
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
GreenMedChem: the challenge in the next decade toward eco-friendly compounds and processes in drug design
Carola
Castiello†
 a,
Pierre
Junghanns†
b,
Annika
Mergel†
ac,
Claus
Jacob
c,
Christian
Ducho
b,
Sergio
Valente
a,
Dante
Rotili
a,
Rossella
Fioravanti
*a,
Clemens
Zwergel
*ac and
Antonello
Mai
ad
a,
Pierre
Junghanns†
b,
Annika
Mergel†
ac,
Claus
Jacob
c,
Christian
Ducho
b,
Sergio
Valente
a,
Dante
Rotili
a,
Rossella
Fioravanti
*a,
Clemens
Zwergel
*ac and
Antonello
Mai
ad
aDepartment of Drug Chemistry and Technologies, Sapienza University of Rome, Piazzale Aldo Moro 5, 00185 Rome, Italy. E-mail: rossella.fioravanti@uniroma1.it; clemens.zwergel@uniroma1.it
bDivision of Pharmaceutical and Medicinal Chemistry, School of Pharmacy, Saarland University, Campus C 2.3, 66123 Saarbrücken, Germany
cDivision of Bioorganic Chemistry, School of Pharmacy, Saarland University, Campus B 2.1, 66123 Saarbrücken, Germany
dPasteur Institute, Sapienza University of Rome, Piazzale Aldo Moro 5, 00185 Rome, Italy
First published on 22nd February 2023
Abstract
Green chemistry has become a hot topic and the focus of not only many companies but also researchers. Green chemistry strives to identify alternative and environmentally friendly reaction conditions and simultaneously aims to increase the rates and reduce the temperature of reactions. Specifically, the goal of green chemistry is to reduce the impact of chemical substances and processes on human health and the effective elimination of environmental pollution through dedicated sustainable prevention programs. Green chemistry is based on innovative scientific solutions to solve environmental problems that arise in the laboratory. In this review, we analyse in detail each of the 12 Principles of Green Chemistry developed by Paul Anastas in 1991, emphasising the aspects of medicinal chemistry carried out in research laboratories for the synthesis of active pharmaceutical ingredients (API). Here, we provide some examples of greener reactions, which can substitute the older strategies carried out by both industries and academia and offer a guide to improve the greenness of reactions. The present review highlights the methods that can be used by medicinal chemists in their daily work not only to improve the yields but also to reduce and prevent pollution, thereby protecting the environment and our health.
 Carola Castiello | Carola Castiello, born in 1996, is in her second year of her PhD course in Pharmaceutical Sciences, focusing on green chemistry applications in medicinal chemistry. She graduated in October 2020 in Medicinal Chemistry and Technologies with honours. Currently, she is carrying out the synthesis of small organic molecules for targeting mainly epigenetic enzymes implicated in a large number of pathologies, in particular cancer and neurodegenerative diseases. |
 Pierre Junghanns | Pierre Junghanns obtained his license to practise as a Pharmacist in 2018, and he is currently working in the research group of Prof. Christian Ducho at Saarland University. He is a fourth-year PhD-student working on innovative antibacterial agents derived from natural products. |
 Annika Mergel | Annika Mergel recently obtained her license to practise as a Pharmacist in Germany. During her final year, she obtained a Master's Degree in Pharmaceutical Sciences within an Erasmus exchange between the Saarland University, Germany and Sapienza University of Rome, Italy. She is seeking for greener applications in drug synthesis, working on dual-targeting epigenetic modulators. |
 Rossella Fioravanti | Rossella Fioravanti graduated in Pharmacy at the Sapienza University of Rome in 1986. She is an Associate Professor of Medicinal Chemistry at the Faculty of Pharmacy and Medicine, Sapienza University of Rome. Her scientific work consists of numerous publications; currently, her research activity is focused on the design and synthesis of small molecules with heterocyclic structure, being potential modulators of enzymes (DNA methyltransferases, HDACs, histone methyltransferases/demethylases) involved in the epigenetic regulation of gene expression with an impact on cancer and tropical diseases. |
 Clemens Zwergel | Dr Clemens Zwergel is an Assistant Professor (RTD-A) for Medicinal Chemistry at the Sapienza University of Rome. In the few last years, he proved to be a true European citizen: he moved after his license to practise as a Pharmacist in Germany to the UK for a Diploma in Pharmaceutical Sciences. Before arriving in Italy, he was a MarieCurie Fellow in France, where he obtained his EuroPhD. Since 2010, his main research interest lies in the design and synthesis of small molecules with potential applications in cancer, neurodegenerative, metabolic, and infectious diseases, resulting in more than 65 publications. |
Introduction
Nowadays, environmental pollution poses a significant threat to the Earth and its inhabitants. Growing soil and groundwater contamination with plastic or chemical substances is one such example.1 Furthermore, due to several anthropogenic processes, carbon emission is increasing at an astonishing rate, leading to global warming, flooding, and extreme climate events.2 Therefore, it is necessary to develop environmentally friendly approaches to meet the demands of society.To achieve this goal, more than 20 years ago, Anastas et al. established a cohesive system consisting of 12 principles as the foundation of green chemistry.3,4 This system is supposed to enhance the overall greenness of chemical strategies in modern research laboratories. In this regard, green chemistry should eliminate the use of hazardous substances and reduce the generation of waste in standard organic synthesis. Moreover, it also involves the use and disposal of substance besides their production with the aim of decreasing their ecological footprint.
Moreover, different sectors such as the pharmaceutical industry are devoting their efforts to developing sustainable chemistry for modern drug manufacturing and optimising the synthesis of many pharmaceuticals in terms of greenness.5 Another important field regarding the production of potential drugs is medicinal chemistry, which represents one of the early processes in fundamental drug research, aiming at the discovery of potential active pharmaceutical ingredients (APIs). In this context, this field is essential for developing novel medicine to successfully treat various diseases. However, most laboratories performing fundamental research still employ older, conventional methodologies for the synthesis of their target compounds.6 Given that the goal of standard medicinal chemistry is the fast and effective production of promising drug candidates, the eco-friendliness of the process is not of prime importance in most cases. Although medicinal chemistry was developed many decades ago,7 vast amounts of waste are still generated daily in academic research labs.
Therefore, from an environmental and ecological perspective, replacing older methods with benign alternatives should be a major priority.3,4 Herein, this review summarises the development of each one of the 12 Principles of Green Chemistry in the last years and demonstrate the possibilities readily available to medicinal chemists. Based on several examples, we show that eco-friendly strategies exist and can be incorporated into modern medicinal drug research. In addition, the cohesiveness of the 12 principles will also be illustrated in this review.
1. Prevention of waste instead of disposal of the created waste
“It is better to prevent waste than to treat or clean up waste after it has been created”.4Waste prevention should be the main goal of chemists, which can be achieved through the application of various strategies, including the efficient use of raw materials, the proper selection of reaction pathways or solvent systems, and efficient separation methods.4 In this case, analytical methods that involve the use of a minimal amount of materials, preventing the generation of waste, and avoids treatment or clean-up after the fact should be chosen. The recent interest in the so-called green analytical chemistry was shown through the development of new methods and analytical methodologies that can reduce and eliminate the use and generation of substances, such as solvents, reagents, products, and by-products, which are hazardous to human health and the environment.4,8 Therefore, green chemistry is an important tool for avoiding waste accumulation, pollution, and reducing the risks to humans and the environment.8 Firstly, the risk has to be defined, which it can be termed as the product of the hazard of a particular substance and the exposure to that substance9
| Risk = hazard × exposure |
Historically, analytical methods have been used as pollutant control approaches for detecting contamination in air, water, and the soil; consequently, the measurement of environmental problems is performed after they were already created. Therefore, over the years, new monitoring and analytical tools have been developed to minimise and contain the exposure, bypassing the upstream problem of pollution creation. At this point, green chemistry is emerging as a useful approach to reduce or eliminate the hazard factor in the risk equation, thereby using chemistry for pollution prevention, deviating the attention from the exposure controls.
Hazards not only include direct and indirect ecological impacts, such as atmospheric damage, plant and animal toxicity, global climate change, and resource depletion, but also chronic toxicity, carcinogenicity, and mutagenicity. Therefore, the consciousness and knowledge of chemists are crucial factors in reducing the hazards and preventing waste. The chosen methods, solvents, reagents, and analysis time are all part of an ecologically conscious way of thinking, which all chemists should contemplate. However, this is not obvious nowadays because, mainly in the academic field, the green approach in medicinal chemistry is not often considered neither at the teaching level nor academic research level, and the awareness at the university level concerning this important topic is still very low. The guidelines available, which will be discussed in the present review, often come from industry, which is a step forward with respect to academia. In 2008, the pharmaceutical company Pfizer developed the first Reagent Selection Guide,10 as a pioneer in advancements in green and sustainable chemistry. In addition, other pharmaceutical companies such as GSK, Sanofi and AstraZeneca outlined Solvent Selection Guides as tools for making the best choice towards greener and more sustainable chemistry,10–13 ranking the most used solvents in terms of their greenness. These guidelines provide important education for medicinal chemists who are approaching this highly relevant topic for the first time. For instance, the use of toxic organic solvents such as acetonitrile and methanol can damage the operator's health if the exposure is frequent, but they are still used because it is ignored that methods without their usage can also be suitable. Moreover, the disposal of these solvents creates more waste and hazards to human health and the environment,14 where although there are processes to neutralise their toxicity, they can still have an effect. For example, the incineration of acetonitrile generates waste that contributes to the creation of acid rain.15 In addition, the reaction time is important, where the longer the reaction proceeds, the longer the operator is required to monitor it, increasing their exposure. Alternatively, devices can be reused, avoiding additional costs and disposal of the created waste.8 Hence, green chemistry also aims to train scientists to consider the environmental impacts of their research. Nevertheless, when it is not possible to achieve these goals, waste production can be decreased through solvent and catalysts recovery and recycling. Closed loop systems can be generated using the waste of a reaction as the starting material for the following reaction, making progress toward a circular economy. Solvents can be reused for cleaning purposes and metal catalysts can be further utilised for other reactions.16 Actually, the elegant concept of “waste valorisation” should be applied, considering the waste as an additional product of the reaction system that can be further utilised, similar to nature.17 Hence, recovery and recycling can have a significant positive effect on the environment, reducing the pollution derived from pharmaceutical ingredients.4,18
As stated above, making the right decisions regarding chemical processes can significantly improve waste reduction and pollution prevention. Through the innovative and strategic design of synthetic routes, chemists can achieve green chemistry goals.4 Continuous processing and process intensification19 are useful analytical methods that can lead to rapid reaction screening and optimisation not only for new reactions but also old processes.20
An example of a reaction performed with continuous processing is the preparation of carbamates described by Grego et al. (Fig. 1).21 This alternative green pathway avoids the use of phosgene by obtaining the desired product methyl phenylcarbamate (3) using zinc carbonate as a catalyst, resulting in high conversions (∼98%) at high temperatures (200 °C) without the significant formation of by-products and catalyst deactivation. The reaction, carried out in an autoclave using a continuous-flow apparatus, showed significant waste prevention according to the first green chemistry principle. On one hand, this procedure is more ecologically friendly as it reduces the number of toxic reagents used; on the other hand, it can be slower than the commonly known reaction procedures consuming more energy, and thus this procedure is not yet fully green.4 Hence, greener proposals have to be further developed.
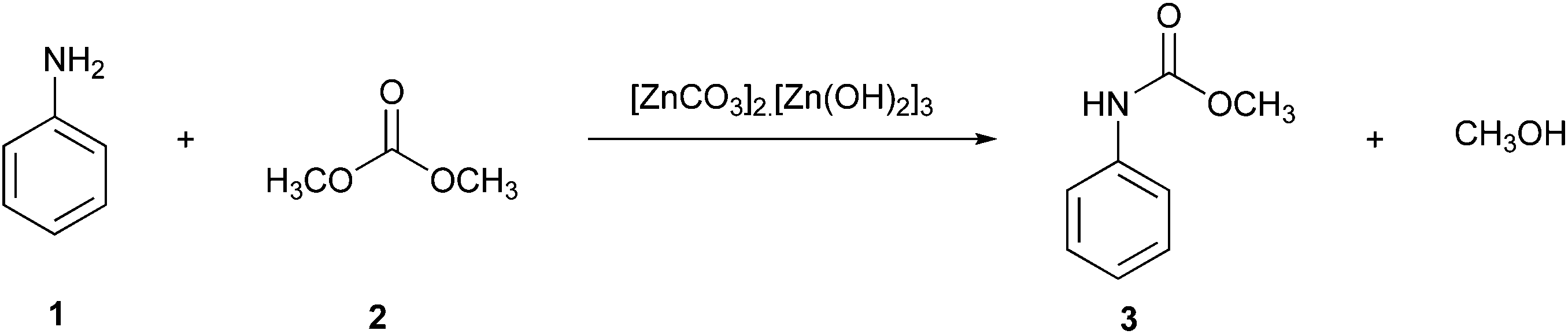 | ||
| Fig. 1 Synthesis of methyl phenylcarbamate using the continuous processing tool.4 | ||
Pedersen et al.22 implemented a heterogeneous Grignard alkylation (Fig. 2) using a continuous stirred-tank reactor (CSTR) and plug-flow reactor (PFR), which resulted in a mini-reactor that reduced the volume of solvent used and difficulties due to the processing of solids in micro-reactors. This process resulted in the transformation of 2-chlorothioxanthen-9-one (4) in the magnesium alkoxide intermediate (5) through allylmagnesium chloride in THF, which was subsequently hydrolysed with dilute acid to produce 9-allyl-2-chloro-9H-thioxanthen-9-ol (6). The advantages of this new process are not only related to the reduction in the solvent volume but also to the superior performance and process safety compared to the traditional processes.20 Indeed, as described by Pedersen et al., using a CSTR bypasses the solubility obstacle, providing an efficient reaction environment and less impurity formation.22,23
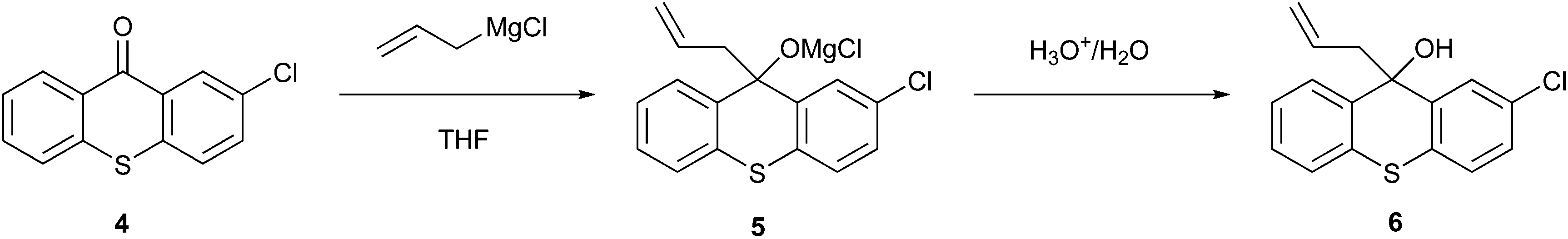 | ||
| Fig. 2 Synthesis of 9-allyl-2-chloro-9H-thioxanthen-9-ol via continuous processing.4 | ||
Hence, continuous processing is a promising approach for reaction optimisation and operational protection, improving the yield of reactions by obtaining cleaner products and avoiding purification steps, therefore improving the greenness of the process (see Principles 6, 8, and 12).
A tool to reduce waste during chemical reactions is the one-pot synthesis approach. Combining all the reagents has some advantages, including avoiding the purification and isolation of intermediates, thereby reducing the amount of solvents used. Consequently, one-pot synthesis can solve many problems in pharmaceutical and synthetic laboratories, among which the exposure time and fewer procedures to follow. Usually, these reactions are performed with the aid of ‘catalysts’.4 The direct α-amination of enolizable carbonyls (ketones, esters, and aldehydes, 7) using secondary aliphatic amines (8) is performed through Cu-catalysis, as reported by Evans et al. (Fig. 3).24 The one-step synthesis between these compounds develops an α-bromo carbonyl intermediate, which subsequently undergoes C–N bond formation, yielding the final product (9).4 This strategy features a green option for α-amination with water as sole benign by-product.24
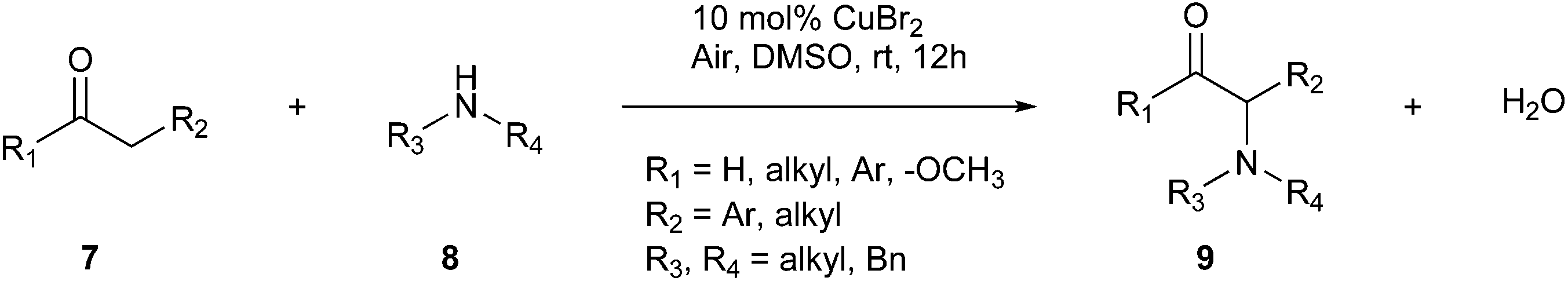 | ||
| Fig. 3 One-step synthesis between α-carbonyls and functionalised amines.4 | ||
1.1. Application of analytical methodologies to improve reaction greenness
Recently, the progress of analytical chemistry and the development of new analytical methodologies and procedures are strictly linked to environmental protection, aiming to design chemical products and project chemical processes that reduce or eliminate the use and generation of harmful substances. To achieve this, analytical chemists can use green chemistry practices, choosing or developing assessment methods that are efficient, generate minimal waste, and employ chemicals that are safe for humans and the environment. During the design of a process or product system, the design team has to consider the environmental impact of their decisions to minimise the waste at the source.9Chemists have devoted their efforts to developing more sustainable and energy-efficient alternatives to replace standard methods.25 Various examples of green analytical methods26,27 are being implemented throughout the analytical lifecycle. Nonetheless, the application of life cycle assessments to product design and development has not been critically addressed.9,28
A valuable and interesting example is the development of automated dilution for standard or sample preparations. The main benefit is that the operator can work with lower quantities of solvent and product to be analysed due to the highly diluted concentration, therefore reducing the exposure and the solvents used.9 The sample preparation technique is a critical method to be further improved by green analytical chemistry, which can definitely increase the green impact on laboratory practices.25,29 To accomplish waste reduction, and thereby the greenness of the process, the sample should be introduced with little or no preparation, reusing sample extraction devices and avoiding toxic organic solvents.25,30 In this case, many sample preparation strategies, such as Soxhlet extraction and liquid–liquid extraction (LLE), have been reconsidered because they do not fully or not at all align with the principles of green analytical chemistry. Here, we summarise some of the greener extraction procedures25 that are outlined in Principle 11.
Ultrasound-assisted extraction (UAE) is a fast extraction process that utilises ultrasound waves to agitate molecules.25 Pressurised fluid extraction (PFE) uses liquid solvents near their supercritical region under high pressure and temperature conditions.31 Compared with the commonly used Soxhlet extraction, these methods exhibit advantages such as a more efficient extraction and less time needed for the process (10 min vs. 16 h for Soxhlet).9 However, UAE and PFE still require a large amount of solvent and high-energy consumption. Solid-phase microextraction (SPME) is a non-exhaustive method that utilises solid material for the extraction phase, and the analytes are partitioned between the sample matrix and the extraction phase. This method utilises little or no solvent, requires little or no sample preparation, is a fast process and is consistent with pollution prevention.25,30
In this context, the term “green analytical chemistry” (GAC) was proposed by J. Namieśnik for the development of new analytic techniques to reduce their environmental impacts.32 A greenness evaluation method called Green Analytical Procedure Index (GAPI) has been developed to determine the agreement of analytical procedures with the Principles of Green Chemistry. Recently, GAPI has been updated to ComplexGAPI (Complementary Green Analytical Procedure Index).33 This evaluation, which considers the entirety of the methods analysed, allows a direct comparison of the greenness of various procedures, while relating the quality of information obtained by each technique simultaneously.25,34
GAC metrics will be further discussed under Principle 11.
1.2. Quantification of chemical waste
Many methods for quantifying chemical waste were developed to optimise waste prevention and reduction. The environmental factor (E-factor or mass efficiency) is one of the oldest parameters to evaluate the waste of a process. It is defined as the ratio between the mass (kg) of waste generated during a process and the mass (kg) of the desired product: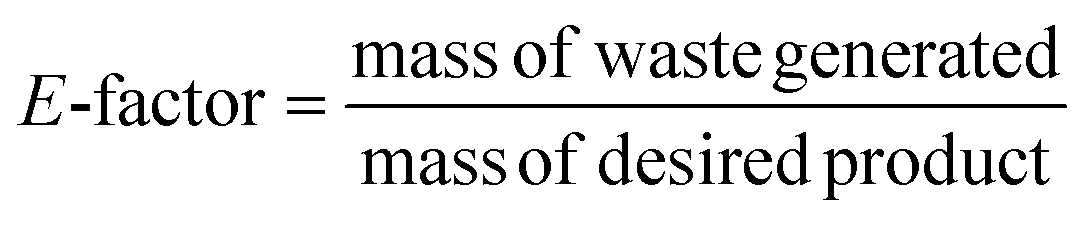
Each substance formed during the process that is not the desired product, such as inorganic salts, is considered waste. The higher the value, the more waste is generated. Lower E-factor values mean that the process produces less substances that are considered undesired waste, and thus the ideal value should be 0. Therefore, theoretical calculation of this value can prevent waste production beforehand.18 The complete E-factor (cEF) considers all the materials involved in the reaction process, such as solvents, starting materials, catalysts, water, and the final product, for a better assessment of the greenness of the process. On the contrary, the simple E-factor (sEF) does not consider water and solvents. Hence, the appropriate metric should be the average between these two values.20 Moreover, E-factor analysis is applied for calculating the waste production in the overall process, therefore providing a general overview aimed at developing cleaner and more sustainable processes.4
2. Atom economy
“Synthetic methods should be designed to maximise the incorporation of all materials used in the process into the final product”.4The second Principle of Green Chemistry analyses all the metrics available that measure the incorporation of as much material as possible into the final product to reduce waste and optimise the process. These metrics measure the performance of a general synthetic process, leading to more sustainable and greener practices.18 To be accepted worldwide, these sustainability metrics should be easy to quantify.35
The concept of atom economy was developed in the last few years as the continuation of common metrics such as product yield,36 which is defined as the ratio between the molecular weight of the final product and the total molecular weight of all the reactants:
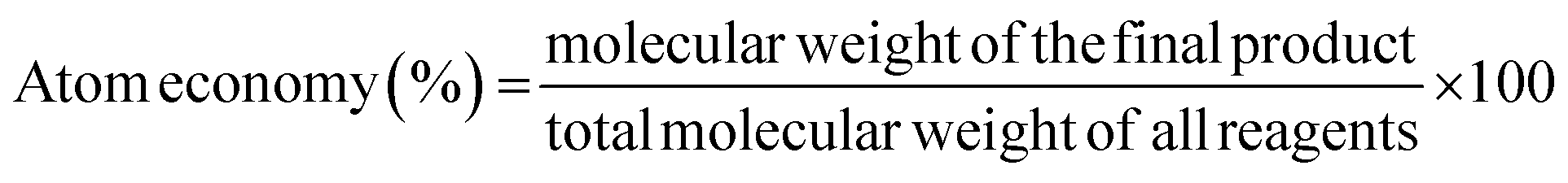
Although AE considers all atoms involved in the formation of the desired product, it does not consider any other substance used in the process, such as solvents and catalysts, and it is engaged only for an individual reaction step.37,38 Since chemists often have to perform many reaction steps, a step-economy strategy can be applied to improve the overall efficiency of the process. It involves minimising the number of reaction steps, thereby allowing chemists to consider a minor number of syntheses to be greener. Computer-aided organic synthesis (CAOS) is a useful computational tool that can not only improve the atom- and step-economy but also optimise the underlying mechanism of the reaction. CAOS works using a retrosynthetic approach to avoid generating unwanted products and improve the reaction networks.4,39
AE is a theoretical value that can be exploited without experimentations, allowing the calculation of the theoretical yield of a product from stoichiometric amounts of reagents.18 For instance, Fig. 4 illustrates two examples of theoretical calculation of AE, in which the reagents (10–12, 14, and 15) are totally converted into the products (13 and 16). Therefore, AE can be a predictive factor of the greenness of a process, evaluating beforehand the waste produced and predicting the efficiency of a chemical process in terms of atoms involved. The ideal AE value is 100% (Fig. 4A).20
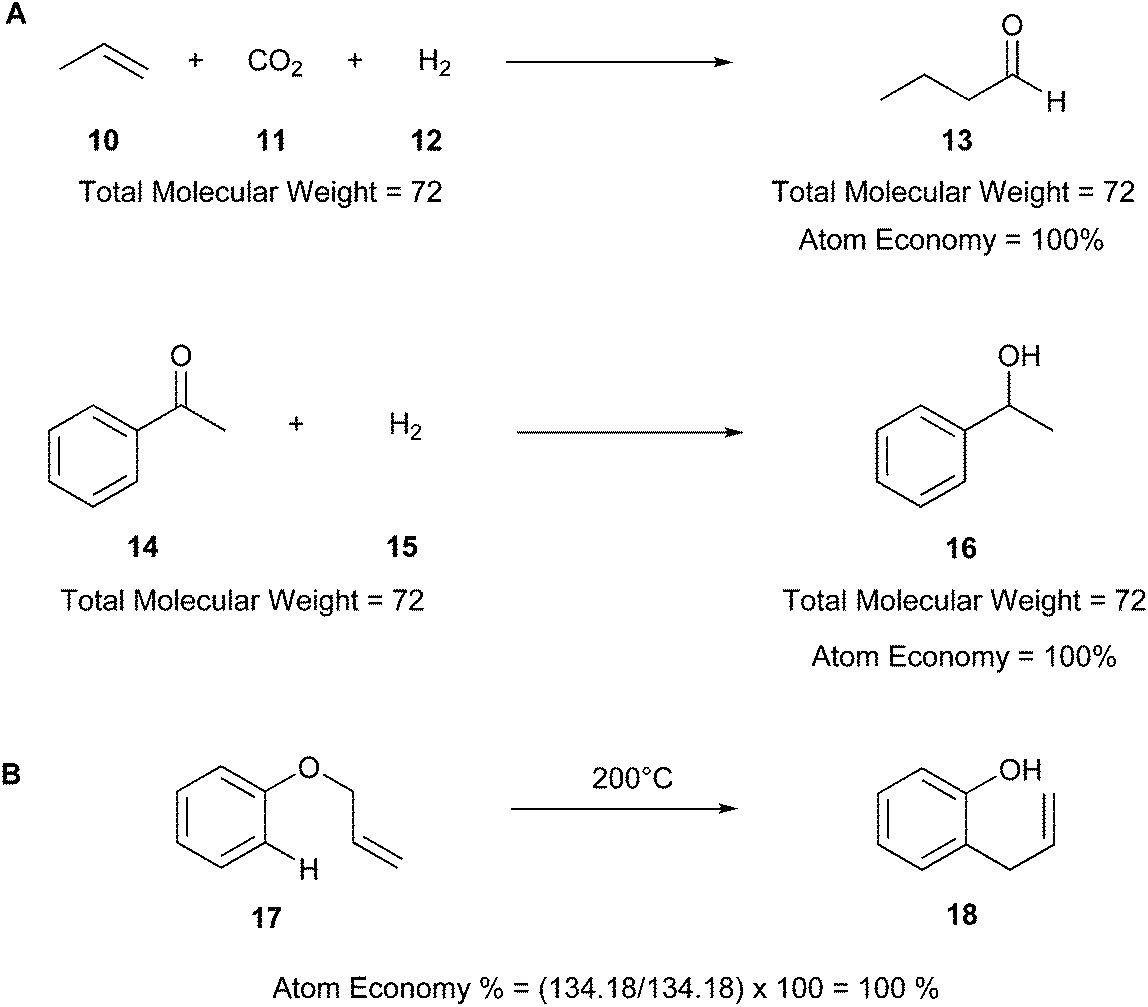 | ||
| Fig. 4 (A) Examples of theoretical calculation of atom economy.20 (B) Atom economy of the Claisen rearrangement reaction.40 | ||
Other examples of reactions with high atom economy include olefin metathesis,41,42 Claisen rearrangement (17 and 18) (Fig. 4B),40 Curtius rearrangement,43 and coupling reactions such as the Suzuki–Miyaura cross-coupling,44 Buchner reaction,45 and Diels–Alder cycloaddition.4,46
Together AE, it is useful to consider another metric, i.e., the reaction mass efficiency (RME), which is a mass balance between the isolated product and the mass of all reagents, as follows:
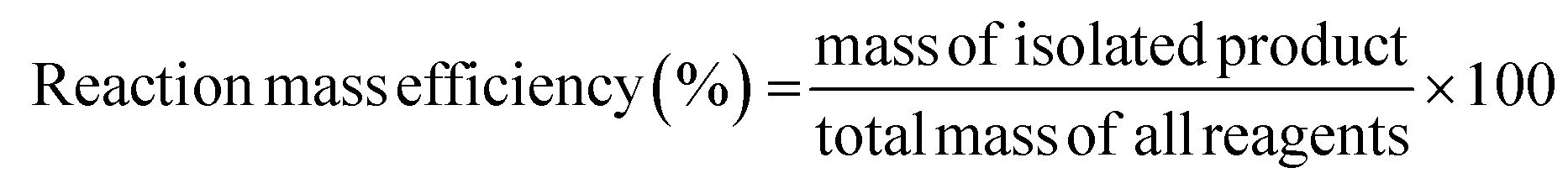
Using this metric, it is possible to evaluate the observed efficiency of the utilisation of all reagents. Similar to AE, RME does not consider the mass of solvents used, and it is particularly useful in research laboratories because working on the mg scale, the efficiency of the process can be masked by the considerable solvent contribution.37
2.1. Green aspiration level (GAL)
The next level of atom economy is the Green Aspiration Level™ (GAL) metric,47 which considers the intricacy of an ideal synthetic route, while simultaneously contemplating the environmental impact of the produced pharmaceutical agent. It represents the first substantial green efficiency goal for any build-up process by defining and quantifying the expected environmental impact, therefore considering the waste reduction and the greenness of the process.48 The GAL measures the green status of a synthetic route through the engagement of the Relative Process Greenness (RPG) metric, the value of which should be greater than 100% for achieving the process greenness goal. Values lower than 100% should be revisited to optimise the process.The beneficial advantage of this metric is that chemists can compare3 the environmental impact between more synthetic routes implicated in manufacturing different pharmaceutical products. The comparison can be done not only at different stages of production but also with commercialised processes. GAL allows the comparison of the production of a new potential drug as well as of an ideal process to that of an existing similar drug to assess the waste (measured with Complete Environmental Impact Factor (cEF) or Process Mass Intensity (PMI)) and the complexity of the process, using the RPG metric. Hence, a chemist can easily compare new technologies with already studied pharmaceutical processes instead of calculating the different and isolated metrics described above.47,48
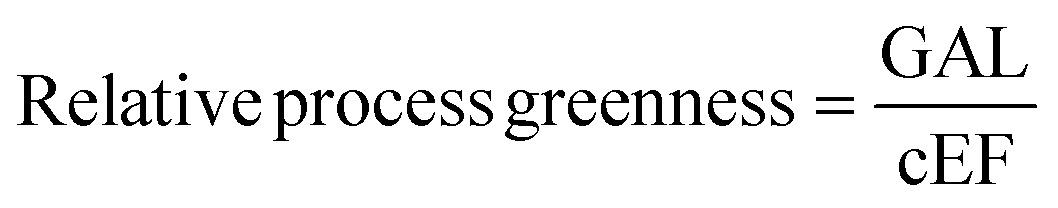
There are two other useful metrics for the greenness quantification of a process: i.e., relative (green) process improvement (RPI), which quantifies the global process progress, and relative (process) complexity improvement (RCI), which is considered in the estimation of the process complexity.20 To evaluate the improvement of a process, RPI can be calculated as the difference between consecutive RPG values, from early development to late development, as follows:
| RPI = RPG(current process) − RPG(early process) |
The higher the value, the better is the process profile. Moreover, RPG values are associated with a percentage that defines the greenness of a process (Table 1).
| Percentile | Colour code | Rating | Minimum RPG for | ||
|---|---|---|---|---|---|
| Early dev | Late dev | Commercial | |||
| 90% |

|
Excellent | 109% | 109% | 248% |
| 70% |

|
Good | 76% | 76% | 197% |
| 40% |

|
Average | 40% | 67% | 93% |

|
Below average | 0% | 0% | 0% | |
The GAL metric can be calculated as follows:
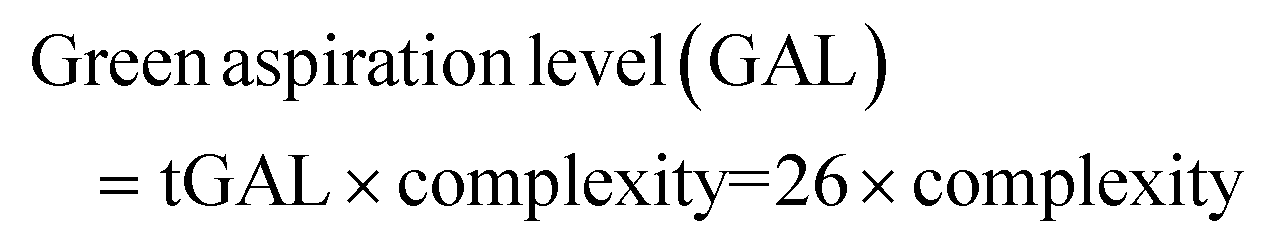
2.2. Process mass intensity (PMI)
The process mass intensity (PMI) is a mass-based green metric that evaluates the efficiency of a process, which can be calculated as follows: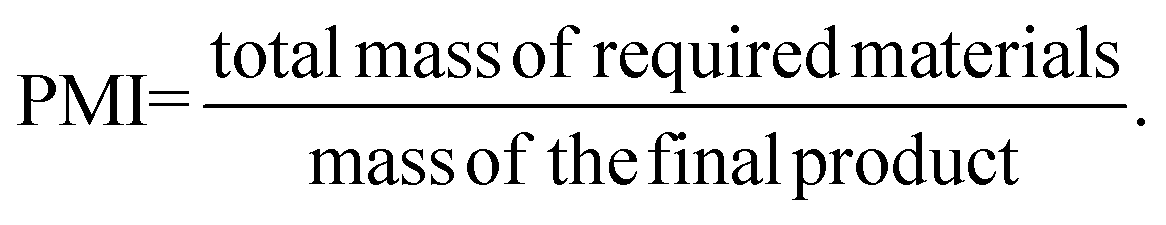
PMI considers all the materials used in the synthetic route, including solvents, co-solvents, reagents, colouring, bulking agents, and surfactants. Accordingly, PMI gives a more precise estimation of the greenness of the process, allowing chemists to transform pharmaceutical synthesis into sustainable, efficient, green ones.18,50 Although PMI can reduce the environmental impact through resource utilisation and optimisation, some reagents may often be more hazardous than the waste they may create.51 Hence, it is necessary to wisely choose the reactants, and when the chemist has no experience, a dataset of multi-kilo scaled-up reactions can be used. The major pharmaceutical and biotech companies, including AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Merck, Novartis, Pfizer, and Roche, created this dataset, containing almost two thousand reactions, to help chemists to choose the best retrosynthetic pathways for product manufacture. This PMI prediction calculator considers the greenness of the components needed for the reactions using PMI historical information and predictive data and helps discriminate among probable efficiencies of plausible routes. Moreover, PMI calculation can provide a useful comparison between different synthetic routes and optimise a proposed synthetic step, always considering the waste management strategies.
This tool can be used to predict the PMI value, virtually screen the available dataset, or at the end of a chemical process, evaluate its greenness.52
Measuring PMI has some advantages, including the ease of use,53 the evaluation of improvements towards more sustainable and green processes, and the tracking of the environmental footprint left by manufacturing processes. Nonetheless, PMI does not measure the possible toxicity and effects of the materials used in the process. Thus, it can be associated with other metrics such as the F-factor (function-factor), which compares different products or different synthetic routes based on the functionality of the products.4
3. Less hazardous chemical syntheses
“Wherever practicable, synthetic methods should be designed to use and generate substances that possess little or no toxicity to human health and the environment”.4Principle 3 merges the previous two green chemistry Principles regarding the rational molecular design of safer chemicals. Using less or non-toxic reagents/substances in the synthetic process prevents waste production, which can be achieved through the aid of the green chemistry metrics discussed above. When chemists project the synthetic route of an API, they should consider the whole life cycle of the future molecule to reduce not only waste, and consequently its environmental impact but also its toxic effects on human health. Designing safer chemicals could play a pivotal role in guaranteeing the effectiveness of the newly produced molecule, which can achieve the desired function, hopefully without toxicity. Thus, the third Principle of Green Chemistry is closely correlated with the fourth one. Utilising all the available information, such as mechanistic studies, empirical methods, hazard data, and computational toxicity models, as well as environmental chemistry and toxicology, chemists can prioritise safety from the beginning of the process.4,18 Moreover, the Environmental Assessment Tool for Organic Synthesis (EATOS) metric has been developed, which quantifies not only the reactants and waste but also their relative toxicity effects.4,54
Another important topic of this Principle is the transformation of waste into energy, fuels, and other reusable materials, always considering the reduction of the impacts on human health and the environment. This “circular economy” is a part of the rational design chemists should perform beforehand because if they create a waste stream that is also reusable as a starting material, they can prevent waste generation (Principle 1).18 However, this circularity has not been realised to date due to the lack of systems to be designed.
Why is achieving this circularity beneficial? It is known that many drugs and APIs, such as antibiotics,55 diclofenac,56 propranolol,57 and gemfibrozil,58 are non-degradable, and thus persist in the environment or bioaccumulate in animals, ultimately having a negative impact on the ecosystem.18 For example, diclofenac has been detected in high amounts in wastewater, affecting the general health of fishes, and consequently human health.56 Therefore, it is imperative to consider the whole product life cycle, including safe degradation and recycling.
In this regard, it is wise to integrate the mechanisms by which active pharmaceutical ingredients can easily degrade once introduced in the environment. Jordan and Gathergood59 as well as Stolte et al.60 worked in the field of biodegradation, especially studying the biodegradation and properties of ionic liquids (ILs). ILs are salts, formed by an anionic and a cationic species, which have a melting point below 100 °C, and therefore liquid and ionic simultaneously. They can be used as solvents and have very good catalytic properties, suggesting their applicability in the area of green chemistry.60 Using ILs in their studies, Gathergood and colleagues61 analysed and predicted biodegradation, also evaluating the various metabolites produced in the process, applying some important rules to improve the biodegradability59 given that API persistence in the environment should be avoided. The so-called “rules of thumb” by Boethling et al.62,63 include some beneficial structural features, such as aromatic rings, long non-substituted alkyl chains, and hydrolysable groups such as alcohols, aldehydes and carboxylic acids, which are related to favorable biodegradation properties.64,65 These important rules will be further discussed under Principle 10.
The so-called “benign by design”66 is the main concept from which the design and synthesis of new molecules should start, and consequently it is necessary to include biodegradation, sustainability, toxicity and life cycle assessment in design criteria to produce safer compounds with a reduced impact on the ecosystem.67 Recently, the GREENER approach has been proposed for developing new APIs with reduced environmental impact following patient use (Fig. 5).68
 | ||
| Fig. 5 GREENER concept for developing new APIs with lower impact on the environment after patient use.68 | ||
This guide provides very useful tips for the design and development of new potential drugs. In particular, it considers the physicochemical properties and solubility of the new molecule, favoring oral administration, and it offers a valuable tool to avoid undesirable functional groups that are known to be toxic and persistent in the environment, such as polyfluorinated moieties. However, several of these moieties are sometimes essential for the pharmacological effect of the potential drug candidate. Therefore, medicinal chemists should consider drug safety and predict the risk to patients, as well as the environmental concerns.69 An interesting example is the difference in the biodegradability of the anti-tumour agents 5-fluorouracil, cytarabine and gemcitabine. These antimetabolites bear the same pyrimidine core scaffold, but cytarabine and gemcitabine contain the sugar arabinose connected to the main ring, which is the moiety favouring their biodegradability. As reported by Kümmerer et al.,70 5-FU and gemcitabine are less degraded due to the presence of fluorine in their pyrimidine ring (for 5-FU) and sugar (in the case of the gemcitabine).
Furthermore, the GREENER concept allows medicinal chemists to incorporate environmental criteria even at early stages of drug development. In this case, medicinal chemists should not be alone in the design of new APIs, where cross-talk with environmental experts is needed to implement the GREENER concept and reach a more sustainable synthetic strategy.68
3.1. Reagent guides to improve greenness
To date, many efforts have been devoted to reducing or eliminating the hazardousness of chemical intermediates or final products. The pharmaceutical companies Pfizer and GSK developed the Reagent Selection Guide as a valuable tool that provides suitable instruments for medicinal chemists to make the best choices for synthesising a product.10,71 If there is a problematic reagent, this guide allows chemists to change it following precise criteria, as depicted in the Venn diagram in Fig. 6.72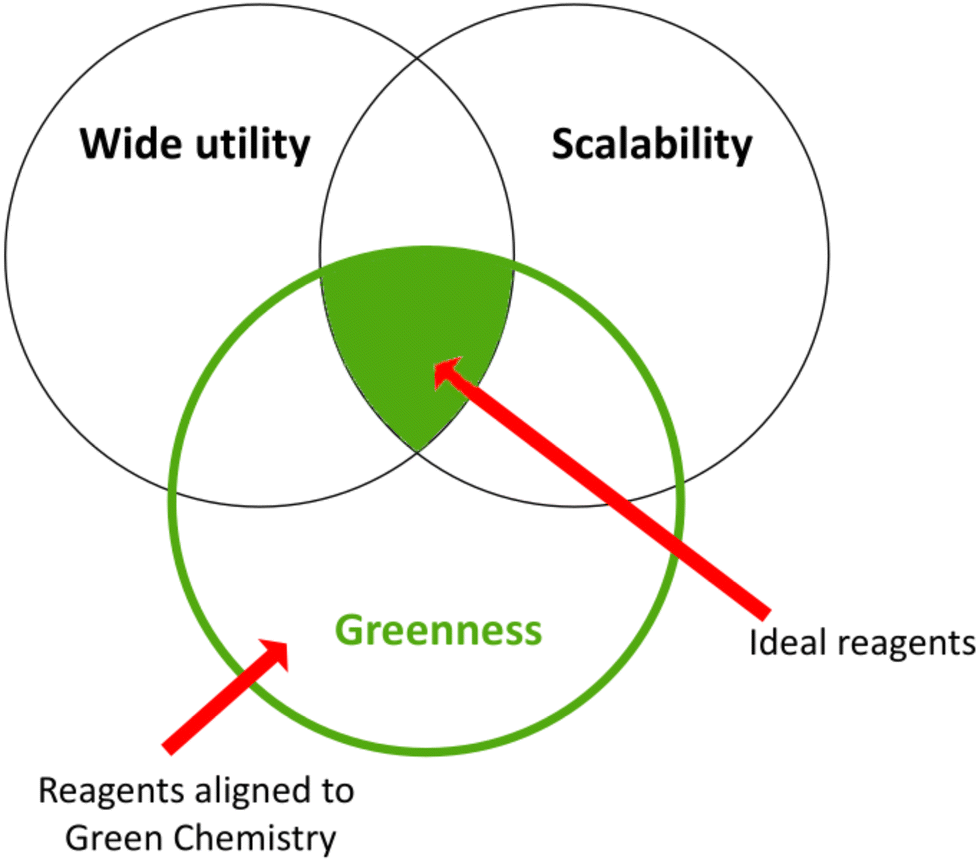 | ||
| Fig. 6 Green criteria for various syntheses: Venn diagram.72 | ||
“Greenness”, “scalability”, and “wide utility” are the criteria employed to rank the well-established reaction conditions. The green area in the middle represents ideal reagents possessing all three characteristics, whereas other reagents can have either two characteristics in common and not the third or only one feature. The placement in every circle is determined by various factors such as solvents, catalysts, and waste management. Furthermore, in the online version of this guide, each reagent is linked to the respective reference with the key literature to raise awareness of newer emerging green methodologies.10 Later on, the guide was donated to the ACS Green Chemistry Institute Pharmaceutical Roundtable (ACS GCIPR) for further development and expansion.73
An example of the Reagent Selection Guide provided by Pfizer is the oxidation of secondary alcohols to ketones (Fig. 7).
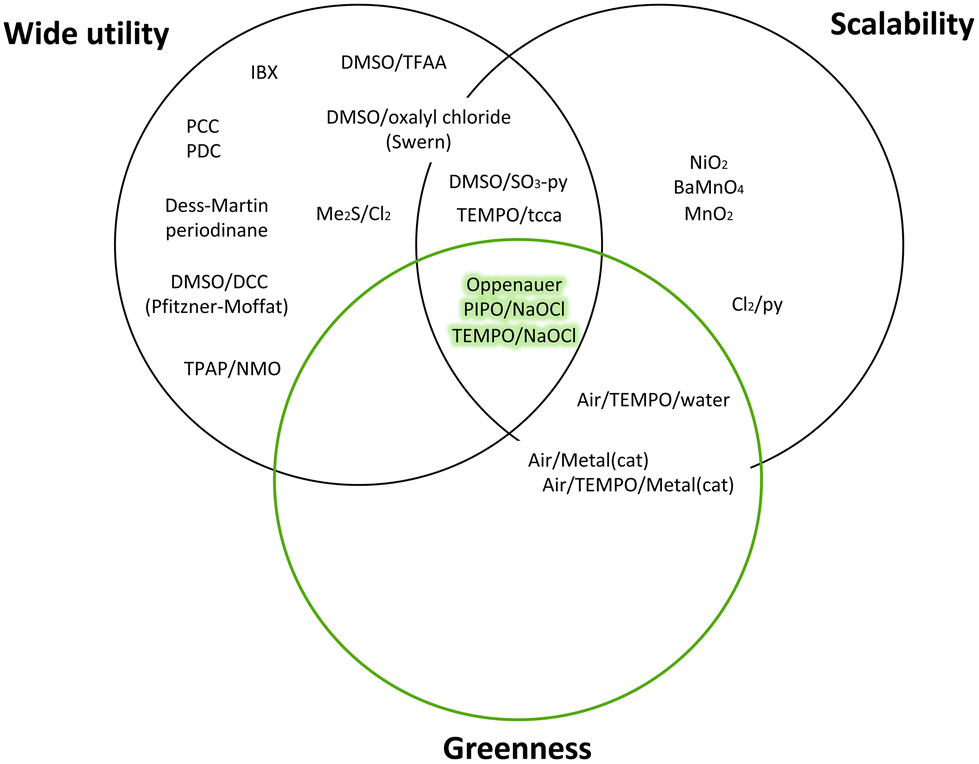 | ||
| Fig. 7 Venn diagram of oxidation of secondary alcohols to ketones following Pfizer reagent guide.10 | ||
As shown in Fig. 8, a green choice for this transformation is the Oppenauer oxidation.74 Although this reaction was discovered in 1937, it is a very green option given that it uses stoichiometric amounts of aluminium catalyst as a base and isopropanol (22) is the only by-product (Fig. 8).
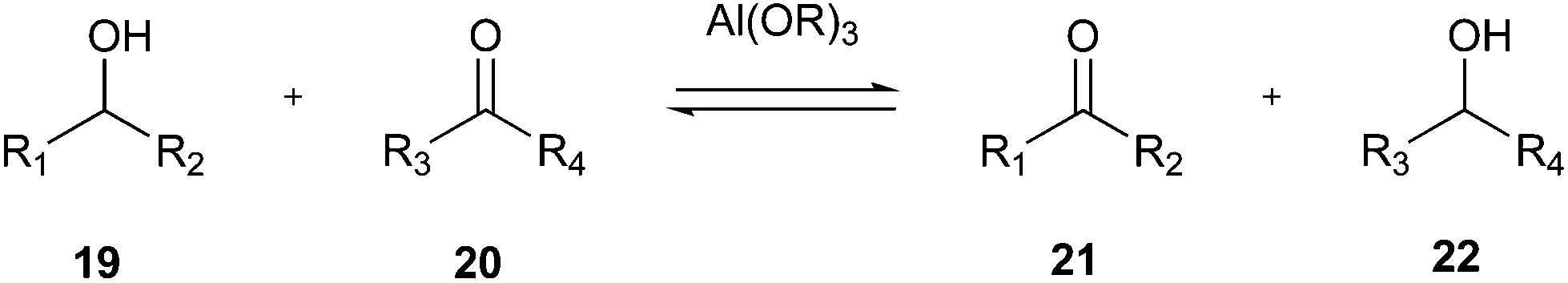 | ||
| Fig. 8 General scheme for Oppenauer oxidation of secondary alcohols.74 | ||
Alternative green methods for this oxidation can be carried out via NaOCl using more environmentally friendly nitroxyl radical catalysts such as TEMPO75 and PIPO.76 Alternatively, although the most common oxidants such as Dess–Martin periodinane,77 Swern oxidation and tetrapropylammonium perruthenate (TPAP),78 have been widely used by Pfizer's medicinal chemists, they have very poor atom economy, and therefore they cannot be used for large-scale reactions.10
A parallel approach was adopted by the pharmaceutical company GSK, which developed a reagent guide covering the 15 most common reactions in drug discovery and development including alkene reduction, amide formation, C–H bromination, C–H chlorination, deoxychlorination, epoxidation, ester formation, ether formation, fluorination, iodination, ketone reduction, nitro reduction, oxidation of alcohols to aldehydes and ketones, reductive amination and sulfur oxidation.71 The aim of this guide is to embed sustainability into everyday practice and provide a greener alternative to the most commonly used reagents. For each transformation, each reagent was assessed based on a number of criteria ranging from 1 (most issues) to 10 (least issues) to provide an Environment, Health and Safety (EHS) score, a Chemistry score (which includes stoichiometry, work-up, considerations of atom efficiency and other issues) and a resulting overall Greenness score. Each of these scores found in the guide resulted from the combination of every variable and chemistry factor considered for the overall score. Notably, a reagent with an overall Greenness score below 4 was scored as “red”, if the overall Greenness value was between 4 and 6 to 7.3 the reagent was classified as “yellow”, and if the Greenness score was above 7.3, the reagent was assessed as “green”. Consequently, to ensure that this guide is readily comprehensible, every reagent is therefore associated with a colour, which stands for few issues (green), some issues (yellow) and major issues (red). An example of this GSK reagent guide is shown in Fig. 9.
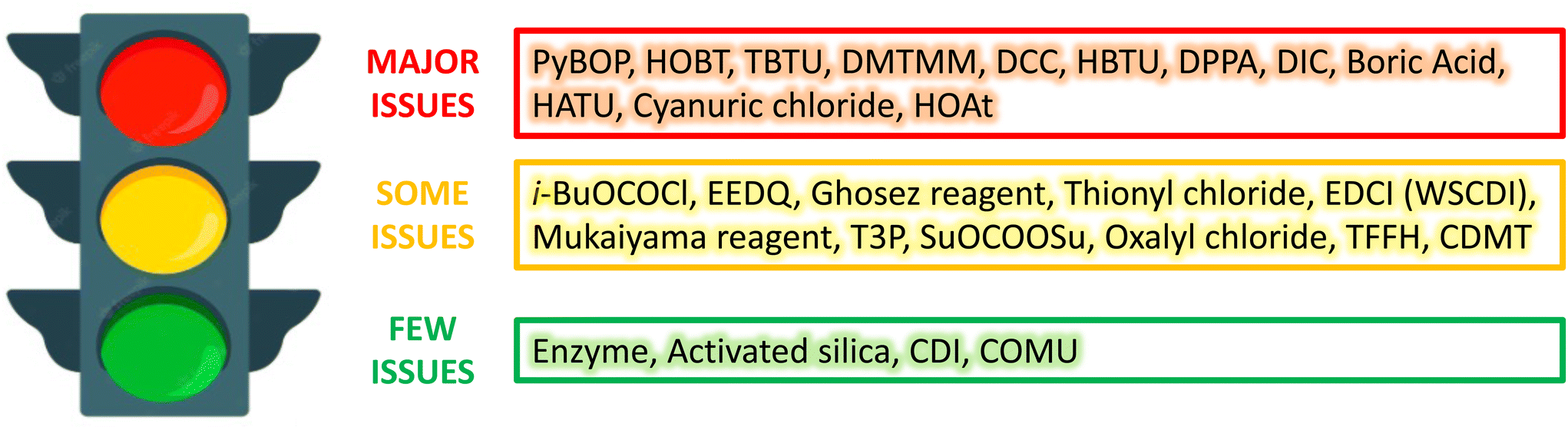 | ||
| Fig. 9 Amide formation following the GSK reagent guide proposed by Adams et al.71 | ||
Hence, good green chemistry requires chemists to consider all available information (reagent guide, safety issues, toxicological profile, atom economy, and efficiency) in the design of synthesis protocols, also choosing the right conditions for a greenness process during construction. Nevertheless, if a green reagent gives a lower yield or results in a longer-step synthesis than a less green reagent, the choice will be the one that provides more benefits, if it is not the greener one.
Here, we present some examples of reactions performed utilising the reagent guide provided by the ACS Pharmaceutical Roundtable.73 In this case, slightly changing a parameter can influence the efficiency and sustainability.18
The high environmental impact of the Suzuki reaction is due to the requirement of undesirable solvents such as N,N-dimethylformamide (DMF) and 1,4-dioxane and the disposal of the palladium catalyst, a high carbon footprint metal, which contributes to the total greenhouse gas (GHG) emission due to its extraction and purification requirements.79
Accordingly, various options have been developed to make the Suzuki reaction greener, for example, using alternative catalysts such as Pd/C, from which Pd can be efficiently recovered, minimising waste production and use of base metals;80 employing greener organic solvents like 2-methyltetrahydrofuran (2-MeTHF) and cyclopentyl methyl ether (CPME), or aqueous systems;81 performing the coupling in flow reactors and one-pot in situ borylation by C–H activation. Regarding the last option, C–H activation occurs via an iridium catalyst (25), thereby in situ forming the boron coupling partner (27). Although iridium is less sustainable than palladium, according to the periodic table of element sustainability,82 various studies demonstrated the advantages of iridium-catalysed C–H borylation. Indeed, Ir borylation is selective, therefore suggesting that Ir by-products may not interfere with subsequent in situ reactions, as in the case of Suzuki–Miyaura coupling,83 and it is an atom economical route.84 Subsequently, the reaction follows the usual Suzuki mechanism via an aryl/heteroaryl halide/Pd catalyst (Fig. 10).85
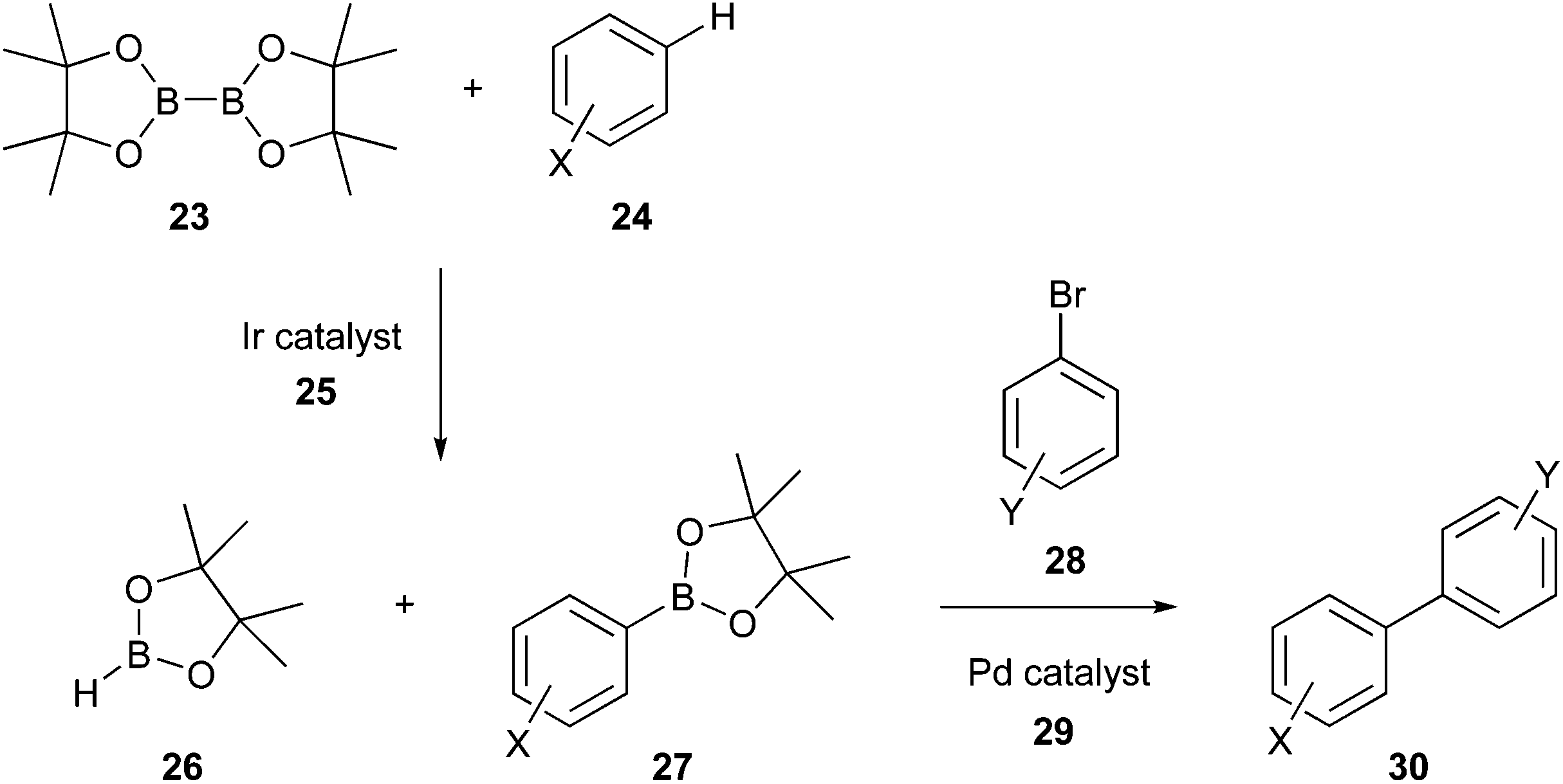 | ||
| Fig. 10 ‘One-pot’ in situ borylation by C–H activation for greener Suzuki process.85 | ||
The intermediate tert-butyl cation 32, which can fragment into potential genotoxic substances (isobutylene, 38), can also react with alkylating nucleophilic sites/substances present in the molecule/reaction environment, leading to the formation of various by-products.87 This alkylation can be avoided by adding scavengers to the reaction (Fig. 11A).88
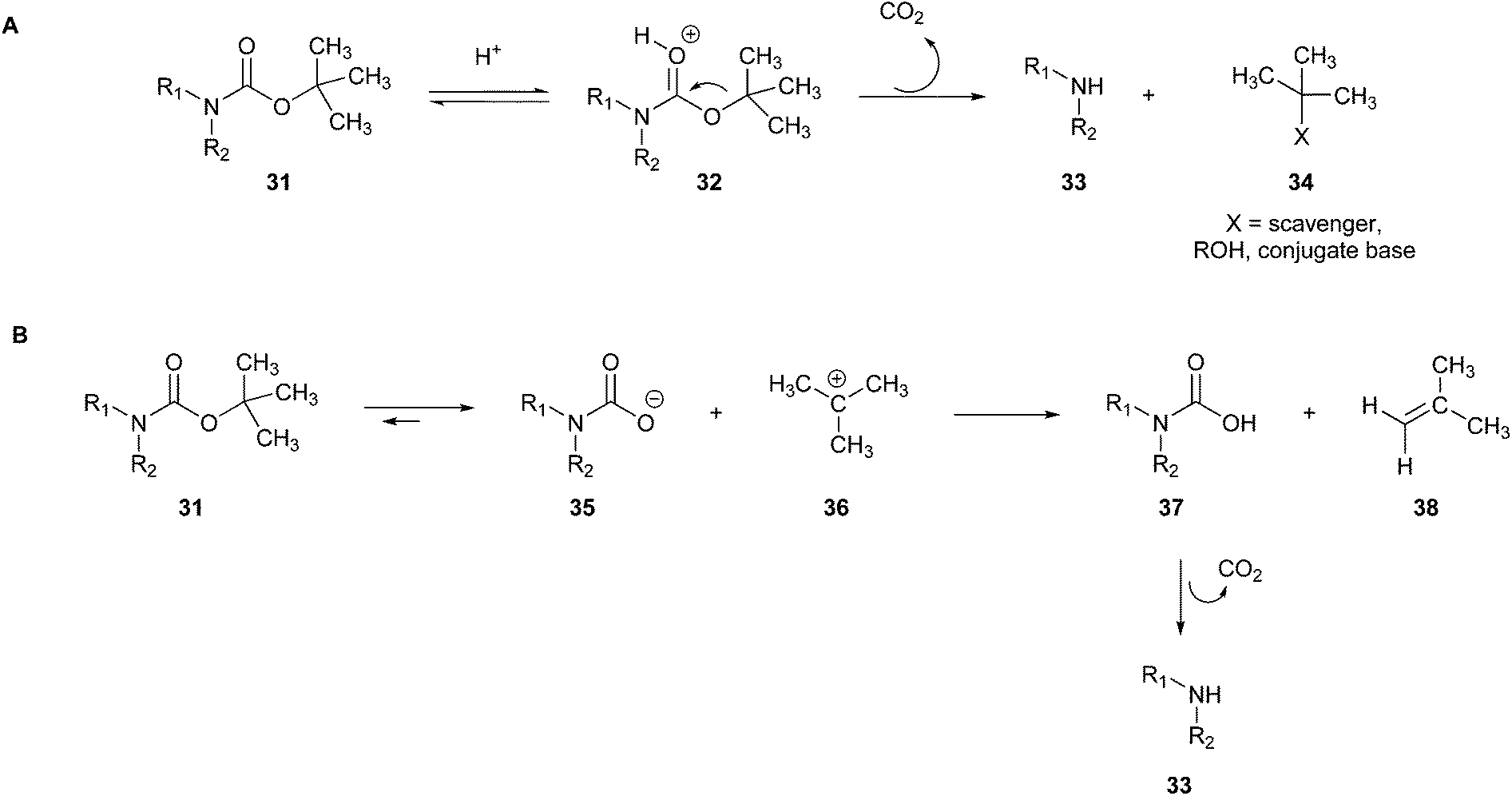 | ||
| Fig. 11 (A) BOC deprotection mechanism with aid of a scavenger.88 (B) Thermal method for BOC deprotection.89 | ||
Moreover, the use of TFA should be avoided because of its toxic and corrosive properties, as well as its environmental persistence. Changing the solvents is also a greener option, for example, replacing dichloromethane or dioxane with ethers such as THF and 2-MeTHF can reduce the aquatic environmental impact of dichloromethane and the hazardousness to human health of the carcinogen 1,4-dioxane, but unfortunately no other green alternative route for this type of deprotection is available to date.89
Another tool for making the process greener is the thermal method, which uses high-temperature heat without any added catalyst (Fig. 11B).89
The mechanism provides the generation of the amine through the previous formation of carbamic acid (37), which is subsequently transformed into the deprotected amine (33) through the release of CO2 and isobutylene (38). Although BOC removal can be very rapid depending on the protected amine especially when using catalysts, in an attempt to make the process greener, a method has been attempted by just heating the reaction vessel. On one hand, this procedure is more ecologically friendly given that it reduces the number of toxic reagents used; on the other hand, it can be slower than the commonly known reaction procedures consuming more energy, and thus this procedure is not yet fully green.89 Hence, greener proposals have to be further developed.
3.2. Towards greener reactions
Green synthetic chemistry is an emerging field of importance; thus, knowledge of different chemical reactions plays a pivotal role in designing greener synthetic routes for both starting materials and APIs.20 Here, we summarise some of the most important reactions that are becoming greener in the last few years.The amide bond is widespread among pharmaceutical compounds and is usually formed via acid chlorides. Recently, other synthetic pathways, which are less efficient and with poor atom economy, such as the activation of carboxylic acids with the aid of coupling reagents90 and the microwave-assisted pathway,91 have been considered to be further improved in terms of green chemistry with respect to sustainability principles.
One-pot synthesis is a valuable strategy to reduce waste and reaction time, which it avoids the intermediate purification issues and increases the chemical yield, according to Principles 1 and 2.92,93
Multicomponent reactions (MCRs) are one-pot syntheses, which provide highly regio- and stereoselective chemical reactions among three or more substrates. The resulting product contains all or almost all the atoms of the starting materials,94 ensuring high atom economy and waste reduction; then, they can be classified as green and sustainable processes to be utilised in the synthesis of pharmaceuticals.20,95 Among the classical MCRs that follow the major Principles of Green Chemistry, we discuss some of them, including the Mannich96,97 Hantzsch98,99 and Gewald100,101 reactions.20 Moreover, we present two examples of established APIs in which the synthetic route was redesigned entirely, including the majority of green chemistry principles.
Lu and colleagues97 implemented a greener Mannich reaction under solvent-free conditions at room temperature (Fig. 12). Benzaldehyde (39), acetophenone (40), and aniline (41) reacted in one-pot to obtain the desired amide derivative 1,3-diphenyl-3- (phenylamino)propan-1-one (42).
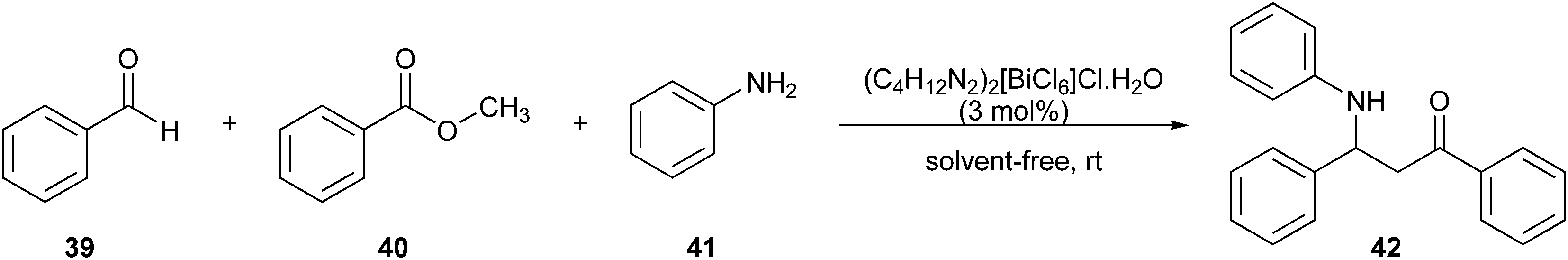 | ||
| Fig. 12 Application of the Mannich reaction in the synthesis of amide derivatives.97 | ||
The reaction was performed using the (C4H12N2)2[BiCl6]Cl·H2O catalyst, which can be reused without loss of efficiency and recycled. Hence, the reaction provided a reduction of hazard and solvent waste and improved atom economy.20
Debache et al. reported a greener one-pot Hantzsch reaction, which was shown to be a more efficient and inexpensive way to obtain 1,4-DHPs (46), yielded by the reaction of aromatic aldehyde (43), ethyl acetoacetate (44), and ammonium acetate (45), using phenylboronic acid as the catalyst (Fig. 13).102
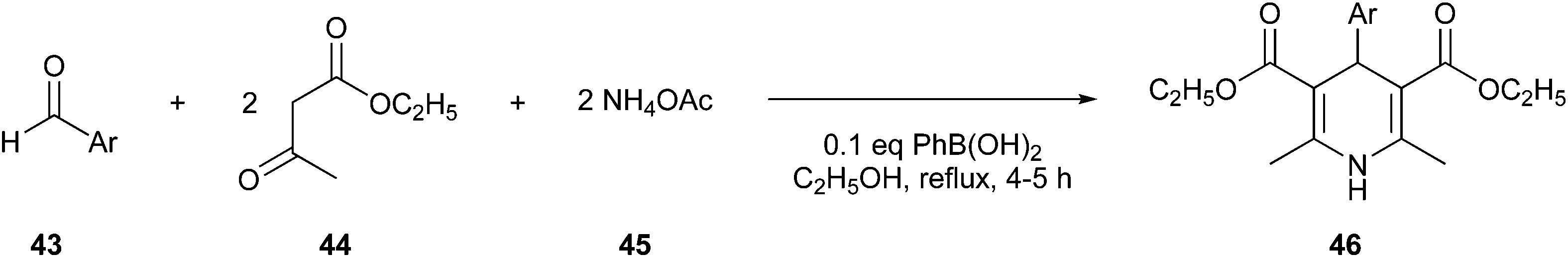 | ||
| Fig. 13 Synthesis of 1,4-DHPs employing phenylboronic acid as a catalyst.102 | ||
Compounds with this scaffold show various bioactive characteristics, including anti-inflammatory, anticonvulsant, and antimicrobial profiles.100
Ma et al. developed a greener Gewald reaction using N-methylpiperazine-functionalized polyacrylonitrile fibres as a catalyst, which could be recycled without loss of catalytic efficiency. Specifically, 2,5-dihydroxy-1,4-dithiane (47) reacts with activated nitriles (48) to synthesize 3-substituted 2-aminothiophenes (49) (Fig. 14).101
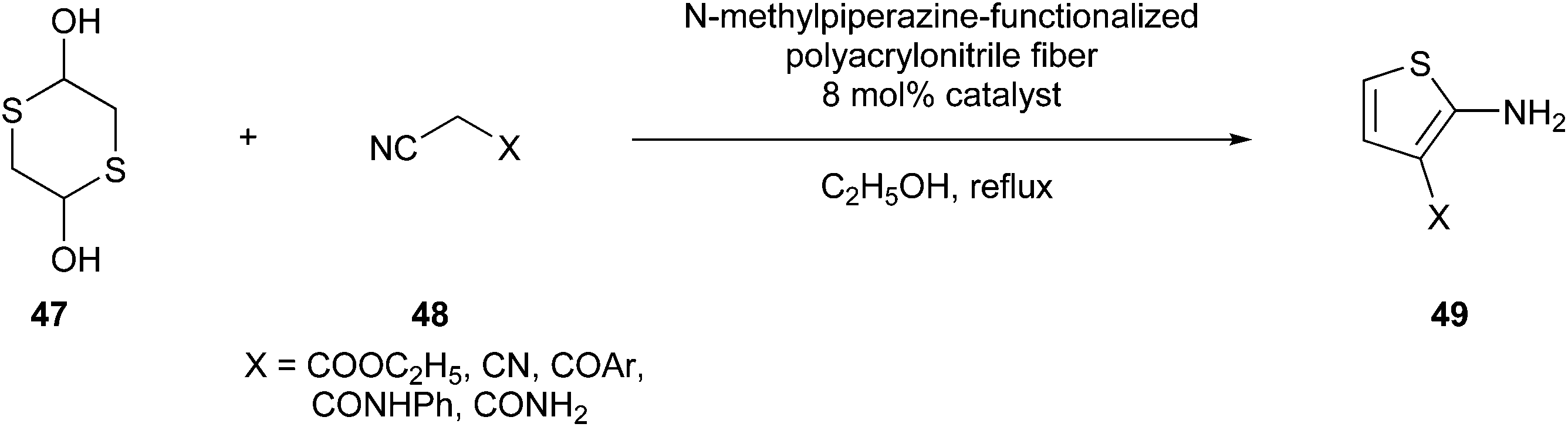 | ||
| Fig. 14 N-Methylpiperazine-functionalized polyacrylonitrile fibre-catalysed Gewald reaction in the synthesis of 2-aminothiophenes.101 | ||
This procedure produced excellent yields of the desired compound with high atom economy and almost no waste generated.20
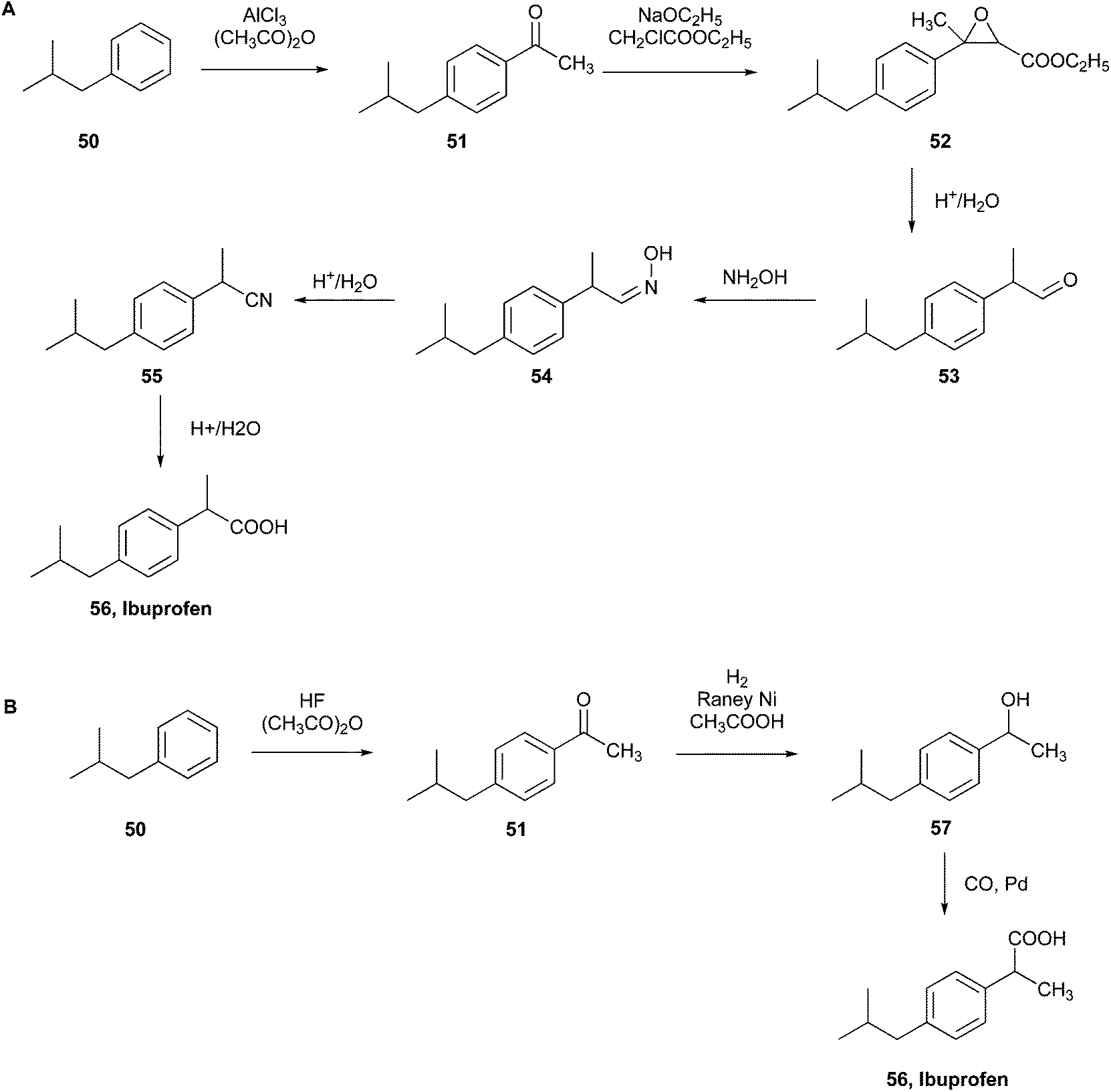 | ||
| Fig. 15 (A) Boots initial commercial synthesis of ibuprofen.103 (B) BHC Company new commercial green synthesis of ibuprofen.104 | ||
According to their patent, the synthesis started with the Friedel–Crafts acetylation of 2-methylpropylbenzene (50) in the presence of AlCl3 and acetic anhydride to obtain 1-(4-isobutylphenyl)-ethenone (51), which was employed in a Darzens reaction with ethyl chloroacetate to give the α,β-epoxy ester (52). In the third step, this intermediate was decarboxylated and hydrolysed to obtain 2-(4-isobutylphenyl)propanal (53), which subsequently reacted with hydroxylamine giving the 2-(4-isobutylphenyl)propanal oxime (54). The last two-step hydrolysis produced the penultimate product 2-(4-isobutylphenyl)propanenitrile (55) and the final product ibuprofen (56), respectively.
After the publication of this patent, the Boots–Hoechst–Celanese (BHC) Company carried out a greener ibuprofen synthesis comprised of only three steps instead of the previous six (Fig. 15B).
Employing the same starting compound, 2-methylpropylbenzene (50), Friedel–Crafts acetylation was performed, producing 1-(4-isobutylphenyl)ethenone (51) using anhydrous hydrogen fluoride as the catalyst instead of AlCl3. The second step was hydrogenation with RANEY® nickel to obtain the alcohol 1-(4-isobutylphenyl)ethanol (57), and the following carbonylation, using cobalt and palladium catalysts, generated ibuprofen (56).
This reaction was performed by recycling and recovering the waste by-products produced, and it demonstrated high atom utilisation (77%) compared to the 40% of the previous six-step process.104 Consequently, it was recognised by the US EPA as a green synthesis model, allowing BHC to receive the Presidential Green Chemistry Challenge Award in 1997.20
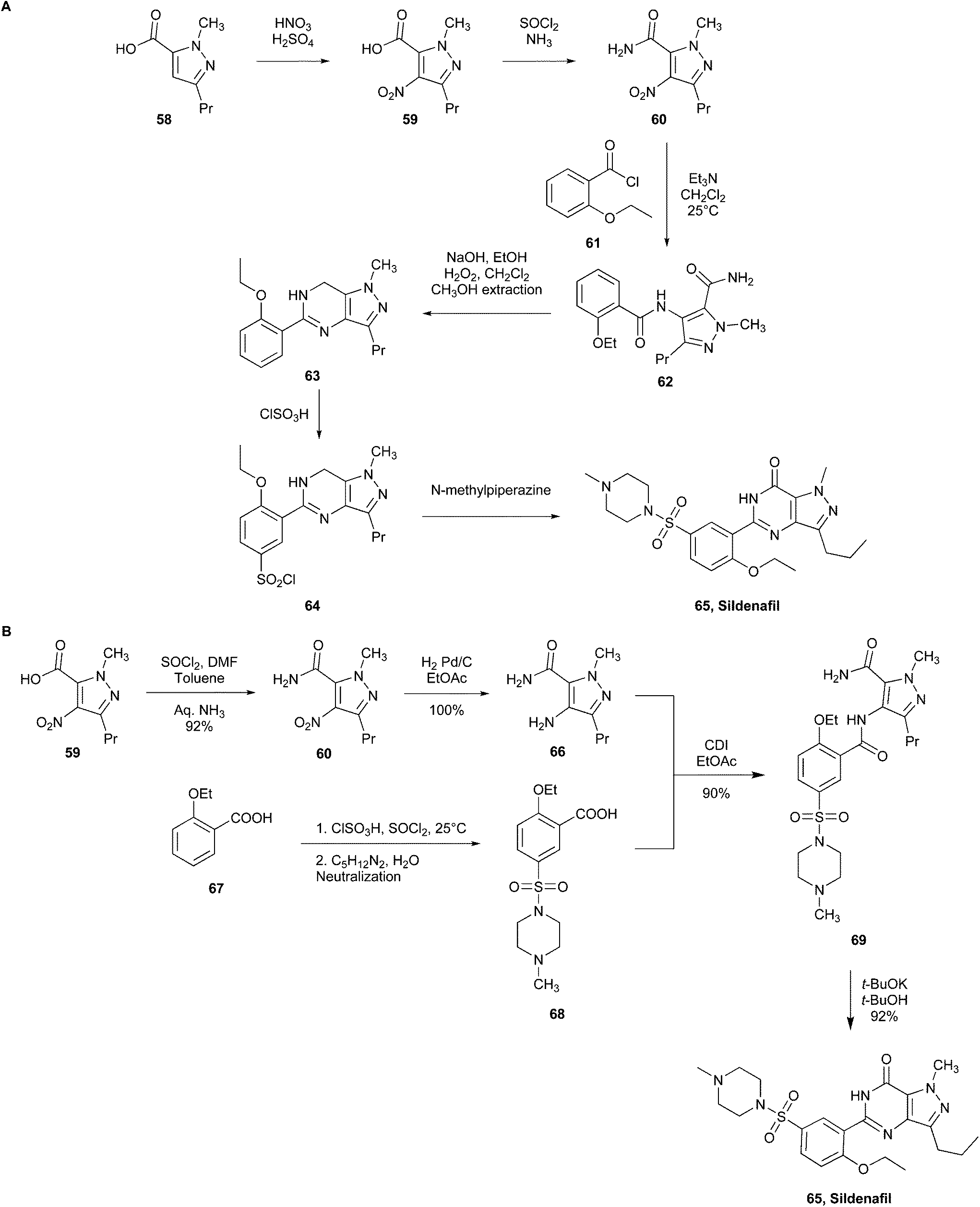 | ||
| Fig. 16 (A) Pfizer's initial commercial synthesis of sildenafil. (B) Pfizer's new commercial green synthesis of sildenafil.105 | ||
Various conditions in the synthetic steps were modified (Fig. 16B)105 to obtain 1-methyl-4-nitro-3-propyl-1H-pyrazole-5-carboxamide (60) from 1-methyl-4-nitro-3-propyl-1H-pyrazole-5-carboxylic acid (59), stoichiometric quantities of thionyl chloride (SOCl2) in toluene were used, rather than thionyl chloride as the solvent, therefore working in safer conditions. This is because toluene helped in the heat sink and reducing the environmental impact and SOCl2 was also replaced in the catalytic hydrogenation step employing H2, Pd/C to convert 1-methyl-4-nitro-3-propyl-1H-pyrazole-5-carboxamide (60) into 4-amino-1-methyl-3-propyl-1H-pyrazole-5-carboxamide (66). The preparation of 2-ethoxybenzoyl chloride (61) using oxalyl chloride was bypassed utilising SOCl2 and chlorosulfonic acid, which avoided CO emissions. Therefore, 2-ethoxy-5-((4-methylpiperazin-1-yl)-sulfonyl)benzoic acid (68) was prepared from 2-ethoxybenzoic acid (67) through the conversion of the intermediate sulfonic acid to sulfonyl chloride, which was subsequently suspended in water to provide 68 by reacting with N-methylpiperazine without the use of solvents. The reaction between 66 and 68, which was performed with N,N-carbonyldiimidazole (CDI), yielded compound 69. The replacement of the hazardous H2O2 with t-BuOK and t-BuOH for the cyclisation of compound 69 yielded sildenafil (65) in high yield and resulted in a green choice for reducing waste.
Comparing the two synthetic pathways, the latter results in 75% overall yield and generates less waste, improving the environmental perspective.20
4. Designing safer chemicals
“Chemical products should be designed to affect their desired function, while minimising their toxicity”.4Every chemical entity possesses toxicity, which should be validated through specific assays. In particular, active pharmaceutical products should be tested for their toxicity to be commercialised and used for their purpose given that they should fulfil their purpose of use in a less toxic way. The unintended biological activities or toxic effects associated with some commercial chemicals can lead to risks to the public health and environment. Therefore, it is urgent to consider possible measures to better ensure human safety and ecological wellbeing. Nonetheless, few chemicals are thoroughly tested because toxicological studies are expensive and require the sacrifice of many laboratory animals, such as rats, mice, and rabbits. Consequently, many chemicals are put on the market, lacking safety data and little or no toxicity testing.106,107 In this case, new in vitro and in silico methods have been developed to evaluate chemical safety and toxicity without animal experiments, at a reasonable cost.
Due to the lack of information regarding the toxicological effects of replacing a substituent with respect to another in a chemical compound, progress in the design of chemical alternatives has been achieved, establishing molecular design guidelines to recognise areas of chemical space with reduced hazard potential,108 while considering purposeful performance.109
The green design of newer and safer chemicals can be carried out with the aid of an adequate toxicity model, which considers all the variables in the chemical structure of the synthesised compounds associated with a particular toxic or non-toxic effect. Accordingly, molecular design guidelines are based on evidence that molecular devices are related to both chemical function and biological effects.110 To build these models, in silico, computational approaches and high-throughput screening (HTS) methods are emerging as useful tools to examine the adverse health outcomes of tested compounds, preventing the synthesis of thousands of unnecessary drugs and chemicals by predicting their toxicity profiles. Other techniques, such as computer-assisted synthesis design (CASD), consider various aspects of green chemistry applied to drug design, with the advantages of cheaper tests, without animal experiments, and reduction of waste. Thus, in silico approaches are important tools in the complex decision-making process, allowing the proper selection of solvents, reaction conditions, and substrates even before practical synthesis within a short period. Testing thousands of compounds and changing various parameters will offer much greater and broader options to synthetic chemists.20
Regarding drug design to achieve reduced toxicity, Campos et al.111 suggested an in silico model of molecular editing, to be employed together with the previously mentioned methods, to modify the chemical structure of a compound by replacing or inserting atoms through various reactions, such as site-selective C–H functionalisation, ring contraction, and ring expansion. Therefore, chemists can hypothetically design new molecules by choosing the best combination of structural devices associated with reduced toxicological effects and avoiding the so-called toxicophores, particularly structural alerts based on chemical structure applied in medicinal chemistry 4.
Also, in silico models aim to study the absorption, distribution, metabolism, and excretion (ADME) properties of chemicals in an organism, the mechanisms of which depend not only on the chemical structural devices but also on the target and individual variability.112 Toxicokinetic and toxicodynamic113 are two diverse but strictly correlated concepts, where the former is defined as the way a chemical species reaches the site of toxic effect, while the latter refers to the biological interaction between the chemical and the target, leading to a toxic effect.114 Thus, understanding these two mechanisms can help improve the performance of models to better design chemicals and the identification of chemical structures and properties relevant to toxic endpoints, as well as the mechanisms of toxic action.4,115
Together with in silico methodologies for toxicity testing, large in vitro databases, such as ToxCast116 and Tox21117 have been specifically designed to help elucidate toxicity mechanisms. Given that they employed the HTS methods to test a vast number of molecules, these large datasets are extremely useful not only to create machine learning predictive models to be applied in the field of green chemistry and find new rules for designing new, safer chemicals but also to predict the toxicity of untested existing molecules. One of the simplest measures of toxicity is provided by high-throughput screening in vitro cytotoxicity assays, which measure how chemicals kill particular types of cells. Furthermore, in vitro cytotoxicity is a good starting point for creating toxicity models with multiple control variables used by chemists to design molecules. Firstly, in vitro cytotoxicity is a valuable method to assess the adverse effects of chemicals, which is usually performed at the beginning of drug development.118 Moreover, it has an extensive dataset valuable for determining changeable variables for safer drug design.
The selection of variables is essential in the first step of the creation of a toxicity model because they represent the particular chemical space associated with a mechanism that leads to a specific adverse effect. Reflection of chemical space toxicological mechanisms can be represented by appropriate chemo-physical theories, which delimit specific variables. The first variable that should be mentioned is the way of interaction between the molecule and the target (toxicodynamic), which can occur, for instance, through covalent modification of the protein119,120 In some cases, molecules need to pass through the membrane to achieve their function; hence, the equilibrium of their lipophilicity and hydrophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) should be considered. In other cases, molecules can destabilise the cell membrane through electrostatic interactions, leading to a lethal effect.121 Polarizability should also be included, due to the capacity of molecules to induce cell stress through non-specific interactions, such as electric dipole moments and dispersion forces.121 Another variable to consider is the tendency for chemicals to form covalent bonds, thereby leading to potential toxic effects (hard–soft acid–base (HSAB) theory).122
P) should be considered. In other cases, molecules can destabilise the cell membrane through electrostatic interactions, leading to a lethal effect.121 Polarizability should also be included, due to the capacity of molecules to induce cell stress through non-specific interactions, such as electric dipole moments and dispersion forces.121 Another variable to consider is the tendency for chemicals to form covalent bonds, thereby leading to potential toxic effects (hard–soft acid–base (HSAB) theory).122
In the context of toxicity, all these variables aid in understanding the related underlying mechanisms of toxicological effects and help create a predictive model based on a variable/toxicity mapping for designing safer molecules.121
Although this plethora of variables affects cytotoxic effects, it is difficult to determine a specific cause that generates the beginning of cytotoxicity due to the simultaneous occurrence of many previously mentioned processes. Nevertheless, this toxicity model is highly predictive, suggesting that cytotoxicity is usually driven by non-specific mechanisms rather than a wide range of specific receptor-mediated mechanisms. Moreover, it can be used for green design, providing chemists with probabilistic diagrams, which help them recognise specific structural tools associated with the onset of adverse events, and then they can change the related design variables. Hence, this model can guide molecular design to reduce the risk of cytotoxicity and obtain safer molecules that fulfil their purpose.4,121
Nonetheless, this field of green chemistry is in its infancy; thus, additional work is required to expand molecular design guidelines and to identify relevant areas of chemical space associated with toxicological mechanisms of action.115
5. Safer solvents and other auxiliaries
Auxiliary substances such as solvents and separation agents make up the largest share of applied chemicals and frequently include a considerable potential of toxicity. Only solvent use accounts for more than 3/4 of the mass utilisation of synthesis in the pharmaceutical industry, as mentioned by Ahsan et al. in ref. 18 their review dealing with the significance of green synthetic chemistry from a pharmaceutical point of view.5.1. Reduction of auxiliaries
One option to improve the safety of chemicals is to reduce their quantity in a synthesis strategy. Porcheddu et al. described an excellent example of improved synthesis using a mechanochemical protocol to achieve Fischer indolisation.123 They drastically reduced the amount of liquid acids such as HCl, H2SO4, and TFA by replacing them with a solid mixture of oxalic acid and dimethylurea. Moreover, this tool minimised the quantity of solvent used and facilitated the purification of the product because it was not necessary to switch or remove any solvent. The solid mixture could be removed easily with water.Another interesting approach not only reducing the quantity of solvents but completely proceeding in their absence is the sonochemistry-based methodology examined by Crawford124 dealing with condensation reactions of solid reagents without any liquid medium. This method is a process that deviates from the classical sonochemistry regarded under the 6th Principle outlined in the next paragraph. The cavitation principle is decisively dependent on solvent, while the above-mentioned method builds on an explanatory approach based on mechanochemistry. This report described the reaction of o-vanillin (70) and 1,2-phenylenediamine (71) to produce a salen ligand (72) (Fig. 17), which is achieved through sonification without using any solvent. In contrast, the conventional reaction requires refluxing for 9 h in ethanol, which is an aspect that also improves the synthesis strategy in terms of the 6th Principle, i.e., the energy efficiency.
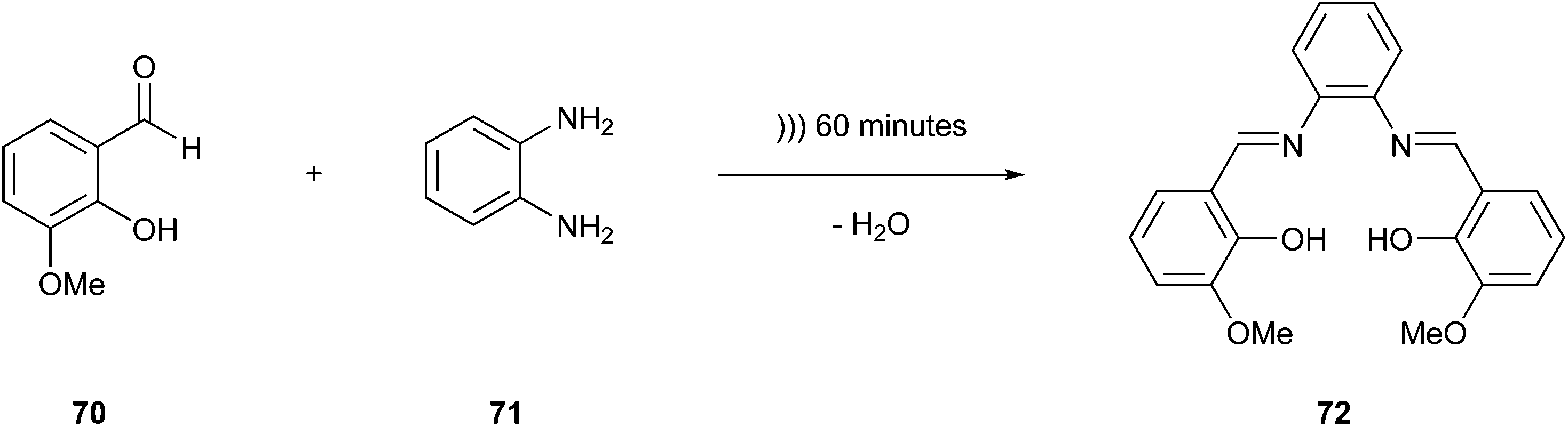 | ||
| Fig. 17 Reaction between o-vanillin and 1,3-phenylenediamine.124 | ||
As another aspect of medicinal chemistry strategies, ultrasonic irradiation can be a handy tool in producing active pharmaceutical compounds. Roy et al.125 developed technology to prepare co-crystals via ultrasound mostly without using solvent, similar to the research group in the previous example. According to the Food and Drug Administration (FDA), co-crystals are crystalline materials containing two or more different molecules.126 These molecules interact via non-covalent interactions such as hydrogen bonds or van der Waals interactions and show, in contrast to the single compounds, improved dissolution, stability, and bioavailability. Mostly, co-crystals are produced using an evaporation technique, including a solvation step followed by evaporation at room temperature to obtain the co-crystals.127
The co-crystallisation process constitutes a promising approach to find new patent drugs or improve the properties of novel compounds. For example, Roy and co-workers worked on the already well-known pharmaceutical compound paracetamol (73).125 They tried to prepare co-crystal structures of paracetamol with caffeine (74), 4,4′-bipyridine (75), and 5-nitroisophthalic acid (76) (Fig. 18) by mixing them in a 1:1 stoichiometric ratio in a sealed glass simple vial, which was sonicated for 60 min in a standard ultrasonic cleaning bath. Furthermore, the group varied the time of sonication in some studies, but complete conversion was achieved only after 60 min. This way, they developed a very efficient strategy that is very easy to perform, inexpensive, and solvent-free simultaneously. Therefore, it is a strongly feasible method to include a green Principle in the laboratory.
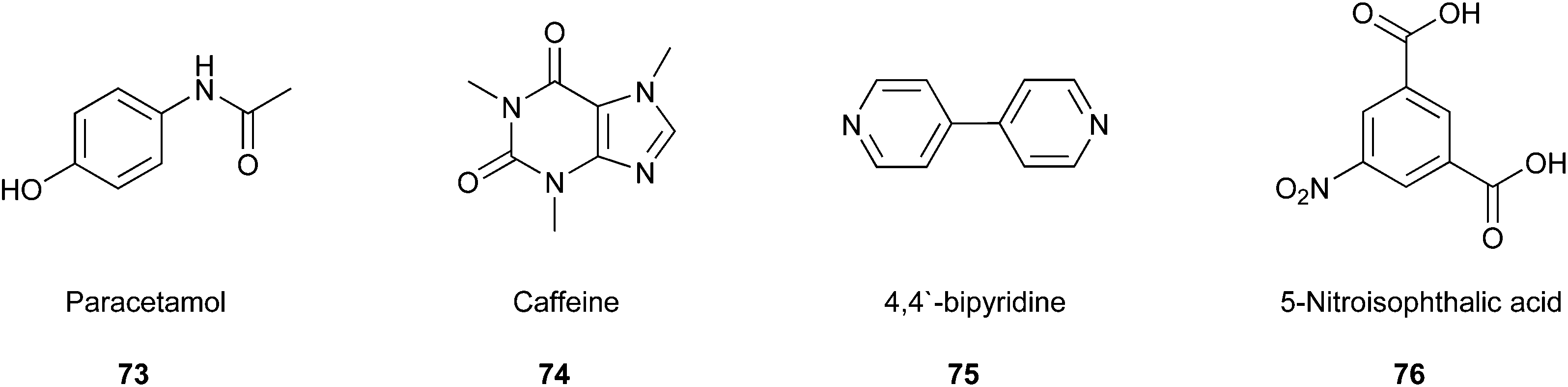 | ||
| Fig. 18 Paracetamol and co-crystal partners.125 | ||
5.2. Alternatives to highly toxic class I and II solvents
The information in review by Ahsan et al.18 mentioned in the introduction is summarized in Table 2, showing the most toxic or environmentally harmful solvents that should be replaced or limited to a minimum whenever possible.| Class | Properties Solvent | Allowable concentration [ppm] |
|---|---|---|
| I | Highly toxic, very harmful environmental impact benzene | 2 |
| Carbon tetrachloride | 4 | |
| 1,2-Dichlorethan | 5 | |
| 1,1,1-Trichloroethane | 1500 | |
| II | Most commonly used, toxicity between class I and III Acetonitrile | 410 |
| Chloroform | 60 | |
| Cyclohexane | 3880 | |
| Ethylene glycol | 620 | |
| Methanol | 3000 | |
| Pyridine | 200 | |
| Tetrahydrofuran | 720 | |
| Trichloroethylene | 80 |
Of course, replacing these highly toxic auxiliaries cannot be achieved by simply exchanging the thus-far used solvents, where the whole chemical implementation must be converted into a process that allows their replacement. One approach to establish this replacement in organic chemistry is the preparation of so-called solvent selection guides. For instance, Jordan and Sneddon et al.128 dealt with this topic using the example of the preparation of thioesters, given that they play an important role in the synthesis of pharmaceutically relevant compounds. This group developed a solvent-reagent selection guide including greener synthetic conditions for the formation of these functional groups. Furthermore, the review elaborated by Byrne et al.129 discusses selection tools for the replacement of conventional organic solvents with greener representatives. Starting from the European regulation ‘Registration, Evaluation, Authorisation, and Restriction of Chemicals’ (REACH), the aim is to restrict the use of toxic solvents such as toluene, DCM and chloroform. Based on this, they discussed the approach of Slater and Savelski, a spreadsheet that aims to show the different options available for a reaction. The two of them developed a method to calculate an overall greenness index, which makes the determination of an alternative and greener process easier to handle. Thereby, the basis of this is a solvent database and the named greenness score.130 The described solvent selection table program generates a so-called index, which enables a quick comparison of the different solvents or procedures. Thereby, the greener the solvent, the smaller the corresponding score. As an example, they compared a solid–liquid filtration using on the one hand acetone, acetonitrile, and methyl isobutyl ketone, and on the other hand side n-butanol, n-pentane, and n-heptane. For the calculation, they assumed the corresponding amounts of corresponding solvents. Consequently, process one including acetone achieved the score of 938, while process 2 had a total score of 429. This means that the second procedure is due to the smaller score, the greener alternative and should be used preferably. Moreover, they included occupational health considerations and safety considerations, thereby incorporating the central aspect of this Principle. Of course, they also thematised the previously discussed selection guides from the industrial side from Pfizer, GSK and Sanofi, given that they can support the improvement of safety and greenness in a reaction efficiently, as stated above.10–13 Furthermore, they mentioned the tool from Capello et al. at the ETH Zürich, which is interesting regarding the deviating starting point of the tool compared to the previously mentioned ones.131 This tool, in contrast to the other ones from industry, goes back to the origin of the solvents, including the aspect of renewability, a crosslink to the 7th Principle dealing with renewable feedstocks, calculating the cumulative energy demand of the solvent production. Unfortunately, it is limited currently to petrochemical solvents. In addition, the authors proposed a comprehensive framework for the environmental performance of a solvent during chemical production and health and safety aspects. In their case study, they considered other aspects and concluded that simple alcohols and alkanes seem to be the “environmentally preferable solvents”.131
Thus, in the past few years, an increasing number of approaches to overcome the problem of the broad application of toxic solvents, ranging from reactions without any solvent to efforts using water as the apparently least toxic variant. These greener, less toxic, and environmentally harmful solvents and reaction strategies will be discussed further based on biodegradability (Principle 7). A successful exchange is possible for tetrahydrofuran (THF) (77), a solvent belonging to the group of cyclic ethers. It is well known that THF tends to form highly explosive peroxides (78) through contact with atmospheric oxygen and upon exposure to light (Fig. 19A).131
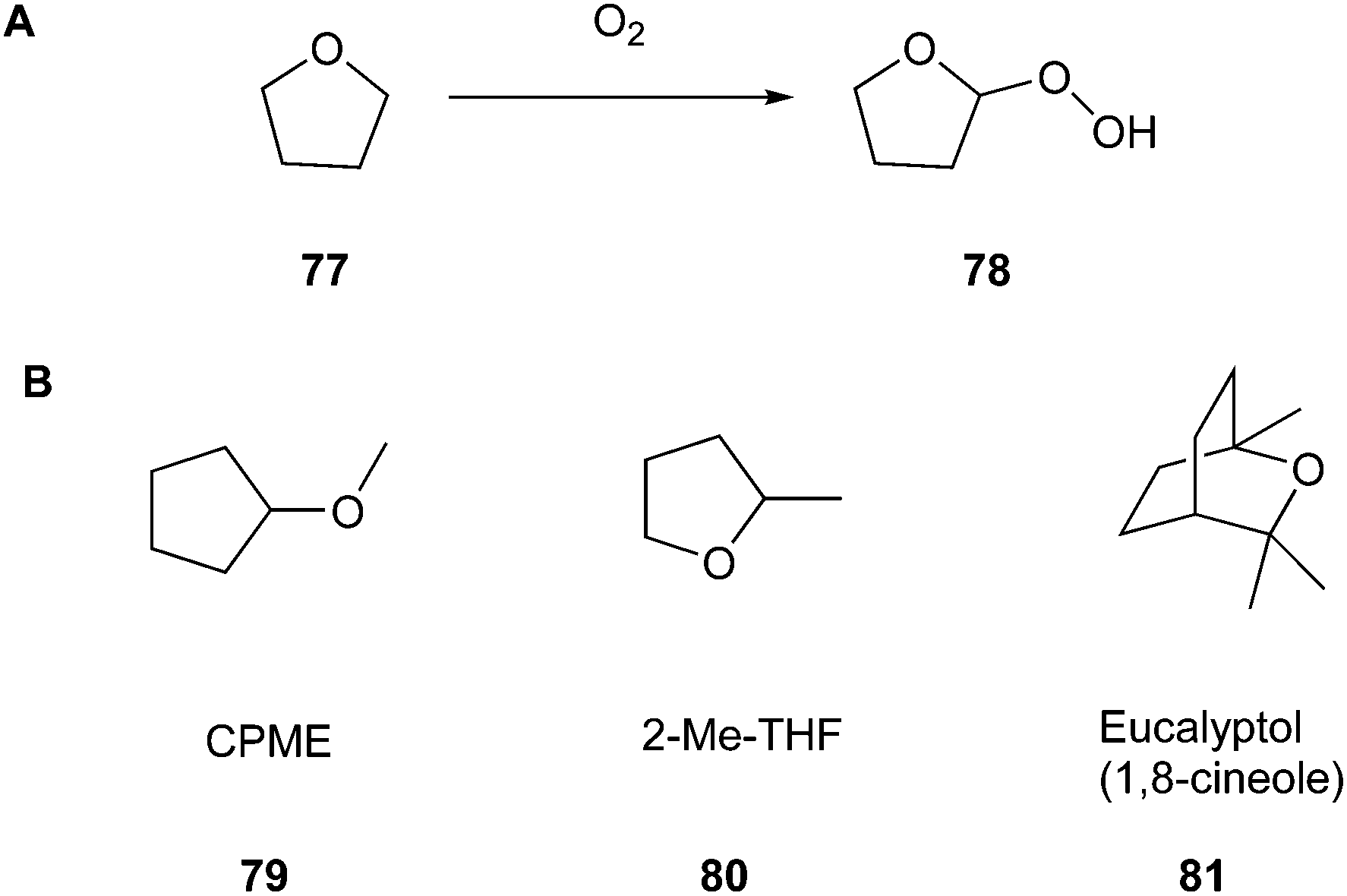
THF can be replaced by the greener alternatives cyclopentyl methyl ether (CPME) (79) and 2-methyltetrahydrofuran (2-Me-THF) (80) (Fig. 19B), showing distinctly less potential for the above-mentioned reaction. Moreover, as reported by de Gonzalo in their review, CPME exhibits great stability in acidic and basic environments and has a comparatively high boiling point (106 °C), where both aspects improve the safety of laboratory work.132 Another advantage that is of interest, especially in terms of practical usage, is that CPME possesses valuable properties, for example, it shows low solubility in water, which facilitates the formation of biphasic systems, e.g., for extractions. Furthermore, it can be dried effectively to make it applicable in reactions that afford water-free conditions.
As a specific example, Campos et al.133 showed in their work the potential of solvents 79–81 to efficiently replace petroleum solvents for the synthesis of O,S,N-heterocyclic compounds. Furthermore, they included eucalyptol (1,8-cineol) (81) (Fig. 19B) as another representative of greener solvents in their studies. They wanted to achieve a synthesis strategy that does not include metal catalysts, reaches optimised yields, and contains safer auxiliars in combination with microwave irradiation. For example, they chose 10H-pyrido[1,2-a]thieno[3,2-d]pyrimidin-10-one (84) to perform the optimization process. They discovered that the best outcome, including their previously mentioned perceptions, could be achieved by using CPME as the solvent, performing the reaction with microwave-assisted heating at 160 °C for one hour, and applying 3 mol% of Pd(OAc)2 and 4 mol% of Xantphos for catalysis (Fig. 20). Consequently, they achieved 71% yield.
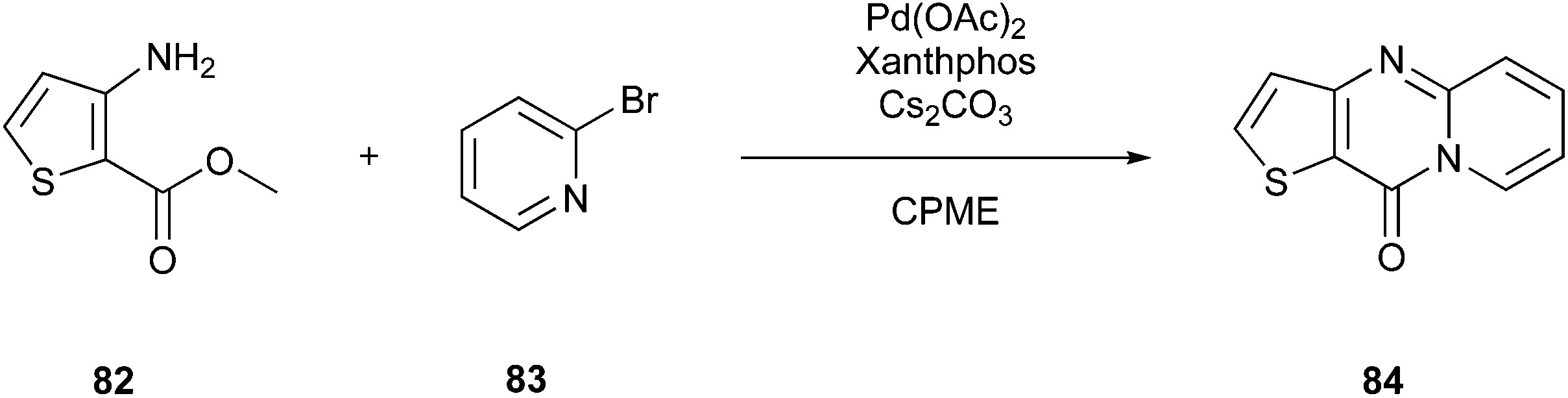 | ||
| Fig. 20 Synthesis of 10H-pyrido[1,2-a]thieno[3,2-d]pyrimidin-10-one.133 | ||
For the sake of completeness, this yield was surpassed by the results achieved with conventional heating with an 11% higher yield but the bare yield in this context plays a subordinate role. In this case, the critical point is to show that it is possible to perform the reaction in this solvent, but obviously, this is an aspect that needs to be further improved to fulfil the demands of medicinal chemistry. Additionally, it needs to be highlighted that the above-mentioned reaction is known to be very robust and can be conducted in almost all media. For example, Gevorgyan et al.134 performed Buchwald–Hartwig amination in vegetable oils. The group set up the reaction in different rapeseed oils, olive oil, sesame oil and some more plant-based oils, extending their study also to triglycerides of animal origin, natural waxes and semisynthetic excipients triacetin and tributyrin. Moreover, all these applied solvents are of (semi)natural origin, and thereby less harmful to the environment, which are related to the 7th Principle, i.e., bio-renewable feedstocks. The reaction is a vivid example for the application of these feedstocks, both with CPME and vegetable oil. The natural origin of the vegetable oils is evident, but to form a connection to CPME, it is necessary to take a look at the building blocks of the solvent, which is formed from furfural, and furfural is derived from pentoses such as xylose, and thus are regenerative raw materials, confirming its seminatural origin. Therefore, a greener approach regarding natural solvents seems feasible at least for some reactions but not yet used in an industrial setting to the best of our knowledge.132
5.3. Supercritical fluids
Another possibility to replace a solvent with a safer, non-toxic, and non-flammable alternative is the application of supercritical fluids (SCF). An SCF is defined as a fluid whose pressure and temperature are above its critical point. In this case, at the upper end of the vapour pressure curve, the border between the fluid and the gaseous phase does not exist, the density will be close to the liquid, and the viscosity is comparable to a gas. Thus, the properties can be classified as between the liquid and gaseous states.135 Among the various SFCs, carbon dioxide (CO2) is used most frequently, while water and other substances can also be applied. CO2 must be pressured above 73 bar and 31 °C to enter the critical phase. This technology has been known for many years and its use realized, e.g., in supercritical fluid chromatography (SFC), supercritical fluid extraction, and solvent in chemical reactions.As shown in the report by Ziqiang Wang,136 especially for SFCs, there are many advantages associated with the use of supercritical CO2 (SCCO2) as a mobile phase, like the ability to dissolve most compounds because of the liquid-like density and even significantly lower costs for the same amount of “solvent”. Moreover, this technique allows high reproducibility and separation speed even for separating complex mixtures.137 To focus on a more specific field of application, we chose an impressive example dealing with chiral analytical method development and preparative separation of enantiomers. This topic was investigated by Michaels et al.,138 whose group worked on an open-access approach using immobilised chiral stationary phases such as CHIRALPAK® IG, a rapid and efficient method to separate chiral compounds. To collect the needed data, they set a core screening process using methanol as a co-solvent for 50 different compounds. A purification was classified as successful only in the case of baseline separation. Accordingly, 60% of the racemates could be separated by applying the previously mentioned CHIRALPAK® IG as the column material. This issue is critical in early drug development regarding the narrow ridge between pharmacological activity and toxicity.
The above-mentioned advantages can also be exploited when using SCCO2 as a reaction medium. Kobata et al. implemented this concept around 20 years ago to form capsaicin analogues within transacylation reactions of capsaicin (85) using lipase as a catalyst (Fig. 21).139 A decisive factor for this strategy was that SCCO2 as a medium is compatible with the temperature range of enzymatic activity.
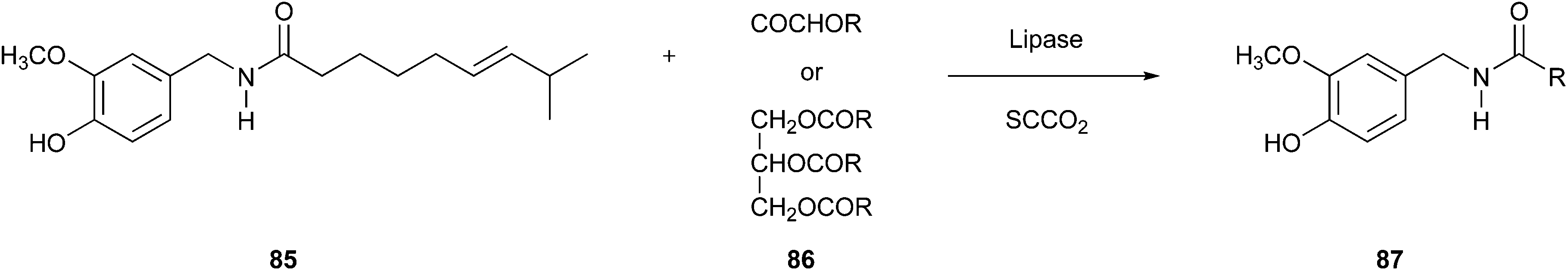 | ||
| Fig. 21 Transacylation of capsaicin.139 | ||
An additional, convenient fact when using SCCO2 as media to perform a reaction is the effortless removal of this “solvent” simply through depressurisation.
Nevertheless, CO2 is not the only supercritical fluid currently in use, where H2O is also applicable in the supercritical status. For water, this status is reached above 373 °C and 220 bar. Of course, there are significant changes in its characteristics as well as in the other examples. Supercritical water (SCW) has small amounts of hydrogen bonds, a low dielectric constant comparably to polar organic solvents, low viscosity, and a high diffusion coefficient. It shows great dissolving ability to organic compounds and becomes, in these terms, a promising reaction medium. SCW is mainly applied in supercritical water oxidation and gasification processes.140
One possible application is in protein hydrolysis to achieve bioactive peptides, a topic Rivas-Vela and co-workers dealt with in their review.141 They reported a particular field in supercritical fluid technology, the subcritical water hydrolysis, as an alternative to the fermentation processes and enzymatic techniques used thus far to gain bioactive peptides or rather drug candidates from protein resources. The protein sources can be, for example, vegetable or animal proteins. Subcritical water is in contrast to the described supercritical status defined in the temperature range between 100–300 °C,142 and thus a temperature above the boiling point but below the supercritical temperature. In this state, there is an increase in the amount of OH− and H3O+ ions, which causes an increase in the reactivity of water. This enables the water to function as a double catalyst for hydrolysis reactions. The conditions of high pressure and temperature support the disruption of weak interactions such as hydrogen bonds, which are essential for forming quaternary, tertiary, and secondary structures. The basic mechanism of the hydrolysis (Fig. 22) starts with the protonation of the N-terminus of the peptide bond (88). After this excitation, the cleavage of the peptide bond occurs. Subsequently, an OH− ion joins the remaining C-terminal carbon cation (89).
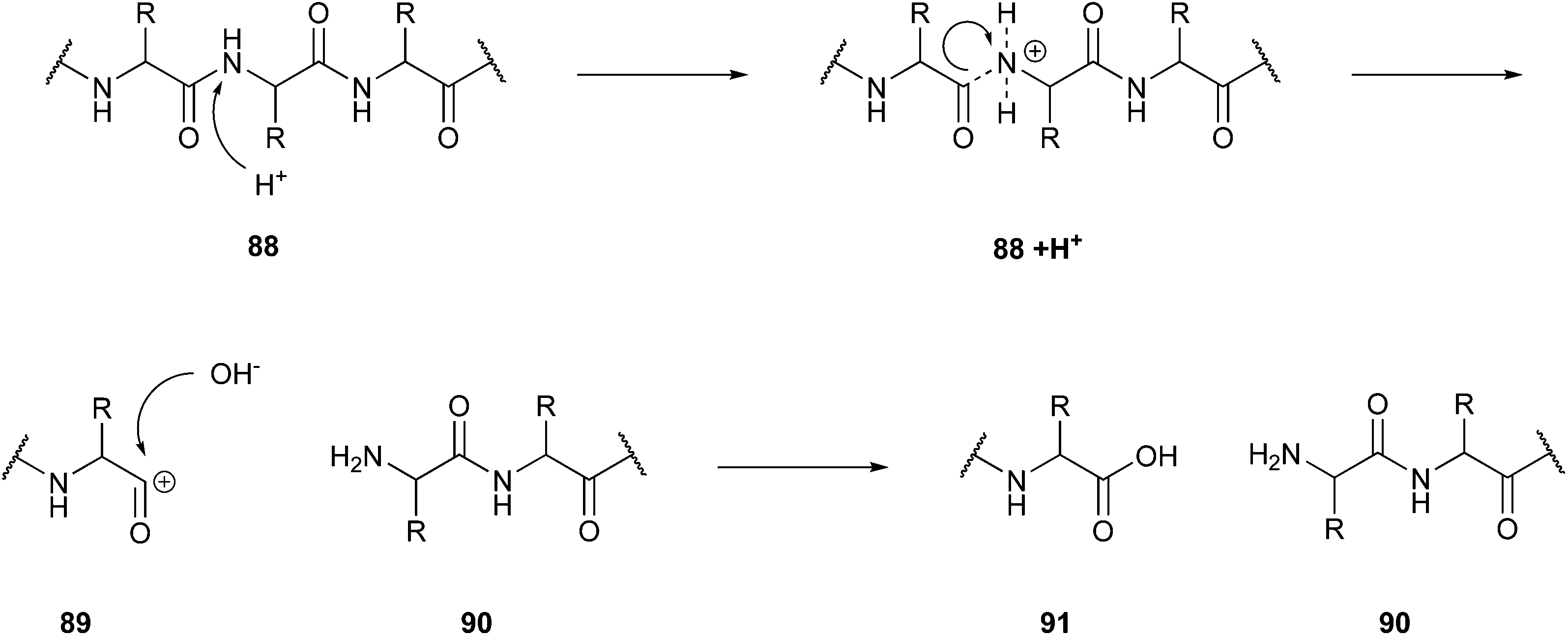 | ||
| Fig. 22 Cleavage mechanism of peptide bonds in subcritical water.142 | ||
A remaining problem of this method is the control and prediction of the location of the cleavage. Compared to the highly specific action of enzymes, subcritical water hydrolysis shows a substantial underperformance in the current state of research. Considering the discussed aspects, this technique is a new promising, and green method to gain peptides from protein materials, but it needs further investigation to overcome the problematically unspecific cleavage reaction.
In conclusion, there are various promising ideas and valuable and innovative possibilities to improve the handling of auxiliaries and solvents in terms of green chemistry. However, considering the scope of this review, we could present only a small insight into the current situation, but there is more possible to work on this topic.
6. Design for energy efficiency
The key aspect of this Principle is to minimise the external energy expenditure of a synthesis strategy whenever and however possible. This includes not only avoiding high temperatures and pressure, where this topic can be approached from various other starting points as well, by reducing the synthesis steps that require a substantial energy input such as chemical separations (especially distillations) or by gaining the needed energy through unconventional, new mechanisms of energy delivery, such as microwave irradiation, sono- and photochemistry.46.1. Lower reaction-temperature
A lower reaction temperature may be the most obvious option to improve a the energy efficiency of a reaction, but it is not as easy as it seems at first sight. Reactions mostly need a precise energy input to proceed correctly and entirely and cannot be simply performed under variable conditions. In this case, the heating can be reduced by finding another path to achieve the desired product. For example, the research group mentioned above,123 which developed a mechanochemical way to perform a Fischer indolisation (Fig. 23), achieved not only an improvement in the reduction of solvents but also in lowering the needed energy for successful implementation. The reaction was performed at room temperature and proceeded without any heating. | ||
| Fig. 23 (A) Flow-chemistry-based implementation of indole synthesis.146 (B) Continuous-flow synthesis of 1-benzyl-3-(2,6-dimethylphenyl)thiourea (97).147 | ||
6.2. Flow chemistry
Another way to apply energy in an intensely focused and specific way is flow chemistry. Here, in contrast to conventional batch chemistry, the underlying Principle is a continuous flowing stream, wherein the reaction process occurs. This is achieved by pumping the reactants into the reactor chambers of the microreactors, which are mainly used for small reaction batches in medicinal chemistry.143 Flow chemistry enables an increase in mass- and heat-transfer processes,144 avoiding temperature gradients, heat-accumulation, or temperature hotspots, which can cause a loss in reaction selectivity. This is also actionable for highly exothermic reactions.145Colella and co-workers,146 similar to the research group in 6.1, amongst other things, investigated Fischer indole synthesis, but with the intention to establishing a protocol for a flow chemistry-based strategy (Fig. 23A). They expanded this knowledge even more by functionalisation reactions of the indole ring (92), in which we will present a closer look here. The group started from the protocols they established for microwave-supported batch reactions, i.e., a reaction using tributylmethylammonium methylcarbonate paired with a supercritical liquid as the catalytic base, a fascinating crosslink to safer auxiliaries. The used reaction conditions were converted into high-temperature and pressure-flow processes, and subsequently further optimised. Finally, they achieved 250–370 mg of the N-methylated indole (93) within 3 min. Thereby, the group used solvents considered toxic from a green chemistry point of view, a fact that should be avoided, as discussed above in Principle 5. However, regarding the fact that the reaction is performed under flow chemistry conditions, in a closed system and with decisively less amount of solvent, we can consider these conditions a slight improvement. Moreover, this is a vivid example of the problem that mostly not all the Principles of Green Chemistry can be fulfilled in an optimal way, but there needs to be a compromise, which can be improved in turn.
Similar to the previous research group, Nemeth et al.147 based their synthesis strategy on flow chemistry. They worked on thioureas, substances that are well represented amongst pharmaceutical compounds and play an essential role as intermediates in synthesising different heterocycles. This group used, amongst other reagents, an N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA)-based aqueous polysulfide solution combined with benzylamine (96) to synthesise thiourea derivatives such as compound 97, which was achieved with a residence time of 42 min in an impressive 96% yield (Fig. 23B). Furthermore, the final compound could be obtained via simple filtration, another fact that makes this strategy regarding solvent usage efficient.
A further approach is the possibility of continuous-flow multi-step synthesis described by Jiao et al.144 This was analysed more closely according to the 8th Principle, focusing on reducing reaction steps, e.g., by avoiding multiple separation and purification steps. As it may be recognised, not least by this crosslink, flow-chemistry offers many possibilities in improving synthesis referring to green chemistry, mainly because of the wide range of Principles that are included in this technique. Regarding this fact, this approach is one of the most promising ones for the future laboratory work.
6.3. Photochemistry
The energy spending system in terms of photochemistry is light. Depending on the technique and needed energy content, it is possible to use ultraviolet, visible, or infrared radiation. A significant advantage of this energy source is its non-toxic, cheap, renewable, and basically limitless character. In contrast to thermal activation, the radiation energy is transferred to molecules by absorption in a very rapid manner. Thereby, the acceptor is transferred to a higher energy level. Overall, it is a process providing enormous potential but should be handled carefully, considering its destructive power.148In medicinal chemistry, photochemistry is often deployed for catalytic issues and broadly used in combination with flow chemistry. This ensemble was examined by Shahbazali et al.149 in their UV-photoisomerization process in a micro-flow system to finally obtain the pure trans-cyclooctene (99). The isomerisation (Fig. 24) did not take place completely, which is why they obtained a mixture of both configurational isomers. To separate them, they used a micro-flow reactor with a fixed bed containing AgNO3/SiO2 powder. The trans-cyclooctene adsorbs at the surface, while the cis-isomer (98) remains in the flow. Finally, they achieved a strategy to convert the cis-into the trans-isomer successfully.
 | ||
| Fig. 24 Configurational isomers of cyclooctene.149 | ||
Another possibility to include photochemistry in organic synthesis is photosensitisation. Yakubov and co-workers investigated this aspect in their review of direct C–H fluorination,148 an innovative functionalisation step useful in drug discovery. The benefit of using this method is that there is no need to introduce other functional groups during the fluorination process. They used visible light as an energy donor for the excitation, and thus finally for the activation of the C–H bond. They introduced so-called photosensitisers or photocatalysts (Fig. 25) to overcome the problem that most small molecules had no structure to absorb the radiation to switch to an excited state. These compounds can absorb radiation, switch into an excited state (triplet state), and then transfer the energy of the excitation to the substrate. Because they do not take part in the reaction, they are comparable to some extent with classic catalysts. Well-known examples of photosensitisers are ruthenium and iridium complexes, but regarding sustainability, they are not attractive. Very interesting representatives are, for example, acetone (100), xanthone (101), and benzophenone (102), which are efficient, affordable, and easily accessible.
 | ||
| Fig. 25 Photosensitiser with triplet state energies (ET) and absorbance (λmax).148 | ||
The choice of an appropriate fluorination agent (Fig. 26A), which refers to the substrate on which the excitation energy of the photosensitiser is transferred, is a complex decision, but not a topic we further explore at this point. The commonly applied fluorination agents are Selectfluor® (1-chloromethyl1-4-fluoro-1,4-diazoniabicyclo [2.2.2]octane bis(tetrafluoroborate)) (103) and Selectfluor® II (104) because of their comparatively easy handling. In the following energy transfer, Selectfluor® abstracts a fluor-radical and forms a radical intermediate. To overcome this unstable condition, a benzylic hydrogen is abstracted, and in reverse, the benzylic position is attacked by the fluor-radical.148
In addition, the group of Xia et al.150 described various fluorination reactions of different compounds, even a well-working benzylic gem-difluorination (Fig. 26B) with xanthone as the photosensitiser and Selectfluor® II (104) as the fluorination agent with 83% yield.
This methodology represents a very attractive opportunity for direct C–H fluorination, which can be integrated easily into laboratory life to make it a bit “greener”.
6.4. Microwave irradiation
Microwave irradiation as an alternative to the conventional heating process is another very effective option to improve the energy efficiency of laboratory work. Additionally, microwave irradiation cannot only be applied as a bare energy source but also to improve a synthesis strategy in terms of duration of the implementation and achieved yield.Ahsan et al.18 described phenyl-1H-pyrazoles and phenyl-1H-pyrazoles-4-carboxylic acid derivatives in an interesting comparison of conventional and microwave-assisted synthesis. The pyrazole heterocycle is, in a pharmaceutical regard, a privileged structure, and an important building block for various ligands for multiple types of targets, thus being an efficient synthesis strategy of high interest in the research of new active compounds.
Regarding phenyl-1H-pyrazoles (e.g., 109), this group applied the synthesis protocol of Finar and Godfrey for the conventional synthesis based on refluxing for 2 h at 75 °C to obtain the desired product starting from phenylhydrazine (107) and 1,1,3,3-tetramethoxypropane (108). For the microwave-assisted method (Fig. 27A), they explored different conditions, but they concluded that the optimal output was achieved with a reaction time of 5 min and a power of 50 W. Temperature variations between 60–80 °C had no significant influence; therefore, they decided to perform the reaction at 60 °C. Finally, they showed that microwave-assisted organic synthesis surpassed the conventional synthesis regarding the duration of the reaction and the achieved yield. For example, phenyl-1H-pyrazoles were synthesised in a yield of 91–98% using microwave heating, while conventional heating only produced 72–90%.
 | ||
| Fig. 27 (A) Microwave-assisted synthesis of phenyl-1H-pyrazole.18 (B) Aldol-condensation of the example of 2′-hydroxyacetophenon under MW.151 (C) Synthesis of (E)-3-styrylflavones.152 | ||
Besides the already mentioned example, Rocha and co-workers151 dealt with the advantages of using microwave irradiation in their project of chalone-synthesis and the isomerisation of this compound into flavones. They worked on optimising the aldol condensation of acetophenone (110) and benzaldehyde (111) into a chalcone (112) (Fig. 27B). This reaction already works quite well at room temperature, but the reaction time can last up to 20 h. Thus, to reduce the reaction times, the authors performed the reaction under microwave irradiation, where depending on the substituents on the acetophenone, with 400 W irradiation for 15 min (R = OH) or 50 W irradiation for 20 min (R = NH2).
It might be quite surprising to heat NaH under microwave conditions, but the authors described their precautions and safety measures taken such as slow addition at low temperatures prior to placing the reaction vessel in the microwave.
This way, they minimised the time the reaction needs to be heated, the most energy requiring aspect of the reaction. Moreover, as the conventional heating process is replaced by a decisively less energy consuming process, i.e., microwave heating, not only the duration of the energy input but also the energy input itself is reduced, resulting in a more energy efficient reaction procedure. Unfortunately, the yields were not improved, a circumstance worth further optimising. In the further optimisation, attention should be paid especially regarding the applied solvent THF. Applying the reagent guides10–13 discussed in section 4.1 will be a very suitable option to enhance the greenness of the reaction procedure.
According to the previously described significant advantages of microwave-assisted heating in medicinal chemistry, Albuquerque et al.152 introduced another fascinating aspect in their review, where they reported on the ability of microwave irradiation to improve a reaction in terms of regioselectivity and stereoselectivity. For instance, they mentioned the synthesis of (E)-3-styrylflavones (e.g., compound 115) (Fig. 27C), which gains the (E)-configuration of the vinylic system and s-cis-stereochemistry, as confirmed by NMR data. This influence of microwave irradiation was also observed for the (E)-3-styrylquinolin-4(1H)-ones.
The use of microwave irradiation offers many significant opportunities to optimise synthesis protocols and is a promising approach in terms of low cost and ease of implementation. It is already increasingly applied in laboratory daily life, but this can be further improved in many cases.
6.5. Sonochemistry
This subject area deals with chemical reactions under energetic ultrasound irradiation (20 kHz to 10 MHz). It unites various advantages such as increased product selectivity, decreased reaction time, and mild reaction conditions. The basic Principle is the so-called acoustic cavitation; precisely, the formation of gas bubbles in the liquid phase, which runs through a continuous process of formation, growth, and collapse, consequently achieving an increase in the reaction surface. This dynamic process causes a rise in the local temperature, which can be converted into the activation energy for the reaction. Thus, ultrasound constitutes an energy-efficient activation technique that can reduce the consumption, and therefore the waste of energy.153Borah and co-workers154 made use of this phenomenon to establish a one-pot synthesis strategy to form amino-substituted 4,8-dihydropyrano[3,2-b] pyran-3-carbonitriles and spiro[indoline-3,40-pyrano[3,2-b] pyran]-3-carbonitrile (120) or carboxylate derivatives (Fig. 28), which are structures with a wide range of application in medicinal chemistry. Additionally, this group wanted to replace the commonly used metal catalyst with L-proline (119), a distinctly more environmentally friendly catalyst, to improve the methodology in terms of “greenness generally”, but this topic is discussed more closely later in this review.
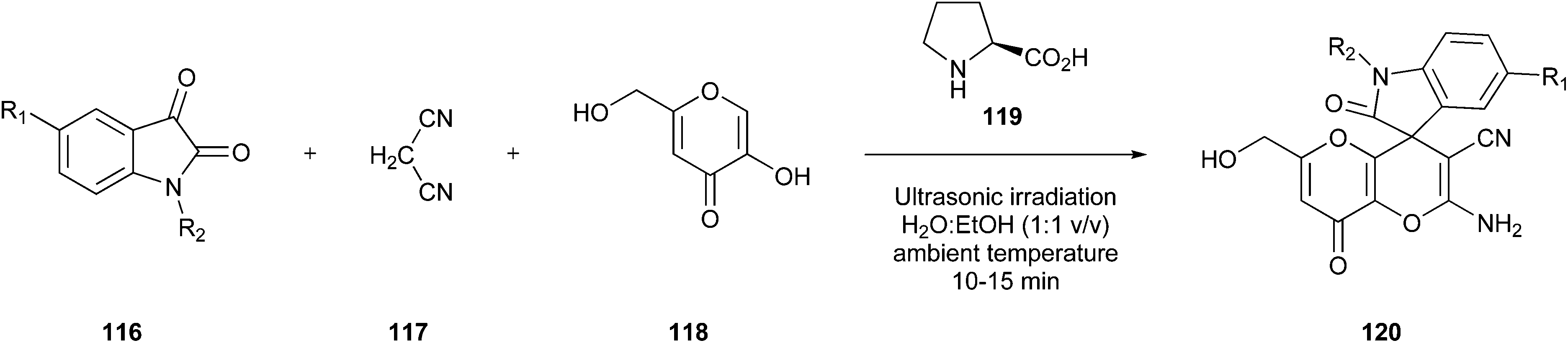 | ||
| Fig. 28 Example for the synthesis of spiro[indoline-3,40-pyrano[3,2-b] pyran]-3-carbonitrile.153 | ||
After synthesising various derivatives and optimisation of processes such as varying the catalyst, solvent-mixtures, or acid/basic environment, they concluded that K2CO3 as the base in a water/ethanol mixture at room temperature and under ultrasound irradiation led to the best output. Moreover, the purification of the product was achieved by filtering, several washing steps with water, and recrystallisation from ethanol, and thus there was no need to perform the elaborate column chromatography. Ultimately, they achieved a synthesis strategy that unites two possibilities for improving the energy efficiency, i.e., an unconventional energy-delivery system and eliminating a highly energetic reaction step.
As previously explained in chapter 5.1, the sonochemistry-based methodology elaborated by Crawford,124 which makes it possible to replace a 9 h refluxing process with one hour of sonification, implements the Principle of energy efficiency strikingly.
As mentioned above, combining these techniques is even more interesting than the different energy delivery methods. There have been many ideas to combine microfluidic systems with reactions using photocatalysis or capillary microreactors, including ultrasound, to take advantage of the resulting synergistic effects. Rashmi et al. addressed this topic vividly in their review,155 where they mentioned multiple advantages such as shorter reaction durations and improved yields, and also some challenges such as the ultrasound-caused streaming or the attenuation of the sound field. However, finally, they presented an example that unites all three methodologies. Nair and co-workers established a way to form ZnO nanoparticles by using the effects of ultrasound in a fluorinated ethylene propylene microtube for the deposition of particles, followed by a selective oxidation-reaction in a microreactor based on photocatalysis.156
7. Use of renewable feedstocks
The usage of renewable feedstocks seems to be one of the most employed topics in an attempt to achieve a more consistent way of life, or in our case, the synthesis of active pharmaceutical compounds. Considering the background of, on the one hand, limited resources, elevating CO2 emissions, and thereby global warming, as a few of the most urgent problems, and on the other hand, the increasing demand of a strongly growing population, this topic has attracted increasing attention, and also in medicinal chemistry.As clearly conveyed in the review “The Green ChemisTREE: 20 Years After Taking Root with the 12 Principles”,4 the most challenging point in replacing conventional with renewable materials is their different basic building blocks. Conventional materials are mainly based on oxidized hydrocarbon species, while renewable substances principally consist of carbohydrates and their polymers. These strongly different C:H:O ratios present us with the need to establish processes that convert the ratio of the building blocks of renewable materials into conventionally used substances. This interesting Principle is already fully actionable in some biobased renewable solvents and will be discussed in the following section.
As the most effective intervention point in the optimisation process of a synthesis of “greenness”, we focus on solvents in the following chapter. This consists merely of their high quantitative amount, where half of the chemicals used in the process of synthesising an active pharmaceutic compound are solvents.20
7.1. Solvent-free reaction conditions
The optimal case is a strategy that does not include solvents, i.e., no consumption of any type of solvent resources. In 2002, the Indian research group of Ranu et al. described a solvent- and catalyst-free pathway to synthesise dihydropyrimidinones (124). These compounds show activity as calcium channel blockers, and therefore of interest in antihypertensive therapy. The synthesis includes a so-called Biginelli condensation (Fig. 29) of 1,3-dicarbonyl (121), aldehyde (122), and oxygen or sulfur-containing urea-derivatives (123), which are converted into the desired ring structure (124) at 100–105 °C within 1 h.157 This team provided the proof of the feasibility of solvent-less catalytic reaction. | ||
| Fig. 29 Synthesis of dihydropyrimidinone derivatives. | ||
A further example of a solvent-free pathway in medicinal chemistry that was already mentioned in chapter 6.3 is the strategy of Roy et al.125 to form co-crystal structures of compounds. They took advantage of the Principle of sonochemistry to achieve this goal.
7.2. Aqueous reaction conditions
Unfortunately, the above-mentioned conditions do not represent a typical case, and thus other solutions are needed. According to the 12 Principles, the most consistent solvent is water. Because of its protic character and the poor water solubility of highly lipophilic compounds, it is impossible to perform every reaction in an aqueous environment. Nevertheless, K. Tandon et al.158 investigated new antifungal and antibacterial compounds. In the synthesis of these compounds, they processed the elimination and cyclisation reactions of nitrogen and sulphur-containing hetero-1,4-naphthoquinones with and without water in the reaction mixture. For various compounds, e.g., 2-(allylamino)-3-chloronaphthalene-1,4-dienon (127), they showed that this green methodology is a promising and well-working alternative. The previously mentioned compound was synthesised from 2,3-dichloro-1,4-naphthoquinone (125) and prop-2-en-1-amine (126) in water within 15 min in 100% yield (Fig. 30). | ||
| Fig. 30 Synthesis of 2-(allylamino)-3-chloronaphthalene-1,4-dienon in aqueous condition.158 | ||
Another idea to circumvent the problematic poor solubility of many compounds or intermediates in water is phase-transfer catalysis (PTC). This technique is mainly based on a two-phase system. Generally, the reactant is not soluble in the phase that hosts the reaction, such as ionic compounds in organic solvents. In this case, it is possible to add a so-called phase transfer catalyst (PTCat). This agent works like a type of detergent that enables the migration of the compounds in the reaction phase. A more specific, and in terms of green chemistry, an interesting possibility is the use of PTCats to enable reactions to be performed in water by “solving” the reactants with PTCats.
Qingyu Zhang et al. showed in 2022 that amphiphilic indoles159 can be used as this type of PTCats and reduce the amount of organic solvent in bromination reactions in this way, a reaction that is commonly used in medicinal chemistry. This research group investigated the hydroxybromination (Fig. 31B) of compounds such as α,β-unsaturated carbonyls, various alkylenes, and aromatic molecules using a previously synthesised sophisticated indole (130) (Fig. 31A). They discovered that it was possible to achieve up to 97% yield with the use of 1 mol% catalyst at 23 °C in 3 mL water, 1 mmol educt (131), and an additional 1.1 mmol N-bromosuccinimide (NBS), a compound that indole reacts with first by forming an intermediate species during the catalytic cycle.
In this case, the application of microwave irradiation conditions is a further chance to enable the use of water as a solvent, and even improve the duration of the reaction process in some cases. Bucciol et al.160 tried to improve the synthesis of α-doxycycline in terms of sustainability, amongst other things, by using water as a solvent in a microwave-assisted strategy. To achieve a successful reaction, they also inserted a rhodium catalyst and an oxytetracycline–cyclodextrin complex to obtain the α-form selectively.
As well as the previously mentioned working groups, Shabalala et al.161 found a way to use water as a solvent. Specifically, they developed a one-pot approach to synthesise pyrazole-derivatives (e.g., 137) in water under ultrasonic irradiation (Fig. 32) starting from aromatic aldehydes (134), hydrazine monohydrate (135), ethyl acetoacetate (133) and malononitrile/ammonium acetate (136). They tried some variations in reaction temperature and based on their results, performed the reaction at 50 °C. Under these reaction conditions, they achieved up to 95% yield for some compounds.
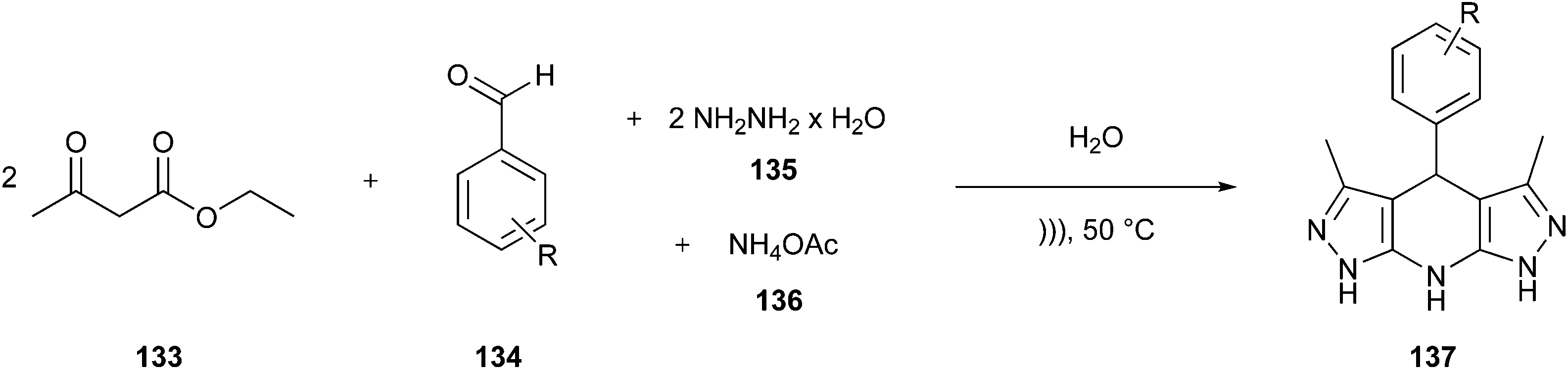 | ||
| Fig. 32 One-pot synthesis of pyrazole-derivatives.161 | ||
The last two named examples are even more of interest, referring to the 6th Principle. Both were already discussed as very attractive methodologies to improve the energy efficiency of a reaction and can now also be seen as valuable possibilities to unite the aims of two different Principles of Green Chemistry.
7.3. Bio-renewable solvents
When all the previously mentioned methods of using a “greener” solvent are not applicable, it is possible to replace a conventional solvent with a biobased one. An example of this replacement is dihydrolevoglucosenone (Cyrene) (139), which can be used instead of dipolar aprotic solvents such as N,N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO) and dimethylacetamide (DMAc).162 As mentioned above, one of the essential aspects of this field is transforming renewable, biological materials into oxygenated raw materials to form alternative solvents. Cyrene is mainly synthesised from levoglucosenone (LGO) (138), an intermediate obtained from the degradation of cellulose (Fig. 33A). To obtain Cyrene from LGO, merely the double bond must be reduced by a hydrogenation reaction, and according to Kudo et al., it is163 possible to achieve the conversion of cellulose to Cyrene in a one-pot reaction.
Peptide synthesis, more accurately, the formation of amides (142), represents an interesting process for the industrial scale and research implementation of this sustainable and green solvent, as examined closely by Wilson and co-workers.164 They carried out the synthesis outgoing from a carboxylic acid (140) and an amine compound (141) with HATU, an activating agent, and excess DIPEA, and obtained yields between 63–100% (Fig. 33B). Additionally, it is a valuable aspect that it was only one washing step with water followed by a trituration to get the pure product, a very time-saving circumstance for the purification of the product.
A further biobased chemical becoming increasingly important is 5-hydroxymethylfurfural (HMF, 144), which is a versatile platform chemical. It is obtained by dehydration of carbohydrates, such as hexoses such as glucose, fructose (143), and xylose, and polysaccharides such as cellulose, chitosan and inulin (see Fig. 34). HMF can be transformed into various other products such as solvents, polymers, liquid fuels, chemicals, and even plastic material. Currently, there are some difficulties regarding the yield obtained by the dehydration reaction, but there are already some promising approaches to overcome this problem, where amongst other things, the application of pristine Nb2O5 as a catalyst achieves significantly improved results.165–167
 | ||
| Fig. 34 Acid-catalysed formation of HMF by dehydrogenation of fructose.165 | ||
Additionally, HMF is fully biodegradable, and the corresponding degradation mechanism by microbes has been studied.168 Overall, HMF is a very encouraging new chemical that needs to be further investigated regarding a future application in medicinal chemistry, for example, as a potential solvent.
Another common example of bio-renewable solvents is the application of ionic liquids (ILs). ILs are analogous to salt ionic substances, which exhibit uncommon low melting points, leading to their liquid appearance at room temperature or somewhat below 100 °C. This class of sustainable solvents unites many favourable properties such as high thermal stability, non-flammability, and wide range of compounds they solve. They consist of unsymmetric, unwieldy organic cations combined with various organic or inorganic anions. Some broadly used cations are N-alkylpyridinium and tetraalkylammonium with [PF6]−, [BF4]− or halogens such as Br−, Cl−, and I− as anions. IL show great potential for use in instrumental analysis, for example, for separation techniques as performed within high-performance liquid chromatography (HPLC) or capillary electrophoresis (CEP).169 They can be applied as stationary phases, additives for the mobile phases, and in CEP as modifiers of the electroosmotic flow.170
For the use of IL as solvents in various reactions, Hallett et al. gave a comprehensive overview in their review.171 They mentioned successful approaches for common reactions such as Knoevenagel reactions, Aldol reactions, and acid-catalysed Mannich reaction. To take a closer look at one of these, here we focus on the Knoevenagel condensation in 1-butyl-3-methylimidazolium hexafluorophosphate ((bmim)PF6) as an IL (Fig. 35), as investigated by Formentín et al.172 This group worked on converting benzaldehyde (145) with malononitrile (146) at room temperature to β,β-dicyanostyrene (147). To improve the solubilisation of the KOH used as the catalyst, they dissolved the hydroxide in ethanol, but this small amount of solvent was removed immediately by heating the reaction mixture. The product was recovered by liquid–liquid extraction with ethyl ether.
 | ||
| Fig. 35 Knoevenagel condensation of benzaldehyde with malononitrile.172 | ||
Further, an interesting aspect of this conversion is that the ionic liquid could be reused five times. They simply heated the IL at 40 °C under reduced pressure for 12 h after the extraction and performed the same reaction under the same conditions, but without the addition of a further amount of base. The second run achieved a significantly higher mass balance than the first run.
One more alternative to the conventionally applied solvents that is appropriate with its simple preparation, tuneable properties, and sustainability is the use of deep eutectic solvents (DES).173 Typically, a DES is a mixture of a quaternary ammonium salt, hydrogen bond acceptor (HBA), and hydrogen bond donor (HBD). In this context choline chloride, glycerol, urea, carboxylic, and amino acids are commonly used.174 The nature of these hydrogen-bonding properties causes the lowering of the melting point and leads to the formation of a eutectic mixture. One effect of this waste-free technique to form a liquid is that there is no need for further purification, which constitutes a decisive advantage compared to the classic ILs.
Piemontese et al.174 used a DES to replace the previously used DMF in some steps of their synthesis strategy for PZ1 (150), a new promising multi-target ligand in the treatment of Alzheimer's disease. For example, they decided to do this for the final reaction step (Fig. 36) using choline chloride/propylene glycol (1:3) as the solvent instead of DMF, improving the yield from 21% to 30%. The reaction duration was the same for both variants, 60 h, but DES required a higher reaction temperature. The reaction step required the presence of N,N′-dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (NHS) for the in situ formation of the amide moiety.
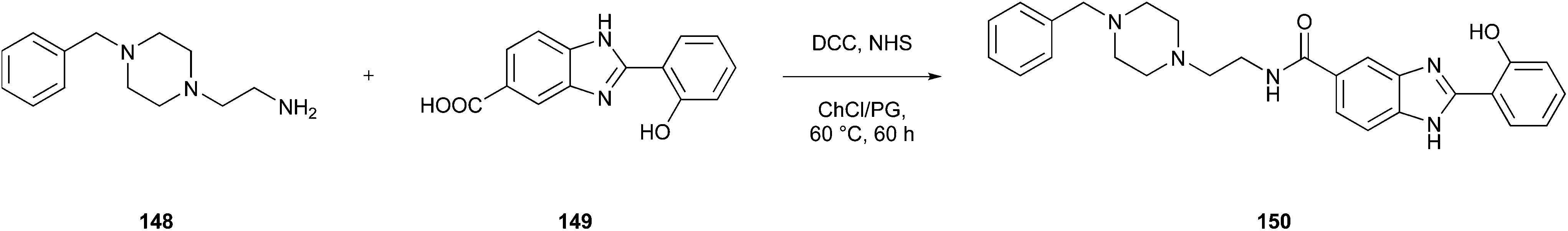 | ||
| Fig. 36 Final reaction step to obtain PZ1 performed in DES.174 | ||
This synthesis is in terms of the used DES of high interest in green chemistry, proving that DES present an applicable and effective reaction media. However, in this case, although the reaction yield can be improved, the application of this new solvent in other synthetic procedures is of high interest.
Nevertheless, considering this reaction strategy encompassing the background of all the Principles of Green Chemistry, we have, amongst other things, a significant issue with energy efficiency in this case. By increasing the reaction temperature and performing a reaction with a duration of 60 h, we obviously cannot see this as a complete success, but this is an excellent approach to start with the process of improving this synthesis in terms of green chemistry.
Thus, this reaction perfectly presents the most challenging trap of green chemistry, i.e., improving the overall greenness of a reaction, not the optimisation of a single aspect at the expense of all the others.
7.4. Supercritical fluids
As already discussed under the fifth Principle, there is also the possibility of replacing conventional solvents with supercritical fluids such as supercritical CO2 and H2O. This strategy is applicable in many cases, as already discussed in the examples by Kobata et al.139 and Rivas-Vela and co-workers.1418. Reducing derivatives
Again, the underlying Principle of Green Chemistry, reducing the amount of chemicals whenever possible, is realised, this time by reducing the steps to the final compound. In our opinion, this needs to be one of the core aspects to be included in the laboratory to achieve a greener way of working.8.1. Protection groups
The use of protection groups is a descriptive example of a strategy involving additional solvents, other auxiliaries, and an increase in the number of reaction steps. Hence, the protection of specific groups should be avoided or replaced. Moreover, this already holds the central problem in most cases because applying protection groups is indisputably necessary to achieve the desired compound without various by-products.One possibility to overcome this problematic point and obtain a specific compound without using a protection group is improving the chemoselectivity of a reaction. Amongst other improvements in terms of “greenness”, the research group of Mudithanapelli et al.175 implemented this method by using N-heterocyclic carbene (NHC) (154) for the chemoselective Morita-Baylis–Hillman (MBH) reaction as well as the aza-Michael addition of isatine (151) (Fig. 37). The so achieved N1- and C3-functionalization was reached after a few optimisations for some derivatives such as 156 with more than 90% yield. This result is even more interesting as isatine is a commonly used building block for various pharmaceutic compounds such as antibiotics and antivirals.
 | ||
| Fig. 37 Reaction scheme for MBH and aza-Michael reactions.175 | ||
As already mentioned, managing a multi-step synthesis without applying any protection group is not possible for every type of reaction. Nevertheless, for the chapter on electrochemistry described below, we found an interesting option to make the use of protection groups “greener”.
8.2. Flow chemistry
This already described method is also, from this Principle's point of view, a very effective option, where it is possible to minimise the number of separate reaction steps and unite them in one continuous process. The intermediates do not have to be separated, dried, or crystallised after each step, which can be performed in a continuous flowing stream.145An innovative and interesting pursuing approach in this field that is focused on the minimisation of necessary reaction steps was described by Jiao et al.,144 presenting a vivid overview of the possibility of continuous-flow multi-step synthesis (Fig. 38). This is a technique that functions without any work-up of the reaction mixture, isolation, and purification steps. Instead of this conventional procedure, they present an integrated synthetic system that is highly efficient, controllable, and independent. Moreover, and in relation to the 5th and 12the Principles, which deal with improving safety in organic synthesis, the contact with highly hazardous reagents can be avoided by using this alternative new method.
 | ||
| Fig. 38 Schematic diagram of continuous-flow multi-step synthesis.144 | ||
One vivid example mentioned was the synthesis of 2-(azidomethyl)oxazoles (160) (Fig. 39), which are recurrent structures in nature, and pharmaceutic compounds, starting from vinyl azides (157) from Rossa and co-workers.176 To explain the synthesis step by step, first, azirine (158) is formed via thermolysis, and subsequently permuted with 2-bromoacetyl bromide (159) to the 2-(bromomethyl)oxazole intermediate, followed by the final reaction with an aqueous NaN3-containing stream to the desired 2-(azidomethyl)oxazole. The whole reaction sequence is performed in a continuous-flow reactor. They explored the optimisation steps in batch and tailored their so-detected insights to the flow-chemistry method. Finally, they achieved an overall yield of up to a 60%.
 | ||
| Fig. 39 Continuous-flow synthesis of 2-(azidomethyl)oxazoles.176 | ||
8.3. Click chemistry
Click chemistry consists basically of small, essential molecule structures collected in combinatory libraries. They can be recombined amongst themselves through selective, simple carbon-heteroatom bonds (C–X–C). This means that it is possible to generate a significant number of novel molecules in a short time in uncomplicated one-step reactions, which is consistent with green chemistry.177 The possibility of using this methodology in a broad range of applications is based on the ability of azide- and alkyne components to be incorporated into multiple substituents. Thus, it is possible in theory to produce the structure of a new final compound by performing only a single reaction, without any intermediate or derivative.An additional interesting application area that is important in developing new compounds is using click-chemistry to label proteins that depict, e.g., a potential target structure. In a biological context, click chemistry is now even more common than in the chemical part of drug development, an aspect that presents an opportunity to improve reactions but will be discussed in this review briefly below.
To start applying click-chemistry in classical chemistry, Rodriguez et al. investigated novel factor Xa inhibitors178 and aimed to implement as many green chemistry approaches as possible. They included a one-pot click chemistry reaction in their strategy to reduce the reaction steps in the synthesis pathway. More precisely, they synthesised the final essential aromatic P1 motif and the triazole linker (163) (Fig. 40). The triazole is generated via the broadly used click-type Huisgen 1,3-dipolar cycloaddition catalysed by copper nanoparticles, followed by hydrolysis immediately. This reaction is a representative of the so-called copper(I)-catalysed azide–alkyne cycloadditions (CuAAC), the “classic” click reaction.
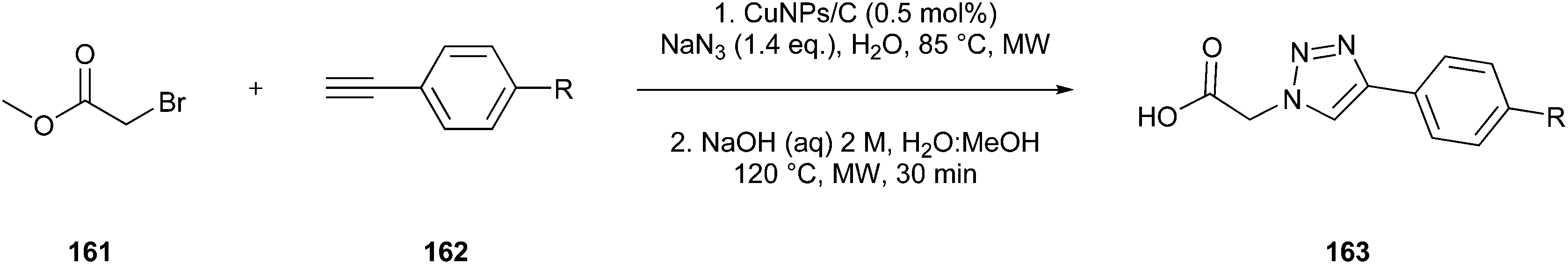 | ||
| Fig. 40 Multi-component click reaction and hydrolysis.178 | ||
They achieved yields of up to 83%, which means, in comparison to the conventional approach, an increase of up to 30%. Also, Tron et al.179 focused on this multifunctional reaction type and substantiated its importance with their given examples.
An alternative option to induce a click reaction is, without applying the cytotoxic copper-catalyst, the strain-promoted alkyne–azide cycloaddition (SPAAC) (Fig. 41), which is another subgroup of click chemistry reactions. Instead of copper, a cyclooctyne (165) is used to promote this bio-orthogonal reaction, yielding compounds such as 167.180
 | ||
| Fig. 41 General mechanism of SPAAC.181 | ||
An improvement of this metal-free approach in click chemistry is the usage of cyclononynes instead of cyclooctynes, an idea investigated by Tummatorn et al.182 more closely. Of course, cyclooctynes show an impressive high reactivity toward azides, but they exhibit problematic sensitivity upon contact with air and a tendency to form polymers. In both cases, the reagent is not fully available in the reaction, and thus overcoming this problem can increase reactivity. They did some variations of the basic cyclononynes, such as including methoxy groups to balance the increase of lipophilicity because of the additional methylene-group to optimise the characteristics of the molecules. Finally, they concluded that they could achieve useful levels of SPAAC reactivity. This group also considered this the missing piece to establish SPAAC as a user-friendly and broadly used click-chemistry reaction.
To also show an example from the biological side, here we look at the work of Li and co-workers,183 where they used a gel-based mass spectrometry method to identify proteins with O-GlcNAc modifications. They included a comparison of CuAAC and SPAAC in terms of labelling efficiency. These modifications are significant in cell signalling and modulation processes. For CuAAC, they applied biotin-diazo-alkyne, while they used biotin-DIBO-alkyne for SPAAC. They concluded that CuAAC outperformed SPAAC in this case because high background activity was detected with a decline in high reactivity against cysteine-containing proteins.
Nevertheless, despite the various advantages of click-chemistry, it should be considered that the variety of produced components is limited by the number of included substituents or rather the linked structures.177 This means that without renewing, the modules combining amongst themselves via click-reaction will not be an endless source of new compounds, while in this case, promising new applications can be expected in the near future.
8.4. Catalysed molecular assembly
Catalysed-assembly or abbreviated “catassembly,” of molecules is an innovative opportunity to unite the well-known chemical principle of catalysis and the relatively novel technique of molecular assembly. This method can assemble small organic compounds that function as building blocks into supramolecular structures. This process is supported by so-called “catassemblers”, which are structures that increase the assembly rate. Similar to conventional catalysts, catassemblers are not part of the final assembly product and work under a constant Gibbs energy. The whole process is based on noncovalent interaction but considers aspects such as configuration or arrangement of the single subunits.184 Because the whole process of building the molecular structure is self-conducted and does not involve several single steps, it is a very efficient way to minimise the number of reaction steps and derivatives.In our opinion, this methodology constitutes a promising opportunity that should be further investigated, established, and employed as soon as possible but is from the current point of view, still a dream for the future.
8.5. Electrochemical synthesis
An up-and-coming and environmentally friendlier alternative to conventional oxidation and reduction reactions with chemical agents is the electrochemical way, which is driven by an electric current generated using renewable sources. This reduces the use of potential hazardous chemicals because it is not necessary to use stoichiometric amounts. This type of chemistry is also considered mild because it is processed under ambient temperature and atmospheric pressure, making it even more interesting for this review.185For example, applying this concept for a Kolbe synthesis or various C–H functionalisation reactions is possible. Jiao et al.186 investigated the latter within a synthesis strategy for selective C–H functionalisation, which combines transition metal catalysis with electrochemistry. They chose Pd, Cu, and Ir catalysts for the different reaction types they studied, and the most successful ones under these circumstances were the C–H oxygenation catalysed by Pd and the C–H annulation catalysed by Ir with asymmetrical alkynes. In the following section, we aim to present a brief insight into their reaction system to oxidise C(sp3)–H bonds by using oximes to guide the oxidation process.
Presumably, the basic mechanism starts from the oxidation of the Pd-catalyst at the anode. This oxidised status of the catalyst allows the oxidation of the substrate via a redox reaction, while the catalyst is reduced again. Depending on the introduced substituent, e.g., an acetyloxylation, there are slight deviations from this basic mechanism, including various intermediates. This group compared their results with conventional reaction pathways using NaNO3/O2 as the oxidant and concluded that the electrochemical method can achieve better yields, especially for mono-functionalisation.
An even more interesting approach is the combination of electrochemical synthesis with flow chemistry to generate a technique that also overcomes the disadvantages of conventional batch electrochemistry, such as mass transfer, selectivity, and ohmic drop.185 Basically, similar to conventional flow chemistry, the solution of reactants is pumped into a flow reactor chamber. However, the difference is that the mass transfer occurs along two electrodes via diffusion, creating a concentration gradient and causing the migration of the ions in a potential field. The electrodes are separated through a spacer, which is important because the distance is proportional to the resistance of the solvent/electrolyte system, and in some cases, this fact even allows the reaction to be performed without any supporting electrolyte. Subsequently, the product is transferred in another reaction chamber if there is a following reaction planned or a collection unit, also enabling to handle highly reactive intermediates or radicals safer and compared to the conventional methods easier.
Green et al.187 used this combination of techniques to deprotect para-methoxybenzyl ethers (168) in methanolic solution to obtain the free alcohol (170) and, as a by-product, the para-methoxybenzaldehyde dimethyl acetate (169) (Fig. 42). The working group established a general protocol for the deprotection, using MeOH solution with the aim of applying a sustainable solvent, and inserting Et4NBF4 as an electrolyte. This electrolyte could be fully recovered and reused. They also stressed the importance of using an appropriate current, balancing the gap between using sufficient current for selectivity and too much current causing overoxidation. Moreover, they demonstrated the ease of scale-up on a laboratory scale by simply varying the flow rate, concentration, and volume of the reaction solution. They performed this anodic cleavage of PMB ethers in 17 different substrates and achieved yields of up to 92%, even if the molecules showed additional protecting groups for alcohols.
 | ||
| Fig. 42 Electrochemical deprotection of para-methoxybenzyl ethers.187 | ||
As mentioned above, the optimal case is to overcome the use of protection groups. However, from the current point of view, this is not applicable in most cases. As explained in the previous section, the reactivity of a specific substituent must be masked to avoid unwanted side products during multi-step synthesis. Nevertheless, the presented technique may be, for some of these cases, a fast, promising alternative to gain the desired final structure without any protection group.
9. Catalysis
A major problem in “green medicinal chemistry” is using stoichiometric amounts of reagents. Numerous organic syntheses such as stoichiometric reductions with metals and metal hydride reagents or oxidations often require an excess of these chemicals, and thereby generate higher amounts of unnecessary by-products.188Switching to catalytic processes represents a suitable alternative, whereby the generation of waste can be reduced in a simple way. Catalysts represent substances that accelerate a chemical reaction by lowering the required activation energy. This matter often allows innovative chemical reactions to be performed under mild conditions such as ambient temperature and pressure without requiring external energy input (Principle 6). They achieve their function without being consumed or inserted into the final products and, by this definition, can be recovered afterwards. As their name suggests, the amounts are usually insignificant and small compared with that employed for stoichiometric methodologies, indicating their superiority over the latter.
Another benefit is the greater product selectivity, facilitating important reactions to be carried out efficiently in medicinal chemistry. Selective acceleration of a chemical process allows a reduction in the number of by-products, and thus reactants are used in the most efficient way and waste generation is minimised. An illustrative example of the advantage of catalyst strategies over stoichiometric is shown in Fig. 43. The conventional reduction of acetophenone to 1-phenylethanol necessitates an excess of sodium borohydride and leads to the production of side products, whereas catalytic hydrogenation achieves the same result without the generation of additional waste.190
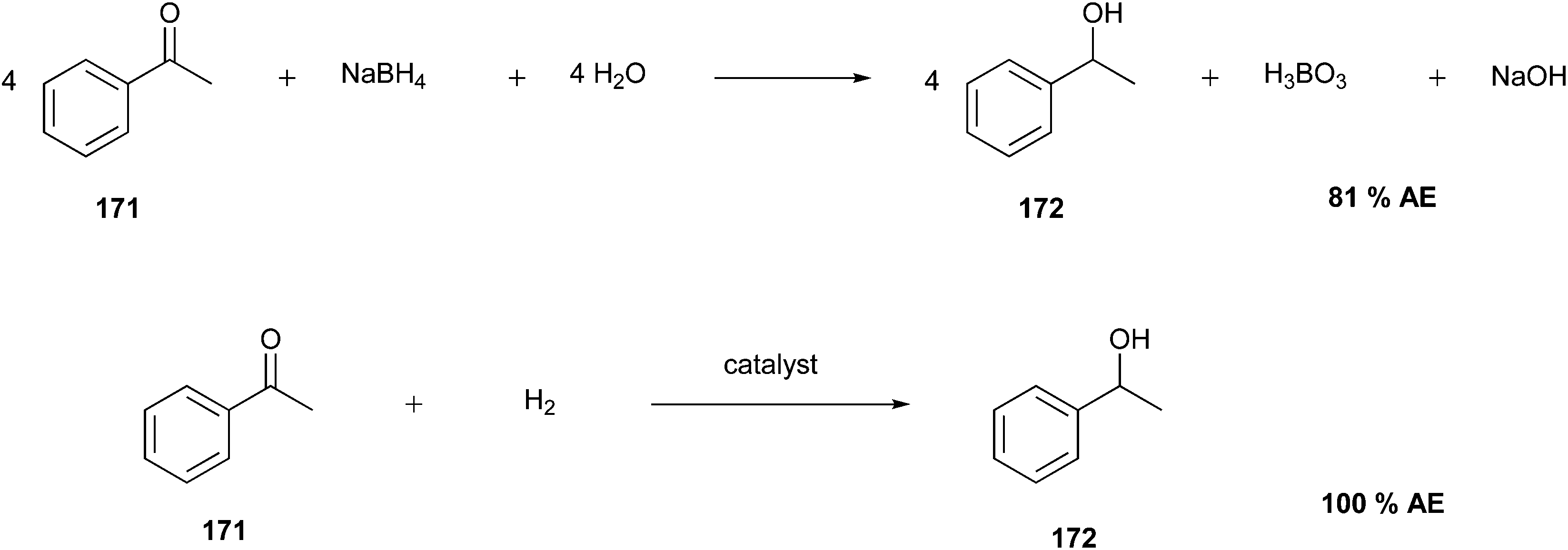 | ||
| Fig. 43 Reduction with metal hydrides and catalytic hydrogenation.189 | ||
Less feedstock leads to a decrease in waste production, resulting in lower E-factors (Principle 1) as well as a higher atom economy (Principle 2). This underlines the correlation between these Principles established by Anastas et al. and shows the cohesiveness of this system.3
Herein, we focus on some greener options available to medicinal chemists in modern drug discovery, including metal catalysis, zeolite catalysis, and biocatalysis. Finally, we want to illustrate the development of catalysis on amide bond formation, one of the most frequent conversions in medicinal chemistry.
9.1. Metal catalysis
Catalytic procedures involving metal and organometallic compounds represent one of the most widely used strategies in medicinal chemistry. The major requirements for reactions to be carried out in medicinal laboratories are a broad reaction scope and high synthetic efficiency. This is accomplished by a large number of heavy metal catalysts, such as platinum, palladium, rhodium, and ruthenium, which are often used in numerous reactions.191 Several research groups greatly enhanced the parameters such as catalyst loading and compatible substrates, reactions, and solvents through the years. For example, Vries et al. developed a method using only “homoeopathic” amounts of ligand-free Pd(OAc)2 for the Suzuki- (0.02–0.05%) and Heck-reaction (0.01–0.1%).192,193The disadvantage of many homogeneous catalysts over their heterogeneous counterparts is the difficulty of their recovery and reuse, requiring purification and isolation steps, and thereby creating excessive amounts of metal-contaminated waste. This problem can be addressed by employing an aqueous biphasic system, where the reactants and products are dissolved in the organic phase, and the catalyst remains in the water phase, facilitating their recovery afterwards. This procedure is nowadays applied for many carbonylations of alcohols employing the water-soluble Pd(tppts) catalyst established by Sheldon and co-workers.194 An illustrative example is the final carbonylation of 1-(4-isobutylphenyl)ethanol (57, IBPE) to the famous analgesic ibuprofen 56 in a two-phase system (Fig. 44).195
 | ||
| Fig. 44 Carbonylation of IBPE to ibuprofen via water-soluble Pd(tppts) catalyst.195 | ||
Gutiérrez et al. employed a heavy metal catalyst for the microwave-assisted one-pot synthesis of pyrazolopyridines, representing structural analogues of purine bases such as 177 in DNA or RNA nucleosides. They used InCl3 in aqueous media for the condensation of a pyrazolo-amine (174), formaldehyde (175), α- and β-diketones (176) under microwave irradiation and short reaction times (<20 min) with good to excellent yields of 67–95% (Fig. 45).196 The conversion represents a safe, efficient green synthesis, applying several Principles of Green Chemistry at once.
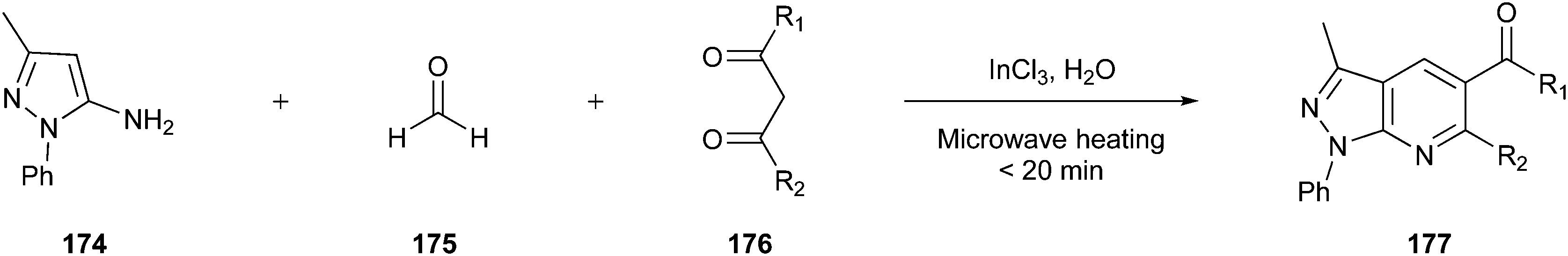 | ||
| Fig. 45 Microwave-assisted synthesis of pyrazolopyridines via InCl3-catalysis in water.196 | ||
In addition, Arcadi and co-workers presented a new green and efficient synthesis of enamines via cold-catalysis of 1,3-dicarbonyl compounds and amines in acetonitrile or ethanol. The catalysis of gold(III) was also extended to the reaction of cyclic 1,3-dicarbonyls with O-, P-, and S-nucleophiles.197 However, heavy metal catalysts nowadays are criticised given that they possess severe environmental toxicity and generate huge amounts of metal waste.198,199 Therefore, applying less hazardous and cheaper metals such as Cu, Zn, Ni, and Fe is highly required.200
Ji et al. proposed a reasonable method for the synthesis of enamines (180) with iron(III) triflate (1 mol%) under solvent-free conditions at room temperature.201 They succeeded at the conversion of β-enamino ketones or esters such as 178 with even less nucleophilic amines such as aniline (179) with excellent yields at an average of 90–99% and concise reaction times (<10 min) (Fig. 46). The low-cost Fe(OTf)3-catalyst could be easily recovered and reused, thereby representing a green alternative to the more toxic heavy metals.
 | ||
| Fig. 46 Catalytic solvent-free synthesis of enamines via Fe(OTf)3 by Ji et al.201 | ||
Bauer and Lenze reported another approach employing an Fe-catalyst in 2013. They showed the rapid and selective oxidation of secondary alcohols in the presence of primary alcohols. The transformation proceeded at room temperature in acetonitrile utilising aqueous H2O2 as the oxidant via catalysis with an organic iron(II)-complex and reaction time of just 15 min.202
9.2. Zeolite catalysis
Zeolites represent solid acidic catalysts consisting of aluminosilicate minerals with a regular pore and cavity structure of molecular dimensions. Zeolites confine substrates in their cavities, which causes changes in their structure and reactivity, where their pore size determines which molecules can access their acidic interior. Therefore, varying their pore sizes (shape selectivity) and tuneable activities enable the application of promising, tailor-made catalysts for various reactions in medicinal chemistry.203Friedel–Crafts acylation is a reaction widely used in medicinal chemistry and needs excess chlorinated compounds such as AlCl3 and acetyl chloride or anhydride, thereby leading to the formation of huge amounts of HCl. A milestone in the latter conversion was achieved with the publication of the first commercial application of a zeolite-catalysed aromatic acylation by researchers of Rhône Poulenc in 1997.188 They performed the acetylation of anisole 181 over zeolite beta with acetic anhydride 182, giving compound 183via simple filtration without needing solvent or a distillation work-up (Fig. 47A).
 | ||
| Fig. 47 (A) Zeolite-catalysed acylation of anisole by Rhône Poulenc.188 (B) N-Alkylation with nanosized zeolite-catalysis.204 | ||
Furthermore, Reddy et al. succeeded at the N-alkylation of various amines (such as 185) with alcohols (such as 186) via nanosized and recyclable zeolite beta catalysis. The reaction involves conditions under air atmosphere, simple zeolite separation by filtration, and water production as the sole by-product (Fig. 47B).204 Slow transport into the zeolite pores, slow reaction rates, and unwanted side-transformations were circumvented through the narrow particle size of nanosized zeolites. Accordingly, conventional methods involving toxic alkyl halides, large amounts of (in)organic bases, and extensive extraction procedures can be avoided. The main disadvantages of this synthesis route are the inconstant yields (10–98%) and high temperatures (135 °C), which are incompatible with heat-labile compounds.
An invaluable transformation nowadays is the Huisgen [3 + 2]-cycloaddition between azides and alkynes, which his often termed “click chemistry”. The latter emerged in the past decades as an important way for triazole synthesis, enabling the discovery of lead compounds through combinatorial chemistry or target structures via proteomic approaches.205 Pale et al. presented a one-pot-synthesis from organic halides (such as 188) with an azide (such as 189) to the corresponding triazoles (such as 190), using cooper-zeolites in water (Fig. 48).206 In addition, the zeolite could be recycled and reused four times without significant yield loss. This strategy lowers the atom economy, avoids the handling of the potentially toxic, explosive azide intermediates (explosives), and represents a safe and green process for click chemistry.
 | ||
| Fig. 48 One-pot-[3 + 2]-cycloaddition.206 | ||
Recently, the one-pot-Baeyer–Villiger oxidation (BVO) of cyclohexanone (192) to ε-caprolactone (194) over Sn-beta zeolites was mentioned in the literature by Luo and co-workers (Fig. 49). The oxidation was performed via in situ-generated hydrogen peroxide from aerobic oxidation of benzhydrol (191) catalysed by N-hydroxyphthalimide (NHPI). In their study, the generated H2O2 performed better and more efficiently than the commercial counterpart (39% > 27%).207
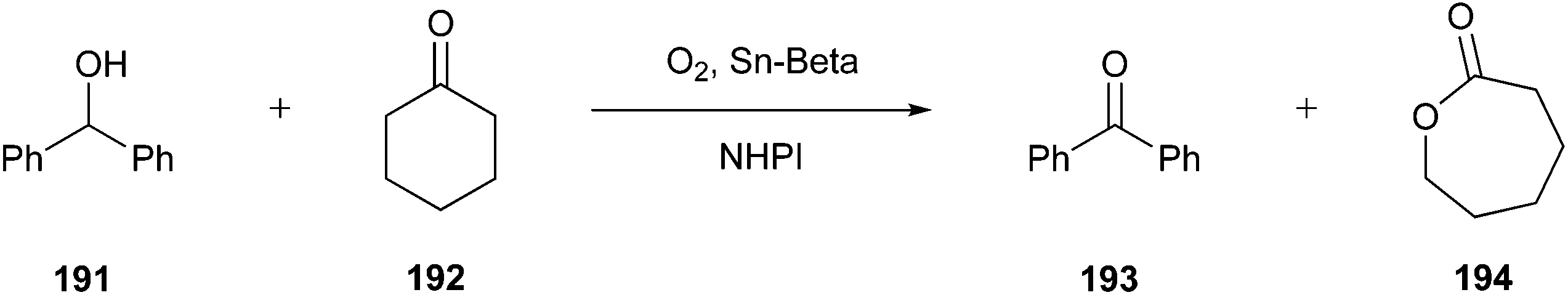 | ||
| Fig. 49 One-pot BVO of cyclohexanone via Sn-beta-zeolite and in situ-generated H2O2.207 | ||
Zeolite-mediated reactions emerged in the past decades as an accepted and emerging field of catalytic procedures in medicinal laboratories. They represent green, heterogeneous catalysts that usually come with simple removability and efficient recyclability without significant activity losses after multiple employment. Nowadays, more than 200 different natural or synthetic zeolites are available to medicinal chemists and allow the green, efficient performance of several organic conversions, leading to many important structural motifs of potential pharmaceuticals. The major drawbacks of zeolites include their insufficient commercial availability, additional procedures for their formation, and poor investigation for numerous reactions, thereby needing optimisation in these aspects.208
9.3. Biocatalysis
Biocatalysis employs nature's catalyst, i.e., enzymes, in chemical processes and represents a highly attractive field in green medicinal chemistry. Biocatalysts have many benefits over their chemical counterparts, given that they are compatible with mild reaction conditions such as ambient pressure and temperature, physiological pH, and water as solvent.Another advantage of enzymes is their chemo-, regio- and stereoselectivities for specific substrates, without any need for additional derivatisation, protection, and deprotection steps. Furthermore, the development of DNA techniques and protein engineering enabled the production and widespread application of tailored enzymes with required substrate specificity, enzyme activity, and stability.189
An illustrative example is given by the enzymatic hydrolysis of 6-aminopenicillanic acid (6-APA, 197), a key precursor for the (semi-)synthetic production of penicillin and cephalosporin antibiotics. The outdated chemical procedure applies a three-step synthesis over the intermediate 196 and requires a hazardous solvent (CH2Cl2) and various chlorinated chemicals (PCl5 and Me3SiCl) at a reaction temperature of −40 °C. However, the enzymatic deacylation of penicillin G (195) proceeds in one step in water at 37 °C, and the liberated phenylacetic acid can be recycled in the fermentation (Fig. 50).209
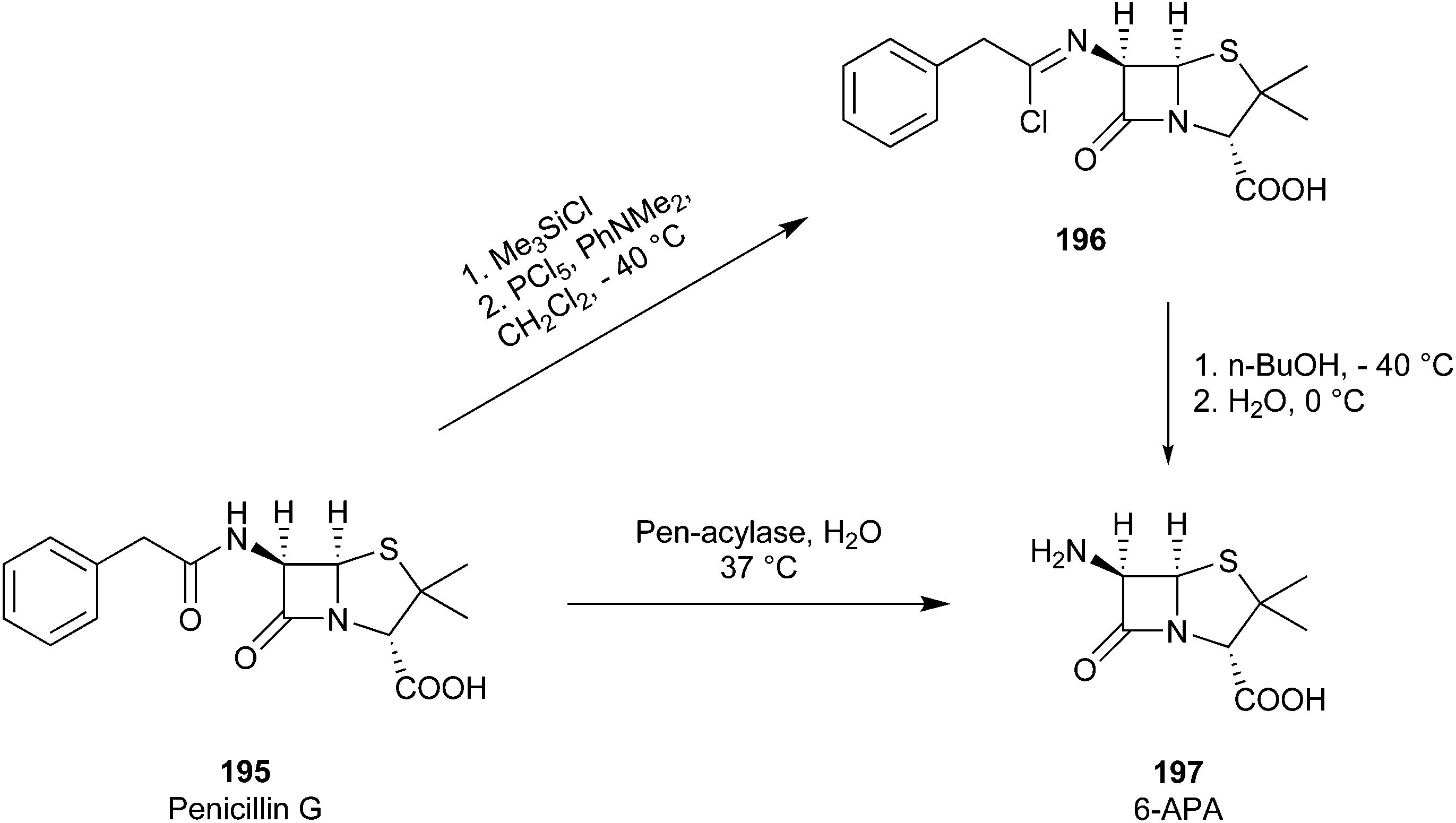 | ||
| Fig. 50 Conventional chemical vs. enzymatic deacylation of penicillin G.209 | ||
Laccases represent fungal enzymes belonging to the oxidase family, which catalyse the oxidation of a wide range of substrates, such as phenols, polyphenols, quinones, hydroxyindoles, and aromatic or aliphatic amines. Laccase-based oxidation involves the formation of radicals under the consumption of molecular oxygen and production of water as the sole by-product, therefore representing a greener alternative to traditional inorganic chemical oxidation.
Compared with standard oxidation techniques, laccase-catalysed reactions show enhanced specificity and a higher oxidation capacity for various substrates and reactions. The use of oxygen as an acceptor makes them a low-priced and effective tool for several types of important oxidative reactions such as aldol addition, Michael reaction, deprotection, cyclisation, and di- and polymerisation (Fig. 51).210–212 Another approach that gained recognition in medicinal laboratories is a chemoenzymatic system involving both laccase and the radical TEMPO in an aqueous buffer as oxidising agents.213 This procedure oxidizes various alcohols to their corresponding aldehydes/ketones or carboxylic acids and avoids the stoichiometric amounts of the chlorinated co-oxidants (NaO2Cl and NaOCl) plus hazardous solvents (CH2Cl2) usually employed in conventional TEMPO-oxidation (Fig. 51).
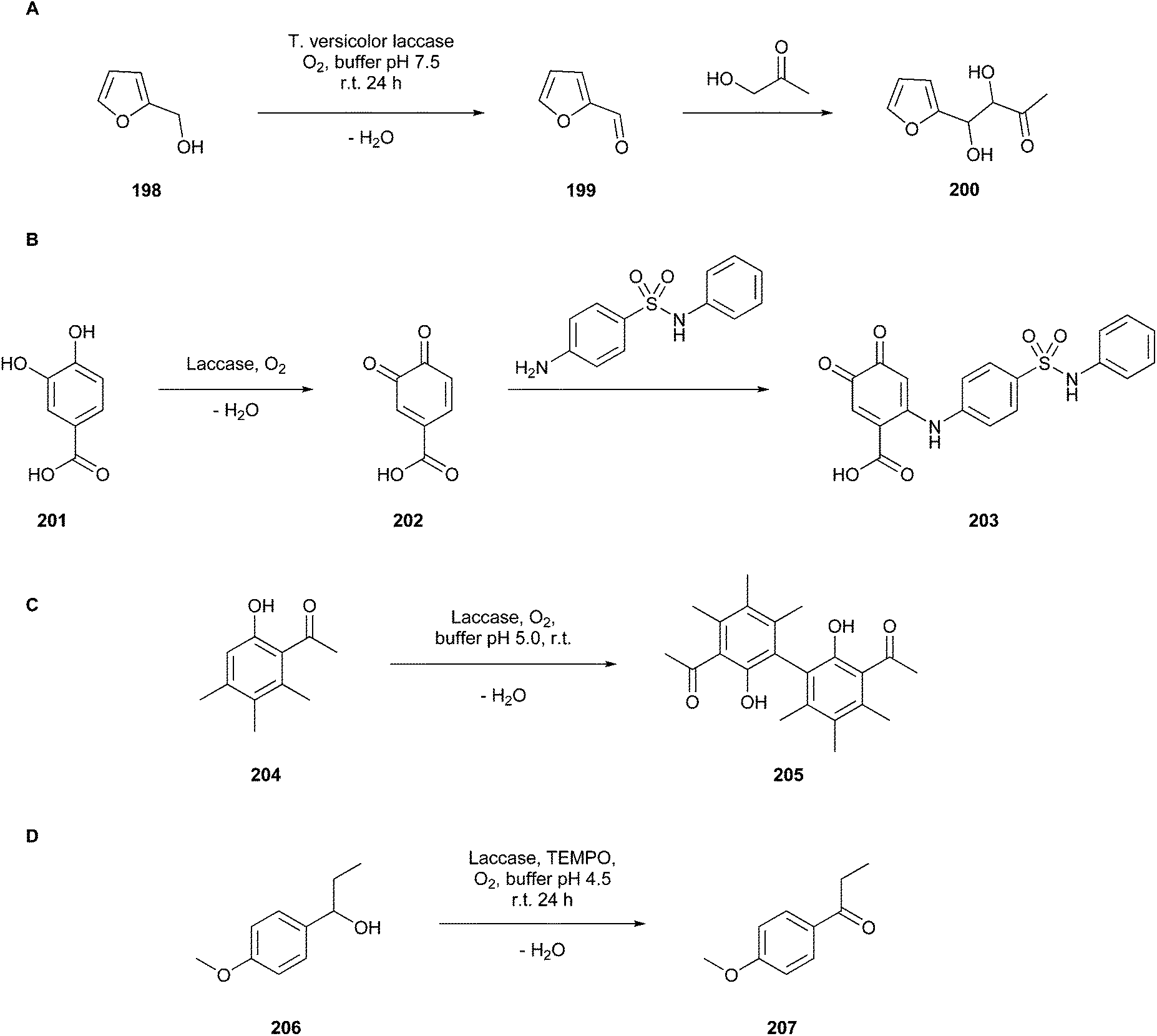 | ||
| Fig. 51 Examples of laccase-catalysed (A) aldol addition, (B) Michael reaction, (C) dimerisation, and (D) chemoenzymatic oxidation with TEMPO.211,214 | ||
Moreover, in 2021, Turner et al. reported the synthesis of N-substituted α-amino esters from the corresponding α-ketoesters (such as 208) via reductive amination catalysed by NAD(P)H-dependent imine reductases (IREDs) in aqueous buffer.215 Chiral, non-natural amino acids are essential building blocks for incorporating into numerous peptides and peptidomimetic molecules such as vancomycin and cyclosporin. Their current synthesis usually proceeds either via reductive amination with an excess of hazardous hydrides or N-alkylation with toxic chemicals. Turner et al. evaluated 12 different IREDs, with 5 of them being S-selective and 7 of them being R-selective. The enantiopreference of the desired N-substituted α-amino ester (210) could be achieved via the application of the required enzyme with high stereoselectivity and moderate to excellent yield, applicable to a wide range of substrates in the scale of grams (Fig. 52).
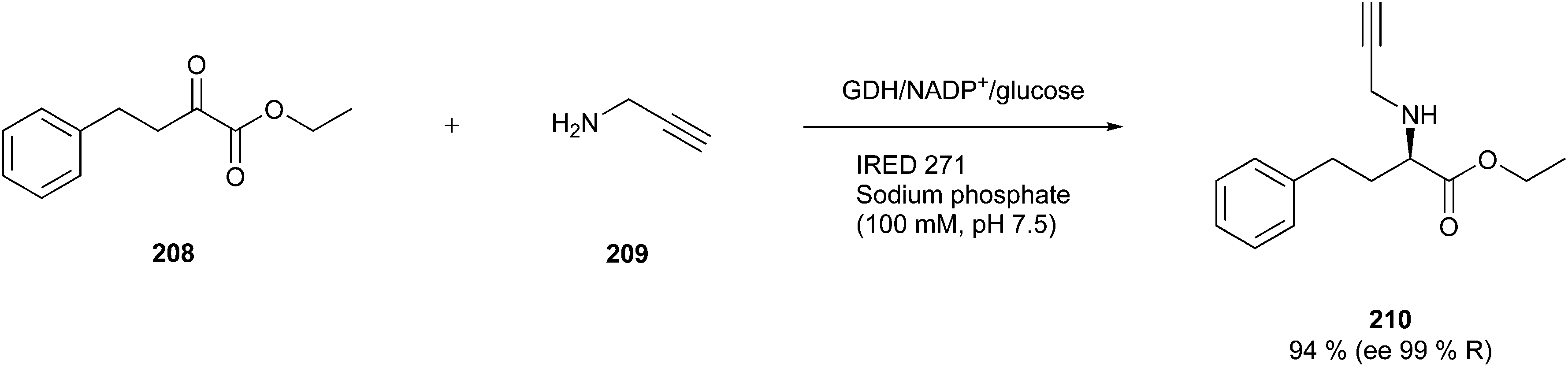 | ||
| Fig. 52 Stereoselective reductive amination of α-ketoesters via IRED-catalysis by Turner et al.215 | ||
The drawbacks of applying enzymes in medicinal chemistry include their limited stability, recovery, reutilization, and high cost. To enhance the stability and recyclability of enzymes, they can get immobilised as cross-linked enzyme aggregates (CLEAs). Therefore, the enzyme gets precipitated (e.g., ammonium sulfate), and the resulting aggregates get connected via cross-linking with a bifunctional reagent (e.g., glutaraldehyde).216 Numerous CLEAs of known enzyme classes such as proteases, aminoacylases, lipases, and many more were reported in manuscripts, increasing the recovery rate and stability of enzymes towards denaturation, proteolysis, and organic solvents.217,218
9.4. Catalytic amide bond formation in medicinal chemistry
Amide bonds are ubiquitous functional groups that appear especially as an essential connection in peptides and proteins, which build the basis of life. Therefore, it should not be surprising that this tendency is also observable in current drug research as numerous pharmaceuticals contain amides such as β-lactams and peptide structures such as the antidiabetic hormone insulin.219,220 It was shown that amidation represents the most significant and widespread reaction of pharmaceutical research in 2014, accounting for 50% of all chemical processes reported in manuscripts and 16% of all reactions performed in medicinal chemistry laboratories.6,7The general method for amide synthesis is the condensation between carboxylic acid (211) and amine (213), which is known as direct amidation. Unfortunately, this reaction is thermodynamically unfavourable, and therefore requires high temperatures (>140 °C) and the presence of molecular sieves (MS) for the removal of water.221 However, because of their instability, these harsh conditions are not feasible for many sensitive functional groups. Therefore, the acid needs to get activated using stoichiometric amounts of a coupling reagent, such as EDC, DCC, and thionyl chloride (Fig. 53A).222
 | ||
| Fig. 53 (A) Direct amidation via acid-activation and following amine-coupling.222 (B) Amide bond formation via Zr-catalysis by Adolfsson et al.223 (C) Large-scale boron-catalysed amide-synthesis in tert-butyl acetate by Sheppard et al. involving Dean–Stark water removal and solid-phase work-up.224 (D) Enantioselective synthesis of chiral (R)-amides via SpL.225 | ||
This necessity of derivatisation, large amounts of coupling reagents, and hazardous solvents (CH2Cl2, DMF, and THF) represent the major drawbacks of common amide bond formation. Several other strategies were already reported in the literature and promote the direct transformation of non-activated carboxylic acids in their corresponding amides.194,219,226,227 However, direct amidation remains the most common approach, although new and greener options are available to medicinal chemists.
Two research groups, namely Adolfsson and Williams, reported the approach of amide bond formation via Zr4+-catalysis.223,228 Williams synthesised different amides starting from the respective acids/amines in toluene under reflux conditions (110 °C). The reaction proceeded without the need for water scavengers, requiring only 5 mol% of the catalyst (ZrCl4 or ZrCp2Cl2), allowing functional groups such as ketone, nitrile, ester, and protecting groups to remain unaffected.228
However, Adolfsson and co-workers applied the latter strategy using mild conditions (THF, 70 °C, 2 mol% ZrCl4), tolerating a significant number of structurally different acid substrates (such as 215) without affecting the functional groups such as (thio-)ethers, acetals, and carbamates. Furthermore, the conversion of N-protected amino acids to their respective amides (such as 216) proceeded without any racemisation. Unfortunately, the addition of a molecular sieve was required for good to excellent yields (60–99%) because of the poor hydrolytic stability of Group (IV) metal chlorides in the presence of water (Fig. 53B).223
The hafnium(IV)-bis(cyclopentadienyl)bichloride-catalyst developed in Et2O at room temperature by the same research group allowed even a broader scope of carboxylic acids and amines to be converted in their corresponding amides. This method could enable modern peptide synthesis via a catalytic synthesis route instead of the applied stoichiometric direct amidation.229 Finally, the same research group also reported a water-tolerant system involving zirconium(IV) bis(cyclopentadienyl)bistriflate, which does not require any dehydrating agents.230
This makes these Lewis acidic metal compounds an exciting class of robust catalysts for further exploration. A disadvantage of many of these methods is the application of unsafe and non-polar solvents, such as aromatic hydrocarbons, chlorinated solvents, and ethers.226,229,231 This fact highlights the limitation of various strategies, which cannot be applied to high polar amines or carboxylic acids.
Sheppard and co-workers addressed this problem by employing a commercially available tris-(trifluoroethyl)borate catalyst, which enables the scalable transformation of polar amines and even unprotected amino acids to their corresponding amides in the polar, green solvent tert-butyl acetate.224 In addition, they developed their approach by applying the more efficient Dean–Stark water removal and a simple purification using a solid-phase work-up procedure with scavenger resins (Amberlyst and Amberlite), removing unreacted reagents and the boron catalyst (Fig. 53C). Therefore, the conventional three-step-amidation of amino acids, including protection and deprotection with various reagents, solvents, and purification, could be avoided.
In 2018, Li and co-workers reported the enantioselective aminolysis of ester and carboxylic acids with the aid of an intracellular lipase from Sphingomonas sp. HXN-200, namely SpL. They expressed the enzyme in E. coli and used either the whole cell as a biocatalyst in the presence of water at 16% (v/v) or the isolated free enzyme with a water concentration of 2–4% (v/v). Both enzymatic systems produced the corresponding amides (such as 221) in good to excellent yield (70–99%), even for weak nucleophiles such as aniline. Moreover, SpL could also catalyse the reactions with racemic ester or racemic amine, providing a new method for the enantioselective synthesis of chiral (R)-amides (221) in high ee, and therefore representing a potentially useful enzyme for the green synthesis of amides in medicinal chemistry (Fig. 53D).225
Besides, various other approaches concerning catalytic amide bond formation were also mentioned in the literature. These strategies employed, for example, solvent-free radiofrequency heating with nickel ferrite nanoparticles,232 sulfated titania nanoparticles,233 low-cost nano-MgO,234 metal-free graphene oxide,235 zeolites236 and enzymes.237,238
In summary, it is apparent that the modern medicinal chemist is not lacking alternatives and has a filled toolbox with efficient and greener options for organic syntheses, such as amide bond formation. These strategies often generate higher yields, higher stereoselectivity, and milder reaction conditions and do not require any activation or protection of functional groups. Thereby, they produce less polluting waste, leading to greener procedures. The respect and the deficient experience for new procedures represent significant issues for modern green synthesis, leading to a rigid adherence to older, common stoichiometric approaches. Therefore, the green medicinal chemist needs to be open to new catalytic alternatives, being environmentally and ecological friendly.
10. Biodegradation
The 10th Principle by Anastas et al. implies that chemical products should be designed so that they break down at the end of their function and do not persist in the aquatic and terrestrial environment.3,4 However, numerous chemical reagents and especially pharmaceuticals and their metabolites show poor biodegradation rates or no biodegradation at all. Thereby, organic pollutants are often classified as biodegradable, persistent, or recalcitrant. The increasing environmental harm due to the accumulation of the latter represents a major problem on Earth and results in negative publicity on medicinal chemistry and drug discovery.239The most important processes for the degradation of chemical compounds include hydrolysis in an aquatic environment, photolysis by sunlight, and aerobic or anaerobic biotransformation via bacteria or fungi, all enabling mineralisation of organic structures. Albert Einstein once said, “Intellectuals solve problems, geniuses prevent them”, and on this account, medicinal chemists need first to incorporate the idea of biodegradability into their structural design. Herein, we want to give a short overview of the tools supporting synthetic chemists concerning environmental degradation.
10.1. Databases containing biodegradation data
Firstly, numerous databases containing information about the known environmental development of molecules are available in medicinal chemistry. Biodegradation data depend highly on many parameters such as substrate structure, substrate concentration, microbial species and activity, temperature, salinity, pH, oxygen concentration, applied biodegradation tests, and others.The University of Minnesota Biocatalysis/Biodegradation Database (UM-BBD) contains information on microbial biocatalytic reactions and biodegradation pathways for primarily xenobiotic chemical compounds. This system presents individual reactions and metabolic pathways and gives information on the starting and intermediate chemical compounds, the microorganisms that transform the latter, the enzymes, and the genes. Furthermore, it includes a Biochemical Periodic Table (UM-BPT) and a rule-based Pathway Prediction System (UM-PPS), which predict the plausible pathways for the microbial degradation of organic compounds.240 In 2014, the rights were transferred to the Swiss water research institute Eawag, which was renamed as EAWAG-BBD, with current information on almost 1400 compounds, almost 1000 enzymes, more than 1500 reactions, and almost 550 microorganism entries.
One of the most extensive available collections of measured biodegradability data, namely the MITI database, covers the results of a single biodegradation test for nearly 900 commercial chemicals.241 The MITI-I test was developed in Japan, which nowadays represents one of the six standardised in vitro biodegradability tests described by OECD regulations (OECD 301C). It involves the inoculation and incubation of a specific concentration of test substance (100 mg L−1) with sludge (30 mg L−1) and the measurement of biological oxygen demand (BOD) during a test period of 28-days. The pass level for biodegradability is reached if the BOD amounts to ≥60% of theoretical oxygen demand (ThOD). Besides, other efficient databases and models for biodegradation design were mentioned in the literature and summarised in other reviews.242–244
10.2. Prediction programmes for biodegradation
If biodegradation data are not available for the chemicals of interest, then the application of prediction programmes at the screening level represents a suitable, low-cost, and fast alternative. These computer programmes rely on data from experimental test results, quantitative structure–activity-relationships (QSAR), or familiar metabolic pathways of structural analogues or substructures.242 The most popular is BIOWIN, a free programme in the EPA's EPIsuite software package. BIOWIN consists of a series of seven group contribution models, which estimate the aerobic and anaerobic biodegradability of organic chemicals and implicate results from the MITI database.245 Other programmes in the EPIsuite software, such as AOPWIN and HYDROWIN, also predict degradation by other pathways such as atmospheric degradation or hydrolysis, thus being an efficient supplement for further insights into the environmental development of a wide range of chemicals.4In addition, the hybrid programme CATABOL consists of both a knowledge-based system for predicting biotransformation pathways and a probabilistic model that calculates probabilities of the individual transformations. Two properties are unique for this model. Firstly, the biodegradation intensity is not purely calculated based on the parent structure but on the individual steps in the entire pathway, and secondly, CATABOL explicitly considers the effect of adjacent fragments.246
The limiting factor here is the variable accuracy of the results given from different databases or prediction models. Generally, they do not pass 80% accuracy, thus making some predictions unsteady and just partly reliable for the design of surely biodegradable compounds. A major reason for this is the poor quality and the lack of knowledge for a great deal of data and structure motifs.245 In this context, prediction accuracy is highly dependent on the current data quality, and the latter needs to be further improved as well as extended for a better understanding and prediction of biodegradation processes in the environment.
10.3. In vitro biodegradability-testing
One possibility to prove the correctness of the mentioned prediction programmes is the application of in vitro tests after synthesising the compound or the different substructures. In 1992 the Organization for Economic Cooperation and Development (OECD) test guidelines were published, and now accepted as the international standard and recommended by the EMA for the risk assessment of chemicals and pharmaceuticals. The OECD guidelines include a series of six tests for aerobic, ready biodegradation and vary in their conditions (inoculum, substrate concentration, light, and oxygen presence) or evaluation method, thus enabling the determination of diverse types of biodegradability.247Presently, the chemical properties (solubility, volatility, adsorption) determine the most suitable test method(s), where poorly soluble compounds can only be analysed by respirometry methods (B, C, D, and F) and highly volatile ones only with the closed bottle test 301 D (Table 3). Each test involves incubation for 28 days in a specific inoculum and mineral medium under aerobic conditions in light or dark conditions. Subsequently, the degradation is measured via the determination of the percentage of DOC (dissolved organic carbon), ThOD (theoretical oxygen demand), or ThCO2 (theoretical carbon dioxide yield).244 In addition, other procedures, including anaerobic biodegradation in digested sludge for 60 days and measurement via gas production (OECD 311), have also been established and reported.248
| Test | Analytical method | For substances that are | Pass at |
|---|---|---|---|
| DOC Die-Away (301 A) | Dissolved organic carbon | (Adsorbing) | 70% DOC |
| CO2 Evolution (301 B) | CO2 evolution | Poorly soluble | 60% ThCO2 |
| Adsorbing | |||
| MITI (I) (301 C) | O2 consumption | Poorly soluble | 60% ThOD |
| (Volatile) | |||
| Adsorbing | |||
| Closed Bottle (301 D) | Dissolved O2 | (Poorly soluble) | 60% ThOD |
| Volatile | |||
| Adsorbing | |||
| Modified OECD Screening (301 E) | Dissolved organic carbon | (Adsorbing) | 70% DOC |
| Manometric Respirometry (301 F) | O2 consumption | Poorly soluble | 60% ThOD |
| (Volatile) | |||
| Adsorbing |
Unfortunately, the reliability of these measurements needs to be taken with caution, considering the wide range of compartments and unclear number of microbial populations in the environment (soil, sludge, and rivers). Thereby, the actual degradation can differ significantly from their experimental or in silico-determined counterparts. Regardless, these tools represent an additional, efficient hint to assess potential breakdown processes, and therefore should be applied more often in the in silico design and in vitro testing of substances in medicinal laboratories.
10.4. Rules of thumb to assess biodegradation properties
The degradation of organic compounds depends on their structural features and functional groups, given that they can either hinder or favour their degradation via hydrolysis, photolysis, and, in particular, microbial biotransformation. Other properties that also play a significant role in the persistence of pharmaceuticals are lipophilicity/hydrophilicity and the size of molecules. The latter affects the uptake into bacterial cells, and therefore their own microbial breakdown pathway. In the last few decades, fundamental rules, known as Rules of Thumb, were established, which can help medicinal chemists in the environmentally friendly design of their substances. Table 4 summarises some structural rules that can enhance or disturb biodegradability.64,65,249,250| Enhancing biodegradability | Hindering biodegradability |
|---|---|
|
|
|
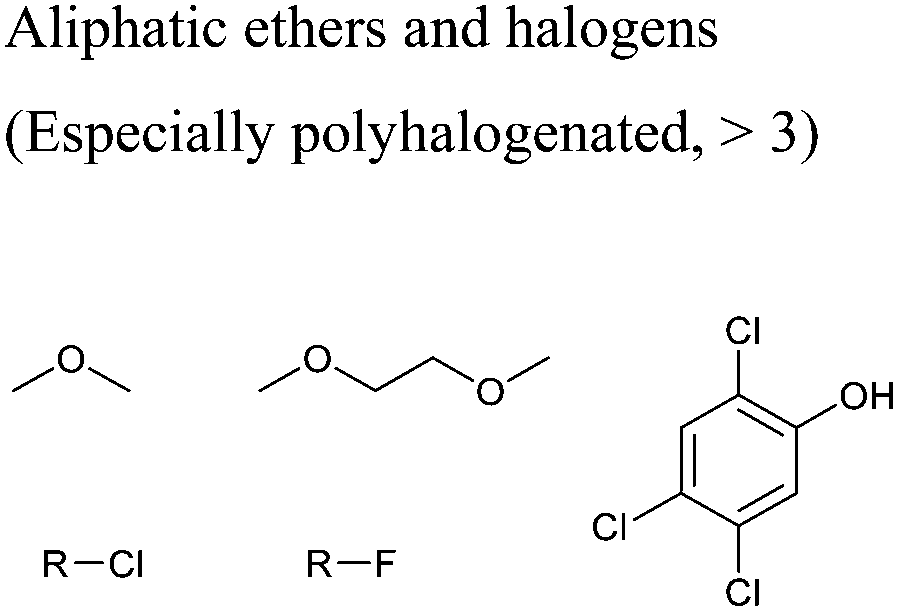
|
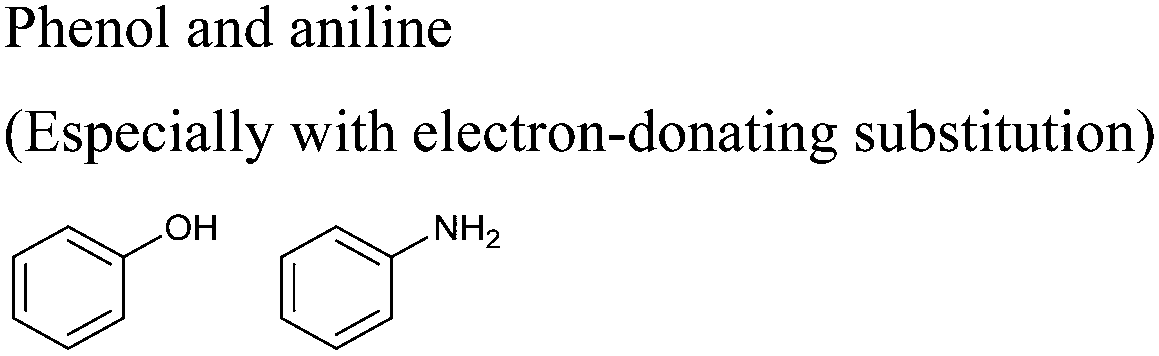
|
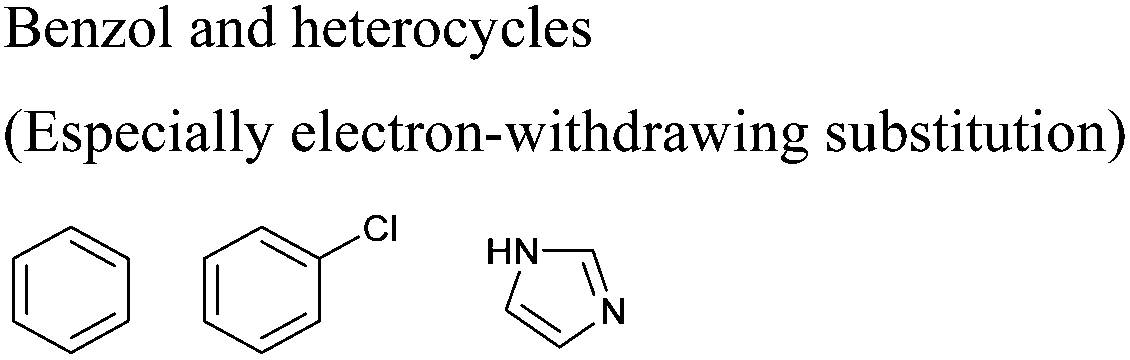
|
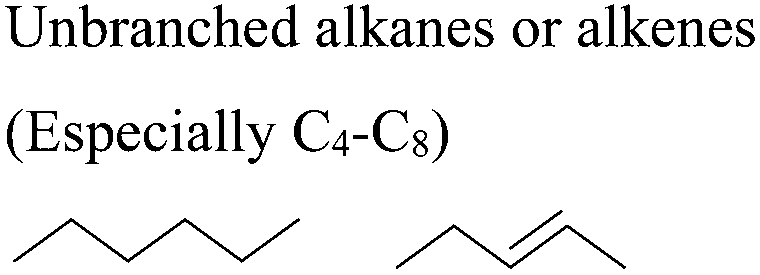
|
|
|
|
|
|
In an aerobic environment, one of the first degradation steps for active pharmaceutical ingredients (APIs) often represents enzymatic oxidation by bacterial oxygenase enzymes through integrating an electrophilic form of oxygen as an oxidant. Consequently, strongly electron-donating substituents such as phenols and anilines are more recommended than their withdrawing counterparts given that they simplify the conversion and often show higher biodegradability.252 However, polycyclic aromatics with more than 3 rings typically lower the biodegradation tendency.
Furthermore, branching, especially quaternary carbons, should also be avoided in the synthesis of non-persistent compounds because steric hindrance can highly hinder enzymatic conversion.64,249 Unsubstituted linear alkanes are degraded by terminal oxidation to a carboxylic acid, followed by β-oxidation to a C2-unit (acetyl-CoA), which often show higher biodegradation than branched alkanes (especially C4–C8).65 This tendency is also visible for nitrogen substitution, given that tertiary or quaternary amines usually have a lower tendency for degradation than their primary or secondary analogues. It is important to note that these rules just give a general trend and have some exceptions, given that numerous substances such as vitamin A, cholesterol, pantothenic acid, and ionic liquids show good biodegradation despite the presence of quaternary carbons/amines.246,249,250
Besides, it was mentioned that thiols typically hinder complete mineralisation, whereas thioethers favour it.251 Functional groups, which enable enzymatic hydrolysis, also play a significant role in a “benign by design” approach. Examples for this purpose are carboxylic and phosphate esters, which easily get degraded by unspecific and widespread esterases, leading to their full or partial breakdown. In comparison, ether, halogenated, and especially polyhalogenated motifs appear to lean towards persistence molecules.64,249
These facts remain a problem in the design of APIs given that the cleavage of labile esters can lead to the deactivation of otherwise active compounds through human metabolic pathways, such as the first pass-effect. In addition, halogens such as fluorine are often integrated into compounds via bioisosteric replacement to increase the metabolic stability. From a pharmaceutical and medicinal point of view, the priority in research laboratories is identifying and synthesising active compounds, showing promising results in both in in vitro and in vivo testing. Therefore, green medicinal chemists need to strike a balance between the bioavailability of the compounds in the targeted organism and their biodegradability after entering the environment.
10.5. Re-design and its application in pharmaceutical research
The possibility of re-designing known and persistent chemicals or pharmaceutical represents another tool for the green design of environmentally biodegradable APIs. For example, the chelating reagent EDTA (223) exists at higher concentrations in wastewaters and has become an environmental concern due to its heavy use and low biodegradability.EDTA (223) contains two tertiary nitrogen atoms, which are known to impede its biodegradability (Rules of Thumb),253 Accordingly, two potential substitutes with greener environmental properties have been reported in several manuscripts. The first is [S,S]-ethylenediamine disuccinate ([S,S]-EDDS, 224), an improved chelator compared to EDTA (223). In [S,S]-EDDS (224), the acetyl groups are moved from the nitrogen to carbon atoms, leading to non-persistent secondary amines. Interestingly, the stereo-isomeric [R,R]-EDDS and mixed isomers are not biodegradable.246 In contrast, all stereoisomers of the second green and biodegradable chelator sodium iminodisuccinate (IDS, 225) show comparable readily biodegradability (Fig. 54). Another green feature of IDS is that, unlike other aminocarboxylates, no hazardous HCN is used for its production.246
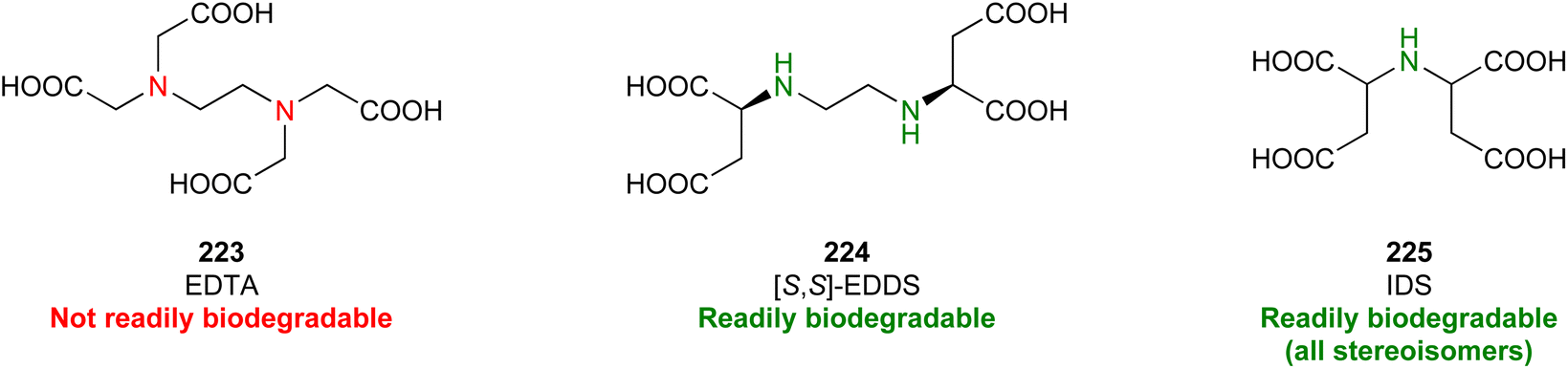 | ||
| Fig. 54 Structures of non-biodegradable EDTA and readily biodegradable [S,S]-EDDS and IDS. | ||
Ionic liquids have gained increasing interest in the last few years as a green alternative to conventional organic solvents. Besides their advantages in green chemistry (high recyclability, low volatility, low flammability, and low toxicity), they sometimes come with negligible biodegradability or persistent transformation products due to their quaternary amines. The biodegradation of numerous ionic liquids was investigated years ago, and it was shown that incorporating groups susceptible to enzymatic hydrolysis could enhance their biodegradability.254,255 The first readily biodegradable ionic liquid (IL) was reported in 2006 by Garcia and co-workers.256 This approach got adopted by Kümmerer et al. in the design of IL 4 (226), where the incorporation of an amide motif led to the favoured biodegradation of the pyridinium-derived IL with non-persistent degradation products (227 and 228) (Fig. 55).257 In this context, it is notable that IL 4 did not fulfil the OECD criteria for ready biodegradability (≤28 days) as complete mineralisation of the substance took an extended time of 42 days. Therefore, this field still requires further development of IL 4 (226) as a promising basic structure for the design of fully biodegradable, green ILs.
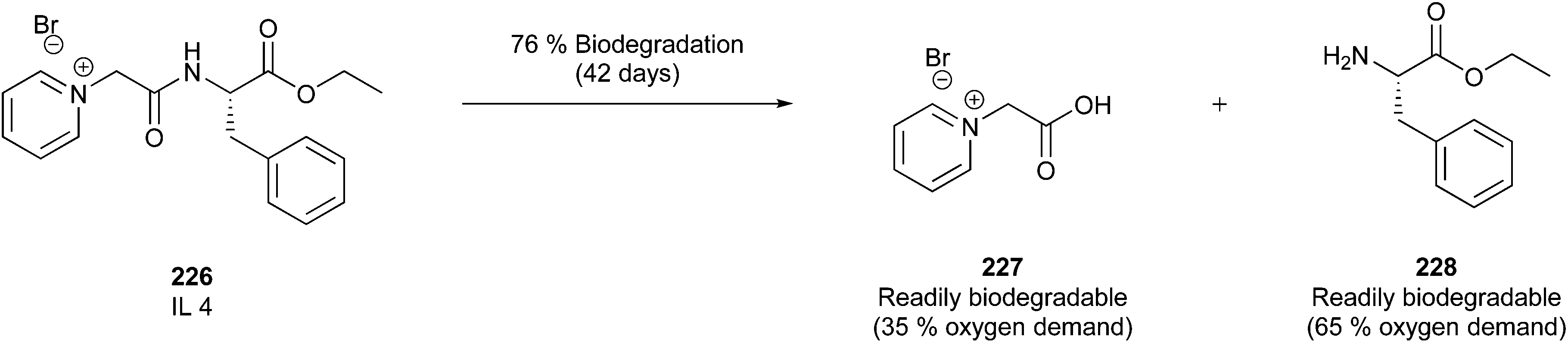 | ||
| Fig. 55 Ready biodegradation of IL 4 by Kümmerer et al.257 | ||
Kümmerer and co-workers also reported the re-design of three types of frequently used and non-readily biodegradable β-blockers, namely atenolol, propranolol, and metoprolol.258–260
The re-design of these drugs started with their photolysis, generating many photo-transformation products (photo-TPs), whose biodegradation was further investigated by aerobic biodegradation tests (301 D and 301 F) to assess the biodegradability of the derivatives in the environment. The ones with improved biodegradability and intact drug moieties (aromatic ring and β-ethanolamine) were selected, and then assessed with in silico molecular docking and in vitro analysis of their pharmacological activity. Final in silico prediction and investigation of the drug-like properties (adsorption, distribution, metabolism, elimination, and toxicity) led to the design of more biodegradable derivatives. The best re-designed candidates of metoprolol, atenolol, and propranolol (229–239) are presented in Fig. 56.
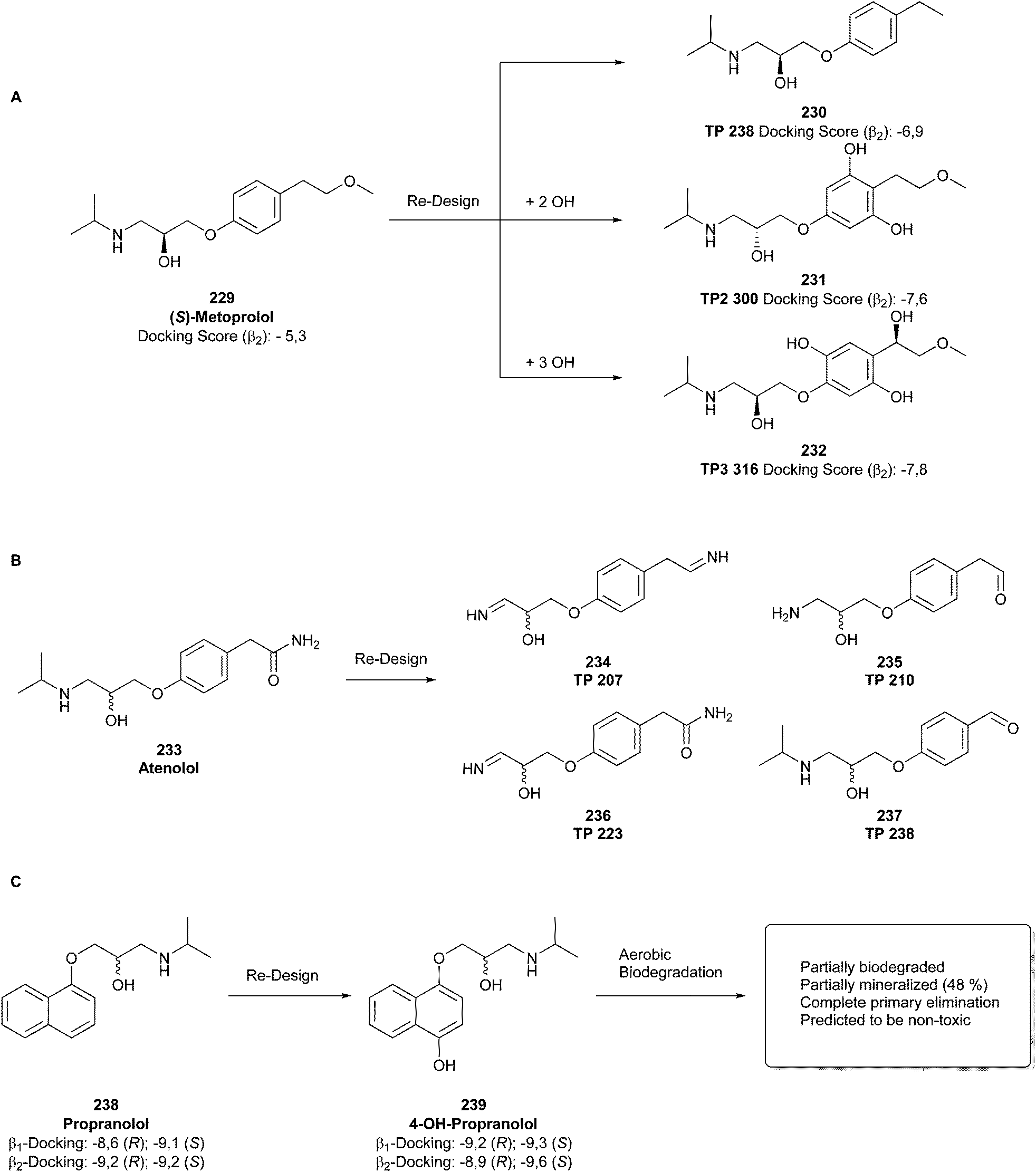 | ||
| Fig. 56 Most promising candidates in the re-design of β-blockers. (A) Metoprolol 229, (B) atenolol 233, and (C) propranolol 238 by Kümmerer et al.258–260 | ||
In the case of metoprolol, the re-design indicated 3 promising and more biodegradable alternatives, namely, TP 238 (230), TP2 300 (231), and TP3 316 (232), which showed even better docking scores than the eutomer of metoprolol (S-Form, 229) (Fig. 56A). However, the results need to be taken with caution given that the in silico tests were only performed using the β2-receptor, whereas metoprolol shows selective nature towards the β1 subtype receptor.258 For atenolol (233), the re-design brought several leading candidates (234–237) with comparable or better biodegradable properties, and they were predicted to be drug-like, orally available, and non-toxic compounds (Fig. 56B).259 Finally, 4-OH-propranolol (239) represented the greener option of propranolol (238) with similar or enhanced docking scores on both β-receptors plus favoured degradation and mineralisation (48%) without any toxicity prediction (Fig. 56C).260
However, even though this approach provides the rational green design of a given pharmaceutical, the structures in these studies represent theoretical designs and require further in vitro testing and experiments in the future.
The spread of persistent antibiotics in the environment causes the formation of an increasing amount of resistant bacterial strains and poses a significant threat to human health. Therefore, the design and synthesis of environmentally friendly antimicrobial agents represent a promising option for preventing antibiotic exposure in wastewater and soils. Ciprofloxacin (CIP, 240) is one of the most active and widely used fluoroquinolone antibiotics, which is known to be non-degradable and persistent.261
When these key data are not available, other types of available information on the degradability of a substance can be used to decide if further testing is needed to assess its potential persistence.
Kümmerer and co-workers occupied themselves with a solution for the latter problem and the re-design of ciprofloxacin. In this context, they re-designed CIP (240) through systematic variation of functional groups on the core structure, using in silico and in vitro methods. The most promising and recently patented candidates, Cipro-Prolin (241) and CIP-Hemi (242), are shown in Fig. 58. The latter showed only slightly inferior activities than CIP against several bacteria strains (MIC in the μg mL−1 range). CIP-Hemi (242) showed favoured biodegradation via hydrolytic cleavage to the persistent fragment CIP-d-CP (243) and a biodegradable linker under acidic conditions (Fig. 57). CIP-d-CP (243), although not readily biodegradable, possesses highly reduced antibacterial activity, thus possibly decreasing selection pressure in the environment. CIP-Hemi (242) maintained its required metabolic stability in numerous tests, and both CIP-Hemi (242) and CIP-d-CP (243) showed weaker cytotoxic, mutagenic or genotoxic effects compared to the parent compound CIP (240).249,262
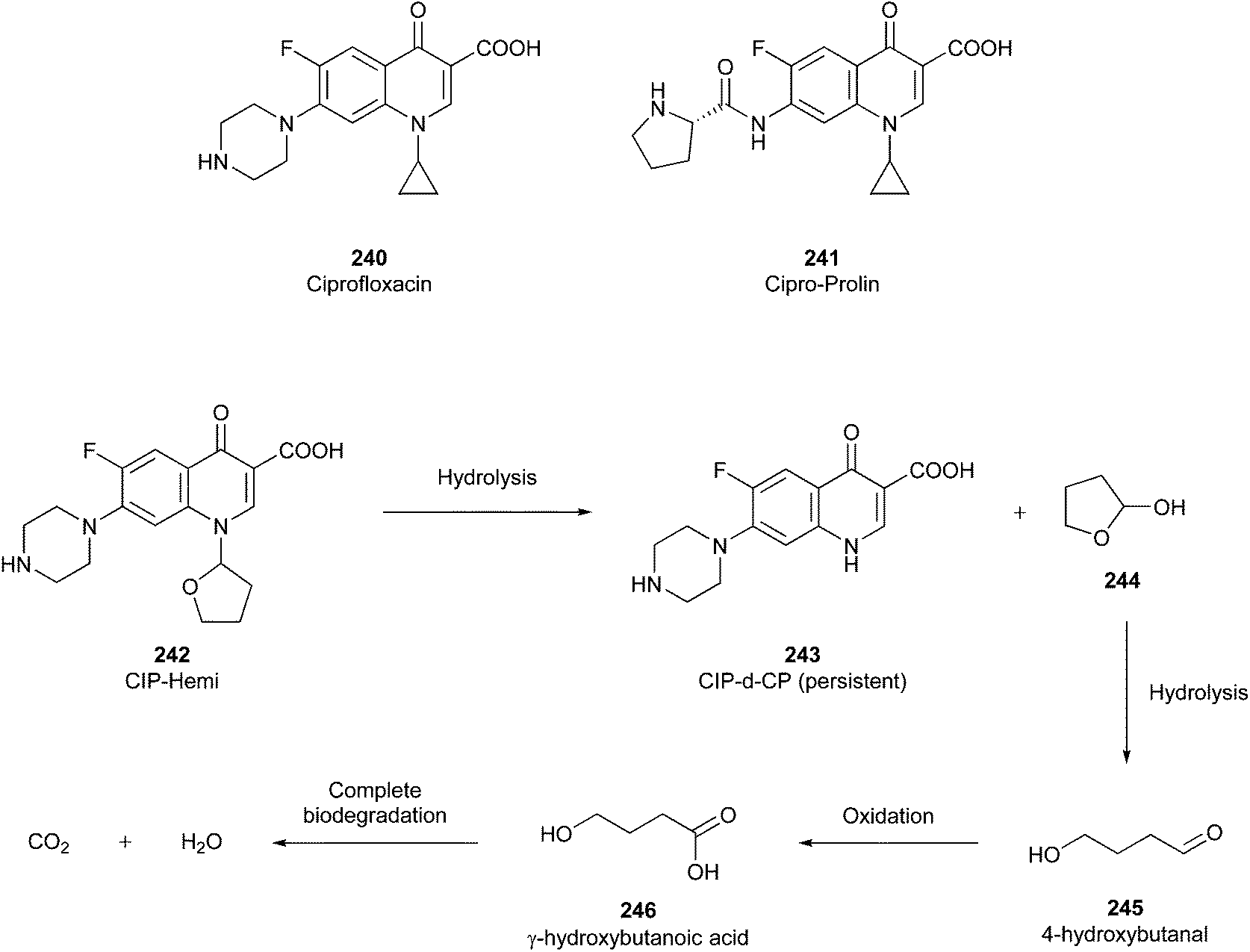 | ||
| Fig. 57 Structures of ciprofloxacin (240) and re-designed Cipro-Prolin (241) and CIP-Hemi (242) including their degradation.249,262 | ||
 | ||
| Fig. 58 12 Principles of GAC with mnemonic SIGNIFICANCE, established by Namieśnik et al.263 | ||
11. Real-time analytical chemistry applications towards greener chemistry
Analytic methods serve to elucidate chemical structures, particularly after their synthesis, and therefore, represent an essential tool in medicinal laboratories. Chemical analysis requires solvents, reagents, energy and generates large amounts of waste. In this context, GAC was coined by J. Namieśnik for the development of new analytic techniques to reduce their environmental impacts.32 The 12 Principles by Anastas et al. are just partially applicable for green analytical chemistry, with the most fitting being the following:• Principle 1: Prevention of waste generation
• Principle 5: Use of safer solvents and auxiliaries
• Principle 6: Energy efficiency
• Principle 8: Reduce Derivatives
• Principle 12: Accident prevention through safer chemistry
In 2013, Namieśnik and co-workers also expanded the latter and established 12 new Principles for GAC with the SIGNIFICANCE mnemonic, as presented in Fig. 58.263 Most of the latter involve elimination or minimising the number of samples, reagents, energy consumption, waste, hazards, and derivatisation required in several analytic procedures. In this context, direct analytical techniques, in situ measurements, or miniaturisation of analytical tools should also be favoured. However, GAC could lead to declining performance parameters such as accuracy, precision, and sensitivity. Therefore, GAC in medicinal chemistry needs to strike a compromise between the reliability of analytic methods and the GAC requirements. Processes that are not monitored cannot be controlled and optimised.
White Analytical Chemistry (WAC) was recently introduced and expanded the common GAC Principles by additionally considering the validation criteria (accuracy, precision, and sensitivity) and practical, economic aspects (cost and speed of analysis and operational simplicity) of the actual analytical process. According to the latter, it is important to strike a balance between greenness, purpose, and functionality of the employed methods.264
Usually, chemical analysis requires several steps for pre-treatment for actual analysis. Accordingly, the generation of analytical waste is mainly linked to the applied analytical method. Herein, the measurement variants depend on the distance of the analysis instrument compared with the sample location (Fig. 59). Firstly, in-line measurements are performed directly during the process through direct contact with the measuring head with the reaction mixture. This is often achieved by sensors, which continuously produce data, and thus highly recommended by the GAC Principles (in situ measurements). Online methods also produce continuous data but are separated from the reaction solution by a bypass, into which the sample is automatically transported. This is usually done when the analytical device cannot withstand harsher temperatures, pressures, or hazardous reagents.
 | ||
| Fig. 59 Measurement variations for analytical investigation.265 | ||
At-line represents a variant where the instrument is near the reaction place, at which samples are taken manually by the operator and brought to the device for further investigation. Offline approaches differ from at-line because the analytical device is not available in the same laboratory but in an external one. To analyse them properly, at-line and offline measurements include sampling and another dilution step with the additional use of potential solvents. Compared with the in-line (seconds) or online (seconds to minutes) variation, the time delay between sampling and the analytical result for at-line (minutes to hours) and offline (hours to days) measurements is highly increased. Therefore, it is not recommended to survey dynamic processes.265 In contrast, manual pre-treatment is sometimes highly required and often enables higher precision for further analysis. Therefore, the ideal measuring method depends on several properties (nature, concentration, and by-products) of the sample itself and cannot be standardised.266
11.1. GAC metrics
Due to the unmanageable amount of analysis options, the green assessment and comparison between them prove to be rather tricky. In this regard, several metrics have been developed for the greenness evaluation of analytical processes. A short overview of the most important ones will be given in this chapter. More details about the following and other GAC metrics can be found in a recently reported review.267 | ||
| Fig. 60 General description of National Environmental Method Index.268 | ||
Modified semi-quantitative NEMI pictograms with a colour scale from red (environmentally unfriendly) over yellow (moderate) to green (benign) coloured fields were also proposed.265
 | ||
| Fig. 61 Composition of GAPI pictogram (left) and evaluation of LLE (right).25 | ||
GAPI covers more aspects than NEMI or AES, but its main drawback is that it does not consider any processes before the extraction, such as the reagents and solvents used in the synthesis. Additionally, the amount of reagents/solvents and the waste generation is assessed as inadequate if more than 10 mL if required or produced. In this context, 11 mL is rated equally bad as 1 litre of waste.267
In 2021, the GAPI pictogram was extended with an additional hexagon, incorporating different aspects of the pre-extraction procedures such as yields, conditions, instrumentation, reagents, and solvents employed in the synthesis. In this context, the tool got renamed ComplexGAPI (complementary green analytical procedure index),33 which differs from the previous GAPI metric because it considers an additional field represented with a supplementary hexagon in the graph, corresponding to the processes performed prior to the sample preparation step and final analysis. These processes include the yield, conditions, reagents, solvents, relation to green economy, instrumentation, work-up and purification, and are evaluated for their environmental impact with colours as in GAPI, where red, yellow or green stand for high, medium or low environmental issues, respectively.33
11.2. Sample preparation
Direct measurements without any sampling or sample treatment represent one of the GAC Principles but are not always employable. In these cases, processes such as offline or at-line collection and sample preparation of the material are required even before the acquisition of the analytical measurements. In some cases, these preparations involve additional treatments such as extraction, filtration, separation, and derivatisation and present an unavoidable step for further analytical investigation. Herein, extraction illustrates the most widely used work-up procedures in numerous reported protocols.Especially, SFE is of high interest in GAC, which uses cheap and harmless supercritical fluids such as supercritical CO2 as green alternatives to common solvents. Supercritical fluids have favourable transport properties because of their high diffusivity through solid samples.275 They also possess the advantage that their density can be altered by changing the temperature or pressure of the analytical system.
Given that density is linked to solubility, the mass transfer during extraction can be modified, and the extraction time is reduced. The major disadvantage of supercritical CO2 is the poor solubility of highly polar compounds, which can be circumvented by adding benign modifiers such as ethanol or water.276
Dispersive liquid–liquid microextraction (DLLME) poses another emerging method for green extraction. DLLME describes a simple, miniaturised LLE-variant with low solvent consumption and waste minimisation. Therefore, it represents a rising, green, automatable, and cost-effective opportunity for GAC in the field of sample treatment.277 The general procedure of DLLME involves the injection of the dispersive and extraction solvent into the aqueous solution containing the analytes. The resulting cloudy solution is then centrifuged, leading to the accumulation of the extraction phase in the form of a single droplet with the required analytical material. Generally, in DLLME, the droplet is sedimented on the bottom of the tube and can easily be removed afterwards. Low-density solvent-based DLLME (LDS-DLLME) represents an alternative method with low-density solvents (hexane, pentane) and flotation of the extraction phase (Fig. 62).278 Furthermore, DLLME procedures with less environmental impact, such as ionic liquids or deep eutectic solvents, connecting several eco-friendly aspects, have been achieved in the last few decades.279,280
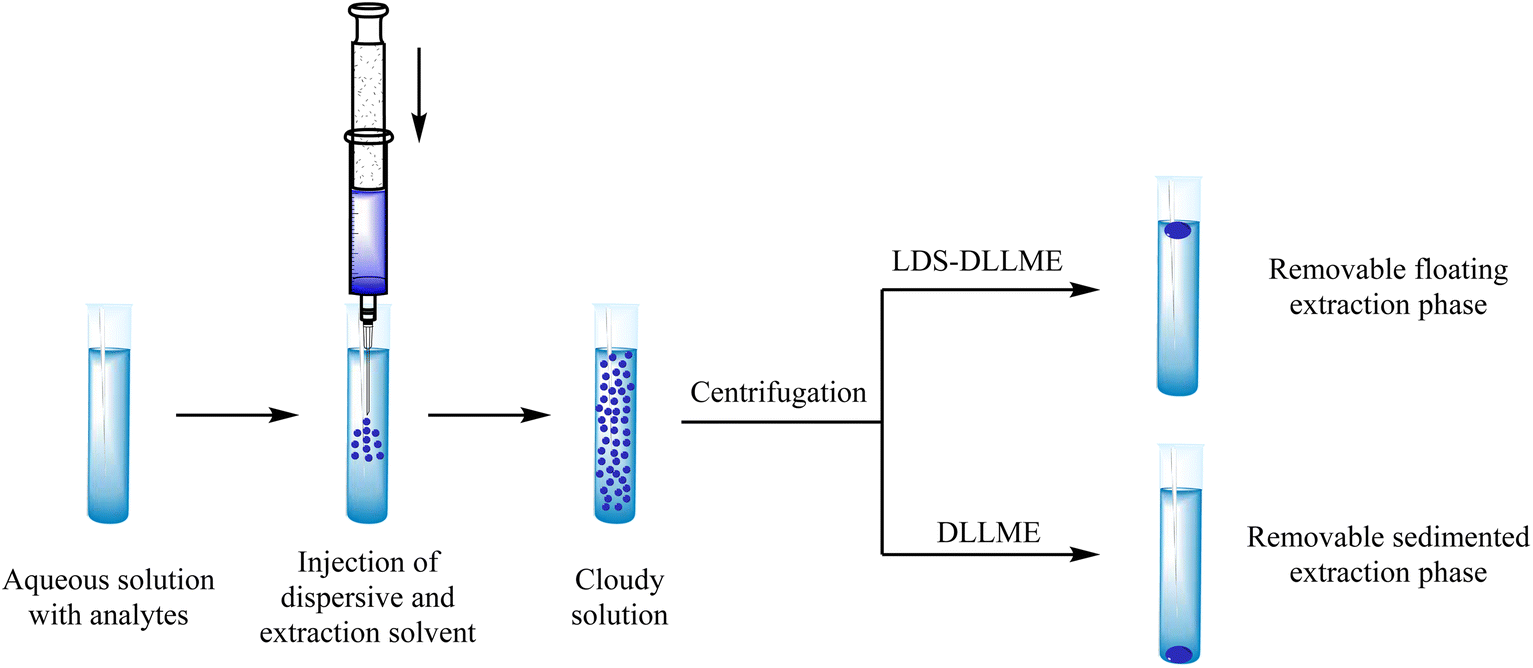 | ||
| Fig. 62 Schematic general process of DLLME and LDS-DLLME.278 | ||
Gas chromatography (GC) represents a greener alternative to standard LC given that it eliminates the use of dangerous organic solvents and employs innocuous, non-flammable, inert gases as mobile phases, decreasing the solvent consumption and increasing the operator safety. Furthermore, recent developments enabled green methods such as micro-GC (μ-GC), two-dimensional GC (GC × GC), and IL-packed GC-columns.282–284 However, GC usually requires the analyte material to be at least semi-volatile and stable to high temperatures, thereby being unsuitable for a wide range of substances.
One possibility to increase the greenness of liquid chromatography is the replacement of common organic solvents with sustainable, benign mobile phases. Similar to extraction, supercritical fluids are applied in modern chromatography, which is known as supercritical fluid chromatography (SFC).285,286 Due to their properties (low density, low viscosity, and high diffusivity), supercritical fluids such as CO2 or water enable higher mass transfer between stationary and mobile phases, thus leading to improvements in terms of efficiency and speed (15 min < 45 min) compared to standard HPLC.275 Fitch et al. compared the greenness and the performance parameters of RP-HPLC and SFC, showing that SFC separations are often favourable. However, regarding energy consumption, SFC necessitates higher requirements than HPLC, as constant heating or cooling of the mobile phase is needed.287
Other eco-friendly methods involve the use of ionic liquids, ethanol mixtures, and superheated water as benign solvents.281,288 Ethanol can be used as a green alternative to hazardous acetonitrile in LC, given that it possesses similar properties to the latter, such as elution strength and low UV cut-off, but is less toxic, less volatile, and low-cost. The only problem is the high viscosity of ethanol mixtures, which is linked with high-back-pressures in the chromatographic system.281,289 Superheated water at a temperature of 80–250 °C is also a cheap and innocuous alternative to common methanol–acetonitrile mixtures because of its tuneable polarities at different temperatures. However, its major drawbacks include incompatibility with thermolabile or highly hydrophobic substances and the necessity of temperature-resistant stationary phases.289,290
Another green approach is represented by micellar liquid chromatography (MLC) with an aqueous mobile phase containing surfactants above their critical micelle concentration (CMC).285 Accordingly, MLC forms a pseudo-stationary phase and enables direct injection of samples, given that micelles can even solubilise complex analytical material such as proteins, whereas surfactant monomers coat the substances, avoiding their agglomeration. This technique shows high reproducibility, low-cost, rapid gradients, and the employment of biodegradable, safe surfactants such as sodium dodecyl sulphate (SDS).291,292
Reducing solvent consumption represents a further strategy for more environmentally friendly analytical separation. The techniques involve miniaturisation of column lengths, shorter run times, and higher pressures. Examples of miniaturised chromatography are capillary-LC (cLC), micro-flow-LC, and nano-LC.291 One of the most addressed is cLC with narrow bore columns (<0.5 mm), enabling high separation efficiency, low flow rates (<10 μL min−1), and small waste production.285 However, smaller columns with decreased particle size usually result in higher back pressures. This drawback can be circumvented by ultra-high performance liquid chromatography (UHPLC), applying high pressures with reduced column lengths, smaller particle sizes, and shorter analysis times. A recent study highlighted the greenness of UHPLC by developing an eco-friendly UHPLC method with benign solvents for determining toxins in cereal products.293 Furthermore, Mohamed et al. compared the greenness profile of three HPLC methods, i.e., RP-HPLC, MLC, and UHPLC, to quantify sulfadiazine and trimethoprim in bovine meat and chicken muscles. Assessment through NEMI, AES, and GAPI metrics demonstrated the superiority of MLC and UHPLC over conventional RP-HPLC in terms of eco-friendliness.294
11.3. Analytical methods
Nowadays, a wide range of monitoring instruments is available for the green analysis of substances in medicinal laboratories. In the case of unavoidable sample treatment, sequentially coupling preparation methods with analytical devices such as UV-VIS- or mass-spectrometers is highly recommended, thus reducing time delays between sampling and analysis. Tang and co-workers established a sensitive analysis method for glycopeptide antibiotics by online coupling an ionic liquid-based microextraction to a cLC and amperometric detection.295 Accordingly, the micro-treatment and analysis proceed consecutively without the need for additional preparation or dilution steps.A reduction in the sample number and size indicates another mentioned strategy in GAC Principles. However, depending on the nature of analytes and synthesis, the correct and frequent collection of samples represents crucial steps in analytical procedures. Sample reduction can lead to significant losses in terms of accuracy, selectivity, sensitivity, precision, and representativeness.
Direct measurements remain one of the greenest strategies, avoiding pre-treatment and transport of the analytical material. Additionally, sample contaminations plus material losses are avoided and the operator safety is improved. In-field analysis significantly reduces the periods between chemical and analytical processes, favouring faster decision-making and prompt action in dynamic processes. Thereby, the application and improvement of direct determination pose a critical, essential approach for medicinal chemists. Examples of direct analysis involve vibrational spectroscopy, such as near-infrared (NIR), mid-infrared, and Raman spectroscopy.296 Non-invasive measurement without sample transformation, integration possibilities into flow-analysis systems, and the availability of portable, miniaturised instruments make vibrational spectroscopy a promising, eco-friendly option for in-line or online determination of pharmaceuticals in medicinal chemistry.297,298 Besides, IR and Raman spectroscopy enable both solid and liquid samples to be investigated without bigger pre-analytical steps. Recent developments resulted in more sensitive methods such as surface-enhanced Raman scattering (SERS), spatially offset Raman spectroscopy (SORS), and attenuated total reflectance-IR (ATR-IR).296 Modern Raman devices with laser emission at 785 nm also circumvent disturbing the sample fluorescence and thermal sample degradation by older instruments.298
X-ray fluorescence (XRF), a method usually related to archeologic analysis, can also be used for non-destructive and direct analysis in pharmaceutical research. Developments in this field resulted in highly accurate systems such as wavelength-dispersive- (WD-XRF) and energy-dispersive X-ray-fluorescence (ED-XRF) measurements. The main advantages of XRF are fast measurements, elemental analysis at trace levels, and the possibility of measuring a wide range of sample types (solids, powders, liquids, metals, etc.). Advances in portable and miniaturised instruments enabled faster in situ determinations, making XRF interesting technology for GAC applications.299,300
UV-VIS and nuclear magnetic resonance (NMR) represent other widely used spectroscopy techniques in medicinal laboratories. Unfortunately, both usually require sample dilution and the employment of hazardous solvents (chloroform, methanol, and benzene). However, through the decades, modern procedures have been established in these areas, offering environmentally friendly methods for drug research.297,300 Recently, the simultaneous UV-detection of a multi-component mixture with benign phosphate buffer as an alternative solvent to common methanol was evaluated. This method showed perfect scores with NEMI and AES and excellent scores with AGREE assessment.301 Recent developments in portable, low-field NMR technology enabled non-invasive analysis, monitoring of flowing material and in situ measurements in contrast to high-fields analogues.302 Soyler et al. reported the online analysis of sucrose hydrolysis with a benchtop NMR instrument in real time. In addition, the setting can be carried out with protonated solvents and tailored water suppression techniques instead of costly deuterated solvents in conventional NMR spectroscopy.303
Nowadays, chemical sensors attract considerable attention in drug research given that they perform real-time analysis, while being cheap, portable, and non-invasive. Furthermore, they are compatible with different detection modes (electrochemical and optical), easy to use, reusable, and reduce the sample size, determination time, and waste generation.304 Carbon dots (CD), carbon nanoparticles (<10 nm) with unique fluorescent properties, need to be especially highlighted. They exhibit the advantages of simple synthesis, low cytotoxicity, high sensitivity, extensive conductivity, and high stability.305 The application of CDs mentioned in several manuscripts is mainly based on their fluorescence, at which CDs coupled with electrochemical detection, especially amperometry, have also been reported.306,307
Other approaches of high interest in GAC involve flow analysis, advanced electrochemistry, and the employment of smartphone technology for colourimetric detection.308–310 Although advances are still required, numerous tools and methods concerning green sample preparation and analysis are already available in medicinal laboratories. They enable a reduction in the amount of solvent used, energy consumption, and waste generation, and in situ measurements, making GAC more friendly in terms of environmental and ecological aspects.
12. Accident prevention
The risk of accidents persists daily in medicinal laboratories due to the hazardous nature of various chemicals. Casualties such as the Bhopal disaster, the Love Canal accident, or the recent explosion in Beirut caused destruction, injuries, and deaths. This should remind scientists that chemicals need to be handled with care, but serious incidents still occur. For example, between 2001 and 2018, more than 120 laboratory accidents were reported in the United States, although the absolute numbers are higher.311Primarily, the first critical safety aspect is the operator themselves, given that the correct handling usually avoids unexpected situations. The assessment of potential risks, hazard identification, and the use of appropriate protective clothing is a mandatory step prior to their employment. A deterrent example is given by the death of Sheri Sanji, suffering from burns caused by working with tert-butyllithium without wearing a lab coat.312 Poor safety culture and compliance are significant reasons for potential incidents. A recent Canadian study demonstrated that more than a quarter of the participants do not conduct any risk assessment before lab work, just 40% wear their protective equipment during lab work, and nearly every tenth participant lacks safety training.313 Thereby, many projects, guides, and approaches encourage sharing of safety learnings, thus creating an information collection and open communication for accident prevention.311,314,315
The assessment of risks starts with the identification of hazardous substances and highly energetic functional groups (HEFG). Herein, functional groups such as acetylenes, azides, perchlorates, peroxides and oximes represent potential, explosives and need to be handled with care when implemented in the substance structure.316,317 These HEFGs can be identified via personal chemistry knowledge or several literatures like Bretherick's Handbook of Reactive Chemical Hazards and Stoessel's Thermal Safety of Chemical Processes.318 The latter provide a detailed summary about hazardous properties of individual substances or in combination with other chemicals. Another theoretical method for the assessment of hazards is the calculation of oxygen balance (OB, OB%), indicating the oxidizability of explosives. Thereby, a negative oxygen balance leads to incomplete decomposition and production of carbon monoxide, whereas a positive oxygen balance induces complete combustion under production of carbon dioxide. Oxygen balances close to zero lead to powerful explosions, often resulting from the mixture of different explosives. The equation for the OB is presented below (where X = number of C-atoms, Y = number of H-atoms, Z = number of O-atoms and M = number of metal-atoms).319 Thanks to online OB calculators, the oxygen balance of compounds is easily generated, serving as a quick tool for the rough evaluation of potential explosive properties.
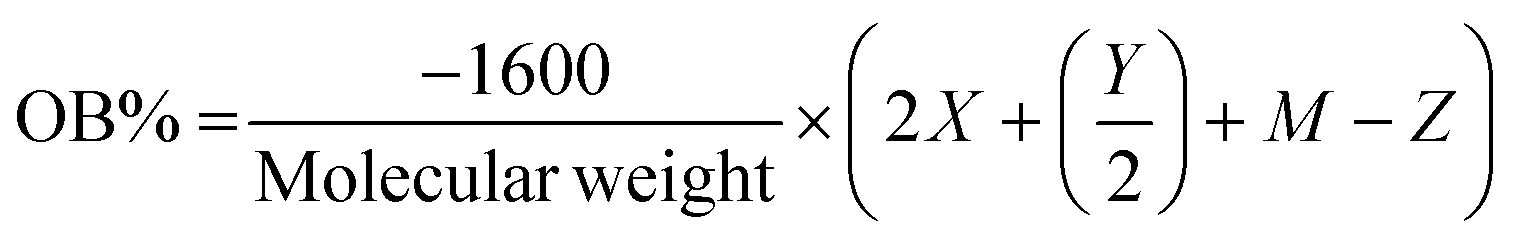
Furthermore, thermal hazard assessments such as differential scanning calorimetry (DSC), thermogravimetric analysis (TGA) and accelerating rate calorimetry (ARC) serve as efficient, practical tools for the safety evaluation of chemicals and pharmaceuticals. For example, diazo compounds represent one of the most important building blocks in organic and medicinal chemistry given that they enable numerous transformations by thermal, metal-catalysed or photoinduced carbene formation. Unfortunately, they often need to be avoided or treated with caution due to their instability and explosive behaviour.320 Green et al. evaluated the thermal stability of several diazo compounds and diazo transfer reagents with TGA, ARC and DSC, providing a useful dataset for academic and industrial research. Some candidates (246–250) and their thermal assessment are presented in Fig. 63.321
 | ||
| Fig. 63 Thermal assessment of selected diazo compounds by Green et al.321 | ||
The data indicated that the stability of the investigated azo substances was enhanced with electron-withdrawing substituents (–CF3 and –NO2), especially in the para position, leading to Tonset of about 120–135 °C. Alternatively, electron-donating groups (–OMe and –Me) showed the opposite, destabilizing effect on the diazo moiety (Tonset = 86 °C), which could be reduced by placing the substituents in the meta-instead of the para position.
The autocatalytic decomposition of DMSO reported in several recent publications is another example for the necessity of risk identification.322–324 Tian et al. reported a rupture disc, happening during a calorimetric test of a hydroamination process (from 251 to 252) with dimethyl sulfoxide as the solvent.324 Herein, the occurring exothermic reaction led to an increase in temperature and pressure with violent gas release, potentially resulting in high energy release and explosions (Fig. 64). In the same year, studies by two other research groups showed that the induction period of DMSO decomposition gets shortened by a wide range of substances including acids, bases, halides, metals, and electrophiles, leading to its decomposition at much lower temperatures.322,323 These findings demonstrate the potential safety concern of dimethyl sulfoxide as a chemical and highlight the need for either thermal profile evaluation of DMSO mixtures or their replacement with greener and safer chemicals.
 | ||
| Fig. 64 Rupture disc burst reported by Tian et al. due to thermal decomposition of DMSO.324 | ||
Guan et al. identified a risky thermal incompatibility between N-bromosuccinimide (NBS) and 2-MeTHF during the safety assessment of a bromination process (253 to 254). The hazardous side reaction between the latter was studied via different calorimeters illustrating a critical, exothermic reaction with low onset temperature (45 °C). Optimization of the reaction conditions (15 °C instead of initial 25 °C) and portion-wise addition of NBS (control of NBS accumulation), allowed the safe and successful scale up (106 kg) of the bromination approach (Fig. 65).325
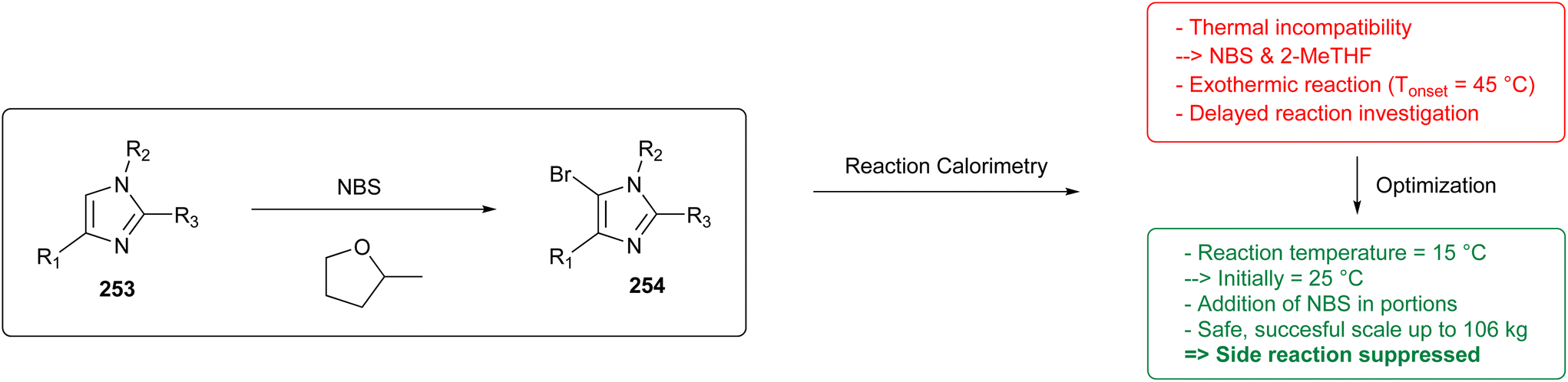 | ||
| Fig. 65 Optimization of NBS/2-MeTHF-incompatibility in bromination process.325 | ||
Other manuscripts regarding safety evaluation of substances have also been reported and again highlight the importance of hazard assessment as a safety precaution for potential accidents in academic research laboratories.326–329
Another approach for safer chemistry is replacing hazardous chemical synthetic reagents with safer substances (Principle 3). In this context, the 12th Principle describes the need for accident prevention through safer chemistry. At this point, it is essential to mention that general green chemistry often also comes with increased safety conditions for the operator. Herein, we want to summarise some already mentioned approaches in this review, which are also beneficial in risk minimisation.
The conventional methylation reaction in organic chemistry is often coupled with higher risks to the laboratory worker given that the applied methods involve methyl iodide, dimethyl sulphate, and formaldehyde. Similar to all potent alkylating agents, the latter is partially toxic, carcinogenic, mutagenic, poisonous, and corrosive. Also, their good skin absorption is critical, calling for safer alternatives.
Zhao and co-workers reported a metal-free system, applying CO2 and recyclable acetylcholine-based ionic liquids (ACH-ILs) for gram-grade N-functionalization (255) at low temperatures. It was fascinating that N-formylation occurred under solvent-free conditions at 30 °C (256), and N-methylation was achieved by adding acetonitrile as a solvent with the reaction temperatures of 50 °C (257) (Fig. 66).330 Another approach described the ligand-controlled N-methylation or -formylation via cooper-catalysis with CO2 and phenyl silane.331
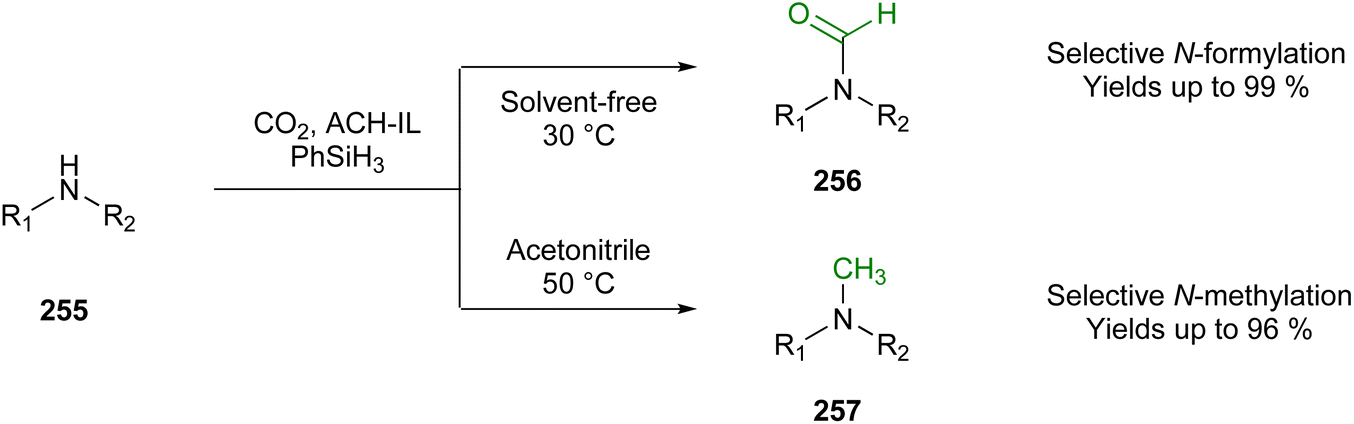 | ||
| Fig. 66 Safe solvent- and temperature-controlled N-functionalization with CO2 and ACH-ILs by Zhao et al.330 | ||
Furthermore, the production of N-oxides (such as 259) of heterocycles using a safe m-CPBA-NH3 system was reported in 2019.332 The synthesis of N-oxides usually requires potential explosophores at higher temperatures such as peracetic acid and hydrogen peroxide mixtures. In this context, meta-chloroperoxybenzoic acid (m-CPBA) offers easier handling and represents a safer option compared to other peroxides. This process enables scalable, efficient N-oxide-formation (259) at up to the kilogram scale with excellent yields over 90% at low temperatures (max. 35 °C). Additionally, the system involved process flow with educt scavenging, safety control with reaction calorimetry, and cautious solvent plus reagent precursor (m-chlorobenzoic acid) recovery. The system is illustrated in Fig. 67.
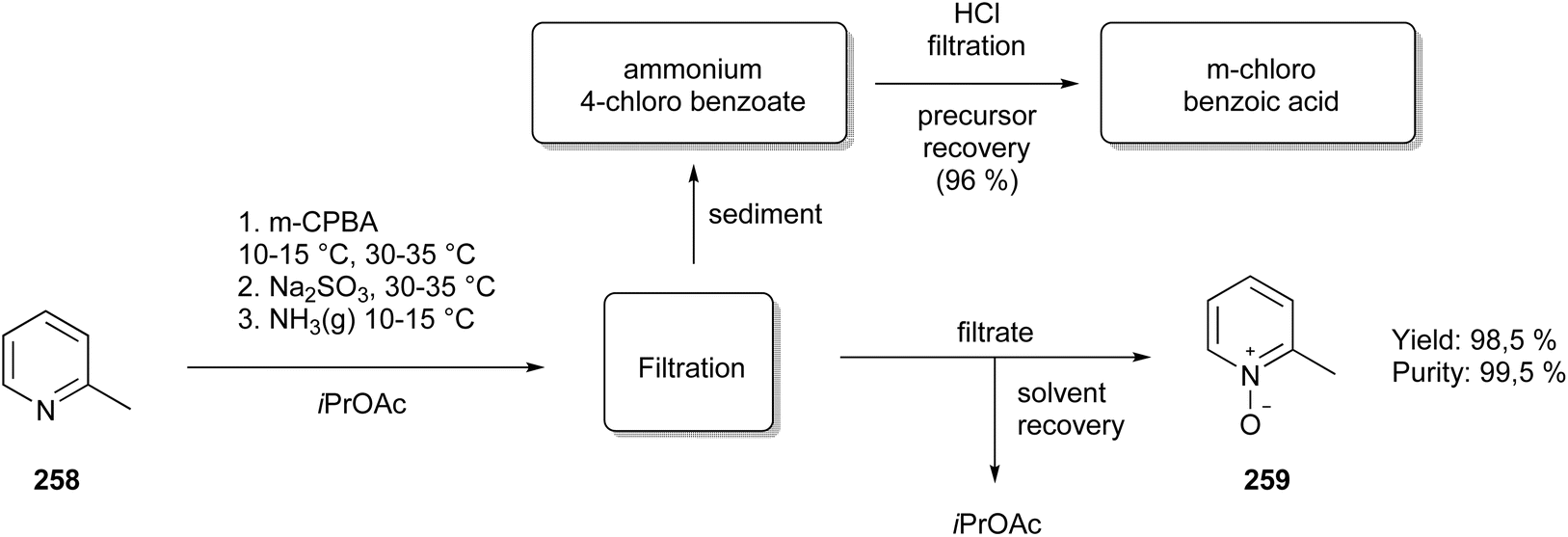 | ||
| Fig. 67 m-CPBA-system for the secure manufacturing of N-oxides (259).332 | ||
If hazardous substances are unavoidable in the synthesis route, then the application of continuous chemical processes represents a valuable tool in organic synthesis. Flow chemistry was already described in an earlier Principle regarding energy efficiency (see Principle 6) but is also highly recommended in accident prevention. The continuous flow enables risky reactions to be carried out in a closed system with strictly controlled reaction conditions (temperature and pressure) without requiring manual hazard, reagent, or intermediate handling. Enhanced safety aspects for operators and reduced reaction times lead to the circumvention of potential casualties and make flow chemistry a green and secure approach for medicinal chemists.333,334
An illustrative example was presented by an Austrian group in 2019, where they applied flow chemistry to the Wolff–Kirshner reaction of ketones (such as 260) and aldehydes (such as 262) (Fig. 68). The conventional method involves high temperatures and the explosive, corrosive, and potentially carcinogenic reducing agent hydrazine. Meanwhile, due to the developed reactor materials such as glass or stainless steel, the performance of this highly corrosive reaction was made possible in continuous manufacturing.333 The flow setup for ketones (such as 260) included a single feed system with low flow rates, heating and cooling modules, and an adjustable back-pressure regulator (BPR). The reduction of aldehyde derivatives (such as 262) required a two-feed approach due to the precipitation of the hydrazone intermediate because of the high reactivity of aldehydes with hydrazine under basic conditions at room temperatures (Fig. 69). The respective methylene products (261 or 263) were obtained with high yields and purity.335
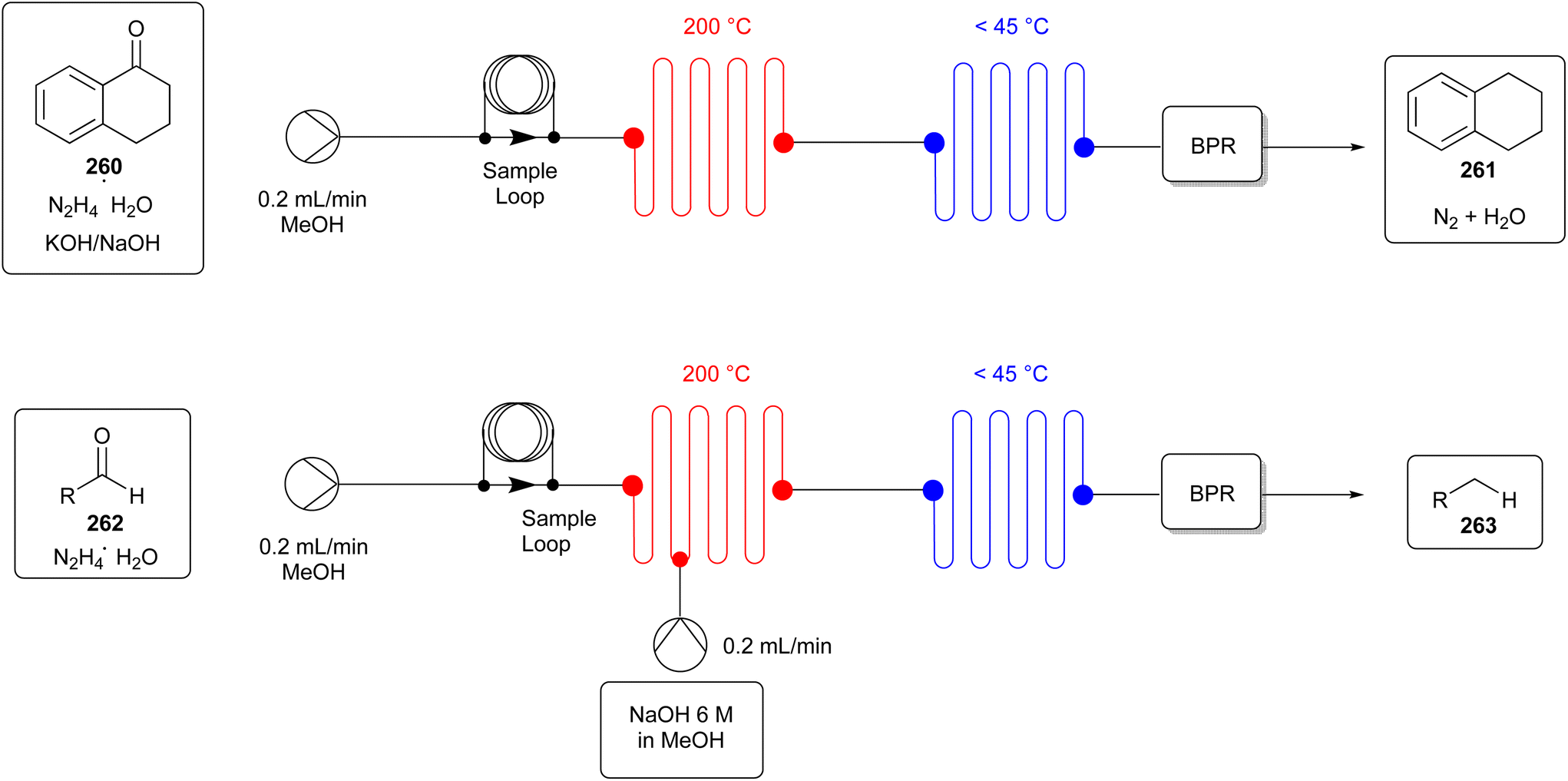 | ||
| Fig. 68 Safer Wolff–Kishner reduction of ketones/aldehydes in flow-chemistry conditions.333,335 | ||
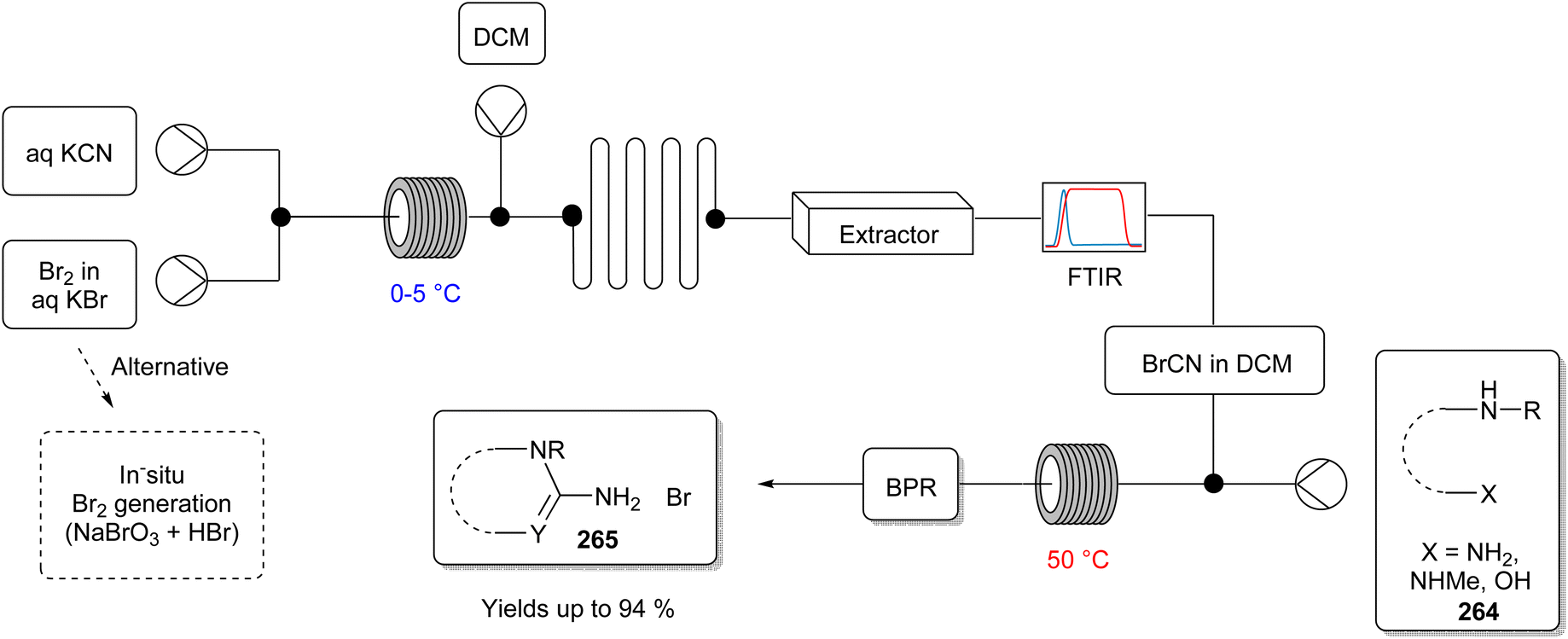 | ||
| Fig. 69 Continuous-flow synthesis of cyclic guanidines (265) with the in situ generation of cyanogen bromide.336 | ||
The in situ generation of high-risk reagents under controlled conditions is a further advantage of flow chemistry, thus avoiding the handling, storage, and transporting of dangerous chemicals. Glotz et al. described an efficient cyclic guanidinylation with the in situ generation of hazardous, volatile cyanogen bromide via a multi-feed system with aqueous KCN and Br2 (Fig. 69).336 Proximate CH2Cl2 addition and in-line extraction enabled the separation of the organic phase containing pure BrCN from the aqueous phase. Subsequent implementation of in-line-FTIR measurement enabled the determination of cyanogen bromide concentration and extraction efficiency due to the characteristic CN and water, OH stretches. The following substrate feed provided 5- and 6-membered guanidines (265) in good yields from variable educts. Furthermore, the successful application of in situ Br2-generation via sym-proportionation by the same research group led to further safety advances. The latter could additionally serve for the secure performance of continuous-flow bromination.
Other approaches in recent years mentioned safer continuous-flow synthesis with hazards such as chemical gases (syngas and CO/O2), phosgene, and organolithiums.334,337–340
Several green chemistry Principles simultaneously lead to safer operations in medicinal laboratories. Secure chemical synthesis (Principle 3) and the design of safer chemicals (Principle 4) result naturally in accident prevention. Replacing common flammable, toxic solvents with benign analogues (Principle 5) or solvent-free methods also increases the general safety. Furthermore, the incorporation of flow chemistry, one-pot procedures, miniaturised in-line analysis (Principle 11), and derivatisation prevention (Principle 8) avoid the treatment of dangerous intermediates. Besides, the application of catalysts (Principle 9) is highly recommended for waste reduction (E-factor, Principle 1) and reaction efficiency (AE, Principle 2), simultaneously preventing stoichiometric amounts of risky reagents. Especially, biocatalysis needs to be mentioned in this aspect, given that it allows efficient conversions to be carried out in water at low temperatures with benign enzymes or whole cells.
Again, this clearly highlights the cohesiveness of the GC system established by Anastas et al. more than 20 years ago.3 In this context, the greenness of medicinal chemistry can be increased by numerous means without losing efficiency and simplicity. Therefore, the employment of GC in common drug research represents a promising approach for making future chemistry more environmentally and economically friendly.
Conclusions
We consider that the main reason why green chemistry is barely applied in medicinal chemistry at present is the firmly rooted belief that green(er) methods bar the way for medicinal chemists to synthesise novel compounds in the fastest and easiest way possible. Consequently, there is little interest in including the aspects of greenness and sustainability in the laboratory. As outlined throughout this review, we would like to make the expert readers in medicinal chemistry aware of this increasingly relevant topic and highlight some of the various interesting, creative, and innovative ideas on how to apply green thinking and methods in drug design. Thus, revisiting our starting point, how can medicinal chemists contribute to fighting pollution and global warming, some of the (if not “the”) most urgent problems of our time, and prove the seemingly conflict between medicinal and green chemistry as disbelief? We found various promising examples for each of the 12 Principles of Green Chemistry3,4 that enable not only an improvement in the greenness of a synthesis pathway but also an enhancement in the duration, yield, safety, or sometimes complicated high demands on reaction conditions. One of the most vivid examples of this in recent times is the rapid expansion of flow chemistry. Within this mode of operation, the reaction steps are reduced, often avoiding various separation and purification steps.145 Additionally, the amount and quantity of toxic or environmentally harmful chemicals such as solvents are distinctly less; thus, the safety is improved significantly.333,334Of course, not every Principle can be realised in every optimisation strategy. Nevertheless, designing one perfect strategy should also not be the demand. It is not the point to work on a perfectly sophisticated strategy that implies every single green aspect and Principle. However, every single and sometimes small step, every approach to improvement, and every innovative idea contribute to making laboratory day life more consistent, less polluting, and safer. Thus, green chemistry starts with each one of us, and step by step, it is a continuous optimisable process that helps us to improve the eventually environmental harmful character of chemistry. This means, in reverse, that future research on drug design should aim to learn from the first ideas and approaches to transform these into future chemical standard procedures.
A further aspect we already mentioned on various occasions above is that the Principles are inseparably connected with each other. Moreover, we discussed some of the Principles thus far to unite them to focus on the main points. Consequently, we revealed the 7 core Principles, as represented in the review graphic including benign chemicals, catalysis, safety, energy efficiency, reduction of derivatives, prevention, and analysis. They, acting as the bold foundation, merge into what we call “green chemistry”. Just like white contains all colours of the rainbow, green chemistry contains all 7 core Principles. Also, what can make a rainbow shine even brighter than the contrast to a dark, black background as we have in the “classical” (medicinal) chemistry?
Thus, now all (medicinal) chemists need to put these Principles of Green Chemistry into action because the ideas are available!
Author contributions
Carola Castiello, Pierre Junghanns, and Annika Mergel: investigation and writing – original draft: Claus Jacob, Christian Ducho, Sergio Valente, Dante Rotili, and Antonello Mai: visualization, writing – review and editing. Rossella Fioravanti and Clemens Zwergel: conceptualization, validation, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by AIRC2021 (IG26172) (S. Valente), PRIN2020 (2020CW39SJ) (S. Valente), Ateneo Sapienza Project 2020 (RG120172B8E53D03) (S. Valente), Ateneo Sapienza Project 2021 (RM12117A61C811CE) (D. Rotili), FISR2019_00374 MeDyCa (A. Mai), Regione Lazio PROGETTI DI GRUPPI DI RICERCA 2020-A0375-2020-36597 (D. Rotili and C. Zwergel), KOHR Aerospace GmbH (C. Zwergel). C. Castiello and C. Zwergel are thankful for the generous funding from FSE REACT-EU within the programme PON “Research and Innovation” 2014–2020, Action IV.6 “Contratti di ricerca su tematiche Green”. The authors thank Ken Rory, Nick Essel and Sodomir Poppojuk for their helpful technical assistance.References
- R. Naidu, B. Biswas, I. R. Willett, J. Cribb, B. Kumar Singh, C. Paul Nathanail, F. Coulon, K. T. Semple, K. C. Jones, A. Barclay and R. J. Aitken, Environ. Int., 2021, 156, 106616 CrossRef CAS PubMed.
- L. H. U. W. Udara Willhelm Abeydeera, J. W. Wadu Mesthrige and T. I. Samarasinghalage, Sustainability, 2019, 11, 3972 CrossRef.
- P. T. Anastas and J. C. Warner, Green chemistry: Theory and practice, Oxford University Press, Oxford, 2000 Search PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- S. Sharma, J. Das, W. M. Braje, A. K. Dash and S. Handa, ChemSusChem, 2020, 13, 2859–2875 CrossRef CAS PubMed.
- D. G. Brown and J. Bostrom, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- B. A. de Marco, B. S. Rechelo, E. G. Totoli, A. C. Kogawa and H. R. N. Salgado, Saudi Pharm. J., 2019, 27, 1–8 CrossRef PubMed.
- P. T. Anastas, Crit. Rev. Anal. Chem., 1999, 29, 167–175 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- L. J. Diorazio, D. R. J. Hose and N. K. Adlington, Org. Process Res. Dev., 2016, 20, 760–773 CrossRef CAS.
- F. Brandão and J. P. Pedroso, Comput. Oper. Res., 2016, 69, 56–67 CrossRef.
- C. National Research, Waste incineration and public health, National Academies Press, Washington, DC, 2000 Search PubMed.
- J. H. Clark and S. J. Tavener, Org. Process Res. Dev., 2006, 11, 149–155 CrossRef.
- P. T. Anastas and J. B. Zimmerman, Green Chem., 2019, 21, 6545–6566 RSC.
- H. Ahsan, S. U. Islam, M. B. Ahmed, Y. S. Lee and J. K. Sonn, Curr. Pharm. Des., 2020, 26, 5767–5782 CrossRef CAS PubMed.
- K. Boodhoo and A. Harvey, in Process Intensification for Green Chemistry, 2013, pp. 1–31, DOI:10.1002/9781118498521.ch1.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef CAS PubMed.
- S. Grego, F. Aricò and P. Tundo, Org. Process Res. Dev., 2013, 17, 679–683 CrossRef CAS.
- M. J. Pedersen, T. L. Holm, J. P. Rahbek, T. Skovby, M. J. Mealy, K. Dam-Johansen and S. Kiil, Org. Process Res. Dev., 2013, 17, 1142–1148 CrossRef CAS.
- M. J. Pedersen, T. Skovby, M. J. Mealy, K. Dam-Johansen and S. Kiil, Org. Process Res. Dev., 2018, 22, 228–235 CrossRef CAS.
- R. W. Evans, J. R. Zbieg, S. Zhu, W. Li and D. W. MacMillan, J. Am. Chem. Soc., 2013, 135, 16074–16077 CrossRef CAS PubMed.
- K. M. Billiard, A. R. Dershem and E. Gionfriddo, Molecules, 2020, 25, 5297 CrossRef CAS PubMed.
- M. de la Guardia and S. Garrigues, in Challenges in Green Analytical Chemistry, The Royal Society of Chemistry, 2020, pp. 1–18, 10.1039/9781788016148-00001.
- H. Liu, N. Sun, P. Ding, C. Chen, Z. Wu, W. Zhu, L. Liu, Z. Wang and R. Pei, Colloids Surf., B, 2020, 191, 110985 CrossRef CAS PubMed.
- G. A. Keoleian, J. Cleaner Prod., 1993, 1, 143–149 CrossRef.
- E. Ibáñez and A. Cifuentes, TrAC, Trends Anal. Chem., 2020, 122, 115698 CrossRef.
- N. Reyes-Garces, E. Gionfriddo, G. A. Gomez-Rios, M. N. Alam, E. Boyaci, B. Bojko, V. Singh, J. Grandy and J. Pawliszyn, Anal. Chem., 2018, 90, 302–360 CrossRef CAS PubMed.
- M. M. Schantz, J. J. Nichols and S. A. Wise, Anal. Chem., 1997, 69, 4210–4219 CrossRef CAS.
- J. Namieśnik, J. Sep. Sci., 2001, 24, 151–153 CrossRef.
- J. Płotka-Wasylka and W. Wojnowski, Green Chem., 2021, 23, 8657–8665 RSC.
- J. Plotka-Wasylka, Talanta, 2018, 181, 204–209 CrossRef CAS PubMed.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- Pharmaceutical Process Development, 2011.
- S. Szymkuc, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed. Engl., 2016, 55, 5904–5937 CrossRef CAS PubMed.
- C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS PubMed.
- S. Kotha, D. Goyal and A. S. Chavan, J. Org. Chem., 2013, 78, 12288–12313 CrossRef CAS PubMed.
- M. J. Koh, T. T. Nguyen, J. K. Lam, S. Torker, J. Hyvl, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 542, 80–85 CrossRef CAS PubMed.
- S. Brase, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed. Engl., 2005, 44, 5188–5240 CrossRef CAS PubMed.
- V. Pandarus, D. Desplantier-Giscard, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2013, 17, 1492–1497 CrossRef CAS.
- S. Reisman, R. Nani and S. Levin, Synlett, 2011, 2437–2442 CrossRef CAS.
- S. Kotha, A. S. Chavan and D. Goyal, ACS Comb. Sci., 2015, 17, 253–302 CrossRef CAS PubMed.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- F. Roschangar, J. Colberg, P. J. Dunn, F. Gallou, J. D. Hayler, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Mergelsberg, J. L. Tucker, R. A. Sheldon and C. H. Senanayake, Green Chem., 2017, 19, 281–285 RSC.
- ACS GCI Pharmaceutical Roundtable, Convergent PMI Calculator, https://www.acs.org/content/dam/acsorg/green-chemistry/industriainnovation/roundtable/Convergent-PMI-Presentation.pptx, (accessed 21.01.2023, 2023).
- A. D. Curzons, D. N. Mortimer, D. J. C. Constable and V. L. Cunningham, Green Chem., 2001, 3, 1–6 RSC.
- J. Li, E. M. Simmons and M. D. Eastgate, Green Chem., 2017, 19, 127–139 RSC.
- A. Borovika, J. Albrecht, J. Li, A. S. Wells, C. Briddell, B. R. Dillon, L. J. Diorazio, J. R. Gage, F. Gallou, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Martinez, M. Olbrich, J. L. Piper, F. Roschangar, E. Sherer and M. D. Eastgate, The PMI Predictor - a Web App Enabling Green-by-Design Chemical Synthesis, 2019, DOI:10.26434/chemrxiv.7594646.v1.
- K. Budzinski, M. Blewis, P. Dahlin, D. D'Aquila, J. Esparza, J. Gavin, S. V. Ho, C. Hutchens, D. Kahn, S. G. Koenig, R. Kottmeier, J. Millard, M. Snyder, B. Stanard and L. Sun, New Biotechnol., 2019, 49, 37–42 CrossRef CAS PubMed.
- M. Eissen and K. Hugngerbühler, Green Chem., 2003, 5, G25–G27 RSC.
- H. J. De Lange, W. Noordoven, A. J. Murk, M. Lurling and E. T. Peeters, Aquat. Toxicol., 2006, 78, 209–216 CrossRef CAS PubMed.
- J. Schwaiger, H. Ferling, U. Mallow, H. Wintermayr and R. D. Negele, Aquat. Toxicol., 2004, 68, 141–150 CrossRef CAS PubMed.
- D. B. Huggett, B. W. Brooks, B. Peterson, C. M. Foran and D. Schlenk, Arch. Environ. Contam. Toxicol., 2002, 43, 229–235 CrossRef CAS PubMed.
- C. Mimeault, A. J. Woodhouse, X. S. Miao, C. D. Metcalfe, T. W. Moon and V. L. Trudeau, Aquat. Toxicol., 2005, 73, 44–54 CrossRef CAS PubMed.
- A. Jordan and N. Gathergood, Chem. Soc. Rev., 2015, 44, 8200–8237 RSC.
- J. Neumann, M. Pawlik, D. Bryniok, J. Thoming and S. Stolte, Environ. Sci. Pollut. Res., 2014, 21, 9495–9505 CrossRef CAS PubMed.
- D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- R. S. Boethling, P. H. Howard, W. Meylan, W. Stiteler, J. Beauman and N. Tirado, Environ. Sci. Technol., 1994, 28, 459–465 CrossRef CAS PubMed.
- J. W. Raymond, T. N. Rogers, D. R. Shonnard and A. A. Kline, J. Hazard. Mater., 2001, 84, 189–215 CrossRef CAS PubMed.
- K. Kümmerer, in Green and Sustainable Medicinal Chemistry, The Royal Society of Chemistry, 2016, pp. 73–81, 10.1039/9781782625940-00073.
- M. Suk, A. Haiß, J. Westphal, A. Jordan, A. Kellett, I. V. Kapitanov, Y. Karpichev, N. Gathergood and K. Kümmerer, Green Chem., 2020, 22, 4498–4508 RSC.
- K. Kümmerer, Sustainable Chem. Pharm., 2019, 12, 100136 CrossRef.
- A. Jordan and N. Gathergood, Antibiotics, 2013, 2, 419–438 CrossRef CAS PubMed.
- C. T. A. Moermond, N. Puhlmann, A. R. Brown, S. F. Owen, J. Ryan, J. Snape, B. J. Venhuis and K. Kummerer, Environ. Sci. Technol. Lett., 2022, 9, 699–705 CrossRef CAS PubMed.
- D. Lagorce, O. Sperandio, H. Galons, M. A. Miteva and B. O. Villoutreix, BMC Bioinf., 2008, 9, 396 CrossRef PubMed.
- K. Kümmerer and A. Al-Ahmad, Acta Hydrochim. Hydrobiol., 1997, 25, 166–172 CrossRef.
- J. P. Adams, C. M. Alder, I. Andrews, A. M. Bullion, M. Campbell-Crawford, M. G. Darcy, J. D. Hayler, R. K. Henderson, C. A. Oare, I. Pendrak, A. M. Redman, L. E. Shuster, H. F. Sneddon and M. D. Walker, Green Chem., 2013, 15, 1542–1549 RSC.
- D. K. Leahy, J. L. Tucker, I. Mergelsberg, P. J. Dunn, M. E. Kopach and V. C. Purohit, Org. Process Res. Dev., 2013, 17, 1099–1109 CrossRef CAS.
- ACS GCI Pharmaceutical Roundtable, https://reagents.acsgcipr.org/reagent-guides, (accessed 21.01.2023, 2023).
- C. R. Graves, B. S. Zeng and S. T. Nguyen, J. Am. Chem. Soc., 2006, 128, 12596–12597 CrossRef CAS PubMed.
- B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- A. E. J. d. Nooy, A. C. Besemer and H. v. Bekkum, Synthesis, 1996, 1153–1176 CrossRef.
- D. B. Dess and J. C. Martin, J. Org. Chem., 2002, 48, 4155–4156 CrossRef.
- S. R. Angle, D. S. Bernier, N. A. El-Said, D. E. Jones and S. Z. Shaw, Tetrahedron Lett., 1998, 39, 3919–3922 CrossRef CAS.
- C. The Carbon Trust Footprinting, Carbon Footprints In The Supply Chain - The Next Step For Business https://www.carbontrust.com/our-work-and-impact/guides-reports-and-tools/carbon-footprints-in-the-supply-chain-the-next-step, (accessed 27.01.2023).
- I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528–7557 CrossRef CAS PubMed.
- A. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- L. Smith, T. Ibn-Mohammed, I. M. Reaney and S. C. L. Koh, Resour., Conserv. Recycl., 2021, 166, 105317 CrossRef CAS.
- J. Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka, Jr. and M. R. Smith, 3rd, Science, 2002, 295, 305–308 CrossRef CAS PubMed.
- I. A. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka, Jr. and M. R. Smith, 3rd, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS PubMed.
- E. A. Englund, H. N. Gopi and D. H. Appella, Org. Lett., 2004, 6, 213–215 CrossRef CAS PubMed.
- I. W. Ashworth, B. G. Cox and B. Meyrick, J. Org. Chem., 2010, 75, 8117–8125 CrossRef CAS PubMed.
- M. Bodanszky and A. Bodanszky, Int. J. Pept. Protein Res., 2009, 23, 565–572 Search PubMed.
- K. E. Krakowiak and J. S. Bradshaw, Synth. Commun., 1996, 26, 3999–4004 CrossRef CAS.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC.
- A. P. Zarecki, J. L. Kolanowski and W. T. Markiewicz, Molecules, 2020, 25, 25081761 CrossRef PubMed.
- L. Mallu, D. Thirumalai and I. V. Asharani, Chem. Biol. Drug Des., 2017, 90, 520–526 CrossRef CAS PubMed.
- T. Song, X. Zhou, X. Wang, J. Xiao and Y. Yang, Green Chem., 2021, 23, 1955–1959 RSC.
- A. Domling and I. I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef CAS PubMed.
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- C. Mannich and W. Krösche, Arch. Pharm., 1912, 250, 647–667 CrossRef CAS.
- Z. Luo, H. Lu, R. Wu, H. Cheng, S. Nie, Y. Tang and Y. Gao, Synthesis, 2015, 1447–1454 CrossRef.
- A. Hantzsch, Liebigs Ann., 1882, 215, 1–82 CrossRef.
- M. Tang and H. Wang, Synthesis, 2017, 4893–4898 CrossRef.
- Y. Huang and A. Domling, Mol. Diversity, 2011, 15, 3–33 CrossRef CAS PubMed.
- W. Zhang, L. Ma, L. Yuan, C. Xu, G. Li and M. Tao, Synthesis, 2012, 45–52 CrossRef.
- A. Debache, R. Boulcina, A. Belfaitah, S. Rhouati and B. Carboni, Synlett, 2008, 509–512 CrossRef CAS.
- J. S. Nicholson and S. S. Adams, US Pat, 3385886A, 1968 Search PubMed.
- A. P. Dicks, Green Organic Chemistry in Lecture and Laboratory, Taylor & Francis, London, 2012 Search PubMed.
- P. J. Dunn, S. Galvin and K. Hettenbach, Green Chem., 2004, 6, 43–48 RSC.
- T. Hartung, Nature, 2009, 460, 208–212 CrossRef CAS PubMed.
- National Research Council Steering Committee on Identification of Toxic Potentially Toxic Chemicals for Consideration by the National Toxicology Program Toxicity Testing: Strategies to Determine Needs and Priorities, National Academy of Sciences, Washington (DC), 1984 Search PubMed.
- A. M. Voutchkova, L. A. Ferris, J. B. Zimmerman and P. T. Anastas, Tetrahedron, 2010, 66, 1031–1039 CrossRef CAS.
- J. A. Tickner, J. N. Schifano, A. Blake, C. Rudisill and M. J. Mulvihill, Environ. Sci. Technol., 2015, 49, 742–749 CrossRef CAS PubMed.
- J. Jaworska and N. Nikolova-Jeliazkova, SAR QSAR Environ. Res., 2007, 18, 195–207 CrossRef CAS PubMed.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS PubMed.
- M. Nendza and A. Wenzel, Environ. Sci. Pollut. Res. Int., 2006, 13, 192–203 CrossRef CAS PubMed.
- J. A. Schwobel, Y. K. Koleva, S. J. Enoch, F. Bajot, M. Hewitt, J. C. Madden, D. W. Roberts, T. W. Schultz and M. T. Cronin, Chem. Rev., 2011, 111, 2562–2596 CrossRef CAS PubMed.
- R. Ashauer and B. I. Escher, J. Environ. Monit., 2010, 12, 2056–2061 RSC.
- K. A. Phillips, J. F. Wambaugh, C. M. Grulke, K. L. Dionisio and K. K. Isaacs, Green Chem., 2017, 19, 1063–1074 RSC.
- D. J. Dix, K. A. Houck, M. T. Martin, A. M. Richard, R. W. Setzer and R. J. Kavlock, Toxicol. Sci., 2007, 95, 5–12 CrossRef CAS PubMed.
- M. S. Attene-Ramos, N. Miller, R. Huang, S. Michael, M. Itkin, R. J. Kavlock, C. P. Austin, P. Shinn, A. Simeonov, R. R. Tice and M. Xia, Drug Discovery, 2013, 18, 716–723 CAS.
- J. A. Kramer, J. E. Sagartz and D. L. Morris, Nat. Rev. Drug Discovery, 2007, 6, 636–649 CrossRef CAS PubMed.
- A. M. Monro, Regul. Toxicol. Pharmacol., 1990, 12, 137–160 CrossRef CAS PubMed.
- J. A. Hinson and D. W. Roberts, Annu. Rev. Pharmacol. Toxicol., 1992, 32, 471–510 CrossRef CAS PubMed.
- L. Q. Shen, R. S. Judson, F. Melnikov, J. Roethle, A. Gudibanda, J. B. Zimmerman and P. T. Anastas, Green Chem., 2016, 18, 4461–4467 RSC.
- R. M. LoPachin and T. Gavin, Chem. Res. Toxicol., 2014, 27, 1081–1091 Search PubMed.
- A. Porcheddu, R. Mocci, M. Brindisi, F. Cuccu, C. Fattuoni, F. Delogu, E. Colacino and M. V. D'Auria, Green Chem., 2022, 24, 4859–4869 RSC.
- D. E. Crawford, Beilstein J. Org. Chem., 2017, 13, 1850–1856 CrossRef CAS PubMed.
- D. Roy, S. L. James and D. E. Crawford, Chem. Commun., 2019, 55, 5463–5466 RSC.
- I. Nugrahani and R. D. Parwati, Molecules, 2021, 26, 3474–3483 CrossRef PubMed.
- A. Raheem Thayyil, T. Juturu, S. Nayak and S. Kamath, Adv. Pharm. Bull., 2020, 10, 203–212 CrossRef PubMed.
- A. Jordan and H. F. Sneddon, Green Chem., 2019, 21, 1900–1906 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- C. S. Slater and M. Savelski, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1595–1605 CrossRef CAS PubMed.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- G. de Gonzalo, A. R. Alcantara and P. Dominguez de Maria, ChemSusChem, 2019, 12, 2083–2097 CrossRef CAS PubMed.
- J. F. Campos, M. Cailler, R. Claudel, B. Prot, T. Besson and S. Berteina-Raboin, Molecules, 2021, 26, 1074 CrossRef CAS PubMed.
- A. Gevorgyan, K. H. Hopmann and A. Bayer, Organometallics, 2021, 41, 1777–1785 CrossRef.
- W. Su, H. Zhang, Y. Xing, X. Li, J. Wang and C. Cai, Nanomaterials, 2021, 11, 1–14 Search PubMed.
- Z. Wang, Bioanalysis, 2012, 4, 1275–1278 CrossRef CAS PubMed.
- A. S. Kaplitz, M. E. Mostafa, S. A. Calvez, J. L. Edwards and J. P. Grinias, J. Sep. Sci., 2021, 44, 426–437 CrossRef CAS PubMed.
- P. Michaels, J. Neef, K. Galyan, C. Ginsburg-Moraff, X. Zhou, D. Dunstan, J. Poirier and J. Reilly, Chirality, 2019, 31, 575–582 CrossRef CAS PubMed.
- K. Kobata, M. Kobayashi, S. Kinpara and T. Watanabe, Biotechnol. Lett., 2003, 25, 1575–1578 CrossRef CAS PubMed.
- N. Wei, D. Xu, B. Hao, S. Guo, Y. Guo and S. Wang, Water Res., 2021, 190, 116634 CrossRef CAS PubMed.
- C. I. Rivas-Vela, S. L. Amaya-Llano, E. Castano-Tostado and G. A. Castillo-Herrera, Molecules, 2021, 26, 6655 CrossRef CAS PubMed.
- A. G. Carr, R. Mammucari and N. R. Foster, Chem. Eng. J., 2011, 172, 1–17 CrossRef CAS.
- F. Fanelli, G. Parisi, L. Degennaro and R. Luisi, Beilstein J. Org. Chem., 2017, 13, 520–542 CrossRef CAS PubMed.
- J. Jiao, W. Nie, T. Yu, F. Yang, Q. Zhang, F. Aihemaiti, T. Yang, X. Liu, J. Wang and P. Li, Chemistry, 2021, 27, 4817–4838 CrossRef CAS PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- M. Colella, L. Degennaro and R. Luisi, Molecules, 2020, 25, 3242 CrossRef CAS PubMed.
- A. G. Nemeth, R. Szabo, G. Orsy, I. M. Mandity, G. M. Keseru and P. Abranyi-Balogh, Molecules, 2021, 26, 303 CrossRef CAS PubMed.
- S. Yakubov and J. P. Barham, Beilstein J. Org. Chem., 2020, 16, 2151–2192 CrossRef CAS PubMed.
- E. Shahbazali, E. M. F. Billaud, A. S. Fard, J. Meuldijk, G. Bormans, T. Noel and V. Hessel, AIChE J., 2021, 67, e17067 CrossRef CAS PubMed.
- J. B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed.
- D. H. A. Rocha, P. Vaz, D. Pinto and A. M. S. Silva, Methods Protoc., 2019, 2, 1–8 Search PubMed.
- H. M. T. Albuquerque, D. Pinto and A. M. S. Silva, Molecules, 2021, 26, 6293 CrossRef CAS PubMed.
- M. Draye, G. Chatel and R. Duwald, Pharmaceuticals, 2020, 13, 23 CrossRef CAS PubMed.
- B. Borah, J. Bora, P. Ramesh and L. R. Chowhan, RSC Adv., 2022, 12, 12843–12857 RSC.
- S. R. Pradhan, R. F. Colmenares-Quintero and J. C. C. Quintero, Molecules, 2019, 24, 315 CrossRef PubMed.
- V. Nair, J. C. Colmenares and D. Lisovytskiy, Green Chem., 2019, 21, 1241–1246 RSC.
- B. C. Ranu, A. Hajra and S. S. Dey, Org. Process Res. Dev., 2002, 6, 817–818 CrossRef CAS.
- V. K. Tandon, H. K. Maurya, M. K. Verma, R. Kumar and P. K. Shukla, Eur. J. Med. Chem., 2010, 45, 2418–2426 CrossRef CAS PubMed.
- Q. Zhang, Y. Xu, X. Liang and Z. Ke, ChemSusChem, 2022, 15, e202200574 CAS.
- F. Bucciol, E. Maffeis, E. Calcio Gaudino, L. Jicsinszky, S. Tagliapietra, A. Barge, C. Prandi and G. Cravotto, Antibiotics, 2021, 10, 1084 0 CrossRef PubMed.
- N. G. Shabalala, R. Pagadala and S. B. Jonnalagadda, Ultrason. Sonochem., 2015, 27, 423–429 CrossRef CAS PubMed.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS PubMed.
- S. Kudo, N. Goto, J. Sperry, K. Norinaga and J.-i. Hayashi, ACS Sustainable Chem. Eng., 2016, 5, 1132–1140 CrossRef.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Org. Biomol. Chem., 2018, 16, 2851–2854 RSC.
- H. Chang, I. Bajaj, A. H. Motagamwala, A. Somasundaram, G. W. Huber, C. T. Maravelias and J. A. Dumesic, Green Chem., 2021, 23, 3277–3288 RSC.
- L. Chen, Y. Xiong, H. Qin and Z. Qi, ChemSusChem, 2022, 15, e202102635 CAS.
- E. I. Garcia-Lopez, F. R. Pomilla, B. Megna, M. L. Testa, L. F. Liotta and G. Marci, Nanomaterials, 2021, 11, 1821 CrossRef CAS PubMed.
- Y. Wang, C. A. Brown and R. Chen, AIMS Microbiol., 2018, 4, 261–273 CAS.
- Y. Huang, S. Yao and H. Song, J. Chromatogr. Sci., 2013, 51, 739–752 CAS.
- S. P. M. Ventura, E. S. F. A. M. V. Quental, D. Mondal, M. G. Freire and J. A. P. Coutinho, Chem. Rev., 2017, 117, 6984–7052 CrossRef CAS PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- P. Formentín, J. Mol. Catal. A: Chem., 2004, 214, 137–142 CrossRef.
- V. Gotor-Fernandez and C. E. Paul, J. Biotechnol., 2019, 293, 24–35 CrossRef CAS PubMed.
- L. Piemontese, R. Sergio, F. Rinaldo, L. Brunetti, F. M. Perna, M. A. Santos and V. Capriati, Molecules, 2020, 25, 174 CrossRef PubMed.
- C. Mudithanapelli, C. S. Vasam, R. Vadde and M. H. Kim, ACS Omega, 2018, 3, 17646–17655 CrossRef CAS PubMed.
- T. A. Rossa, N. S. Suveges, M. M. Sa, D. Cantillo and C. O. Kappe, Beilstein J. Org. Chem., 2018, 14, 506–514 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., 2001, 113, 2056–2075 CrossRef.
- D. F. Rodriguez, F. Duran-Osorio, Y. Duarte, P. Olivares, Y. Moglie, K. Dua and F. C. Zacconi, Pharmaceutics, 2021, 14, 33 CrossRef PubMed.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- B. Albada, J. F. Keijzer, H. Zuilhof and F. van Delft, Chem. Rev., 2021, 121, 7032–7058 CrossRef CAS PubMed.
- D. Svatunek, N. Houszka, T. A. Hamlin, F. M. Bickelhaupt and H. Mikula, Chemistry, 2019, 25, 754–758 CrossRef CAS PubMed.
- J. Tummatorn, P. Batsomboon, R. J. Clark, I. V. Alabugin and G. B. Dudley, J. Org. Chem., 2012, 77, 2093–2097 CrossRef CAS PubMed.
- S. Li, H. Zhu, J. Wang, X. Wang, X. Li, C. Ma, L. Wen, B. Yu, Y. Wang, J. Li and P. G. Wang, Electrophoresis, 2016, 37, 1431–1436 CrossRef CAS PubMed.
- Y. Wang, H. X. Lin, L. Chen, S. Y. Ding, Z. C. Lei, D. Y. Liu, X. Y. Cao, H. J. Liang, Y. B. Jiang and Z. Q. Tian, Chem. Soc. Rev., 2014, 43, 399–411 RSC.
- N. Tanbouza, T. Ollevier and K. Lam, iScience, 2020, 23, 101720 CrossRef CAS PubMed.
- K. J. Jiao, Y. K. Xing, Q. L. Yang, H. Qiu and T. S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed.
- R. A. Green, K. E. Jolley, A. A. M. Al-Hadedi, D. Pletcher, D. C. Harrowven, O. De Frutos, C. Mateos, D. J. Klauber, J. A. Rincon and R. C. D. Brown, Org. Lett., 2017, 19, 2050–2053 CrossRef CAS PubMed.
- R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233–1246 CrossRef CAS.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- K. Bowden and M. Hardy, Tetrahedron, 1966, 22, 1169–1174 CrossRef CAS.
- M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed.
- A. Alimardanov, L. Schmieder-van de Vondervoort, A. H. M. de Vries and J. G. de Vries, Adv. Synth. Catal., 2004, 346, 1812–1817 CrossRef CAS.
- A. H. de Vries, J. M. Mulders, J. H. Mommers, H. J. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS PubMed.
- G. Papadogianakis, L. Maat and R. A. Sheldon, J. Chem. Soc., Chem. Commun., 1994, 23, 2659–2660 RSC.
- G. Papadogianakis, L. Maat and R. A. Sheldon, J. Chem. Technol. Biotechnol., 1997, 70, 83–91 CrossRef CAS.
- E. Polo, K. Ferrer-Pertuz, J. Trilleras, J. Quiroga and M. Gutiérrez, RSC Adv., 2017, 7, 50044–50055 RSC.
- A. Arcadi, G. Bianchi, S. D. Giuseppe and F. Marinelli, Green Chem., 2003, 5, 64–67 RSC.
- P. Sobrova, J. Zehnalek, V. Adam, M. Beklova and R. Kizek, Open Chem., 2012, 10, 1369–1382 CrossRef CAS.
- K. S. Egorova and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 12150–12162 CrossRef CAS PubMed.
- J. R. Dunetz, D. Fandrick and H.-J. Federsel, Org. Process Res. Dev., 2015, 19, 1325–1326 CrossRef CAS.
- C.-L. Feng, N.-N. Chu, S.-G. Zhang, J. Cai, J.-Q. Chen, H.-Y. Hu and M. Ji, Chem. Pap., 2014, 68, 1097–1103 CAS.
- M. Lenze and E. B. Bauer, Chem. Commun., 2013, 49, 5889–5891 RSC.
- B. M. Weckhuysen and J. Yu, Chem. Soc. Rev., 2015, 44, 7022–7024 RSC.
- M. M. Reddy, M. A. Kumar, P. Swamy, M. Naresh, K. Srujana, L. Satyanarayana, A. Venugopal and N. Narender, Green Chem., 2013, 15, 3474–3483 RSC.
- H. C. Kolb and K. B. Sharpless, Drug Discovery, 2003, 8, 1128–1137 CAS.
- V. Bénéteau, A. Olmos, T. Boningari, J. Sommer and P. Pale, Tetrahedron Lett., 2010, 51, 3673–3677 CrossRef.
- J. Luo, H. Yuan, H. Liu, J. Li, Y. Wang, Y. Wang, J. Yao and H. Li, GreenChE, 2021, 2, 294–300 Search PubMed.
- S. Chassaing, V. Bénéteau and P. Pale, Curr. Opin. Green Sustainable Chem., 2018, 10, 35–39 CrossRef.
- M. A. Wegman, M. H. A. Janssen, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2001, 343, 559–576 CrossRef CAS.
- H. M. Bialk, C. Hedman, A. Castillo and J. A. Pedersen, Environ. Sci. Technol., 2007, 41, 3593–3600 CrossRef CAS PubMed.
- S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187–1209 CrossRef CAS.
- I. Bassanini, E. E. Ferrandi, S. Riva and D. Monti, Catalysts, 2021, 11, 26 CrossRef CAS.
- O. V. Morozova, G. P. Shumakovich, S. V. Shleev and Y. I. Yaropolov, Appl. Biochem. Microbiol., 2007, 43, 523–535 CrossRef CAS.
- M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS.
- P. Yao, J. R. Marshall, Z. Xu, J. Lim, S. J. Charnock, D. Zhu and N. J. Turner, Angew. Chem., Int. Ed., 2021, 60, 8717–8721 CrossRef CAS PubMed.
- R. A. Wahab, N. Elias, F. Abdullah and S. K. Ghoshal, React. Funct. Polym., 2020, 152, 104613 CrossRef CAS.
- T. Nuijens, J. A. W. Kruijtzer, C. Cusan, D. T. S. Rijkers, R. M. J. Liskamp and P. J. L. M. Quaedflieg, Tetrahedron Lett., 2009, 50, 2719–2721 CrossRef CAS.
- M.-Q. Xu, S.-S. Wang, L.-N. Li, J. Gao and Y.-W. Zhang, Catalysts, 2018, 8, 460 CrossRef.
- N. Martín and F. G. Cirujano, Catal. Commun., 2022, 164, 106420 CrossRef.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- L. J. Gooßen, D. M. Ohlmann and P. P. Lange, Synthesis, 2009, 160–164 CrossRef.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chemistry, 2012, 18, 3822–3826 CrossRef CAS PubMed.
- C. E. Coomber, V. Laserna, L. T. Martin, P. D. Smith, H. C. Hailes, M. J. Porter and T. D. Sheppard, Org. Biomol. Chem., 2019, 17, 6465–6469 RSC.
- S. Zeng, J. Liu, S. Anankanbil, M. Chen, Z. Guo, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2018, 8, 8856–8865 CrossRef CAS.
- K. Wang, Y. Lu and K. Ishihara, Chem. Commun., 2018, 54, 5410–5413 RSC.
- M. Todorovic and D. M. Perrin, Pept. Sci., 2020, 112, e24210 CAS.
- C. L. Allen, A. R. Chhatwal and J. M. Williams, Chem. Commun., 2012, 48, 666–668 RSC.
- H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS.
- H. Lundberg, F. Tinnis and H. Adolfsson, Appl. Organomet. Chem., 2019, 33, e5062 CrossRef.
- M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed.
- T. K. Houlding, K. Tchabanenko, M. T. Rahman and E. V. Rebrov, Org. Biomol. Chem., 2013, 11, 4171–4177 RSC.
- S. M. Patil, S. A. Vanalakar, S. A. Sankpal, S. P. Deshmukh and S. D. Delekar, Results Chem., 2021, 3, 100102 CrossRef CAS.
- V. K. Das, R. R. Devi and A. J. Thakur, Appl. Catal., A, 2013, 456, 118–125 CrossRef CAS.
- K. P. Patel, E. M. Gayakwad and G. S. Shankarling, ChemistrySelect, 2020, 5, 8295–8300 CrossRef CAS.
- M. Amirsoleimani, M. A. Khalilzadeh and D. Zareyee, React. Kinet., Mech. Catal., 2020, 131, 859–873 CrossRef CAS.
- A. Goswami and S. G. Van Lanen, Mol. BioSyst., 2015, 11, 338–353 RSC.
- M. Petchey, A. Cuetos, B. Rowlinson, S. Dannevald, A. Frese, P. W. Sutton, S. Lovelock, R. C. Lloyd, I. J. S. Fairlamb and G. Grogan, Angew. Chem., Int. Ed., 2018, 57, 11584–11588 CrossRef CAS PubMed.
- K. Kummerer, J. Environ. Manage., 2009, 90, 2354–2366 CrossRef PubMed.
- L. B. Ellis and L. P. Wackett, Microb. Inf. Exp., 2012, 2, 1 CrossRef PubMed.
- J. Tunkel, P. H. Howard, R. S. Boethling, W. Stiteler and H. Loonen, Environ. Toxicol. Chem., 2000, 19, 2478–2485 CrossRef CAS.
- A. Lombardo, A. Manganaro, J. Arning and E. Benfenati, Sci. Total Environ., 2022, 838, 156004 CrossRef CAS PubMed.
- W. Tang, Y. Li, Y. Yu, Z. Wang, T. Xu, J. Chen, J. Lin and X. Li, Chemosphere, 2020, 253, 126666 CrossRef CAS PubMed.
- S. Vorberg and I. V. Tetko, Mol. Inf., 2014, 33, 73–85 CrossRef CAS PubMed.
- C. Rücker and K. Kümmerer, Green Chem., 2012, 14, 875–887 RSC.
- R. S. Boethling, E. Sommer and D. DiFiore, Chem. Rev., 2007, 107, 2207–2227 CrossRef CAS PubMed.
- OECD, OECD Guideline for Testing of Chemicals - Ready Biodegradabilty, https://www.oecd.org/chemicalsafety/risk-assessment/1948209.pdf, (accessed 28.01.2023).
- OECD, Revised Introduction to the OECD Guidelines for Testing of Chemicals, Section 3, OECD, Paris, 2006 Search PubMed.
- N. Puhlmann, R. Mols, O. Olsson, J. C. Slootweg and K. Kümmerer, Green Chem., 2021, 23, 5006–5023 RSC.
- X. D. Hou, Q. P. Liu, T. J. Smith, N. Li and M. H. Zong, PLoS One, 2013, 8, e59145 CrossRef CAS PubMed.
- C. Rucker, W. M. M. Mahmoud, D. Schwartz and K. Kummerer, Environ. Sci. Pollut. Res., 2018, 25, 18393–18411 CrossRef PubMed.
- P. H. Howard and R. S. Boethling, Designing for Non–Persistence, Wiley, Stuttgart, 2012 Search PubMed.
- K. Kümmerer, Green Chem., 2007, 9, 899–907 RSC.
- N. Gathergood, M. T. Garcia and P. J. Scammells, Green Chem., 2004, 6, 166–175 RSC.
- M. T. Garcia, N. Gathergood and P. J. Scammells, Green Chem., 2005, 7, 9–14 RSC.
- N. Gathergood, P. J. Scammells and M. T. Garcia, Green Chem., 2006, 8, 156–160 RSC.
- A. Haiß, A. Jordan, J. Westphal, E. Logunova, N. Gathergood and K. Kümmerer, Green Chem., 2016, 18, 4361–4373 RSC.
- T. Rastogi, C. Leder and K. Kummerer, Chemosphere, 2014, 111, 493–499 CrossRef CAS PubMed.
- T. Rastogi, C. Leder and K. Kümmerer, RSC Adv., 2015, 5, 27–32 RSC.
- T. Rastogi, C. Leder and K. Kummerer, Environ. Sci. Technol., 2015, 49, 11756–11763 CrossRef CAS PubMed.
- P. Kovalakova, L. Cizmas, T. J. McDonald, B. Marsalek, M. Feng and V. K. Sharma, Chemosphere, 2020, 251, 126351 CrossRef CAS PubMed.
- C. Leder, M. Suk, S. Lorenz, T. Rastogi, C. Peifer, M. Kietzmann, D. Jonas, M. Buck, A. Pahl and K. Kümmerer, ACS Sustainable Chem. Eng., 2021, 9, 9358–9368 CrossRef CAS.
- A. Gałuszka, Z. Migaszewski and J. Namieśnik, TrAC, Trends Anal. Chem., 2013, 50, 78–84 CrossRef.
- P. M. Nowak, R. Wietecha-Posłuszny and J. Pawliszyn, TrAC, Trends Anal. Chem., 2021, 138, 116223 CrossRef CAS.
- M. de la Guardia and S. Armenta, Elsevier Science, Elsevier Science, 2010 Search PubMed.
- C. L. Gargalo, I. Udugama, K. Pontius, P. C. Lopez, R. F. Nielsen, A. Hasanzadeh, S. S. Mansouri, C. Bayer, H. Junicke and K. V. Gernaey, J. Ind. Microbiol. Biotechnol., 2020, 47, 947–964 CrossRef PubMed.
- M. Sajid and J. Plotka-Wasylka, Talanta, 2022, 238, 123046 CrossRef CAS PubMed.
- L. H. Keith, H. J. Brass, D. J. Sullivan, J. A. Boiani and K. T. Alben, Environ. Sci. Technol., 2005, 39, 173A–176A CrossRef CAS PubMed.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
- M. Tobiszewski, Anal. Methods, 2016, 8, 2993–2999 RSC.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS PubMed.
- S. Armenta, S. Garrigues and M. de la Guardia, TrAC, Trends Anal. Chem., 2015, 71, 2–8 CrossRef CAS.
- S. Armenta, S. Garrigues, F. A. Esteve-Turrillas and M. d. l. Guardia, TrAC, Trends Anal. Chem., 2019, 116, 248–253 CrossRef CAS.
- O. Filippou, D. Bitas and V. Samanidou, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1043, 44–62 CrossRef CAS PubMed.
- R. P. F. F. da Silva, T. A. P. Rocha-Santos and A. C. Duarte, TrAC, Trends Anal. Chem., 2016, 76, 40–51 CrossRef CAS.
- C. Da Porto, D. Decorti and A. Natolino, J. Supercrit. Fluids, 2014, 87, 1–8 CrossRef CAS.
- L. Mousavi, Z. Tamiji and M. R. Khoshayand, Talanta, 2018, 190, 335–356 CrossRef CAS PubMed.
- A. Quigley, W. Cummins and D. Connolly, J. Chem., 2016, 2016, 1–12 CrossRef.
- I. Rykowska, J. Ziemblińska and I. Nowak, J. Mol. Liq., 2018, 259, 319–339 CrossRef CAS.
- G. Li and K. H. Row, TrAC, Trends Anal. Chem., 2019, 120, 115651 CrossRef CAS.
- M. A. Korany, H. Mahgoub, R. S. Haggag, M. A. A. Ragab and O. A. Elmallah, J. Liq. Chromatogr. Relat. Technol., 2017, 40, 839–852 CrossRef CAS.
- B. P. Regmi and M. Agah, Anal. Chem., 2018, 90, 13133–13150 CrossRef CAS PubMed.
- H. D. Bahaghighat, C. E. Freye and R. E. Synovec, TrAC, Trends Anal. Chem., 2019, 113, 379–391 CrossRef CAS.
- A. Gholizadeh, M. Chowdhury and M. Agah, Anal. Chem., 2020, 92, 10635–10642 CrossRef CAS PubMed.
- P. I. Napolitano-Tabares, I. Negrín-Santamaría, A. Gutiérrez-Serpa and V. Pino, Curr. Opin. Green Sustainable Chem., 2021, 32, 100536 CrossRef CAS.
- C. West, Anal. Bioanal. Chem., 2018, 410, 6441–6457 CrossRef CAS PubMed.
- B. N. Fitch, R. Gray, M. Beres, M. B. Hicks, W. Farrell, C. Aurigemma and S. V. Olesik, Green Chem., 2022, 24, 4516–4532 RSC.
- L. C. Chen, T. Naito, S. Ninomiya and K. Hiraoka, Analyst, 2018, 143, 5552–5558 RSC.
- M. Yabre, L. Ferey, I. T. Some and K. Gaudin, Molecules, 2018, 23, 1065 CrossRef PubMed.
- O. Claux, C. Santerre, M. Abert-Vian, D. Touboul, N. Vallet and F. Chemat, Curr. Opin. Green Sustainable Chem., 2021, 31, 100510 CrossRef CAS.
- H. Shaaban, Anal. Bioanal. Chem., 2016, 408, 6929–6944 CrossRef CAS PubMed.
- R. N. El-Shaheny, M. H. El-Maghrabey and F. F. Belal, Open Chem., 2015, 13, 877–892 Search PubMed.
- M. A. Gab-Allah, I. F. Tahoun, R. N. Yamani, E. A. Rend and A. B. Shehata, J. Food Compos. Anal., 2022, 107, 104395 CrossRef CAS.
- D. Mohamed and M. M. Fouad, Microchem. J., 2020, 157, 104873 CrossRef CAS.
- Y. Tang, N. Zhang, B. Zhang, X. Lei and X. Wu, J. Chromatogr. A, 2018, 1556, 10–20 CrossRef CAS PubMed.
- R. Deidda, P.-Y. Sacre, M. Clavaud, L. Coïc, H. Avohou, P. Hubert and E. Ziemons, TrAC, Trends Anal. Chem., 2019, 114, 251–259 CrossRef CAS.
- S. Garrigues and M. de la Guardia, TrAC, Trends Anal. Chem., 2013, 43, 161–173 CrossRef CAS.
- B. Deng, X. Luo, M. Zhang, L. Ye and Y. Chen, Colloids Surf., B, 2019, 173, 286–294 CrossRef CAS PubMed.
- A. R. Chowdhury, N. Maheshwari, J. Soni, M. Kapil, T. Mehta and A. Mukharya, J. Pharm. Biomed. Anal., 2020, 189, 113292 CrossRef CAS PubMed.
- S. Garrigues and M. de la Guardia, in Challenges in Green Analytical Chemistry, The Royal Society of Chemistry, 2020, pp. 19–54, 10.1039/9781788016148-00019.
- K. S. Kokilambigai and K. S. Lakshmi, Green Chem. Lett. Rev., 2020, 14, 99–107 CrossRef.
- M. Grootveld, B. Percival, M. Gibson, Y. Osman, M. Edgar, M. Molinari, M. L. Mather, F. Casanova and P. B. Wilson, Anal. Chim. Acta, 2019, 1067, 11–30 CrossRef CAS PubMed.
- A. Soyler, D. Bouillaud, J. Farjon, P. Giraudeau and M. H. Oztop, LWT–Food Sci. Technol., 2020, 118, 108832 CrossRef CAS.
- M. del Valle, in Challenges in Green Analytical Chemistry, The Royal Society of Chemistry, 2020, pp. 55–91, 10.1039/9781788016148-00055.
- J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6, 2179–2195 CrossRef CAS PubMed.
- H. Liu, J. Ding, K. Zhang and L. Ding, TrAC, Trends Anal. Chem., 2019, 118, 315–337 CrossRef CAS.
- M. Yasa, A. Deniz, M. Forough, E. Yildirim, O. Persil Cetinkol, Y. A. Udum and L. Toppare, J. Polym. Sci., 2020, 58, 3336–3348 CrossRef CAS.
- P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. D. Williams and C. O. Kappe, Angew. Chem., Int. Ed., 2021, 60, 8139–8148 CrossRef CAS PubMed.
- R. Th. El-Eryan, S. S. Toubar, A. A. Ashour and M. S. Elshahed, Microchem. J., 2022, 178, 107347 CrossRef.
- Y. Fan, J. Li, Y. Guo, L. Xie and G. Zhang, Measurement, 2021, 171, 108829 CrossRef.
- K. Fukuoka and M. Furusho, Sci. Rep., 2022, 12, 3185 CrossRef CAS PubMed.
- R. Huising and S. S. Silbey, Ann. Am. Acad. Pol. Soc. Sci., 2013, 649, 157–177 CrossRef.
- H.-R. Ayi and C.-Y. Hon, J. Chem. Health Saf., 2018, 25, 6–12 CrossRef.
- N. J. O'Neil, S. Scott, R. Relph and E. Ponnusamy, J. Chem. Educ., 2020, 98, 84–91 CrossRef.
- A. J. M. Miller and I. A. Tonks, Organometallics, 2018, 37, 3225–3227 CrossRef CAS.
- C. A. Tickner, P. M. Gillespie and M. Hoyle, Screening Protocol to Identify Potentially Explosive Compounds in Early Stage Development, Institution of Chemical Engineers, 25th IChemE Symposium on Hazards 2015 (Hazards 25), 2015.
- A. D. Allian, N. P. Shah, A. C. Ferretti, D. B. Brown, S. P. Kolis and J. B. Sperry, Org. Process Res. Dev., 2020, 24, 2529–2548 CrossRef CAS.
- P. Urben, Bretherick's handbook of reactive chemical hazards, Elsevier, Amsterdam, 2017 Search PubMed.
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, Berlin, 2015 Search PubMed.
- L. W. Ciszewski, K. Rybicka-Jasinska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432–448 RSC.
- S. P. Green, K. M. Wheelhouse, A. D. Payne, J. P. Hallett, P. W. Miller and J. A. Bull, Org. Process Res. Dev., 2020, 24, 67–84 CrossRef CAS PubMed.
- Q. Yang, M. Sheng, X. Li, C. Tucker, S. Vásquez Céspedes, N. J. Webb, G. T. Whiteker and J. Yu, Org. Process Res. Dev., 2020, 24, 916–939 CrossRef CAS.
- Y. Deguchi, M. Kono, Y. Koizumi, Y.-i. Izato and A. Miyake, Org. Process Res. Dev., 2020, 24, 1614–1620 CrossRef CAS.
- Y. Tian, X. Zhang, B. Yu, Y. Bai, L. Guan, S. Teng, J. Li, C. Huang, M. Lanz and P. Hoehn, Org. Process Res. Dev., 2020, 24, 2927–2934 CrossRef CAS.
- M. Guan, Y. Tian, J. Zhao, X. Gu, X. Jiang, X. Wang, Y. Zhang and X. Zhang, Org. Process Res. Dev., 2021, 25, 1375–1382 CrossRef CAS.
- M. M. Al-Tabakha, S. A. Khan, A. Ashames, H. Ullah, K. Ullah, G. Murtaza and N. Hassan, Journal, 2021, 14, 234 CAS.
- S. H. Liu, C. R. Cao, W. C. Lin and C. M. Shu, J. Hazard. Mater., 2019, 365, 164–177 CrossRef CAS PubMed.
- M. K. Trivedi, P. Panda, K. K. Sethi, M. Gangwar, S. C. Mondal and S. Jana, J. Pharm. Anal., 2020, 10, 334–345 CrossRef PubMed.
- H. Yao, L. Ni, P. Wu, J. Jiang, Y. Ni and X. Yao, J. Anal. Appl. Pyrolysis, 2021, 159, 105299 CrossRef CAS.
- W. Zhao, X. Chi, H. Li, J. He, J. Long, Y. Xu and S. Yang, Green Chem., 2019, 21, 567–577 RSC.
- X.-D. Li, S.-M. Xia, K.-H. Chen, X.-F. Liu, H.-R. Li and L.-N. He, Green Chem., 2018, 20, 4853–4858 RSC.
- A. Palav, B. Misal, A. Ernolla, V. Parab, P. Waske, D. Khandekar, V. Chaudhary and G. Chaturbhuj, Org. Process Res. Dev., 2018, 23, 244–251 CrossRef.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- C. A. Hone, P. Lopatka, R. Munday, A. O'Kearney-McMullan and C. O. Kappe, ChemSusChem, 2019, 12, 326–337 CrossRef CAS PubMed.
- D. Znidar, A. O'Kearney-McMullan, R. Munday, C. Wiles, P. Poechlauer, C. Schmoelzer, D. Dallinger and C. O. Kappe, Org. Process Res. Dev., 2019, 23, 2445–2455 CrossRef CAS.
- G. Glotz, R. Lebl, D. Dallinger and C. O. Kappe, Angew. Chem., Int. Ed., 2017, 56, 13786–13789 CrossRef CAS PubMed.
- A. Bonner, A. Loftus, A. C. Padgham and M. Baumann, Org. Biomol. Chem., 2021, 19, 7737–7753 RSC.
- A. Sivo, R. d, S. Galaverna, G. R. Gomes, J. C. Pastre and G. Vilé, React. Chem. Eng., 2021, 6, 756–786 RSC.
- H. Yasukouchi, A. Nishiyama and M. Mitsuda, Org. Process Res. Dev., 2018, 22, 247–251 CrossRef CAS.
- M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814–1838 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |