
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic photo-battery with high operating voltage using a multi-junction organic solar cell and an organic redox-polymer-based battery†
Rodrigo
Delgado Andrés‡
 af,
Robin
Wessling‡
abcf,
Jan
Büttner
ad,
Leonie
Pap
e,
Anna
Fischer
adf,
Birgit
Esser
*abcf and
Uli
Würfel
*aef
af,
Robin
Wessling‡
abcf,
Jan
Büttner
ad,
Leonie
Pap
e,
Anna
Fischer
adf,
Birgit
Esser
*abcf and
Uli
Würfel
*aef
aCluster of Excellence livMatS @ FIT – Freiburg Center for Interactive Materials and Bioinspired Technologies, Freiburg Center for Interactive Materials and Bioinspired Technologies (FIT), University of Freiburg, Georges-Köhler-Allee 105, 79110 Freiburg, Germany. E-mail: birgit.esser@uni-ulm.de; uli.wuerfel@fmf.uni-freiburg.de
bInstitute of Organic Chemistry II and Advanced Materials, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany. E-mail: birgit.esser@uni-ulm.de; Web: https://www.esserlab.com
cInstitute for Organic Chemistry and Biochemistry, University of Freiburg, Albertstr. 21, 79104 Freiburg, Germany
dInstitute for Inorganic and Analytical Chemistry, University of Freiburg, Albertstr. 21, 79104 Freiburg, Germany
eFraunhofer Institute for Solar Energy Systems (ISE), Heidenhofstr. 2, 79110 Freiburg, Germany. E-mail: uli.wuerfel@ise.fraunhofer.de
fFreiburg Materials Research Center (FMF), University of Freiburg, Stefan-Meier-Str. 21, 79104 Freiburg, Germany
First published on 21st September 2023
Abstract
The demand for small energy devices capable of powering consumer electronics in the frame of the Internet of Things is ever increasing. The constraints placed on the size of these devices requires the design of power sources in the most compact way, for which a viable solution is the combination of energy harvesting and storage in one power pack. Herein, we report on a fully integrated monolithic organic photo-battery, consisting of an organic polymer-based battery, powered by a multi-junction organic solar cell capable of charging up to voltages as high as 4.2 V under varying illumination conditions. The full device demonstrated fast photo-charging within minutes and supplied on-demand discharge capacities of up to 22 mA h g−1 (regarding battery active material) with average discharge potentials of 3.6 V vs. Li/Li+. The combined device constitutes the first monolithically integrated photo-battery made from organic building blocks capable of reaching competitive voltages sufficiently high for use in small, mobile power devices.
Broader contextOne of the simplest strategies to address the increasing energy consumption and combat climate change is enhancing the energy efficiency of consumer goods and buildings. This approach can be readily implemented through real-time monitoring of various influential factors and stands as a fundamental aspect of the Internet of Things (IoT). Achieving this requires the autonomous operation of small electronic devices, like sensors and actuators, which rely on a dependable energy source, ideally, independent of the grid. As the number of connected devices is projected to increase significantly, the practicality of replacing or recharging batteries diminishes rapidly. Consequently, an alternative solution gaining attention involves integrated devices, capable of both harnessing energy from surrounding sources and storing it. For harvesting, solar energy is well suited for a wide range of situations, while the storage can be generally realized through the utilization of a battery. Ensuring no critical materials are used in the fabrication to increase their sustainability and limit the cost of such devices might be critical for their widespread adoption. In this context, organic functional materials are becoming a focus of intense research. Here, we present a fully integrated monolithic organic photo-battery, consisting of an organic polymer-based battery that is powered by a multi-junction organic solar cell. This unique design enables the battery to charge under varying illumination conditions and release stored energy when needed. |
Introduction
The worldwide increase in energy consumption and the associated carbon emissions present a major challenge and therefore call for the development of new and cleaner energy harvesting and storage devices.1 These ideally rely on renewable energy sources and have a minimized carbon footprint.2 Intense efforts have been and are currently directed to develop new, better performing and more sustainable devices that can both harvest and store electrical energy. The most straightforward approach is a grid-based combination of physically and geographically separate harvesting and storage units. Apart from this method, there is also a growing interest in interconnected systems that can combine energy conversion from their surrounding and storage of the resulting electrical energy. The vast majority of this type of devices rely on a wired connection of the harvesting and storage unit which are controlled and compatibilized using electronics.3,4 As such combined devices are mainly relevant for low-power applications, such as the so-called Internet of Things (IoT),5 there are strict inherent limitations for size. This creates a specific demand for a maximized integration of the harvesting and storage unit into a single, autonomous device.Among the renewable energy sources available, photovoltaics are best suited for harvesting energy in many scenarios combined with a compact design without any moving parts. Some examples show storage of solar energy in systems, but with designs that make integration and usability by small electronic devices complex, such as chemical discharge,6 photothermal applications,7 beaker-cell based systems8,9 or other proof-of-concept reports based on new materials.10,11 Furthermore, a number of solar flow batteries have been reported, but the addition of electrolyte reservoirs make them incompatible with the size constrains.12,13 Therefore, the best alternatives for the energy storage part of such a device are either (super)capacitors or batteries. Some attempts have already been made to combine solar cells with these storage devices in monolithic and compact device architectures,14–20 and even to identify materials with the ability to perform both tasks.21–24 While capacitors can deliver stored energy with high power, batteries are capable of providing higher energy densities at more constant voltages, which makes them the better option for the operation of small electronic devices. Thus, the ideal, fully integrated device combines solar cell-based energy harvesting with battery-based energy storage. This will be referred to as “photo-battery” (PB) in the following.
However, most solar cells deployed in this regard are single-junction cells and therefore usually have a limited voltage (<1 V).14,15,18,25,26 Hence they are unable to charge most battery types, as higher voltages are required, and can only assist in the charging process (“photo-assisted charging”), thusrequiring an additional power supply.27–36 While some systems have been reported that circumvent this limitation by using tandem or multi-junction solar cells,16,37 the voltages are still insufficient in most cases, and batteries with a lower voltage (up to 1.6 V) must be selected,16 thereby decreasing the energy density of the device.
Due to their high customizability and sustainability, organic materials represent an ideal basis for the design of PBs. In the field of photovoltaics, organic solar cells (OSCs) have received much attention due to their low cost, light weight, mechanical flexibility and possibility for roll-to-roll fabrication.38,39 The power conversion efficiency of OSCs has significantly improved over the last years through the synthesis of new materials, with values now exceeding 19%.40,41 Further, their application is expanding to low light intensity or indoor illumination conditions.42 Similarly, the development of batteries based on organic materials has seen intense efforts and shows immense progress, especially in comparison to their more common inorganic counterparts.43–48 In this field, the design flexibility of organic compounds offers access to electrode materials with a wide range of fine-tunable redox potentials and charge storage mechanisms.33,49–51
We herein present a monolithic, organic PB composed of an organic multi-junction solar cell in conjunction with an organic battery in a single device. This PB is capable of photocharging, reaching voltages up to 3.9 V under reduced illumination intensity and delivering discharge capacities up to 22 mA h g−1 (13 mA h cm−2) by tailoring the illumination time and discharge current. The combined device is the first monolithically integrated PB made out of organic active materials capable of reaching competitive voltages for use in small power devices through photocharging (see Table S7, ESI† for comparison with the literature).37
The charge storage part of the PB is based on a dual-ion organic battery boasting a high cell voltage of 3.7 V vs. Li/Li+, which is comparable to commercial lithium-ion batteries. This is achieved by using a phenothiazine-based conjugated redox polymer as cathode-active material vs. lithium metal as anode material. The redox polymer P(PT-T2) is a copolymer of an N-arylphenothiazine (PT) and a bithiophene (T2) unit and has already shown outstanding performance in previous studies with a theoretical specific capacity of 36.5 mA h g−1 and excellent rate performance.43,48,52 The high rate capability makes P(PT-T2) an optimal candidate for this type of application, as no additional electronics are in place that will mediate the high peak currents the solar cell will supply during the first instances after illumination.
This battery design requires a solar cell that provides an open-circuit voltage (VOC) larger than 3.7 V to be able to photocharge the battery without any additional external voltage or current, enabling an energy autonomous device. Hence, we chose a multi-junction design consisting of several OSCs interconnected in series. While the module technology is better established38,53 and can be combined with storage units,54,55 it generally results in larger areas and is thus less suited for small electronics. On the other hand, multi-junction cells will result in more compact device geometries, but challenges such as spectral matching of the different sub cells and the losses in the interconnection layer (ICL, i.e., the layer between sub cells where an electron and a hole recombine isoenergetically) have to be carefully addressed.56–58 For tandem (2-junction) cells, these problems have been extensively investigated and some solutions are reported.59–61 Nevertheless, for the selected dual-ion organic battery, a 2-junction cell is still insufficient in voltage, and the only alternative is to stack more cells in order to obtain a sufficiently high VOC.37,62–65 Therefore, we fabricated a 5-junction OSC, resulting in a high VOC of about 4.2 V, which places it among the highest voltage, solution-processed multi-junction OSCs reported to date (see also Fig. S14 and Table S6, ESI†),62,66 and monolithically combined them with the organic battery as depicted in Fig. 1.
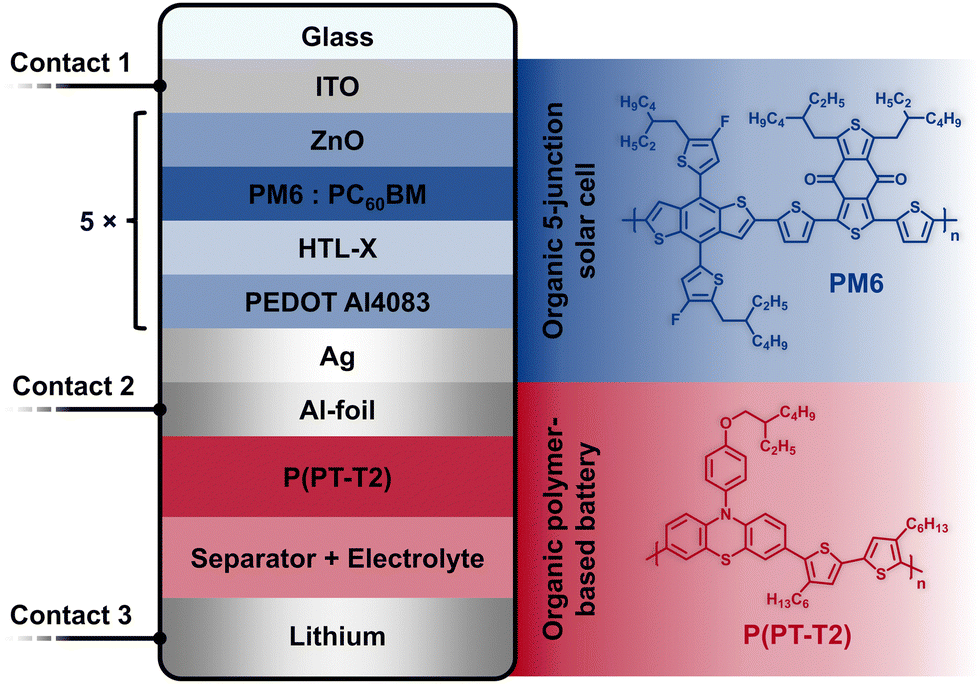 | ||
Fig. 1 Schematic of the components of the organic photo-battery. The multi-junction solar cell is prepared on an ITO-(contact 1) coated glass. The repeating unit in the solar cell follows the structure ZnO/PM6:PC60BM/HTL-X/PEDOT AI4083, i.e., the ICL consists of the electron transport layer (ETL) ZnO and the hole transport layer (HTL) HTL-X/PEDOT AI4083 and the solar cell part is completed with an Ag contact. The organic battery is directly in contact with the positive contact (Ag) of the solar cell through the Al-foil (contact 2) on which the composite film (P(PT-T2)/Super C65/PVDF 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:10) (positive battery electrode) is coated. An electrolyte-soaked separator separates this from the lithium metal (negative battery electrode), which constitutes contact 3. The chemical structures of PM6 and P(PT-T2) are shown on the right. Legend: ITO = indium tin oxide, PM6 = PBDB-T-2F, poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)]; chemical structure shown in blue, PC60BM = phenyl-C61-butyric acid methyl ester, HTL-X = PEDOT-based HTL formulation, PEDOT AI4083 = poly(3,4-ethylenedioxythiophene) polystyrene sulfonate, P(PT-T2) = poly[(3,7-10-(4-((2-ethylhexyl)oxy)phenyl)-10H-phenothiazine)-alt-(5,5′-4,4′-dihexyl-2,2′-bithiophene)]; chemical structure shown in red. :30:10) (positive battery electrode) is coated. An electrolyte-soaked separator separates this from the lithium metal (negative battery electrode), which constitutes contact 3. The chemical structures of PM6 and P(PT-T2) are shown on the right. Legend: ITO = indium tin oxide, PM6 = PBDB-T-2F, poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)]; chemical structure shown in blue, PC60BM = phenyl-C61-butyric acid methyl ester, HTL-X = PEDOT-based HTL formulation, PEDOT AI4083 = poly(3,4-ethylenedioxythiophene) polystyrene sulfonate, P(PT-T2) = poly[(3,7-10-(4-((2-ethylhexyl)oxy)phenyl)-10H-phenothiazine)-alt-(5,5′-4,4′-dihexyl-2,2′-bithiophene)]; chemical structure shown in red. | ||
Methods and materials
Materials
For the organic solar cells (OSCs), PM6 (PBDB-T-2F, poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)]) was used as donor in the active layer as purchased from 1-Material. As acceptor, PC60BM ([6,6]-phenyl-C61-butyric acid methyl ester, 99%) from Luminescence was used. As electron transport layer (ETL), a ZnO nanoparticles solution (2.5 wt% in a mixture of alcohols) was used (N11 from Avantama) after further dilution to 1 wt% by addition of isopropanol. As hole-transport layer (HTL), PEDOT AI4083 from Heraeus, which was filtered through a 2.7 μm filter, further diluted in a ratio of 1:2 with isopropanol, and 1 vol% of the surfactant Capstone FS31 were added, as well as the PEDOT formulation HTL-X (HTL-XC and HTL-XD mixed in a ratio of 1:4) purchased from Raynergy Tek. Molybdenum(VI) oxide (MoO3) 99.97% trace metals basis was purchased from Sigma-Aldrich.
Organic battery fabrication
For the organic battery, P(PT-T2) was synthesized as previously reported.43 Composite electrodes were fabricated by mixing 60 wt% P(PT-T2) with 30 wt% carbon black (Super C65, Timical; dried by heating to 80 °C under vacuum, <10−3 mbar, overnight) and 10 wt% PVdF (Sigma-Aldrich) and dispersing this mixture in 1-methyl-2-pyrrolidone (NMP, 99.5%, extra dry, AcroSeal™, Acros Organics). For homogenizing the respective slurries, a planetary centrifugal mixer (Thinky Mixer ARM-310) was used (30 min, 1500 rpm). The resulting slurries were blade-coated onto KOH-etched aluminum foil (1235 aluminum foil, H18 hard state, 20 μm, 99.8% purity by Gelon LIB). After coating, the films were first dried in a regular drying oven at 60 °C to obtain a “visually dry” film, before being transferred into a vacuum drying oven, where the films were further dried at 80 °C under vacuum (10−2 mbar) over night. Circular electrodes (14 mm diameter) were punched out of the so-obtained films using a high precision electrode cutter (EL-Cut by EL-Cell). Average active mass loadings of 0.6 mg cm−2 were achieved.Organic solar cell fabrication
For the experiments described herein, two types of solar cell geometries were used with different active areas: small (0.0925 cm2) and large (1.2 cm2), which will be referred to by these names. OSCs were fabricated in inverted architecture, i.e., light enters the device through the electron contact, onto 25 × 25 mm2 large ITO-covered glass slides (Visiontek Systems LTD, Rsq = 15 Ohms sq−1). Prior to fabrication, the substrates were cleaned by sequential dipping in ultrasonic baths with acetone, isopropanol, and water, twice in each solvent for 5 min, followed by drying with a nitrogen gun. To ensure a clean and organic-free surface, the substrates were further purified in a UV ozone-plasma reactor for 20 min. Finally, for substrates with larger cell areas, a support structure made of Cr/Ag (5/100 nm) was thermally evaporated to act as current collector around the ITO-electrode, i.e., outside the active area.For single junction OSCs, on top of the ITO glass, ZnO as ETL was statically spin-coated at 4000 rpm for 60 s, resulting in a 30 nm thick layer, followed by annealing at 130 °C for 10 min. Afterwards, the PM6:PC60BM active layer was prepared by dynamically spin-coating the solution at 2000 rpm for 60 s and annealing at 130 °C for 2 min. Next, a double PEDOT layer was added to the stack. This started with a 40 nm thick HTL-X layer, prepared by statically spin-coating the solution at 4000 rpm for 60 s and a drying step of 5 min on a hot plate at 130 °C. Next, the PEDOT AI4083 layer (40 nm) was prepared using identical processing parameters as for the underlying HTL-X layer. All the layers were processed inside a nitrogen-filled glovebox. To complete the device, a 100 nm thick top Ag contact was thermally evaporated (pressure <10−5 mbar) with a rate of 1 nm s−1.
For multi-junction cells, the procedure followed the same basic pattern as for the single-junction cells. The repeating unit (ZnO/active layer/HTL-X/PEDOT AI4083) was coated several times, with the same parameters as for the single-junction cells. For the active layer, the thickness for each junction was adjusted according to the optical simulations. The spin coating speeds were thus reduced for each subsequent BHJ layer, and for the 5-junction OSC they were 2000, 1250, 1250, 1000 and 850 rpm, respectively, resulting in the layer thicknesses listed in Table S5 (ESI†). Again, as for the single junction solar cells, the devices were finished with an Ag top contact as described above.
Organic photo-battery assembly
The organic PB was assembled using a custom-made prototype cell (Fig. S6, ESI†), which facilitates the process and improves reproducibility. Also, given the volatile nature of the solvents used for the battery electrolyte, and the sensitivity of Li metal to ambient air, this holder provides protection and prevents fast degradation.Firstly, the OSC was placed in the holder with the glass side pointing outwards and sealed with a VITON seal. The two solar cell electrodes were then connected to the holder electrodes using adhesive Al tape. An insulator film was placed on top of the bottom electrode (Cr/Ag support structure) to avoid shunting the solar cell during the placement of the battery part. The P(PT-T2) composite electrode was then secured onto the Ag top electrode of the OSC and surrounded by a Teflon sealing. Within the confines of the Teflon sealing, three Whatman GF/C separators were stacked on top of this positive battery electrode. Subsequently, 150 μL of electrolyte (1.0 M LiPF6 in EC/DMC (50:50, v/v), were added onto the separator. Finally, a solid piece of Li metal (14 mm diameter, 0.2 mm thick round electrode) was placed on top, and the holder was closed. The adjustable top steel electrode on the holder was then used to apply moderate pressure to improve the contact between all components. Nevertheless, precaution had to be taken as excessive pressure can cause shunts in the solar cell by perforation of the layers or shatter the glass substrate on which the solar cell is coated.
Characterization techniques
Photocharge was carried out using two separate channels of the potentiostat. The first channel monitored the voltage of both the OSC and the organic battery using a 3-electrode configuration, where contact 1 was used as reference (see Fig. S7, ESI†). The second channel measured the current flow between the OSC and the battery (electrodes 1 and 3, see Fig. S7, ESI†) through a 1 Ohm resistor with a two-point configuration. The measurements of photocharge were performed as follows: firstly, the battery was discharged through contacts 2 and 3 to 3.2 V (while keeping contacts 1 and 3 disconnected). For the photocharge, the solar simulator was turned on, leading to photocurrent generation in the solar cell. The connection between contacts 1 and 3 was short-circuited and, consequently, the voltage of the battery increased until it reached a value close to the solar cell open-circuit voltage or until a set voltage limit. In this last case, the first potentiostat channel monitored the battery voltage through contacts 2 and 3, and when the limit was reached, disconnected contact 1 and 3, so no more current flow was possible. Since the second channel (measuring the current flow between 1 and 3) is not controlled by the voltage limit, a time limit had to be set to stop the recording in this channel, which was set to 1 min. In all other experiments, the illumination times were fixed and varied between 5 and 30 min. Afterwards, the light was turned off and contacts 1 and 3 were disconnected. The PB was then galvanostatically discharged to 3.2 V between contacts 2 and 3 (Fig. 5) with the respective current. To evaluate the operation stability of the device, the photocharge/dark discharge cycles were repeated several times.
Results
Multi-junction organic solar cell
A single-junction OSC fabricated with the PM6:PC60BM blend shows good characteristics for its integration with the organic battery.67 Its high open-circuit voltage of almost 950 mV ensures that a comparably low number of cells is needed to obtain a total voltage above 4 V. For performing first tests with this blend, MoO3/Ag was used as top electrode, which requires thermal evaporation of MoO3 as HTL. As this is time-consuming, we investigated suitable solution-processable materials as alternative HTLs. One of the most common materials for this task is PEDOT:PSS. Out of many PEDOT:PSS formulations and compositions available, HTL-X and AI4083 were selected as they showed the best hole selectivity and thus reduced the voltage loss at the blend/HTL interface. The results for the HTL substitution in single-junction cells can be seen in Fig. S1 (ESI†). Next, to assess the suitability of these HTL materials as part of the ICL layer, tandem cells were fabricated with two sub-cells of different active layer thicknesses using ZnO as ETL (Fig. 2). | ||
| Fig. 2 Characterization results of tandem (2-junction) cells (active area = 0.0925 cm2) with different HTL materials in the ICL. (a) Comparison of the VOC values of tandem cells with different HTL materials used for the ICL together with ZnO. Black triangles represent the mean value, dots represent actual measurements, the black lines represent the median value. (b) Current voltage curves (JV-curves) of tandem cells with different HTL for the ICL. As evident from these figures, some voltage loss is observed for cells using MoO3. While using HTL-X, the voltage only amounted to a single-junction cell. This is explained by an incompatibility between HTL-X and ZnO, as it was observed that coating the latter on top of the former caused some decomposition of the HTL-X layer. | ||
The results for PEDOT formulation AI4083, even without any thickness optimization, were very promising, as shown in Fig. 2. In this case, the voltage of the tandem cells is close to the sum of the voltages of single cells, with less than 100 mV loss. The short-circuit current is smaller in the tandem cells, since the photogeneration is divided by the number of sub-cells. In addition, the current of any serially interconnected multi-junction solar cell is limited by the sub cell with the lowest current. Only the fill factor (FF) seems to be reduced compared to the single cells, as can be seen in Fig. S1 (ESI†) which could be a consequence of inefficient current matching. For detailed values for JSC, VOC, FF and efficiency, please refer to Table S1 (ESI†).
With the material for the HTL chosen, it was imperative to optimize the thickness of the different sub-cells in the stack to maximize the current generation. While the thickness of all interlayer materials was fixed (30 nm for ZnO and 40 nm for the PEDOT:PSS layers), that of the active PM6:PC60BM layer was iteratively changed using optical simulation as guideline (additional information in ESI†), with the goal of maximizing the current in the least generating cell. Experimentally, changes in the active layer thickness were achieved by varying the spin coating speed. With the resulting thickness values from the optical simulations, multi-junction cells with up to 5 sub cells were manufactured. The results and the comparison with the simulations can be found in Fig. 3.
 | ||
| Fig. 3 Results from multi-junction solar cells and comparison with simulations (active area = 0.0925 cm2). (a) JV-curves recorded for multiple solar cells with increasing number of junctions using AI4083/ZnO as ICL. (b) Open-circuit voltage as a function of the number of junctions comparing the experimental values (blue) with values calculated from the photogenerated current as optically simulated (red) and calculated from experimentally measured photogenerated current (yellow). While for tandem cells all the values are in good agreement, for higher number of junctions the voltage difference between experiment and theory deviates more strongly. (c) Short-circuit current density vs. number of junctions. | ||
From the results plotted in Fig. 3(b), a voltage loss is apparent, which becomes more severe with an increasing number of junctions. Since the short-circuit current and measured thickness closely match the results of the optical simulations (see ESI† for details) and further, the open-circuit voltage does only logarithmically depend on the photogenerated current, this observation cannot be explained by an optical effect such as e.g. a strongly reduced absorption in the sub cell(s) farthest away from the light source (Fig. S4, ESI†). For detailed values for JSC, VOC, FF and efficiency, please refer to Table S2 (ESI†).
Hence, two alternatives were considered to explain this voltage deficit. The first hypothesis was that the solution-based deposition of layers can damage underlying layers that were deposited before. To test this, different combinations of interconnection and blend layers were tested in single-junction solar cells to investigate potential damage on underlying layers. However, none of these experiments could reproduce the voltage losses observed for the multi-junction cells (see Fig. S2, ESI†).
Our second hypothesis was that the processing of the ICL did not result in pin-hole free, full-area coverage, leading to local shunts. We addressed this by processing a combination of HTL-X/AI4083 as the p-conducting part of the ICL. From the previous experiments it was noted that the wetting properties on top of the active layer were superior for HTL-X compared to PEDOT AI4083. Indeed, as shown in Fig. 4, the results for the ICL comprising HTL-X/AI4083 and ZnO are very close to that of the simulations, with only around 200 mV loss for the whole 5-junction OSC, i.e., less than 50 mV per cell. With this layer stack, the VOC of the multi-junction solar cell is larger than 4 V, which is enough to fully charge the organic battery.
 | ||
| Fig. 4 Multi-junction parameter overview and cell geometry comparison. (a) Open-circuit voltage (left) and short-circuit current density (right) for the two HTL materials used in the ICL. In this case, the simulation results are calculated for a stack with only AI4083. The dots represent the mean value of 12 devices, with the error bars indicating the standard deviation (active area = 0.0925 cm2). (b) JV-curves comparing the two different cell areas. Large area cells are advantageous for the battery integration, as it facilitates the process of integration with the positive battery electrode and provides space for connections. | ||
The last challenge left before integration into the PB was to scale up the active area of the multi-junction solar cells, as the results discussed previously were achieved for cells with an area ≪1 cm2. After some optimization, the new cell layout (Fig. S3, ESI†) resulted in an active area of 1.2 cm2, while almost retaining the same performance (see Fig. 4(c)).
In summary, by carefully selecting the components of the ICL and optimizing the active layer thickness with the help of optical simulations, we developed a 5-junction organic solar cell with minimal voltage losses. To the best of our knowledge, this is the first report of a solution-processed 5-junction organic solar cell with an open-circuit voltage of more than 4 V, which also ranks among the highest voltage multi-junction solar cells for any number of junctions (Table S6, ESI†).
Organic photo-battery
For the PB application (Fig. 5), the assembly of both the battery and OSC are different from the common measurement setups for the respective separate devices. Thus, it was imperative to first investigate both the harvesting- and the storage unit parts independently inside the newly designed cell prototype used for the PB measurements (Fig. S6, ESI†). Since the resulting data showed good agreement with the respective conventional measurement setups (see Fig. S8 and S9, ESI†),43 we proceeded to assemble the PB as explained in the methods and materials section, combining the 5-junction OSC with the organic battery. | ||
| Fig. 5 Working principles of the organic photo-battery during (a) photocharge and (b) discharge in the dark. The solar cell and battery are simplified to only represent the most important layers. The numbering of the contacts is referred to in the description of the experiments in the experimental part. (a) During photocharge, electrodes 1 and 3 are shorted, and the polymer P(PT-T2) is oxidized by the holes generated in the solar cell. The resulting positive charges are balanced by anions inserting from the electrolyte. Simultaneously, Li ions from the electrolyte are reduced at the negative battery electrode by the electrons originating from the negative electrode of the solar cell (contact 1) to be deposited as elemental Li. (b) During the discharge process in the dark, contacts 2 and 3 are connected through an external load and the electrochemical processes are reversed (for more details, see Fig. S7, S12 and S13, ESI†). This then completes a full photocharge-dark discharge cycle. | ||
The measurement procedure (described in more detail in the ‘materials and methods’ section) consists of two parts: photocharge is achieved by illuminating the device from the solar cell ITO-side and recording the resulting voltage in the battery and solar cell, as well as the current flow. This is then followed by a galvanostatic discharge of just the battery part in the dark. As the solar cell provides a VOC of about 4.2 V, the charging protocol for these first tests was configured with an upper battery voltage limit of 3.9 V, at which the charging process would be terminated, in order to avoid damaging the battery due to overcharging.
As visible from Fig. 6(a), the voltage measured in the organic battery part rises sharply after illumination and reaches the cut-off limit of 3.9 V within a few seconds. This is due to the relatively high current delivered by the solar cell, about 0.8 mA at 3.2 V, the onset of photocharge, as inferred from the JV-curve at 1 sun (Fig. S8c, ESI†), corresponding to a C-rate of 23C for the battery charge. The following discharge in the dark is carried out at a constant current of 25.3 mA g−1 (corresponding to a rate of approximately 0.7C), which completes a full cycle and was repeated ten more times. It is apparent, that the discharge capacities achieved with this initial charging protocol are much below the theoretical specific capacity of the P(PT-T2) composite electrode of 36.5 mAh g−1. This incomplete charge is mostly related to the protocol itself which cuts the charge after few seconds because the voltage limit is rapidly reached with the charging photocurrents delivered at 1 sun, and partially due to kinetic limitations, which are described by Peukert's law,68 and they are especially effective at these high charge rates.
 | ||
| Fig. 6 Results of photocharge with voltage limitation. (a) Voltage profile of the photo-battery (contacts 2 and 3) during several cycles of photocharge and dark discharge with a fixed upper potential limit (3.9 V) and at a fixed discharge current of 0.7C. (b) Detailed view of the first photocharge and discharge cycle. Note the time axis interruption at 4 seconds. The time between the end of photocharge and onset of discharge is a result of the measurement protocol, since only the channel recording the voltage could be set with a voltage limit. For the current measurement, a time limit (1 minute) was placed, resulting in a waiting time. (c) Specific discharge capacity for several cycles of photocharge-dark discharge with a fixed cut-off voltage. (d) Energy input during illumination, output during dark discharge and resulting cycle efficiency, i.e. the ratio of both. Note the axis interruption for the energy and the difference between input and output as a result of incomplete charge. | ||
Nevertheless, the calculated cycle efficiency (Fig. 6(d)),25 which is the ratio of output to input energy (see eqn S3 in ESI†), remains constant during these eleven cycles. During these cycles, the charging times get shorter (from 3.6 s in cycle 1 to 1.6 s in cycle 11), which means that the input energy is reduced, and hence less discharge capacity (energy) can be extracted afterwards, but their ratio remains constant.
While these measurements provide proof that the 5-junction organic PB is functional, the next goal was to find an alternative charging protocol to maximize the useable capacity of the battery part. In order to reduce the current flowing into the battery during illumination and to set a more compatible cut-off voltage at the same time, the illumination intensity was reduced, such that the VOC of the solar cell closely matched the 3.9 V voltage limit. In practical terms, this meant a reduction of the light irradiation by approximately 60%, so that the impinging power was 370 W m−2, following the JV-curves in Fig. S8c (ESI†). The photocharge-discharge process was then repeated for different illumination times (5 and 10 min) with a discharge current of 25.3 mA g−1 corresponding to a rate of approximately 0.7C.
With these illumination times of 5 and 10 min under reduced light intensity, the photocharge was not fully completed (Fig. 7(a)), not making full use of the available battery capacity. In order to maximize the capacity, a longer (15 min) illumination time was chosen, and the discharge current reduced to 0.3C (12.7 mA g−1).
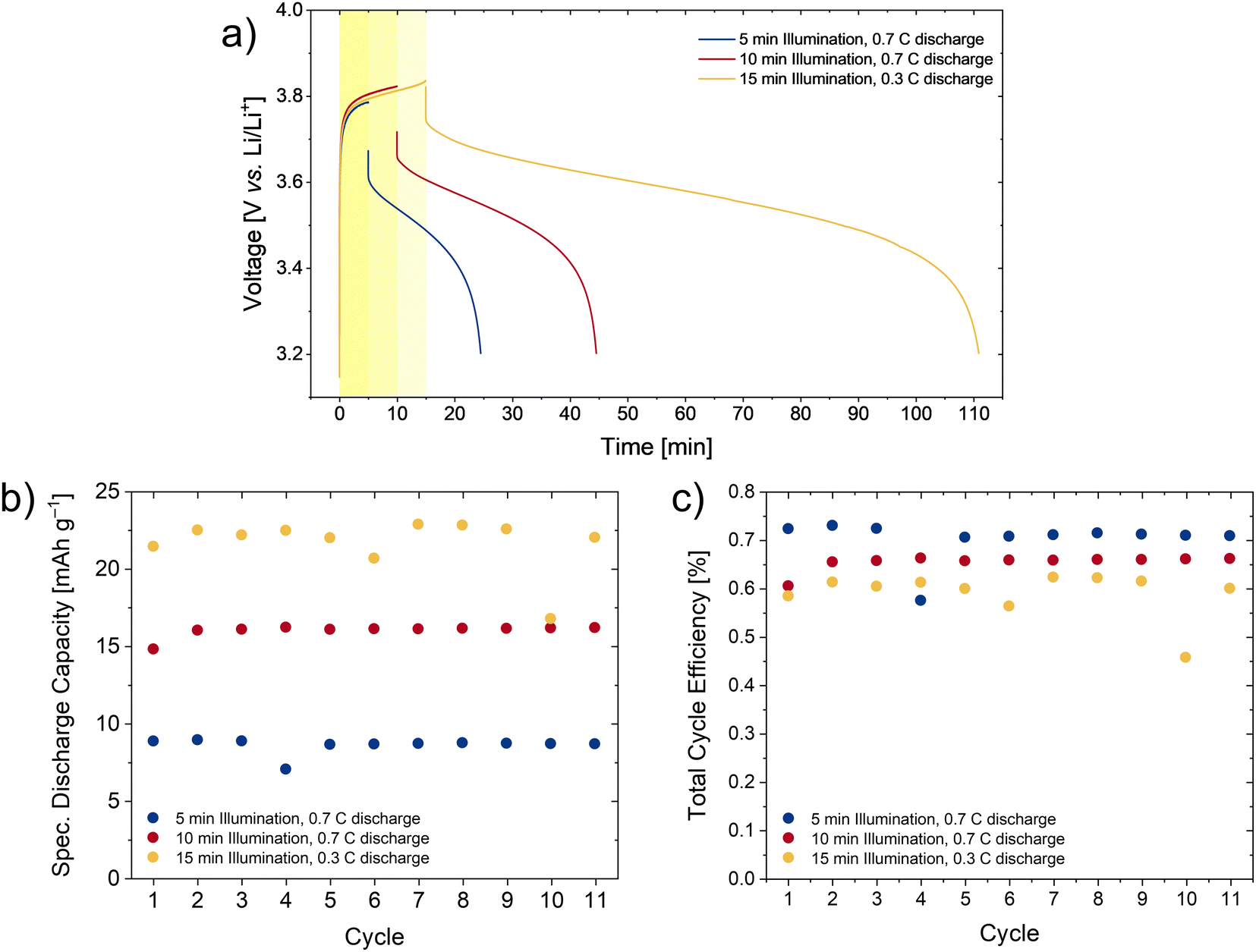 | ||
| Fig. 7 Results from photocharge with time limit and reduced light intensity. (a) Solar battery voltage profile for the first cycle of photocharge-dark discharge with different illumination times and discharge currents. The sharp rise in voltage at the start is a consequence of the high currents delivered by the solar cell with respect to the battery capacity. The influence of the illumination time can be seen, as for longer times the charging plateau is more complete. (b) Discharge capacity progression for several cycles of photocharge-dark discharge. (c) Cycle efficiency for the same number of cycles. | ||
With these parameters, the voltage plateau representing the battery redox reaction is more clearly seen, and suggesting a more complete use of the available battery capacity. In all cases, the current delivered by the solar cell peaked at 0.13 mA cm−2, and followed the previously measured JV-curves (Fig. S10, ESI†), thus being heavily influenced by solar cell FF. In order to assess the reproducibility, additional photocharge-dark discharge cycles were performed for each combination of illumination times and discharge currents. During this number of cycles no degradation of the capacity is observed, and the extracted energy remains rather constant (Fig. 7(b)). Similar to before, the cycle efficiency (Fig. 7(c)) can be evaluated by comparing the ratio between energy input during illumination and output in the dark. While at first sight it may appear striking that the cycle efficiency is lower than in the voltage-limited photocharge at 1 sun illumination, this is just an effect of the longer illumination times. While the discharge capacity is larger, for the 15 min illumination at reduced light intensity, the energy input also scales with the illumination time, and this is much higher in these measurements. Also, it is worth noting that since the battery's coulombic efficiency is ≫95% (see Fig. S8, ESI†), the solar cell efficiency will be the limiting factor, and thus the maximum cycle efficiency will approach the solar cell efficiency, which under these illumination conditions amounts to 1.3%. Finally, analyzing the energy and power density output and plotting these values in a classical Ragone plot (Fig. S11, ESI†), it can be seen that for higher dark discharge currents, higher values of power density are achieved, close to 95 mW g−1 (58 μW cm−2). On the other hand, to reach larger energy density values, a lower discharge current and larger illumination time is beneficial. In our case, for 15 min illumination and a discharge rate of 0.3C, an average energy density of 69 mW h g−1 (43 μW h cm−2) is obtained.
In summary, the PB device provides the best results under reduced illumination conditions, where sufficient time is given for the organic battery to fully charge. Under these conditions and illumination times of 15 min, reversible discharge capacities of the PB of up to 22 mA h g−1 (13 mA h cm−2) can be obtained during repeated cycling at an average discharge potential of 3.6 V. Future development will involve further improving the technical aspects of the prototype cell housing in order to provide long term cycling stability.
Conclusions
In conclusion, we have presented the first example of a photochargeable, organic-based, monolithically integrated photo-battery. For this, we developed a simple and scalable solution process to manufacture 5-junction organic solar cells with an open-circuit voltage of 4.2 V, high enough to fully charge a commercial battery and among the highest values ever achieved for solution-processed solar cells. This was realized by vertically stacking five organic sub-cells with the absorber material PM6:PC60BM on active areas >1 cm2. By means of optical modelling, the thickness of all five photoactive layers was optimized to maximize charge carrier generation. By monolithically combining these 5-junction cells with a dual-ion lithium organic battery consisting of the high rate capability redox polymer P(PT-T2) as cathode-active material, a device capable of storing charge in situ under illumination could be realized. With careful control of the illumination times and discharge rates, this device is able to achieve fast charge in less than 15 min and discharge capacities of up to 22 mA h g−1. Combined with the high average discharge potential of 3.6 V, which, to the best of our knowledge, is the highest of all examples of fully integrated photochargeable devices, the device is able to deliver energy and power densities of 69 mW h g−1 and 95 mW g−1, respectively. The photo-battery reported herein, while significantly optimizable, represents a large step forward in the development of compact energy sources for integrated, low power applications and will pave the way for future developments of such devices using organic active materials.Author contributions
The manuscript was written through the contribution of all authors and all authors have approved the final version of the manuscript. B. E., A. F., U. W.: supervision, discussion, writing – review and editing. R. D., R. W.: experiments, synthesis, design of the test cell, characterization, data analysis, writing original draft. J. B.: design of the test cell, experiments and discussion, writing – review and editing. L. P.: optical simulations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC-2193/1 – 390951807 (livMatS Cluster of Excellence). Special thanks to Pascal Acker for his help in the development of some of the measurement methods and Taisiia Berestok for her contribution to the design of the cell prototype which allowed for the fabrication of photo-batteries.Notes and references
- S. Dhakal, J. C. Minx, F. L. Toth, A. Abdel-Aziz, M. J. Figueroa, K. Hubacek, I. G. Jonckheere, K. Yong-gun, G. Nemet, S. Pachauri and others, in Climate Change 2022: Mitigation of Climate Change, IPCC: Intergovernmental Panel on Climate Change, 2022.
- L. Clarke, Y.-M. Wei, A. de la Vega Navarro, A. Garg, A. N. Hahmann, S. Khennas, I. M. Azevedo, A. Löschel, A. K. Singh, L. Steg and others, Climate Change 2022: Mitigation of Climate Change. Working Group III Contribution to the IPCC Sixth Assessment Report, Cambridge University Press, 2022.
- Y. Li and R. Shi, EURASIP J. Wirel. Commun. Netw., 2015, 2015, 179 CrossRef.
- O. Astakhov, T. Merdzhanova, L.-C. Kin and U. Rau, Sol. Energy, 2020, 206, 732–740 CrossRef.
- C. Perera, C. H. Liu, S. Jayawardena and M. Chen, IEEE Access, 2014, 2, 1660–1679 Search PubMed.
- P. M. Stanley, F. Sixt and J. Warnan, Adv. Mater., 2023, 35, 2207280 CrossRef CAS PubMed.
- J. Sun, E. Zhao, J. Liang, H. Li, S. Zhao, G. Wang, X. Gu and B. Z. Tang, Adv. Mater., 2022, 34, 2108048 CrossRef CAS PubMed.
- C.-T. Wang and H.-H. Huang, J. Non-Cryst. Solids, 2008, 354, 3336–3342 CrossRef CAS.
- K. Shimokawa, S. Matsubara, A. Okamoto and T. Ichitsubo, Chem. Commun., 2022, 58, 9634–9637 RSC.
- A. Paolella, C. Faure, G. Bertoni, S. Marras, A. Guerfi, A. Darwiche, P. Hovington, B. Commarieu, Z. Wang, M. Prato, M. Colombo, S. Monaco, W. Zhu, Z. Feng, A. Vijh, C. George, G. P. Demopoulos, M. Armand and K. Zaghib, Nat. Commun., 2017, 8, 14643 CrossRef CAS PubMed.
- V. Chakrapani, F. Rusli, M. A. Filler and P. A. Kohl, J. Power Sources, 2012, 216, 84–88 CrossRef CAS.
- W. Li, H.-C. Fu, Y. Zhao, J.-H. He and S. Jin, Chem, 2018, 4, 2644–2657 CAS.
- W. Li, J. Zheng, B. Hu, H.-C. Fu, M. Hu, A. Veyssal, Y. Zhao, J.-H. He, T. L. Liu, A. Ho-Baillie and S. Jin, Nat. Mater., 2020, 19, 1326–1331 CrossRef CAS PubMed.
- T. Berestok, C. Diestel, N. Ortlieb, J. Buettner, J. Matthews, P. S. C. Schulze, J. C. Goldschmidt, S. W. Glunz and A. Fischer, Sol. RRL, 2021, 5, 2100662 CrossRef CAS.
- T. Berestok, C. Diestel, N. Ortlieb, S. W. Glunz and A. Fischer, Adv. Mater. Technol., 2022, 7, 2200237 CrossRef CAS.
- S. F. Hoefler, R. Zettl, D. Knez, G. Haberfehlner, F. Hofer, T. Rath, G. Trimmel, H. M. R. Wilkening and I. Hanzu, ACS Sustainable Chem. Eng., 2020, 8, 19155–19168 CrossRef CAS.
- Y.-X. Tan, X. Zhang, J. Lin and Y. Wang, Energy Environ. Sci., 2023, 16, 2432–2447 RSC.
- R. Liu, M. Takakuwa, A. Li, D. Inoue, D. Hashizume, K. Yu, S. Umezu, K. Fukuda and T. Someya, Adv. Energy Mater., 2020, 10, 2000523 CrossRef CAS.
- A. Gouder, F. Podjaski, A. Jiménez-Solano, J. Kröger, Y. Wang and B. V. Lotsch, Energy Environ. Sci., 2023, 16, 1520–1530 RSC.
- W. Guo, X. Xue, S. Wang, C. Lin and Z. L. Wang, Nano Lett., 2012, 12, 2520–2523 CrossRef CAS PubMed.
- R. Wessling, R. Delgado Andrés, I. Morhenn, P. Acker, W. Maftuhin, M. Walter, U. Würfel and B. Esser, Macromol. Rapid Commun., 2022, 2200699 CrossRef PubMed.
- J. Büttner, T. Berestok, S. Burger, M. Schmitt, M. Daub, H. Hillebrecht, I. Krossing and A. Fischer, Adv. Funct. Mater., 2022, 32, 2206958 CrossRef.
- B. Deka Boruah, A. Mathieson, S. K. Park, X. Zhang, B. Wen, L. Tan, A. Boies and M. De Volder, Adv. Energy Mater., 2021, 11, 2100115 CrossRef CAS.
- B. D. Boruah and M. D. Volder, J. Mater. Chem. A, 2021, 9, 23199–23205 RSC.
- R. Delgado Andrés, T. Berestok, K. Shchyrba, A. Fischer and U. Würfel, Sol. RRL, 2022, 6, 2200614 CrossRef.
- B. D. Boruah, A. Mathieson, B. Wen, S. Feldmann, W. M. Dose and M. De Volder, Energy Environ. Sci., 2020, 13, 2414–2421 RSC.
- N. Yan and X. Gao, Energy Environ. Mater., 2021, eem2.12182 Search PubMed.
- X. Zhang, K. Su, A. G. A. Mohamed, C. Liu, Q. Sun, D. Yuan, Y. Wang, W. Xue and Y. Wang, Energy Environ. Sci., 2022, 15, 780–785 RSC.
- R. Fan, Y. Wu, H. Xie, Y. Gao, L. Wang, B. Zhao, D. Li, S. Liu, Y. Zhang, H. Kong, Y. Li, Q. Chen, A. Cao and H. Zhou, ChemSusChem, 2022, 15, e202201473 CrossRef CAS PubMed.
- J. Wang, S. Li, Y. Chen, L. Dong, M. Liu, J. Shi, S. Li and Y. Lan, Adv. Funct. Mater., 2022, 2210259 CrossRef CAS.
- K. Kato, A. B. Puthirath, A. Mojibpour, M. Miroshnikov, S. Satapathy, N. K. Thangavel, K. Mahankali, L. Dong, L. M. R. Arava, G. John, P. Bharadwaj, G. Babu and P. M. Ajayan, Nano Lett., 2021, 21, 907–913 CrossRef CAS PubMed.
- X. Cao, Q. Shi, Y. Feng, X. Zhang, E. Zhou, L. Zhu and Y. Wang, Mater. Today Chem., 2022, 26, 101082 CrossRef CAS.
- X. Chen, X. Yin, J. Aslam, W. Sun and Y. Wang, Electrochem. Energy Rev., 2022, 5, 12 CrossRef CAS.
- C. Xu, X. Zhang, L. Duan, X. Zhang, X. Li and W. Lü, Nanoscale, 2020, 12, 530–537 RSC.
- A. Gurung, K. M. Reza, S. Mabrouk, B. Bahrami, R. Pathak, B. S. Lamsal, S. I. Rahman, N. Ghimire, R. S. Bobba, K. Chen, J. Pokharel, A. Baniya, M. A. R. Laskar, M. Liang, W. Zhang, W.-H. Zhang, S. Yang, K. Xu and Q. Qiao, Adv. Funct. Mater., 2020, 30, 2001865 CrossRef CAS.
- J. Lv, Y.-X. Tan, J. Xie, R. Yang, M. Yu, S. Sun, M.-D. Li, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2018, 130, 12898–12902 CrossRef.
- P. Sullivan, S. Schumann, R. Da Campo, T. Howells, A. Duraud, M. Shipman, R. A. Hatton and T. S. Jones, Adv. Energy Mater., 2013, 3, 239–244 CrossRef CAS.
- R. Søndergaard, M. Hösel, D. Angmo, T. T. Larsen-Olsen and F. C. Krebs, Mater. Today, 2012, 15, 36–49 CrossRef.
- A. Gambhir, P. Sandwell and J. Nelson, Sol. Energy Mater. Sol. Cells, 2016, 156, 49–58 CrossRef CAS.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, (accessed 4 May 2023).
- D. Müller, L. Campos Guzmán, E. Jiang, B. Zimmermann and U. Würfel, Sol. RRL, 2022, 6, 2200175 CrossRef.
- P. Acker, L. Rzesny, C. F. N. Marchiori, C. M. Araujo and B. Esser, Adv. Funct. Mater., 2019, 29, 1906436 CrossRef CAS.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef.
- B. Esser, F. Dolhem, M. Becuwe, P. Poizot, A. Vlad and D. Brandell, J. Power Sources, 2021, 482, 228814 CrossRef CAS.
- M. D. Hager, B. Esser, X. Feng, W. Schuhmann, P. Theato and U. S. Schubert, Adv. Mater., 2020, 32, 2000587 CrossRef CAS PubMed.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed.
- B. Esser, Org. Mater., 2019, 01, 63–70 CrossRef CAS.
- C. Ding, C. Li, H. Tian, Y. Tong, W. Huang and Q. Zhang, Batteries Supercaps, 2022, 5, e202200160 CrossRef CAS.
- P. Poizot, F. Dolhem and J. Gaubicher, Curr. Opin. Electrochem., 2018, 9, 70–80 CrossRef CAS.
- C. Friebe, A. Lex-Balducci and U. S. Schubert, ChemSusChem, 2019, 12, 4093–4115 CrossRef CAS PubMed.
- P. Acker, J. S. Wössner, G. Desmaizieres and B. Esser, ACS Sustainable Chem. Eng., 2022, 10, 3236–3244 CrossRef CAS.
- M. Fernández Castro, E. Mazzolini, R. R. Sondergaard, M. Espindola-Rodriguez and J. W. Andreasen, Phys. Rev. Appl., 2020, 14, 034067 CrossRef.
- Y. Jin, L. Sun, L. Qin, Y. Liu, Z. Li, Y. Zhou and F. Zhang, Appl. Phys. Lett., 2021, 118, 203902 CrossRef CAS.
- G. Dennler, S. Bereznev, D. Fichou, K. Holl, D. Ilic, R. Koeppe, M. Krebs, A. Labouret, C. Lungenschmied, A. Marchenko, D. Meissner, E. Mellikov, J. Meot, A. Meyer, T. Meyer, H. Neugebauer, N. S. Sariciftci and S. Taillemite, Sol. Energy, 2007, 11 Search PubMed.
- D. Di Carlo Rasi, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Adv. Energy Mater., 2017, 7, 1701664 CrossRef.
- B. Minnaert and P. Veelaert, Materials, 2012, 5, 1933–1953 CrossRef.
- X. Du, O. Lytken, M. S. Killian, J. Cao, T. Stubhan, M. Turbiez, P. Schmuki, H.-P. Steinrück, L. Ding, R. H. Fink, N. Li and C. J. Brabec, Adv. Energy Mater., 2017, 7, 1601959 CrossRef.
- D. Di Carlo Rasi, K. H. Hendriks, G. H. L. Heintges, G. Simone, G. H. Gelinck, V. S. Gevaerts, R. Andriessen, G. Pirotte, W. Maes, W. Li, M. M. Wienk and R. A. J. Janssen, Sol. RRL, 2018, 2, 1800018 CrossRef.
- Z. Zheng, J. Wang, P. Bi, J. Ren, Y. Wang, Y. Yang, X. Liu, S. Zhang and J. Hou, Joule, 2022, 6, 171–184 CrossRef CAS.
- J. Wang, Z. Zheng, Y. Zu, Y. Wang, X. Liu, S. Zhang, M. Zhang and J. Hou, Adv. Mater., 2021, 33, 2102787 CrossRef CAS PubMed.
- J. Tong, S. Xiong, Y. Zhou, L. Mao, X. Min, Z. Li, F. Jiang, W. Meng, F. Qin, T. Liu, R. Ge, C. Fuentes-Hernandez, B. Kippelen and Y. Zhou, Mater. Horiz., 2016, 3, 452–459 RSC.
- D. Di Carlo Rasi, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2018, 30, 1803836 CrossRef PubMed.
- R. Meerheim, C. Körner, B. Oesen and K. Leo, Appl. Phys. Lett., 2016, 108, 103302 CrossRef.
- S. Esiner, H. van Eersel, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2013, 25, 2932–2936 CrossRef CAS PubMed.
- D. Di Carlo Rasi and R. A. J. Janssen, Adv. Mater., 2019, 31, 1806499 CrossRef PubMed.
- M.-A. Pan, T.-K. Lau, Y. Tang, Y.-C. Wu, T. Liu, K. Li, M.-C. Chen, X. Lu, W. Ma and C. Zhan, J. Mater. Chem. A, 2019, 7, 20713–20722 RSC.
- Q. Zhang, N. Cui, Z. Zhou, Y. Shang and B. Duan, Int. J. Energy Res., 2022, 46, 23808–23823 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee01822a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |