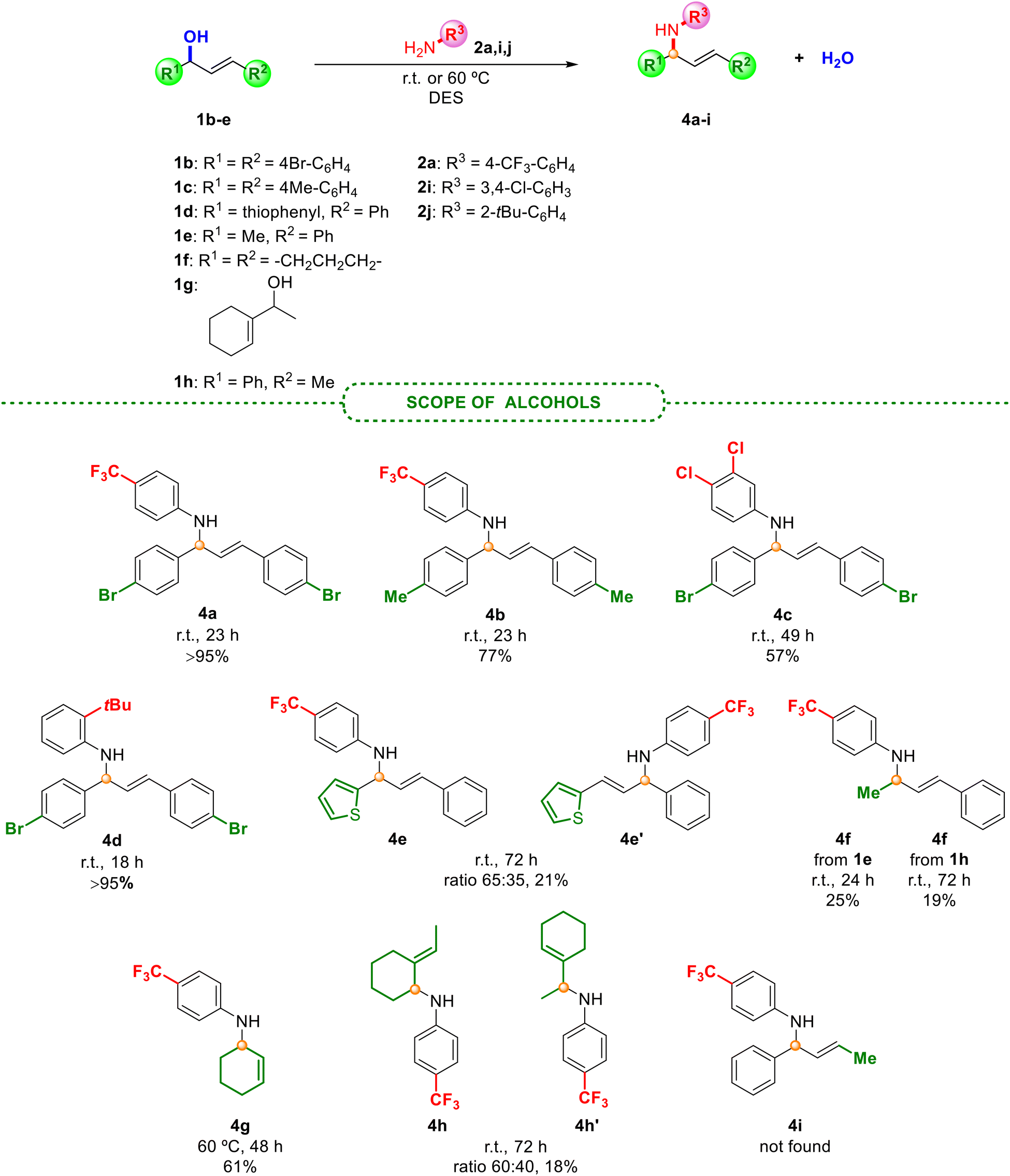 | ||
| Scheme 3 Scope of the allylic amination employing different allyl alcohols 1b–h under the optimised reaction conditions. | ||
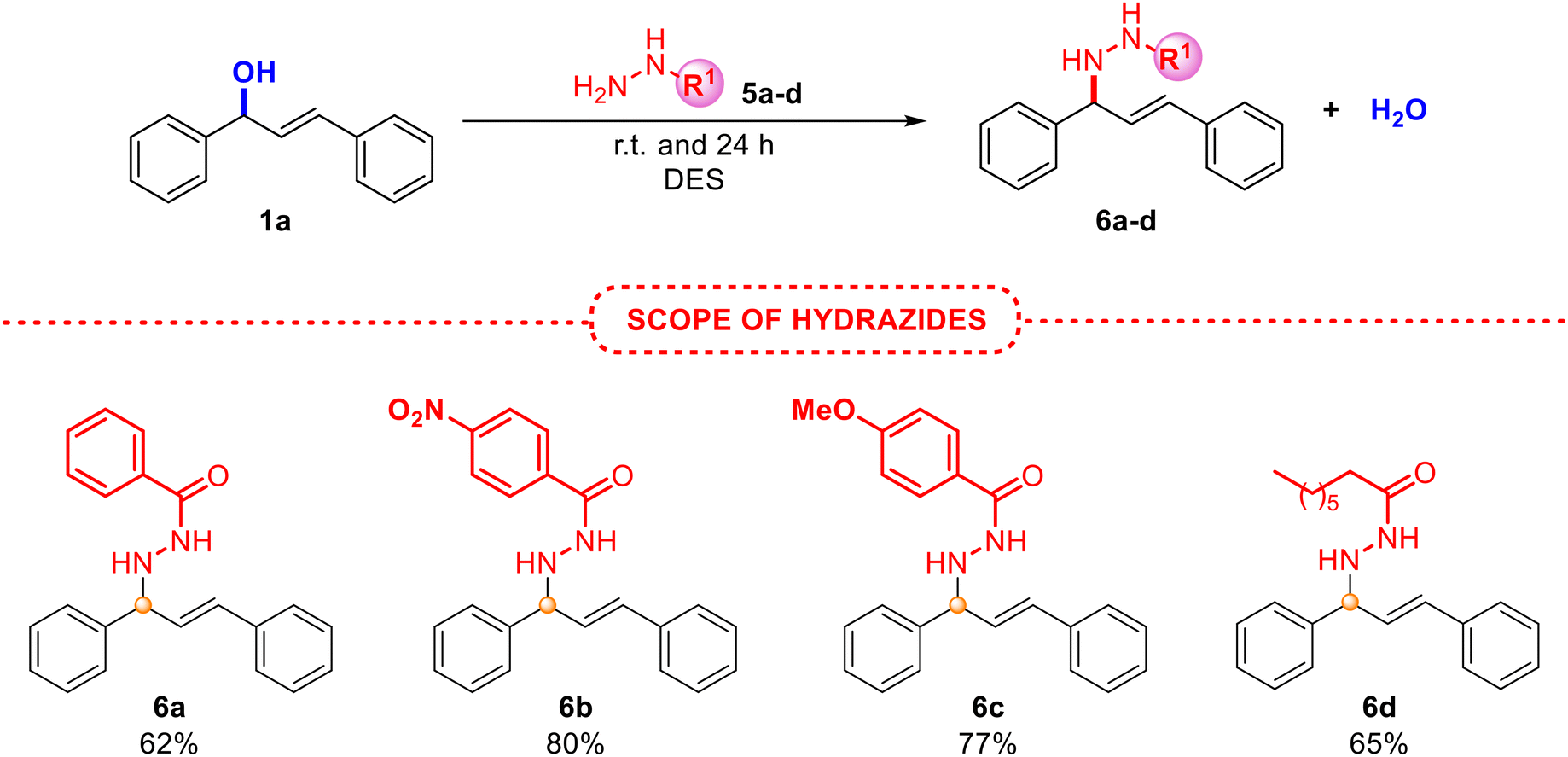 | ||
| Scheme 4 Scope of the allylic amination employing allyl alcohol 1a using hydrazides 5a–d. | ||
As presented in Scheme 2, halogenated anilines smoothly underwent N-allylation with excellent isolated yields and selectivity, obtaining in all cases an E isomer. Surprisingly, less active anilines (with electron-withdrawing groups) afforded the best results. Unfortunately, the reaction in the presence of electron-donating groups (4-OMe- or 4-tBu-) in the aniline did not provide the product formation, but an excellent yield was obtained between the hindered aniline 2j (with a tBu group in the ortho position) and alcohol 1b (Scheme 3). This unexpected behaviour, since anilines with electron-donating groups are expected to be better nucleophiles than those with electron-withdrawing groups, was also observed by Pastor and co-workers,20 and other authors.19 Based on these findings, the protonation of the alcohol could be inhibited due to the interaction with a more basic aniline (those with electron-donating groups). However, steric hindrance in aniline 2j prevents such protonation, favouring a nucleophilic attack instead. Moreover, due to the increased nucleophilicity of the monoalkylated product compared to the initial amine, the nitrogen centre could undergo additional alkylation, which may become a drawback in some synthetic approaches. The process was also accomplished at a higher scale with a very good yield of product 3a, although longer reaction times were required. With the aim of broadening the scope of the reaction and shedding light on the mechanism involved in this process, different alcohols, including non-symmetric ones, were also explored (Scheme 3).
