
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermal modulation of reaction equilibria controls mass transfer in CO2-binding organic liquids†
Thomas
Moore
 *a,
Anthony J.
Varni
a,
Simon H.
Pang
a,
Sneha A.
Akhade
a,
Sichi
Li
a,
Du T.
Nguyen
a and
Joshuah K.
Stolaroff
b
*a,
Anthony J.
Varni
a,
Simon H.
Pang
a,
Sneha A.
Akhade
a,
Sichi
Li
a,
Du T.
Nguyen
a and
Joshuah K.
Stolaroff
b
aLawrence Livermore National Laboratory, 7000 East Avenue, Livermore, CA 94550, USA. E-mail: moore260@llnl.gov
bMote Hydrogen, Los Angeles, CA, USA
First published on 23rd December 2022
Abstract
CO2-Binding organic liquids (CO2BOLs) are non-aqueous solvents which may reduce the parasitic energy of carbon capture processes. These solvents exhibit surprising mass transfer behavior: at fixed pressure driving force, the flux of CO2 into CO2BOLs decreases exponentially with increased temperature, a phenomenon not observed in aqueous amines. Here, we demonstrate that this phenomenon is primarily driven by a shift in reaction equilibrium, which reduces the degree to which chemical reactions enhance the CO2 flux. First-principles surface renewal models quantitatively reproduce mass transfer data for CO2 absorption into 2-EEMPA, IPADM-2-BOL and DBU:Hexanol across a range of temperatures. Density functional theory calculations are used to identify structural modifications likely to improve the CO2 flux. These findings reveal a fundamental trade-off between CO2 flux and the energy required for solvent regeneration, and provide a theoretical foundation for rational solvent design and the development of physics-informed mass transfer models.
Broader contextThe capture of CO2 from dilute gas streams, and its subsequent storage or utilization, is a critical technology for reducing global CO2 emissions. The best-established carbon capture technologies use aqueous amine solutions, though non-aqueous solvents, such as CO2-Binding Organic Liquids, have attracted increased attention, as they may allow reductions in process energy requirements. However, the mechanism by which these solvents absorb CO2 has remained a fundamental mystery since they were first synthesized. When CO2BOLs are heated, CO2 absorption rates decrease exponentially – an unintuitive behavior opposite what is observed in aqueous systems. The lack of understanding of basic mass transport mechanisms hampers the development of these solvents, as it rules out the use of physics-based mass transfer models routinely used by process modeling software, and complicates the rational design of new solvent systems. In this work, we explain the surprising mass transport behavior in CO2BOLs, and develop first-principles mass transport models in excellent agreement with experimental data. |
1 Introduction
CO2-binding organic liquids (CO2BOLs) are a promising class of non-aqueous solvent for carbon capture and storage (CCS).1,2 These solvents reversibly react with CO2, and exhibit CO2 capacities and uptake rates comparable with state-of-the-art aqueous amines.3,4 Several modeling studies predict CO2BOLs may improve the energy efficiency of CCS processes,5–7 though this is a matter of active debate in the CCS community.8–12An enduring mystery in the CO2BOL literature pertains to the unexpected temperature dependence of the CO2 absorption rate. The flux of CO2 into a wide range of CO2BOLs exponentially decreases with increasing temperature at constant pressure driving force: the opposite trend to what is typically observed in aqueous amines. Formally, the rate of absorption into a chemical solvent may be quantified by the liquid-film mass transfer coefficient, which (in the absence of gas-side resistance) is given by:
 | (1) |
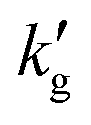 is experimentally observed to decrease rapidly with temperature, while in more traditional, aqueous solvents
is experimentally observed to decrease rapidly with temperature, while in more traditional, aqueous solvents  is usually a weakly increasing function of temperature. In spite of multiple experimental1,3,4,13 and molecular modeling studies,14–19 no adequate explanation of this phenomenon has been discovered. Some authors have speculated that this surprising behaviour may be caused by a decrease in the physical solubility of CO2, however fundamental studies utilizing non-reactive CO2 mimics such as N2O did not show similar thermal effects (Fig. 1c).4,13 This has led several authors to speculate that novel film theories may be required to explain mass transport phenomena in CO2BOLs,3,13 possibly involving mesoscopic structures which can spontaneously form during CO2 absorption.20 Understanding mass transport mechanisms within CO2BOLs is essential for molecular design of solvents, for reliable modeling of industrial-scale processes, and is also of basic scientific importance.
is usually a weakly increasing function of temperature. In spite of multiple experimental1,3,4,13 and molecular modeling studies,14–19 no adequate explanation of this phenomenon has been discovered. Some authors have speculated that this surprising behaviour may be caused by a decrease in the physical solubility of CO2, however fundamental studies utilizing non-reactive CO2 mimics such as N2O did not show similar thermal effects (Fig. 1c).4,13 This has led several authors to speculate that novel film theories may be required to explain mass transport phenomena in CO2BOLs,3,13 possibly involving mesoscopic structures which can spontaneously form during CO2 absorption.20 Understanding mass transport mechanisms within CO2BOLs is essential for molecular design of solvents, for reliable modeling of industrial-scale processes, and is also of basic scientific importance.
 | ||
| Fig. 1 Liquid-film mass transfer coefficients as a function of temperature. (a) Wetted Wall column data for four CO2BOLs: IPADM-2-BOL,3 DBU:Hexanol,3 GAP-1/TEG,4 and EEMPA1 (b) Wetted wall column data for two aqueous amines: piperazine and monoethanolamine; 80 °C and 100 °C data interpolated.21 (c) Wetted wall column data for absorption of CO2 and N2O into DBU:Hexanol3,13 and GAP-1/TEG,4 at various CO2 loadings (mol CO2/mol amine × 100%). GAP-1/TEG CO2 data interpolated from data at CO2 loadings of 4% and 13%. | ||
In this article, we demonstrate that the decrease in the rate of CO2 absorption into CO2BOLs at increased temperature is not primarily driven by decreases in the physical solubility of CO2, but by a thermally-driven shift in reaction equilibrium, which decreases the extent of reaction of CO2 with the solvent in the vicinity of the gas–liquid interface. This in turn decreases the enhancement in gas flux due to chemical reaction. The CO2 flux into CO2BOLs is typically limited by diffusion of reaction products away from the gas–liquid interface, and due to shifts in reaction equilibrium, the rate of this diffusion decreases sharply with increased temperature. Surface renewal models based on this hypothesis are in excellent agreement with available kinetic data for multiple CO2BOLs. This work demonstrates that novel film theories are not required to explain the surprising thermal effects in CO2BOLs. The interplay between mass transport and Vapor–Liquid Equilibria (VLE) suggests several non-obvious design criteria for the molecular design of CO2BOLs, and we use DFT simulations to identify structural modifications which may improve absorption kinetics.
2 CO2 transport in CO2BOLs
2.1 Mass transport mechanism
The hypothesized mechanism by which the flux of CO2 into CO2BOLs decreases with increased temperature is demonstrated by an idealized example sketched in Fig. 2. Fig. 2a shows VLE data for the CO2BOL 2-EEMPA (to avoid confusion between molecular names and stoichiometric coefficients, we denote this as EEMPA throughout the manuscript) which reacts with CO2 as follows:1| CO2 + 2EEMPA ⇌ EEMPACOO− + EEMPAH+ | (2) |
 | ||
Fig. 2 Mechanism for decrease in  with temperature. (a) Vapor–Liquid Equilibrium data for CO2 absorption into EEMPA. Data points taken from Zheng et al.1 Solid lines regressed to data based on methodology of Gabrielson et al.,22 see ESI,† Section S3. (b and c) Concentration profiles within static EEMPA solution in the vicinity of the gas–liquid interface, 0.1 s and 1 s after it has been exposed to pCO2 = 10 kPa, at (b) 70 °C, and (c) 30 °C. Calculated using surface renewal model under assumption of instantaneous reaction. with temperature. (a) Vapor–Liquid Equilibrium data for CO2 absorption into EEMPA. Data points taken from Zheng et al.1 Solid lines regressed to data based on methodology of Gabrielson et al.,22 see ESI,† Section S3. (b and c) Concentration profiles within static EEMPA solution in the vicinity of the gas–liquid interface, 0.1 s and 1 s after it has been exposed to pCO2 = 10 kPa, at (b) 70 °C, and (c) 30 °C. Calculated using surface renewal model under assumption of instantaneous reaction. | ||
Two points are marked on this plot: both points are at a CO2 partial pressure of 10 kPa (a pressure typically found in a CCS absorber), but point (1) is at 30 °C, while point (2) is at 70 °C. At point (1), the equilibrium CO2 loading, α, is around 37% on a mol CO2:mol EEMPA basis, while at point (2), the equilibrium CO2 loading is only about 3.5%. This decrease in equilibrium CO2 loading is responsible for the decrease in solvent kinetics shown in Fig. 1a; the mechanism is illustrated in Fig. 2b and c. These figures show concentration profiles of CO2, EEMPA and EEMPACOO− within an EEMPA solution with bulk CO2 loading of α = 0, 0.1 s and 1s after it has been exposed to a gas with pCO2 = 10 kPa. These profiles were calculated using surface renewal theory models discussed below. In Fig. 2c (which corresponds to point (1), with system temperature of 30 °C) CO2 reacts strongly with EEMPA, leading to a large concentration of EEMPACOO− at the gas–liquid interface, which carries bound CO2 into the liquid at a fast rate. On the other hand, at 70 °C (Fig. 2b), due to equilibrium effects, the extent of Rxn (2) is far smaller, being unable to exceed an equilibrium CO2 loading of ∼3.5%, resulting in a much smaller EEMPACOO− concentration gradient, and slower CO2 absorption at this higher temperature. In both examples, pCO2 = 10 kPa and  , so by eqn (1) the decrease in CO2 flux with increased temperature corresponds to a proportional decrease in
, so by eqn (1) the decrease in CO2 flux with increased temperature corresponds to a proportional decrease in 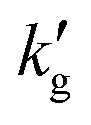 .
.
Formally, the liquid-film mass transfer coefficient may be factorized into three terms,
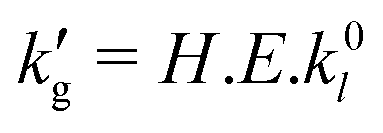 | (3) |
 with temperature (Fig. 1a) is primarily driven by a decrease in the enhancement factor, E, caused by a thermally-driven shift in reaction equilibrium which decreases the extent of the CO2–CO2BOL reaction, and in turn the magnitude of the concentration gradient of CO2-bound-CO2BOL adjacent to the gas–liquid interface. Decreases in the physical CO2 solubility, H, are of secondary importance. The reason this phenomena is observed in CO2BOLs, but not commonly used aqueous amines, is that for the latter the reaction with CO2 is more irreversible under absorber conditions, resulting in much smaller shifts in reaction equilibrium. For example, between 30 °C and 60 °C, and at a CO2 partial pressure of pCO2 = 10 kPa, the equilibrium CO2 loading in 30 wt% monoethanol amine decreases from 0.49 mol CO2/mol amine to 0.46 mol CO2/mol amine (for reference, the maximum chemical loading based on a 1
with temperature (Fig. 1a) is primarily driven by a decrease in the enhancement factor, E, caused by a thermally-driven shift in reaction equilibrium which decreases the extent of the CO2–CO2BOL reaction, and in turn the magnitude of the concentration gradient of CO2-bound-CO2BOL adjacent to the gas–liquid interface. Decreases in the physical CO2 solubility, H, are of secondary importance. The reason this phenomena is observed in CO2BOLs, but not commonly used aqueous amines, is that for the latter the reaction with CO2 is more irreversible under absorber conditions, resulting in much smaller shifts in reaction equilibrium. For example, between 30 °C and 60 °C, and at a CO2 partial pressure of pCO2 = 10 kPa, the equilibrium CO2 loading in 30 wt% monoethanol amine decreases from 0.49 mol CO2/mol amine to 0.46 mol CO2/mol amine (for reference, the maximum chemical loading based on a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry is 0.5 mol CO2/mol amine).22 Under the same conditions, the equilibrium CO2 loading into EEMPA decreases from 0.37 mol CO2/mol amine to 0.07 mol CO2/mol amine: a much more significant shift in reaction extent (see Fig. S1, ESI†). The effect may also be seen in Fig. S2 (ESI†), which shows results of simulations under identical conditions to those in Fig. 2b and c, for a 30 wt% MEA solution, a 30 wt% K2CO3 solution, and an EEMPA solution with finite reaction kinetics. It is clear that the concentration profiles which form adjacent to the gas–liquid interface are far more sensitive to temperature in the CO2BOL system than in the aqueous systems. Because the equilibrium CO2 loading is not strongly temperature dependent in the aqueous solvents under these conditions, shifts in reaction equilibrium with T do not significantly affect
:2 stoichiometry is 0.5 mol CO2/mol amine).22 Under the same conditions, the equilibrium CO2 loading into EEMPA decreases from 0.37 mol CO2/mol amine to 0.07 mol CO2/mol amine: a much more significant shift in reaction extent (see Fig. S1, ESI†). The effect may also be seen in Fig. S2 (ESI†), which shows results of simulations under identical conditions to those in Fig. 2b and c, for a 30 wt% MEA solution, a 30 wt% K2CO3 solution, and an EEMPA solution with finite reaction kinetics. It is clear that the concentration profiles which form adjacent to the gas–liquid interface are far more sensitive to temperature in the CO2BOL system than in the aqueous systems. Because the equilibrium CO2 loading is not strongly temperature dependent in the aqueous solvents under these conditions, shifts in reaction equilibrium with T do not significantly affect  ; instead, increases in diffusion coefficient and reaction kinetics cause an increase in
; instead, increases in diffusion coefficient and reaction kinetics cause an increase in  .
.
2.2 Model development
The example in the previous section was idealized in several ways. It considered an unloaded solvent ( ) with infinitely fast reaction kinetics, on which a fixed CO2 pressure was applied. While this example provides useful intuition for why
) with infinitely fast reaction kinetics, on which a fixed CO2 pressure was applied. While this example provides useful intuition for why  should decrease with T, more sophisticated models are required to capture the interplay between reaction kinetics, VLE and
should decrease with T, more sophisticated models are required to capture the interplay between reaction kinetics, VLE and  in solvents at arbitrary loading and temperature. For this reason, models for CO2 absorption into several agitated CO2BOLs were constructed based on the surface renewal theory of Danckwerts.23,24 Surface renewal theory has been widely used to simulate mass transfer into agitated chemical solvents,25,26 and is generally considered more accurate than alternative theories, such as the penetration or film theory.27 In this model, partial differential equations governing reactive absorption into a static liquid are solved, however at random intervals the concentration profiles in the liquid are ‘reset’ or ‘renewed’ to bulk conditions, mimicking a random eddy carrying fresh liquid to the surface. These surface renewals are modelled as a Poisson process,28 so the time between renewals, θ, is governed by an exponential probability distribution with probability density function:
in solvents at arbitrary loading and temperature. For this reason, models for CO2 absorption into several agitated CO2BOLs were constructed based on the surface renewal theory of Danckwerts.23,24 Surface renewal theory has been widely used to simulate mass transfer into agitated chemical solvents,25,26 and is generally considered more accurate than alternative theories, such as the penetration or film theory.27 In this model, partial differential equations governing reactive absorption into a static liquid are solved, however at random intervals the concentration profiles in the liquid are ‘reset’ or ‘renewed’ to bulk conditions, mimicking a random eddy carrying fresh liquid to the surface. These surface renewals are modelled as a Poisson process,28 so the time between renewals, θ, is governed by an exponential probability distribution with probability density function:| f(θ) = se−sθ | (4) |
The actual gas flux is then taken to equal the long-time average gas flux for this stochastic process. The mean surface renewal rate, s, is the only parameter in the model, and it may be calculated via:23
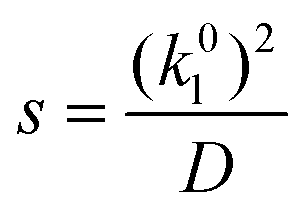 | (5) |
In order to use surface renewal theory to estimate CO2 flux into a CO2BOL, species diffusivities, reaction rates, and solubilities must be estimated, along with VLE data and k0l in the experimental system. Models were developed for three CO2BOLs: EEMPA, IPADM-2-BOL, and DBU:Hexanol. Diffusivities were estimated from available literature data or, if no data were available, from the correlation of Wilke and Chang,29 using temperature- and loading-dependent viscosity data from the literature for each CO2BOL modeled.1,3 The Henry's constants for CO2 were estimated from measurements in comparable organic liquids (for reasons discussed in Section S3 of the ESI,† the predicted gas fluxes were only weakly dependent on the choice of Henry's constant.) k0l for wetted-wall column data was estimated at experimental conditions from the correlation of Vivian and Peaceman.30 Experimental VLE data has previously been reported for each CO2BOL above,1,3 and this data was fit to models of the kind discussed by Gabrielson22 (see Fig. S3–S5, ESI†). Regarding reaction rate constants: for DBU:Hexanol, the kinetic data of Ozturk et al.31 was used. They observed relatively slow kinetics in DBU:Hexanol, with a second order rate constant about 10 times smaller than for MEA.32 On the other hand, several molecular modeling studies14,16 suggest the reaction of IPADM-2-BOL with CO2 is instantaneous, though this has not been verified experimentally. Hence, for both IPADM-2-BOL and EEMPA, two reaction rates were considered: an instantaneous reaction, and a reaction with finite kinetics equal to those found in DBU:Hexanol. All reactions were treated as reversible, with equilibrium constants calculated from the VLE data and CO2 Henry's constant. Further details on the implementation of the surface renewal model are provided in the ESI,† Section S3. The Julia code used to generate all figures is also included in the ESI.†
3 Results and discussion
In Fig. 3, experimentally measured liquid-film mass transfer coefficients in a range of CO2BOLs and over a range of temperatures are compared with the predictions of the surface renewal theory. We stress that the surface renewal models are not fit to the experimental mass transport data, but are instead physics-based predictions, based on independently measured or estimated VLE, reaction kinetics, diffusivity and solubility data. Not only does the surface renewal theory correctly predict trends in with temperature and between different CO2BOLs, but the match is quantitatively reasonable.
with temperature and between different CO2BOLs, but the match is quantitatively reasonable.
 | ||
Fig. 3 Comparison between surface renewal theory models (lines and shared areas) and wetted wall column data (points), for IPADM-2-BOL, EEMPA and DBU:Hexanol. For IPADM-2-BOL and EEMPA, an instantaneous reaction (solid lines) and a finite reaction rate (DBU:Hexanol kinetics, dashed line) were modelled; the range of intermediate values is colored in. Dash-dot lines refer to simulations in which the reaction equilibrium constant is artificially fixed at its value at 30 °C (EEMPA) and 40 °C (IPADM-2-BOL), so that  is not influenced by changes in reaction extent. CO2 loading and sources as follows: EEMPA data from Zheng et al.1 at 0.01 mol CO2/mol CO2BOL; IPADM-2-BOL data from Mathias et al.3 at 0.0106 mol CO2/mol CO2BOL; DBU:Hexanol data from Mathias et al.3 at 0.0146 mol CO2/mol CO2BOL. is not influenced by changes in reaction extent. CO2 loading and sources as follows: EEMPA data from Zheng et al.1 at 0.01 mol CO2/mol CO2BOL; IPADM-2-BOL data from Mathias et al.3 at 0.0106 mol CO2/mol CO2BOL; DBU:Hexanol data from Mathias et al.3 at 0.0146 mol CO2/mol CO2BOL. | ||
The surface renewal models correctly predict the decrease of  with increased temperature. That this decrease is primarily driven by a thermally-driven shift in reaction equilibrium, as discussed above, and not by a decrease in physical solubility of CO2, is shown by the dash-dotted lines in Fig. 3. These show the results of simulations in which the reaction equilibrium constant is fixed at a constant value, while all other parameters vary with temperature. When the temperature-dependence of the reaction equilibrium constant is ignored,
with increased temperature. That this decrease is primarily driven by a thermally-driven shift in reaction equilibrium, as discussed above, and not by a decrease in physical solubility of CO2, is shown by the dash-dotted lines in Fig. 3. These show the results of simulations in which the reaction equilibrium constant is fixed at a constant value, while all other parameters vary with temperature. When the temperature-dependence of the reaction equilibrium constant is ignored,  increases with temperature, as it does in aqueous amines (Fig. 1b). In Fig. S6 (ESI†), we factorize the surface renewal model's prediction of
increases with temperature, as it does in aqueous amines (Fig. 1b). In Fig. S6 (ESI†), we factorize the surface renewal model's prediction of  for IPADM-2-BOL into contributions from H, E and k0l (see eqn (2)), and plot the relative change in each parameter as T varies from 40 °C to 100 °C. It is clear that it is primarily the thermally-driven decrease in reactive enhancement factor, E (caused by the shift in reaction equilibrium) which drives the decrease in
for IPADM-2-BOL into contributions from H, E and k0l (see eqn (2)), and plot the relative change in each parameter as T varies from 40 °C to 100 °C. It is clear that it is primarily the thermally-driven decrease in reactive enhancement factor, E (caused by the shift in reaction equilibrium) which drives the decrease in  ; the smaller decrease in Henry's constant, H, with T is largely canceled out by the increase in k0l with T, as seen in other chemical solvents.21 This may also be seen in Fig. S7 and S8 (ESI†), which show modelled changes in temperature for both
; the smaller decrease in Henry's constant, H, with T is largely canceled out by the increase in k0l with T, as seen in other chemical solvents.21 This may also be seen in Fig. S7 and S8 (ESI†), which show modelled changes in temperature for both  and also kl = Ek0l. As changes in E drive the decrease in mass transfer coefficient, both
and also kl = Ek0l. As changes in E drive the decrease in mass transfer coefficient, both  and kl exhibit similar trends with temperature.
and kl exhibit similar trends with temperature.
The relative insignificance of changes in physical CO2 solubility with temperature is supported by several other lines of evidence. First, when N2O, a non-reactive CO2-mimic,33,34 is absorbed by CO2BOLs,  is much smaller, and does not decrease with temperature, suggesting changes in chemical reaction equilibrium constants, rather than physical solubility, drive this phenomenon (see Fig. 1c). Second, if we make the plausible assumption that the CO2 flux is at most proportional to the physical CO2 solubility (i.e. there are no phenomena such as autocatalytic reactions which cause a higher-order dependence of the flux on the CO2(aq) concentration) then the 10-fold decrease in
is much smaller, and does not decrease with temperature, suggesting changes in chemical reaction equilibrium constants, rather than physical solubility, drive this phenomenon (see Fig. 1c). Second, if we make the plausible assumption that the CO2 flux is at most proportional to the physical CO2 solubility (i.e. there are no phenomena such as autocatalytic reactions which cause a higher-order dependence of the flux on the CO2(aq) concentration) then the 10-fold decrease in  for IPADM-2-BOL between 40 °C to 100 °C (Fig. 1a) would require at minimum a commensurate decrease in CO2 solubility. This in turn would imply (via the Gibbs-Helmholtz equation35–37) that the enthalpy of physical dissolution of CO2 was on the order of −70 kJ mol−1: much larger than the value of −10 to −15 kJ mol−1 typically observed for physical CO2 dissolution.38 Finally, we note that several authors have hypothesized that thermal effects in CO2BOLs may be related to the relatively large equilibrium mole fraction of physically dissolved CO2 in organic liquids as compared to aqueous solutions.1,3 However, the small mole fraction of CO2 in water is predominantly driven by the small molecular weight of water, rather than differences in CO2 concentration. As shown in Fig. S9 (ESI†), when CO2 solubility is calculated in reference to volumetric concentration, rather than mole fraction, (the former being the more appropriate driving force for mass flux39) the CO2 solubility in a range of organic liquids is only modestly larger than in water.
for IPADM-2-BOL between 40 °C to 100 °C (Fig. 1a) would require at minimum a commensurate decrease in CO2 solubility. This in turn would imply (via the Gibbs-Helmholtz equation35–37) that the enthalpy of physical dissolution of CO2 was on the order of −70 kJ mol−1: much larger than the value of −10 to −15 kJ mol−1 typically observed for physical CO2 dissolution.38 Finally, we note that several authors have hypothesized that thermal effects in CO2BOLs may be related to the relatively large equilibrium mole fraction of physically dissolved CO2 in organic liquids as compared to aqueous solutions.1,3 However, the small mole fraction of CO2 in water is predominantly driven by the small molecular weight of water, rather than differences in CO2 concentration. As shown in Fig. S9 (ESI†), when CO2 solubility is calculated in reference to volumetric concentration, rather than mole fraction, (the former being the more appropriate driving force for mass flux39) the CO2 solubility in a range of organic liquids is only modestly larger than in water.
These models explain the trends observed between different solvents. In particular, that EEMPA absorbs CO2 more slowly than IPADM-2-BOL at low loadings is primarily a consequence of the difference in VLE between the solvents. For example, at 60 °C and pCO2 = 5 kPa, the CO2 loading in IPADM-2-BOL and EEMPA are ∼18% (see Fig. S4, ESI†) and ∼3% (see Fig. 2a), respectively. Hence, the reaction of IPADM-2-BOL with CO2 proceeds much further than the equivalent reaction with EEMPA, resulting in much larger concentration gradients of reaction products and a greater reactive enhancement factor.
The surface renewal model also correctly predicts trends with CO2 loading, as shown in Fig. S10 (ESI†), which compares the surface renewal model with  data for EEMPA collected by Zheng et al.1 At higher loadings, the mechanism discussed in Section 2.1 for a solvent at zero CO2 loading must be expanded upon. In general, when temperature changes cause an increase in the gradient of the absorption isotherm,
data for EEMPA collected by Zheng et al.1 At higher loadings, the mechanism discussed in Section 2.1 for a solvent at zero CO2 loading must be expanded upon. In general, when temperature changes cause an increase in the gradient of the absorption isotherm, 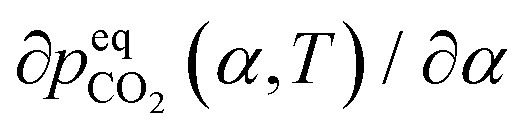 , this causes the loading, α, to be less sensitive to changes in pressure. If diffusion of product species from the surface to the bulk is rate controlling, this will result in a smaller change in α between the interface and the bulk for a fixed pressure driving force, which can cause a reduction in gas flux and also (as the pressure driving force is fixed) a reduction in
, this causes the loading, α, to be less sensitive to changes in pressure. If diffusion of product species from the surface to the bulk is rate controlling, this will result in a smaller change in α between the interface and the bulk for a fixed pressure driving force, which can cause a reduction in gas flux and also (as the pressure driving force is fixed) a reduction in 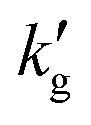 with increased temperature. Of course, in general such equilibrium phenomena occur alongside and compete with kinetic effects, and it is only through the development of rigorous models that their relative impact on
with increased temperature. Of course, in general such equilibrium phenomena occur alongside and compete with kinetic effects, and it is only through the development of rigorous models that their relative impact on  can be assessed.
can be assessed.
Finally, we note that reductions in reactive enhancement factor due to shifts in reaction equilibria are not limited to CO2BOLs. For example, under stripper conditions the equilibrium loading of MEA solutions becomes highly temperature dependent, and this causes a reduction in  with increased T (see Fig. S11, ESI†). This effect is not observed in K2CO3 solutions, (Fig. S12, ESI†) as the reaction kinetics are too slow for diffusion of product species from the interface to the bulk to be rate limiting. In general, reductions in
with increased T (see Fig. S11, ESI†). This effect is not observed in K2CO3 solutions, (Fig. S12, ESI†) as the reaction kinetics are too slow for diffusion of product species from the interface to the bulk to be rate limiting. In general, reductions in  may be expected in solvents systems with fast reaction kinetics operated in a regime where the VLE is highly temperature-dependent.
may be expected in solvents systems with fast reaction kinetics operated in a regime where the VLE is highly temperature-dependent.
3.1 Implications for CO2BOL development
These results demonstrate that the primary factor influencing CO2 uptake rates in CO2BOLs is the extent of the reversible chemical reaction. Hence, in order to maximize the absorption rate, CO2BOLs should be designed to have large reaction conversion under absorber conditions. Improving the diffusivity of the liquid-phase species (i.e. the CO2BOL and the CO2-bound-CO2BOL) will also improve the CO2 uptake rate. Increasing reaction kinetics helps to a degree, but the benefit plateaus once diffusion of unreacted CO2BOL and CO2-bound-CO2BOL to and from the bulk becomes rate-limiting.As CO2BOL CO2 uptake kinetics are strongly influenced by the VLE, it is likely they can be tuned via structural changes which shift the reaction equilibrium. To investigate this, quantum chemical simulations were used to explore the enthalpy of CO2 absorption (ΔHabs) for selected CO2BOL species. Initially, CO2 binding in IPADM-2-BOL and EEMPA was examined to identify key binding modes and inform chemical modifications that may increase the magnitude of ΔHabs, which in turn would shift the reaction equilibrium towards reaction products, increasing E at higher temperatures.
Molecular dynamic simulations were performed on each compound to identify an initial series of structures corresponding to local energy minima, which were then further optimized via DFT calculations at the M06L/6-31++G(d,p) level. Implicit solvent corrections to energies were applied using the Polarizable Continuum Model using the integral equation formalism variant (IEFPCM). While the data presented was calculated using the dielectric constant of water, calculations using additional solvents (as well as gas phase) were performed to evaluate the impact of dielectric constant on CO2 binding enthalpy (Table S1, ESI†). Additional computational details are provided in the ESI† (Section S4).
Two different CO2 bound species emerged as the most thermodynamically favorable for both IPADM-2-BOL and EEMPA: an intramolecularly H-bonded monomolecular species, and intermolecularly H-bonded bimolecular species (Fig. 4a and b). Interestingly, for both CO2BOLs, the bimolecular species was calculated to have markedly lower ΔHabs than the monomolecular species, in agreement with the predicted stoichiometry from modeling of the VLE data. Additionally, the bimolecular binding enthalpies of around −52 kJ mol−1 and −77 kJ mol−1 for IPADM-2-BOL and EEMPA, respectively, are in reasonable agreement with those predicted by the VLE data.
 | ||
| Fig. 4 Enthalpies of monomolecular and bimolecular CO2 absorption for (a) EEMPA, (b) IPADM-2-BOL, and (c) phosphazene based CO2BOL structures. | ||
Given the prominence of H-bonding in these CO2 bound structures, we hypothesized that increasing the strength of the basic moiety might be a reasonable approach towards increasing the magnitude of ΔHabs. Inspired by the use of guanidine in IPADM-2-BOL, we were particularly interested in the effect of incorporating a phosphazene organic superbase, which is a stronger base than guanidine and highly tunable (Fig. 4c). Compared to the structurally similar IPADM-2-BOL, the examined phosphazene-based structure was calculated to have stronger ΔHabs for both the monomolecular and bimolecular species, by around 15 kJ mol−1 and 25 kJ mol−1, respectively. A shift of this magnitude could improve absorption kinetics by increasing the extent of the CO2–CO2BOL reaction under absorber conditions. For example, if the partial molar entropy of CO2 absorption is not affected by the structural changes, then decreasing ΔHabs by 20 kJ mol−1 would increase the equilibrium CO2 loading into EEMPA at 60 °C, pCO2 = 5 kPa from its actual value of ∼3% to close to the stoichiometric limit of 50%. This would in turn be expected to increase the CO2 flux into an unloaded solvent by about an order of magnitude. However the impact is likely to be lower at higher solvent loadings. Furthermore, these structural modifications may also affect other properties of the solvent, including the entropy of absorption,40 reaction constants, diffusivities, viscosity, volatility, and how easily it may be manufactured. A more complete in silico solvent design study could systematically screen for solvents with favorable physical, thermodynamic and kinetic properties. Classical mass transfer theories such as surface renewal models could also be incorporated into such a framework. As demonstrated in this work, they are a powerful tool for understanding novel solvent systems, and are capable of deconvoluting kinetic, equilibrium and hydrodynamic phenomena in a computationally efficient manner.
System-scale tradeoffs must also be considered. Shifting CO2BOL VLE closer to the relatively irreversible VLE of aqueous amines could improve absorption kinetics, especially at lower CO2 loadings, but it will also increase the magnitude of the temperature swing required for solvent regeneration. Aqueous amines must typically be heated to >100 °C during regeneration, and similar thermal swings may be required if CO2BOLs are designed with the sole aim of maximizing mass transfer rates in the absorber. In practice, there is likely to be a trade-off between enhanced mass flux and ease-of-regeneration, which must be assessed on an application-dependent basis.
4 Conclusion
The mechanism by which the CO2 flux into CO2BOLs decreases with temperature has remained a central, unanswered question since CO2BOLs were first developed. In this study, we have demonstrated that this phenomena is not driven by decreases in physical CO2 solubility, as has been previously hypothesized, but is instead caused by thermal shifts in chemical equilibrium, which decrease the enhancement factor for gas flux. Surface renewal models based on this hypotheses were in excellent agreement with wetted wall column data for a range of CO2BOLs. The mechanism directly connects VLE data and the CO2 absorption rate, and reveals a fundamental tradeoff between maximizing CO2 flux under absorption conditions, and minimizing the temperature swing required for solvent regeneration, which will inform rational design of CO2BOLs. This work also provides the necessary theoretical foundation for the development of CFD and process-scale simulations based on fundamental, physics-based models.Author contributions
Thomas Moore: conceptualization, methodology, software, formal analysis, writing – original draft. Anthony J. Varni: methodology, software, formal analysis, writing – original draft. Simon H. Pang: conceptualization, writing – review & editing. Sneha A. Akhade supervision. Sichi Li writing – review & editing, supervision. Du T. Nguyen: writing – review & editing, supervision. Joshuah K. Stolaroff writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Funding was provided by the US Department of Energy, Office of Fossil Energy and Carbon Management under FWP-FEW0225. Release number: LLNL-JRNL-840252.References
- R. F. Zheng, D. Barpaga, P. M. Mathias, D. Malhotra, P. K. Koech, Y. Jiang, M. Bhakta, M. Lail, A. V. Rayer and G. A. Whyatt, et al. , Energy Environ. Sci., 2020, 13, 4106–4113 RSC.
- P. M. Mathias, K. Afshar, F. Zheng, M. D. Bearden, C. J. Freeman, T. Andrea, P. K. Koech, I. Kutnyakov, A. Zwoster and A. R. Smith, et al. , Energy Environ. Sci., 2013, 6, 2233–2242 RSC.
- P. M. Mathias, F. Zheng, D. J. Heldebrant, A. Zwoster, G. Whyatt, C. M. Freeman, M. D. Bearden and P. Koech, ChemSusChem, 2015, 8, 3617–3625 CrossRef PubMed.
- G. A. Whyatt, A. Zwoster, F. Zheng, R. J. Perry, B. R. Wood, I. Spiry, C. J. Freeman and D. J. Heldebrant, Ind. Eng. Chem. Res., 2017, 56, 4830–4836 CrossRef.
- D. Heldebrant, Y. Jiang, R. Zheng, D. Barpaga, C. Freeman, P. K. Koech, D. Malhotra, G. Whyatt and A. Zwoster, Available at SSRN 3813876, 2021.
- Y. Jiang, P. M. Mathias, C. J. Freeman, J. A. Swisher, R. F. Zheng, G. A. Whyatt and D. J. Heldebrant, Int. J. Greenhouse Gas Control, 2021, 106, 103279 CrossRef.
- Y. Jiang, P. M. Mathias, G. Whyatt, C. Freeman, F. Zheng, V.-A. Glezakou, R. Rousseau, P. K. Koech, D. Malhotra and D. Heldebrant, Development of an Advanced Water-Lean Capture Solvent From Molecules to Detailed Process Design (April 29, 2019), 2019 Search PubMed.
- R. R. Wanderley, D. D. Pinto and H. K. Knuutila, Sep. Purif. Technol., 2020, 260, 118193 CrossRef.
- Y. Yuan and G. T. Rochelle, Int. J. Greenhouse Gas Control, 2019, 84, 82–90 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, R. Rousseau, V.-A. Glezakou, D. Cantu, D. Malhotra, F. Zheng, G. Whyatt, C. J. Freeman and M. D. Bearden, Energy Procedia, 2017, 114, 756–763 CrossRef CAS.
- N. Mac Dowell, D. Heldebrant, P. Brandl and J. Hallett, Available at SSRN 3379760, 2019.
- G. A. Whyatt, C. J. Freeman, A. Zwoster and D. J. Heldebrant, Ind. Eng. Chem. Res., 2016, 55, 4720–4725 CrossRef CAS.
- D. C. Cantu, J. Lee, M.-S. Lee, D. J. Heldebrant, P. K. Koech, C. J. Freeman, R. Rousseau and V.-A. Glezakou, J. Phys. Chem. Lett., 2016, 7, 1646–1652 CrossRef CAS PubMed.
- D. C. Cantu, D. Malhotra, P. K. Koech, D. J. Heldebrant, F. R. Zheng, C. J. Freeman, R. Rousseau and V.-A. Glezakou, Green Chem., 2016, 18, 6004–6011 RSC.
- D. C. Cantu, D. Malhotra, P. K. Koech, D. J. Heldebrant, R. F. Zheng, C. J. Freeman, R. Rousseau and V.-A. Glezakou, Energy Procedia, 2017, 114, 726–734 CrossRef CAS.
- D. C. Cantu, D. Malhotra, M.-T. Nguyen, P. K. Koech, D. Zhang, V.-A. Glezakou, R. Rousseau, J. Page, R. Zheng and R. J. Perry, et al. , ChemSusChem, 2020, 13, 3429–3438 CrossRef CAS.
- D. Malhotra, P. K. Koech, D. J. Heldebrant, D. C. Cantu, F. Zheng, V.-A. Glezakou and R. Rousseau, ChemSusChem, 2017, 10, 636–642 CrossRef CAS.
- D. Malhotra, D. C. Cantu, P. K. Koech, D. J. Heldebrant, A. Karkamkar, F. Zheng, M. D. Bearden, R. Rousseau and V.-A. Glezakou, ACS Sustainable Chem. Eng., 2019, 7, 7535–7542 CrossRef CAS.
- X.-Y. Yu, J. Yao, D. B. Lao, D. J. Heldebrant, Z. Zhu, D. Malhotra, M.-T. Nguyen, V.-A. Glezakou and R. Rousseau, J. Phys. Chem. Lett., 2018, 9, 5765–5771 CrossRef CAS PubMed.
- R. E. Dugas and G. T. Rochelle, J. Chem. Eng. Data, 2011, 56, 2187–2195 CrossRef CAS.
- J. Gabrielsen, M. L. Michelsen, E. H. Stenby and G. M. Kontogeorgis, Ind. Eng. Chem. Res., 2005, 44, 3348–3354 CrossRef CAS.
- P. V. Danckwerts, Gas-liquid reactions, McGraw-Hill, 1970 Search PubMed.
- P. Danckwerts, Ind. Eng. Chem., 1951, 43, 1460–1467 CrossRef CAS.
- M. Saidi, J. Taiwan Inst. Chem. Eng., 2017, 80, 301–313 CrossRef CAS.
- H. Huang and S. G. Chatterjee, Chem. Eng. Sci., 2021, 234, 116449 CrossRef CAS.
- D. A. Glasscock and G. T. Rochelle, AIChE J., 1989, 35, 1271–1281 CrossRef CAS.
- G. Grimmett and D. Stirzaker, Probability and random processes, Oxford University Press; 2020 Search PubMed.
- C. Wilke and P. Chang, AIChE J., 1955, 1, 264–270 CrossRef CAS.
- J. Vivian and D. Peaceman, AIChE J., 1956, 2, 437–443 CrossRef CAS.
- M. Ç. Öztürk, C. S. Ume and E. Alper, Chem. Eng. Technol., 2012, 35, 2093–2098 CrossRef.
- G. Versteeg, L. Van Dijck and W. P. M. van Swaaij, Chem. Eng. Commun., 1996, 144, 113–158 CrossRef CAS.
- Q. Chen, S. P. Balaji, M. Ramdin, J. J. Gutiérrez-Sevillano, A. Bardow, E. Goetheer and T. J. Vlugt, Ind. Eng. Chem. Res., 2014, 53, 18081–18090 CrossRef.
- J. G.-S. Monteiro and H. F. Svendsen, Chem. Eng. Sci., 2015, 126, 455–470 CrossRef.
- J. Oexmann and A. Kather, Int. J. Greenhouse Gas Control, 2010, 4, 36–43 CrossRef.
- T. C. R. Moore, PhD thesis, University of Melbourne: 2019.
- P. M. Mathias, Ind. Eng. Chem. Res., 2016, 55, 1076–1087 CrossRef.
- X. Gui, Z. Tang and W. Fei, J. Chem. Eng. Data, 2011, 56, 2420–2429 CrossRef.
- R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena revised 2nd Edition, 2006 Search PubMed.
- G. Puxty, W. Conway, Q. Yang, R. Bennett, D. Fernandes, P. Pearson, D. Maher and P. Feron, Int. J. Greenhouse Gas Control, 2019, 83, 11–19 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SI.pdf contains supplementary figures and tables, numerical methodology. CO2BOLScripts.zip contains code for generation of plots in Fig. 1–3. See DOI: https://doi.org/10.1039/d2ee03237f |
| This journal is © The Royal Society of Chemistry 2023 |