
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking nanotubular bismuth oxyiodide toward carbon-neutral electrosynthesis†
Peng-Fei
Sui
 a,
Min-Rui
Gao
a,
Meng-Nan
Zhu
a,
Chenyu
Xu
a,
Yi-Cheng
Wang
a,
Subiao
Liu
b and
Jing-Li
Luo
*ac
a,
Min-Rui
Gao
a,
Meng-Nan
Zhu
a,
Chenyu
Xu
a,
Yi-Cheng
Wang
a,
Subiao
Liu
b and
Jing-Li
Luo
*ac
aDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 1H9, Canada. E-mail: jingli.luo@ualberta.ca; Tel: +1 780 492 2232
bSchool of Minerals Processing and Bioengineering, Central South University, Changsha, Hunan 410083, P. R. China
cShenzhen Key Laboratory of Polymer Science and Technology, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, Guangdong, China
First published on 18th February 2023
Abstract
The electrochemical CO2 reduction reaction (CO2RR) to formate is one of the attractive strategies for achieving carbon-neutral sustainability, but synthesizing highly active catalytic materials capable of selectively forming formate, especially under industrially relevant conditions, remains challenging. We herein report ultrathin one-dimensional nanotubular Bi5O7I with substantial amounts of nanopores and oxygen vacancies for the CO2RR. A desirable selectivity of over 93% and high current density for formate are achieved in a potential window of 1000 mV in a flow cell. More importantly, a stable performance of 140 h with faradaic efficiency over 90% in a membrane electrode assembly system demonstrates great potential for industrial CO2RR application. The abundant defects within the ultrathin nanotubular structure not only improve the CO2 adsorption and charge transfer capabilities that facilitate the reaction kinetics, but also modulate the electronic structure and optimize the energy barrier toward formate as revealed by the theoretical calculations. Concurrently, the local high pH caused by the structural defects and hydrophobic surface could remarkably suppress the hydrogen evolution reaction and in turn, accelerate formate formation. These advances demonstrate an effective structure and defect engineering to unlock one-dimensional electrocatalysts to boost the CO2RR toward target products.
Broader contextThe conversion of greenhouse gas CO2 into value-added chemicals and fuels through electrochemical CO2 reduction reaction (CO2RR) holds promising potential to reduce the CO2 emission level and simultaneously store renewable energy to achieve sustainable carbon neutrality. Given the commercial viability of different reduced products, reducing CO2 to formate is one of the most economically viable pathways based on the recent technoeconomic analysis. Nevertheless, achieving high selectivity and current density for formation in a wide potential window still remains challenging when implementing practical CO2RR technology applications. In this study, we present an effective strategy by introducing abundant defects into the one-dimensional ultrathin nanotubular structure of Bi5O7I for efficient CO2RR to formate at an industrial-level current density in both flow cell and membrane electrode assembly. The experimental and theoretical studies comprehensively unravel the origins of the enhanced catalytic activity and selectivity resulting from the joint effort of structure and defect merits. These findings underscore the effectiveness of the combination of structure and defect engineering as an attractive strategy for designing advanced electrocatalysts for achieving carbon-neutral sustainability. |
1. Introduction
The ever-increasing accumulation of CO2 in the atmosphere has caused severe climate changes that negatively impact the natural environment and sustainable development of human society.1–3 The electrochemical CO2 reduction reaction (CO2RR) is regarded as an attractive technology to lower the CO2 emission level when powered by the electricity generated from renewable energy sources (e.g., wind, solar or tidal energy), and it can eventually achieve sustainable carbon neutrality by converting CO2 to valuable chemicals.4–6 However, the inertness of CO2 molecules and multi-electron transfer process during the CO2RR usually lead to high thermodynamic energy barriers and sluggish reaction kinetics.7,8 The low solubility of gaseous CO2 in aqueous media and broad distributions of target products also account for the unsatisfactory CO2RR performance.9 In addition, the hydrogen evolution reaction (HER) always competes with the CO2RR in aqueous media and can significantly deteriorate the selectivity for specific products.10 Thus, it is of great importance to design highly active and selective electrocatalysts for efficient CO2RR.As one of the typical liquid products with two-electron reduction of CO2, formate can be utilized in fuel cells as hydrogen carriers, and in the textile and pharmaceutical industries.11,12 So far, various metal-based electrocatalysts (e.g., Pd,13,14 Sn,15,16 In,17,18 Pb19,20 and Bi21–24) have been demonstrated to form formate, among which Bi-based materials show great potential due to their low toxicity and earth abundance. Various studies on Bi-based electrocatalysts, including Bi nanoparticles,25 Bi dendrites26 and two-dimensional Bi nanosheets,27–30 have demonstrated the effectiveness of structure engineering in improving CO2RR performance toward formate formation, but still fail to meet the requirements of industrial applications, e.g., high current densities (j) and wide potential window with high faradaic efficiencies (FEs) to lower the capital investment for the desired production rate.31,32 However, most of the reported formate faradaic efficiencies (FEformate) on Bi-based materials show an increased peak of over 90% at moderate potential, while such high selectivity could only be maintained in a narrow potential window.33 Although some reported electrocatalysts demonstrated wider potential windows with higher FEformate, the achieved j is still far from the one for practical implementations. Therefore, it is imperative to develop efficient electrocatalysts for CO2RR-to-formate conversion with high j and FE over a wide potential window to realize practical applications at an industrial scale.
It has been well recognized that the structure modulation of electrocatalysts is an effective approach to improving CO2RR performance. Among numerous nanomaterials with various morphologies and structures, one-dimensional nanomaterials are regarded as promising candidates for the CO2RR.34,35 The large surface area guarantees a substantial amount of active sites for the CO2RR, while the unique channel structure of the nanotubes could significantly improve the charge transfer ability and the mass transport in aqueous media. Nonetheless, electrocatalysts with nanotubular structures are challenging the position to address the issues of low CO2 solubility and sluggish multi-electron transfer kinetics. As another critical factor, defects on as-prepared electrocatalysts can introduce massive uncoordinated sites, thereby ensuring desirable CO2RR performance. Zhang et al.36 demonstrated that defects could enrich the reaction intermediates and OH−, which could consequently prompt the C–C bond formation and improve the selectivity of the target product. This is further confirmed by Yang et al.37 who showed that defective indium/indium oxide heterostructures exhibit a high selectivity for C1 products with FE approaching 100% in a broad potential range. Presently, among various electrocatalysts, defects are mostly introduced on bulk or two-dimensional materials but there are almost no studies on one-dimensional materials.38,39 In light of the advantages of low-dimensional materials and structure defects, introducing defects into the nanotubular Bi-based architecture holds great potential to drive highly active and selective CO2RR to formate.
In this work, ultrathin Bi5O7I nanotubes (NTs) with nanopores and abundant oxygen vacancies are synthesized and demonstrated as a highly active, selective and stable electrocatalyst for the CO2RR toward formate formation. The as-obtained Bi5O7I NTs show a desirable FEformate within a wide potential range as well as a high formate formation rate of 1145 μmol h−1 cm−2 at a moderate potential. Moreover, the CO2RR performance is boosted in a flow cell that delivers a high current density of 225 mA cm−2 at −1.0 V versus reversible hydrogen electrode (vs. RHE) and achieves a good stability of 7 h with negligible degradation. The Bi5O7I NTs are further evaluated in a membrane electrode assembly system at a current density of 200 mA cm−2 and show negligible changes of cell voltage and FE over 140 h for CO2RR. Benefitting from the nanotubular morphology and defective structure, the massive uncoordinated active sites can not only strengthen the CO2 adsorption and charge transfer, thereby speeding up the CO2RR reaction kinetics, but also provide a hydrophobic surface with a higher local pH to effectively inhibit the competing HER. Density functional theory (DFT) calculations reveal that the defective structure of the electrocatalyst can effectively modulate the electronic structure and optimize the adsorption of the reaction intermediate, thereby promoting CO2RR performance. All of the above merits collectively contribute to the enhanced electrocatalytic performance for CO2-to-formate conversion.
2. Results and discussion
2.1 Structural characterizations of Bi5O7I nanotubes
The Bi5O7I NTs were synthesized via the hydrothermal method. As characterized by the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), the prepared catalyst shows an ultrathin nanotubular structure with a uniform diameter of about 5 nm, where a substantial amount of nanopores can be observed on the side wall (Fig. 1a and b). The magnified HAADF-STEM image further clearly shows the wall thickness of about 1 nm with the distribution of nanopores along the wall (Fig. 1c). It is expected that the abundant defects on ultrathin porous nanotubes are beneficial for gas adsorption and charge transfer during the electrochemical reactions. Energy dispersive X-ray spectroscopy (EDS) elemental mapping (Fig. 1d–g) confirms the uniform distribution of Bi, O and I along the nanotubes, indicating a successful preparation of the Bi5O7I NTs. Moreover, Bi5O7I NTs were further characterized by powder X-ray diffraction (XRD), as shown in Fig. 1h. The three peaks of the XRD patterns ranging from 25 to 35° are assigned to the (312), (004) and (600) planes of Bi5O7I (JCPDS 40-0548), suggesting a high purity of the prepared Bi5O7I NTs. For comparison, Bi5O7I nanobelts (NBs) were obtained under the same conditions except that the pH was increased to 12.5. The scanning electron microscopy (SEM) and TEM images (Fig. S1, ESI†) clearly show the structure of belts with the width of around a micrometer, and the lattice fringe of 0.32 nm is assigned to the (312) plane of Bi5O7I, in agreement with the XRD result (Fig. S2, ESI†).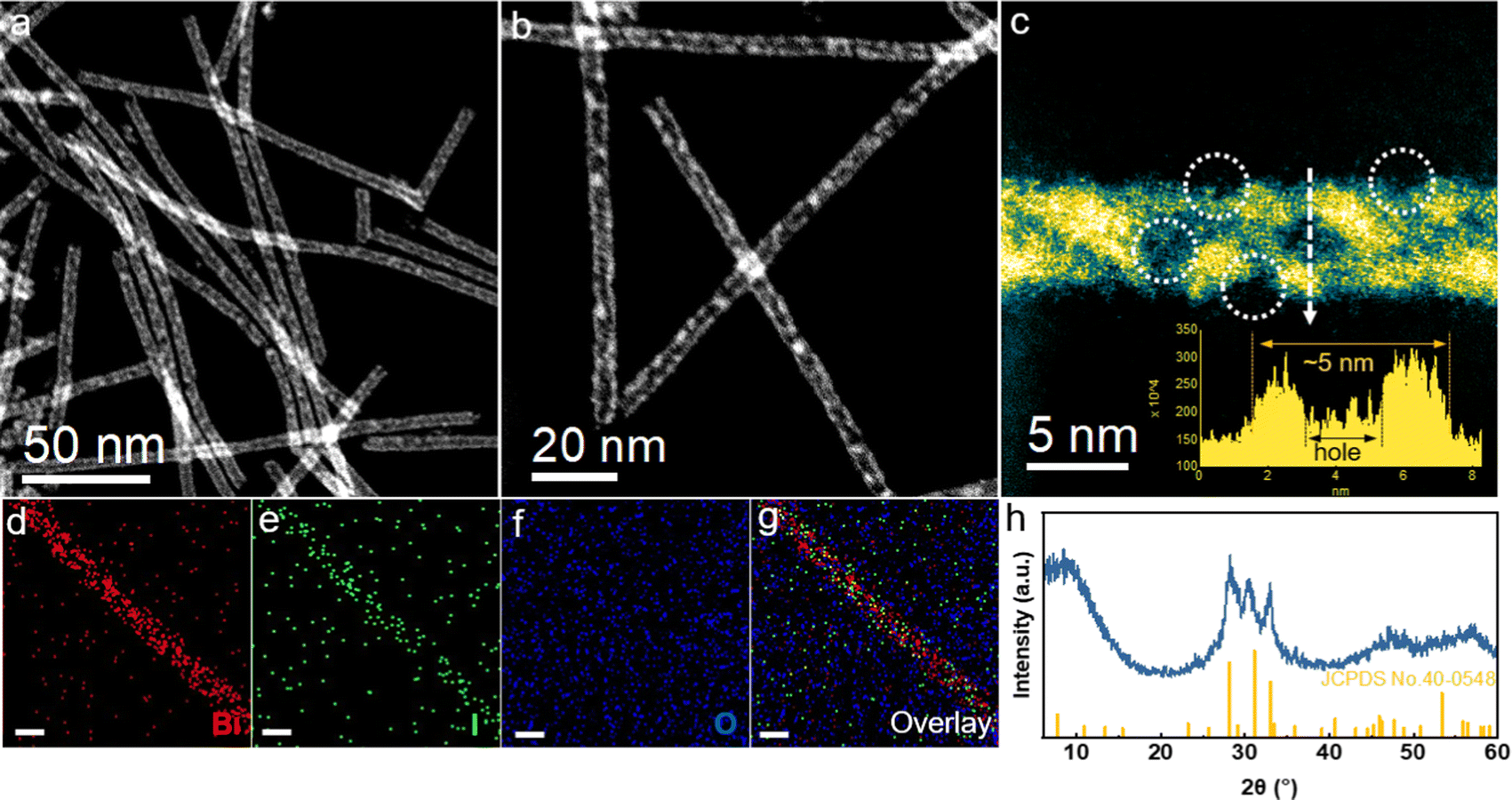 | ||
| Fig. 1 Catalyst characterizations. (a–c) HADDF-STEM images of Bi5O7I NTs (inset is the line scan of the depth); (d–g) the corresponding EDS elemental mappings and overlay image of Bi5O7I NTs (scale bar: 5 nm); (h) XRD pattern of Bi5O7I NTs. | ||
2.2 Electrocatalytic performance for the CO2RR
The CO2RR electrocatalytic activities of Bi5O7I NTs and NBs were first evaluated in an H-type cell containing 0.5 M CO2-saturated KHCO3 with all the potentials referred to the reversible hydrogen electrode (RHE) unless specified otherwise. Firstly, linear sweep voltammetry (LSV) curves were measured in Ar- and CO2-saturated electrolytes, respectively, to evaluate the electrocatalytic activities toward the CO2RR. As shown in Fig. 2a, the Bi5O7I NTs deliver a comparably higher total current density (j) in CO2-saturated electrolyte than that in Ar, implying the higher preference to the CO2RR. A similar result is also obtained on Bi5O7I NBs, while the higher j and more positive onset potential of the Bi5O7I NTs suggest their better electrocatalytic activity for CO2 conversion. To exclude the preferential occurrence of the HER and identify the reduction products of the CO2RR, controlled electrolysis was carried out (Fig. S3, ESI†) at different potentials. The gas-phase products were detected by online gas chromatography (GC), and the liquid products were analyzed by ion chromatography (IC). The results show that formate is the dominating product, together with minor amounts of CO and H2. As displayed in Fig. 2b, the Bi5O7I NTs exhibit higher selectivity for formate formation and the FEformate maintains over 92% in a wide potential range from −0.8 to −1.3 V, whereas Bi5O7I NBs show the inferior selectivity with the maximum FEformate of about 80% under the same applied potentials. Specifically, the maximum FEforamte of 95.5% is achieved at −1.0 V where the HER is significantly inhibited with the FEH2 of about 1%. Moreover, the FEs for the C1 products (i.e., formate and CO) are approaching near-unity within a wide potential gap of 500 mV (Fig. S4, ESI†), demonstrating the high preference to the CO2RR over the competing HER for Bi5O7I NTs.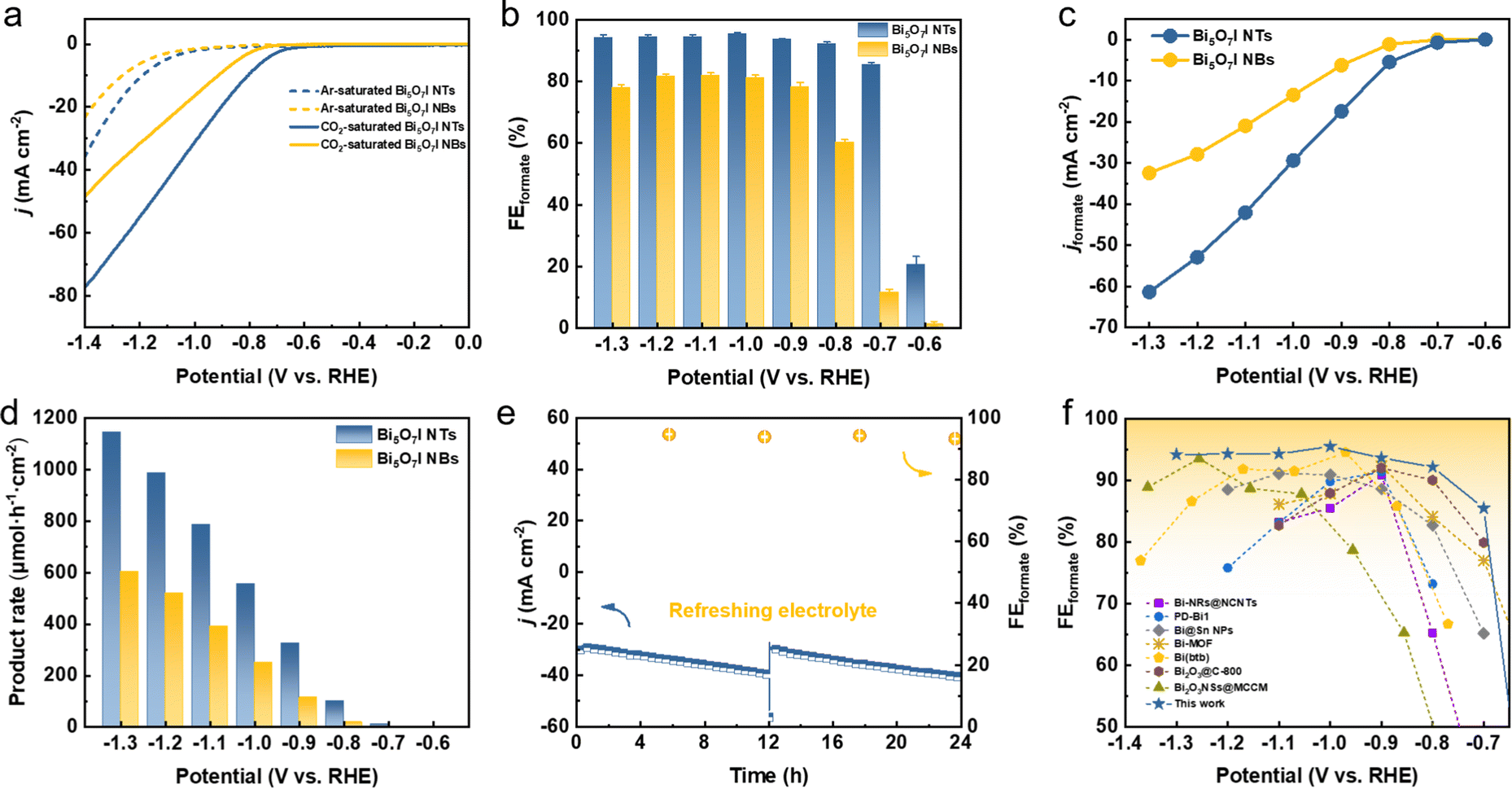 | ||
| Fig. 2 CO2RR performance in a H-type cell. (a) LSV curves in 0.5 M CO2- or Ar-saturated KHCO3 electrolyte, (b) FEs of formate, (c) jformate and (d) formate formation rates of Bi5O7I NTs and NBs; (e) stability test of Bi5O7I NTs at −1.0 V in 0.5 M CO2-saturated KHCO3 electrolyte; (f) comparison of FEformate of this work with recently reported Bi-based catalysts. | ||
To reveal the electrocatalytic activity for CO2RR to formate, the partial current densities of formate (jformate) were derived from the j and FEformate, as shown in Fig. 2c. Clearly, Bi5O7I NTs possess better electrocatalytic activity for CO2-to-formate conversion, where a jformate of about 61 mA cm−2 was reached at a moderate potential of −1.3 V, about two times higher than that of Bi5O7I NBs (32 mA cm−2). Fig. 2d shows the formate formation rate, in which Bi5O7I NTs exhibit a rate of 1145 μmol h−1 cm−2 at −1.3 V, comparably higher than Bi5O7I NBs (605 μmol h−1 cm−2). Furthermore, a potentiostatic electrolysis measurement was conducted at −1.0 V on Bi5O7I NTs to evaluate the stability. As shown in Fig. 2e, the j shows a slight increment after electrolysis for 12 h, which is mainly attributed to the accumulation of products in electrolyte. The j could go back to the original value after refreshing the electrolyte, indicating the good stability of the electrocatalyst. It is worth noting that the average FEformate was maintained at about 92% over 24 h electrolysis, and this further confirms the remarkable durability of Bi5O7I NTs for formate formation. Further post-catalysis characterizations were performed to identify the structure and composition changes of the catalyst after the CO2RR. The Bi5O7I NTs are fully reduced to metallic Bi after electrolysis as confirmed by the XRD result (Fig. S5, ESI†). It is worth noting that the morphologies of the nanotubular structures with surface defects are well maintained under the reduction conditions, as shown in the TEM images (Fig. S6, ESI†). To better demonstrate the reduction of Bi5O7I NTs to defective metallic Bi NTs, an electrochemical activation measurement was conducted using fresh electrode at −1.0 V vs. RHE in 0.5 M CO2-saturated KHCO3 for 10 min. The XRD results show that Bi5O7I NTs are already fully reduced to metallic Bi in 10 min (Fig. S7, ESI†). From the HRTEM images, some nanopores can be clearly identified on the nanotube walls. The Fourier transform pattern of the selected area belongs to the zone axis [001] where the lattice fringe of 0.23 nm is well indexed to the (110) facet of metallic Bi (Fig. S8, ESI†). The good structure durability of defective nanotubes accounts for the negligible degradation of the electrocatalytic performance as well as the good stability. To the best of our knowledge, the electrocatalytic performance of Bi5O7I NTs outperforms the recently reported Bi-based electrocatalysts for the CO2RR toward formate (Fig. 2f and Table S1, ESI†).23,28,40–44
Given the reduction of Bi5O7I NTs to defective metallic Bi NTs during the CO2RR, the similar nanotubular Bi NTs without defects were further prepared to compare the CO2RR performance. As shown in Fig. S9 (ESI†), the high purity of metallic Bi is synthesized using the hydrothermal method. The nested Bi NTs can be clearly observed by SEM and TEM images and the lattice distance of 0.33 nm is well indexed to the (012) facet of metallic Bi (Fig. S10, ESI†). The EDS result further confirms the successful preparation of Bi NTs. The CO2RR measurement was conducted under the same conditions as Bi5O7I NTs and NBs. As shown in Fig. S11 (ESI†), the selectivity for formate formation is less than 90% in all measured potentials and the maximum FEformate of 86.6% is achieved at −1.3 V vs. RHE. In addition, the jformate of Bi NTs is much lower compared to that of Bi5O7I NTs, demonstrating the inferior electrocatalytic activity for formate formation.
2.3 High-throughput CO2RR performance
It is suggested that the economically compelling application of the CO2RR toward formate formation requires a minimum j of 200 mA cm−2 with the selectivity of 90% to promote the practical CO2 electrolysis technology.32,45 However, the low CO2 gas solubility in aqueous media (∼33 mM) strongly restricts the CO2 mass transfer, which results in the limited j. To this end, the CO2RR performance of Bi5O7I NTs on a gas diffusion electrode (GDE) was evaluated in a flow cell with 1.0 M KOH (Fig. S12 and S13, ESI†). As shown in Fig. 3a, the onset potential of −0.3 V was reached in the flow cell, much more positive than −0.6 V in the H-type cell. Notably, the Bi5O7I NTs show a high j of 390 mA cm−2 at −1.4 V that decreases to about 225 mA cm−2 at −1.0 V, demonstrating the considerably high electrocatalytic activity for CO2RR. The shortened gas diffusion pathway on the GDE facilitates the greater access to CO2, resulting in better performance than in the H-type cell. In addition, the reduced ohmic resistance and charge transfer resistance (Fig. S14, ESI†) in the flow cell system further promote the enhanced CO2RR electrocatalytic performance. The potential controlled electrolysis measurements were also conducted, and the results of product analysis suggest that the FEformate exceeds 93% at all the applied potentials from −0.3 to −1.3 V with corresponding jformate increasing from 4.0 to 296 mA cm−2 (Fig. 3b). Meanwhile, the Bi5O7I NTs also exhibit a good stability with a high j of over 200 mA cm−2 and an average FEformate of about 94% in the flow cell system for 7 h, as shown in Fig. 3c. The high FEformate and j of Bi5O7I NTs in a wide potential window make it stand out from the recently reported electrocatalysts for formate production in flow cell systems (Table S2, ESI†).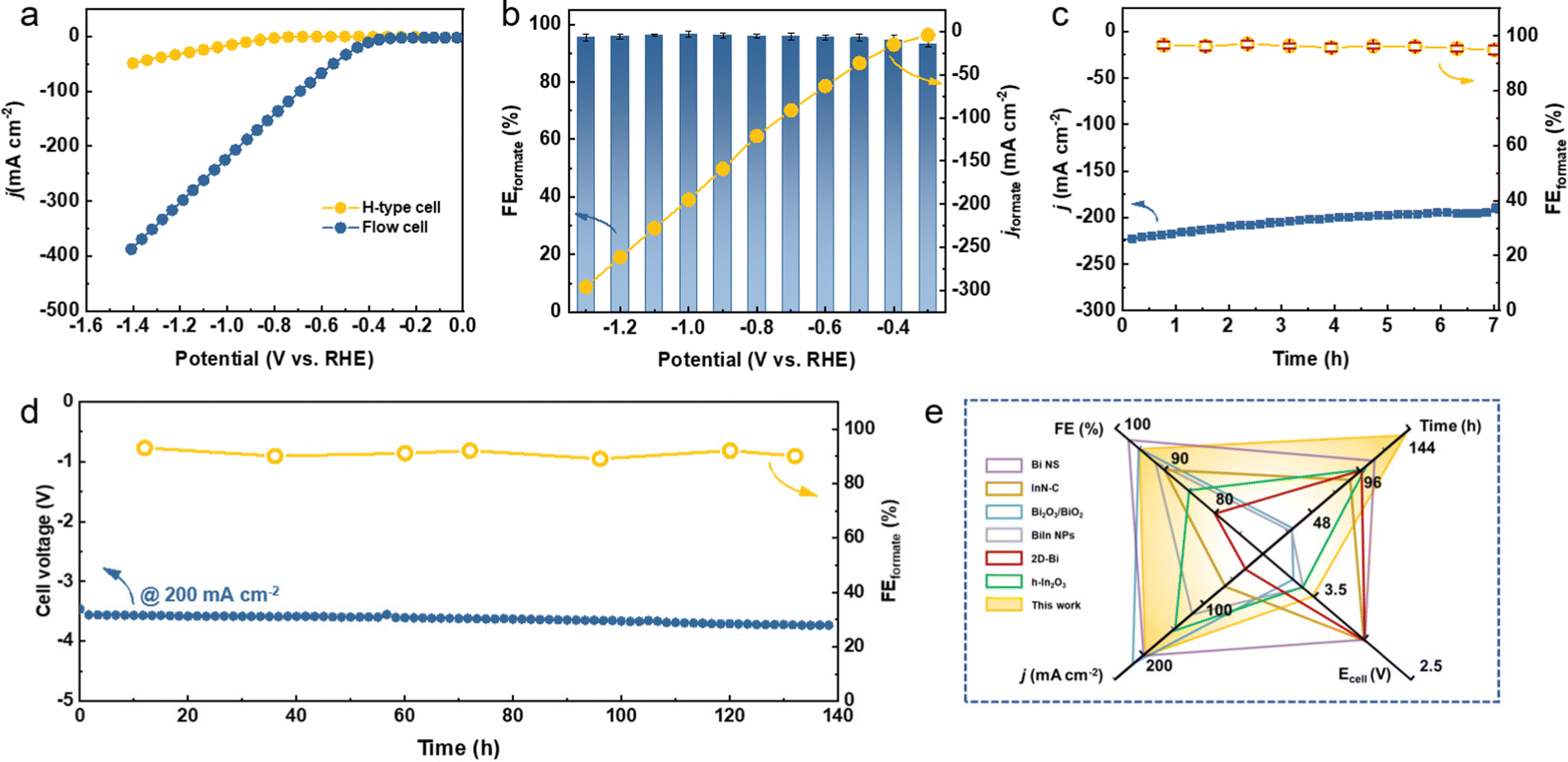 | ||
| Fig. 3 High-throughput CO2RR performance. (a) LSV curves of Bi5O7I NTs in the H-type cell and flow cell, (b) FEformate and jformate of Bi5O7I NTs at all potentials in the flow cell, (c) stability test of Bi5O7I NTs at −1.0 V vs. RHE in the flow cell, (d) the CO2RR performance of Bi5O7I NTs at 200 mA cm−2 in the MEA system, and (e) comparison of the FEformate, j, Ecell and stability of recently reported electrocatalysts for the CO2RR to formate in the MEA system. | ||
To evaluate the feasibility of the actual application of Bi5O7I NTs in the CO2RR process, a custom-built two-electrode membrane electrode assembly (MEA) system was used to perform the stability test (Fig. S15, ESI†). As shown in Fig. 3d, the electrolysis can be stably operated at 200 mA cm−2 for 140 h with negligible fluctuation of cell voltage, suggesting the good stability of the electrocatalyst under the MEA working conditions. Moreover, the FEformate was maintained over 90% in the period of 140 h, demonstrating the superb CO2RR performance of Bi5O7I NTs. Such results make it outperform many recently reported electrocatalysts for CO2 to formate conversion in MEA systems (Fig. 3e and Table S3, ESI†). Conclusively, the Bi5O7I NTs possess strengthened electrocatalytic activity, selectivity and durability for CO2-to-formate conversion in both the flow cell and MEA systems, thereby demonstrating the promising future for their practical applications in the field of CO2RR technology.
2.4 Reaction kinetics and mechanistic studies for CO2RR to formate
To investigate the reaction kinetics for CO2RR to formate formation, Tafel plots were derived based on static state j and FEformate (Fig. 4a). Compared with Bi5O7I NBs (70.2 mV dec−1), its smaller Tafel slope of 63.4 mV dec−1 indicates the faster reaction kinetics process and better stabilization for the *CO2− intermediate on Bi5O7I NTs, which ensures the improved CO2RR performance. It is generally recognized that the rate-determining step (RDS) of formate formation could be identified by matching the theoretical Tafel slopes and experimentally derived values.46–48 The theoretical Tafel slope of 118 mV dec−1 suggests the RDS of the initial electron transfer to the adsorbed CO2, i.e., * + CO2 + e− → *CO2−, where (*) corresponds to the active site for adsorption species, while the value of 59 mV dec−1 corresponds to the RDS of proton (H+) transfer served by bicarbonate (HCO3−), i.e., *CO2− + HCO3− → OCHO* + CO32−. It is worth noting that the RDS for the CO2RR toward formate formation is identical for Bi5O7I NTs and NBs, where the Tafel slopes are close to the theoretical value of 59 mV dec−1. Therefore, the electroreduction of CO2 to formate on Bi5O7I follows the steps below.11| * + CO2 + e− → *CO2− | (1) |
| *CO2− + HCO3− → OCHO* + CO32− (RDS) | (2) |
| OCHO* + e− → HCOO−* | (3) |
| HCOO−* → HCOO− + * | (4) |
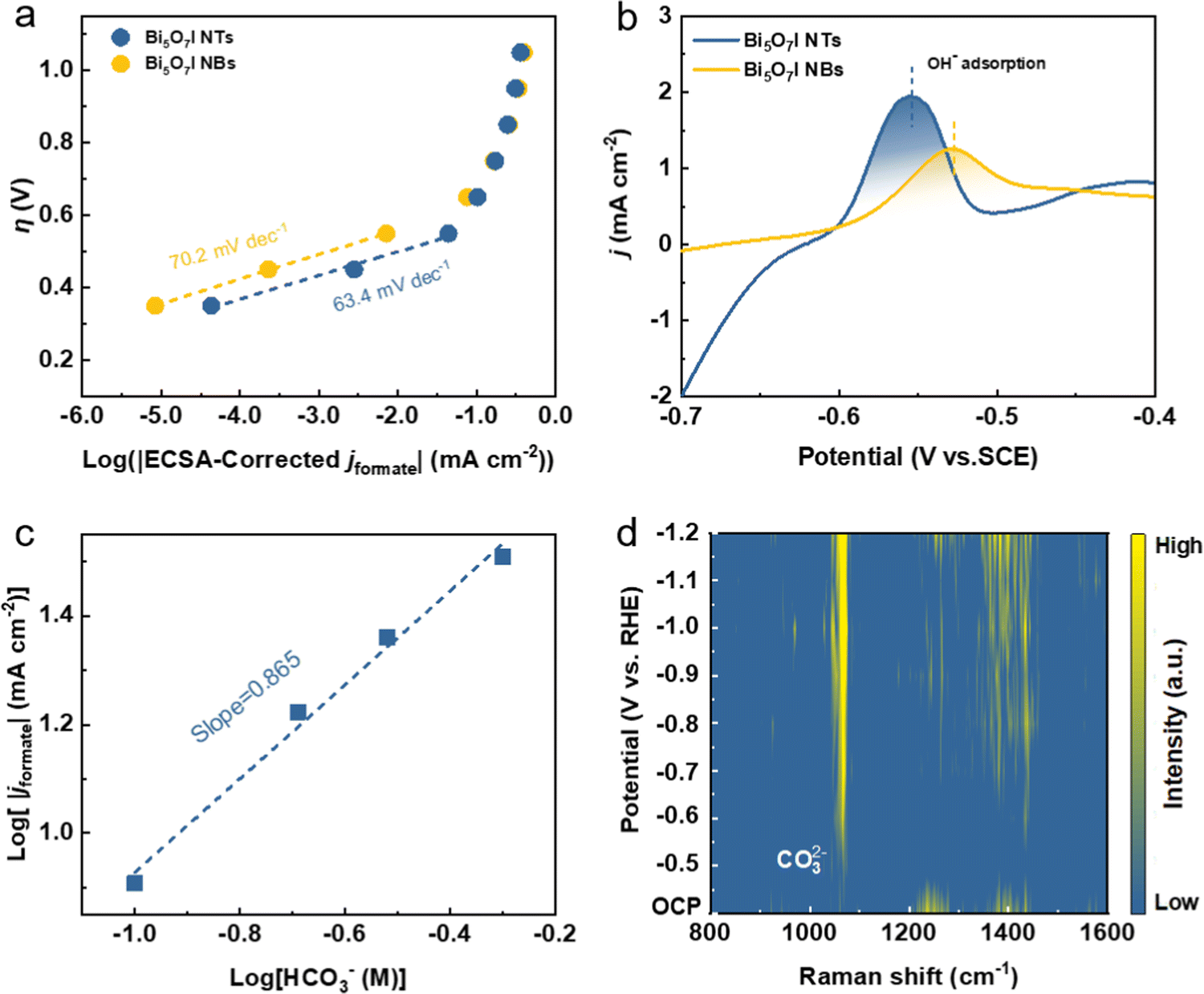 | ||
| Fig. 4 Kinetic and mechanistic studies. (a) ECSA-corrected Tafel plots for formate production, (b) oxidative LSV curves in N2-saturated 0.1 M KOH, (c) jformate of Bi5O7I NTs in different concentrations of HCO3− at −1.0 V vs. RHE, and (d) potential-dependent Raman spectra on Bi5O7I NTs in 0.5 M CO2-saturated KHCO3. | ||
The reaction mechanism for the CO2RR to formate formation was further validated by the in situ electrochemical Raman spectroscopy measurement (Fig. S17, ESI†). Fig. 4d shows the Raman spectra on Bi5O7I NTs at different applied potentials in 0.5 M CO2-saturated KHCO3 electrolyte, in which two obvious peaks are observed at the applied negative potentials. As stated before, the initial electron transfer to form *CO2− is the first reaction step during the CO2RR. The Raman peaks at 450 to 550 cm−1 (Fig. S17c, ESI†) start to appear when the applied potential reaches −0.6 V vs. RHE, which are assigned to the peak of carboxylate δ(CO2−).49 The increased peak intensity suggests the first stage of reaction (1) during the CO2RR. Moreover, the peak located at about 1065 cm−1 is ascribed to the symmetric C–O stretching vibration v1(CO32−),50 and the surface weakly adsorbed monodentate carbonate η1-CO32− (Fig. S17d, ESI†). It is noted that the peak intensity gradually increases as the applied potential shifts to a more negative range, while no peak is observed when the applied potential is lower than −0.6 V vs. RHE. This demonstrates the occurrence of reaction step (2) during the CO2RR when the intermediate is formed and accompanied by the production of carbonate ions. Notably, the peaks of δ(CO2−) and v1(CO32−) disappear when the applied potential is stopped, further proving that the carbonate intermediate comes from the reaction process (Fig. S18, ESI†). All these results provide solid evidence to confirm the reaction mechanism on Bi5O7I NTs for the electroreduction of CO2 to formate.
Density functional theory (DFT) calculations were performed to deepen our understanding on the origins of the enhanced catalytic activity and selectivity toward the CO2RR. Given the full reduction of Bi5O7I NTs to defective metallic Bi NTs during the CO2RR, the three models of perfect Bi and Bi with different defects were investigated (Bi-V1 means Bi surface with a mono vacancy and V2 means di-vacancy). As depicted in Fig. 5a, the charge density difference between Bi and the adsorbate *OCHO on perfect Bi shows relatively low interaction compared to those on Bi-V1 and Bi-V2. The apparent charge transfer between Bi and O atoms on defective Bi demonstrates the stronger interaction of the adsorbate on the Bi surface, which is beneficial for the formation of the *OCHO intermediate. Given the structure difference of Bi, the electronic local functions were further studied (Fig. 5b). The local charge distribution of adjacent Bi atoms can be clearly observed with the existence of defects, which is attributed to the increased orbital overlap caused by the shortened bond length between two adjacent atoms (Fig. S19, ESI†), thus resulting in electron delocalization. The p orbital delocalization of the Bi atoms leads to orbital hybridization between the Bi atoms and the adsorbed *OCHO. The more delocalized electrons near the Bi atom with vacancies can effectively promote the formation of the intermediate *OCHO by donating electrons to the reaction, enhancing the interaction between *OCHO as evidenced by the electron density difference. In addition, the adsorbed intermediate *OCHO shift to the electron delocalized region on Bi atoms, as shown in Fig. S20 (ESI†), further confirming the favorable formation of intermediates over the electron delocalized region.
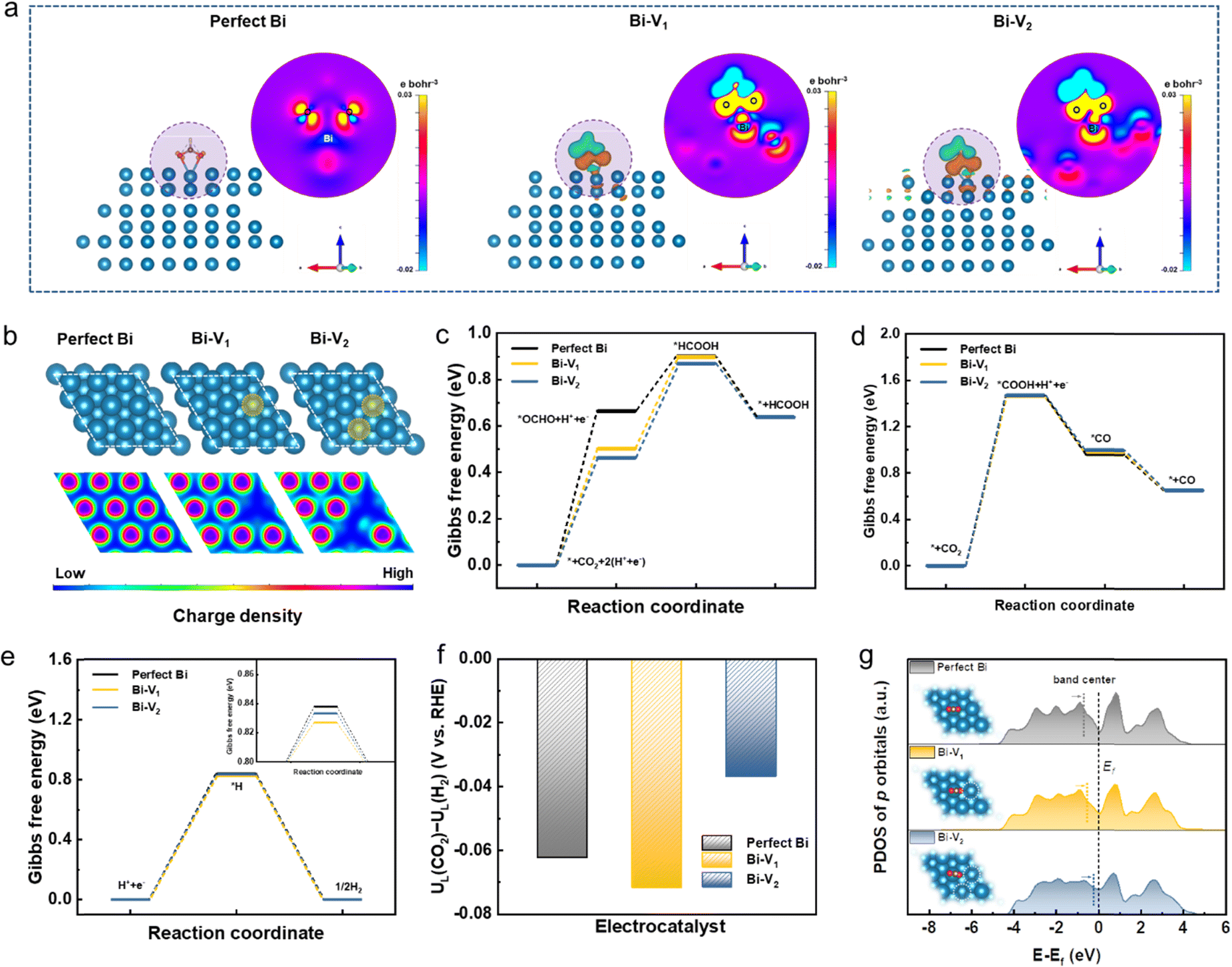 | ||
| Fig. 5 DFT calculation results. (a) The electron density difference with *OCHO adsorbates (2D view generated from the plane derived by Bi–O–O atoms), (b) electron local functions, Gibbs free energy of the CO2RR to (c) HCOOH pathway and (d) CO pathway, (e) Gibbs free energy of HER, (f) difference in limiting potentials for the CO2RR and HER, and (g) the Bi p-PDOS with *OCHO adsorbates of perfect Bi, Bi-V1 and Bi-V2. | ||
Furthermore, the Gibbs free energies for CO2 to HCOOH formation were calculated over the optimized structures shown in Fig. 5c and Fig. S21 (ESI†). The formation energy of intermediate *OCHO decreases from 0.66 to 0.46 eV when introducing a di-vacancy on the Bi, suggesting the lower energy barrier compared to the perfect Bi. The stronger charge interaction between Bi and O atoms and higher local charge distribution near Bi atoms synergistically contribute to better adsorption ability of *OCHO, leading to the decreased energy barrier in the rate determining step. In addition, the Gibbs free energies for CO2 to CO formation were also calculated (Fig. 5d and Fig. S22, ESI†). It can be found that the rate determining step is the formation of intermediate *COOH on perfect Bi and defective Bi where no obvious energy difference is observed. The high energy barrier of about 1.47 eV for *COOH formation is significantly higher than that for *OCHO formation, leading to the unfavorable CO formation. Given the competing reaction of the HER during the CO2RR, the DFT calculation was further conducted on the HER, as shown in Fig. 5e and Fig. S23 (ESI†). When introducing defects on the Bi structure, the Gibbs free energies of the formed *H display less difference compared to that of perfect Bi. The energy barrier of over 0.8 eV on *H formation is higher than the rate determining step of *OCHO formation, indicating the suppression of the HER during the CO2RR. Furthermore, the limiting potential difference (ΔUL) between the CO2RR and HER was also determined to reveal the selectivity toward the CO2RR. As depicted in Fig. 5f, Bi-V2 has a more positive potential value (−0.036 V) than perfect Bi (−0.062 V) and Bi-V1 (−0.072 V), demonstrating the higher selectivity for CO2 to formate conversion. The projected density of states (PDOS) for Bi p orbitals with adsorbed *OHCO were plotted where the band center shifts upwards to the Fermi level with increased vacancies, indicating the stronger interaction between *OCHO and Bi that facilitates the HCOOH formation that also accounts for the high selectivity for CO2-to-formate conversion (Fig. 5g). Thus, the vacancies present on Bi can effectively modulate the electronic structure and lower the energy barrier, leading to the superior catalytic performance for the CO2RR.
2.5 Structural merits of Bi5O7I nanotubes towards the enhanced CO2RR performance
To uncover the merits of the defective nanotubular structure, the electrochemical double-layer capacitance (Cdl) was measured (Fig. S24, ESI†) to determine the electrochemically active surface area (ECSA). As shown in Fig. S25a (ESI†), the ultrathin Bi5O7I NTs possess a higher Cdl (6.81 mF cm−2) than Bi5O7I NBs (3.26 mF cm−2), suggesting that the defective nanotubular structure offers a substantial number of active sites and thereby improves the electrocatalytic activity for the CO2RR. Moreover, the previous study51–53 also manifests the benefits of the ultrathin nanostructure for CO2RR performance. The thinner thickness of the electrocatalyst provides higher surface area and more active sites. Compared to the nanobelt structure, the ultrathin nanotube structure of Bi5O7I endows it with larger surface area and more exposed electrocatalytic active sites for the CO2RR to proceed, which improves both the electrocatalytic activity and selectivity for the conversion of CO2 to formate. To exclude the effect of morphology on the CO2RR performance, the jformate was further normalized by the ECSA. As shown in Fig. S25b (ESI†), the Bi5O7I NTs still deliver a higher jformate compared to Bi5O7I NBs, suggesting the higher intrinsic electrocatalytic activity of the Bi5O7I NTs for the CO2RR to formate conversion. In addition, the electrochemical impedance spectroscopy (EIS) measurement was conducted to evaluate the charge transfer process between the electrocatalyst and electrolyte. Apparently, the Bi5O7I NTs show comparably smaller interfacial charge transfer resistance compared to NBs (Fig. S26, ESI†), indicating a better charge transfer ability to facilitate the occurrence of the CO2RR. As stated before, the defective nanotubular structure is conducive to gas adsorption, and volumetric CO2 adsorption isotherms prove the superb CO2 capture ability of Bi5O7I NTs that is three times higher than that of Bi5O7I NBs (Fig. 6a). The enhanced CO2 adsorption capability originates from the abundant nanopores on the ultrathin nanotube wall. This ensures the good mass transfer in aqueous media and benefits the reaction processes on the active sites and consequently, promotes the electrocatalytic performance for formate formation. Aside from the observed nanopores, the result of electron paramagnetic resonance (EPR) measurement displays an obvious signal peak at g = 2.001, which is ascribed to the oxygen vacancies in Bi5O7I NTs (Fig. 6b). Previous results have demonstrated that the spare electrons at the defect sites of oxygen vacancies could serve as the electron donor to ease the activation of electron-deficient CO2 molecules, which is beneficial for the CO2 activation to form the *CO2−.38,39,54 This also accounts for the improved electrocatalytic activity over Bi5O7I NTs for CO2-to-formate conversion.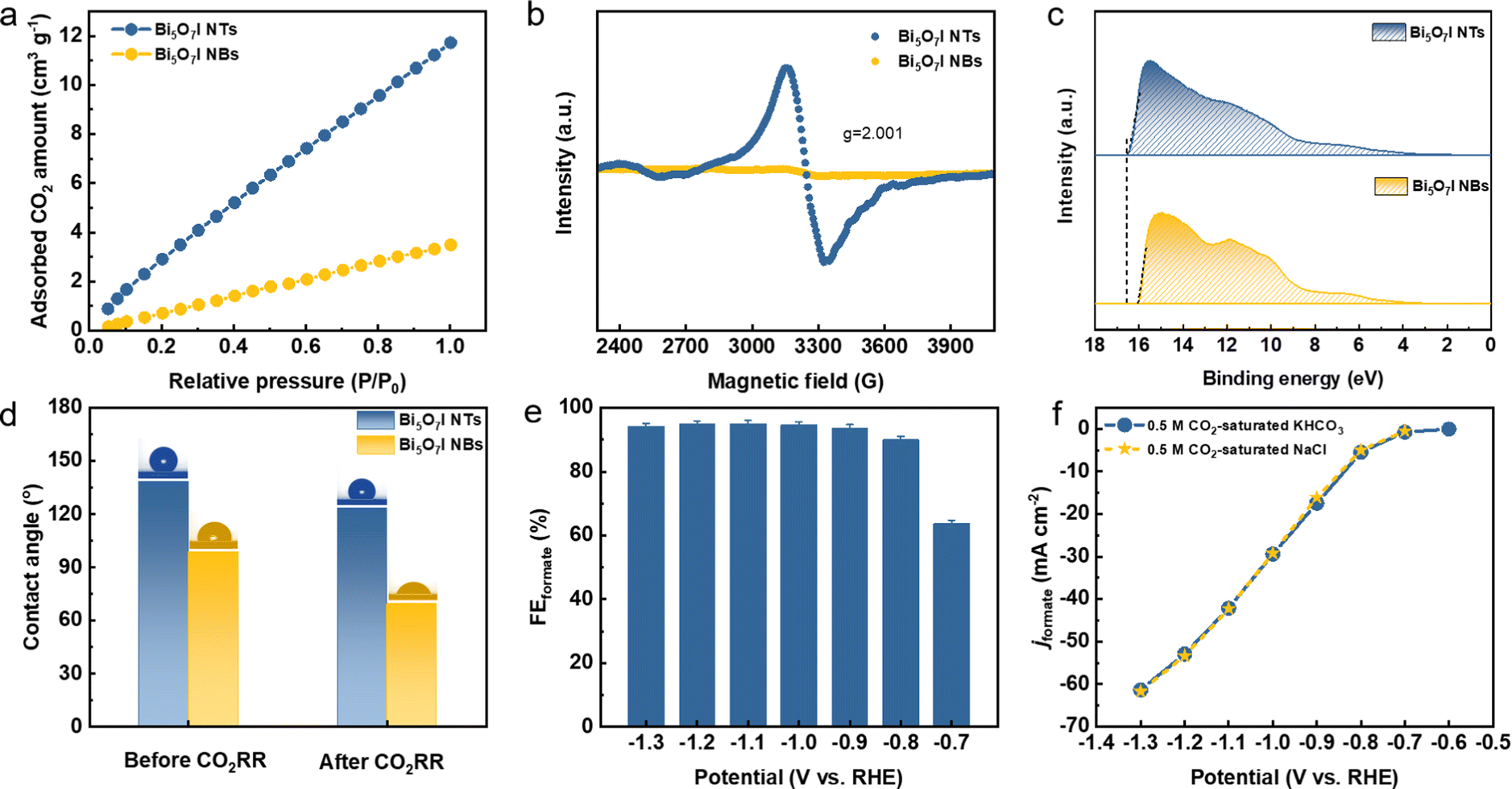 | ||
| Fig. 6 (a) CO2 adsorption isotherms, (b) EPR spectra, (c) UPS spectra and (d) contact angle measurements before and after the CO2RR on Bi5O7I NTs and NBs, (e) FEformate and (f) jformate of Bi5O7I NTs in CO2-saturated 0.5 M NaCl. | ||
Since the CO2RR is a proton-coupled electron transfer (PCET) process and the complicated multi-electron transfer steps occur on the catalyst surface, it is important to take the charge transfer ability into consideration to achieve higher activity and selectivity. Work function (WF) is regarded as an effective indicator to describe the electron transfer ability over the catalyst surface. A lower WF corresponds to a higher tunneling probability of electrons, i.e., the electrons are easier to transfer from the catalyst to the reactant and participate in the subsequent reactions of the CO2RR.55,56 It can be inferred that the unique channel structure of the nanotubes with abundant nanopores and oxygen vacancies can serve as the charge transport pathway to ensure the faster electron transfer on the catalyst. To this end, the WFs of Bi5O7I NTs and NBs were measured by ultraviolet photoelectron spectroscopy (UPS) as shown in Fig. 6c. Apparently, the defective Bi5O7I NTs possess a lower WF of about 4.75 eV compared to NBs (i.e., 5.25 eV), demonstrating the enhanced electron transfer ability (Fig. S27, ESI†). The fast electron transfer on the catalyst surface facilitates the quick pre-equilibrium of CO2 to *CO2−, thereby ensuring the formate formation from the highly selective CO2RR.
It is widely accepted that the CO2RR is a gas involved reaction and takes place at the triple-phase boundaries. The wettability of the catalyst plays an indispensable role in the reaction process and can consequently tune the product selectivity. Previous studies have suggested that the hydrophobic interface is favorable for formate formation owing to the limited supply of protons to participate in the competitive HER.57–59 In light of the substantial amount of nanopores on the defective Bi5O7I NTs, the hydrophobicity is expected across the catalyst surface. Thus, the influence of wettability on CO2RR performance was further studied (Fig. S28, ESI†). As shown in Fig. 6d, the defective Bi5O7I NTs present a hydrophobic surface with the contact angle of 138.4°, while a lower hydrophobicity is observed on NBs. After CO2RR measurement, the defective nanotubular structure still maintains the hydrophobicity with a slight decrement of the contact angle to 123.5°. Nevertheless, the surface of Bi5O7I NBs turns out to be hydrophilic with an obvious change of contact angle (Fig. S28d, ESI†). The hydrophobic Bi5O7I NTs can effectively limit the proton supply to suppress the HER, which is consistent with the product distribution result. Concurrently, the enhanced CO2 adsorption capability can promote sufficient proton donors of HCO3− to enhance the proton-depleted formate formation, thus improving the selectivity of the CO2RR. Moreover, the parasitic HER during the CO2RR process usually deteriorates the reaction selectivity toward the target product. The research results suggest that the higher local pH near the electrode surface can change the concentrations of proton-donating species and effectively suppress the HER during the CO2RR.36,60,61 It is speculated that a higher local pH could be achieved on the catalyst surface considering the superior hydrophobicity of the Bi5O7I NTs. To better observe the local pH effect, the phenolphthalein color transition experiment was performed on the prepared catalysts (Fig. S29, ESI†). It is found that the defective Bi5O7I NTs show more significant color change to pink in comparison to the NBs, demonstrating the higher local pH around its surface. This higher pH can lead to a local conversion of CO2 to HCO3−, i.e., CO2 + OH− → HCO3−, which facilitates the reaction process by providing more proton-donating species and simultaneously suppressing the HER. Based on the abovementioned, the defective nanotubular structure of Bi5O7I demonstrates the advantages of creating massive uncoordinated active sites, ensuring the CO2 adsorption capability and improving the charge transfer capability. Concurrently, the hydrophobic surface with a higher local pH effectively inhibits the HER, thus achieving the selective formation of formate.
2.6 CO2RR performance in a simulated seawater-based system
It has been shown that most CO2RR performance is measured in bicarbonate electrolyte, typically potassium or sodium bicarbonate. To some extent, using low-carbon electrolyte is a promising approach to reducing the carbon source for the CO2RR. Natural seawater is considered as an alternative to substitute the bicarbonate-based electrolyte owing to its abundance and negligible cost.62,63 The chlorine evolution reaction at the anode can simultaneously produce higher-value products rather than oxygen gas when directly using seawater as the electrolyte for the CO2RR. The CO2RR performance of defective Bi5O7I NTs was further evaluated in a 0.5 M NaCl solution that mimics seawater (Fig. S30, ESI†). Remarkably, the high FEformate is achieved over a wide potential range with a maximum value of 95% that is comparable to the result by using KHCO3 electrolyte (Fig. 6e). Furthermore, the derived jformate shows a similar value compared to the measurement in bicarbonate solution, implying the good intrinsic electrocatalytic activity of the Bi5O7I NTs toward formate formation (Fig. 6f) in a 0.5 M NaCl solution. Therefore, the highly active and selective Bi5O7I NTs also demonstrate promising prospects for CO2RR applications in future seawater-based electrolysis systems.3. Conclusions
Ultrathin Bi5O7I NTs with abundant nanopores and oxygen vacancies are successfully synthesized and exhibit excellent electrocatalytic performance for the CO2RR toward formate with good selectivity and stability, which is further improved in the flow cell and MEA systems by overcoming the limitation of CO2 mass transport in aqueous media. The high selectivity and current density of formate over a wide potential window, as well as the good stability, make this electrocatalyst a possible candidate for CO2RR applications at an industrial scale. The experimental results prove that the nanopores and oxygen vacancies can serve as massive uncoordinated active sites and facilitate the adsorption of CO2, which promotes the charge transfer and stabilizes the reaction intermediates toward the CO2RR. Theoretical calculations unveil that the existence of defects can effectively modulate the electronic structure, thus benefiting the formation of intermediate *OHCO toward HCOOH. Furthermore, the hydrophobic catalyst surface with higher local pH effectively suppresses the HER and results in the very efficient performance of the CO2RR. These findings highlight the importance of structure and defect engineering as an attractive strategy for designing advanced electrocatalysts for achieving carbon-neutral sustainability.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada, Discovery Grant (GRPIN-2016-05494) and China Scholarship Council. As a part of the University of Alberta's Future Energy Systems research initiative, this research was made possible in part thanks to funding from the Canada First Research Excellence Fund (CFREF-2015-00001). Thanks to Jiannan Lin for the guidance on materials fabrication. The computation sources are provided by the Digital Research Alliance of Canada (the Alliance).References
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- C. Xu, J. Hong, P. Sui, M. Zhu, Y. Zhang and J.-L. Luo, Cell Rep. Phys. Sci., 2020, 1, 100101 CrossRef.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J. L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- P.-F. Sui, S. Liu, C. Xu, J. Xiao, N. Duan, R. Feng and J.-L. Luo, Chem. Eng. J., 2022, 427, 131654 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- N. Han, P. Ding, L. He, Y. Li and Y. Li, Adv. Energy Mater., 2019, 1902338 Search PubMed.
- M. F. Philips, G.-J. M. Gruter, M. T. M. Koper and K. J. P. Schouten, ACS Sustainable Chem. Eng., 2020, 8, 15430–15444 CrossRef CAS.
- T. Gunji, H. Ochiai, T. Ohira, Y. Liu, Y. Nakajima and F. Matsumoto, Chem. Mater., 2020, 32, 6855–6863 CrossRef CAS.
- N. Han, M. Sun, Y. Zhou, J. Xu, C. Cheng, R. Zhou, L. Zhang, J. Luo, B. Huang and Y. Li, Adv. Mater., 2021, 33, 2005821 CrossRef CAS PubMed.
- Z. Chen, T. Fan, Y.-Q. Zhang, J. Xiao, M. Gao, N. Duan, J. Zhang, J. Li, Q. Liu, X. Yi and J.-L. Luo, Appl. Catal., B, 2020, 261, 118243 CrossRef CAS.
- S. Liu, J. Xiao, X. F. Lu, J. Wang, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2019, 58, 8499–8503 CrossRef CAS PubMed.
- Y. Huang, X. Mao, G. Yuan, D. Zhang, B. Pan, J. Deng, Y. Shi, N. Han, C. Li, L. Zhang, L. Wang, L. He, Y. Li and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 15844–15848 CrossRef CAS PubMed.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal, J. Ma, Y. Huang, X. Sun, S. Liu and B. Han, J. Am. Chem. Soc., 2021, 143, 6877–6885 CrossRef CAS PubMed.
- M. J. W. Blom, V. Smulders, W. P. M. van Swaaij, S. R. A. Kersten and G. Mul, Appl. Catal., B, 2020, 268, 118420 CrossRef CAS.
- C. H. Lee and M. W. Kanan, ACS Catal., 2014, 5, 465–469 CrossRef.
- P. Deng, H. Wang, R. Qi, J. Zhu, S. Chen, F. Yang, L. Zhou, K. Qi, H. Liu and B. Y. Xia, ACS Catal., 2019, 10, 743–750 CrossRef.
- C. W. Lee, J. S. Hong, K. D. Yang, K. Jin, J. H. Lee, H.-Y. Ahn, H. Seo, N.-E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 CrossRef CAS.
- S. Liu, X. F. Lu, J. Xiao, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2019, 58, 13828–13833 CrossRef CAS PubMed.
- P. F. Sui, C. Xu, M. N. Zhu, S. Liu, Q. Liu and J. L. Luo, Small, 2021, e2105682 Search PubMed.
- Y. Qiu, J. Du, W. Dong, C. Dai and C. Tao, J. CO2 Util., 2017, 20, 328–335 CrossRef CAS.
- J. H. Koh, D. H. Won, T. Eom, N.-K. Kim, K. D. Jung, H. Kim, Y. J. Hwang and B. K. Min, ACS Catal., 2017, 7, 5071–5077 CrossRef CAS.
- F. P. Garcia de Arquer, O. S. Bushuyev, P. De Luna, C. T. Dinh, A. Seifitokaldani, M. I. Saidaminov, C. S. Tan, L. N. Quan, A. Proppe, M. G. Kibria, S. O. Kelley, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, e1802858 CrossRef PubMed.
- Y. Wang, Y. Li, J. Liu, C. Dong, C. Xiao, L. Cheng, H. Jiang, H. Jiang and C. Li, Angew. Chem., Int. Ed., 2021, 60, 7681–7685 CrossRef CAS PubMed.
- J. Yang, X. Wang, Y. Qu, X. Wang, H. Huo, Q. Fan, J. Wang, L. M. Yang and Y. Wu, Adv. Energy Mater., 2020, 10, 2001709 CrossRef CAS.
- W. Ma, J. Bu, Z. Liu, C. Yan, Y. Yao, N. Chang, H. Zhang, T. Wang and J. Zhang, Adv. Funct. Mater., 2020, 2006704 Search PubMed.
- Y. Cheng, P. Hou, X. Wang and P. Kang, Acc. Chem. Res., 2022, 55, 231–240 CrossRef CAS PubMed.
- R. I. Masel, Z. Liu, H. Yang, J. J. Kaczur, D. Carrillo, S. Ren, D. Salvatore and C. P. Berlinguette, Nat. Nanotechnol., 2021, 16, 118–128 CrossRef CAS PubMed.
- Y. Guan, M. Liu, X. Rao, Y. Liu and J. Zhang, J. Mater. Chem. A, 2021, 9, 13770–13803 RSC.
- A. Guan, C. Yang, Y. Quan, H. Shen, N. Cao, T. Li, Y. Ji and G. Zheng, Chem. – Asian J., 2019, 14, 3969–3980 CrossRef CAS PubMed.
- S. P. Paula, M. Stefano, E. L. S. Ifan and E. E. María, ChemCatChem, 2019, 11, 1–21 CrossRef.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- W. Yang, Y. Zhao, S. Chen, W. Ren, X. Chen, C. Jia, Z. Su, Y. Wang and C. Zhao, Inorg. Chem., 2020, 59, 12437–12444 CrossRef CAS PubMed.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- W. Zhang, S. Yang, M. Jiang, Y. Hu, C. Hu, X. Zhang and Z. Jin, Nano Lett., 2021, 21, 2650–2657 CrossRef CAS PubMed.
- Y. Xing, X. Kong, X. Guo, Y. Liu, Q. Li, Y. Zhang, Y. Sheng, X. Yang, Z. Geng and J. Zeng, Adv. Sci., 2020, 1902989 CrossRef CAS PubMed.
- P. Deng, F. Yang, Z. Wang, S. Chen, Y. Zhou, S. Zaman and B. Y. Xia, Angew. Chem., Int. Ed., 2020, 59, 10807–10813 CrossRef CAS PubMed.
- F. Li, G. H. Gu, C. Choi, P. Kolla, S. Hong, T.-S. Wu, Y.-L. Soo, J. Masa, S. Mukerjee, Y. Jung, J. Qiu and Z. Sun, Appl. Catal., B, 2020, 277, 119241 CrossRef CAS.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu, K. Daasbjerg, T. Skrydstrup and N. Lock, Adv. Funct. Mater., 2020, 1910408 CrossRef CAS.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- C. W. Lee, N. H. Cho, S. W. Im, M. S. Jee, Y. J. Hwang, B. K. Min and K. T. Nam, J. Mater. Chem. A, 2018, 6, 14043–14057 RSC.
- A. Wuttig, Y. Yoon, J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2017, 139, 17109–17113 CrossRef CAS PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS PubMed.
- C. He, S. Chen, R. Long, L. Song and Y. Xiong, Sci. China: Chem., 2020, 63, 1721–1726 CrossRef CAS.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261–E9270 CrossRef CAS PubMed.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed.
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed.
- P. Su, W. Xu, Y. Qiu, T. Zhang, X. Li and H. Zhang, ChemSusChem, 2018, 11, 848–853 CrossRef CAS PubMed.
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2020, 2009070 Search PubMed.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- P. F. Sui, M. R. Gao, S. Liu, C. Xu, M. N. Zhu and J. L. Luo, Adv. Funct. Mater., 2022, 2203794 CrossRef CAS.
- P. Yue, Q. Fu, J. Li, L. Zhang, L. Xing, Z. Kang, Q. Liao and X. Zhu, Chem. Eng. J., 2021, 405, 126975 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- Z. Cai, Y. Zhang, Y. Zhao, Y. Wu, W. Xu, X. Wen, Y. Zhong, Y. Zhang, W. Liu, H. Wang, Y. Kuang and X. Sun, Nano Res., 2018, 12, 345–349 CrossRef.
- M. Alfath and C. W. Lee, Catalysts, 2020, 10, 859 CrossRef CAS.
- Y. Yoon, A. S. Hall and Y. Surendranath, Angew. Chem., Int. Ed., 2016, 55, 15282–15286 CrossRef CAS PubMed.
- A. Chen and B.-L. Lin, Joule, 2018, 2, 594–606 CrossRef CAS.
- X. Tan, C. Yu, X. Song, C. Zhao, S. Cui, H. Xu, J. Chang, W. Guo, Z. Wang, Y. Xie and J. Qiu, Adv. Energy Mater., 2021, 2100075 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00034f |
| This journal is © The Royal Society of Chemistry 2023 |