The use of different substituted alcohols 1 also yielded the desired products 4a–d with very good results. Although compounds 4e–h were obtained in lower yields, possibly due to the lower stability of the formed carbocation, derivatives of 4e, 4f and 4h have been isolated for the first time in the literature. Additionally, product 4g had previously only been achieved by hydroamination of 1,3-dienes by using palladium,26 ruthenium27 or zeolite catalysts.28 The reaction with alcohols 1d and 1g gave rise to a mixture of both products 4e and 4e′ and 4h and 4h′, respectively, suggesting the presence of an allylic carbocation intermediate. Unfortunately, product 4i was not found, as the reaction with alcohol 1h afforded the same final product 4f, providing proof of a plausible SN1 mechanism, as previously observed by Nájera and co-workers.29 These findings indicate that the regiochemistry of the process could be related to the stabilisation of the resulting allylic carbocation intermediate, which would afford the most thermodynamically stable intermediate prior to the nucleophilic attack, depending on the substrate. Furthermore, as an additional case of allylic amination, representative hydrazides with electronically and differently substituted aryls and an alkyl derivative also provided the corresponding allylated adducts 6a–d with excellent isolated yields and selectivity, as depicted in Scheme 4.
In all cases, only the product E was cleanly obtained and no mixture of the Z isomer was observed (Schemes 2–4). Similar compounds were previously obtained using palladium catalysis,30 which makes our approach as an interesting synthetic alternative. These good results encouraged us to test the utility of our developed protocol with another important structural core, the indole, as a representative example of a pivotal structure. We have extensive experience in the field of the Friedel–Crafts alkylation reaction using nitroalkenes, aldehydes and β,γ-unsaturated-α-ketoestes,31 since indole is considered a key structural core present in many biologically active compounds.32 Therefore, we tried to trap the in situ generated allylic carbocation with different indole derivatives as reported in Scheme 5.
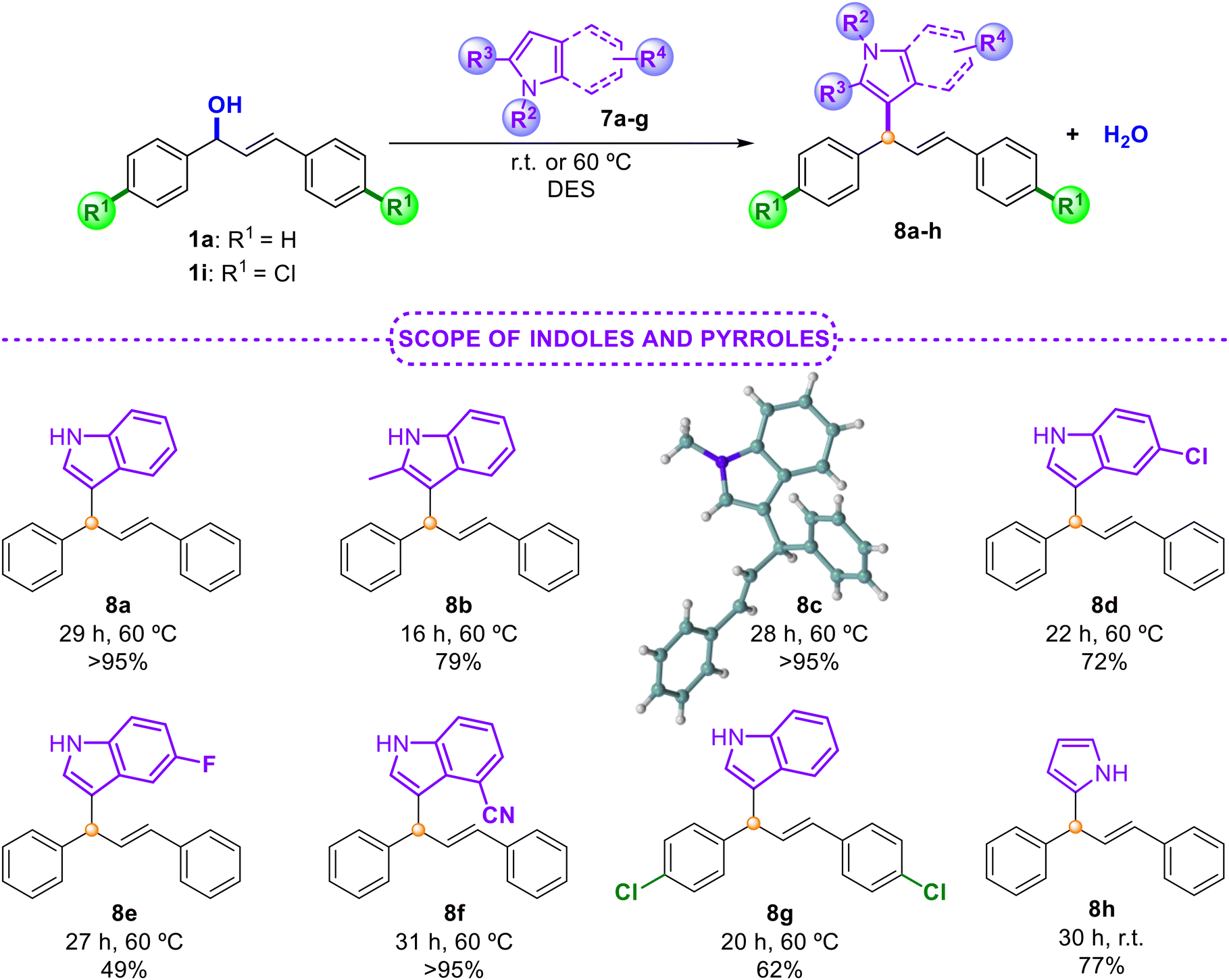 | ||
| Scheme 5 Scope of the allylic alkylation of indoles 7a–g and pyrrole 7h. | ||
It is worth noting that this protocol has been overlooked in the literature so far for the preparation of these products.4 Therefore, our methodology represents the first example for trapping allylic carbocation intermediates with these model molecules. In all cases, high isolated yields with indoles bearing electron-donating or electron-withdrawing groups 7a–f and pyrrole 7g were obtained (Scheme 5). The crude products of these reactions were clean and, in some cases, the final products were achieved after a simple extraction from the DES medium (75% for 8a). The structure of the final derivative 8c has also been confirmed by X-ray diffraction.33 Finally, in order to define the scope and limitations of this methodology, we turned our attention to nucleophiles of different nature (Scheme 6). For this purpose, alcohol 9a, carbamates 9b and 9c, trimethylsilylated nucleophile 9d, azide 9e, amide 9f, sulfonamide 9g and N,N-dimethylaniline 9h were successfully evaluated. In all cases, the final products 10a–h were isolated in high yields. Challenging nucleophiles, such as allyltrimethylsilane 9d, or sulfonamide 9g gave rise to the corresponding diene 10d or derivative 10g, respectively, in excellent yields. Moreover, 9h also led to the corresponding product of a Friedel–Crafts alkylation reaction with remarkable results after a simple extraction.33 Interestingly, we also tested the possibility of recycling the same DES medium in successive runs for the reaction between alcohol 1a and aniline 9h (ratio 1:1). After 10 runs, the same excellent results were obtained with a simple extraction using hexane (>90% of product 10h). These findings suggest that this process could continue with more rounds without impairing the reactivity of the process. These examples support the utility of this procedure as a simple and straightforward protocol that enables the use of diverse nucleophiles under very mild reaction conditions, resulting in the corresponding allylic adducts in high yields. This provides an alternative greener option to those previously reported.
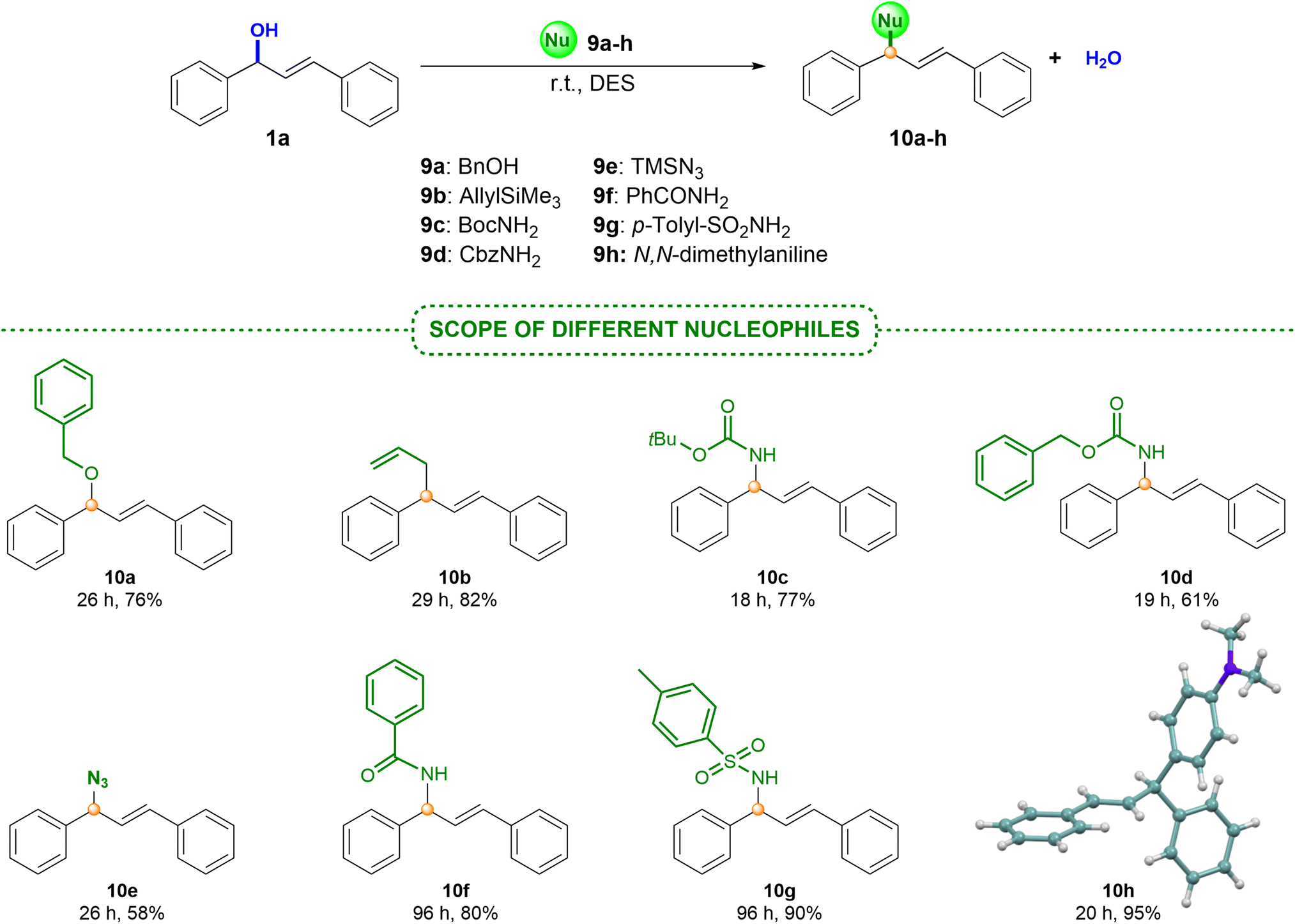 | ||
| Scheme 6 Scope of the methodology employing allyl alcohol 1a and nucleophiles 9a–h. | ||
To shed light on the reaction mechanism, beyond the above-mentioned observations, we also synthesised unsymmetrically substituted alcohols 1j and 1k and we analysed the results of their reaction with aniline 2a (Scheme 7).
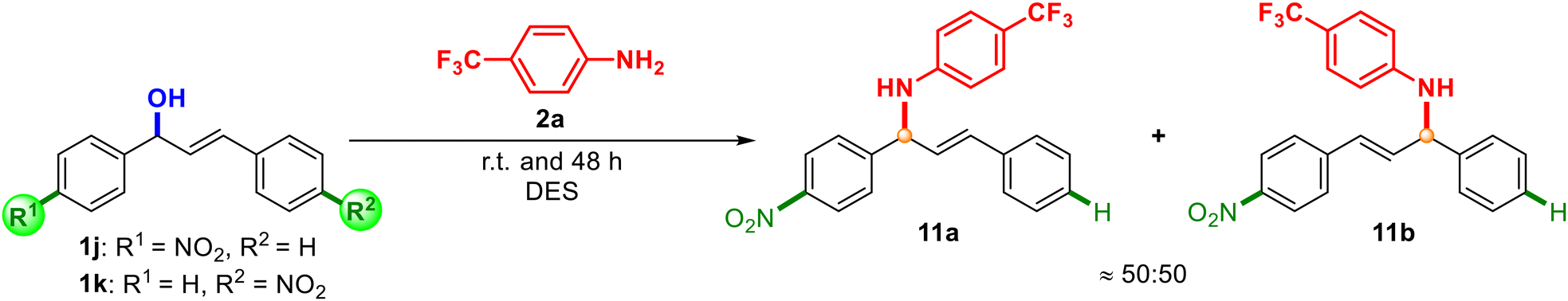 | ||
| Scheme 7 Mechanistic assays using alcohols 1j and 1k. | ||
In both cases, a mixture of products 11a:11b was observed in a ratio around 50:50, which supported the generation of an allylic carbocation intermediate in the medium and a SN1-type mechanism.34,35 Different reactivity was observed, since starting from alcohol 1j, 36% of the final mixture was obtained, while starting from 1k, a 66% yield was achieved. However, it is worth noting that with alcohol 1e (Scheme 3), with the possibility of isomerisation of the carbocation affording two different products, only one product (4f) was detected, which suggested that the reaction with the carbocation was faster than the isomerisation of the carbocation. Based on the experimental results, a plausible reaction mechanism is depicted in Scheme 8. In this proposal, the hydroxyl group could be activated with the assistance of ChCl and lactic acid as a source of proton [H+], which would act as a Brønsted acid catalyst (A). The release of a molecule of H2O would generate allylic carbocation B. Finally, the attack of the corresponding amine or nucleophile would trap the formed carbocation, giving rise to a protonated intermediate C. Final deprotonation of the product would recover the initial source of [H+] to initiate the cycle again. Besides, the effect of the stability of the carbocations is supported when comparing alcohol 1a with 1e–g, having obtained better results with 1a (benzylic and allylic carbocations) against 1e–g, which are only allylic carbocations (Schemes 2 and 3). As previously mentioned in this work, the stabilisation of the resulting allylic carbocation intermediate, prior to the nucleophilic attack, can influence the regiochemistry of the reaction with other non-symmetric alcohols such as 1d, 1g, and 1h.
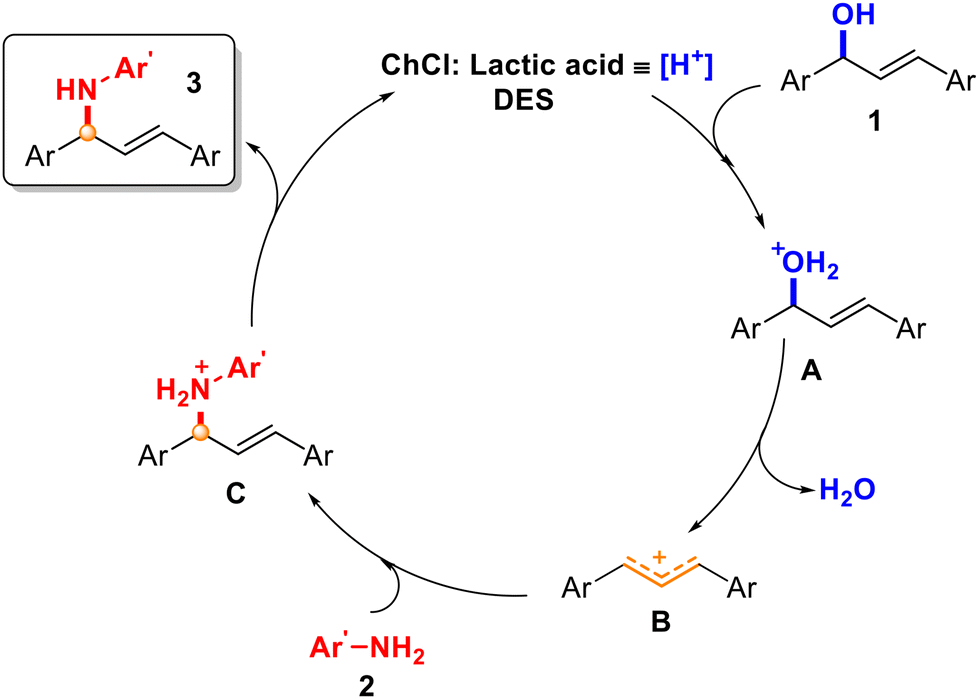 | ||
| Scheme 8 Plausible catalytic cycle. | ||
As a result, this protocol is complementary to those previously reported and an interesting alternative to the use of metal catalysts for the activation of these types of substrates.
Conclusions
In this work, we have shown that the allylation of anilines, hydrazides, indole derivatives and several interesting nucleophiles can be performed using a new approach under very mild conditions, including room temperature and a green medium. This means a great improvement in comparison with most of the examples described in the literature so far, where high temperatures or stronger reaction conditions are usually necessary. In addition, we show that a simple mixture of ChCl and lactic acid provides the best results for a wide substrate scope with moderate to quantitative isolated yields. These features make our approach an attractive green and interesting alternative compared with other examples reported in the literature on alkylation of amines by allylic alcohols. Moreover, preliminary mechanistic experiments support that the reaction should proceed via an SN1 pathway. This novel methodology can not only be considered as an alternative to the conventional metal or Brønsted acid-catalysed allylic substitution reactions, but also as a complementary method, as it enables the successful use of certain nucleophiles that frequently fail in previously reported procedures.Experimental
General experimental methods and instrumentation
Analytical thin-layer chromatography was performed on 0.25 mm silica gel 60-F plates. ESI and a micrOTOF-Q mass analyser were used for HRMS measurements. NMR spectroscopy was conducted using a Bruker AVANCE-II spectrometer. 1H NMR spectra were recorded at 300 and 400 MHz; 13C{1H}-APT NMR spectra were recorded at 75 and 101 MHz; CD3CN or CD3COCD3 were used as the deuterated solvents. Chemical shifts were reported in the δ scale relative to the residual CH3CN (1.94 ppm) and CH3COCH3 (2.05 ppm) for 1H-NMR and the central line of CD3CN (1.32 ppm) and CD3COCD3 (29.84 ppm) for 13C{1H}-APT NMR.All commercially available solvents and reagents were used as received. Spectral data for many of the synthesised compounds were consistent with the values previously reported in the literature: 3a,363e,373f,20a3h,19b4g,26a6a,388a,398b,398c,398d,408e,418g,418h,4210a,4310b,4410c,19b10d,19b10e,19b10f,19b10g19b and 10h.45 For all known products, we have also provided the 1H NMR spectra. The synthesis of alcohols 1b–d and 1g–k was performed following the described procedures in the literature.46
General procedure for the preparation of DES mixtures
Choline chloride (1 equiv., 83.3 mg) and lactic acid (2 equiv., 162.2 μL) were mixed in a 5 mL vial. The mixture was stirred and heated for 5 min at 60 °C until a clear homogeneous solution appeared. Then, the DES was cooled down in some cases prior to setting up the reaction.General procedure for the synthesis of allylic anilines 3a–h and 4a–i
In a vial with a magnetic stirring bar and a cap, a DES (250 mg) was prepared in advance. Then, alcohols 1a–h (0.1 mmol) and anilines 2a–j (0.2 mmol) were added to the vessel at room temperature. When the reaction was over, neutralisation was done with a microspatula of potassium carbonate. Then, 5 mL of water was added to the crude reaction mixture and it was extracted with ethyl acetate (3 × 5 mL). The organic layer was dried over MgSO4, the crude mixture was filtered, and the organic phase was evaporated under vacuum. The final product was purified by short flash column chromatography (silica gel and toluene), and if the crude product was clean, a Pasteur pipette could also be used for fast purification.47General procedure for the synthesis of allylic hydrazides 6a–d
In a vial with a magnetic stirring bar and a cap, the DES (250 mg) was prepared in advance. Alcohol 1a (0.1 mmol) and hydrazides 5a–d (0.2 mmol) were added to the vessel at 60 °C. When the reaction was over, neutralisation was done with a microspatula of potassium carbonate. Then, 5 mL of water was added to the crude reaction mixture and it was extracted with ethyl acetate (3 × 5 mL). The organic layer was dried over MgSO4, the crude mixture was filtered, and the organic phase was evaporated under vacuum. The final product was purified by short flash column chromatography (silica gel and heptane:AcOEt), and if the crude product was clean, a Pasteur pipette could also be used for fast purification.47
General procedure for the allylic alkylation of indoles 7a–f and pyrrole (7g)
In a vial with a magnetic stirring bar and a cap, the DES (250 mg) was prepared in advance. Alcohols 1a and 1g (0.1 mmol), indoles 7a–f (0.1 mmol) and pyrrole (7g) (0.1 mmol) were added to the vessel at 60 °C and at room temperature, respectively. When the reaction was over, neutralisation was done with a microspatula of potassium carbonate. Then, 5 mL of water was added to the crude reaction mixture, and it was extracted with ethyl acetate (3 × 5 mL). The organic layer was dried over MgSO4, the crude mixture was filtered, and the organic phase was evaporated under vacuum. The final product was purified by short flash column chromatography (silica gel and toluene), and if the crude product was clean, a Pasteur pipette could also be used for fast purification.47General procedure for the synthesis of allylic derivatives 10a–h
In a vial with a magnetic stirring bar and a cap, the DES (250 mg) was prepared in advance. Alcohol 1a (0.1 mmol) and nucleophiles 9a–h (0.5 mmol) were added to the vessel at room temperature. When the reaction was over, neutralisation was done with a microspatula of potassium carbonate. Then, 5 mL of water was added to the crude reaction mixture and it was extracted with ethyl acetate (3 × 5 mL). The organic layer was dried over MgSO4, the crude mixture was filtered, and the organic phase was evaporated under vacuum. The final product was purified by short flash column chromatography (silica gel and heptane:AcOEt), and if the crude product was clean, a Pasteur pipette could also be used for fast purification.
Author contributions
M. C. G. and R. P. H. conceptualised the idea and supervised the project. S. Z.-R. performed the experiments. All the authors discussed the results, wrote, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Agencia Estatal de Investigación (AEI), project PID2019-104379RB-C21 and PID2020-117455GB-I00/AEI/10.13039/501100011033, and Gobierno de Aragón-Fondo Social Europeo (Research Group E07_23R). S. Z.-R. thanks Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico) for a predoctoral fellowship (Grant 739848).References
- (a) A. Seayad, M. Ahmed, H. Klein, R. Jackstell, T. Gross and M. Beller, Science, 2002, 297, 1676–1678 CrossRef CAS PubMed; (b) S. A. Lawerence, Amines: Synthesis Properties and Applications, Cambridge University, Cambridge, 2004 Search PubMed; (c) B. R. Brown, The Organic Chemistry of Aliphatic Nitrogen Compounds, Cambridge University, Cambridge, 2004 Search PubMed.
- (a) L.-M. Wang, Y. Morioka, K. Jenkinson, A. E. H. Wheatley, S. Saito and H. Naka, Sci. Rep., 2018, 8, 1–7 Search PubMed; (b) N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed; (c) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- (a) Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH, 2008 Search PubMed; (b) Methodologies in Amine Synthesis: Challenges and Applications, ed. A. Ricci and L. Bernardi, Wiley-VCH, 2021 Search PubMed.
- (a) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS; (b) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC; (c) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed; (d) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef; (e) C. Gunanathan and D. Milstein, Science, 2013, 341, 249–260 CrossRef CAS PubMed; (f) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC; (g) K.-I. Shimizu, Catal. Sci. Technol., 2015, 5, 1412–1427 RSC; (h) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed; (i) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- L. Zhang, Y. Zhang, Y. Deng and F. Shi, Catal. Sci. Technol., 2015, 5, 3226–3234 RSC.
- M. Bandini, Angew. Chem., Int. Ed., 2011, 50, 994–995 CrossRef CAS PubMed.
- M. Wang, Y. Deng, C. Zhou, D. Yang, X. Hui, H. Su and J. Gao, WO2006125397A1, 2006.
- A. Kasa, R. Varala, P. M. Swami and P. Zubaidha, Chem. J., 2013, 3, 66–74 CAS.
- B. C. Van Wagenen, S. T. Moe, M. F. Balandrin, E. G. DelMar and E. F. Nemeth, US6211244B1, 2001.
- P. Jessop, Green Chem., 2020, 22, 13–15 RSC.
- For selected examples, see: (a) S. E. Lyubimov, V. A. Davankov and K. N. Gavrilov, Tetrahedron Lett., 2006, 47, 2721–2723 CrossRef CAS; (b) Z. S. Qureshi, K. M. Deshmukh, P. J. Tambade, K. P. Dhake and B. M. Bhanage, Eur. J. Org. Chem., 2010, 6233–6238 CrossRef CAS; (c) A. Zhu, L. Li, J. Wang and K. Zhuo, Green Chem., 2011, 13, 1244–1250 RSC; (d) F. Han, L. Yang, Z. Li and C. Xia, Adv. Synth. Catal., 2012, 354, 1052–1060 CrossRef CAS.
- For selected examples, see: (a) Y. Uozumi, H. Tanaka and K. Shibatomi, Org. Lett., 2004, 6, 281–283 CrossRef CAS PubMed; (b) T. Nishikata and B. H. Lipshutz, Org. Lett., 2009, 11, 2377–2379 CrossRef CAS PubMed; (c) J. Niemeier, R. V. Engel and M. Rose, Green Chem., 2017, 19, 2839–2845 RSC; (d) G. N. Vaidya, R. H. Choudhary, M. Nagpure, S. K. Lokhande, P. Rana and D. Kumar, Green Chem., 2022, 24, 3977–3984 RSC.
- J.-J. Yu, L.-M. Wang, F.-L. Guo, J.-Q. Liu, Y. Liu and N. Jiao, Synth. Commun., 2011, 41, 1609–1616 CrossRef CAS.
- S. Khandelwal, Y. K. Tailor and M. Kumar, J. Mol. Liq., 2016, 215, 345–386 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- Y. Chen and T. Mu, Green Chem. Eng., 2021, 2, 174–186 CrossRef.
- (a) C. Ruß and B. König, Green Chem., 2012, 14, 2969–2982 RSC; (b) J. A. Sirviö, M. Visanko and H. Liimatainen, Green Chem., 2015, 17, 3401–3406 RSC; (c) N. Fanjul-Mosteirín and V. del Amo, Tetrahedron, 2021, 84, 131967 CrossRef; (d) D. A. Alonso, S.-J. Burlingham, R. Chinchilla, G. Guillena, D. J. Ramón and M. Tiecco, Eur. J. Org. Chem., 2021, 4065–4071 CrossRef CAS.
- I. Gill and E. Vulfson, Trends Biotechnol., 1994, 12, 118–122 CrossRef CAS PubMed.
- (a) R. Sanz, A. Martínez, D. Miguel, J. M. Álvarez-Gutiérrez and F. Rodríguez, Adv. Synth. Catal., 2006, 348, 1841–1845 CrossRef CAS; (b) K. Motokura, N. Nakagiri, T. Mizugaki, K. Ebitani and K. Kaneda, J. Org. Chem., 2007, 72, 6006–6015 CrossRef CAS PubMed; (c) P. Trillo, A. Baeza and C. Nájera, J. Org. Chem., 2012, 77, 7344–7354 CrossRef CAS PubMed; (d) Z. Wang, H. Mo, D. Cheng and W. Bao, Org. Biomol. Chem., 2012, 10, 4249–4255 RSC.
- (a) M. Albert-Soriano, L. Hernández-Martínez and I. M. Pastor, ACS Sustainable Chem. Eng., 2018, 6, 14063–14070 CrossRef CAS; (b) M. Albert-Soriano and I. Pastor, Adv. Synth. Catal., 2020, 362, 2494–2502 CrossRef CAS.
- A. M. Martínez-Gualda, R. Cano, L. Marzo, R. Pérez-Ruiz, J. Luis-Barrera, R. Mas-Ballesté, A. Fraile, V. A. de la Peña O'Shea and J. Alemán, Nat. Commun., 2019, 10, 2634 CrossRef PubMed.
- For another interesting example, see: L. Zhang, Y. Zhang, Y. Deng and F. Shi, Catal. Sci. Technol., 2015, 5, 3226–3234 RSC.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- R. Amoroso, F. Hollmann and C. Maccallini, Molecules, 2021, 26, 6286 CrossRef CAS PubMed.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- (a) O. Löber, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4366–4367 CrossRef PubMed; (b) D. Banerjee, K. Junge and M. Beller, Org. Chem. Front., 2014, 1, 368–372 RSC.
- D. Intrieri, A. Caselli, F. Ragaini, P. Macchi, N. Casati and E. Gallo, Eur. J. Inorg. Chem., 2012, 569–580 CrossRef CAS.
- O. Jimenez, T. E. Müller, W. Schwieger and J. A. Lercher, J. Catal., 2006, 239, 42–50 CrossRef CAS.
- P. Trillo, A. Baeza and C. Nájera, Eur. J. Org. Chem., 2012, 2929–2934 CrossRef CAS.
- (a) M. Lotz, G. Kramer and P. Knochel, Chem. Commun., 2002, 2546–2547 RSC; (b) D. Popa, R. Marcos, S. Sayalero, A. Vidal-Ferran and M. A. Percàs, Adv. Synth. Catal., 2009, 351, 1539–1556 CrossRef CAS; (c) H. Yuan, Z. Zhou, J. Xiao, L. Liang and L. Dai, Tetrahedron: Asymmetry, 2010, 21, 1874–1884 CrossRef CAS.
- (a) G. Dessole, R. P. Herrera and A. Ricci, Synlett, 2004, 2374–2378 CAS; (b) R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576–6579 CrossRef CAS PubMed; (c) E. Marqués-López, A. Alcaine, T. Tejero and R. P. Herrera, Eur. J. Org. Chem., 2011, 3700–3705 CrossRef; (d) D. Roca-López, E. Marqués-López, A. Alcaine, P. Merino and R. P. Herrera, Org. Biomol. Chem., 2014, 12, 4503–4510 RSC; (e) J. Beltrá, M. C. Gimeno and R. P. Herrera, Beilstein J. Org. Chem., 2014, 10, 2206–2214 CrossRef PubMed; (f) E.-M. Schön, E. Marqués-López, R. P. Herrera, C. Alemán and D. D. Díaz, Chem. – Eur. J., 2014, 20, 10720–10731 CrossRef PubMed; (g) V. Juste-Navarro, E. Marqués-López and R. P. Herrera, Asian J. Org. Chem., 2015, 4, 884–889 CrossRef CAS; (h) J. V. Alegre-Requena, A. Valero-Tena, I. G. Sonsona, S. Uriel and R. P. Herrera, Org. Biomol. Chem., 2020, 18, 1594–1601 RSC.
- (a) U. Pindur and T. Lemster, Curr. Med. Chem., 2001, 8, 1681–1698 CrossRef CAS PubMed; (b) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed; (c) P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Álvarez, Chem. – Eur. J., 2011, 17, 1388–1408 CrossRef CAS PubMed; (d) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed.
- CCDC 2218009 (8c) and 2252454 (10h) contain the supplementary crystallographic data for this paper.†.
- (a) A. Baeza and C. Nájera, Synthesis, 2014, 25–34 Search PubMed; (b) R. Ortiz, A. Koukouras, E. Marqués-López and R. P. Herrera, Arabian J. Chem., 2020, 13, 1866–1873 CrossRef CAS.
- Other authors have also proposed a plausible SN1 mechanism to explain this reactivity. Among them, the authors of ref. 21 supported their example with computational calculations.
- T. Ohshima, Y. Nakahara, J. Ipposhi, Y. Miyamoto and K. Mashima, Chem. Commun., 2011, 47, 8322–8324 RSC.
- Y. Wu, F. Y. Kwong, P. Li and A. S. C. Chan, Synlett, 2013, 24, 2009–2013 CrossRef CAS.
- H. Yuan, Z. Zhou, J. Xiao, L. Liang and L. Dai, Tetrahedron: Asymmetry, 2010, 21, 1874–1884 CrossRef CAS.
- M. Bandini, A. Melloni and A. Umani-Ronchi, Org. Lett., 2004, 6, 3199–3202 CrossRef CAS PubMed.
- H. Y. Cheung, W.-Y. Yu, F. L. Lam, T. T.-L. Au-Yeung, Z. Zhou, T. H. Chan and A. S. C. Chan, Org. Lett., 2007, 9, 4295–4298 CrossRef CAS PubMed.
- Z. Cao, Y. Liu, Z. Liu, X. Feng, M. Zhuang and H. Du, Org. Lett., 2011, 13, 2164–2167 CrossRef CAS PubMed.
- J. Le Bras and J. Muzart, Tetrahedron, 2007, 63, 7942–7948 CrossRef CAS.
- M. Kato, T. Nakamura, K. Ogata and S.-I. Fukuzawa, Eur. J. Org. Chem., 2009, 5232–5238 CrossRef CAS.
- B.-L. Yang and S.-K. Tian, Chem. Commun., 2010, 46, 6180–6182 RSC.
- P. Trillo and I. M. Pastor, Adv. Synth. Catal., 2016, 358, 2929–2939 CrossRef CAS.
- (a) C.-K. Chan, Y.-L. Tsai and M.-Y. Chang, Tetrahedron, 2017, 73, 3368–3376 CrossRef CAS; (b) J. R. M. Ferreira, R. N. da Silva, J. Rocha, A. M. S. Silva and S. Guieu, Synlett, 2020, 31, 632–634 CrossRef CAS.
- The use of toluene was considered the best option in comparison with the alternative hexane-diethyl ether to purify the final products (the products are very non-polar substrates), in agreement with the classification of the solvent sustainability guides: (a) C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC; (b) P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC; (c) C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2218009 and 2252454. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc01017a |
| This journal is © The Royal Society of Chemistry 2023 |