
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Seawater-degradable, tough, and fully bio-derived nonwoven polyester fibres reinforced with mechanically defibrated cellulose nanofibres†
Miyu
Yamagata
a,
Yoshiyasu
Nagakawa
 b,
Mizuki
Irie
c,
Shin-ichiro
Suye
acd and
Satoshi
Fujita
*acd
b,
Mizuki
Irie
c,
Shin-ichiro
Suye
acd and
Satoshi
Fujita
*acd
aDepartment of Frontier Fiber Technology and Sciences, Graduate School of Engineering, University of Fukui, 3-9-1, Bunkyo, Fukui 910-8507, Japan
bBiotechnology Group, Tokyo Metropolitan Industrial Technology Research Institute, 2-4-10, Aomi, Koto-ku, Tokyo 135-0064, Japan
cDepartment of Materials Science and Biotechnology, Faculty of Engineering, University of Fukui, 3-9-1, Bunkyo, Fukui 910-8507, Japan
dOrganization for Life Science Advancement Programs, University of Fukui, 3-9-1, Bunkyo, Fukui 910-8507, Japan
First published on 14th November 2022
Abstract
Developing nonwoven fabrics using sustainable materials is necessary to alleviate marine plastic pollution. Poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHBH) has attracted attention because it undergoes natural degradation, particularly in the ocean. Electrospun PHBH nonwoven fabrics have limited applications owing to poor mechanical properties. Although the use of cellulose nanofibres (CNFs) as fillers can potentially result in enhancement of the mechanical properties of PHBH nonwoven fabrics, their low dispersibility in CHCl3, a good solvent for PHBH, hinders the direct electrospinning of CNF-containing PHBH. Herein, nonwoven fibres were fabricated directly by electrospinning Pickering emulsions stabilized with mechanically defibrated CNFs. The CNFs were found to be encapsulated in the core of the PHBH fibres. CNF-reinforced fibres showed excellent toughness, approximately 11 and 22 times greater than those of PHBH films and CNF-free PHBH fibres, respectively. These also underwent complete and very rapid degradation in seawater (7–10 d), compared to the PHBH film (14–40 d). The proposed CNF-reinforced PHBH is a seawater-degradable bioplastic containing complete biomass-derived components.
Environmental significanceNonwoven fabrics widely used in various industries are mainly made of synthetic polymers, which cause marine pollution by breaking down to microplastics. Seawater-degradable nonwoven fabrics are highly desired but have not yet been put to practical use. Herein, PHBH, which has seawater-degradability but low toughness, was used in the manufacture of nonwoven nanofibrous fabrics by blending with CNFs. Electrospinning via Pickering emulsion yielded CNF-containing PHBH fibres, which exhibited excellent toughness despite using only a small amount of CNF. This entirely biomass-derived, seawater-degradable bioplastic nonwoven fabric would be a “green” alternative to conventional polyester nonwoven fabrics. This approach would also be a promising method to fabricate CNF-hybrid plastics. |
1. Introduction
In response to COVID-19, nonwoven fabrics have emerged as an expanding field in textile products for various applications including clothing, medical devices, hygiene products, and other industries.1 Most nonwoven products are made of petrochemical-based thermoplastics, such as polyethylene terephthalate, polypropylene, and polyethylene, which are non-biodegradable.2 Such disposable nonwoven products with a relatively short service life can cause serious environmental issues. For example, discarded surgical masks with a release rate of 396 billion microplastics per day contribute to marine microplastic pollution.1 The global inclination toward sustainability has driven a strong interest in the use of biodegradable renewable bio-based materials for nonwoven fabrics.1–3 Polyhydroxyalkanoates, which are polyesters produced by microorganisms, have attracted attention as a potential solution to these issues. In particular, poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) (PHBH) is expected to mitigate marine microplastic pollution efficiently because it is completely converted into CO2 and water upon enzymatic degradation by microorganisms in the ocean.4–7 PHBH has been extensively used to fabricate plastic bags and disposable straws,8 and has been investigated for fiberization into micrometre-sized fibres using melt-spun blowing.9,10 Electrospinning, an efficient method for obtaining nonwoven fabrics with nanometre-sized fibres, can also be applied to PHBH, but electrospun nonwoven PHBH fibres exhibit poor mechanical properties.11–13 Fillers such as nanosilica,14 montmorillonite,15 carbon nanotubes,16 and glass fibres17 have been used to enhance the mechanical characteristics of PHBH. However, challenges remain in the use of fillers for sustainable biodegradable polymers.Cellulose nanofibres (CNFs) are prepared by finely disassembling plant fibres, such as pulp and wood chips, into nanosized fibres. As these are bio-based biodegradable materials, CNFs are attracting attention as a next-generation renewable organic resource that can facilitate the transition toward a sustainable society with minimal environmental impact.18 CNFs have also received considerable attention as alternative fillers to carbon and glass fibres because of their high strength, lightweight, and low cost.19–21 CNFs do not cause any environmental problems even if they are released into the ocean, which demonstrates their potential to be used as an ideal filler for seawater-degradable plastics. Our group has found that electrospun nanofibres can be strengthened via the addition of CNFs.22 CNF-reinforced composite PHBH nonwoven fabrics are expected to be extensively applied as novel, completely bio-based, and seawater-degradable materials. However, as CNFs are hydrophilic and polymers including PHBH are usually hydrophobic, the low dispersibility of CNFs in polymers poses a challenge in their use as a filler. CNF surfaces have been chemically treated to disperse CNFs as fillers in polymers. The dispersibility of CNFs can be improved by introducing hydrophobic functional groups, such as 3-isocyanatopropyl triethoxysilane23 and benzoic acid anhydride,24 and ionic functional groups, such as tannic acid25 and epoxypropyltrimethylammonium chloride.26 However, a simple chemical-treatment-free method for blending CNFs with polymers is still desired to avoid the routinely adopted chemical treatments of CNFs in these investigations. Here, Pickering emulsions containing CNFs were used to achieve a thorough blend of CNF with PHBH. Pickering emulsions, which are stabilized by the adsorption of solid particles on the interface between the two phases in the emulsion,27 were targeted in the present study. Owing to their amphiphilicity, mechanically defibrated CNFs can stabilize oil-in-water (o/w) emulsions by forming a network-like fibre structure on the interface.28,29 It has previously been demonstrated that simple casting of Pickering emulsions does not result in sufficient mechanical strength because of the agglomeration of CNFs.30–32 To the best of our knowledge, electrospinning of PHBH incorporated CNF has never been reported. This study focused on the electrospinning of Pickering emulsion to prepare CNF-incorporated fibres.
It is expected that the direct electrospinning of Pickering emulsions containing hydrophobic polymers and mechanically defibrated untreated CNFs can yield CNF-containing nonwoven fabrics via a simple manufacturing process. In this study, CNF-reinforced PHBH-nanofibre-based nonwoven fibres were fabricated using Pickering emulsions, and their superior mechanical properties and seawater degradability were demonstrated. These results highlight the potential of PHBH as a sustainable nonwoven material.
2. Experimental
2.1. Preparation of samples
PHBH powder (containing 10.6 mol% poly-3-hydroxyhexanoate; Kaneka, Osaka, Japan) was dissolved in CHCl3 (Fujifilm Wako, Osaka, Japan) and stirred overnight to prepare a 20% PHBH solution. To investigate the effects of the length and concentration of CNFs on the mechanical properties, long and short CNFs (Ima-10005 and Fma-10005, respectively; BiNFi-s; Sugino Machine, Toyama, Japan) were suspended in water, vortexed, and sonicated (40 kHz, 110 W, 10 min) to prepare 1.0%, 2.0%, 3.0%, and 4.0% CNF suspensions. The PHBH/CHCl3 solution and CNF/water suspensions were mixed at a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Three cycles of vortexing (30 s) and sonication (5 min) were executed to prepare the Pickering emulsions. Table S1† presents the compositions of the prepared emulsions and fabricated fibres. The S- and L-prefixes in the sample notations indicate short and long CNFs, respectively, and the numbers represent the final composition of the CNFs in the synthesized materials. For example, the L-6.3 fibres contain 6.3% long CNFs dispersed in the PHBH matrix.
:1. Three cycles of vortexing (30 s) and sonication (5 min) were executed to prepare the Pickering emulsions. Table S1† presents the compositions of the prepared emulsions and fabricated fibres. The S- and L-prefixes in the sample notations indicate short and long CNFs, respectively, and the numbers represent the final composition of the CNFs in the synthesized materials. For example, the L-6.3 fibres contain 6.3% long CNFs dispersed in the PHBH matrix.
Fibres were obtained by electrospinning (NANON; MECC, Fukuoka, Japan), which involves spraying of a polymer solution through application of a high voltage to a grounded collector.11,12 Electrospinning was performed by injecting the emulsions through a 22G spinneret at a flow rate of 0.4 mL h−1 and an electric field of 1.25 kV cm−1, which yielded fibre sheets on a drum collector rotating at a linear velocity of 3.1 or 12.6 m s−1. For comparison, cast films were prepared by spreading the emulsion over a φ 100 mm glass dish and drying overnight at 25 °C.
2.2. Characterization of fabricated materials
| S = 2〈cos2θ〉 − 1 = 〈cos2θ〉 | (1) |
θ, where θ is the scattering angle, was calibrated using silver behenate. One-dimensional (1D) profiles were acquired by sector-averaging the 2D SAXS patterns within a sector angle of 45° with the meridional axis. Structures with a long period (2π/q) were evaluated according to Bragg's law, where q is the value of the peak (qmax) in the q2I(q) − q plot.33
2.3. Evaluation of degradability
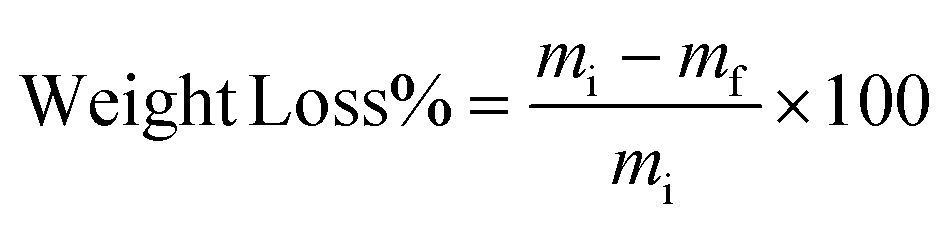 | (2) |
3. Results and discussion
3.1. Fabrication of CNF-incorporated PHBH fibres
The PHBH/CHCl3 solutions and CNF/water suspensions were homogenized at a volume ratio of 3:1 to prepare Pickering emulsions. CNF-containing nonwoven fibres were obtained by electrospinning each emulsion onto a rotating collector (Fig. 1a). Mechanically defibrated CNFs with two different lengths were used. The shorter CNFs with an average length (estimated from microscopic observation of dispersion) of 722 ± 370 nm (Fig. S1b†) stood straight (Fig. 1b), whereas the longer CNFs, with an average length of 54 ± 26 μm (Fig. S1a†), were sufficiently long and flexible to entangle with each other (Fig. 1c). The small peak in the L-6.3 emulsion (Fig. S1a†) can be attributed to the presence of a small fragment of CNF in long CNF because a similar peak appears in a dilute suspension of long CNF. Well-stabilized emulsions were obtained for all the examined compositions (Table S1†). SEM analysis of the lyophilized cloudy L-6.3 emulsion revealed that the particle surfaces were covered with a fibrous structure (Fig. 1a). This observation suggests that the continuous layer of the emulsion was aqueous and that the particles were o/w emulsions consisting of a PHBH/CHCl3 phase surrounded by CNFs. Dynamic light scattering (DLS) analysis indicated that the L-6.3 and S-6.3 emulsions had wide diameter distributions, with averages of approximately 1160 and 3410 nm, respectively (Fig. S2†). Mechanically defibrated CNFs are amphiphilic and their hydrophilicity is governed by the degree of mechanical fibrillation.34 Since the formation of Pickering emulsion is affected by the hydrophilicity of CNF, the emulsion size would be affected by the CNF length because it contributed to the different hydrophilicity of CNF.
 | ||
Fig. 1 (a) Preparation of CNF-containing PHBH fibres. A CNF/water suspension and PHBH/CHCl3 solution are homogenized to form a Pickering emulsion, in which particles are enveloped by CNFs. Images of the L-6.3 emulsion are shown. Highly aligned CNF-containing PHBH fibres are obtained via direct electrospinning of the emulsion onto a rotating collector. (b–g) Morphological analysis. SEM images of (b and e) CNFs and (c and f) electrospun aligned sheets of CNF-containing fibres collected at a high speed (12.6 m s−1). (d and g) TEM image of a single CNF-containing fibre. (b–d) and (e–g) show short and long CNFs, respectively. (h) WCAs of CNF cast film and the S-6.3, L-6.3, and CNF-free fibre sheets. (i) ATR-FTIR spectra of PHBH, S-6.3 fibres, L-6.3 fibres, and CNFs. (j) Enlarged region highlighting the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O-derived peaks. O-derived peaks. | ||
Fibre sheets were prepared by electrospinning the emulsions. Highly oriented fibres with a smooth surface and no beading were obtained by rotating the collector at a high speed (12.6 m s−1) (Fig. 1c and f). Regardless of the length and concentration of the added CNFs, the average diameter obtained was ∼1.2 μm and the second-order parameter, S, was always greater than 0.8, indicating that the electrospun fibres were well-oriented (Fig. S3†). Thinner fibres were obtained by stretching during electrospinning onto the high-speed rotating collector. Electrospinning performed by rotating the collector at a low speed (3.1 m s−1) yielded randomly oriented fibres (S = 0.257) with smooth surfaces and minimal beading (Fig. S4†). The average diameter of these randomly oriented fibres (∼1.7 μm) was greater than that of the oriented fibres. The sizes of the emulsions and the diameters of the prepared fibres did not appear to be directly related, which was expected because the emulsions collapsed in the Taylor cones during electrospinning.
3.2. Characterization of electrospun fibres
To directly observe the internal structure of the fibres, these were frozen in liquid nitrogen and then sectioned. The cross sections were observed using SEM. A spike-like structure was observed in the core of L-6.3 fibres (Fig. S6a†), indicating that the L-6.3 fibres contained CNF in their cores, whereas the CNF-free fibres observed for comparison were smooth and homogeneous (Fig. S6b†).
O str.)38 red-shifted upon the addition of CNFs (Fig. 1j), suggesting that CO could be involved in the hydrogen bonding between PHBH and the CNFs.
 | ||
| Fig. 2 (a–e) 2D SAXS profiles. (a) Aligned CNF-free fibres, (b) aligned L-6.3 fibres, (c) CNF-free film, (d) L-6.3 film, and (e) CNF cast film. Arrows indicate the direction of fibre alignment. (f–h) 1D SAXS profiles. (f) CNF-free PHBH fibres, (g) CNF-containing aligned fibres, and (h) films with and without CNFs. | ||
 | ||
| Fig. 3 (a) Stress–strain curves and (b) toughness data of aligned L-6.3 and S-6.3 fibres, CNF-free fibres, and L-6.3-based and CNF-free films (brown, green, yellow, blue, and cyan profiles, respectively). Asterisks represent a significant difference (p < 0.05). | ||
The fabricated films were also analysed for comparison. The elastic modulus, tensile strength, and toughness of the pristine film decreased from 29.1 ± 13.1 to 5.8 ± 1.7 MPa, 3.5 ± 0.9 to 0.6 ± 0.4 MPa, and 0.4 ± 0.1 to 0.03 ± 0.02 MPa, respectively, upon the addition of CNFs. This behaviour could be due to the nonuniform aggregation of CNFs during film fabrication, which contributes to breakage in the thin PHBH region of the film. In contrast, the CNFs did not locally aggregate inside the electrospun fibres, which instead displayed a continuous core–shell-like structure that presumably enabled their high toughness. The effects of the CNF length and concentration as well as the orientation of the fibre sheet were also examined (Fig. S8 and S9†).
The stress–strain curves indicate that the stress remained constant, whereas the strain increased after the yield point, without any grip slippage or fibre breakage. These results were obtained presumably because of the presence of a high-density CNF core inside the fibres, entangled CNFs that hindered fibre breakage, and a CNF core that stretched as the fibre elongated. The intense interfacial interactions between the CNFs and PHBH might also have contributed to the toughness.
A SEM image of the fracture surface of S-6.3 fibre after tensile testing shows that no CNF protrusions were observed in the cross-section image of the fracture surface of S-6.3 fibre (Fig. S6c†). This indicates that the rupture during elongation may have occurred in the PHBH rather than the CNF section.
3.3. Evaluation of seawater degradability
 | ||
| Fig. 4 Seawater degradation tests of fibre sheets and films with/without CNFs: (a and b) in situ (pump room) and (c and d) in vitro experiments. (a and c) and (b and d) show apparent morphology and weight loss data of the specimens, respectively. | ||
A similar experiment conducted in a water tank, which was not influenced by tides, mud, sand, and seaweed (Fig. S10b†), showed similar results but with increased degradation (Fig. S11†). This observation suggests that degradation was not induced by physical influences such as waves and sand but by microbes. Degradation was promoted in a fresh-seawater-based environment suitable for aerobic microorganisms, and differences in the microbial growth environments affected the degradation rates. The reason for the faster degradation of the fibre in comparison to the film would be the larger specific surface area of the fibre, which allowed more microorganisms to adhere.43,44
 | ||
| Fig. 5 SEM images of specimens before and after degradation tests (scale bar = 5 μm). | ||
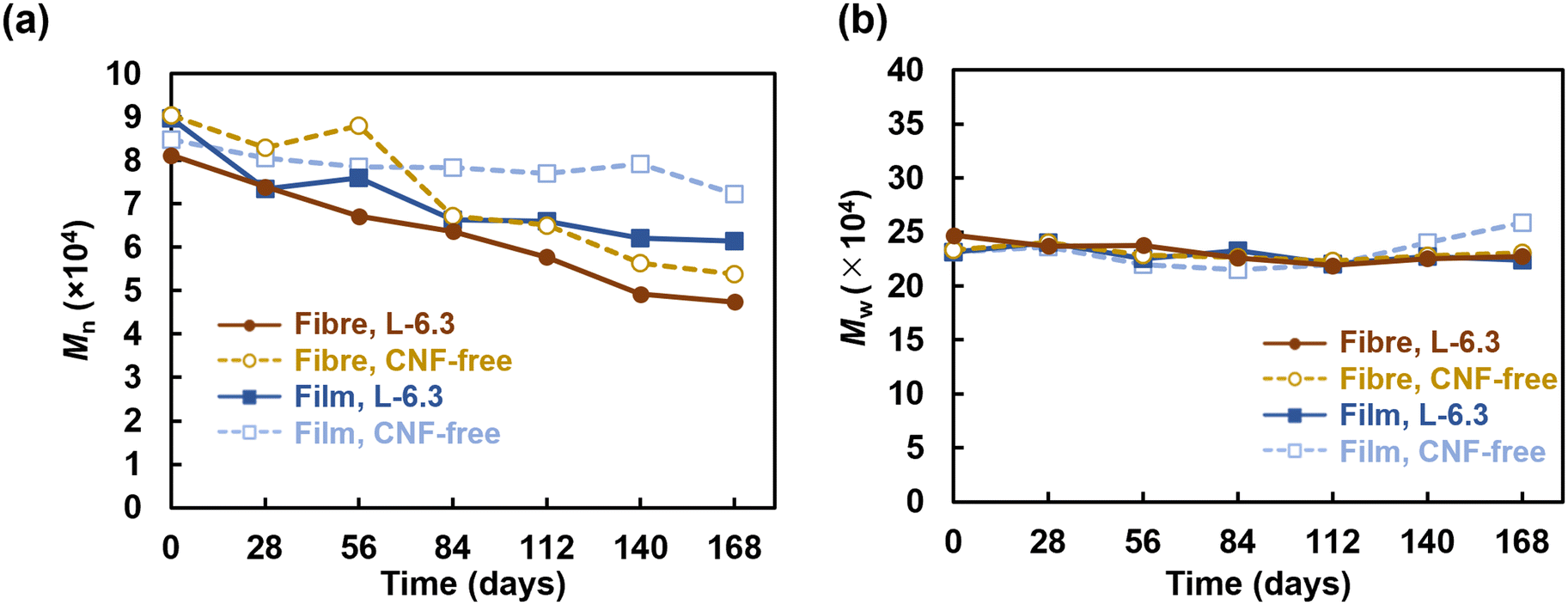 | ||
| Fig. 6 GPC analysis results: (a) number-average molecular weight, Mn, (b) weight-average molecular weight, Mw, and (c) polydispersity index, Mw/Mn. | ||
 | ||
| Fig. 7 (a) Crystal violet staining of specimens before and after degradation tests. SEM images of degraded aligned L-6.3 fibres after (b) in situ immersion (pump room) for 7 d, (c) in situ immersion (water tank) for 7 d, and (d) in vitro immersion for 112 d. | ||
Regarding biodegradability, PHBH has been reported to be degraded by Alteromonadaceae in seawater.48 Volant et al. reported that PHBH microbeads enclosed in a container with seawater and sand degraded by 80% in 250 d.49 Sashiwa et al. found that PHBH powder degraded by 31% in 28 d.5 The film fabricated in the present study degraded by 50% in 14–40 d in a natural environment, which is comparable to the degradation behaviour observed in previous studies.5 In contrast, more than 50% of the fibre sheets degraded in 7–10 d and almost completely degraded within ∼2 weeks. The high degradability of the fibres can be explained by the enhancement of hydrolysis owing to their larger specific surface area.50 Microbial biofilms did not readily form on the surface of PHBH film because of its hydrophobicity (Fig. 7); however, the fibres provided a porous microenvironment suitable for bacterial adhesion and growth, resulting in high biodegradation. In situ degradation was remarkably faster than its in vitro equivalent. The influences of water currents and mud were considered. However, rapid degradation was also observed in the water tank without these influences, suggesting that microbial growth was promoted in the environment where seawater was constantly supplied. The in vitro degradation tests showed that neither the fibre sheets nor the films degraded significantly after approximately 1 month of immersion; however, rapid degradation was realized after 3 months of immersion. This observation is consistent with the exponential increase in the microorganism content. The degradation of the CNF-containing fibres was accelerated, particularly during the later period. This behaviour could be due to the encapsulation of the CNFs in the fibres during the early degradation stage. However, certain CNFs disintegrated as degradation progressed, and the microorganisms could readily penetrate the voids created by the loss of these CNFs, thereby accelerating degradation in both the exterior and interior parts of the fibres in the later period. The formation of microbial flora on the fibre sheets would enable the sheets to be used as a platform for the culture of polymer-degrading bacteria, thus facilitating investigations into the bacteria and enzymes involved in degradation.
4. Conclusions
CNF-containing PHBH nonwoven fibres exhibited a higher elastic modulus, tensile strength, and uniform elongation than the PHBH fibres and CNF-free film. In particular, the oriented L-6.3 fibres showed high toughness, which was 11-times and 22-times greater than that of the PHBH films and CNF-free fibres, respectively. These properties can be attributed to the densely entangled CNF core within the fibres and the hydrogen bonds at the CNF–PHBH interface. The CNF-containing fibres were degraded by microorganisms in seawater within 7–10 d, which was much more rapid than the degradation of the PHBH films and CNF-free fibres. The chemical treatment of CNF surfaces is a common approach for dispersing CNFs in hydrophobic polymer matrices; however, the electrospinning of Pickering emulsions is a simpler method for mass production because mechanically defibrated CNFs can be directly used. Moreover, the CNF-reinforced nonwoven PHBH fibres are a fully bio-derived plastic, that is, born from and returned to nature, because both the CNFs and PHBH are derived from biomass and are biodegradable. This material can potentially contribute to breaking away from a petroleum-based society and solving marine plastic pollution.Author contributions
Miyu Yamagata: data curation, methodology, writing–original draft, and visualization. Yoshiyasu Nagakawa: data curation, methodology, and investigation. Shin-ichiro Suye: writing–reviewing and editing. Satoshi Fujita: conceptualization, methodology, investigation, validation, writing–original draft, reviewing, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a JSPS KAKENHI grant (No. 21K04686) and research grants from University of Fukui (FY 2021). The authors are grateful to Dr. Shunsuke Sato for fruitful discussions; Mr. Takashi Suzuki and Dr. Seiji Sasai (Echizen Matsushima Aquarium) for their cooperation with the degradation tests; and Editage (https://www.editage.jp) for English proofing.References
- J. Sun, S. Yang, G.-J. Zhou, K. Zhang, Y. Lu, Q. Jin, P. K. S. Lam, K. M. Y. Leung and Y. He, Environ. Sci. Technol. Lett., 2021, 8, 1065–1070 CrossRef CAS.
- G. Bhat and D. V. Parikh, in Applications of Nonwovens in Technical Textiles, Elsevier, 2010, pp. 46–62 Search PubMed.
- A. S. Santos, P. J. T. Ferreira and T. Maloney, Cellulose, 2021, 28, 8939–8969 CrossRef CAS.
- Y. Doi, S. Kitamura and H. Abe, Macromolecules, 1995, 28, 4822–4828 CrossRef CAS.
- H. Sashiwa, R. Fukuda, T. Okura, S. Sato and A. Nakayama, Mar. Drugs, 2018, 16, 34 CrossRef PubMed.
- S. Wang, K. A. Lydon, E. M. White, J. B. Grubbs, E. K. Lipp, J. Locklin and J. R. Jambeck, Environ. Sci. Technol., 2018, 52, 5700–5709 CrossRef CAS PubMed.
- C. Kato, A. Honma, S. Sato, T. Okura, R. Fukuda and Y. Nogi, High Pressure Res., 2019, 39, 248–257 CrossRef CAS.
- E. Amasawa, T. Yamanishi, J. Nakatani, M. Hirao and S. Sato, Environ. Sci. Technol., 2021, 55, 3380–3388 CrossRef CAS PubMed.
- T. Kabe, C. Hongo, T. Tanaka, T. Hikima, M. Takata and T. Iwata, J. Appl. Polym. Sci., 2015, 132, 41258 CrossRef.
- F. Selli, R. Hufenus, A. Gooneie, U. H. Erdoğan and E. Perret, Polymers, 2022, 14, 200 CrossRef CAS PubMed.
- W.-Y. Huang, S. Suye and S. Fujita, ACS Appl. Bio Mater., 2021, 4, 7456–7466 CrossRef CAS PubMed.
- W.-Y. Huang, N. Hashimoto, R. Kitai, S. Suye and S. Fujita, Polymers, 2021, 13, 2273 CrossRef CAS PubMed.
- A. M. Díez-Pascual, Polymers, 2021, 13, 2233 CrossRef.
- D. Li, J. Fu and X. Ma, Polym. Compos., 2020, 41, 381–390 CrossRef CAS.
- J. Dong, W. Zhou, Y. Su and X. Ma, Polym. Compos., 2020, 41, 4538–4549 CrossRef CAS.
- L.-P. Wu, M. You, D. Wang, G. Peng, Z. Wang and G.-Q. Chen, Polym. Chem., 2013, 4, 4490 RSC.
- W. Arifin and T. Kuboki, Polym. Compos., 2018, 39, 491–503 CrossRef CAS.
- Q.-F. Guan, H.-B. Yang, Z.-M. Han, L.-C. Zhou, Y.-B. Zhu, Z.-C. Ling, H.-B. Jiang, P.-F. Wang, T. Ma, H.-A. Wu and S.-H. Yu, Sci. Adv., 2020, 6, eaaz1114 CrossRef PubMed.
- A. Iwatake, M. Nogi and H. Yano, Compos. Sci. Technol., 2008, 68, 2103–2106 CrossRef CAS.
- K. Benhamou, H. Kaddami, A. Magnin, A. Dufresne and A. Ahmad, Carbohydr. Polym., 2015, 122, 202–211 CrossRef CAS.
- K.-Y. Lee, Y. Aitomäki, L. A. Berglund, K. Oksman and A. Bismarck, Compos. Sci. Technol., 2014, 105, 15–27 CrossRef CAS.
- M. Yamagata, H. Uematsu, Y. Maeda, S. Suye and S. Fujita, J. Fiber Sci. Technol., 2021, 77, 223–230 CrossRef.
- A. Anžlovar, A. Krajnc and E. Žagar, Cellulose, 2020, 27, 5785–5800 CrossRef.
- A. Anžlovar, I. Švab, A. Krajnc and E. Žagar, Cellulose, 2021, 28, 7813–7827 CrossRef.
- S. Shrestha, R. A. Chowdhury, M. D. Toomey, D. Betancourt, F. Montes and J. P. Youngblood, Cellulose, 2019, 26, 9631–9643 CrossRef CAS.
- M. Rincón-Iglesias, E. Lizundia, D. M. Correia, C. M. Costa and S. Lanceros-Méndez, Cellulose, 2020, 27, 3821–3834 CrossRef.
- J. Wu and G.-H. Ma, Small, 2016, 12, 4633–4648 CrossRef CAS PubMed.
- J. L. Sanchez-Salvador, A. Balea, M. C. Monte, A. Blanco and C. Negro, Appl. Sci., 2019, 9, 359 CrossRef CAS.
- F. Cherhal, F. Cousin and I. Capron, Biomacromolecules, 2016, 17, 496–502 CrossRef CAS PubMed.
- X. Liu, X. Qi, Y. Guan, Y. He, S. Li, H. Liu, L. Zhou, C. Wei, C. Yu and Y. Chen, Carbohydr. Polym., 2019, 224, 115202 CrossRef.
- Y. He, X. Liu, X. Qi, Y. Guan, S. Li, H. Liu, L. Zhou, C. Wei and C. Yu, Polym. Adv. Technol., 2020, 31, 2676–2686 CrossRef CAS.
- Y. Zhang, L. Cui, H. Xu, X. Feng, B. Wang, B. Pukánszky, Z. Mao and X. Sui, Int. J. Biol. Macromol., 2019, 137, 197–204 CrossRef CAS.
- Y. Nagakawa, S. Fujita, S. Yunoki, T. Tsuchiya, S. Suye, K. Kinoshita, M. Sasaki and T. Itoi, J. Mater. Chem. B, 2022, 10, 4375–4385 RSC.
- T. Tsuji, K. Tsuboi, S. Yokota, S. Tagawa and T. Kondo, Biomacromolecules, 2021, 22, 620–628 CrossRef CAS.
- C. Xiong, Z. Quan, H. Zhang, L. Wang, X. Qin, R. Wang and J. Yu, Appl. Surf. Sci., 2020, 532, 147400 CrossRef CAS.
- Y. Wakuda, S. Nishimoto, S. Suye and S. Fujita, Sci. Rep., 2018, 8, 6248 CrossRef.
- S. Fujita, Y. Wakuda, M. Matsumura and S. Suye, J. Mater. Chem. B, 2019, 7, 6556–6563 RSC.
- R. A. Rebia, N. S. B. Sadon and T. Tanaka, Nanomaterials, 2019, 9, 1665 CrossRef CAS.
- B. Hammouda and D. L. Ho, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 2196–2200 CrossRef CAS.
- A. K. Maurya, L. Weidenbacher, F. Spano, G. Fortunato, R. M. Rossi, M. Frenz, A. Dommann, A. Neels and A. Sadeghpour, Nanoscale, 2019, 11, 7176–7187 RSC.
- L. Wang, D. J. Gardner and D. W. Bousfield, Polym. Eng. Sci., 2018, 58, 793–801 CrossRef CAS.
- W. Leng, J. Li and Z. Cai, Polymers, 2017, 9, 597 CrossRef PubMed.
- L. G. Greca, M. Rafiee, A. Karakoç, J. Lehtonen, B. D. Mattos, B. L. Tardy and O. J. Rojas, ACS Nano, 2020, 14, 12929–12937 CrossRef PubMed.
- P. Hughes, S. Fujita, T. Satomura and S. Suye, Mater. Lett., 2012, 72, 88–91 CrossRef CAS.
- R. Arjmandi, A. Hassan, M. K. M. Haafiz and Z. Zakaria, in Natural Fiber-Reinforced Biodegradable and Bioresorbable Polymer Composites, Elsevier, 2017, pp. 111–136 Search PubMed.
- F. Li, X. Xu, J. Yu and A. Cao, Polym. Degrad. Stab., 2007, 92, 1053–1060 CrossRef CAS.
- I. Taniguchi, S. Nakano, T. Nakamura, A. El-Salmawy, M. Miyamoto and Y. Kimura, Macromol. Biosci., 2002, 2, 447–455 CrossRef CAS.
- T. Morohoshi, K. Ogata, T. Okura and S. Sato, Microbes Environ., 2018, 33, 19–25 CrossRef PubMed.
- C. Volant, E. Balnois, G. Vignaud, A. Magueresse and S. Bruzaud, J. Polym. Environ., 2022, 30, 2254–2269 CrossRef CAS.
- W.-Y. Huang, T. Hibino, S. Suye and S. Fujita, RSC Adv., 2021, 11, 5703–5711 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2en00441k |
| This journal is © The Royal Society of Chemistry 2023 |