
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MBene promoted Zn peroxide chemistry in rechargeable near-neutral Zn–air batteries†
Yue
Hou‡
a,
Ze
Chen‡
a,
Xinliang
Li
e,
Yiqiao
Wang
a,
Pei
Li
a,
Huilin
Cui
a,
Rong
Zhang
a,
Shuo
Yang
a,
Shaoce
Zhang
a and
Chunyi
Zhi
 *abcd
*abcd
aDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, China. E-mail: cy.zhi@cityu.edu.hk
bHong Kong Institute for Advanced Study, City University of Hong Kong, Kowloon, Hong Kong 999077, China
cHong Kong Institute for Clean Energy, City University of Hong Kong, Kowloon 999077, Hong Kong
dCentre for Functional Photonics, City University of Hong Kong, Kowloon, Hong Kong
eKey Laboratory of Material Physics, Ministry of Education, School of Physics and Microelectronics, Zhengzhou University, Zhengzhou 450001, China
First published on 8th June 2023
Abstract
Highly reversible Zn–air batteries (ZABs) in near-neutral aqueous electrolytes can suppress the formation of Zn dendrites and carbonates. However, near-neutral ZABs (NNZABs) based on Zn peroxide (ZnO2) chemistry are challenging to build due to the air circulation requirement and the Swagelok structure. To achieve cost-effective NNZABs in a coin-cell configuration, it is essential to explore air-breathing cathodes with high catalytic activity for near-neutral systems. Previous studies focused on catalysts for alkaline ZABs, which are usually not suitable for near-neutral batteries. Here, Mo4/3B2−xTz MBene with ordered vacancies is for the first time exploited as a high-performance catalyst for NNZABs. Interestingly, a stable lifecycle for 380 h under a current density of 2 mA cm−2 and a 5 h cycling period was achieved, which is attributed to the high O2-adsorbability and air stock capacity of Mo4/3B2−xTz MBene. We also demonstrate that the vacancies at the Y sites of Mo4/3B2−xTz MBene are active sites for boosting the sluggish kinetics of ZnO2 chemistry. This work highlights the significance of introducing highly efficient cathode catalysts for NNZABs and the high efficiency of MBenes as catalysts in near-neutral systems.
Broader contextNear-neutral ZABs (NNZABs) based on the super-concentrated zinc trifluoromethanesulfonate (Zn(OTf)2) electrolyte and Zn–O2/ZnO2 chemistry through a 2e−/O2 process support a stable ZAB operation in ambient air and present higher reversibility than their alkaline counterparts. However, a previous study has achieved Zn–O2/ZnO2 chemistry in the Swagelok-type cell configuration, which is bulky and costly. To achieve a highly reversible ZnO2 electrochemical process in the cost-effective coin- and pouch-type NNZABs, a superior electrocatalyst is a crucial factor. The traditional best PtC and IrO2 would vastly decrease the probability of the combination between O2 and Zn ions, unsuitable for near-neutral electrolyte-based NNZABs. To boost the reversible ZnO2 electrochemical process, we first employed an efficient 2D transition metal boride (Mo4/3B2−xTz MBene) with ordered vacancies as the cathode catalyst. The rate performance and cycling endurance of the NNZAB-MBene are significantly enhanced compared with NNZAB–PtC–IrO2. Introducing catalysts to achieve highly reversible NNZABs in a coin-cell configuration is of high necessity. The comprehensive study in this work could provide different insights for a deeper understanding of NNZABs and enable higher applicability in portable devices powered by NNZABs. |
Introduction
Zn–air batteries (ZABs) are deemed some of the most promising candidates for the next-generation sustainable energy-storage systems with remarkable electrochemical performance, intrinsic safety, high cost-efficiency, and eco-friendly byproducts.1–6 To date, alkaline ZABs with a high discharge plateau remain the research mainstream. Various protocols have been developed to alleviate Zn anode passivation and suppress dendrites, including bifunctional air-breathing catalyst design,7–13 electrolyte modification,14–16 and electrode architecture optimization.7,17,18 However, ZABs in alkaline electrolytes still suffer from irreversible Zn consumption at the anode and intrinsic sluggish kinetics of the 4e− oxygen reduction reaction (ORR) pathway involving H2O on the cathode side.19–26 A near-neutral electrolyte is expected to solve the inherent problems of alkaline electrolytes and improve the reversibility of ZABs. This statement has been verified in a recent report by Wang et al.,1 in which near-neutral ZABs based on super-concentrated zinc trifluoromethanesulfonate (Zn(OTf)2) electrolytes can achieve Zn–O2/ZnO2 chemistry through a 2e−/O2 process, support stable ZAB operation in ambient air and present higher reversibility than their alkaline counterparts.1Nevertheless, near-neutral ZABs (NNZABs) are based on home-built Swagelok-type cells and require air circulation systems to accelerate the reaction between Zn and O2, the schematic illustration of which is presented in Fig. 1a. It has high requirements for operating conditions with a constant air-flow rate. To enhance the portability and cut the costs of NNZABs, we have explored the electrochemistry process in conventional coin cells, making a step toward the accessible applications of NNZABs. Firstly, pure carbon cloth with or without ketjen black as the active material was employed as the air cathode which however achieved unsatisfactory performances. As shown in Fig. 1b and c, the NNZABs perform several cycles with a limited specific capacity of only 10 mA h cm−2 at a current density of 2.0 mA cm−2. Even though ketjen black is used as the active material for the cathode, the coin-cell type NNZAB still shows poor cyclability, which is attributed to the limited physical contact between the cathode and air, and low air stock capacity. It reveals that the electrocatalytic activities in the air cathode are insufficient for NNZABs. To tackle this problem, the use of catalysts with abundant active sites27–29 and oxygen buffer ability30 as air-breathing cathodes is necessary. Related studies on near-neutral electrolytes have been missed so far.
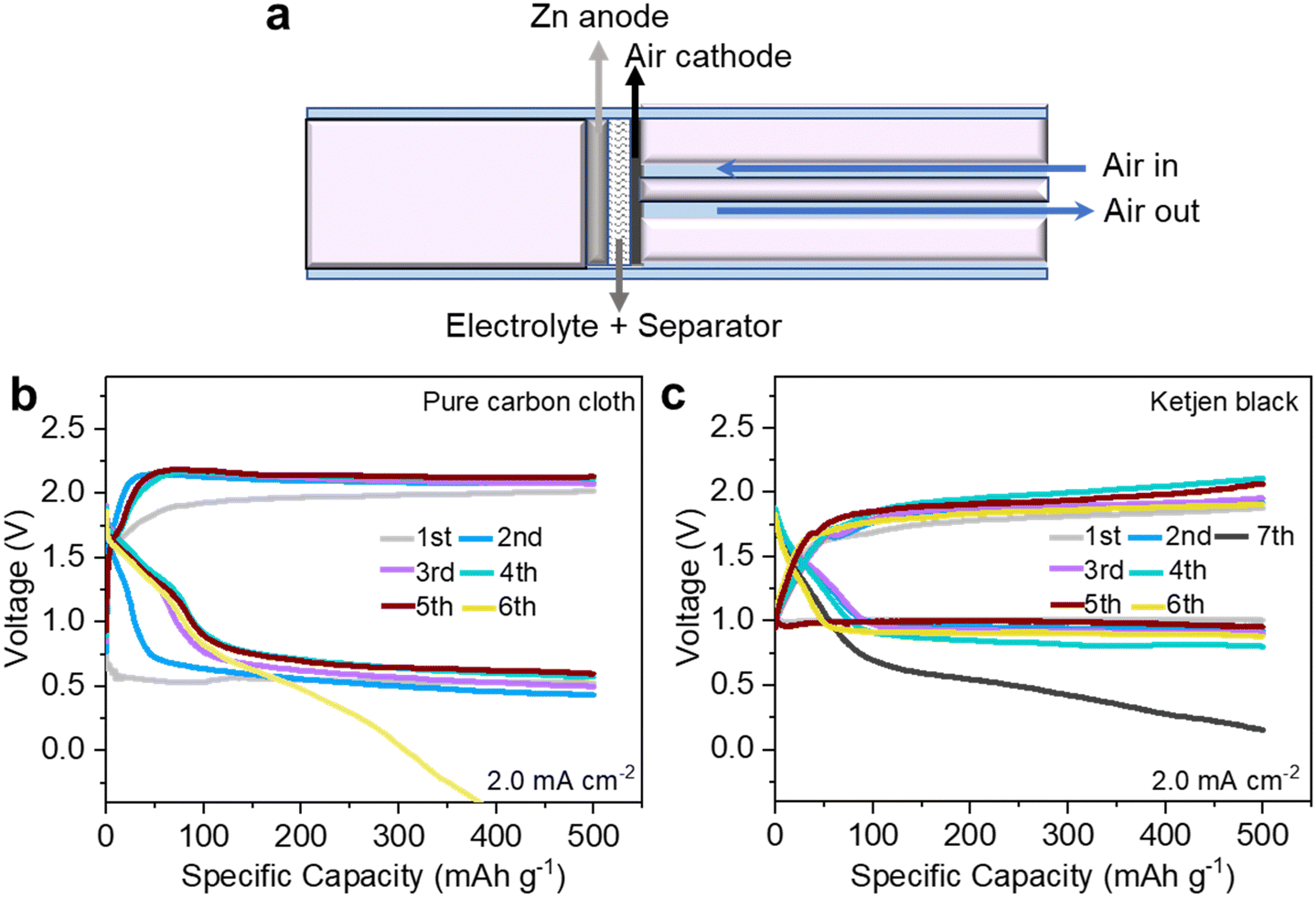 | ||
| Fig. 1 Schematic illustration and photograph of (a) the bulky Swagelok-type NNZAB; the cycling performance of NNZABs with pure carbon cloth (b) and ketjen black (c) as the cathode. | ||
Conventional catalysts such as metallic Pt and Ir atoms were copiously researched.31 Pt and Ir atoms would be oxidized to PtO and IrOx during the ORR. These oxides could offer the active phases for the oxygen evolution reaction (OER).32,33 However, the oxidation reaction of such catalysts would vastly decrease the probability of the combination between oxygen and Zn ions, unsuitable for the catalysts for NNZABs with the Zn–O2/ZnO2 chemistry. Defect engineering is a critical strategy to boost the activity of electrocatalysts for metal–air batteries.34–37 Exotic bonding states exist in Mo4/3B2−xTz MBene because of the electron-deficient character of boron compared with carbon, enabling unique geometrical and electronic structures of Mo4/3B2−xTz MBene.38,39 Mo4/3B2−xTz MBene with ordered Y vacancies and deficient occupation of the B sites, potentially serving as highly active sites for the ORR and OER, has excellent potential to be applied in NNZABs.
Herein, we advance NNZABs with a coin-cell configuration with a reversible Zn–O2/Zn peroxide (ZnO2) chemistry based on the innovation of a two-dimensional (2D) transition metal boride for the first time, referred to as Mo4/3B2−xTz MBene. We found that the Y vacancies have a priority to adsorb O2 than the exposed Mo or B sites, even if they all present O2-adsorbability. Based on experimental and theoretical evidence, the Y vacancies favor reversible 2e− ORR and OER processes, further presenting proof to the Zn–O2/Zn peroxide (ZnO2) chemistry. Mo4/3B2−xTz MBene gives rise to the substantially enhanced electrochemical performance of NNZABs. As a result, the resulting NNZAB incorporating a well-designed Mo4/3B2−xTz catalyst delivers a gravimetric energy density of 730 mA h g−1 and exhibits a long-lasting stability of 380 h at a current density of 2 mA cm−2.
Results and discussion
The synthesis process of Mo4/3B2−xTz MBene is illustrated in Fig. 2a. The (Mo2/3Y1/3)2AlB2 (MAB) phase was firstly etched with HF aqueous solution, triggering an exothermal reaction between the HF and the three-dimensional (3D) MAB phase.39 As shown in Fig. S1a (ESI†), the (Mo2/3Y1/3)2AlB2 precursor shows highly packed structure morphology as characteristic MAX phases. The EDX spectra in Fig. S1b (ESI†) suggest that the laminated (Mo2/3Y1/3)2AlB2 is composed of Mo, Y, Al, and B. The HF etching can selectively remove the Al layers and the in-plane ordered Y atoms in the MAB phase.39–41 Moreover, as observed from the schematic atomic arrangement in Fig. 2a, the ordered Y vacancies and point defects would be introduced after HF treatment, potentially leading to additional electrochemically active sites to the NNZABs.42–44 A multi-layered crystal morphology has been evidenced by scanning electron microscopy (SEM) in Fig. S2 (ESI†). Next, the etched multilayer products are treated with bath sonication, spontaneously delaminating in water under handshaking. Then a colloidal suspension of the delaminated MBene can be acquired, showing an obvious Tyndall effect45 through illuminating light on the MBene dispersed in water, as presented in Fig. S3a (ESI†). It reveals that the MBene can highly homogeneously disperse in water, contributing to the contact between the MBene cathode and the Zn(OTf)2-in-water electrolyte. A few-layer MBene flake is observed from the overview SEM image shown in Fig. S3b (ESI†). The corresponding energy dispersive X-ray (EDX) spectra are shown in Fig. S3c and d (ESI†), exhibiting an atomic molar ratio of Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Y of 22.82:1, which evidences the additional removal of the Y atoms. Furthermore, transmission electron microscopy (TEM) (Fig. S4a, ESI†) was performed to observe the final delaminated Mo4/3B2−xTz, which exhibits a few-layer sheet morphology with a 500 nm width. A d-spacing value (d) of 0.755 nm is calculated from the high-resolution transmission electron microscopy (HRTEM) image (Fig. S4b, ESI†), confirming the exposure of (1000) planes and consistent with the space group R
:Y of 22.82:1, which evidences the additional removal of the Y atoms. Furthermore, transmission electron microscopy (TEM) (Fig. S4a, ESI†) was performed to observe the final delaminated Mo4/3B2−xTz, which exhibits a few-layer sheet morphology with a 500 nm width. A d-spacing value (d) of 0.755 nm is calculated from the high-resolution transmission electron microscopy (HRTEM) image (Fig. S4b, ESI†), confirming the exposure of (1000) planes and consistent with the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (no. 166) of hexagonal symmetry.39
m (no. 166) of hexagonal symmetry.39
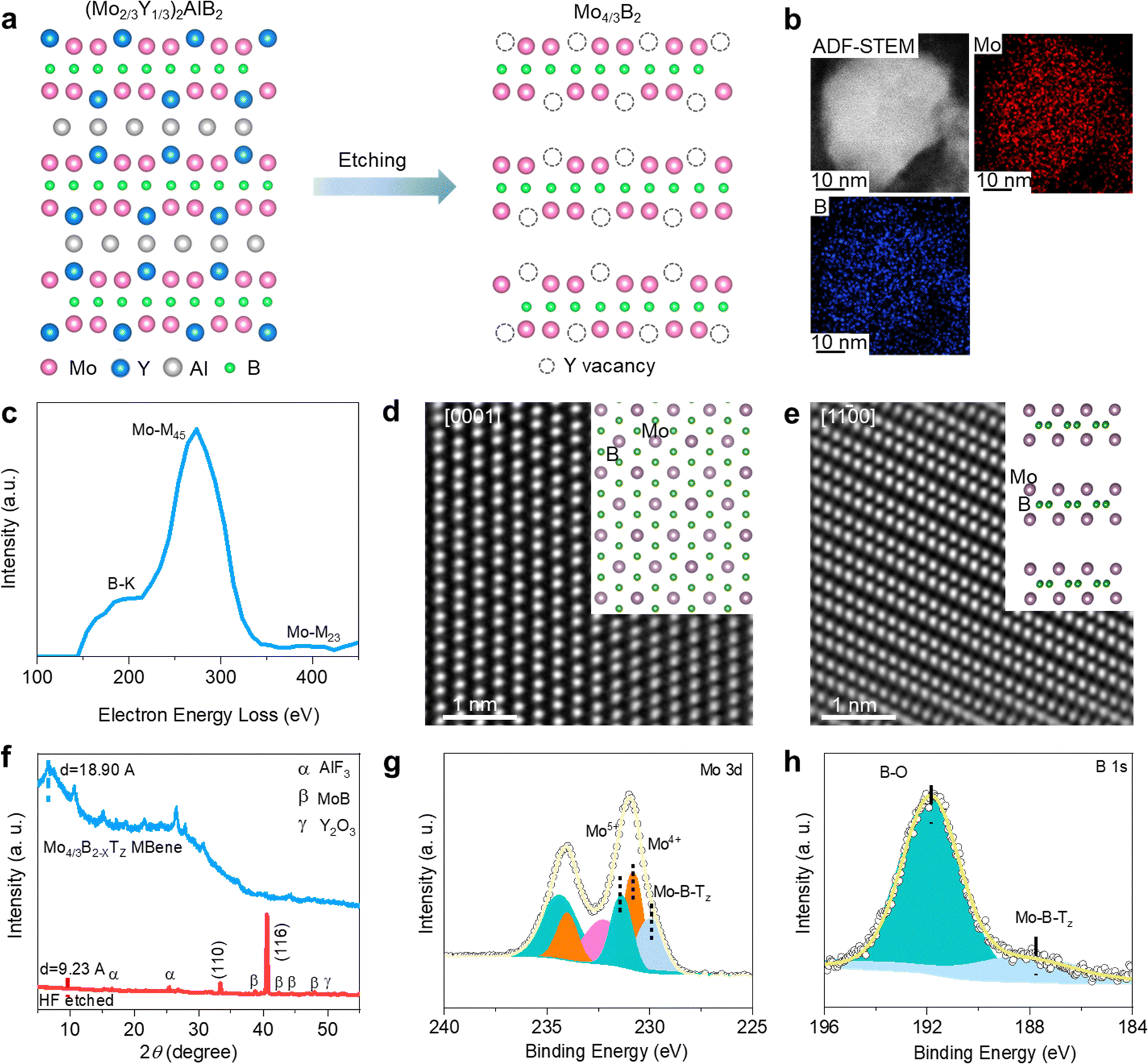 | ||
| Fig. 2 Fabrication and characterization of the 2D Mo4/3B2−xTz MBene. (a) The schematic atomic structure of (Mo2/3Y1/3)2AlB2 before etching (left) and after etching (right). (b) EDX mapping images, (c) EELS spectra, and (d and e) high-resolution HAADF-STEM images with their atomic arrangement (insets) of the few-layer Mo4/3B2−xTz MBene nanosheets. (f) XRD pattern of (Mo2/3Y1/3)2AlB2 after (red) HF etching and after delamination (blue); high-resolution XPS spectra of Mo4/3B2−xTz with peak fittings for the Mo 3d (g) and B 1s (h) regions. | ||
The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Mo4/3B2−xTz MBene is presented in Fig. 2b. The few-layer structure guarantees sufficient contact between the charged species and active sites. The electron energy loss spectroscopy (EELS) mapping images shown in Fig. 2b further demonstrate the homogeneous distribution of Mo and B in the Mo4/3B2−xTz MBene layer. As shown in Fig. 2c, the corresponding EELS spectra in Fig. 2b present the existence of Mo and B after the removal of the C signal. What is more, the Y signal cannot be detected, confirming the absence of Y and the presence of the resulting ordered Y vacancies. We acquired the HAADF-STEM images of Mo4/3B2−xTz MBene along the [0001] (Fig. 2d) and [1–100] zone axes (Fig. 2e), respectively. The brightest atoms are Mo atoms because B is so light that it cannot be detected in imaging situations. An obvious close-packed (hexagonal) structure is observed from the top view in Fig. 2d. From the direction of the [1–100] zone axis, Mo atoms and Y vacancies are overlapped. They thus display an ordered atomic arrangement without prominent vacancies at Y sites in the HAADF-STEM image in Fig. 2e.
The corresponding X-ray diffraction (XRD) results of Mo4/3B2−xTz MBene before and after delamination are shown in Fig. 2f, in which the (000l) peak becomes broader and downshifts from 2θ = 9.35° to a lower angle of 2θ = 4.74°, in correspondence with the increased d from 9.23 Å to 18.90 Å triggered by the intercalation process. This enlarged d value of the (000l) plane is a common phenomenon in MXenes prepared by HF etching of a MAX phase precursor.25,45,46 In addition, the corresponding low-intensity peaks belonging to the AlF3, MoB, and Y2O3 impurities also appear in the XRD results, illustrating their existence in the final Mo4/3B2−xTz MBene, which can be attributed to the incomplete reaction between the MAB phase and HF. The X-ray photoelectron spectroscopy (XPS) spectrum of Mo 3d (Fig. 2g) shows the presence of Mo5+ (232 eV), Mo4+ (231 eV), and Mo–B–Tz (229 eV). Similarly, Fig. 2h reveals the signals from B–O (191.6 eV) and Mo–B–Tz (187.5 eV). Therefore, the XPS results in Fig. 2g and h further confirmed the delaminated 2D material as Mo4/3B2−xTz. The few-layer Mo4/3B2−xTz MBene was prepared successfully and ordered vacancies can be formed after removing Y atoms, exhibiting high potential to improving the catalytic activity of Mo4/3B2−xTz.
The Brunauer–Emmett–Teller (BET) method was employed to determine the specific surface area and pore size from the nitrogen adsorption and desorption isotherm (Fig. S5, ESI†). Fig. S5a and c (ESI†) present the BET surface areas of (Mo2/3Y1/3)2AlB2 MAB and Mo4/3B2−xTz MBene, which were calculated to be 1.90 and 14.32 m2 g−1, respectively. The pore size distribution plots of (Mo2/3Y1/3)2AlB2 MAB and Mo4/3B2−xTz MBene are displayed in Fig. S5b and d (ESI†), among which the average adsorption pore diameters are 22.67 nm and 11.08 nm, respectively. The larger specific surface area of Mo4/3B2−xTz MBene is ascribed to the removal of the Al layer of (Mo2/3Y1/3)2AlB2. Thus, Mo4/3B2−xTz MBene with a porous structure and large specific surface area can supply vast amounts of ion transport channels and plenty of active sites for the ORR and OER at the surface of the electrodes.
To evaluate the electrocatalytic properties of Mo4/3B2−xTz MBene as the NNZAB cathode, a commercial catalyst, PtC + IrO2, was employed as the reference cathode of the NNZAB (denoted as NNZAB-PtC + IrO2; details are provided in the ESI†). As presented in Fig. 3a, there is a significant reduction peak in the cyclic voltammograms (CVs) of both samples assembled in the coin-cell NNZABs with Zn(OTf)2 electrolyte. The peak potential and current density of the MBene cathode are obviously larger than those of the PtC + IrO2 cathode, demonstrating the higher oxygen reduction activity of the MBene and further proving the low polarization and fast kinetics of the MBene-catalyzed NNZAB. Electrochemical impedance spectroscopy (EIS) measurements of the NNZAB-MBene and NNZAB-PtC + IrO2 suggest that the charge transfer resistance (Rct) of the prepared Mo4/3B2−xTz MBene is closer to that of the commercial PtC + IrO2, which are 45 and 60 Ω, respectively (Fig. S6, ESI†). At room temperature (25 °C), the NNZAB-MBene stably maintains an open-circuit voltage (OCV) of 0.997 V for 2500 seconds (Fig. 3b), approximately the theoretical value of NNZABs (1.16 V). In contrast, the OCV of NNZAB-PtC + IrO2 exhibits a slight decrease to 0.94 V due to self-discharge. The excellent stability of the NNZAB-MBene can be concluded when compared to the OCV plots of the NNZAB-MBene and NNZAB-PtC + IrO2.
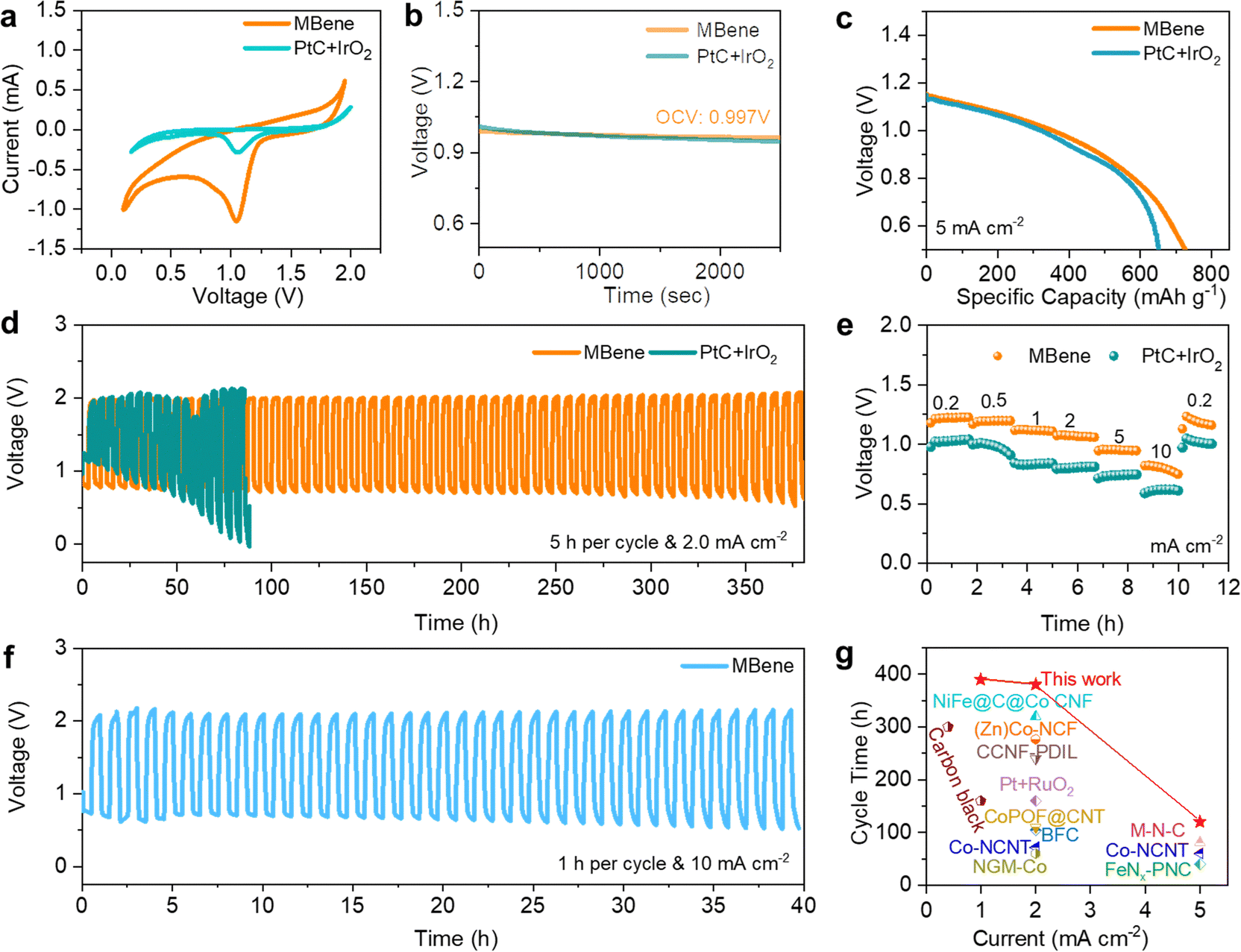 | ||
| Fig. 3 Electrochemical performance of Mo4/3B2−xTz MBene. (a) CV curves of the NNZAB-MBene and NNZAB-PtC + IrO2 between 0.1 and 2.0 V at a scan rate of 0.1 mV s−1; (b) the OCV plot with an inset showing a stable voltage output of the NNZAB-MBene and NNZAB-PtC + IrO2; (c) discharge capacity plots of the NNZAB-MBene and NNZAB-PtC + IrO2 recorded at a current density of 5 mA cm−2; (d) the cycling performance of the NNZAB-MBene and NNZAB-PtC + IrO2 at a current density of 2 mA cm−2 with a 5 h charging and discharging duration in each cycle; (e) rate performance of the NNZAB-MBene and NNZAB-PtC + IrO2 at various current densities; (f) the cycling performance of the NNZAB-MBene at a current density of 10 mA cm−2 with a 1 h cycling period; (g) the comparison of the cycling stability of the MBene-catalyzed NNZAB in this work and other reported catalysts in various electrolytes, including Co-nitrogen doped carbon nanotubes (denoted as Co-NCNT), bamboo-shaped fibrous catalysts (denoted as BFC), highly graphitic 2D porous nitrogen-doped carbon (PNC) embedded with Fe–Nx species (denoted as FeNx-embedded PNC), Co catalytic units with a porphyrin covalent organic framework (POF) embedded with carbon nanotubes (denoted as CoPOF@CNT), Ni–Fe–N–C@C@Co–N–C carbon nanofibers (denoted as NiFe@C@Co CNF), dual-cation ionic liquid (PDIL) within a 3D porous cationic cellulose nanofiber matrix (CCNF) (denoted as CCNF-PDIL), Co/N/O tri-doped graphene mesh (denoted as NGM-Co), Zn/Co–N–C-based freestanding catalyst (denoted as (Zn)Co–NCF), and metal–nitrogen complex carbon (denoted as M–N–C). Detailed references are given in Table S1 (ESI†). | ||
Fig. 3c shows the first galvanostatic discharge profiles of the NNZAB-PtC + IrO2 and the NNZAB-MBene at a current density of 5 mA cm−2 and a cut-off discharge voltage limited to 0.5 V versus Zn2+/Zn. The NNZAB-MBene delivers a capacity of 730 mA h gzn−1, higher than the value of 652 mA h gzn−1 for NNZAB-PtC + IrO2, implying the superior electrocatalytic activity of Mo4/3B2−xTz MBene. The coin-cell NNZABs were also electrically cycled to test the rechargeability of the NNZAB-MBene and NNZAB-PtC + IrO2. The NNZAB-MBene maintained a stable operation in ambient air for 380 hours at a current density of 2 mA cm−2 over a 5 h cycling period, presenting a four times longer cycling lifespan than that of NNZAB-PtC + IrO2 (Fig. 3d). The lower overpotential of NNZAB-PtC + IrO2 in the first 5 cycles (Fig. 3d) is attributed to the good conductivity of PtC + IrO2 and fast infiltration of electrolytes on the PtC + IrO2 cathode. However, Pt and Ir atoms would be oxidized to PtO and IrOx during the ORR. These oxides could offer the active phases for the OER. The oxidation reaction of such catalysts would vastly decrease the probability of the combination between oxygen and Zn ions, which makes them unsuitable as catalysts for NNZABs with Zn–O2/ZnO2 chemistry. Thus, NNZAB-PtC + IrO2 shows a lower overpotential in the first 5 cycles but achieves a shorter cycling lifespan. Fig. 3e displays that the NNZAB-MBene also shows higher discharging voltage plateaus when tested under different current densities and superior resilience capability when compared with NNZAB-PtC + IrO2. In addition, as shown in Fig. S7a (ESI†), a stable cycle life up to nearly 400 hours with an ultralow overpotential of 0.78 V has been achieved for the NNZAB-MBene under 1 mA cm−2 with a 10 min charge and discharge duration per cycle. As the NNZAB-MBene operates at higher current densities of 3 and 5 mA cm−2 with an ultralong 20 hour cycle period, the NNZAB-MBene still keeps stable cycle lifespans of 340 hours and 120 hours, respectively, as shown in Fig. S7b and c (ESI†). Additionally, the NNZAB-MBene shows a stable lifespan of 40 hours when cycled at an increased current density of 10 mA cm−2 (Fig. 3f). As the current density increases, the performance of the NNZAB-MBene is predominantly controlled by mass transfer at the interface. Note that there is no significant increase in the overpotential of the NNZAB-MBene when large current densities were applied (e.g., 0.78 V @ 1.0 mA cm−2 and 1.1 V @ 5.0 mA cm−2, and 1.2 V @ 10.0 mA cm−2), suggesting its high electrocatalytic performance even at large current densities. To compare, the cycling lifespans of other ZABs and the MBene-catalyzed NNZAB are summarized in Fig. 3g. Evidently, the as-fabricated NNZAB-MBene delivered the most stable long cycling lifespan compared to those in other works,7,29 demonstrating the successful application of the Mo4/3B2−xTz MBene catalyst and proving the considerable potential of the NNZAB-MBene cell for a sustainable energy output. Interestingly, the developed pouch cell assembled by the MBene cathode can power an electronic timer even under bending conditions (Fig. S8, ESI†), demonstrating the enormous potential of our quasi-solid-state NNZAB-MBene as a reliable power supply system.
For Zn–air cells, the impedance consists of solid electrolyte impedance (RSEI), charge transfer resistance (Rct) and Zn ion transfer resistance (Wzn). The value of RSEI mainly comes from the ionic impedance of the electrolyte and the electronic impedance of the electrode. As shown in Fig. S9 (ESI†), there is no obvious variation of RSEI during various discharge and charge stages, indicating that the formation of the discharge product ZnO2 had negligible effects on the electronic impedance of the electrode. The maintenance of good contact between the electrode and electrolyte contributes to a satisfactory cycling lifespan of the NNZAB-MBene. The electrocatalytic role of Mo4/3B2−xTz MBene in the cycling stability and polarization of the NNZAB is also validated by performing ex situ measurements on the disassembled MBene cathodes with the control of discharged and charged states (Fig. 4a). Ex situ XRD measurements on Mo4/3B2−xTz MBene-based cathodes under pivotal stages are carried out during discharging and charging after 5 cycles at 2 mA cm−2. As illustrated in Fig. 4b, the intensities of the XRD peaks at 32.4° and 38.1° consistent with the (110) and (200) crystal planes of ZnO2 (PDF 76-1364)47 gradually become stronger from the pristine state to the completed discharged state c, then show a decreasing trend and finally disappear after being fully charged at state f. It is firmly evidenced that Mo4/3B2−xTz MBene could enable the high reversibility of ZnO2-based conversion chemistry when cycled in the Zn(OTf)2 electrolyte.
 | ||
| Fig. 4 Exploring the electrocatalytic reaction mechanisms of NNZABs at a current density of 2.0 mA cm−2. The charge–discharge curves (a) of the NNZAB-MBene at 2.0 mA cm−2 and the corresponding ex situ XRD patterns (b) of the Mo4/3B2−xTz MBene cathode; Zn 2p (c) and O 1s (d) XPS spectra of the MBene cathodes obtained from the NNZABs in the pristine state, after the 1st discharged state, and after the 1st charged state; SEM image of the MBene cathode in the pristine state (e), after the 1st discharged state (f), and after the 1st charged state (g). | ||
The XPS survey spectra presented in Fig. S10a (ESI†) also prove the formation/decomposition of ZnO2 on the MBene cathode. The high-resolution XPS spectra of Zn 3d (Fig. 4c), O 1s (Fig. 4d) and Mo 3d (Fig. S10b and c, ESI†) for the MBene cathodes obtained from the NNZABs in the pristine state, after the 1st discharged state, and after the 1st charged state show results consistent with the XRD data. The dominant peak at 532 eV shown in Fig. 4d belongs to the well-crystallized ZnO2, which further proves highly reversible ZnO2 generation and decomposition chemistry for NNZABs.48 As shown in the high-resolution XPS spectra of Mo 3d in Fig. S10b and c (ESI†), the peak at 235 eV which belongs to Mo4+ shows an obvious intensity decay after the cathode discharging and recovers after charging. This is because the Zn2+ and O2 prefer to first react on the vacancy sites in Mo4/3B2−xTz. As demonstrated in the SEM image in Fig. 4e, the pristine cathode shows a 2D nanosheet structure of the MBene. Upon discharge, the discharge product ZnO2 adsorbed on the active sites of MBene nanosheets (Fig. 4f), showing a particle decorated morphology. Upon recharge, the ZnO2 completely disappeared and the cathode recovers the 2D nanosheet Mo4/3B2−xTz MBene structure (Fig. 4g), indicating the complete decomposition of ZnO2 during charging. To sum up, all the collected experimental data can strongly support that our rationally designed MBene catalysts could serve as reliable and highly efficient O2-breathing cathodes for Zn(OTf)2-based ZABs, shedding light on MBenes as ORR and OER electrocatalysts in NNZAB systems.
To explore the catalytic activity of the Mo4/3B2−xTz MBene and further determine the formation/decomposition kinetics of ZnO2 during the ORR/OER process, DFT calculations are performed with an emphasis on the adsorption energies of reaction species and possible reaction pathways.49–51Fig. 5a illustrates the ideal interaction sites between the discharged species (O2, Zn2+, ZnO2) and the Mo4/3B2−xTz MBene host. There are three different locations above B, Mo atoms, and Y vacancies, respectively. It is well known that inherent mutual attraction can be defined as a spontaneous behavior if the adsorption energies (Ead) between the interaction sites and hosts are negative. As shown in Fig. 5b, the Ead values of ZnO2 on the B, Mo, and etched Y sites are −8.90, −8.14, and −9.42 eV, respectively. Among all the possible interaction sites, the vacancies at Y sites are the lowest in energy. Hence, the possible reaction pathways were conducted on the vacancies at Y sites. As displayed in Fig. 5c, we also used the calculation of density of states (DOS) near the Femi energy (Ef) around Mo atoms to explore the advantages of the fabricated MBene catalysts, which further proved the metallic characteristics of the MBene featuring competent electron conduction.
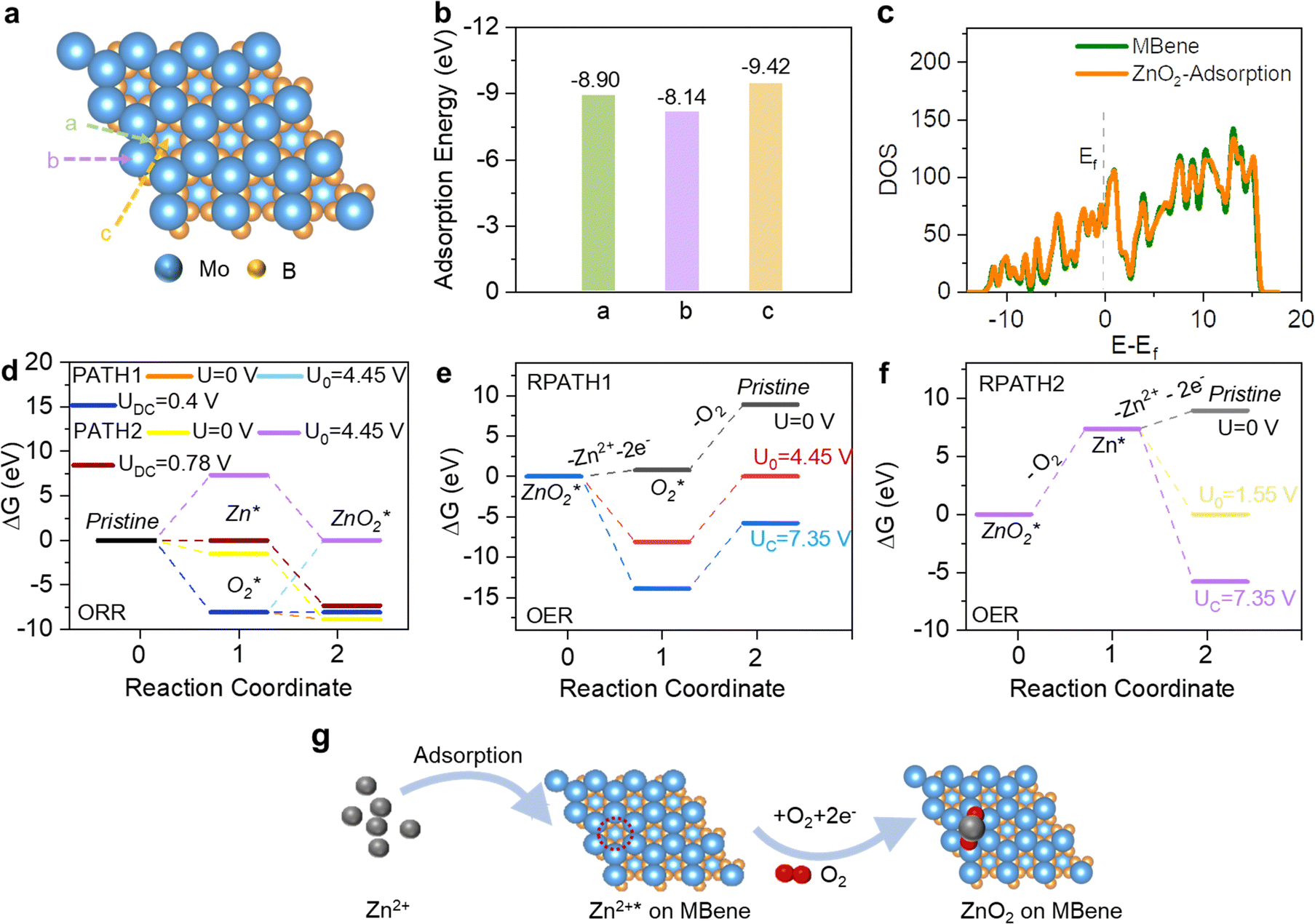 | ||
| Fig. 5 Reaction pathways elucidated by theoretical calculations. (a) Top-view crystal structure of Mo4/3B2−xTz MBene nanosheets and the ideal interactive sites. (b) The summarized adsorption energy of ZnO2 on Mo4/3B2−xTz MBene flakes. (c) Total density of states (DOS) profiles of the Mo4/3B2−xTz MBene before and after ZnO2 species adsorption. Calculated energy diagram of (d) the (0001) plane of the Mo4/3B2−xTz MBene for the ORR via PATH1 and PATH2. Calculated energy diagram of RPATH1 (e) for OER processes. Calculated energy diagram of RPATH2 (f) for OER processes. (g) Schematic illustration of the ORR pathways for ZnO2 generation on the Mo4/3B2−xTz MBene surface. | ||
According to the ex situ XRD and XPS analyses of the charge/discharge processes, the ORR/OER reactions occur on the vacancies at the metal sites of the MBene, which are investigated by DFT simulations. Fig. 5d–f illustrates the energy diagrams of different reaction pathways. The formation of the discharge product starts from the adsorption of Zn2+ (Ead = −1.55 eV) and O2 (Ead = −8.10 eV) on the cathode surface via a two-electron step to form ZnO2, which has been evidenced by the final product in the cathode reaction. Two ORR reaction pathways occurring on the Mo4/3B2−xTz MBene surface are elucidated as follows:
| PATH1: pristine → O2* → ZnO2* |
| PATH2: pristine → Zn2+* → ZnO2* |
As observed in Fig. 5d, an overpotential ηORR = 3.675 V is acquired for PATH2, revealing that the ORR process originates from the vacancies adsorbing Zn2+. The phase diagram in Fig. 5d reveals that the formation of ZnO2via a two-electron step is thermodynamically favored above 3.675 V. When the voltage drops to 3.675 V, the nucleation of ZnO2 would take the lead in the ORR step. We also calculate the OER reaction pathway based on reversing PATH1 and PATH2, referring to RPATH1 (Fig. 5e) and RPATH2 (Fig. 5f), respectively. RPATH1 and RPATH2 have the same equilibrium potential U0 = 4.45 V, but they are different in free energy variation and the minimum charging potential. The first step (ZnO2* – O2 → Zn2+*) of RPATH2 presents a high positive free energy of 7.35 eV (U0 = 4.45 V). It is difficult to achieve this step thermodynamically. In contrast, the decomposition process of ZnO2* on the Y sites of the MBene surface is at a greater probability to occur owing to the lower free energy of 4.45 eV.
The formation/decomposition mechanism of ZnO2 on the Mo4/3B2−xTz MBene electrode is summarized in Fig. 5g. During the ORR process, generation of ZnO2 starts from the adsorption of Zn2+ on the vacancies at the Y sites of the MBene, which then acquires electrons and absorbs O2 stored in the vacancies on the electrode surface to form ZnO2. Conversely, the discharge product ZnO2 would decompose following a stepwise reaction pathway during the OER process, which is attributed to the superior stabilizing effects and high adsorption ability of MBene catalysts toward O2 and ZnO2 species.
Conclusions
In summary, the highly reversible NNZABs based on ZnO2 chemistry are promising and previous work has only achieved Zn–O2/ZnO2 chemistry in the Swagelok-type cell configuration, which is bulky and costly. This work proposes to achieve a highly reversible ZnO2 electrochemical process in coin-type and pouch cell-type NNZABs by designing an efficient 2D transition metal boride catalyst with ordered vacancies at the etched metal sites as the cathode. The Y vacancies of Mo4/3B2−xTz MBene obtained after HF etching were investigated to have the adsorption priority toward O2 and ZnO2 species, mainly enhancing the formation/decomposition of ZnO2 during the ORR and OER. Consequently, the rate capability and cycling endurance of the NNZABs significantly are optimized when compared to commercial PtC–IrO2 catalysts. The NNZAB-MBene supported a stable ZAB operation in ambient air and presented higher reversibility for 380 hours at a current density of 2 mA cm−2 with a 5 hour cycling duration and delivered decent cycle life at higher current densities, including achieving a lifespan of 340 hours and 120 hours at 3 and 5 mA cm−2, respectively. The obtained outstanding electrochemical performance is ascribed to the superior stabilizing effects and high adsorption ability of Mo4/3B2−xTz MBene catalysts toward O2 and ZnO2. We clearly elucidate the formation/decomposition mechanism of ZnO2 on the Mo4/3B2−xTz MBene electrode. This work can inspire researchers to explore highly efficient catalysts for NNZABs in the coin or pouch cell configuration, showing applicability in portable devices powered by NNZABs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key R&D Program of China under Project 2019YFA0705104 and Guangdong Basic and Applied Basic Research Foundation under Project 2022B1515120019.References
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46 CrossRef CAS PubMed.
- C.-X. Zhao, J.-N. Liu, J. Wang, N. Yao, X.-Y. Li, X. Chen, B.-Q. Li and Q. Zhang, Angew. Chem., 2022, 134, 1 Search PubMed.
- Z. Song, J. Ding, B. Liu, Y. Shen, J. Liu, X. Han, Y. Deng, C. Zhong and W. Hu, Chem. Eng. J., 2022, 429, 132331 CrossRef CAS.
- H. Jiang, J. Xia, L. Jiao, X. Meng, P. Wang, C.-S. Lee and W. Zhang, Appl. Catal., B, 2022, 310, 121352 CrossRef CAS.
- L. Yang, X. Zhang, L. Yu, J. Hou, Z. Zhou and R. Lv, Adv. Mater., 2022, 34, e2105410 CrossRef PubMed.
- W. Sun, F. Wang, P. Bieker and M. Winter, Angew. Chem., 2022, 134, 1 Search PubMed.
- Z. Pei, C. Wang, S. Zhao, J. Fei, L. Wei, J. Chen, C. Wang, R. Qi, Z. Liu and Y. Chen, Angew. Chem., 2020, 132, 4823 CrossRef.
- X. Zhang, J. Wang, C. Dai, X. Jin, Y. Zhao and L. Qu, J. Power Sources, 2022, 538, 231563 CrossRef CAS.
- J. N. Liu, C. X. Zhao, D. Ren, J. Wang, R. Zhang, S. H. Wang, C. Zhao, B. Q. Li and Q. Zhang, Adv. Mater., 2022, 34, e2109407 CrossRef PubMed.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W. C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- Z. Li, L. Lv, X. Ao, J.-G. Li, H. Sun, P. An, X. Xue, Y. Li, M. Liu, C. Wang and M. Liu, Appl. Catal., B, 2020, 262, 118291 CrossRef CAS.
- H. Pang, P. Sun, H. Gong, N. Zhang, J. Cao, R. Zhang, M. Luo, Y. Li, G. Sun, Y. Li, J. Deng, M. Gao, M. Wang and B. Kong, ACS Appl. Mater. Interfaces, 2021, 13, 39458 CrossRef CAS PubMed.
- J. Cao, H. Gong, L. Xie, Y. Li, N. Zhang, W. Tian, R. Zhang, J. Zhou, T. Wang, Y. Zhai, N. Li, M. Luo, K. Liang, P. Chen and B. Kong, Mater. Today Energy, 2021, 20, 100682 CrossRef CAS.
- J. Yi, P. Liang, X. Liu, K. Wu, Y. Liu, Y. Wang, Y. Xia and J. Zhang, Energy Environ. Sci., 2018, 11, 3075 RSC.
- S. Hosseini, S. Masoudi Soltani and Y.-Y. Li, Chem. Eng. J., 2021, 408, 127241 CrossRef CAS.
- C. Shao, Y. Zhao and L. Qu, SusMat, 2022, 2, 142 CrossRef CAS.
- Z. Pei, L. Ding, C. Wang, Q. Meng, Z. Yuan, Z. Zhou, S. Zhao and Y. Chen, Energy Environ. Sci., 2021, 14, 4926 RSC.
- M. Zhu, Nano Res. Energy, 2023, 2, e9120038 CrossRef.
- J.-N. Liu, C.-X. Zhao, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Energy Environ. Sci., 2022, 15, 4542 RSC.
- T. W. S. Chen, L. Ma, B. Zhou, J. Wu, D. Zhu, Y. Yang Li, J. Fan and C. Zhi, Chem, 2023, 9, 1 Search PubMed.
- A. R. Mainar, O. Leonet, M. Bengoechea, I. Boyano, I. de Meatza, A. Kvasha, A. Guerfi and J. A. Blázquez, Int. J. Energy Res., 2016, 40, 1032 CrossRef CAS.
- X. Yang, X. Zheng, H. Li, B. Luo, Y. He, Y. Yao, H. Zhou, Z. Yan, Y. Kuang and Z. Huang, Adv. Funct. Mater., 2022, 32, 2208994 CrossRef.
- Q. Wei, Y. Fu, G. Zhang and S. Sun, Curr. Opin. Electrochem., 2017, 4, 45 CrossRef CAS.
- Z. Zhao, X. Fan, J. Ding, W. Hu, C. Zhong and J. Lu, ACS Energy Lett., 2019, 4, 2259 CrossRef CAS.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33, e2007406 CrossRef PubMed.
- S.-M. Lee, Y.-J. Kim, S.-W. Eom, N.-S. Choi, K.-W. Kim and S.-B. Cho, J. Power Sources, 2013, 227, 177 CrossRef CAS.
- Y. Liang, D. McLaughlin, C. Csoklich, O. Schneider and A. S. Bandarenka, Energy Environ. Sci., 2019, 12, 351 RSC.
- D. Malko, A. Kucernak and T. Lopes, Nat. Commun., 2016, 7, 13285 CrossRef CAS PubMed.
- Z. Pei, Y. Huang, Z. Tang, L. Ma, Z. Liu, Q. Xue, Z. Wang, H. Li, Y. Chen and C. Zhi, Energy Storage Mater., 2019, 20, 234 CrossRef.
- Y. Ma, Z. Guo, X. Dong, Y. Wang and Y. Xia, Angew. Chem., 2019, 58, 4622 CrossRef CAS PubMed.
- Z. C. J. Zhu, M. Xie, Z. Lyu, M. Chi, M. Mavrikakis, W. Jin and Y. Xia, Angew. Chem., 2019, 131, 7322 CrossRef.
- V. P. V. J. D. Cooper and D. L. Whalen, J. Am. Chem. Soc., 1971, 93, 6296 CrossRef.
- L. T. R. L. Zhang, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412 CrossRef CAS PubMed.
- F. Guo, M. Zhang, S. Yi, X. Li, R. Xin, M. Yang, B. Liu, H. Chen, H. Li and Y. Liu, Nano Res. Energy, 2022, 1, e9120027 CrossRef.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- S. Peng, X. Han, L. Li, S. Chou, D. Ji, H. Huang, Y. Du, J. Liu and S. Ramakrishna, Adv. Energy Mater., 2018, 8, 1800612 CrossRef.
- M. Wu, G. Zhang, L. Du, D. Yang, H. Yang and S. Sun, Small Methods, 2021, 5, e2000868 CrossRef PubMed.
- A. Biswas, S. Bhardwaj, T. Boruah and R. S. Dey, Mater. Adv., 2022, 3, 5207 RSC.
- J. Zhou, J. Halim, M. Dahlqvist, Q. Tao, I. Persson, L. Hultman, P. O. Å Persson and J. Rosen, Science, 2021, 373, 801 CrossRef CAS PubMed.
- M. Dahlqvist, Q. Tao, J. Zhou, J. Palisaitis, P. O. A. Persson and J. Rosen, J. Am. Chem. Soc., 2020, 142, 18583 CrossRef CAS PubMed.
- K. Kim, C. Chen, D. Nishio-Hamane, M. Okubo and A. Yamada, Chem. Commun., 2019, 55, 9295 RSC.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., 2015, 54, 7399 CrossRef CAS PubMed.
- Y. Hou, J. Wang, J. Liu, C. Hou, Z. Xiu, Y. Fan, L. Zhao, Y. Zhai, H. Li, J. Zeng, X. Gao, S. Zhou, D. Li, Y. Li, F. Dang, K. Liang, P. Chen, C. Li, D. Zhao and B. Kong, Adv. Energy Mater., 2019, 9, 1901751 CrossRef CAS.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807 CrossRef CAS PubMed.
- Z. Chen, X. Li, D. Wang, Q. Yang, L. Ma, Z. Huang, G. Liang, A. Chen, Y. Guo, B. Dong, X. Huang, C. Yang and C. Zhi, Energy Environ. Sci., 2021, 14, 3492 RSC.
- X. Li, X. Ma, Y. Hou, Z. Zhang, Y. Lu, Z. Huang, G. Liang, M. Li, Q. Yang, J. Ma, N. Li, B. Dong, Q. Huang, F. Chen, J. Fan and C. Zhi, Joule, 2021, 5, 2993 CrossRef CAS.
- M. Sun, W. Hao, C. Wang and T. Wang, Chem. Phys. Lett., 2007, 443, 342 CrossRef CAS.
- Q. Guo, Q. Zhang, H. Wang and Z. Zhao, Catal. Commun., 2018, 103, 24 CrossRef CAS.
- B. He, G. Li, J. Li, J. Wang, H. Tong, Y. Fan, W. Wang, S. Sun and F. Dang, Adv. Energy Mater., 2021, 11, 2003263 CrossRef CAS.
- G. Li, N. Li, S. Peng, B. He, J. Wang, Y. Du, W. Zhang, K. Han and F. Dang, Adv. Energy Mater., 2020, 11, 2002721 CrossRef.
- G. Zhang, C. Liu, L. Guo, R. Liu, L. Miao and F. Dang, Adv. Energy Mater., 2022, 12, 2200791 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee01297b |
| ‡ Y. H. and Z. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |