
Electrochemical primary amination of imidazopyridines with azidotrimethylsilane under mild conditions†
Yan
Zhu‡
,
Qiao
Chu‡
,
Heng
Li
,
Ping
Liu
* and
Peipei
Sun
 *
*
School of Chemistry and Materials Science, Jiangsu Provincial Key Laboratory of Material Cycle Processes and Pollution Control, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, Nanjing Normal University, Nanjing 210023, China. E-mail: sunpeipei@njnu.edu.cn; pingliu@njnu.edu.cn
First published on 23rd November 2022
Abstract
A straightforward protocol involving electrochemical primary amination of imidazopyridines under mild conditions is described. This transformation utilizes TMSN3 as the nitrogen source and a trace amount of H2O as the hydrogen source. A wide range of imidazopyridines were found to be compatible, providing the corresponding C3-amino substituted imidazopyridines in moderate to good yields.
Introduction
Arylamines, especially heteroarylamines, are an important class of organic compounds that are widely found in a variety of natural products,1 pharmaceuticals,2 agrochemicals,3 dyes,4etc. (Scheme 1). The amino group is easily converted to amides, imines, and diazo compounds, and it is also a strongly activating and ortho- and para-directing group in electrophilic aromatic substitution reactions. Thus, explosive progress has been made in the development of synthetic methods for the introduction of an amino group to arenes or heteroarenes during the past decades. One traditional method for the preparation of this class of molecules involved nitration of arenes or heteroarenes, followed by the reduction of the nitro group via metal catalytic hydrogenation or other methods in the presence of stoichiometric reducing agents (Scheme 2a).5 An alternative route for the construction of primary anilines was the cross-coupling reaction of aryl halides, aromatic boronic acids or heteroaryl halides with an amino source such as aqueous ammonia (Scheme 2b). However, this process suffers from some drawbacks like the prefunctionalization of substrates and the use of expensive ligands and transition-metals such as Cu,6 Pd,7 Ni,8etc. Therefore, the development of direct C–H amination under environmentally benign, atom-efficient and mild conditions is still highly desired.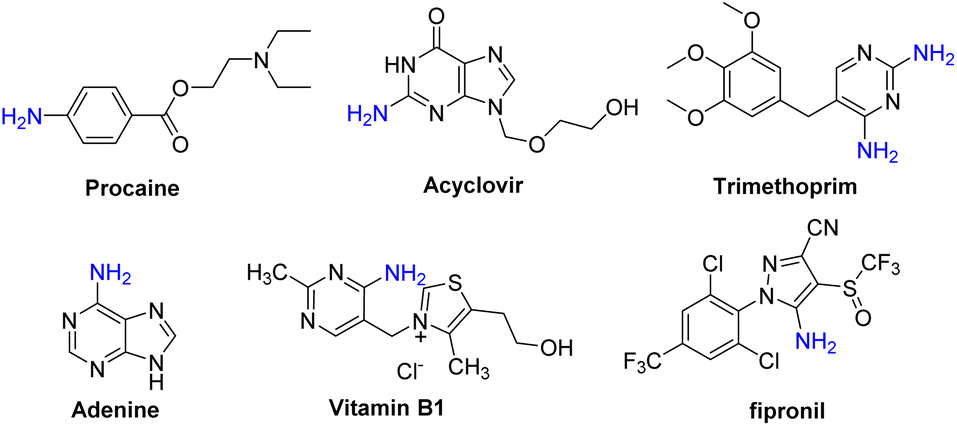 | ||
| Scheme 1 Some amino group-containing pharmaceuticals and agrochemicals. | ||
 | ||
| Scheme 2 Strategies for the amination of arenes or heteroarenes. | ||
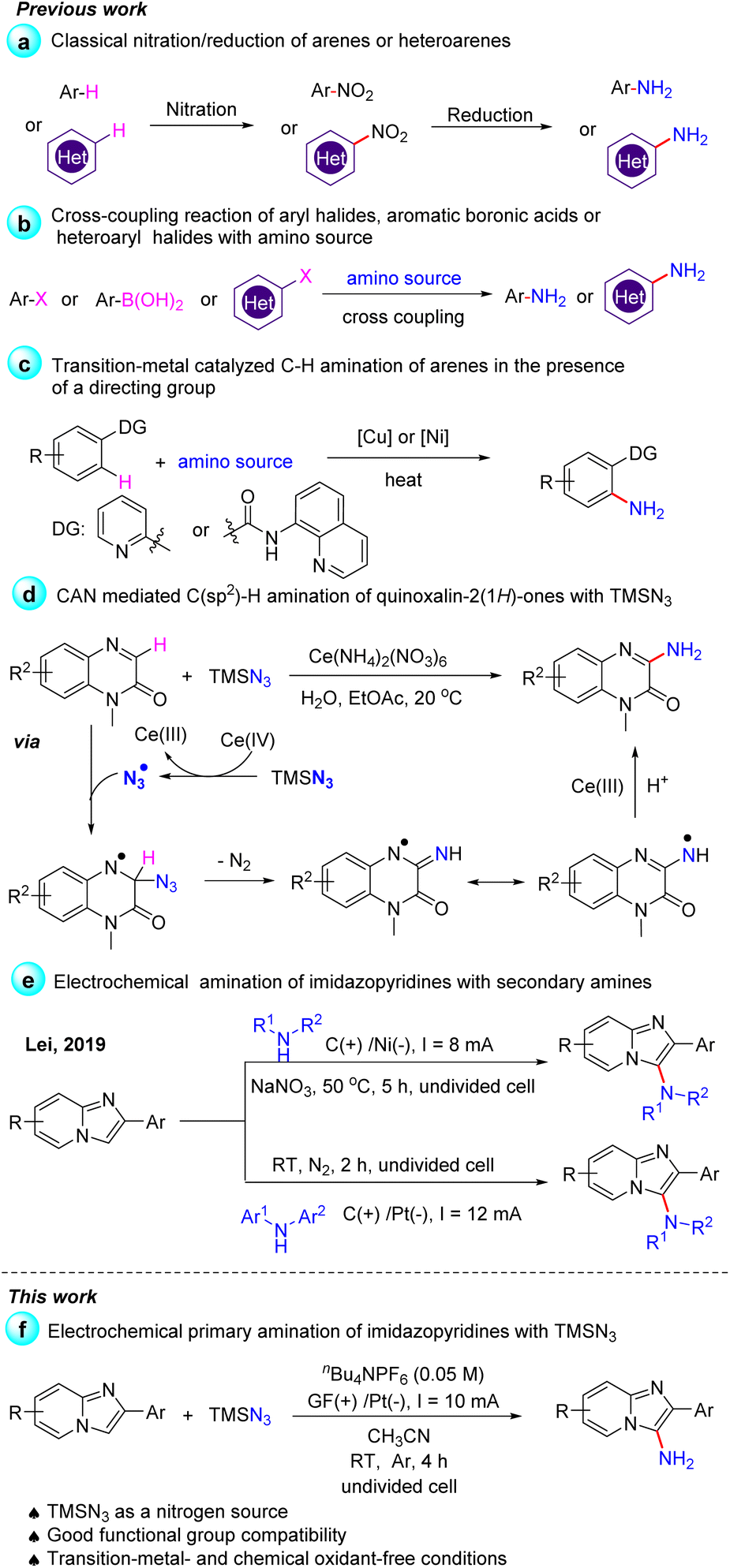
Although transition-metal catalyzed C–H bond activation has been developed for the direct amination of arenes with good regioselectivity, this strategy often requires the preinstallation of a directing group (Scheme 2c).9 In 2019, Zhang and co-workers demonstrated a convenient method for the C–H amination of quinoxalin-2(1H)-ones with TMSN3 using ceric ammonium nitrate (CAN) as the oxidant.10 The key step of the reaction was the interaction of TMSN3 and CAN to generate the azido radical, followed by the addition of an azido radical to quinoxalin-2(1H)-ones (Scheme 2d). The C3-functionalization of imidazo-[1,2-a]pyridine is synthetically attractive since many pharmaceuticals such as Zolpidem, Alpidem, Minodronic acid, etc. bear a substituent at this position.11 Imidazo[1,2-a]pyridin-3-amines are of great value in drug synthesis, for example, as synthetic precursors of imidazo[1,2-a]pyridin-3-amides, which were applied as γ-aminobutyric acid type A (GABAA) receptors.12 However, in early studies, the synthesis of imidazo[1,2-a]pyridin-3-amines could only be achieved in multiple synthetic steps.13
Chemists disclose that the electrochemical method can enable many reactions that cannot occur under conventional thermal conditions or in the absence of oxidants or reducing agents, thereby making organic synthesis green and sustainable.14 Compared to other C3-functionalized imidazopyridines, the C3-amination of imidazo[1,2-a]pyridines under electrochemical conditions has been less explored.15 In 2019, Lei and co-workers developed an efficient electrochemical C–H/N–H oxidative cross-coupling of imidazo[1,2-a]pyridines with azoles, secondary amines or diarylamines to access C3-aminated imidazo[1,2-a]pyridines (Scheme 2e).16 When we tried to perform the primary amination of imidazopyridines with TMSN3 according to Lei's work, no C3-amino substituted imidazopyridines were obtained. Azidation involving an azide anion or azido radical under electrochemical conditions has also gained much attention in recent years.17 Inspired by the above excellent works and based on our previous research on the electrochemical C–H functionalization of imidazo-[1,2-a]pyridine and other heteroarenes,18 herein, we demonstrated the first electrochemical primary amination of imidazopyridines with TMSN3 under mild conditions in the absence of external chemical oxidants such as CAN (Scheme 2f). The interaction of the substrates with TMSN3 was also much different from the process proposed in Scheme 2d because the azido radical was not the reactive intermediate in our electrochemical amination reaction.
Results and discussion
Initially, we selected 2-phenylimidazo[1,2-a]pyridine (1a) and TMSN3 as the model substrates to evaluate the electrochemical reaction conditions (Table 1). We carried out the reaction under a constant current (10 mA) in an undivided cell equipped with a graphite felt (GF) anode and a platinum plate cathode containing the electrolyte nBu4NPF6 (0.05 M) in CH3CN (10 mL) at room temperature under an Ar atmosphere. After electrolysis for 4 h, the desired product 2-phenylimidazo[1,2-a]pyridin-3-amine (2a) was obtained in 89% yield (entry 1). If NH3·H2O (1.0 mL) was used as the amino source instead of TMSN3, no desired product 2a was obtained. Experiments revealed that other electrolytes such as nBu4NBF4 and LiClO4 led to lower yields (entries 2 and 3). The use of electrode materials had an obvious influence on the reaction outcome. Changing the arrangement of the graphite felt and platinum plate did not improve the yields of 2a (entries 4–6). When other solvents including CH2Cl2, acetone, CH3OH and N,N-dimethylacetamide (DMA) were screened, lower yields were obtained (entries 7–10). Increasing the reaction current to 15 mA resulted in an inferior yield (75%, entry 11). Reducing the reaction current to 5 mA did not improve the yield of 2a, either (entry 12). When the reaction was performed under air, a lower yield of 71% was obtained (entry 13). The current was essential to this transformation and the reaction did not occur in the absence of continuous energy (entry 14).|
|
||||
|---|---|---|---|---|
| Entry | Anode/cathode | Electrolyte | Solvent | Yieldb (%) |
| a Standard conditions: 1a (0.5 mmol), TMSN3 (1.0 mmol, 2 equiv.), nBu4NPF6 (0.05 M), CH3CN (10 mL), GF(+)/Pt(−), RT, Ar, 4 h. b Isolated yield based on 1a. c I = 15 mA. d I = 5 mA. e Under air. f No current. | ||||
| 1 | GF(+)/Pt(−) | n Bu4NPF6 | CH3CN | 89 |
| 2 | GF(+)/Pt(−) | n Bu4NBF4 | CH3CN | 65 |
| 3 | GF(+)/Pt(−) | LiClO4 | CH3CN | 30 |
| 4 | GF(+)/GF(−) | n Bu4NPF6 | CH3CN | 83 |
| 5 | Pt(+)/GF(−) | n Bu4NPF6 | CH3CN | 81 |
| 6 | Pt(+)/Pt(−) | n Bu4NPF6 | CH3CN | 52 |
| 7 | GF(+)/Pt(−) | n Bu4NPF6 | CH2Cl2 | n.d. |
| 8 | GF(+)/Pt(−) | n Bu4NPF6 | Acetone | 73 |
| 9 | GF(+)/Pt(−) | n Bu4NPF6 | CH3OH | 41 |
| 10 | GF(+)/Pt(−) | n Bu4NPF6 | DMA | 68 |
| 11c | GF(+)/Pt(−) | n Bu4NPF6 | CH3CN | 75 |
| 12d | GF(+)/Pt(−) | n Bu4NPF6 | CH3CN | 85 |
| 13e | GF(+)/Pt(−) | n Bu4NPF6 | CH3CN | 71 |
| 14f | GF(+)/Pt(−) | n Bu4NPF6 | CH3CN | n.d. |
With the optimized reaction conditions in hand, we next examined the scope of the primary amination of various imidazopyridines, and the results are summarized in Scheme 3. The effect of the substituent and its position on the C2-benzene ring was firstly investigated. The substrates 1 bearing electron-withdrawing groups (F, Cl, Br, CN, CF3, SO2CH3 and Ph) or electron-donating groups (Me and OMe) at the para-position of the C2-phenyl were compatible for this reaction and afforded the desired C3-aminated products 2b–2j in good yields (78–88%). Then the halogen atom (F, Cl or Br) or MeO at the ortho- or meta-position of the benzene ring of 2-phenylimidazo[1,2-a]pyridines was also tolerated under the reaction conditions and the corresponding products 2k–2p were obtained in 52–87% yields. The substrate 1q substituted with a 2-thienyl group at the C2-position was transformed into the corresponding aminated product 2q in 85% yield. Besides, reactions of imidazopyridines decorated with dichlorophenyl (1r) or 2-naphthyl (1s) smoothly proceeded as well, providing products 2r and 2s in 61% and 87% yields, respectively.
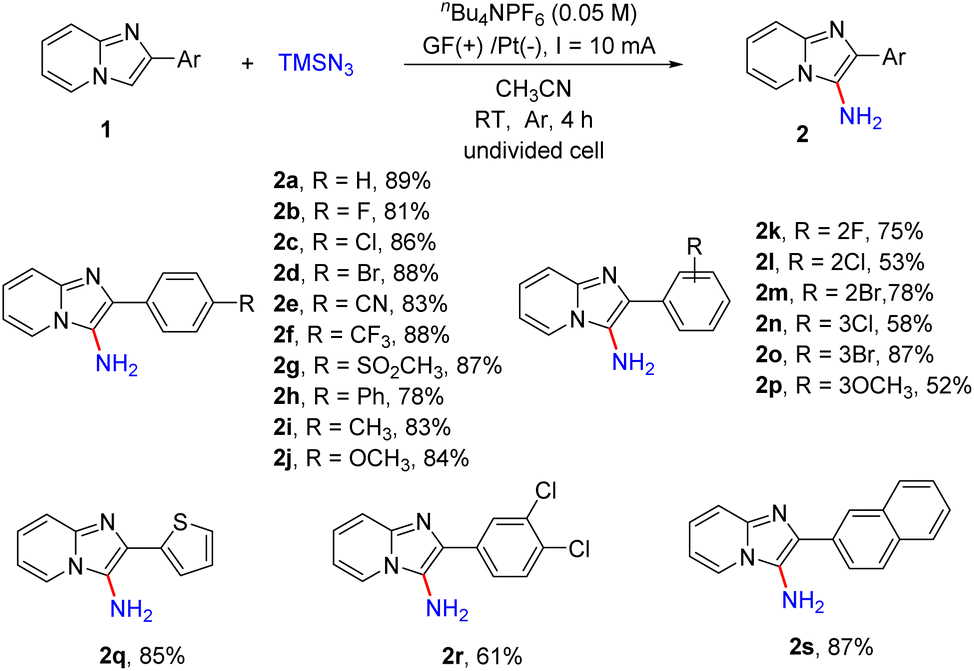 | ||
| Scheme 3 Substrate scope of imidazo[1,2-a]pyridines 1. Reaction conditions: 1 (0.5 mmol), TMSN3 (1.0 mmol, 2 equiv.), nBu4NPF6 (0.05 M) and CH3CN (10 mL) in an undivided cell were electrolyzed (I = 10 mA) at room temperature under Ar for 4 h. Isolated yields based on 1. | ||
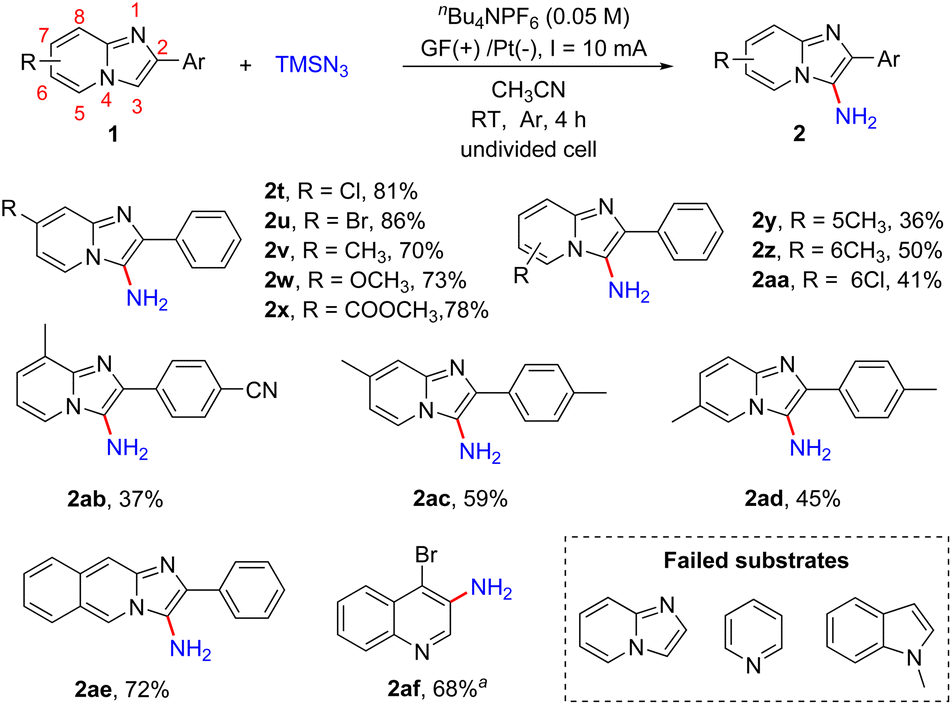
Subsequently, we explored the effect of the substituents on the pyridine ring of 2-phenylimidazo[1,2-a]pyridines (Scheme 4). The substrates 1 with an electron-donating (Me, OCH3) or electron-withdrawing (CO2Me) group or a halogen atom (Cl, Br) at the C7-position of 2-phenylimidazo[1,2-a]pyridines were well tolerated, affording the desired products 2t–2x in 70–86% yields. The reaction was also compatible with different substituents including Me and Cl at the C5- or C6-position of imidazo[1,2-a]pyridines and provided the corresponding products in moderate yields (2y, 2z, 2aa). In addition, imidazopyridines with two substituents on both the pyridine ring and 2-phenyl were also shown to be suitable for this reaction (2ab–2ad). Encouraged by these results, 2-phenylimidazo[1,2-b]isoquinoline was also tested and product 2ae was obtained in 72% yield. Moreover, we tested other nitrogen-containing heterocycles including imidazo[1,2-a]pyridine, pyridine, 1-methyl-1H-indole and 4-bromoquinoline. 4-Bromoquinoline gave the desired product 4-bromoquinolin-3-amine (2af) in 68% yield after prolonging the reaction time to 12 h. Unsubstituted imidazo[1,2-a]pyridine and pyridine were unreactive maybe because of the instability of the intermediates, and most of the substrates were recovered. For 1-methyl-1H-indole, the overoxidation of this heterocycle led to the generation of complex compounds.
 | ||
| Scheme 4 Substrate scope of other imidazo[1,2-a]pyridines 1. Reaction conditions: 1 (0.5 mmol), TMSN3 (1.0 mmol, 2 equiv.), nBu4NPF6 (0.05 M) and CH3CN (10 mL) in an undivided cell were electrolyzed (I = 10 mA) at room temperature under Ar for 4 h. Isolated yields based on 1. Electrolysis for 12 h. | ||
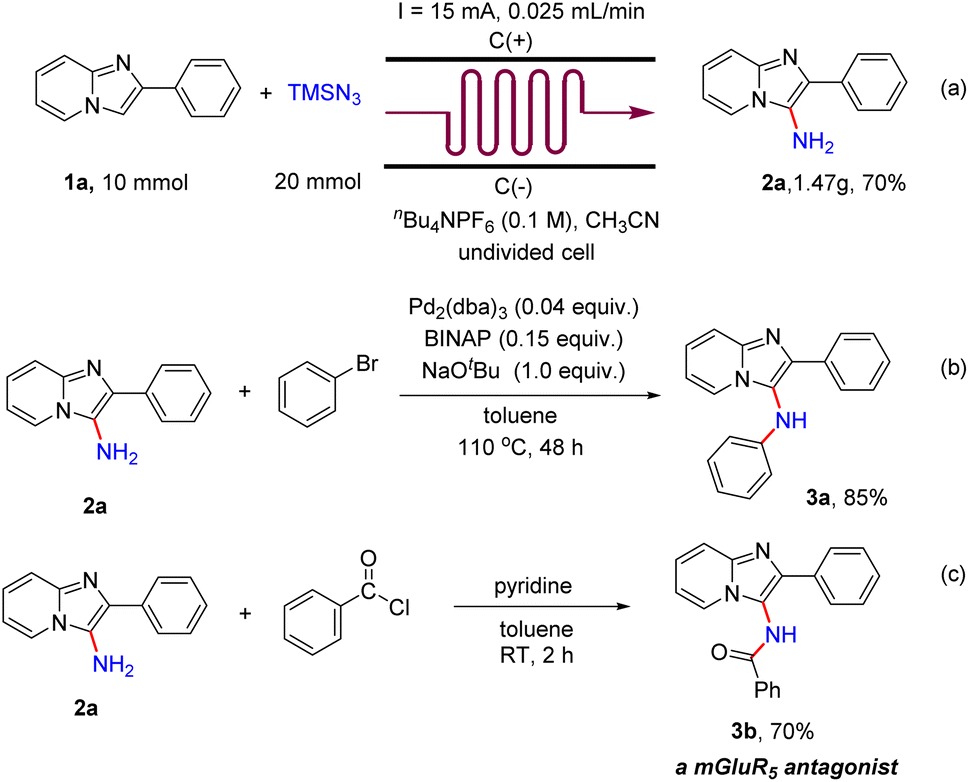
To evaluate the practicality of this electrochemical primary amination protocol, a scaled-up experiment was accomplished in continuous-flow electrochemical microreactors. A 70% yield was afforded when 10 mmol 1a was used (Scheme 5a). To illustrate the application of the products, a palladium-catalyzed cross coupling reaction of 2a with bromobenzene was conducted at 110 °C for 48 h, and the product N,2-diphenylimidazo[1,2-a]pyridin-3-amine (3a) was obtained in 85% yield (Scheme 5b). In addition, when 2a was treated with benzoyl chloride (1.5 equiv.) in toluene (4 mL) and pyridine (2 mL) at room temperature for 1 h, the metabotropic glutamate 5 receptor (mGluR5) antagonist N-(2-phenylimidazo[1,2-a]pyridin-3-yl)benzamide (3b)19 was obtained in 70% yield (Scheme 5c).
 | ||
| Scheme 5 Gram-scale experiment and further transformation of product 2a. Reaction conditions of the scaled-up experiment: carbon rod anode, carbon rod cathode, nBu4NPF6 (0.1 M), CH3CN, I = 15 mA, 0.025 mL min−1, 1a (0.1 M), TMSN3 (0.2 M), 3.67 F mol−1, RT, 66 h. | ||
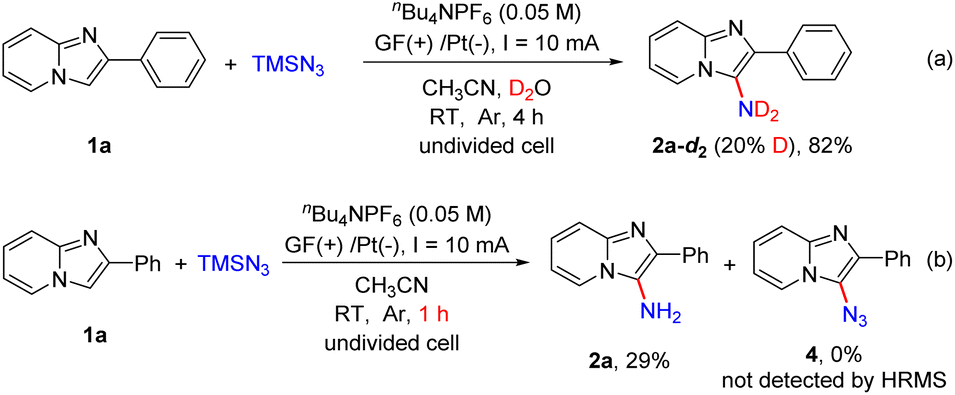
In order to understand this direct C–H amination reaction in detail, several control experiments were conducted (Scheme 6). When this electrochemical reaction was carried out in the presence of D2O in dry CH3CN, product 2a-d2 with partial deuteration of the amino group was obtained in 82% yield (Scheme 6a). This result revealed that the hydrogen of the amino group in the product was derived from H2O. When the model reaction was performed for 1 h, 2a was obtained in 29% yield, and 3-azido-2-phenylimidazo[1,2-a]pyridine (4) could not be detected by HRMS (Scheme 6b).
 | ||
| Scheme 6 Control experiments. | ||
Next, cyclic voltammetry (CV) experiments were performed to detect the oxidation potential of substrates 1a and TMSN3 in this reaction. As shown in Fig. 1, 2-phenylimidazo[1,2-a]pyridine (1a) had a lower oxidation potential (1.40 V vs. SCE) than TMSN3 (no distinct oxidation peak) in the range of 0–2.5 V, indicating that the initial step may be the oxidation of imidazopyridine 1a.
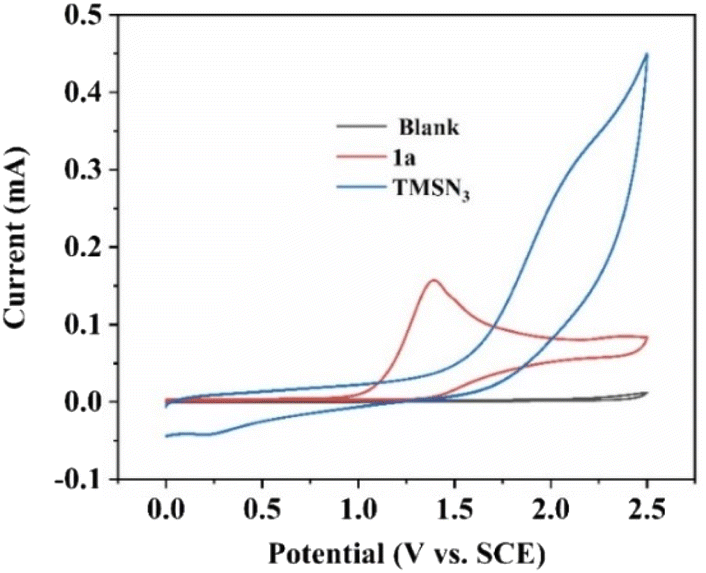 | ||
| Fig. 1 CV scans (scan rate 100 mV s−1) of substrates. Blank (nBu4NPF6 (0.02 M) in MeCN); 2-phenylimidazo[1,2-a]pyridine (1a, 0.01 M, red curve) in the blank. TMSN3 (0.01 M, blue curve) in the blank. | ||
Based on the above experimental and previous studies, a possible reaction pathway of the electrochemical C–H amination is proposed in Scheme 7. Initially, 2-phenylimidazo[1,2-a]pyridine (1a) underwent an oxidation process at the anode to afford the radical cation intermediate A.16 Next, the addition of radical cation A to TMSN3 generated the radical intermediate B.20 The protonation of B and the following release of N2 and deprotonation gave radical D or D′.10 Finally, cathodic reduction of D or D′ and subsequent protonation of E delivered the desired product 2a.
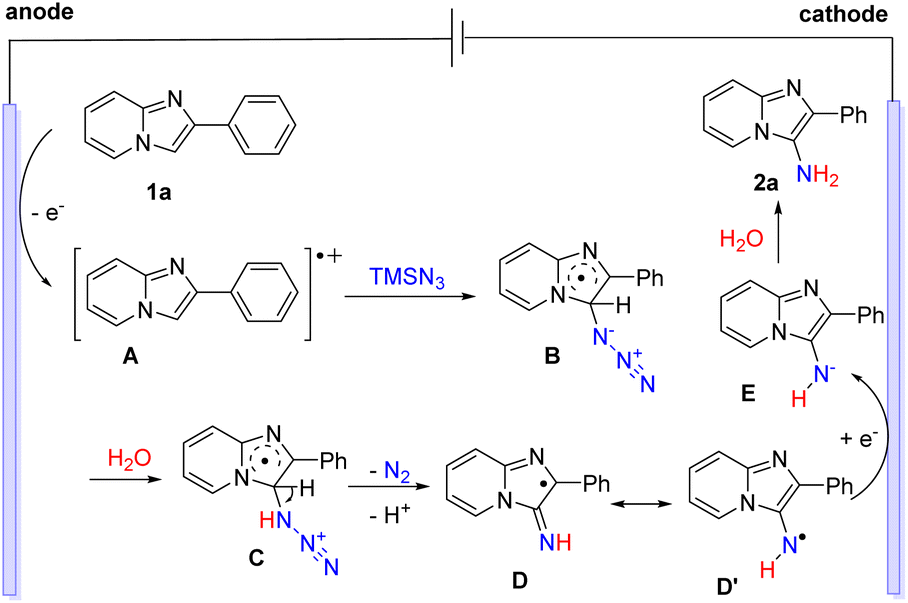 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In summary, we have developed an electrochemical primary amination of imidazopyridines using TMSN3 as the nitrogen source and H2O as the hydrogen source. The reaction scope was broad and a variety of C3-aminated imidazo[1,2-a]pyridines were obtained in moderate to good yields. The remarkable merits of this protocol include transition-metal-free, chemical oxidant-free and mild conditions, and easy transformation. Further synthetic application of this strategy to the primary amination of other heteroarenes is ongoing in our laboratory.Author contributions
Y. Zhu and Q. Chu studied the reaction conditions, investigated the substrate scope and probed the reaction mechanism. H. Li prepared some imidazopyridines. P. Liu and P. Sun contributed to manuscript writing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Project 21672104, 21502097) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- P. Fu, M. Johnson, H. Chen, B. A. Posner and J. B. MacMillan, J. Nat. Prod., 2014, 77, 1245–1248 CrossRef CAS.
- (a) R. Romagnoli, P. G. Baraldi, T. Sarkar, M. D. Carrion, C. L. Cara, O. Cruz-Lopez, D. Preti, M. A. Tabrizi, M. Tolomeo, S. Grimaudo, A. D. Cristina, N. Zonta, J. Balzarini, A. Brancale, H.-P. Hsieh and E. Hamel, J. Med. Chem., 2008, 51, 1464–1468 CrossRef CAS; (b) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (c) C.-Y. Chen, C.-M. Lin, H.-C. Lin, C.-F. Huang, C.-Y. Lee, T.-C. S. Tou, C.-C. Hung and C.-S. Chang, Eur. J. Med. Chem., 2017, 125, 1023–1035 CrossRef CAS.
- A. S. Gunasekara, T. Truong, K. S. Goh, F. Spurlock and R. S. Tjeerdema, J. Pestic. Sci., 2007, 32, 189–199 CrossRef CAS.
- R. Gong, Y. Sun, J. Chen, H. Liu and C. Yang, Dyes Pigm., 2005, 67, 175–181 CrossRef CAS.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
- (a) F. Lang, D. Zewge, I. N. Houpis and R. P. Volante, Tetrahedron Lett., 2001, 42, 3251–3254 CrossRef CAS; (b) N. Xia and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 337–339 CrossRef CAS; (c) H. Xu and C. Wolf, Chem. Commun., 2009, 45, 3035–3037 RSC; (d) M. Fan, W. Zhou, Y. Jiang and D. Ma, Org. Lett., 2015, 17, 5934–5937 CrossRef CAS.
- (a) Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 10028–10029 CrossRef CAS PubMed; (b) D. S. Surry and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 10354–10355 CrossRef CAS PubMed; (c) G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049–11061 CrossRef CAS; (d) F. Bourriquen, A. Bruneau-Voisine, A. Jeandin, E. Stihle and S. Fantasia, Chem. – Eur. J., 2019, 25, 9006–9011 CrossRef CAS PubMed.
- (a) R. A. Green and J. F. Hartwig, Angew. Chem., Int. Ed., 2015, 54, 3768–3772 CrossRef CAS PubMed; (b) R. T. McGuire, J. F. J. Paffile, Y. Zhou and M. Stradiotto, ACS Catal., 2019, 9, 9292–9297 CrossRef CAS.
- (a) J. Peng, M. Chen, Z. Xie, S. Luo and Q. Zhu, Org. Chem. Front., 2014, 1, 777–781 RSC; (b) L. Yu, X. Chen, D. Liu, L. Hu, Y. Yu, H. Huang, Z. Tan and Q. Gui, Adv. Synth. Catal., 2018, 360, 1346–1351 CrossRef CAS.
- Q. Yang, Z. Yang, Y. Tan, J. Zhao, Q. Sun, H.-Y. Zhang and Y. Zhang, Adv. Synth. Catal., 2019, 361, 1662–1667 CrossRef CAS.
- For selected reviews, please see: (a) C. Enguehard-Gueiffier and A. Gueiffier, Mini-Rev. Med. Chem., 2007, 7, 888–899 CrossRef CAS; (b) Z. Tashrifi, M. Mohammadi-Khanaposhtani, B. Larijani and M. Mahdavi, Eur. J. Org. Chem., 2020, 269–284 CrossRef CAS; (c) A. K. Bagdi and A. Hajra, Org. Biomol. Chem., 2020, 18, 2611–2631 RSC.
- K. Yakoub, S. Jung, C. Sattler, H. Damerow, J. Weber, A. Kretzschmann, A. S. Cankaya, M. Piel, F. Rösch, A. S. Haugaard, B. Frølund, T. Schirmeister and H. Lüddens, J. Med. Chem., 2018, 61, 1951–1968 CrossRef CAS PubMed.
- (a) C. Blackburn and B. Guan, Tetrahedron Lett., 2000, 41, 1495–1500 CrossRef CAS; (b) Y. Sandulenko, A. Komarov, K. Rufanov and M. Krasavin, Tetrahedron Lett., 2008, 49, 5990–5993 CrossRef CAS; (c) K. Monir, M. Ghosh, S. Jana, P. Mondal, A. Majee and A. Hajra, Org. Biomol. Chem., 2015, 13, 8717–8722 RSC; (d) R. C. Cioc, H. D. Preschel, G. van der Heijden, E. Ruijter and R. V. A. Orru, Chem. – Eur. J., 2016, 22, 7837–7842 CrossRef CAS PubMed.
- For partial reviews, please see: (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS; (b) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS; (c) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS; (d) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120–1152 CrossRef CAS.
- D. Ghosh, S. Ghosh and A. Hajra, Adv. Synth. Catal., 2021, 363, 5047–5071 CrossRef CAS.
- (a) Y. Yu, Y. Yuan, H. Liu, M. He, M. Yang, P. Liu, B. Yu, X. Dong and A. Lei, Chem. Commun., 2019, 55, 1809–1812 RSC; (b) K. Liu, J. Wu, Y. Deng, C. Song, W. Song and A. Lei, ChemElectroChem, 2019, 6, 4173–4176 CrossRef CAS.
- (a) N. Chen and H.-C. Xu, Green Synth. Catal., 2021, 2, 165–178 CrossRef; (b) S. Guo, L. Liu, K. Hu, Q. Sun, Z. Zha, Y. Yang and Z. Wang, Chin. Chem. Lett., 2021, 32, 1033–1036 CrossRef CAS.
- (a) T. Cui, Y. Zhan, C. Dai, J. Lin, P. Liu and P. Sun, J. Org. Chem., 2021, 86, 15897–15905 CrossRef CAS PubMed; (b) T. Cui, X. Zhang, J. Lin, Z. Zhu, P. Liu and P. Sun, Synlett, 2021, 267–272 CAS; (c) Y. Zhan, Y. Li, J. Tong, P. Liu and P. Sun, Eur. J. Org. Chem., 2021, 2193–2197 CrossRef CAS.
- T. de Paulis, K. Hemstapat, Y. Chen, Y. Zhang, S. Saleh, D. Alagille, R. M. Baldwin, G. D. Tamagnan and P. J. Conn, J. Med. Chem., 2006, 49, 3332–3344 CrossRef CAS PubMed.
- Y. Zhu, C. Jiang, H. Li, P. Liu and P. Sun, J. Org. Chem., 2022, 87, 11031–11041 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03703c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |