 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring electrolyte effects on metal–alkyl bond stability: impact and implications for electrosynthesis
Dylan G.
Boucher
 ,
Zachary A.
Nguyen
and
Shelley D.
Minteer
*
,
Zachary A.
Nguyen
and
Shelley D.
Minteer
*
Department of Chemistry, University of Utah, Salt Lake City, UT, USA. E-mail: minteer@chem.utah.edu
First published on 11th May 2023
Abstract
Transition metal catalysis hinges on the formation of metal–carbon bonds during catalytic cycles. The stability and reactivity of these bonds are what determine product chemo-, regio-, and enantioselectivity. The advent of electrosynthetic methodologies has placed the current understanding of these metal–alkyl bonds into a new environment of charged species and electrochemically induced reactivity. In this paper, we explore the often neglected impact of supporting electrolyte on homogeneous electrocatalytic mechanisms using the catalytic reduction of benzyl chlorides via Co and Fe tetraphenylporphyrins as a model reaction. The mechanism of this reaction is confirmed to proceed through the formation of the metal–alkyl intermediates. Critically, the stability of these intermediates, in both the Co and Fe systems, is found to be affected by the hydrodynamic radius of the supporting electrolyte, leading to differences in electrolyte–solvent shell. These studies provide important information for the design of electrosynthetic reactions, and provide a starting point for the rational design of functional supporting electrolytes.
1. Introduction
The formation and reactivity of metal–carbon bonds are foundational to the fields of catalysis and organometallic chemistry.1 An important part of any catalytic cycle, the formed organometallic intermediates offer a wide array of reactivity and chemical transformations. As such, understanding the molecular and chemical design principles that determine the specific reactivity of these intermediates can unlock the path to new and useful chemical transformations. For example, the Suzuki cross-coupling reaction, which is a palladium-catalyzed method of forming C–C bonds and one of the most used organic methods, proceeds via alkyl–metal bond formation from aryl and alkyl halides and boronic acids.2 Nature utilizes these species as well, with the reversible metal–organic bond homolysis of vitamin B12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3–5 and radical SAM enzymes.6 These species use metal–alkyl bond homolysis to provide access to highly unstable organic radicals by providing stability via the reversible formation of organometallic intermediates to increase the effective lifetime of the organic radical.7 Understanding the myriad of ways the intermediate metal–carbon bonds can be stabilized or destabilized is thus crucial to promoting the desired reactivity.
3–5 and radical SAM enzymes.6 These species use metal–alkyl bond homolysis to provide access to highly unstable organic radicals by providing stability via the reversible formation of organometallic intermediates to increase the effective lifetime of the organic radical.7 Understanding the myriad of ways the intermediate metal–carbon bonds can be stabilized or destabilized is thus crucial to promoting the desired reactivity.
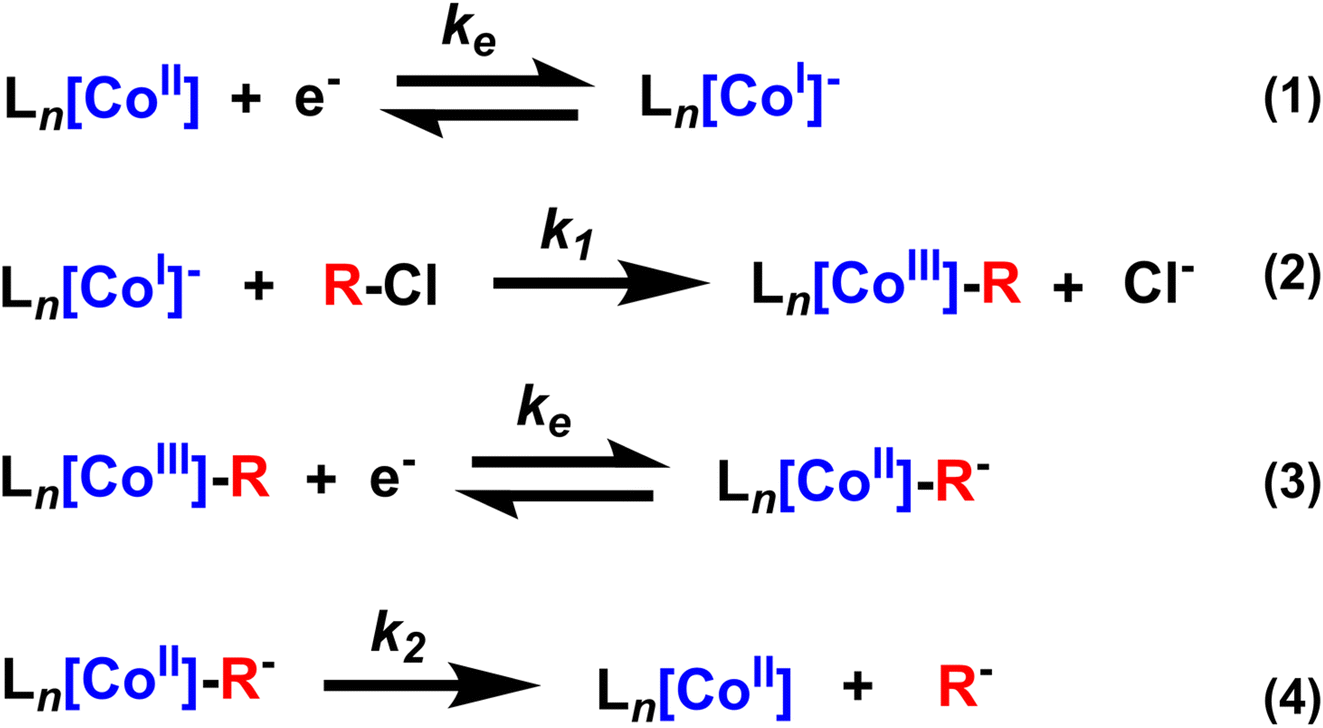
Metal–carbon bonds have also proven central to the burgeoning field of electrosynthesis.8 As many organometallic catalytic cycles involve changes in the oxidation state of the metal or utilize chemical reductants or oxidants to turn over catalytic cycles,9 electrochemical methods are uniquely suited to providing this reactivity in a more sustainable and controllable way (Scheme 1a).10 In fact, electrochemical reduction or oxidation of metal complexes can provide access to high-energy intermediates with unique reactivity not typically accessible by chemical means.11In situ generation of reduced or oxidized intermediates has been studied via electrochemical methods for decades. One of the most well-studied examples is the reduction of M(II) species to M(I) which can act as a potent nucleophile with alkyl halides.5,12–16 In the case of vitamin B12 mimics like cobalt or iron tetraphenylporphyrin (TPP), an established SN2 reaction yields a quasi-stable metal–alkyl intermediate that is observable via electrochemistry and isolable to study via other methods as well (Scheme 1b). Thus, these complexes have provided a wealth of information on the stability and reactivity of organometallic intermediates, information that has enabled a new generation of electrochemical organometallic methodologies.
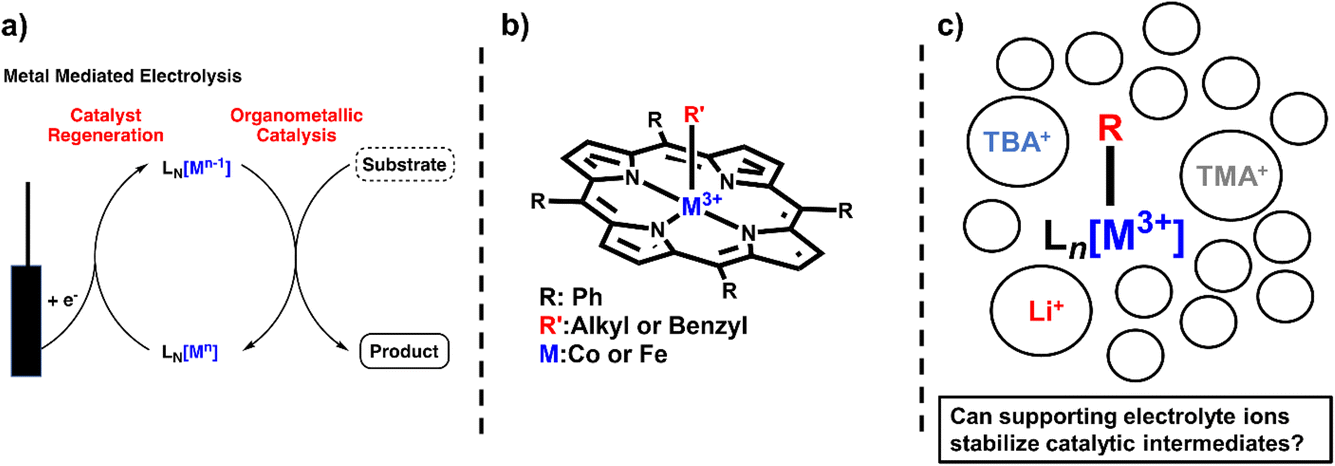 | ||
| Scheme 1 (a) Illustration of electrocatalysis mediated by a metal complex. (b) Structure of the alkyl–metal porphyrin catalysts used in this study. (c) Illustration of the ion and solvent shell surrounding catalytic intermediates. | ||
However, not all of the ways electrochemical methods have been adapted to organic transformations are well understood. One prime example of this is the inclusion of supporting electrolyte. In electrochemical experiments, supporting electrolyte plays the crucial role of increasing solution conductivity by providing a high concentration of charged species.17 However, many of these supporting electrolyte systems are comprised of common counter ions (coordinating and non-coordinating) for charged organometallic species and, thus, are likely to provide charge stabilization to catalytic intermediates (Scheme 1c). The energetic landscape of these homogeneous electrochemical transformations is likely quite different than their chemical counterparts, with certain intermediates stabilized versus others and thereby impacting product chemo-, regio-, and enantioselectivity, depending on the specific reactions. Many studies have noted the impact supporting electrolyte can have on electroorganic transformations. For example, the Lin group noted the marked decrease in enantioselectivity of a hydrocyanation reaction when using a smaller supporting electrolyte (LiClO4) versus a larger supporting electrolyte (tetrabutylammonium tetrafluoroborate, TBABF4),18 citing differences in the polarizability of the electrical double layer at the electrode surface.19 In another example, the Reisman group showed the clear dependence of yield and enantioselectivity of an alkenyl coupling reaction.20 NaI was used both as a supporting electrolyte and an iodide source, which is postulated to enhance electron transfer or facilitate the formation of organo-iodide electrophiles.21 These reactions show the distinct possibilities of rationally designing electrolytes to accomplish a number of tasks beyond solution conductivity, and to actually provide distinct reaction selectivity. Unfortunately, a useful quantitative understanding of the impact of the electrolyte on electrochemical organic reactions with established electroanalytical techniques has yet to be realized.
Here, we investigate the impact of supporting electrolyte on the stability of electrochemically generated organometallic species using cyclic voltammetry (CV).22 Monitoring the catalytic reduction of benzyl chloride via Co and Fe TPP complexes using CV, we observed the formation of metal–alkyl intermediates. The scan rate dependence of these voltammograms revealed that the metal–organic species decays upon further electrochemical reduction of the organometallic intermediate. We illustrate that this decay is dependent on electrolyte hydrodynamic radius, with enhanced stability in diffuse, weakly coordinating electrolytes (tetrabutylammonium counter ions) and decreased stability (faster decay) with hard, strongly coordinating electrolytes (like Li+ cations). These effects were observed for both Co(II) and Fe(III)Cl TPP complexes, highlighting the generality of the results for this reaction. These results show the critical role the supporting electrolyte can play in electroorganic reactions and provide a new rationale for choosing a particular supporting electrolyte species in screening organic reactions. We anticipate these studies will help enable the rational design of new, functional supporting electrolytes for organic transformations.
2. Experimental
All experiments were carried out in an mBraun glovebox with O2 and moisture levels below 0.5 ppm. Electrochemical techniques were performed on a Biologic VSP potentiostat routed into the glovebox. Electrochemistry was performed using a three-electrode setup with a 0.07 cm2 glassy carbon working electrode (CH Instruments, CHI 104), a platinum mesh counter electrode (0.5 cm2, Strem, 99.99%), and a silver wire pseudo-reference electrode (CH Instruments, CHI 112) with a Teflon frit and glass tube filled with electrolyte. In all cases, extra dry dimethylformamide was used as the solvent (DMF, Acros 99.95% Extra Dry) and the supporting electrolytes tetrabutylammonium hexafluorophosphate (TBAPF6, Sigma-Aldrich, >99.0% for electrochemical analysis), tetramethylammonium hexafluorophosphate (TMAPF6, Sigma-Aldrich, >99.0% for electrochemical analysis), and lithium perchlorate (LiClO4, Sigma-Aldrich, >99.0% for electrochemical analysis) were all recrystallized and dried before use. All measurements were referenced to ferrocene immediately after the experiment. For cyclic voltammetry experiments, cobalt(II) tetraphenylporphyrin (Strem, 99%) or iron(III) tetraphenylporphyrin chloride (Strem, 99%) were dissolved in electrolyte to afford a 1 mM solution of the analyte of interest. 2 mL of this solution was added to a glass scintillation vial for measurements. Another 2 mL of this solution was used to dissolve the substrate 1-(chloroethyl)benzene (TCI America, 97+%) to afford a solution that was 1 M in substrate. From this solution, aliquots were taken and added via micropipette to the electrochemical cell. Thus substrate was added while keeping the catalyst concentration constant. Concentrations were corrected to account for dilution of the substrate for analysis. Voltammetry was performed at multiple scan rates, 10, 25, 50, and 100 mV s−1, and all peak and current plateau analysis was done with the current of the first scan. Solution was agitated between experiments to ensure a refresh of the electrode surface.3. Results and discussion
3.1 Cobalt tetraphenylporphyrin
First we investigated the cyclic voltammetry of Co(II)(TPP). The complex exhibited a reversible wave centered at −1.28 V vs. Fc+/0 associated with the Co(II)/Co(I) redox couple over several scan rates (Fig. 1a and b, blue traces). With the addition of a benzyl chloride substrate, 1-(chloroethyl)benzene (PhEtCl), a second wave appears, with a cathodic peak centered around −1.6 V vs. Fc+/0, and the return wave of the Co(II)/Co(I) couple begins to disappear, indicating consumption of Co(I) in a chemical process. At moderate scan rates (100 mV s−1), the new peak is quasi-reversible and proportional to the concentration of PhEtCl added (Fig. 1a). At low scan rates (10 mV s−1), this wave loses its peak-like behavior and becomes a plateau, indicative of catalytic behavior. The magnitude of this plateau current is also proportional to the concentration of substrate PhEtCl, increasing dramatically as more was added. The current at −1.7 V vs. Fc+/0 (roughly where the steady-state plateau current is achieved for all scan rates) as a function of substrate concentration was normalized to the current at this potential when no substrate is present to compare between scan rates. Analysis of the normalized plateau current revealed a linear dependence of the current with respect to square root substrate concentration at all scan rates, indicative of a homogeneous chemical process (Fig. 1c). The current normalization revealed that the greatest current increases were observed at slow scan rates and decreased steadily as the scan rate was increased, indicative of an EC′ catalytic mechanism.23,24 | ||
| Fig. 1 (a) Cyclic voltammetry of 1 mM Co(TPP) with increasing concentrations of phenylethylchloride at 100 mV s−1, (b) at 10 mV s−1. (c) Normalized plateau currents (taken to be around −1.7 V vs. Fc+/0, normalized to the current at this potential when no substrate was present) for several scan rates, showing that a chemical process contributes to current increases at low scan rates. Working electrode was a 0.07 cm2 glassy carbon electrode in a solution of 0.1 M TBAPF6 in DMF. | ||
These voltammetry studies reveal a number of important features of Co(TPP)'s reaction with benzyl chloride. First, the decrease of the Co(I) return oxidative wave with PhEtCl concentration with no substantial increase in the forward reductive wave is indicative of an EC process, wherein Co(II) is reduced to Co(I) (E step), which reacts subsequently with the benzyl chloride (C step). The chemical step consumes Co(I) at the electrode surface, and thus leaves none for the return wave. In this context, the appearance of a new peak at −1.6 V vs. Fc+/0 as the return wave disappears can be attributed to the reduction of the product of the C step, the alkylated metal complex, Co(III)–R(TPP) to Co(II)–R(TPP).12 At slower scan rates, this quasi-reversible peak changes shape to an irreversible current plateau. This sigmoidal shape is indicative of a catalytic process,24 presumably the decay of the reduced alkylated Co species to the original Co(II) state and the carbanion. In the kinetic zone diagram formalism developed by Savéant, this transition from quasi-reversible peak to plateau corresponds to the transition from zone KD to KS. In other words, at low concentrations the chemical step is slow relative to the voltammetric time-scale, and increasing concentration or decreasing scan rate gives the catalytic response. The normalized peak current was correlated to the square root of substrate concentration, as expected for a catalytic process.23 The linear correlation is in keeping with the expectation of a homogeneous process under kinetic control (zone K). The overall reaction can be summarized as an ECEC′ process as outlined in Scheme 2.
 | ||
| Scheme 2 Overall mechanism of Co(TPP) reduction of a benzyl chloride. | ||
Owing to our ability to resolve the decay of the metal–alkyl species via the current plateau at slow scan rates, we resolved to study this process in a number of supporting electrolytes. We envisioned that the hydrodynamic radius of the dissolved metal complex was indicative of the solvation environment of the metal–alkyl intermediate. Essentially, a smaller hydrodynamic radius is indicative of a harder, more compact coordination environment (Li+), while a softer, more diffuse coordination environment is expected for the larger hydrodynamic radius (TBA+). The supporting electrolytes used, ordered from largest to smallest hydrodynamic radius (solvated ion size), were tetrabutylammonium hexafluorophosphate (TBAPF6), tetramethylammonium hexafluorophosphate (TMAPF6), and lithium perchlorate (LiClO4). This was verified from the diffusion constant of Co(TPP) which was calculated in each supporting electrolyte (when no substrate was present) according to the Randles–Sevcik equation.17 The diffusion constant varied with electrolyte as 3.8 × 10−6 cm2 s−1 for TBAPF6, 6.3 × 10−6 cm2 s−1 for TMAPF6, and 6.9 × 10−6 cm2 s−1 for LiClO4, where a lower diffusion constant indicates a smaller (compacted) hydrodynamic radius in keeping with changes in solution viscosity. The same plateau behavior was observed for all electrolytes at slow scan rates of 10 mV s−1. The normalized current plateau at −1.7 V vs. Fc+/0 as a function of square-root PhEtCl concentration is outlined in Fig. 2, with different electrolytes providing different increases in the magnitude of the plateau current. The difference in the current increases is maximized at high concentrations of PhEtCl. The largest current increases were seen for LiClO4, intermediate increases were observed for TMAPF6 and the smallest current increases were observed for TBAPF6.
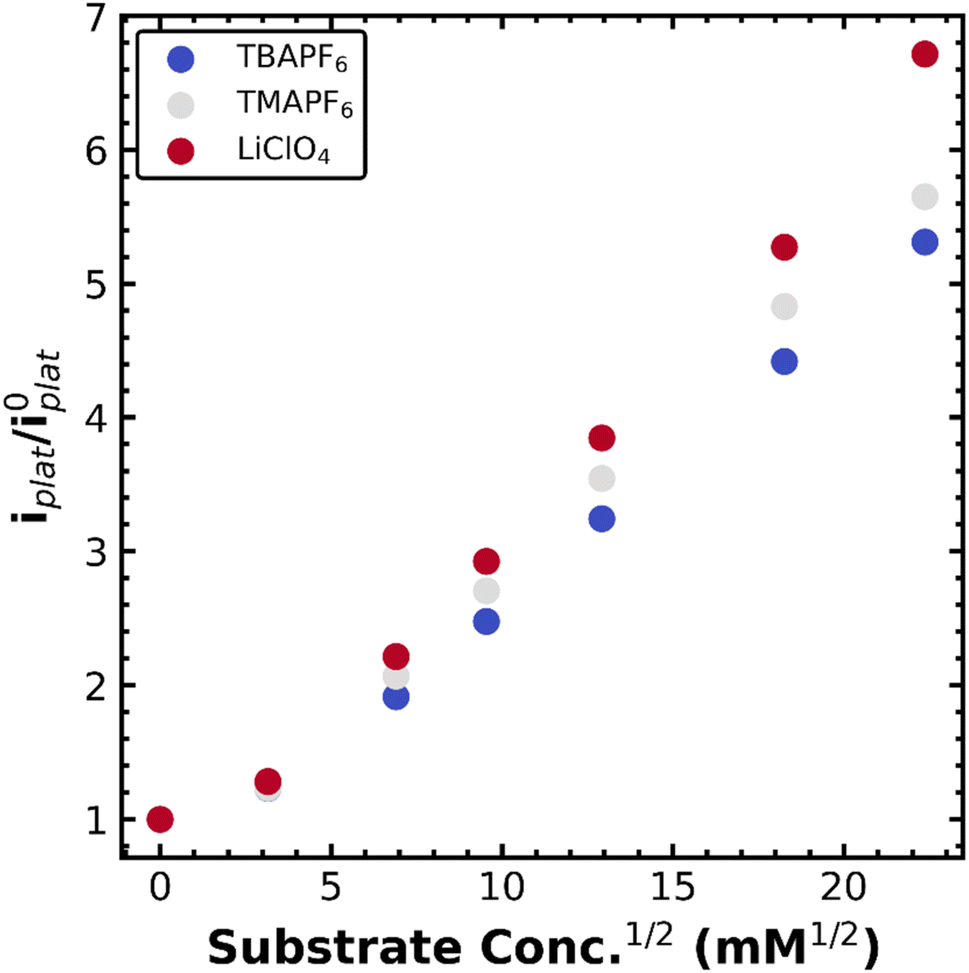 | ||
| Fig. 2 Normalized plateau currents at −1.7 V vs. Fc+/0 for 1 mM Co(TPP) in a DMF solution containing 0.1 M TBAPF6 (blue), TMAPF6 (grey), or LiClO4 (red) for increasing amounts of the substrate PhEtCl. Scan rate was 10 mV s−1 with a 0.07 cm2 glassy carbon working electrode. | ||
The differences in current increases between different supporting electrolytes clearly show the impact supporting electrolyte has on the catalytic process. As established above, the current increase of the plateau is attributed to the decay of the metal–alkyl species to return to the original oxidation state of Co(II), where it can undergo the catalytic process again and thereby increasing the current. The differences in current increases between the different electrolytes can be interpreted as an increase or decrease in the rate of the catalytic step, decay of the metal alkyl species. Differences in diffusion can also impact the current plateau24 (and electrolyte does impact diffusion as seen above) but normalization to the current when no substrate is present cancels out this effect. Interference from capacitive charging can be discounted, for the same reason. Importantly, the increase in rate correlates to the hydrodynamic radius of the supporting electrolyte: the largest cationic supporting electrolyte ion is TBA+ which gives the smallest current increases and the smallest cationic supporting electrolyte is Li+ which gives the largest current increases. We attribute this to stabilization (or destabilization) of the charged metal–alkyl intermediate Co(II)–R− due to the hardness of the supporting electrolyte solvation shell. In the case of the large, diffuse cationic species, TBA+, the solvent–ion shell surrounding the intermediate can better stabilize the charged organometallic species, owing to its softer, more organic nature. In contrast, the small cation Li+ likely destabilizes the organometallic intermediate with its harder ion shell, promoting catalysis via the decay of the metal–alkyl bond. These results indicate a method to stabilize or destabilize useful organometallic intermediates during electroorganic reactions via supporting electrolyte choice.
3.2 Iron chloride tetraphenylporphyrin
Given the supporting-electrolyte dependence of Co(TPP) on the reduction of benzyl chlorides, we wanted to extend these results to a related, but more complicated system. FeCl(TPP) appeared as an interesting candidate as it is reported to do similar chemistry but requires an extra reduction (as it begins in the Fe(III) state) and the dissociation of the coordinating chloride counter ion. We reasoned these features might reveal more ways that the supporting electrolyte can impact the mechanism of catalysis. Cyclic voltammetry of a 1 mM solution of Fe(III)Cl(TPP) showed two successive reductive waves (Fig. 3a), one centered at −0.65 V vs. Fc+/0 associated with the reduction of Fe(III) to Fe(II), and another at −1.5 V vs. Fc+/0 associated with the reduction of Fe(II) to Fe(I). The second of these peaks, the Fe(II)/Fe(I) couple is reversible, but the first peak, the Fe(III)/Fe(II) couple exhibits a scan rate dependent feature on the return wave associated with re-coordination of the chloride ion. Upon the addition of PhEtCl, the second peak begins to increase in magnitude and the reverse peak associated with reoxidation of Fe(I) begins to disappear. Further addition at high scan rates shows the emergence of a second oxidative wave in the return scan that lies positive of the reoxidation of Fe(I). At slower scan rates, the increase in the peak current is much greater (Fig. 3b). In addition, the formation of a shoulder wave near the second reductive wave (evident at 100 mV s−1 but much clearer at slow scans) becomes observable, and the emergence of the second oxidative wave is much less pronounced. The current increase of the second reductive wave (normalized to the current of the second reductive wave when no substrate is present) revealed a linear correlation with the square root substrate concentration (Fig. 3c). Slower scan rates exhibited larger normalized current increases than the faster scan rates, indicating a slow chemical process taking place.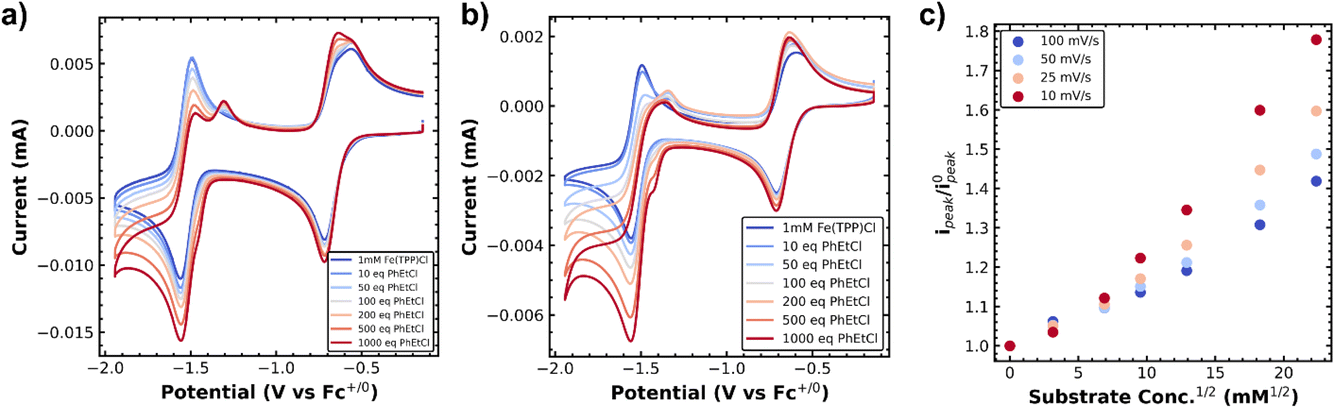 | ||
| Fig. 3 (a) Cyclic voltammetry of 1 mM FeCl(TPP) with increasing concentrations of phenylethylchloride at 100 mV s−1, (b) at 10 mV s−1. (c) Normalized peak currents (of peak at −1.55 V vs. Fc+/0, normalized to the current when no substrate was present) for several scan rates, showing that a slow chemical process contributes to current increases at low scan rates. Working electrode was a 0.07 cm2 glassy carbon electrode in a solution of 0.1 M TBAPF6 in DMF. | ||
As with the Co(TPP) complex, this voltammetry reveals critical info about the mechanism of benzyl chloride reduction with FeCl(TPP). First, the current increases upon the addition of PhEtCl are restricted to the Fe(II)/Fe(I) couple, indicating that Fe(I) is the species that reacts with the benzyl chloride, a fact further supported by the decrease in the return oxidation of Fe(I). The emergence of a second oxidative wave on the return scan is indicative of the oxidation of the product of the reaction between Fe(I) and the benzyl chloride, confirmed to be the metal–alkyl intermediate.16 As such, the formation of the shoulder to the Fe(II) reduction is likely associated with the reduction of the same product, meaning the peaks of the Fe(II)/Fe(I) couple, and the formed Fe(III)–R/Fe(II)–R couple overlap significantly. However, the scan rate dependence suggests that the current increase is not only due to the emergence of a new redox active species. The current increases at slow scan rates are greater than at faster scan rates, which could be due to a slow catalytic process, but could also result from more Fe(III)–R being produced over the longer scan (and subsequently being reduced to produce higher current). Crucially, the new return wave is much less pronounced at slower scan rates, indicating that the reduction of Fe(III)–R to Fe(II)–R is not wholly reversible, and at least some of the reduced Fe(II)–R decays before it can be reoxidized on the return scan. Another interesting note is that the return wave of the Fe(III)/Fe(II) couple is also affected by PhEtCl concentration, presumably because the reaction of Fe(I) with PhEtCl produces chloride ions, which in turn impacts the magnitude of the ion-coupled wave. The overall chemical process can be summarized as EECEC′ and is illustrated schematically in Scheme 3.
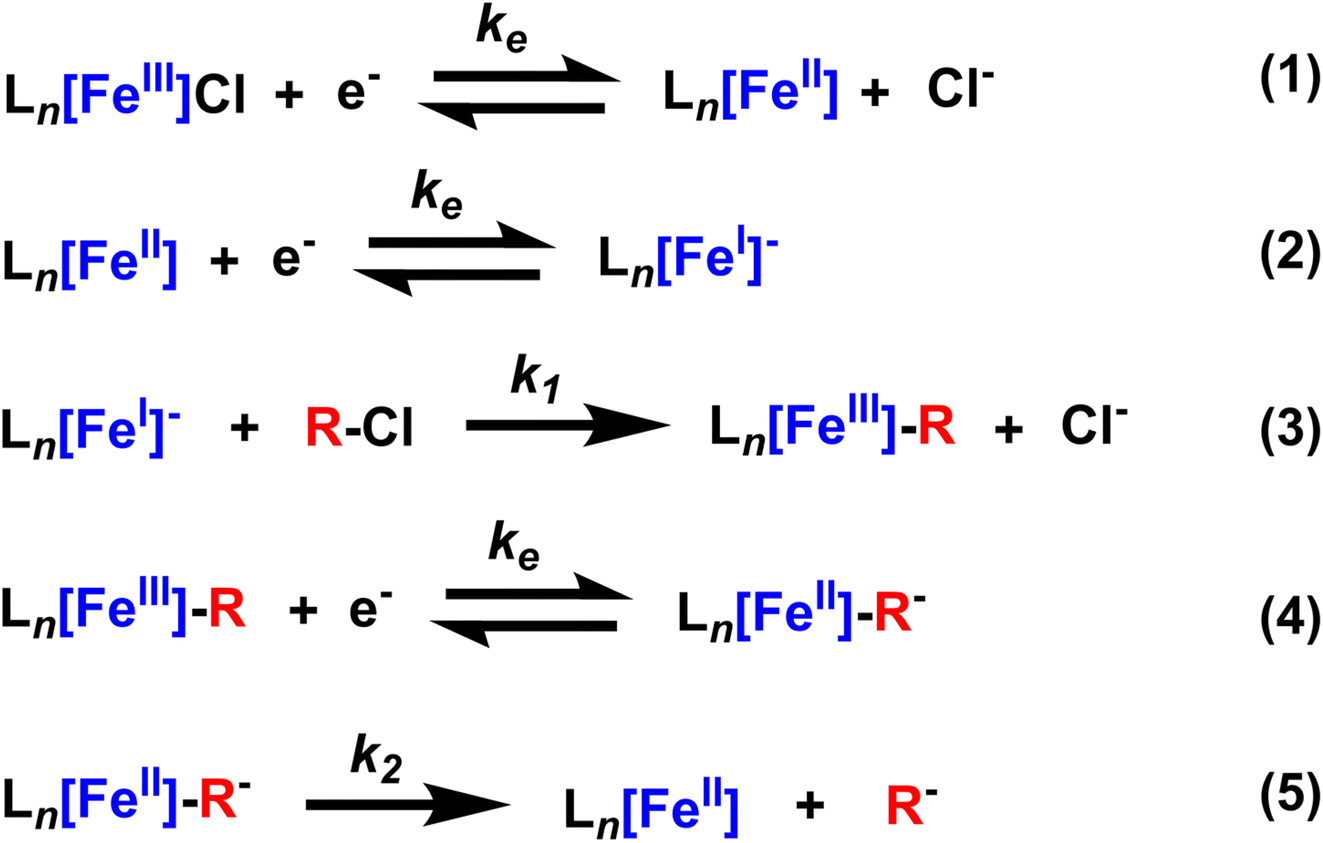 | ||
| Scheme 3 Overall mechanism of FeCl(TPP) reduction of a benzyl chloride. | ||
With this mechanistic assignment, we moved to again assess the stability of the metal–alkyl species in several supporting electrolytes (TBAPF6, TMAPF6, and LiClO4). As with the Co(TPP) system, electrolyte choice impacts the diffusion constant according to changes in electrolyte viscosity: 2.7 × 10−6 cm2 s−1 for TBAPF6, 4.7 × 10−6 cm2 s−1 for TMAPF6, and 4.8 × 10−6 cm2 s−1 for LiClO4 as calculated by the Randles–Sevcik equation. Fig. 4 depicts the normalized peak current (not plateau) as a function of PhEtCl concentration for several different supporting electrolytes. Generally, the smaller supporting electrolyte once again gave higher current increases, though clearly, the dependence is less straightforward than in the case of Co(TPP). Namely, TBAPF6, the largest, provided the smallest current increases until the highest PhEtCl concentration, where it surpassed TMAPF6. The increase in current between PhEtCl concentrations for TBAPF6 looks mostly linear, while it appears that the TMAPF6 increases begin to saturate, allowing the TBAPF6 to surpass it. LiClO4 remained the highest in current increases at all concentrations.
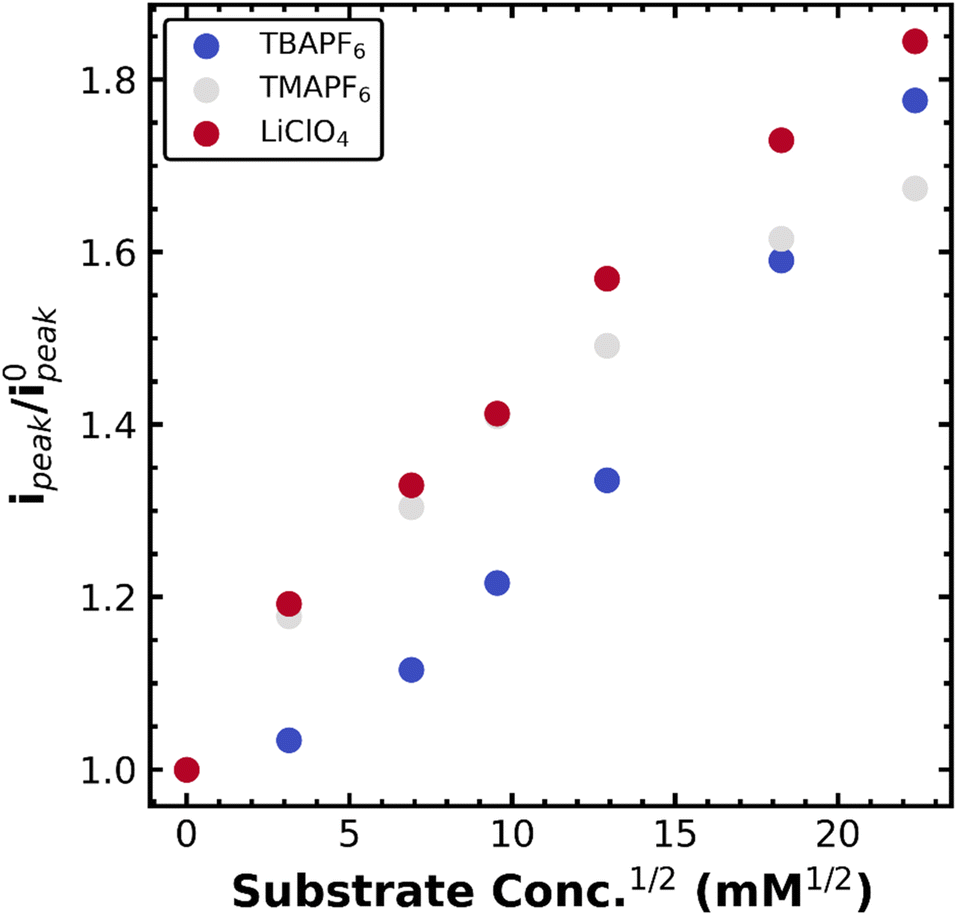 | ||
| Fig. 4 Normalized peak currents at −1.55 V vs. Fc+/0 for 1 mM FeCl(TPP) in a DMF solution containing 0.1 M TBAPF6 (blue), TMAPF6 (grey), or LiClO4 (red) for increasing amounts of the substrate PhEtCl. Scan rate was 10 mV s−1 with a 0.07 cm2 glassy carbon working electrode. | ||
As with the Co(TPP) system, the softness or hardness of the cation of the supporting electrolyte appears to have a pronounced effect on the stability of the iron–alkyl bond, with hard (Li+) supporting electrolytes promoting catalysis via decay of the metal alkyl species, presumably for the same reasons as outlined above. These current increases are much smaller than those observed for the Co(TPP) system as well, highlighting the relative stability of the iron–alkyl species versus the Co–alkyl species. One important caveat is that at high PhEtCl concentrations, the TMAPF6 begins to saturate, highlighting a more complicated mechanism at play. The saturation may be due to differences in solubility or interference with the ion-coupled movement of the iron chloride species. Regardless, these results indicated the generality of the electrolyte effects for metal porphyrin catalyzed benzyl chloride reduction. Extension of these concepts to other metal complexes with different mechanisms is underway. Systems that undergo reversible bond homolysis similar to vitamin B12 are of particular interest, as this analysis may provide a way to quantify longstanding questions regarding radical cage collapse mechanisms in organometallic catalysis.
4. Conclusions
In this study, we have outlined the impact of supporting electrolyte on metal porphyrin catalyzed benzyl chloride reduction. Cyclic voltammetry studies revealed an ECEC′ mechanism where the final step was the decay of the metal–alkyl intermediate, thus providing a handle on the stability of the metal–alkyl bond stability via the current plateau. Plateau current correlation with substrate concentration in different supporting electrolytes revealed a dependence on the hydrodynamic radius of the supporting electrolyte, where the smallest electrolyte (LiClO4) provided the largest current increase. These results are interpreted as supporting electrolyte altering the stability of the metal–alkyl intermediate via the hardness or softness of the electrolyte–solvation shell. However, there is also a possibility that current increases are driven by the stabilization of the carbanion, in particular the case of Li+ cations which are known to form organoalkali compounds with aryl and benzylic carbanions.25,26 Studies identifying the chemical intermediates of these reactions are ongoing, and will no doubt inform electrolyte choice and design in the future. A similar mechanism was explored for FeCl(TPP), albeit with an additional reduction and ion-coupled movement of the chloride. Analysis showed that the same general trend in the current increase and supporting electrolyte hydrodynamic radius could be observed for the Fe–alkyl intermediate. We anticipate these supporting electrolyte effects can be extended to a number of electrochemical reactions. While the precise dependence will depend on the exact mechanism, there are likely a number of unexplored avenues for electrolyte-mechanism interactions. Most importantly, these results have major implications for the design of electrosynthetic reactions. The impact of electrolyte on metal–carbon bond stability means that electrolytes can be chosen rationally, if the mechanism is well understood, to stabilize or destabilize particular intermediates and promote product specificity. Thus, these results are an important first step in the realization of designed supporting electrolytes, that may perform multiple functions beyond the traditional role.Data availability
All data is made available in the main text.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the NSF CCI Center for Synthetic Organic Electrochemistry (CHE-2002158).References
- J. D. Atwood, Inorganic and Organometallic Reaction Mechanisms, VCH Publishers, New York, 2nd edn, 1997 Search PubMed.
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95(7), 2457–2483 CrossRef CAS.
- R. G. Finke and B. D. Martin, Coenzyme AdoB12 vs. AdoB12.− homolytic Co-C cleavage following electron transfer: a rate enhancement ≥1012, J. Inorg. Biochem., 1990, 40(1), 19–22 CrossRef CAS PubMed.
- J. Halpern, Mechanisms of coenzyme B12-dependent rearrangements, Science, 1985, 227(4689), 869–875 CrossRef CAS PubMed.
- D. Lexa and J. M. Saveant, Electrochemistry of vitamin B12 I. Role of the base-on/base-off reaction in the oxidoreduction mechanism of the B12r–B12s system, J. Am. Chem. Soc., 1976, 98(9), 2652–2658 CrossRef CAS PubMed.
- J. B. Broderick, et al., Radical S-adenosylmethionine enzymes, Chem. Rev., 2014, 114(8), 4229–4317 CrossRef CAS PubMed.
- H. Fischer, The persistent radical effect: a principle for selective radical reactions and living radical polymerizations, Chem. Rev., 2001, 101(12), 3581–3610 CrossRef CAS PubMed.
- C. A. Malapit, et al., Advances on the Merger of Electrochemistry and Transition Metal Catalysis for Organic Synthesis, Chem. Rev., 2022, 122(3), 3180–3218 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96(2), 877–910 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117(21), 13230–13319 CrossRef CAS PubMed.
- R. Francke and R. D. Little, Redox catalysis in organic electrosynthesis: basic principles and recent developments, Chem. Soc. Rev., 2014, 43(8), 2492–2521 RSC.
- D. Lexa, J. M. Savéant and J. P. Soufflet, Chemical catalysis of the electrochemical reduction of alkyl halides, J. Electroanal. Chem. Interfacial Electrochem., 1979, 100(1–2), 159–172 CrossRef CAS.
- G. B. Maiya, B. C. Han and K. M. Kadish, Electrochemical studies of cobalt–carbon bond formation. A kinetic investigation of the reaction between (tetraphenylporphinato)cobalt(I) and alkyl halides, Langmuir, 1989, 5(3), 645–650 CrossRef CAS.
- R. L. Birke, et al., Electroreduction of a series of alkylcobalamins: mechanism of stepwise reductive cleavage of the Co-C bond, J. Am. Chem. Soc., 2006, 128(6), 1922–1936 CrossRef CAS PubMed.
- C. Costentin, et al., Concertedness in proton-coupled electron transfer cleavages of carbon–metal bonds illustrated by the reduction of an alkyl cobalt porphyrin, Chem. Sci., 2013, 4(2), 819–823 RSC.
- D. Lexa, J. Mispelter and J. M. Saveant, Electroreductive alkylation of iron in porphyrin complexes. Electrochemical and spectral characteristics of σ-alkylironporphyrins, J. Am. Chem. Soc., 1981, 103(23), 6806–6812 CrossRef CAS.
- A. J. Bard, L. F. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, Wiley, 3rd edn, 2022, p. 1104 Search PubMed.
- L. Song, et al., Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes, Nat. Chem., 2020, 12(8), 747–754 CrossRef CAS PubMed.
- A. Redden and K. D. Moeller, Anodic coupling reactions: exploring the generality of Curtin-Hammett controlled reactions, Org. Lett., 2011, 13(7), 1678–1681 CrossRef CAS PubMed.
- T. J. DeLano and S. E. Reisman, Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis, ACS Catal., 2019, 9(8), 6751–6754 CrossRef CAS PubMed.
- A. H. Cherney and S. E. Reisman, Nickel-catalyzed asymmetric reductive cross-coupling between vinyl and benzyl electrophiles, J. Am. Chem. Soc., 2014, 136(41), 14365–14368 CrossRef CAS PubMed.
- J. M. Savéant and C. Costentin, Elements of Molecular and Biomolecular Electrochemistry, John Wiley & Sons Inc, 2nd edn, 2019 Search PubMed.
- C. Costentin and J.-M. Savéant, Multielectron, Multistep Molecular Catalysis of Electrochemical Reactions: Benchmarking of Homogeneous Catalysts, ChemElectroChem, 2014, 1(7), 1226–1236 CrossRef CAS.
- J. M. Saveant and E. Vianello, Potential-sweep chronoamperometry: Kinetic currents for first-order chemical reaction parallel to electron-transfer process (catalytic currents), Electrochim. Acta, 1965, 10(9), 905–920 CrossRef CAS.
- H. Gilman and G. L. Schwebke, Improved Method for the Preparation of Benzyllithium, J. Org. Chem., 1962, 27(12), 4259–4261 CrossRef CAS.
- R. Waack, L. D. McKeever and M. A. Doran, The nature of carbon–lithium bonding in benzyl-lithium and its variation with solvent, J. Chem. Soc. D, 1969,(3), 117b–118 RSC.
| This journal is © The Royal Society of Chemistry 2023 |