
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacet-engineered photoelectrochemical nanocatalysts toward fast kinetic lithium–air batteries†
Yiqiao
Wang
abcd,
Siyuan
Pan
abc,
Huan
Li
e,
Dewang
Li
abc,
Yong
Guo
abc,
Sijia
Chi
abc,
Chuannan
Geng
abc,
Shichao
Wu
 *abc and
Quan-Hong
Yang
*abc
*abc and
Quan-Hong
Yang
*abc
aNanoyang Group, Tianjin Key Laboratory of Advanced Carbon and Electrochemical Energy Storage, State Key Laboratory of Chemical Engineering, School of Chemical Engineering and Technology, and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin University, Tianjin, 300072, China. E-mail: qhyangcn@tju.edu.cn; wushichao@tju.edu.cn
bNational Industry-Education Integration Platform of Energy Storage, Tianjin University, Tianjin, 300072, China
cHaihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192, China
dDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, China
eSchool of Chemical Engineering and Advanced Materials, The University of Adelaide, Adelaide, SA 5005, Australia
First published on 13th March 2023
Abstract
Despite lithium–air batteries having unprecedented theoretical energy supremacy, rambling Li2O2 growth typically impedes O2 diffusion and electron transfer, causing low capacity, poor charging kinetics and short cycle life. Herein, we propose synergistic catalysis in the cathode by engineering the crystal facet of Fe2O3 nanoplates and introducing a visible light-excited photochemical effect. The growth of Li2O2 is regulated with a uniform distribution, mitigating local product aggregation and thus reducing the barriers of O2 diffusion and electron transfer. The improved reaction kinetics lead to ∼420% capacity improvement, low charge potential, superior rate performance, and long-term stability (300 cycles) in actual air. We reveal a mechanism that photoelectrons around each catalyst node and the resultant homogeneous internal electric field increase charge transfer, decrease soluble intermediates, and enable high electrochemical reversibility of amorphous Li2O2 with an ordered process. This study provides inspiring insights for future research into controlling product ordering in Li–air batteries.
Broader contextLi–air batteries with ultra-high energy density promise the next-generation batteries for long-range electric vehicles. However, their actual energy density is far below the theoretical value because the diffusion transfer of the O2 reactant from air to the cathode interiors is largely hindered by the locally accumulated solid discharge products outside cathodes. Here, we demonstrate that introducing visible light energy is an effective strategy to enable batteries with largely enhanced discharge capability and cycling stability in actual air conditions. We reveal an intriguing mechanism that a photoelectron-induced internal micro-electric field can change the product morphology and more importantly homogenize product growth within the cathodes, accelerating electron conduction and O2 diffusion. This simple but efficient approach shows great potential for advancing the practicality of Li–air batteries. |
1. Introduction
Li–air batteries with an ultrahigh theoretical energy density (∼3500 W h kg−1) promise next-generation energy storage solutions.1 Its energy superiority derives from the reactants that are O2 gas in ambient air and the redox reaction of O2 → Li2O2.2 During discharge, O2 first transfers from ambient air to active sites in porous cathodes through diffusion.3,4 After that, O2 receives one electron and is reduced to O2− which can be adsorbed on the cathode surface followed by a further one-electron electrochemical reduction or dissolved in the electrolyte followed by a disproportionation reaction, leading to the formation of a Li2O2 film or particles determined by the current densities,5,6 electrolytes,7,8 cathode structures,9,10 and catalysts.11–14 Nevertheless, premature failure happens when the discharge capacity is far from the high theoretical value of Li–air batteries, hence greatly plagueing their practicability.15–17 The culprit of this problem is the uncontrolled deposition of Li2O2 that exhibits considerably low electronic conductivity (only 10−10–10−9 S cm−1 at 100 °C)18 and insolubility (4 × 10−20 mol L−1 in ether-based electrolytes),19 hindering the electron and oxygen transfer during the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) process. Undoubtfully, promoting fast mass (Li+, O2) and electron transfer processes is an indispensable route towards a high-energy-density Li–air battery with remarkable battery lifespan.20To realize the high energy density of Li–air batteries, air cathodes need to achieve a high mass loading of active materials, and batteries are required to operate in ambient air rather than O2 tanks.21 Compared with the large capacity of Li–O2 batteries (LOBs) in pure O2 gas, Li–air batteries always show an apparent capacity decrease due to the slower O2 transfer in ambient air (∼21% O2 content).22–24 Besides, more than 65% capacity loss was observed when the O2 pressure decreased from 21 to 14 psi.25 Notably, the O2 concentration and electronic conductivity are further subject to the limited diffusion space and high mass loading in a practical pouch cell configuration, leading to much more severe performance deterioration.26 Such a vast gradient generated inside the thick electrode may correlate with the discharging voltage that reached the termination without fully utilizing the active sites of the air electrode. With the increase of the cell thickness, the energy density of the pouch cell decreased from 700 to <200 W h kg−1. Moreover, the local nucleation and growth of Li2O2 particles together with byproducts (Li2CO3, etc.) and their inhomogeneous accumulation not only clog the oxygen-transport pores27,28 but also cause cathode surface passivation.29,30 These local depositions mean that O2 and e− cannot transport deep inside the thick air cathodes, leaving plenty of unavailable “dead” active sites and leading to premature battery failure (Fig. 1b), which seriously compromises the primary superiority (high energy density) of Li–air batteries. In addition, the particle Li2O2 products are usually more difficult to decompose, leading to excessive charging polarization and further impairing the energy efficiency and long-term cycling stability of the batteries.31
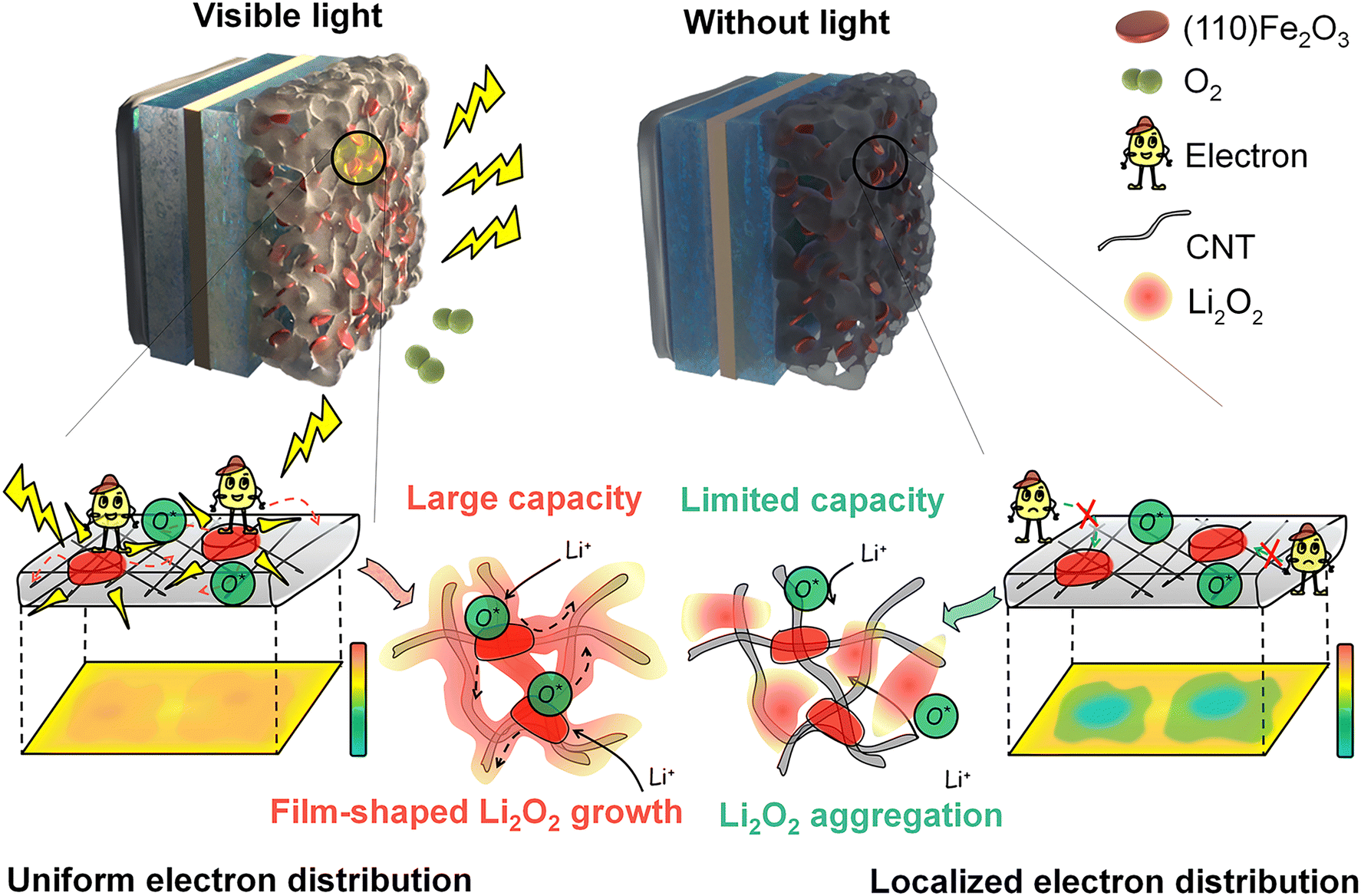 | ||
| Fig. 1 Schematic of visible-light-induced photoelectron-rich microdomains and uniform Li2O2 deposition in the (110) Fe2O3 catalyst-based cathodes. (a) Instantaneously generated photoelectrons on each catalyst particle junction and improved catalysis of Fe2O3 with light modify Li2O2 formation, and the uniform deposition gives large discharge capacities. (b) The gradient distribution of electrons from electrode tabs to active sites and the unfavorable catalysis of Fe2O3 without light result in the local accumulation of large Li2O2 particles, pore-clogging, oxygen transfer obstruction and limited capacities. | ||
In this work, we initiated a photoelectrochemical effect to guide Li2O2 nucleation, growth and reversible decomposition in air cathodes containing the Fe2O3 catalyst with high numbers of (110) facets (referred to as (110) Fe2O3) (Fig. 1a). The (110) facet of Fe2O3 possess superior visible light response and oxygen adsorption effect, enabling uniform distribution of photoelectrons, and promoting fast charge transport inside the electrode. In addition, the enhanced adsorption of oxygen species leads to a homogeneous nucleation and growth process of the Li2O2 products, avoiding local pore-clogging and premature battery failure. This accelerated O2 and charge transfer effect not only facilitates the enhancement of the discharge capacity but also promotes product decomposition during the charge process, especially in the air. (110) Fe2O3-based electrodes delivered a significantly increased capacity (∼260% in O2 and more promisingly ∼420% in air) and extended cycle life (over 300 cycles) in the presence of visible light. The visible light-regulated Li2O2 formation provides an innovative but effective way to improve the energy density, power density and cycling stability of Li–air batteries for practical use.
2. Results and discussion
2.1. Material preparation and characterization
(110) Fe2O3 particles were synthesized by a cation-assisted template method, and the crystal phase was α-Fe2O3 based on the X-ray diffraction (XRD) patterns (Fig. 2a). The crystals are nanoplates with an average diameter of about 80 nm (Fig. 2b). In contrast, the shapes of referenced pristine Fe2O3 (pFe2O3) particles are irregular (Fig. S1, ESI†). The ratio of integrated XRD peak intensities I(110)/I(104) increased from 0.77 for pFe2O3 to 1.24 for (110) Fe2O3. The transmission electron microscope (TEM) images in Fig. S2 (ESI†) show that the thickness of the nanoplates is less than 10 nm, and the high-resolution TEM (HRTEM, Fig. 2c) image of the atomic planes shows lattice fringes with a spacing of 0.25 nm, corresponding to the (110) planes of Fe2O3. These results confirm the synthesis of (110) Fe2O3.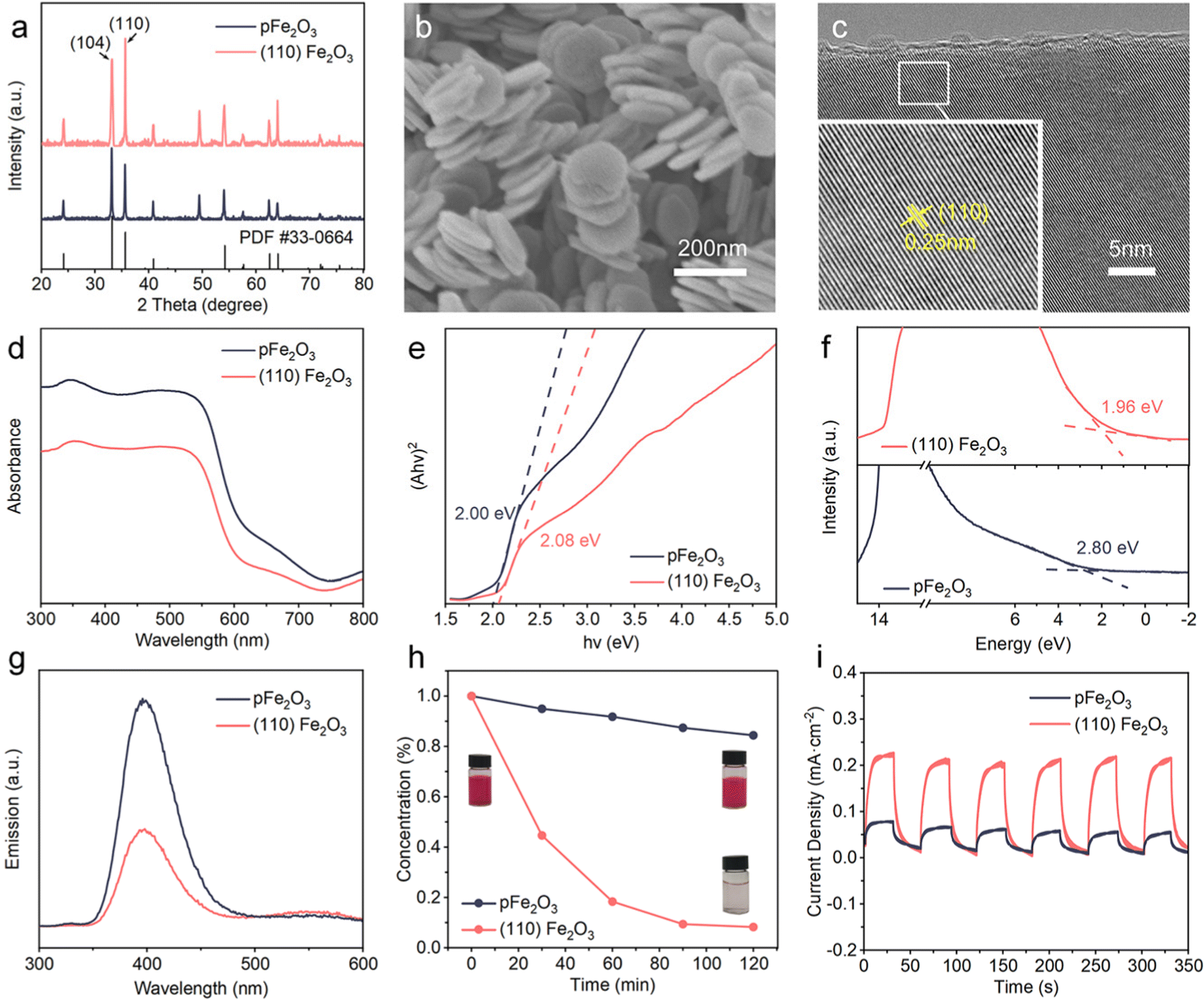 | ||
| Fig. 2 Physicochemical and photoelectrochemical properties of faceted Fe2O3. (a) XRD patterns of (110) Fe2O3, pFe2O3, and PDF#33-0664 indexed to α-Fe2O3. (b) SEM image of the (110) Fe2O3 morphology. (c) HRTEM image showing the lattice fringes of the α-Fe2O3 particles assigned to the (110) facet. (d) UV-Vis absorption spectra of (110) Fe2O3 and pFe2O3. (e) Tauc plot based on the UV-vis absorbance data. The intercept of the tangent to the linear part of each curve and the X-axis represents the bandgap. (f) XPS valence band spectra of (110) Fe2O3. (g) PL spectra of (110) Fe2O3 and pFe2O3. (h) Photocatalytic degradation of RhB. (i) I–t curves at 0.898 V (vs. SHE). | ||
The effect of crystal facets on the photocatalytic activity of Fe2O3 was shown by ultraviolet-visible (UV-vis), X-ray photoelectron (XPS) and photoluminescence (PL) spectra. The UV-vis results (Fig. 2d) indicate that (110) Fe2O3 has a blue shift in the absorption edge compared to pFe2O3. The fitted Tauc plot (Fig. 2e) shows an increase in the bandgap from 2.00 eV of pFe2O3 to 2.08 eV of (110) Fe2O3. The valence band (VB) positions of (110) Fe2O3 and pFe2O3 analyzed by XPS valence band spectroscopy are at 1.96 eV and 2.80 eV, respectively (Fig. 2f). Accordingly, the relative band positions of (110) Fe2O3 and pFe2O3 were determined and shown in Fig. S3 (ESI†). The faceting lowers the conduction band (CB) position of Fe2O3 by 0.92 eV, making it below the redox potential of O2/Li2O2. Thus the generated photoelectrons on (110) Fe2O3 can be transferred spontaneously and react with Li+ and O2 in the discharge process.32 In addition, the PL spectra in Fig. 2g show that the luminescence intensity of (110) Fe2O3 is apparently lower than that of pFe2O3, indicating that the lifetime of photogenerated electrons is prolonged. This phenomenon is favorable to the photocatalytic performance, as shown by experimental observation of the photo-Fenton reaction degradation of rhodamine (RhB) on both materials (Fig. 2h). The degradation efficiency on (110) Fe2O3 (91.8%) is much higher than that on pFe2O3 (15.6%) after 2 h of light exposure. The photoelectrochemical properties of the samples were also investigated using amperometric I–t tests (Fig. 2i). The photocurrent density of (110) Fe2O3 is 0.22 mA cm−2, much higher than that of pFe2O3 (0.08 mA cm−2). This enhanced photocatalytic performance confirms the effect of faceting on the improvement of the visible-light response.
2.2. Discharge capability in Li–O2 batteries and actual Li–air batteries
To demonstrate the effect of light and catalysts towards the Li–oxygen-based reactions, LOBs were assembled with different cathodes, namely (110) Fe2O3-loaded CNTs, pFe2O3-loaded CNTs or unloaded CNT-based cathodes. The LiTFSI/tetraglyme based electrolytes and Li metal anodes were coupled with these cathodes. Cyclic voltammetry (CV) was performed without (Fig. 3a) and with visible light (Fig. 3b) to clarify the reactions during discharge and charge. The electrocatalytic activities of the ORR and OER are enhanced. In the discharging process, compared to pFe2O3 which shows a reduction peak at 2.15 V, the onset potential of the reduction reaction in the (110) Fe2O3 battery was increased to 2.77 V and two reduction peaks at 2.52 V and 2.34 V could be distinctly found, which may indicate the two-step electron ORR process. In the charging process, two oxidation peaks at 3.20 and 3.39 V appeared in the presence of (110) Fe2O3, while pFe2O3-based batteries showed no obvious oxidation occurring under 4.0 V. With the presence of light (Fig. 3b), due to the lowered band position of (110) Fe2O3, the photoelectrons could be involved in the Li–O2 reactions to facilitate their kinetics, especially for the reduction process. The peak current density reaches 0.46 mA cm−2 in (110) Fe2O3-based batteries, while it is only 0.20 mA cm−2 in pFe2O3-based ones. The Tafel slopes for the (110) Fe2O3 and pFe2O3 cases under light (Fig. S5, ESI†) were 146 mV dec−1 and 194 mV dec−1, respectively, which indicates that faceted (110) Fe2O3 has a better electrocatalytic performance as well as a significant photogenerated current that promotes the ORR process and enables an improved discharge ability and rate performance, especially with light illumination.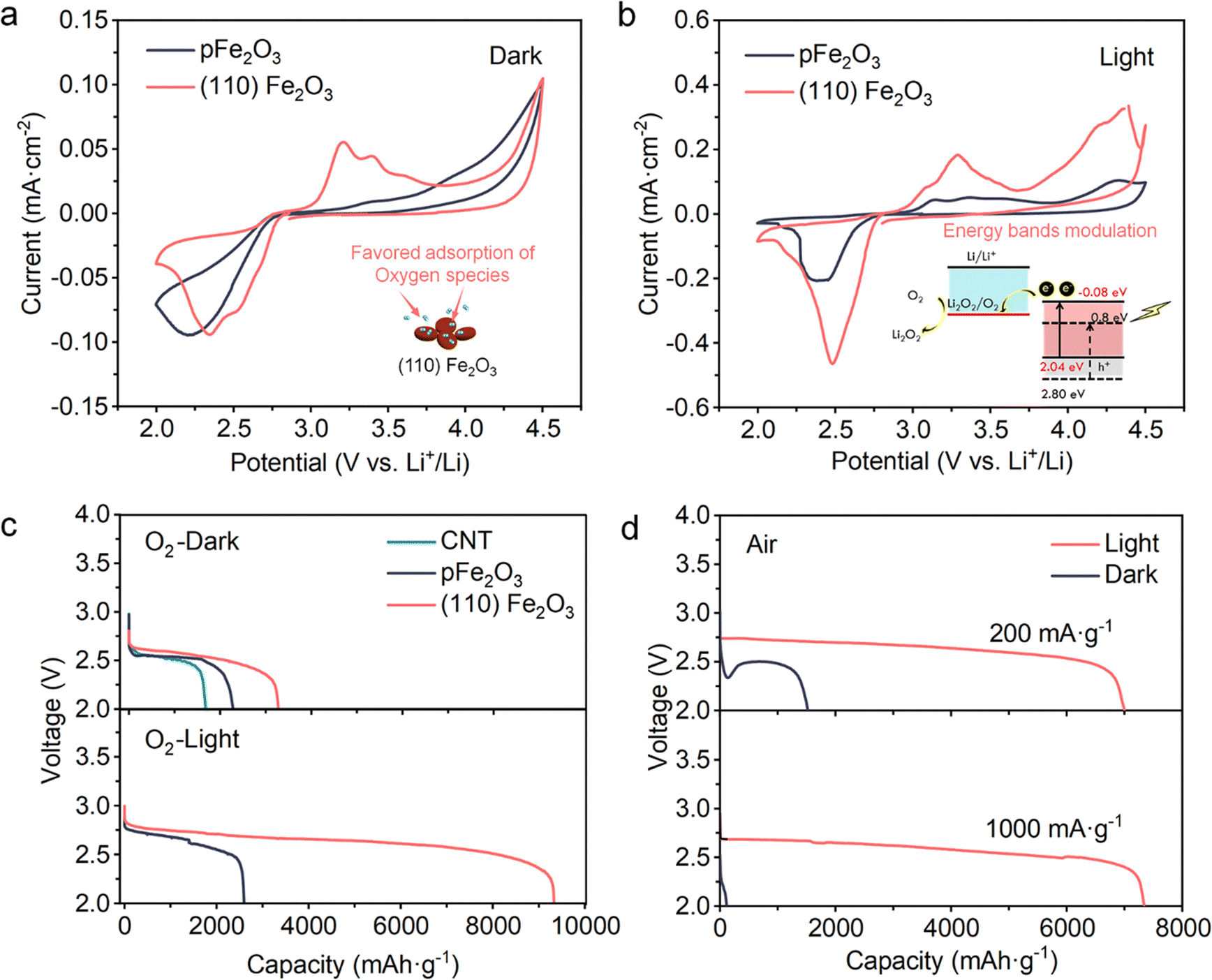 | ||
| Fig. 3 (110) Fe2O3 induced photo-enhancement of the ORR processes in LOBs. (a and b) CV curves of the LOBs (a) without and (b) with light at 0.2 mV s−1; the inset schematics illustrate the electrocatalytic and photocatalytic effects for Li–O2-based reactions. (c) Galvanostatic discharge profiles of (110) Fe2O3, pFe2O3, and CNT samples at 200 mA g−1 in O2. (d) Galvanostatic discharge profiles of (110) Fe2O3 samples at 200 mA g−1 and 1000 mA g−1 in air. | ||
In a pure oxygen environment, the (110) Fe2O3-based cathodes exhibited an increased specific capacity of 3276 mA h g−1 without light (Fig. 3c). In comparison, the unloaded CNT and pFe2O3 cases were 1678 mA h g−1 and 2275 mA h g−1, respectively. An improvement is further observed in the presence of light. After the addition of light, the discharge specific capacity of the (110) Fe2O3-based cathodes was unexpectedly increased to 9329 mA h g−1, in sharp comparison to only 2596 mA h g−1 of the pFe2O3 case. Considering that both materials have similar specific surface areas, 183.84 m2 g−1 for (110) Fe2O3/CNT and 183.42 m2 g−1 for pFe2O3/CNT (Fig. S6 and S7, ESI†), this apparent increase is attributed to the capacity provided by (110) Fe2O3 under light. Notably, the difference in capacity was more significant when the batteries were operated in the ambient air, as illustrated in Fig. 3d. Compared to the small capacity (1670 mA h g−1 at 200 mA g−1) in the dark conditions, the light-assisted discharge process exhibits reduced polarization (∼0.25 V) and much increased capacity (over 7000 mA h g−1), which retained no degradation of capacity even under high current densities of 1000 mA g−1. Overall, the (110) Fe2O3-based electrode produced a ∼420% capacity increase even in the air, indicating the excellence of the photochemical effect on (110) Fe2O3-based cathodes.
2.3. Light-induced surface deposition of discharge products
Changes in the composition and morphology of the discharge products on the (110) Fe2O3-based cathodes in dark/light conditions were investigated by XRD, SEM and XPS. The XRD pattern in Fig. 4a shows a characteristic peak at 34.8° corresponding to the (101) plane of crystalline Li2O2 generated by discharge in dark conditions, while the products are amorphous when the discharge process is performed in the presence of light, which is indicated by the absence of characteristics peaks of Li2O2. These amorphous products were verified to be mainly Li2O2 by Raman (Fig. S8, ESI†) and XPS spectra (Fig. 4f and g). The SEM images (Fig. 4b and c) indicate the differences in Li2O2 morphology. In dark conditions, micron-sized Li2O2 spherical particles are randomly deposited on the cathode, while film-like Li2O2 is formed consistently on the surface of CNTs and Fe2O3 nanorods. SEM images captured at different discharge stages were applied to further clarify the photo-organized Li2O2 formation process (Fig. 4d). At the discharge depths of 500, 1500, 3000 and 5000 mA h g−1 and full discharge at 2.0 V, Li2O2 first nucleates on the nanoplate-shaped (110) Fe2O3 surface, then grows along the electrode surface, and finally covers the electrode surface uniformly.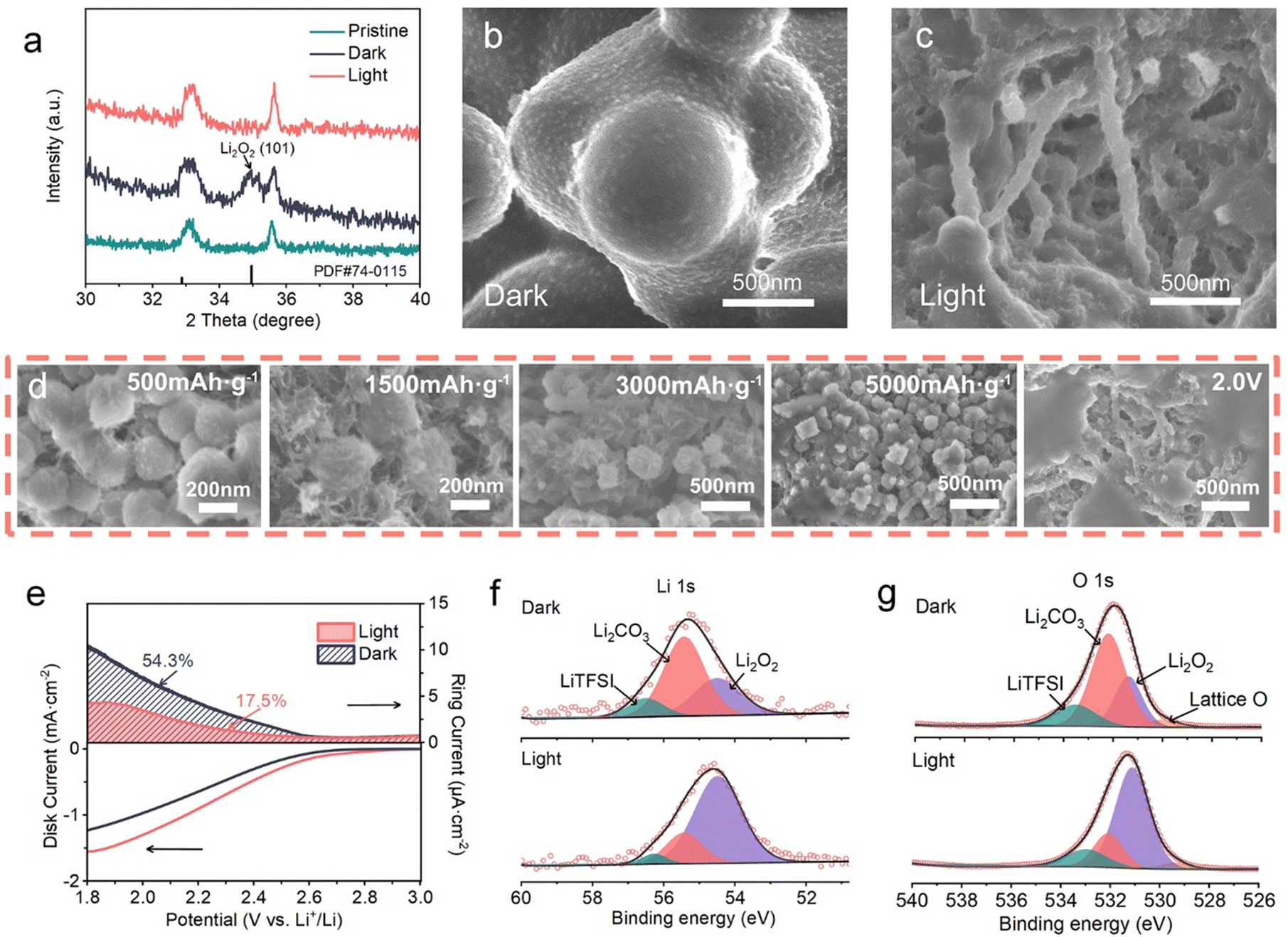 | ||
| Fig. 4 Surface nucleation and growth process of products under visible light. (a) XRD pattern of the (110) Fe2O3 based cathodes after discharge. (b and c) Morphologies of the fully discharged (110) Fe2O3-based cathodes (b) without and (c) with light. (d) SEM images at different discharge depths at 200 mA g−1. (e) RRDE measurements at 800 rpm. The disc potential increases from open circuit potential to 1.8 V vs. Li+/Li. Ering was held at 3.5 V vs. Li+/Li. (f, g) XPS spectra of (f) Li 1s and (g) O 1s for the discharged cathode in dark/light conditions. | ||
As a rule, the reason for the Li2O2 deposition difference can be attributed to the existence of discharge intermediates, LiO2, whether formed in electrolyte (LiO2,sol) or adsorbed on the cathode (LiO2,ads).33–35 And the relative content of LiO2,sol can be monitored by the rotating ring-disk electrode (RRDE) test34,36,37 (Fig. 4e and Fig. S9, ESI†). The disc electrode was loaded with (110) Fe2O3 and pFe2O3, respectively, and the ring electrode potential was set at 3.5 V (vs. Li+/Li) to detect the soluble intermediates. The disc and ring currents were monitored during a negative sweep from the equilibrium potential. The linear sweep voltammetry (LSV) curves obtained from the disk currents show an increase of the onset potential (2.85 V vs. 2.69 V) and a larger current density (1.56 mA cm−2vs. 0.98 mA cm−2) in the presence of light, showing that the illumination on (110) Fe2O3 greatly accelerates the ORR process. It is worth noting that the ring current decreased from 10.8 to 4.2 μA cm−2 in the presence of light, and the weight of solution-phase reaction (Qring/QORR) is revealed by this value. The Qring/QORR of the (110) Fe2O3/CNT cathode decreased from 54.3% in the dark to 17.5% in light conditions, indicating that the production of soluble intermediates was suppressed. This phenomenon was analyzed by Raman spectroscopy (Fig. S8, ESI†) which is generally conducted to detect soluble LiO2 intermediates.38 In dark conditions, the discharged cathode showed obvious LiO2 peaks at 1135 and 1480 cm−1, while these peaks almost disappeared with light illumination. These experiment results suggest that the effect of light on (110) Fe2O3 decreases the amount of solvated LiO2 intermediates in the electrolyte and favors the ORR process on the electrode rather than in the electrolyte. This transformation has a positive impact toward the suppression of side products like Li2CO3, which may be attributed to less 1O2 generated during the disproportionation reactions, and they would further react with the carbon substrate at the cathode.39–41 Changes in the compositions of the discharge products on (110) Fe2O3-based cathodes in dark/light conditions were investigated by XPS (Fig. 4f, g and Fig. S10, ESI†). Li2O2 and Li2CO3 species were observed on the (110) Fe2O3 based cathode, both in dark conditions and in light conditions, while the relative content of Li2CO3 decreased considerably after light discharge.
2.4. Boosted electron and oxygen transfer by the (110) Fe2O3 catalyst
The intrinsic reason for light-induced surface deposition of Li2O2 is revealed through the theoretical calculation and experimental method. The adsorption of oxygen and intermediate species on the (110) and (104) facets of Fe2O3 were simulated by DFT calculations (Fig. S11, ESI†), and the decreases in free energy (ΔG) corresponding to the formation of LiO2 on the electrode surface and its later reduction were also calculated (Fig. 5a and b). The negative values of adsorption energy (Eads) indicate that the thermodynamic process is favorable in both facets. The lower Eads (−1.37 eV) on (110) than on (104) (−0.84 eV) indicates stronger oxygen adsorption on the former. ΔG(LiO2)* and ΔG(Li2O2)* are respectively −3.76 eV and −3.69 eV for the (110) facets, whereas these values for (104) are −3.30 eV and −3.51 eV, respectively. Owing to the favored adsorption of oxygen species on the (110) facet, (110) Fe2O3 is more favorable for the electrode surface proceeded ORR process.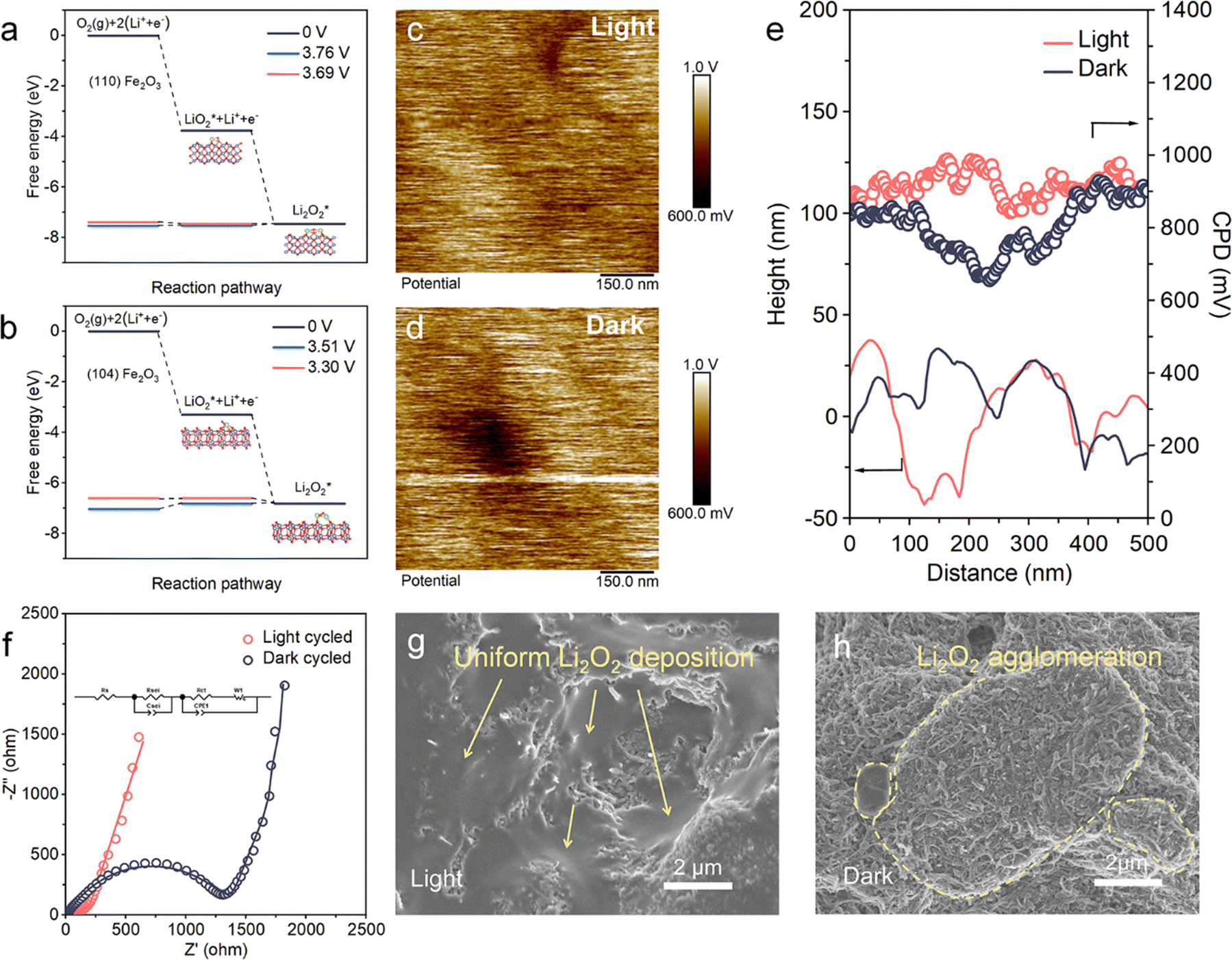 | ||
| Fig. 5 Enhanced adsorption of oxygen intermediates and construction of a photoelectric field by (110) Fe2O3 to regulate Li2O2 deposition. (a and b) Calculated free energy diagrams for the ORR reactions on (a) (110) and (b) (104) facets. (c and d) CPD maps of the (110) Fe2O3 based cathode in (c) light and (d) dark conditions. (e) CPD and height curves. (f) Nyquist plots and equivalent circuit of the (110) Fe2O3 electrode after ten cycles with the current density at 300 mA g−1 and cycling specific capacity of 600 mA h g−1. (g and h) Low magnification pictures of the morphologies of the fully discharged (110) Fe2O3-based cathodes (b) with and (c) without light. | ||
Except for oxygen-related reactants, the photo-induced morphology transformation is closely related to the charge distribution inside the porous cathode, which is determined by mapping the nanometer-resolution surface potential (Fig. 5c and d) using a Kelvin probe force microscope (KPFM). The values of linear contact potential difference (CPD) and height in dark and light conditions are shown in Fig. 5e. In dark conditions, the CPD minimum is 655 mV and increases to 842 mV under light. The CPD values show larger variations in dark conditions than those in light conditions. In addition, the surface potential in light conditions is more evenly distributed than that in dark conditions (Fig. 5c and d). This difference suggests that light induces an internal-built electric field, which increases the kinetics for charge transfer, as shown by the electrochemical impedance spectra (EIS), both of fresh and cycled cathodes (Fig. 5f and Fig. S13, ESI†). The obtained charge transfer resistance (Rct) and Warburg resistance (Rw) acquired from the equivalent circuits of EIS indicate the kinetics of electron transfer and oxygen transfer in the cathode, respectively.42,43 The apparently lowered Rct and Rw of the (110) Fe2O3 based cathode under light illustrates the unimpeded transfer process of oxygen and electrons, especially for the cycled conditions.
These results show that the light on (110) Fe2O3 plays a critical role in regulating the initial nucleation and subsequent growth of Li2O2 by anchoring intermediates on the cathode and homogenizing the charge distribution. As shown in Fig. 5g and h, Li2O2 produced under light is uniformly distributed on the whole surface, whereas the products produced without light accumulated on cathode domains (marked by the yellow dotted dash line). The local aggregation of products and their further obstruction of oxygen and electron-transport pathways leave plenty of unavailable “dead” active sites, leading to premature battery failure and limited capacity. This photo-assisted effect enables the smooth evolution of Li2O2 and the unimpeded transfer of reactants, and gives the cathodes the power to produce and store products associated with significantly improved capacities.
2.5. Cycling stability in actual Li–air batteries
The photo-assisted effect on (110) Fe2O3 significantly improves the cycling performance in O2 and the actual air environment. In light conditions, the discharge plateau with reduced polarization is above 2.70 V and remains stable during cycling (Fig. 6a). This constant capacity has high reversibility and cycling stability accompanied by a very slight increase of Rct (Fig. 5f and Fig. S13, ESI†). In sharp contrast, in dark conditions, the initial discharge plateau was lower (around 2.5 V), and during cycling, the polarization increased rapidly (Fig. S13, ESI†) and the battery failed after only 10 cycles accompanied by a significant increase of Rct, which may result from byproduct accumulation (XPS spectra in Fig. 4f and g) caused by the large amounts of super-nucleophile LiO2 intermediates (Fig. 4e).44 An improvement in the pFe2O3 cathode was also observed under light (Fig. S14, ESI†), but it is less obvious than that on the faceted (110) Fe2O3 due to the lower photocatalytic activity (Fig. 2h, i and Fig. 3a). A Li–O2 pouch cell was performed under dark and light conditions, respectively (Fig. 6d). Compared to the dark discharged profile, the light-assisted discharge plateau increased from 2.58 V to 2.80 V and the discharge capacity soared from 21 mAh to 53 mAh at a cut off voltage of 2.50 V. As for the cycling process conducted in the air environment, compared to only several cycles and fast failure of the battery performed without light, the discharge plateau of the light-assisted battery remained steady for over 300 cycles, which exhibits better reversibility and stability against the potential side-reactions present in ambient air.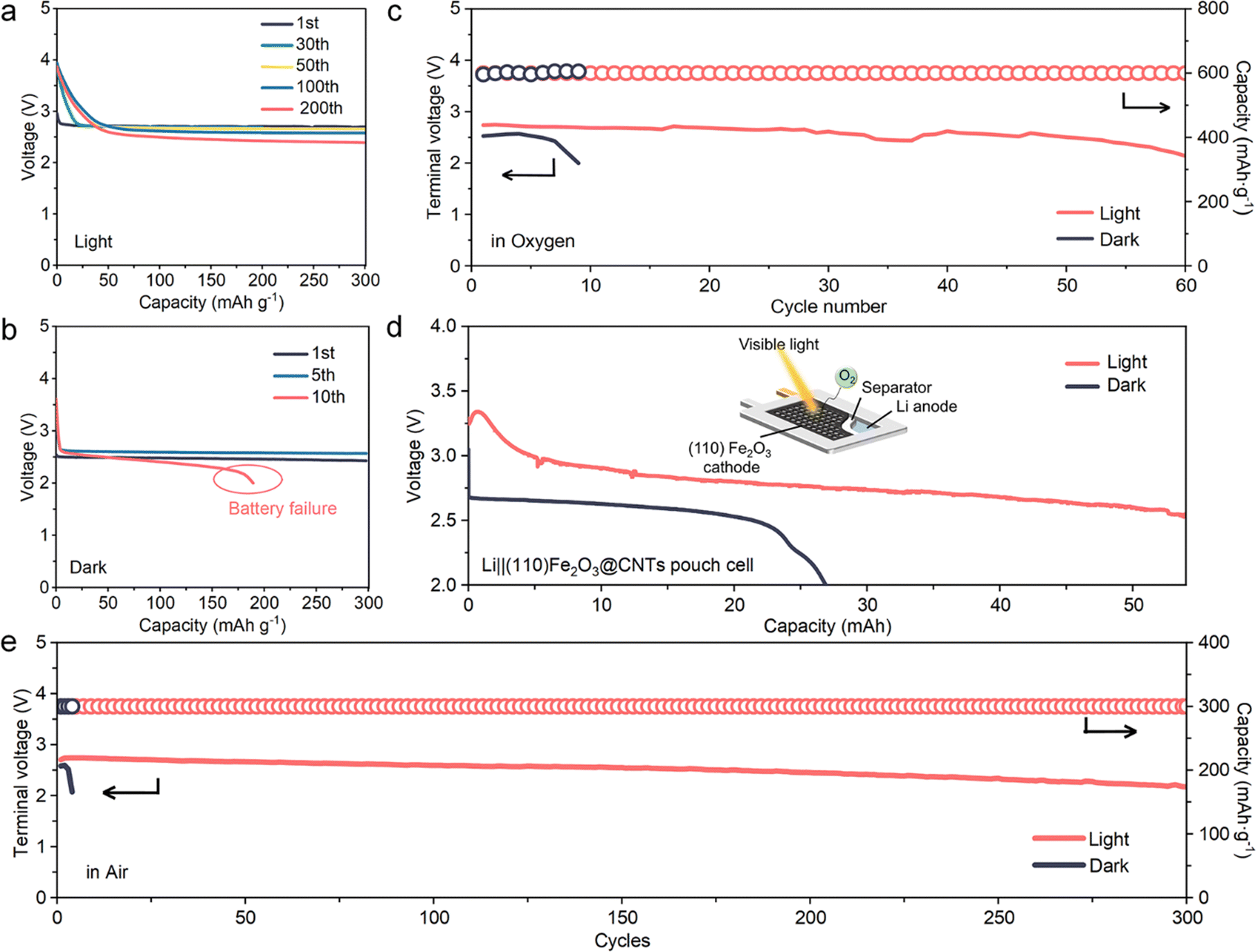 | ||
| Fig. 6 Improved charge transfer kinetics and cycling stability induced by visible light on (110) Fe2O3 based cathodes. (a and b) Discharge profiles of the (110) Fe2O3 based cathode discharged in ambient air under (a) light and (b) dark conditions at 300 mA g−1 for different cycles. (c) The terminal discharge potential of each cycle for (110) Fe2O3 electrodes discharged in O2 under dark and light conditions with the current density at 300 mA g−1. (d) Discharge profiles for the (110) Fe2O3 based pouch cell at 0.05 mA cm−2 in dark/light conditions. (e) The terminal discharge potential of each cycle for the (110) Fe2O3 electrodes discharged in ambient air under dark and light conditions with the current density at 300 mA g−1. | ||
3. Conclusion
In summary, we demonstrated an efficient photochemical strategy to regulate Li2O2 nucleation and growth behavior under visible light. An exceptional capacity increase and extended cycle life were realized, both in O2 (∼260%, over 60 cycles) and more promisingly in the actual air environment (∼420%, over 300 cycles). This effect derives from two aspects. The (110) facet engineering of α-Fe2O3 improves its adsorption ability toward oxygen intermediates, suppressing the disproportionation reactions and byproduct generation. Additionally, the improved photocatalytic activities of (110) Fe2O3 cause rapid generation of photoelectrons and synchronous construction of a uniform photo-electric field throughout the entire electrode under visible light. The preferential nucleation of Li2O2 on (110) Fe2O3 surfaces and its subsequent consistent growth with the assistance of the photo-electric field induce the homogeneous distribution of solid Li2O2 depositions, preventing the local product accumulation and accelerating the O2 diffusion and electron transfer, thus prohibiting premature battery failure and promoting the reversible decomposition of Li2O2, achieving a long-term stable Li–air battery working in the actual air environment. This novel distributed electron transfer induced by visible light provides both material and conceptual breakthroughs for developing many other high-performance open battery systems that could promise the synergistic coordination of electric and light power.4. Experimental section
4.1. Preparation of the cathode materials
All reagents in this work are analytically pure and used directly without further purification. Multiwalled Carbon Nanotubes (CNT, φ10–20 nm) were purchased from Time Nano Co. Ltd, and used as a conducting matrix in Li–O2 cathodes. FeCl3·6H2O (Aladdin, 99%) and CH3COONa·3H2O (Aladdin, 99%) were used as precursors for hydrothermal synthesis of (110) Fe2O3. 2.76 g FeCl3·6H2O and 5.25 g CH3COONa·3H2O were dissolved in 35 mL anhydrous ethanol and stirred vigorously for 10 minutes. The solution was transferred into a 50 mL Teflon-lined stainless-steel autoclave and heated in an oven at 200 °C for 24 h. After being cooled down, the resulting powder was vacuum-filtrated and then dried at 80 °C.4.2. Characterization
X-Ray Diffraction (XRD, Rigaku MiniFlex-600) patterns were recorded to confirm the successful synthesis of (110) Fe2O3. Transmission electron microscopy (TEM, JEM 2100-F) was utilized to identify the microstructure and exposed facets of a single (110) Fe2O3 nanoparticle. Scanning Electron Microscopy (SEM, Regulus-8100) is used for morphology observation of Fe2O3 materials and the cathode. N2 adsorption–desorption (Belsorp-mini) was conducted and the specific area of pFe2O3, (110) Fe2O3, pFe2O3/CNT and (110) Fe2O3/CNT could be calculated according to the Brunauer–Emmett–Teller (BET) mode. X-Ray photoelectron spectroscopy (XPS, Thermal Fisher Scientific Escalab-Xi +) was analyzed to identify the surface species via each detected element for the (110) Fe2O3 cathode being discharged. The UV-visible diffuse reflectance spectrum (DRS, Shimadzu UV-3600 Plus) was measured to characterize the light absorbance (a) of the samples in the range of 300 < λ < 800 nm, and then the Tauc plot could be converted according to the Kubelka–Munk method. XPS valence band spectroscopy is conducted to determine the VB potential of the Fe2O3 sample by a VersaProbe III (Scanning ESCA Microprobe) SCA (Spherical Analyzer). Photoluminescence (PL) spectra were measured by a Steady state/Lifetime Spectrofluorometer (Edinburgh FLS1000) to characterize the lifetime of photoelectrons. Confocal Raman Microscopy (LabRAM HR Evolution) with an excitation wavelength of 532 nm was used to measure and collect Raman spectra.4.3. Electrochemical and photoelectrochemical performance
The photocatalytic activity of Fe2O3 was investigated by decomposing Rhodamine B (RhB) via irradiation with visible light. An amount of the Fe2O3 powders was dispersed in 100 mL of the RhB solution (3 × 10−5 M). The prepared solution was stirred for 30 min in the dark to achieve an adsorption–desorption equilibrium. Before the suspension was irradiated with 300 W Xeon lamps, 3 mL of H2O2 (30%) was added. After a given time interval, 4 mL of the mixture was collected, filtered and analyzed using the U-2910 single-beam spectrometer (Hitachi). Chronoamperometry measurement was tested at 0.87 V (vs. SHE) on an electrochemical workstation (Autolab) using a three-electrode system, in which the platinum electrode and Ag/AgCl electrode were used as a counter electrode and reference electrode, respectively. The contact potential difference (CPD) measurement was conducted with a Kelvin Probe Force Microscope (KPFM, Bruker Dimension icon) under an ambient atmosphere with Xeon arc light irradiation (400 nm < λ < 800 nm). The alternating current voltage was set to 0.5 V to avoid a high bias-induced band bending.The prepared Fe2O3 powder, CNT and PVDF (poly(vinylidene fluoride)) were mixed with a mass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:1, ground for 30 min to blend uniformly. After that, the slurry was spread on clean Carbon Paper (Avcarb P50T) with a diameter of 8 mm and dried in a vacuum oven at 100 °C for 24 h, and the Fe2O3/CNT cathode was obtained with the total average loading of Fe2O3 and CNT of 0.4–0.6 mg cm−2. For a typical process for assembling a CR2032 coin Li–O2 battery, all the operations were conducted in an Argon atmosphere filled glove-box with both H2O and O2 contents below 0.5 ppm, in which the Fe2O3/CNT cathode was an oxygen electrode and photoelectrode, Li foil (12 mm in diameter and 0.5 mm in thickness) and glass fiber (Whatman, GF/A) were used as the counter electrode and the separator, respectively, and 80 μL of 1 M LiTFSI dissolved in TEGDME (DoDoChem, LK-001) was used as the electrolyte and dropped on the separator. The assembled Li–O2 battery was stored in a volume capacity of 250 mL sealed glass test device, which was filled with O2 and tested on a LAND CT2001A multi-channel battery system. The Li–O2 pouch cell assembled by a (110) Fe2O3/CNT cathode (2 cm × 5 cm), lithium foil (3 cm × 6 cm) and glass fiber (Whatman, GF/A), Carbon paper and Cu-mesh were employed as a current collector for the cathode and anode, respectively. The pouch cell was packed by a plastic-sealed Al-film with openings at the cathode side. The Li–air battery was assembled according to the above-mentioned process and operated under actual air. All the batteries were performed at room temperature (∼25 degrees). An Autolab multichannel workstation was used to evaluate the cyclic voltammetry (CV) performance and electrochemical impedance spectra (EIS) in a frequency range from 106 Hz to 10−2 Hz of the coin cell. For photo-assisted discharge, the Xe lamp (CEAULIGHT-HXF300) (400 nm < λ < 800 nm) was opened at the power of 130 W to irradiate the coin cell through the vessel and porous cathode shells.
:6:1, ground for 30 min to blend uniformly. After that, the slurry was spread on clean Carbon Paper (Avcarb P50T) with a diameter of 8 mm and dried in a vacuum oven at 100 °C for 24 h, and the Fe2O3/CNT cathode was obtained with the total average loading of Fe2O3 and CNT of 0.4–0.6 mg cm−2. For a typical process for assembling a CR2032 coin Li–O2 battery, all the operations were conducted in an Argon atmosphere filled glove-box with both H2O and O2 contents below 0.5 ppm, in which the Fe2O3/CNT cathode was an oxygen electrode and photoelectrode, Li foil (12 mm in diameter and 0.5 mm in thickness) and glass fiber (Whatman, GF/A) were used as the counter electrode and the separator, respectively, and 80 μL of 1 M LiTFSI dissolved in TEGDME (DoDoChem, LK-001) was used as the electrolyte and dropped on the separator. The assembled Li–O2 battery was stored in a volume capacity of 250 mL sealed glass test device, which was filled with O2 and tested on a LAND CT2001A multi-channel battery system. The Li–O2 pouch cell assembled by a (110) Fe2O3/CNT cathode (2 cm × 5 cm), lithium foil (3 cm × 6 cm) and glass fiber (Whatman, GF/A), Carbon paper and Cu-mesh were employed as a current collector for the cathode and anode, respectively. The pouch cell was packed by a plastic-sealed Al-film with openings at the cathode side. The Li–air battery was assembled according to the above-mentioned process and operated under actual air. All the batteries were performed at room temperature (∼25 degrees). An Autolab multichannel workstation was used to evaluate the cyclic voltammetry (CV) performance and electrochemical impedance spectra (EIS) in a frequency range from 106 Hz to 10−2 Hz of the coin cell. For photo-assisted discharge, the Xe lamp (CEAULIGHT-HXF300) (400 nm < λ < 800 nm) was opened at the power of 130 W to irradiate the coin cell through the vessel and porous cathode shells.
The rotating ring-disk electrode (RRDE) test was performed with a similar procedure described by Sankarasubramanian et al.37 The working electrode was a Φ 5 mm glassy carbon (GC) disk and Pt ring made by Phychemi (Hongkong) Co., Ltd. The slurry coated on the GC electrode was prepared with 2 mg Fe2O3 powder and carbon black blended with 15–20 μL Nafion dispersion (Alfa) was dispersed into isopropanol. The average loading is about 0.2 mg. The Pt electrode and partially delithiated LiFePO4 electrode were used as the counter electrode and reference electrode, respectively. The 0.2 M solution of lithium bis-trifluoromethanesulfonimidate (LiTFSI) (Sigma-Aldrich) in tetraethylene glycol dimethyl ether (DME) (Sigma-Aldrich) was prepared in an Argon filled glovebox after a further water removal process and used as the electrolyte. The water content was 36.3 ppm, measured by a Karl Fischer titration analyzer (Metrohm 899 Coulometer).
4.4. Computational section
All calculations in this work were carried out using the density functional theory (DFT) method as implemented in the VASP code. The electronic exchange–correlation energy was modeled using the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA). The projector augmented wave (PAW) method was used to describe the ionic cores. For the plane-wave expansion, a 450 eV kinetic energy cut-off was used after testing a series of different cut-off energies. A Monkhorst–Pack 3 × 3 × 1 k-point grid was used to sample the Brillouin zone. The convergence criterion for the electronic structure iteration was set to be 10−4 eV, and that for geometry optimizations was set to be 0.01 eV Å−1 on force. Gaussian smearing of 0.1 eV was applied during the geometry optimization and for the total energy computations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate support from the National Key Research and Development Program of China (No. 2021YFF0500600), the National Natural Science Foundation of China (No. 52272231, U2001220 and 51902223), the Haihe Laboratory of Sustainable Chemical Transformations, the Fundamental Research Funds for the Central Universities, the Natural Science Foundation of Tianjin (No. 20JCYBJC00850), and the National Key Research and Development Program of China (No. 2019YFE0118800). DFT computations in this research were undertaken with the assistance of resources and services from the National Computational Infrastructure (NCI) and Phoenix High Performance Computing, which are supported by the Australian Government and the University of Adelaide.References
- X.-D. Lin, Y. Gu, X.-R. Shen, W.-W. Wang, Y.-H. Hong, Q.-H. Wu, Z.-Y. Zhou, D.-Y. Wu, J.-K. Chang, M.-S. Zheng, B.-W. Mao and Q.-F. Dong, Energy Environ. Sci., 2021, 14, 1439–1448 RSC.
- Y. Dou, Z. Xie, Y. Wei, Z. Peng and Z. Zhou, Natl. Sci. Rev., 2022, nwac040 CrossRef PubMed.
- K. Chen, G. Huang and X.-B. Zhang, Chin. J. Chem., 2021, 39, 32–42 CrossRef CAS.
- Y. Liu, J. Cai, J. Zhou, Y. Zang, X. Zheng, Z. Zhu, B. Liu, G. Wang and Y. Qian, eScience, 2022, 2, 389–398 CrossRef.
- L. D. Griffith, A. E. S. Sleightholme, J. F. Mansfield, D. J. Siegel and C. W. Monroe, ACS Appl. Mater. Interfaces, 2015, 7, 7670–7678 CrossRef CAS PubMed.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772–1778 RSC.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 1–11 Search PubMed.
- C.-L. Li, G. Huang, Y. Yu, Q. Xiong, J.-M. Yan and X. Zhang, J. Am. Chem. Soc., 2022, 144, 5827–5833 CrossRef CAS PubMed.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 RSC.
- J. Shui, F. Du, C. Xue, Q. Li and L. Dai, ACS Nano, 2014, 8, 3015–3022 CrossRef CAS PubMed.
- A. Liu, X. Liang, X. Ren, W. Guan and T. Ma, Electrochem. Energy Rev., 2022, 5, 112–144 CrossRef CAS.
- G. Zhang, G. Li, J. Wang, H. Tong, J. Wang, Y. Du, S. Sun and F. Dang, Adv. Energy Mater., 2022, 12, 2103910 CrossRef CAS.
- Q. Zhang, C. Wang, Z. Xie and Z. Zhou, Energy Environ. Mater., 2022, 5, 1103–1116 CrossRef CAS.
- L. Guo, L. Tan, A. Xu, G. Li, G. Zhang, R. Liu, J. Wang, Y. Du and F. Dang, Energy Storage Mater., 2022, 50, 96–104 CrossRef.
- X. Gao, Y. Chen, L. R. Johnson, Z. P. Jovanov and P. G. Bruce, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- M. Balaish, X. Gao, P. G. Bruce and Y. Ein-Eli, Adv. Mater. Technol., 2019, 4, 1800645 CrossRef.
- F. Z. Wang and X. L. Li, ACS Appl. Mater. Interfaces, 2018, 10, 26222–26232 CrossRef CAS PubMed.
- V. Viswanathan, K. S. Thygesen, J. S. Hummelshøj, J. K. Nørskov, G. Girishkumar, B. D. McCloskey and A. C. Luntz, J. Chem. Phys., 2011, 135, 214704 CrossRef CAS PubMed.
- L. Cheng, P. Redfern, K. C. Lau, R. S. Assary, B. Narayanan and L. A. Curtiss, J. Electrochem. Soc., 2017, 164, E3696 CrossRef CAS.
- S. Zhang, M. Chen, X. Zhao, J. Cai, W. Yan, J. C. Yen, S. Chen, Y. Yu and J. Zhang, Electrochem. Energy Rev., 2021, 4, 336–381 CrossRef CAS.
- K. Chen, D.-Y. Yang, G. Huang and X.-B. Zhang, Acc. Chem. Res., 2021, 54, 632–641 CrossRef CAS PubMed.
- D. Geng, N. Ding, T. S. A. Hor, S. W. Chien, Z. Liu, D. Wuu, X. Sun and Y. Zong, Adv. Energy Mater., 2016, 6, 1502164 CrossRef.
- E. J. Nemanick and R. P. Hickey, J. Power Sources, 2014, 252, 248–251 CrossRef CAS.
- A. K. Thapa, Y. Hidaka, H. Hagiwara, S. Ida and T. Ishihara, J. Electrochem. Soc., 2011, 158, A1483 CrossRef CAS.
- C. Tran, X.-Q. Yang and D. Qu, J. Power Sources, 2010, 7 Search PubMed.
- H. C. Lee, J. O. Park, M. Kim, H. J. Kwon, J.-H. Kim, K. H. Choi, K. Kim and D. Im, Joule, 2019, 3, 542–556 CrossRef CAS.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2009, 113, 20127–20134 CrossRef CAS.
- Z. Lyu, Y. Zhou, W. Dai, X. Cui, M. Lai, L. Wang, F. Huo, W. Huang, Z. Hu and W. Chen, Chem. Soc. Rev., 2017, 46, 6046–6072 RSC.
- J. Wang, Y. Zhang, L. Guo, E. Wang and Z. Peng, Angew. Chem., Int. Ed., 2016, 55, 5201–5205 CrossRef CAS PubMed.
- H. Gong, T. Wang, K. Chang, P. Li, L. Liu, X. Yu, B. Gao, H. Xue, R. Ma, J. He and J. Ye, Carbon Energy, 2022, 4, 1169–1181 CrossRef CAS.
- M. Wang, J. Chen, Z. Tian, W. Dai, B. Cui, X. Cui, D. Wang, Y. Xiao, X. Lian, C. Jiang, H. Yang, Y. Wang, Z. Sun, Y. Ding, Y.-Y. Sun, J. Zhang and W. Chen, Energy Environ. Sci., 2023, 16, 523–534 RSC.
- D. Du, Z. Zhu, K.-Y. Chan, F. Li and J. Chen, Chem. Soc. Rev., 2022, 51, 1846–1860 RSC.
- N. B. Aetukuri, B. D. McCloskey, J. M. Garcia, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed.
- Y. Wang, N.-C. Lai, Y.-R. Lu, Y. Zhou, C.-L. Dong and Y.-C. Lu, Joule, 2018, 2, 2364–2380 CrossRef CAS.
- T. Liu, J. T. Frith, G. Kim, R. N. Kerber, N. Dubouis, Y. Shao, Z. Liu, P. C. M. M. Magusin, M. T. L. Casford, N. Garcia-Araez and C. P. Grey, J. Am. Chem. Soc., 2018, 140, 1428–1437 CrossRef CAS PubMed.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J.-M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- S. Sankarasubramanian, J. Seo, F. Mizuno, N. Singh and J. Prakash, J. Phys. Chem. C, 2017, 121, 4789–4798 CrossRef CAS.
- J. Wang, L. Ma, J. Xu, Y. Xu, K. Sun and Z. Peng, SusMat, 2021, 1, 345–358 CrossRef CAS.
- S. Dong, S. Yang, Y. Chen, C. Kuss, G. Cui, L. R. Johnson, X. Gao and P. G. Bruce, Joule, 2022, 6, 185–192 CrossRef CAS.
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 1–9 Search PubMed.
- Y. K. Petit, E. Mourad, C. Prehal, C. Leypold, A. Windischbacher, D. Mijailovic, C. Slugovc, S. M. Borisov, E. Zojer, S. Brutti, O. Fontaine and S. A. Freunberger, Nat. Chem., 2021, 1–7 Search PubMed.
- J. Huang, B. Tong, Z. Li, T. Zhou, J. Zhang and Z. Peng, J. Phys. Chem. Lett., 2018, 9, 3403–3408 CrossRef CAS PubMed.
- X. Luo, R. Amine, K. C. Lau, J. Lu, C. Zhan, L. A. Curtiss, S. Al Hallaj, B. P. Chaplin and K. Amine, Nano Res., 2017, 10, 4327–4336 CrossRef CAS.
- V. S. Bryantsev, V. Giordani, W. Walker, M. Blanco, S. Zecevic, K. Sasaki, J. Uddin, D. Addison and G. V. Chase, J. Phys. Chem. A, 2011, 115, 12399–12409 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00022b |
| This journal is © The Royal Society of Chemistry 2023 |