
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-high-rate CO2 reduction reactions to multicarbon products with a current density of 1.7 A cm−2 in neutral electrolytes†
Asato
Inoue
a,
Takashi
Harada
 ab,
Shuji
Nakanishi
*ab and
Kazuhide
Kamiya
*ab
ab,
Shuji
Nakanishi
*ab and
Kazuhide
Kamiya
*ab
aResearch Center for Solar Energy Chemistry, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: nakanishi.shuji.es@osaka-u.ac.jp; kamiya.kazuhide.es@osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 3rd November 2022
Abstract
CO2 electrolysis to value-added products is a promising technology to close the carbon cycle and sequester anthropogenic CO2 into chemical feedstocks; an increase of the current density for multicarbon products is one of the requirements for practical implementation. We have successfully increased the partial current density for gaseous CO2 reduction reactions to multicarbon products (C2+) over Cu nanoparticles on gas diffusion electrodes in neutral electrolytes to a record value of 1.7 A cm−2. The faradaic efficiency for multicarbon products increased with the current density below total current density of 2000 mA cm−2 and reached 76% at a total current density of 1600 mA cm−2. The turnover frequency for the production of C2+ per Cu atoms exceeded 1.1 s−1. Optimizing the standard components and their assembly as the cathode elicits the high-turnover frequency of oxide-derived Cu catalysts, resulting in the record partial current density for C2+. Especially, we demonstrated that the thickness of catalyst layers was one highly sensitive factor in determining the maximum current density for C2+.
Broader contextThe electrochemical reduction of carbon dioxide (CO2) has attracted considerable attention to maintain the carbon cycle. An increase in the current density for value-added multicarbon products (C2+), such as ethylene and ethanol, is required for practical implementation of this technology. Herein, we successfully obtained the partial current density for C2+ over cupric oxide (CuO) nanoparticles on gas diffusion electrodes in neutral electrolytes to a value of 1.7 A cm−2. Additionally, the faradaic efficiency for multicarbon products increased with the current density below Jtotal = 2000 mA cm−2 and reached up to 76%. Our ultra-high-rate CO2 electrolysis was achieved by optimizing the ordinary components, including oxide-derived Cu nanoparticles and carbon-based gas-diffusion electrodes, and their assembly as the cathode. In the other word, the present study succeeded in eliciting the potential performance of an ordinary combination. Although more detailed analyses are needed, our electrodes will teach us essential requirements for ultra-high-rate electrolysis. Additionally, the obtained knowledge through the detailed analyses of our electrodes would be widely utilized to develop novel materials, such as catalysts and electrodes. |
Introduction
The electrochemical carbon dioxide reduction reaction (CO2RR) has attracted considerable attention as a promising strategy for the conversion of anthropogenic CO2 into value-added products.1–10 One advantage of this method is that this greenhouse gas can be reduced under mild conditions (close to ambient temperature and pressure).11 However, the operative efficiency, product selectivity and rate must be improved for practical implementation.12,13 Among them, it has been reported that the current density for the production of high value-added products is correlated to the capital cost of the electrodes employed.14 Therefore, improvement of the current density would significantly affect the feasibility of this technology.The use of gas diffusion electrodes (GDEs) that allow the CO2RR to occur at the solid-catalyst/liquid-electrolyte/gaseous-CO2 interface, effectively accelerate the CO2RR by solving the problem of the mass transport limitation due to the inherently low diffusion and solubility of CO2 in water.15,16 A catalyst layer (CL) composed of metal Cu particles and ionomers on carbon-based microporous layers has been widely studied as the standard cathode for gaseous CO2RR to produce C2+ organics, such as C2H4, C2H5OH, C3H7OH and CH3COOH.8,17–19 Although the partial current density for C2+ (jC2+) has still remained below 500 mA cm−2 in many studies,18,20–27 a few studies have developed novel cathode assemblies or components toward an ultra-high-rate (UHR-) CO2RR with over jC2+ = 1 A cm−2. Sargent et al. successfully achieved a jC2+ over 1.2 A cm−2 in 7 M KOH using an electrode fabricated by dropping Cu nanoparticles on a sputtered Cu film/porous polytetrafluoroethylene (PTFE).28 Additionally, in this work, the bulk heterojunction of catalysts and ionomers decoupled the pathways for gases, electrons and ions, which resulted in a jC2+ over 1 A cm−2. Wang and co-authors reported that a fluorine-modified copper catalyst deposited on GDEs exhibited a total current density (Jtotal) of 1600 mA cm−2 with a faradaic efficiency for C2+ (FEC2+) of over 80% (i.e., jC2+ = over 1.2 A cm−2) in 0.75 M KOH.29 Very recently, Zheng et al. obtained jC2+ = 0.91 A cm−2 using nitrogen-engineered Cu nanoparticles in 1 M KOH.10
In these reports, UHR-CO2RR has been achieved using high alkaline solutions to suppress H2 evolution, which competes with the CO2RR. However, in addition to the toxicity and corrosivity, alkaline solutions quickly absorb CO2 and convert it to electrochemically inert (bi)carbonate.30–33 Based on these drawbacks, UHR electrolysis in neutral electrolytes is thus required from a practical viewpoint. Furthermore, these previous studies succeeded in achieving UHR-CO2RR by developing novel components in the three-phase interfaces (i.e., catalysts or electrodes). However, the obtainable maximum jC2+ value is still unknown when optimizing the standard components; CuO nanoparticle catalysts (CuONPs), which are known to serve as oxide-derived Cu catalysts for effective C2+ production,34–36 and carbon-based gas diffusion layers. An electrochemical system that can elicit the maximum jC2+ using only standard components will become the primary platform for future studies to design materials and interfaces for UHR-CO2RR. In other words, the design guidelines obtained from the UHR-CO2RR with only ordinary materials would be quite general, and thus, they are expected to be widely applicable to novel materials and systems.
In the present work, we attempt to pursue a high partial current density for C2+ (jC2+) using neutral electrolytes by optimizing the standard components and properly assembling them as the cathode. In particular, we achieved a record current density of 1.7 A cm−2 for multicarbon products in neutral electrolytes using synthetic or commercial CuONPs and carbon-based gas diffusion layers. Firstly, we will explain the details in characterizations and CO2RR activity of our optimized electrode, and let us discuss the mechanism of UHR-CO2RR. Then, the requirements for our UHR electrolysis will be discussed with varying components and conditions.
Results and discussion
UHR-CO2RR over 1.7 A cm−2 for C2+ products obtained with CuONPs
In this first section, let us show the detailed characterizations and electrochemical properties of our optimized electrode for UHR-CO2RR, which is composed of the GDE (GDL 34BC) carrying our synthesized CuONPs. CuONP catalysts were synthesized by wet-chemical reduction using NaBH4 as a reductant. Transmission electron microscopy (TEM) observations (Fig. 1a and Fig. S1, ESI†) show that the particle morphology and size is spherical and 20 nm, respectively. X-ray diffraction (XRD) patterns (Fig. S2, ESI†) indicate peaks at 35.5° and 38.7°, which are attributed to CuO(002) and CuO(111), respectively. This oxidation state of the as-prepared sample was also confirmed by narrow scan X-ray photoelectron spectroscopy (XPS) and X-ray absorption near edge structure (XANES), as shown in Fig. S3 and S4 (ESI†), respectively. Note that the Cu(II) oxidation state was reduced to Cu(I)/Cu(0) during electrolysis (Fig. S5 and S6, ESI†). Morphological characterization of the CuONPs/GDEs (Fig. S7, ESI†) was conducted using scanning electron microscopy (SEM). Fig. 1b–d and Fig. S8 (ESI†) show top- and cross-sectional SEM images of CuONPs-1.7/GDE (where the loading amount of CuONPs is 1.7 mg cm−2 on GDL 34BC), respectively. CuONPs completely covered the electrode surface based on the top-view SEM images, and were deposited on the microporous layer (MPL) composed of carbon nanoparticles. The cross-sectional SEM images reveal that the thickness of the MPL and the CL were ca. 80 and 2 μm for CuONPs-1.7/GDE, respectively. When the loading amount of CuONPs was varied to 0.34 mg cm−2 (Fig. S9, ESI†) and 3.1 mg cm−2 (Fig. S10, ESI†), the CL thicknesses changed to ca. 0.5 μm and 4 μm, respectively. The roughness factors of the CL were obtained using a surface profilometer. When increasing CL thickness, the surface becomes rougher (Fig. S11 and Table S1, ESI†). | ||
| Fig. 1 (a) Representative TEM image of the synthesized CuONPs. (b) Top-view and (c and d) cross-sectional SEM micrographs of CuONPs-1.7/GDE. | ||
We measured the CO2RR activity of the Cu-based GDE catalysts. An in-house-built three-compartment electrochemical cell was used for CO2RR evaluation (Fig. S12, ESI†).37,38Jtotalvs. potential (U) curves were obtained in 1 M KCl under CO2 and argon conditions, as shown in Fig. S13 (ESI†). The onset potential of the cathodic current for bare and Cu-modified GDEs were −1.6 and −1.2 V vs. Ag/AgCl under CO2, respectively. When the Cu amount was increased from CuONPs-0.34/GDE to CuONPs-3.1/GDE, the cathodic current under CO2 gradually increased.
The CO2RR products in outlet gas streams from the gas chamber and electrolytes from the catholyte room were analysed using gas chromatography (Fig. S14, ESI†) and 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S15, ESI†) after constant-current electrolysis, respectively. Fig. 2a, b and Fig. S16 (ESI†) show the FE and partial current density of CO2RR products against the applied potential for CuONPs-1.7/GDE in 1 M KCl, respectively. Under low current conditions (Jtotal = 200 mA cm−2) at −1.36 V vs. Ag/AgCl electrode, CO was the main product (FECO > 50%), and the FEC2H4 was below 20%. The FE for C2+ products increased and reached 76% at −1.69 V vs. Ag/AgCl electrode (Jtotal = 1600 mA cm−2) when the applied potential was shifted to the negative side. In particular, the FEs for C2H4 and C2H5OH were 46% and 22% at −1.76 V vs. Ag/AgCl electrode, respectively. The partial current density for C2+ products (C2H4, C2H5OH, C3H7OH, and CH3COOH) was over jC2+ = 1.7 A cm−2 at −1.76 V vs. Ag/AgCl electrode. On the contrary, the FEs for C1 products (CO, CH4 and HCOOH) and H2 were 8.8 and 14% at −1.76 V vs. Ag/AgCl electrode, respectively. Fig. 2c and Table S2 (ESI†) show a comparison of jC2+ and FEC2+ in neutral electrolytes among the reported values. The partial current density for C2+ production (jC2+ = over 1.7 A cm−2) is a record value, and the FEC2+ is also one of the highest. Therefore, the present study succeeded in eliciting the potential performance of an ordinary combination for UHR-CO2RR (i.e., CuO-derived Cu nanoparticles and carbon-based GDEs). Significantly, such a high jC2+ value for our assembly was remarkably reproducible even using different batches of electrodes, as shown in Fig. S17 and Table S3 (ESI†). When the applied potential was changed to −1.79 V vs. Ag/AgCl electrode (Jtotal = 2800 mA cm−2), the FE for C2+ products suddenly decreased, and the FE for H2 increased. The total FEs at −1.79 V vs. Ag/AgCl electrode did not reach 100% (ca. 80%) because the bubbles contained CO2 substrate, CO2RR products, and H2 formed at the cathode surface and diffused into the catholyte chamber. The applied potential was compensated using the current interrupt method as written in the experimental section39–41 (see Fig. S18 for the values versus reversible hydrogen electrode, ESI†). However, as accurate potential corrections are difficult under ultra-high-current conditions, we must note that the applied potentials are still referenced values.
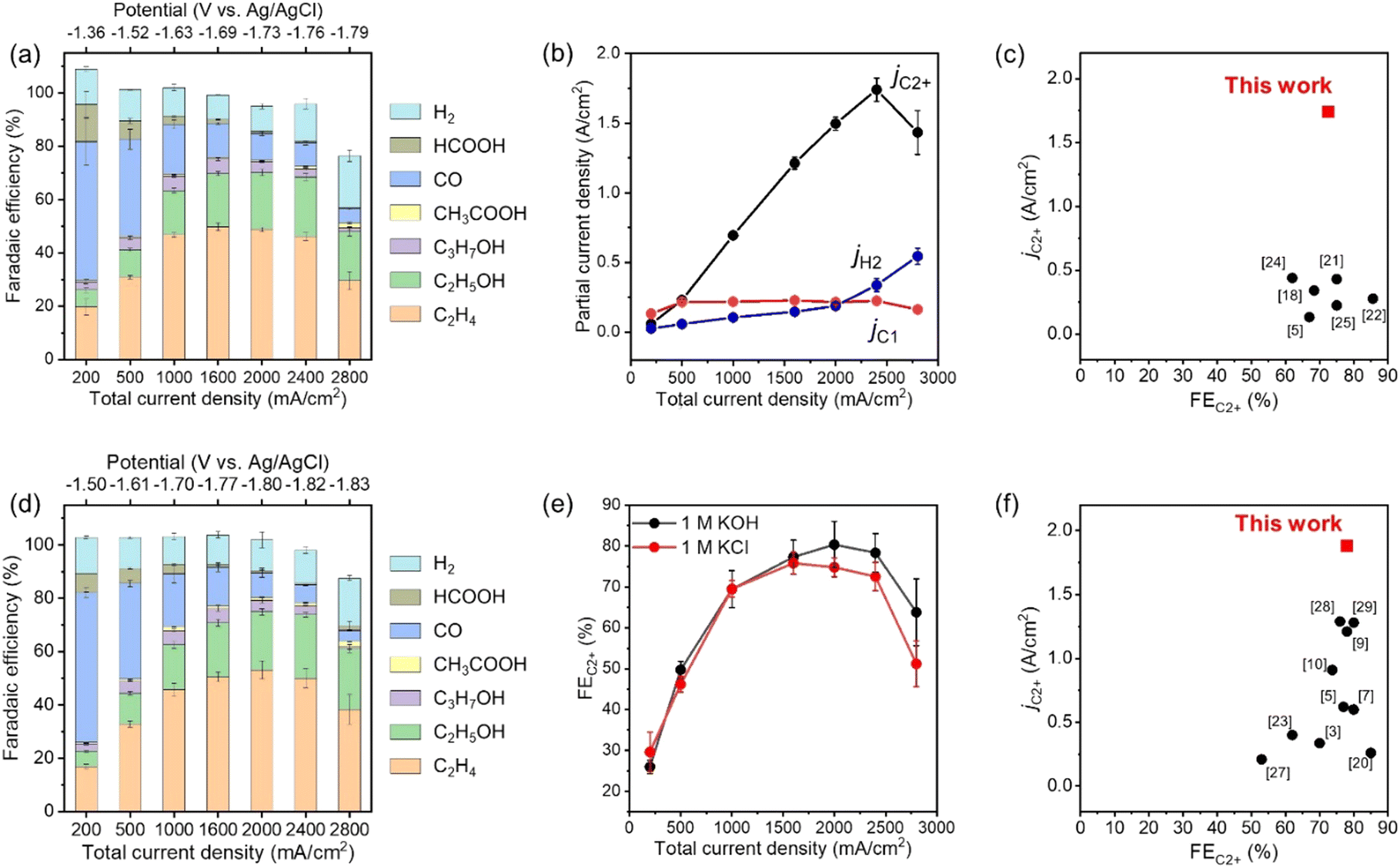 | ||
| Fig. 2 (a) FEs of the CO2RR products under various applied potentials on CuONPs-1.7/GDE in 1 M KCl. Reaction time: 30 min. (b) Partial current density for C2+, C1 and H2 on CuONPs-1.7/GDE in 1 M KCl. (c) Comparison of FEC2+ and jC2+ for CuONPs-1.7/GDE with reported Cu-based catalysts in neutral solution (for the detailed references, see Table S2, ESI†). (d) FEs for the CO2RR products under various applied potentials on CuONPs-1.7/GDE in 1 M KOH. Reaction time: 30 min. (e) Comparison of FEC2+ in 1 M KOH and in 1 M KCl under various Jtotal over CuONPs-1.7/GDE. (f) Comparison of FEC2+ and jC2+ for CuONPs-1.7/GDE with reported Cu-based catalysts in alkaline solution (for the detailed references, see Table S5, ESI†). | ||
Even when using neutral solutions, the local pH at the surface of the catalyst increases significantly during UHR-CO2RR due to the consumption of protons with an increase in the CO2RR current density. Our rough calculation and previous reports showed that the local pH becomes almost the same in neutral and alkaline electrolytes during high-current electrolysis (Fig. S19 and Table S4, ESI†). Therefore, the cathode reaction proceeded under alkaline environments for UHR-CO2RR even with neutral solutions; thus, the fabricated cathode was expected to be suitable for UHR-CO2RR even in bulk alkaline solutions. Fig. 2d shows the FEs of CO2RR products against the applied potential for CuONPs-1.7/GDE in 1 M KOH alkaline electrolyte, and Fig. 2e shows a comparison of the FEs of C2+ products for CuONPs-1.7/GDE in 1 M KCl and 1 M KOH solutions. The FEs for CO2RR activity in alkaline and neutral solutions were almost the same in the range of Jtotal = 200–2400 mA cm−2, which indicates that the local pH around the CL in 1 M KOH and 1 M KCl solutions was not dependent on the bulk pH during electrolysis at Jtotal = 200 mA cm−2. The partial current density for C2+ products in alkaline solutions reached jC2+ = 1.8 A cm−2, which is a record value in alkaline solution, as compared and listed in Fig. 2f and Table S5 (ESI†) respectively, i.e., this electrode can exhibit a record jC2+ in both neutral and alkaline solutions. The mass activity, turnover frequency (TOF) and energy efficiency values are also summarized in Fig. S20 and Table S6 (ESI†) as reference data.
The volcano-type relation between FEC2+and current density was observed in the present UHR-CO2RR system. Especially, the FEC2+ increased with increasing current density below Jtotal = 2000 mA cm−2, and thus, a trade-on relationship between rate and selectivity was exhibited in this current region, as shown in Fig. 2a. Let us discuss the mechanism for the increase of FEC2+ with the current density. It should be noted here that it is difficult to strictly distinguish whether the selectivity toward the product depends on the current density (i.e., reaction rate) or on the applied potential because we cannot control each parameter independently. We here explain the product selectivity from the viewpoint of current density: the effect of (1) CO partial pressure, (2) local pH,42 and (3) in situ formation of the three-phase interface. The CO partial pressure in the CL became high with an increase in the current density, which facilitated CO dimerization. Jun Li et al. controlled local CO availability at the catalyst–electrolyte interface and demonstrated that a high concentration of CO accelerated CO dimerization, and caused a higher current density with C2+ production.43 Jing Li and co-workers reported that a high CO partial pressure suppresses H* adsorption, which leads to lowering of the hydrogen evolution reaction (HER).44 It should also be noted here that the FE for C2H5OH against C2H4 increased with increasing Jtotal (Fig. S21, ESI†). It has been reported that high CO pressure on a Cu surface favours C2H5OH production over C2H4 because the *CHCHOH intermediate for C2H5OH becomes more stable than *CCH for C2H4 under high CO coverage.43,45 This is because CCH* and *CHCHOH are adsorbed on Cu sites with side-on and end-on configurations, respectively, so that CCH* requires a larger occupied molecular area than *CHCHOH. This change in selectivity between C2H4 and C2H5OH supports that the CO local pressure depended on the current density. The increase in the local pH at the surface of the CL due to proton consumption by the CO2RR suppressed HER (vide supra). The pH at the electrode surfaces is known to reach up to 11.9 under electrolysis at just Jtotal = 50 mA cm−2 in neutral electrolytes.15,46 Considering that the almost same product selectivity was obtained in neutral (1 M KCl) and alkaline (1 M KOH) solutions between Jtotal = 200–2400 mA cm−2, the local pH in KCl solution became similar to that in KOH solution under electrolysis with Jtotal of just Jtotal = 200 mA cm−2. Further increase in the current density (Jtotal > 1000 mA cm−2) would lead to a pH of over 15 at the surface of the catalyst.7 Moreover, we propose that the in situ formation of the gas-transport pathway depending on the current density occurs during electrolysis. Evolved gases, such as H2, would migrate through gas-transport channels in the CL; therefore, the area of the CO2RR active interface (three-phase interfaces) could be expanded with an increase in the current density (see Fig. S22, ESI†).
It is important to identify the origin of the decrease in the CO2RR activity by application of Jtotal = 2800 mA cm−2 for CuONPs-1.7/GDE (Fig. 2a and d) toward a further increase in the production rate. It was assumed that the three-phase interface was broken over Jtotal = 2800 mA cm−2, which resulted in a severe decrease in the FE for C2+ products. Fig. 3 shows cross-sectional SEM images after application of Jtotal = 1000, 2000, and 2800 mA cm−2 (30 min) in 1 M KCl, respectively. The particle size of the CuONPs became about 10% smaller after electrolysis due to the reduction of CuONPs to Cu(I)/Cu(0)NPs (Fig. S23, ESI†). Cross-sectional elemental energy dispersive X-ray spectroscopy (EDX) mapping show K+ derived from electrolytes entered the MPL layer. One-tenth and one-fourth of the MPL layers were flooded after application of Jtotal = 1000 mA cm−2 and 2000 mA cm−2, respectively. In contrast, after the degradation with application of Jtotal = 2800 mA cm−2 (30 min), the K+ distribution was shown to reach half of the MPL layer (Fig. 3e). In addition, a residue of K+ crystals (KHCO3 and K2CO3, KCl and KOH) was observed throughout the MPL after application of Jtotal = 2800 mA cm−2 (Fig. S24, ESI†). Electrowetting effects, which decrease the solid–liquid interfacial tension and facilitate electrolyte penetration into the CL and MPL, are known to be emphasized under application of highly negative potentials.47–49 Although the evolved gases may push out and expand the gas-transport pathways (as the in situ formation of the three-phase interface), flooding of the MPL due to electrowetting may exceed the rate of pushing out the electrolyte. The flooded CL (and MPL) produced only H2 due to a lack of CO2 transportation, and thus, H2 production should become dominant at Jtotal = 2800 mA cm−2. This assumption is also supported by the diffusion of evolved gases into the catholyte chamber at Jtotal = 2800 mA cm−2 (vide supra).
 | ||
| Fig. 3 (a, c and e) Cross-sectional SEM images of CuONPs-1.7/GDE and (b, d and f) K+ distribution (purple) obtained by EDX after application of Jtotal = (a and b) 1000 mA cm−2, (c and d) 2000 mA cm−2, and (e and f) 2800 mA cm−2 for 30 min under CO2RR conditions in 1 M KCl. | ||
UHR-CO2RR with varying components and conditions
Here, let us attempt to reveal which factors are sensitive for our UHR-CO2RR by varying several components and conditions.Fig. S25 and Table S7 (ESI†) show the FE of CO2RR products under Jtotal = 2000 mA cm−2 when changing catalysts, the thickness of CL, electrolytes, and GDEs. One can notice that several kinds of commercial GDEs are available for UHR-CO2RR. Additionally, the high jC2+ of over 1.3 A cm−2 was obtained using 1 M KHCO3, which has been widely used as a neutral electrolyte for CO2RR. The slight decrease of jC2+ in 1 M KHCO3 is likely due to an acceleration of salt precipitation in MPL in the bicarbonate-rich electrolytes.
Surprisingly, UHR-CO2RR with jC2+ of over 1.4 A cm−2 was realized even when using commercial CuONPs with the size of 50 nm. These results indicated that the GDE and catalyst used in this study are not unique, and the optimized assembly elicits their undermined potential. Further studies are currently in progress to determine the detailed requirements for catalysts in our laboratory.
Among these parameters, Fig. S25 (ESI†) reveals that the thickness of CL is one high-sensitivity factor in determining maximum jC2+. The optimal thickness of CL was required for UHR-CO2RR, not too thin and not too thick. Thus, we investigated the detail of the dependence of CL thickness. The CO2RR was investigated for CuONPs-3.1/GDE and CuONPs-0.34/GDE (detailed product selectivities are shown in Fig. S26 and S27, ESI†) and the standard CuONPs-1.7/GDE (see the FEs in Fig. 2a) in 1 M KCl. The maximum jC2+ for CuONPs-3.1/GDE was achieved at Jtotal = 2400 mA cm−2, which was the same as that for CuONPs-1.7/GDE. The FEs for C2+ over CuONPs-3.1/GDE were 7% lower than those over CuONPs-1.7/GDE, although there was a similar tendency of the potential dependence (Fig. 4a). The FEH2 over CuONPs-3.1/GDE was higher than that over CuONPs-1.7/GDE for all current density regions (Fig. S28, ESI†). These results indicated that a lack of Cu active sites did not cause a decrease in FEC2+ over Jtotal = 2400 mA cm−2 for CuONPs-1.7/GDE. It was presumed that the Cu sites immersed in the electrolyte cannot serve as CO2RR sites but only as HER sites, and thus, selectivity toward H2 production increased with the amount of Cu (Fig. 4d). Next, the Cu amount on the GDE was decreased to 0.34 mg cm−2. Although the FEC2+ over CuONPs-0.34/GDE was greater than 80% at Jtotal = 1000 mA cm−2, it decreased to below 60% at Jtotal = 1600 mA cm−2. In contrast, when Jtotal was varied from 1000 mA cm−2 to 1600 mA cm−2, the FEs for CH4 and H2 increased from 1.6% to 14%, and from 5.4% to 17%, respectively. This is because the lack of catalytic Cu sites would result in relative enhancement of the HER on carbon particles that compose the MPL. In other words, the three-phase interface formed in CuONPs-0.34/GDE was thinner than that in CuONPs-1.7/GDE, resulting that the current density with the highest FEC2+ was smaller than the optimal for CuONPs-1.7/GDE (Fig. 4b and c). A significant increase in FE for CH4 with an increase in current density was observed on CuONPS-0.34/GDE. This tendency for CH4 was basically consistent with the reported potential dependence of the selectivity of the CO2RR on Cu-based catalysts.50–52 The same trends in the catalytic activity under various amounts of catalyst loadings were observed in 1 M KOH electrolyte (Fig. S29–S31, ESI†).
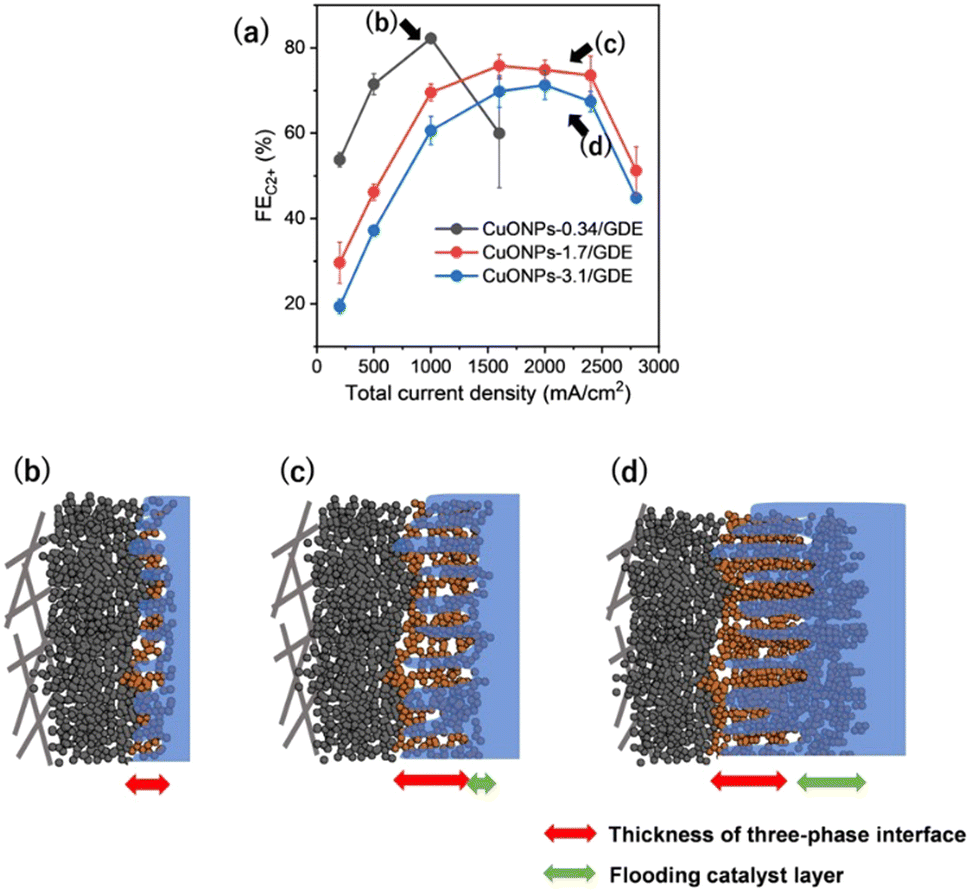 | ||
| Fig. 4 (a) FEC2+ against Jtotal for various amounts of Cu loading (CuONPs-0.34, -1.7 and -3.1/GDE) in 1 M KCl. (b–d) Schematic image of the three-phase interfaces at each position which are indicated in (a). The red and green arrows represent the thickness of three-phase interface and flooding catalyst layer, respectively. | ||
Fig. S32 (ESI†) shows the FEs depending on CL thickness at 500, 1000, 1600 and 2000 mA cm−2, to further demonstrate the importance of CL thickness for maximum jC2+. The thin CL (CuONPs-0.34) showed the best FE for C2+, and FEC2+ monotonically decreased with increasing the thickness of CL at Jtotal = 500 and 1000 mA cm−2. In contrast to FEC2+, the total FE for CO2RR was almost constant (85–90%) at these low current density regions, even when varying CL thickness. This is likely because CO partial pressure was not enough to form C–C bonds for thicker CL at the low current density regions. When increasing current density to Jtotal = 1600 mA cm−2, the FEC2+ for CuONPs-1.7 became the best in these three electrodes. Thus, our results clearly indicated that an optimal CL thickness for C2+ evolution depends on Jtotal.
Although our study is not the first paper to optimize and discuss each parameter, including the size of catalysts, porosity, and thickness of CL, we think that these interdependent parameters were optimized simultaneously, likely resulting in the highest jC2+ in this work. However, we must note that some requirements, especially for catalysts, toward UHR-CO2RR are still veiled. This work is focused on demonstrating the potential of ordinary catalysts; providing more detailed requirements will be left for a future paper.
Long-term stability dependence on current density
Long-term stability might be a challenging issue in the practical implementation of UHR-CO2RR. For example, in ref. 29 the authors reported that the FEC2+ started to decrease within one hour at Jtotal = 1200 mA cm−2 (jC2+ = 1 A cm−2) in 1 M KOH. In this time scale, the decrease in the CO2RR activity on GDE is not due to the chemical degradation of components, but due to the flooding of the CL and MPL. However, there is no systematic knowledge about the long-term stability of UHR-CO2RR; especially, a durability test of UHR-CO2RR in neutral solutions has not been conducted at all. Therefore, we analysed the change in the CO2RR activity for long-term electrolysis under ultra-high-rate electrolysis conditions in neutral solutions.Fig. 5b and Fig. S33 (ESI†) show the FEs for CO2RR products at Jtotal = 1600 mA cm−2 in 1 M KCl as a function of the electrolysis time. The change in the FEs for CO2RR products under electrolysis at Jtotal = 1600 mA cm−2 was almost negligible for over 2 h. Although the FE for C2+ and H2 then began to decrease and increase from 2 h, respectively, the FEC2+ production remained over 75% for 2.5 h and 60% for over 6 h. The electrode was analysed after the decrease of FEs for C2+ to clarify the origin of activity decrease. SEM observation (Fig. S34, ESI†) after electrolysis for 6 h at 1600 mA cm−2 showed that the morphology of CLs did not significantly vary, and most of the MPL was flooded in a similar manner to that described in previous reports.53,54 The recycling test was also conducted. Fig. S35 (ESI†) shows that the FEC2+ value was recovered by washing the electrode with pure water after the decrease. This result also indicates that the decrease in FEC2+ was due not to the degradation of catalysts but to the salt precipitation induced by flooded MPL. The stability dependence on the applied current density was investigated next (Fig. 5a–c and Fig. S33, ESI†). The FEC2+ of 70–80% almost unvaried for over 3 h and 1.5 h at Jtotal = 1000 mA cm−2 and Jtotal = 2000 mA cm−2, respectively. These results indicated a trade-off relationship between stability and current density; however, the decrease in FEC2+ within a short timescale was due to flooding of the CL and MPLs. Therefore, we can expect that the stability could be improved by the addition of hydrophobic materials and modulation of the porosity in the CL and MPL. The present electrochemical system and conditions will become one of the standard platforms toward these future studies.
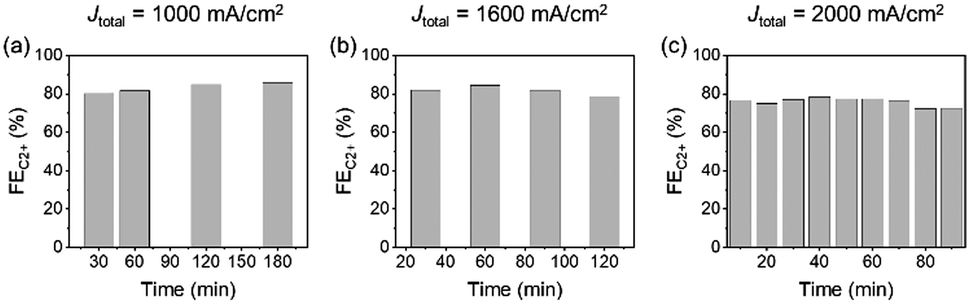 | ||
| Fig. 5 FEC2+ stability tests with respect to the current density over CuONPs-1.0/GDE in 1 M KCl under (a) Jtotal = 1000 mA cm−2, (b) 1600 mA cm−2 and (c) 2000 mA cm−2. | ||
Conclusions
In this work, our electrode exhibited partial current densities for CO2 reduction to C2+ of 1.7 A cm−2 in neutral solutions by optimizing the standard components and their assembly. When the total current density was increased, the FEs for C2+ increased monotonically below Jtotal = 2000 mA cm−2. Therefore, the trade-on relationship between the reaction rate and selectivity was shown in the present UHR-CO2RR under those Jtotal regions. Although the cathode of this electrochemical system was composed of typical CuO-derived Cu nanoparticles, a carbon-based GDE, we successfully maximized the potential of the catalytic activity of the Cu surface by constructing a CL with optimal porosity and thickness. Especially, we revealed that the thickness of CL was directly correlated to jC2+. The optimal thickness of CL was required for UHR-CO2RR. It is noteworthy that this electrode allowed us to achieve UHR-CO2RR even in neutral solutions, which is more practical than alkaline solutions. The present system is expected to become one of the standards when pursuing high-current density for the CO2RR by the design of novel components or assemblies. Although more detailed analyses are needed, our electrodes will teach us essential requirements for UHR electrolysis. Furthermore, the obtained knowledge through the detailed analyses of our electrodes would be widely utilized to develop novel materials, such as catalysts and electrodes because our electrodes are composed of only standard components. Systematic stability tests were also conducted at over jC2+ = 1 A cm−2. Although the stability under ultra-high-current conditions was still far from a practical level, SEM observations revealed that flooding of the CL and MPL is a problem that occurs within the shortest time scale. The electrochemical conditions and systems in this study can serve as a platform for improvement of the stability of the UHR-CO2RR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was based on the Integrated Electrochemical Systems for Scalable CO2 Conversion to Chemical Feedstocks project performed as part of the Moonshot Research and Development Program funded by the New Energy and Industrial Technology Development Organization (grant no. 20001627-0). This work was also supported by a JSPS KAKENHI Program (20H02568) and CREST (grant no. JPMJCR18R3) of the Japan Science and Technology Agency (JST). TEM measurement was carried out by using a facility in the Research Center for Ultra-High Voltage Electron Microscopy, Osaka University, and TEM observation was supported by Dr Takao Sakata. Synchrotron radiation experiments were performed using the BL01B1 beamline of SPring-8 with approval of the Japan Synchrotron Radiation Research Institute (Proposals 2020A1426, 2021A1294, 2021B1204, and 2022A1165). Mr Ryo Kurihara supported the local pH calculation.References
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef PubMed.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- G. Zhang, Z. J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang, G. A. Ozin, T. Wang and J. Gong, Nat. Commun., 2021, 12, 5745 CrossRef CAS PubMed.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- T. H. M. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- C. T. Dinh, T. Burdyny, G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. Pelayo García De Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- C. Chen, X. Yan, Y. Wu, S. Liu, X. Zhang, X. Sun, Q. Zhu, H. Wu and B. Han, Angew. Chem., Int. Ed., 2022, 61, e2022026 Search PubMed.
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936–14944 CrossRef CAS PubMed.
- K. Kamiya, K. Fujii, M. Sugiyama and S. Nakanishi, Chem. Lett., 2021, 50, 166–179 CrossRef CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav.3506 CrossRef PubMed.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- S. Verma, B. Kim, H. R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. A. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef CAS.
- Y. C. Tan, K. B. Lee, H. Song and J. Oh, Joule, 2020, 4, 1104–1120 CrossRef CAS.
- P. P. Yang, X. L. Zhang, F. Y. Gao, Y. R. Zheng, Z. Z. Niu, X. Yu, R. Liu, Z. Z. Wu, S. Qin, L. P. Chi, Y. Duan, T. Ma, X. S. Zheng, J. F. Zhu, H. J. Wang, M. R. Gao and S. H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. García de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O’Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef PubMed.
- T. Möller, T. Ngo Thanh, X. Wang, W. Ju, Z. Jovanov and P. Strasser, Energy Environ. Sci., 2021, 14, 5995–6006 RSC.
- Z. Z. Wu, X. L. Zhang, Z. Z. Niu, F. Y. Gao, P. P. Yang, L. P. Chi, L. Shi, W. S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M. R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS PubMed.
- X. Zhang, J. Li, Y. Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, J. Am. Chem. Soc., 2021, 143, 3245–3255 CrossRef CAS PubMed.
- T. T. Zhuang, Z. Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P. N. Chen, X. L. Zheng, H. Liang, W. N. Ge, B. J. Ye, D. Sinton, S. H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- F. P. García de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 RSC.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- R. Xia, J. J. Lv, X. Ma and F. Jiao, J. Catal., 2021, 398, 185–191 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- D. Cheng, Z.-J. Zhao, G. Zhang, P. Yang, L. Li, H. Gao, S. Liu, X. Chang, S. Chen, T. Wang, G. A. Ozin, Z. Liu and J. Gong, Nat. Commun., 2021, 12, 395 CrossRef CAS PubMed.
- X. Wang, K. Klingan, M. Klingenhof, T. Möller, J. Ferreira de Araújo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- S. Kato, K. Iwase, T. Harada, S. Nakanishi and K. Kamiya, ACS Appl. Mater. Interfaces, 2020, 12, 29376–29382 CrossRef CAS PubMed.
- Y. Wu, K. Iwase, T. Harada, S. Nakanishi and K. Kamiya, ACS Appl. Nano Mater., 2021, 4, 4994–5003 CrossRef CAS.
- A. R. Heenan, J. Hamonnet and A. T. Marshall, ACS Energy Lett., 2022, 7, 2357–2361 CrossRef CAS.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 CrossRef CAS PubMed.
- M. Hussainy and D. W. Agar, Chem. Eng. Technol., 2016, 39, 2135–2141 CrossRef CAS.
- J. Chen and L. Wang, Adv. Mater., 2021, 34, 2103900 CrossRef PubMed.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C. T. Dinh, T. T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- J. Li, K. Chang, H. Zhang, M. He, W. A. Goddard, J. G. Chen, M. J. Cheng and Q. Lu, ACS Catal., 2019, 9, 4709–4718 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D. H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S. F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C. T. Dinh, Y. Wang, T. T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- X. Lu, C. Zhu, Z. Wu, J. Xuan, J. S. Francisco and H. Wang, J. Am. Chem. Soc., 2020, 142, 15438–15444 CrossRef CAS PubMed.
- E. R. Cofell, U. O. Nwabara, S. S. Bhargava, D. E. Henckel and P. J. A. Kenis, ACS Appl. Mater. Interfaces, 2021, 13, 15132–15142 CrossRef CAS PubMed.
- K. Junge Puring, D. Siegmund, J. Timm, F. Möllenbruck, S. Schemme, R. Marschall and U. P. Apfel, Adv. Sustainable Syst., 2021, 5, 2000088 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 33–40 CrossRef CAS.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 CrossRef CAS PubMed.
- Y. Kong, H. Hu, M. Liu, Y. Hou, V. Kolivoška, S. Vesztergom and P. Broekmann, J. Catal., 2022, 408, 1–8 CrossRef CAS.
- Y. Wu, S. Garg, M. Li, M. N. Idros, Z. Li, R. Lin, J. Chen, G. Wang and T. E. Rufford, J. Power Sources, 2022, 522, 230998 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00035k |
| This journal is © The Royal Society of Chemistry 2023 |