
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Manganese catalysed enantioselective hydrogenation of in situ-synthesised imines: efficient asymmetric synthesis of amino-indane derivatives†
Conor L.
Oates
,
Alister S.
Goodfellow
 ,
Michael
Bühl
and
Matthew L.
Clarke
*
,
Michael
Bühl
and
Matthew L.
Clarke
*
School of Chemistry, University of St Andrews, EaSTCHEM, St Andrews, Fife, KY16 9ST, UK. E-mail: mc28@st-andrews.ac.uk
First published on 3rd May 2023
Abstract
A manganese catalyst of a facially coordinating P,N,N ligand catalyses the hydrogenation of imines derived from indanone derivatives with high enantioselectivity. There is no requirement for an activating group and imines can be generated in situ. The selectivity can be rationalised by DFT calculations.
Since chiral amines are commonly found functional groups in many pharmaceuticals, agrochemicals and biologically active molecules, there has been intense interest in their asymmetric synthesis for decades. Enantioselective hydrogenation of ketimines is one of the most efficient and scalable methods for making enantiomerically enriched amines. Related methods such as Direct Asymmetric Reductive Amination, where the ketimine is generated in situ, are appealing since some ketimines are difficult to isolate and avoiding an isolation step reduces waste.1
The majority of the many studies on asymmetric hydrogenation of ketimines have utilised precious metal catalysts, especially Ir, Ru, Rh and Pd.1a These reactions continue to represent efficient methodology that often has a low E-factor and low cost. However, it would be desirable if more catalytic processes made use of earth abundant metals since there will be no issues with sustainability of supply, and the environmental impact of obtaining earth abundant metal precursors should be lower. In the case of metals like Mn, Fe and Cu, which have low toxicity concerns, it is likely to be less energetically demanding to remove trace metals from organic products. The development of earth abundant metal catalysts is consequently attracting a global effort to develop and refine applications and map out their reactivity. Despite this, the use of any earth abundant metals in hydrogenation of ketimines is not extensively developed. An iron catalyst combined with an enantiomerically pure organocatalyst have been used with high enantioselectivity for hydrogenation of a range of acyclic N-aryl imines.2b Another key publication reports excellent enantioselectivity using 3 mol% of an iron catalyst to reduce isolated imines bearing a diphenylphosphinyl activating group; swapping the activating substituent on the nitrogen for a non-activating aryl group causes the reaction to fail.2a This paper also reports greater difficulty (lower enantiomer ratio (er)) with the industrially significant reduction of an indanone derived imine (even with Ph2P(O) activating group).
Another paper reports the use of activated imines derived from indanone that contain an enantiomerically pure chiral auxiliary; in this way Fe catalysed diastereoselective hydrogenation of an indanone derived imine was achieved.2c While the use of stoichiometric amounts (1.5 equivalents) of an enantiomerically pure auxiliary is an obvious weak point, this paper is significant since enantiomerically pure amino indanes are significant building blocks for pharmaceuticals,2c,3 and accessing them using earth abundant metal promoted hydrogenation is attractive and hadn't been done before. The enantioselective hydrogenation of in situ synthesised imines using earth abundant metals4 seems to be very scarce indeed; a recent review article predicted this to be a future direction for catalytic reductive amination research.1c
There is a lot of current interest in using manganese as a sustainable hydrogenation catalyst,5–7 but there seem to be no publications on asymmetric hydrogenation of simple ketimines-whether they be isolated or made in situ. The nearest precedents are three papers reporting C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond reduction in quinolines6a,8a and 3-H indoles6b (enantioselective hydrogenation) and very recently hydrazones (by enantioselective transfer hydrogenation).8b Given these examples are structurally quite different to an exocyclic ketimine, the question of whether enantioselective ketimine hydrogenation would occur needed to be answered; our focus was to produce the valuable 1-amino-indane chiral building blocks by Mn catalysed hydrogenation of either an isolated imine or, if possible, an in situ synthesised imine. Here we show that this can be achieved with high enantioselectivity-bringing what is essentially an asymmetric reductive amination process‡ into the arena of Mn catalysis.
N bond reduction in quinolines6a,8a and 3-H indoles6b (enantioselective hydrogenation) and very recently hydrazones (by enantioselective transfer hydrogenation).8b Given these examples are structurally quite different to an exocyclic ketimine, the question of whether enantioselective ketimine hydrogenation would occur needed to be answered; our focus was to produce the valuable 1-amino-indane chiral building blocks by Mn catalysed hydrogenation of either an isolated imine or, if possible, an in situ synthesised imine. Here we show that this can be achieved with high enantioselectivity-bringing what is essentially an asymmetric reductive amination process‡ into the arena of Mn catalysis.
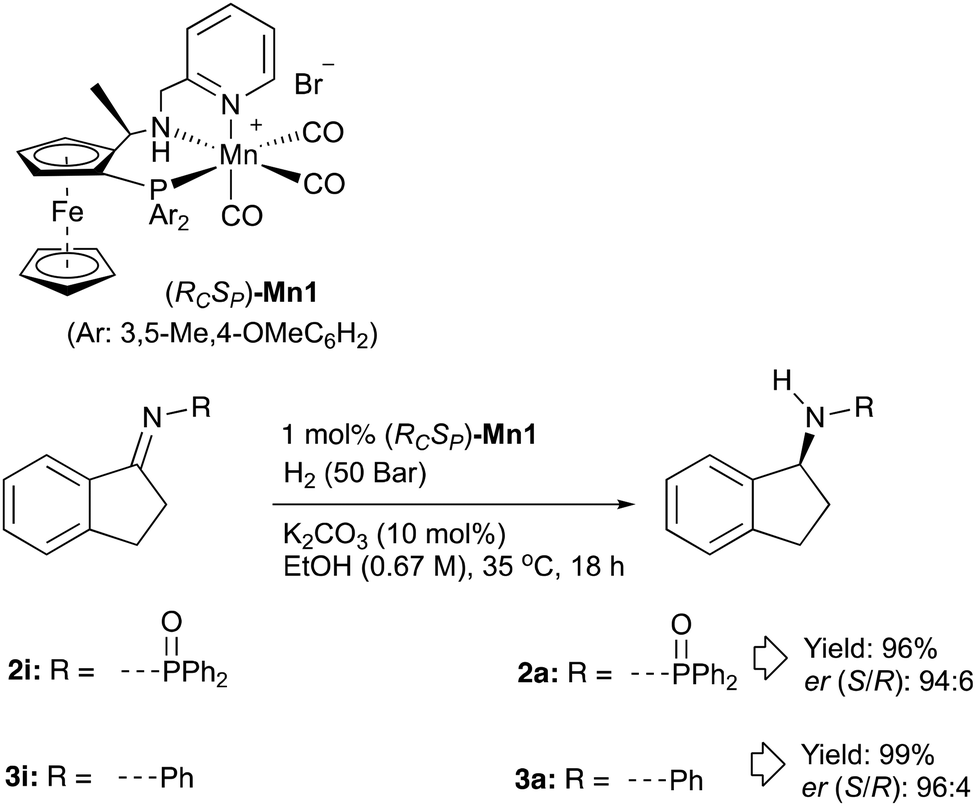
We have recently been exploring the use of a family of Mn catalysts related to Mn1 (Scheme 1) for the hydrogenation of esters and ketones to produce commercially valuable examples of alcohol products. We were motivated by the report in ref. 2c for amino-indane synthesis discussed earlier. This seemed a worthwhile target with an obvious significant improvement being the use of an enantiomerically pure earth abundant metal catalyst instead of a chiral auxiliary. Since there was no precedent for Mn catalysed ketimine hydrogenation and ref. 2a suggested that activated imines might be a requirement, we started out hydrogenating substrate 2i. The reaction proceeded smoothly using catalyst Mn1 and gave a synthetically useful er of 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.
:6.
 | ||
| Scheme 1 Earth abundant metal catalysts used in the hydrogenation of CN bonds. | ||
Encouraged by this result, we were interested to see if the conditions could be applied to unactivated ketimines. Compound 3i (derived of indanone and aniline) was subsequently synthesised and subjected to the hydrogenation conditions. Pleasingly, the reaction proceeded with high conversion and gave amine product 3a with high er (Scheme 2). We then investigated whether the imines could be produced in situ and therefore avoid the need to isolate pure imines – a step which can often be difficult. It was found that stirring a 1:1 ratio of ketone to amine in toluene in the presence of molecular sieves leads to near complete conversion to imine in 4 hours at 70 °C. A protocol where indanone and aniline were heated at 70 °C in the presence of molecular sieves before being decanted into the pressure vessel and subjected to the hydrogenation conditions used on isolated imine 3i gave amine 3a with similar enantioselectivity and yield. The reaction protocol was then applied to anisidine as starting material to form imine 4iin situ. The para-methoxybenzene protecting group is known to be removable to give the widely used chiral primary amine, 1-amino-indane. Similarly high enantioselectivity and good yield for amine 4a was possible.
 | ||
| Scheme 2 Manganese catalysed enantioselective hydrogenation of an activated and unactivated ketimine. Yields are determined by 1H NMR using 1,4-dimethoxybenzene as internal standard. | ||
For benzyl protected amine, 5a, we experienced considerable difficulty with product isolation and with finding a suitable measurement of enantioselectivity; the latter requiring direct conversion to a N-Boc derivative (see ESI†). None-the-less, it is promising that N-alkyl imines can also be reduced, even if this specific example could not be carried out in a practical way. The most likely cause of the isolation problems was traced to 5i only reaching 80% conversion, with 20% imine remaining. It is therefore likely that N-benzyl imines are less easy to reduce than N-aryl ones, in agreement with their relative electrophilicity. A recent publication suggests that 4-bromoindanone imines could also be valuable intermediates to another drug,3d,e so we also established that model substrate 6i would also undergo reduction without issues with C–Br cleavage, which it did to give 6a with a high enantiomer ratio (Table 1, entry 4).
|
|
||||
|---|---|---|---|---|
| Entrya | Substrate | Product | Conversion of imine to amine {yield} | Enantiomer ratioc |
| a For reaction conditions, see equation. Conversions of ketone were determined by 1H NMR with yields of isolated pure products in parentheses unless stated. b Isolated material contained impurity. Yield is determined against internal standard. c Determined by chiral HPLC on either amine or N-Boc derivative: see ESI† for details. | ||||
| 1 | 3k | 3a | >99% {73%} | 96:4 |
| 2 | 4k | 4a | >99% {71%} | 95:5 |
| 3 | 5k | 5a | 80% {75}b | 98:2 |
| 4 | 6k | 6a | >99% {89%} | 95:5 |

This project was focussed on amino-indane synthesis and finding out if the fundamental asymmetric amination procedure could be catalysed selectively by manganese at all; while a study on asymmetric hydrogenation of a range of structurally diverse ketimines was outside our scope, it would be a welcome and important advance for the future. One very preliminary observation worth noting is, that using this catalyst and typical conditions, it is possible to hydrogenate an acyclic imine derived from acetophenone and aniline in 93% yield and with some enantioselectivity but the latter is lower (er = 82:18) than the levels reported for the indanone imines (Scheme 3). Some catalyst tuning or new catalyst development may be necessary to achieve very high enantioselectivity with acetophenone-derived imines.
 | ||
| Scheme 3 Manganese catalysed enantioselective hydrogenation of in situ synthesises acetophenone derived ketimine. a>99% conversion with 93% isolated yield. | ||
DFT computations have been performed to rationalise these findings (PBE0-D3PCM//RI-BP86PCM level, benchmarked against first-row transition metal hydricities9 and validated in our previous work on ketone reduction7c). Using the ketimine substrate, 3i we have examined the barrier heights for the stereo-defining step of hydride transfer and have been able to accurately model the experimental selectivity of 96:4 (S). As expected, the lowest barrier involves a favourable π-stacking interaction between the aromatic indanone ring system and the pyridine ring of the catalyst (Scheme 4). We calculate a difference in barrier heights of the diastereomeric transition states of ΔΔ‡G = 1.57 kcal mol−1, corresponding to an er of 92:8 (S). This is a noticeably stronger selectivity than for the analogous ketone with this catalyst, arising from a stronger interaction energy, with larger dispersive contributions from the N-phenyl substituent (ΔΔD3int(imine–ketone) = −5.39 kcal mol−1, Fig. S2†). Experimentally, amination of the acyclic substrate 7k (via7i), occurs with reduced enantioselectivity of 82:18 (S). In silico evaluation reveals that the selectivity is reduced as a consequence of the extra conformational flexibility of the acyclic system compared to the more rigid cyclic indanone ring system (ΔΔ‡G = 1.03 kcal mol−1, er of 83:17 (S) [Fig. S4†]).
 | ||
| Scheme 4 Comparison of the stereodefining transition states for substrates 2i and 3i. Areas with attractive non-covalent interactions shown in green. Atoms are colour-coded as: C – grey, N – blue, P – orange, Mn – magenta, Fe – pink, H – white, O – red. | ||
Comparing the reduction of aryl imines and ketones, we calculate that hydride transfer is more challenging for the former than for the latter, e.g. by 3.51 kcal mol−1-for 3k relative to 3i, which may explain why these non-activated ketimines cannot be reduced by the iron catalyst of the Morris group.2a The more reactive activated imines 2i can bind through the N-phosphinoyl oxygen acting as a proton acceptor, forming stereo-defining eight-membered ring transition states (see Scheme 4 with further detail in the ESI†). We calculate this to proceed through the re-face, with ΔΔ‡G = 1.75 kcal mol−1 corresponding to an er of 94:6 (S). This eight-membered ring transition state for forming (S)-2a is favoured by 0.78 kcal mol−1 over the transition state with a “normal” six-membered ring where the imine nitrogen acts as a proton acceptor (both favour (S)-2a). The activated imine 2i has a significant reduction in barrier compared to the non-activated imine 3i (e.g. by ΔΔ‡G = 5.58 kcal mol−1 for forming (S) amine products as shown in Fig. S7 in the ESI†). In contrast to iron catalysis,2a we calculate that for manganese reduction of the activated imine 2i, hydrogen activation remains rate determining (see Fig. S7 in the ESI†).
Conclusions
It has proven possible to use a chiral manganese catalyst to hydrogenate N-aryl or N-phosphinyl imines to give protected amines with good enantiomeric ratio, and also to form the imine in situ with no need for purification. A detailed comparison of the sustainability of the research scale reactions described here with other research scale alternatives including a ‘potential environmental impact matrix’ is available in the ESI.† However, the protocol in ref. 2c was the closest inspiration for this work so is summarised here; it can be seen this new process overcomes the primary disadvantages of this route which are: (i) the need for a super-stoichiometric amount of a chiral auxiliary – which will also be more energetic to produce than aniline or anisidine, and (ii), not having a separate purification step which uses chemicals, energy and results in product loss (the yield for the imine synthesis is 60% in ref. 2c). There are other advantages to the manganese catalysed procedure in terms of activator and catalyst loading. The new procedure also compares favourably with published precious-metal-catalysed hydrogenations that produce amino-indane products and other routes we are aware of. This has been analysed and is discussed in detail in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the University of St Andrews, and the EPSRC Centre for Doctoral Training in Critical Resource Catalysis (CRITICAT) for financial support [Ph.D. studentship to CO; Grant code: EP/L016419/1], and the EaSI-CAT Programme PhD studentship to AG. We thank Solvias for the gift of chiral chemicals. Calculations were performed on a local HPC cluster maintained by Dr H. Früchtl.References
- (a) R. A. A. Abdine, G. Hedouin, F. Colobert and J. Wencel-Delord, ACS Catal., 2021, 11, 215–247 CrossRef CAS; (b) K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC; (c) Y. Tian, L. Hu, Y.-Z. Wang, X. Zhang and Q. Yin, Org. Chem. Front., 2021, 8, 2328–2342 RSC; (d) J. Barrios-Rivera, Y. Xu, M. Wills and V. K. Vyas, Org. Chem. Front., 2020, 7, 3312 RSC.
- (a) C. S. Seo, T. Tannoux, S. A. Smith, A. J. Lough and R. H. Morris, J. Org. Chem., 2019, 84, 12040–12049 CrossRef CAS PubMed; (b) S. Zhou, S. Fleiccher, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120–5124 CrossRef CAS PubMed; (c) D. Brenna, S. Rossi, F. Cozzi and M. Benagalia, Org. Biomol. Chem., 2017, 15, 5685–5688 RSC.
- (a) Y. Akao, W. Maruyama, H. Yi, M. Shamoto-Nagai, M. B. Youdim and M. Naoi, Neurosci. Lett., 2002, 326, 105–108 CrossRef CAS PubMed; (b) N. Pinterova, R. R. Horsley and T. Palenicek, Front. Phychiatry, 2017, 8, 236 CrossRef PubMed; (c) J. A. Shimshoni, I. Winkler, N. Edery, E. Golan, R. van Wettum and D. Nutt, Toxicol. Appl. Pharmacol., 2017, 319, 59–68 CrossRef CAS PubMed; (d) C. Cianferotti, G. Barreca, V. Bollabathini, L. Carcone, D. Grainger, S. Staniland and M. Taddei, Eur. J. Org. Chem., 2021, 1924 CrossRef CAS and ref's therein; (e) F. Uthoff, J. Löwe, C. Harms, K. Donsbach and H. Gröger, J. Org. Chem., 2019, 84, 4856–4866 CrossRef CAS PubMed.
- (a) S. Zhou, S. Fleiccher, H. Jiao, K. Junge and M. Beller, Adv. Synth. Catal., 2014, 356, 3451–3455 CrossRef CAS; (b) P. Yang, L. Hui Lim, P. Chuanprasit, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2016, 55, 12083–12087 CrossRef CAS PubMed; (c) X. Zhao, H. Xu, X. Huang and J. S. Zhou, Angew. Chem., Int. Ed., 2019, 58, 292–296 CrossRef CAS PubMed.
- (a) S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed; (b) N. A. Espinosa-Jalapa, A. Nerush, L. J. W. Shimon, G. Leitus, L. Avram, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2017, 23, 5934–5938 CrossRef CAS PubMed; (c) M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed; (d) A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS PubMed; (e) M. Glatz, B. Stoger, D. Himmelbauer, L. F. Veiros and K. Kirchner, ACS Catal., 2018, 8, 4009–4016 CrossRef CAS PubMed; (f) V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS PubMed; (g) L. Zhang, Y. Tang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2019, 58, 4973–4977 CrossRef CAS PubMed; (h) A. Kumar, T. Janes, N. A. Espinosa-Jalapa and D. Milstein, Angew. Chem., Int. Ed., 2018, 57, 12076–12080 CrossRef CAS PubMed; (i) A. Kaithal, M. Hölsher and W. Leitner, Angew. Chem., Int. Ed., 2018, 57, 13449 CrossRef CAS PubMed; (j) D. Wang, A. Bruneau-Voisine and J.-B. Sortais, Catal. Commun., 2018, 105, 31–36 CrossRef CAS; (k) C. S. G. Seo, B. T. H. Tui, M. V. Gradiski, S. A. M. Smith and R. H. Morris, Catal. Sci. Technol., 2021, 11, 3153–3163 RSC; (l) L. Zeng, H. Yang, M. Zhao, J. Wen, J. H. R. Tucker and X. Zhang, ACS Catal., 2020, 10, 13794 CrossRef CAS; reviews on Mn-catalysed reduction: (m) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed; (n) B. Maji and M. K. Barman, Synthesis, 2017, 49, 3377–3393 CrossRef CAS; (o) Homogeneous Hydrogenation with Non-Precious Metal Catalysts, ed. J. F. Teichert, Wiley-VCH, Weinheim, 2019 Search PubMed.
- (a) C. Liu, M. Wang, Y. Wang, Y. Peng, Y. Lan and Q. Liu, Angew. Chem., Int. Ed., 2021, 60, 5108 CrossRef CAS PubMed; (b) C. Liu, M. Wang, Y. Xu, Y. Li and Q. Liu, Angew. Chem., Int. Ed., 2022, 61, e202202814 CAS; (c) J. M. Pérez, R. Postolache, M. Castiñeira Reis, E. G. Sinnema, D. Vargová, F. deVries, E. Otten, L. Ge and S. R. Harutyunyan, J. Am. Chem. Soc., 2021, 143, 20071 CrossRef PubMed; (d) F. Ling, H. Hou, J. Chen, S. Nian, X. Yi, Z. Wang, D. Song and W. Zhong, Org. Lett., 2019, 21, 3937 CrossRef CAS PubMed.
- (a) M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS PubMed; (b) M. B. Widegren and M. L. Clarke, Org. Lett., 2018, 20, 2654–2658 CrossRef CAS PubMed; (c) C. L. Oates, A. S. Goodfellow, M. Bühl and M. L. Clarke, Angew. Chem., Int. Ed., 2022, e202212479 Search PubMed; (d) M. B. Widegren and M. L. Clarke, Catal. Sci. Technol., 2019, 9, 6047 RSC.
- (a) V. Papa, J. Fessler, F. Zaccaria, J. Hervochon, P. Dam, C. Kubis, A. Spannenberg, Z. Wei, H. Jiao, C. Zuccaccia, A. Macchioni, K. Junge and M. Beller, Chem. – Eur. J., 2022, 28, e202202774 Search PubMed; (b) L. Wang, J. Lin, C. Xia and W. Sun, J. Catal., 2022, 412, 487–497 CrossRef.
- A. S. Goodfellow and M. Bühl, Molecules, 2021, 26, 4072 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details and characterisation data, Cartesian coordinates and other computational data, and analysis of potential environmental impacts. See DOI: https://doi.org/10.1039/d3gc00399j. The research data underpinning this publication can be accessed at: https://doi.org/10.17630/cf0c7769-e031-4bb2-8d88-b1636282e4a1. |
| ‡ We have used the term in situ synthesised imines throughout rather than reductive amination, since reductive amination is generally defined as needing all the reagents and catalysts present from the beginning. However, the main practical advantage comes from the lack of an isolation step, and this is accomplished irrespective of when the catalyst first contacts the substrate. |
| This journal is © The Royal Society of Chemistry 2023 |