
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimolecular sinks of Criegee intermediates derived from hydrofluoroolefins – a computational analysis†
Nathan A. I.
Watson
 *ab and
Joseph M.
Beames
ac
*ab and
Joseph M.
Beames
ac
aSchool of Chemistry, Main Building, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: nathan.watson@manchester.ac.uk
bDepartment of Earth and Environmental Sciences, Simon Building, University of Manchester, Manchester, M13 9PS, UK
cSchool of Chemistry, Main Building, Edgbaston, Birmingham, B15 2TT, UK
First published on 17th August 2023
Abstract
A novel range of stabilised Criegee intermediate (sCI) species with halogenated substituent groups have been identified as products to the reaction between with gaseous ozone and hydrofluoroolefins (HFOs), a series of recently-developed and increasingly prevalent haloalkene refrigerants. The bimolecular chemistry of this group of hydrofluoroolefin-derived sCIs (HFO-sCIs) has yet to be explored in any significant detail so this work evaluates the reaction chemistry of common tropospheric gaseous species with the following group of HFO-sCIs: syn- & anti-CF3CHOO & syn- & anti-CF3CFOO. Using high-level theoretical calculations (DF-HF/DF-LCCSD(T)-F12a//B3LYP/aug-cc-pVTZ), this study demonstrates that HFO-sCIs will deplete many pollutants (e.g. HCHO, SO2 & H2S) but also act as a source of other atmospheric contaminants (e.g. SO3 & TFA). The bimolecular reactivity of the HFO-sCIs were compared against CH2OO, the most frequently studied sCI, for which the general reactivity trend has been identified: kTHEO (syn-CF3CHOO) < kTHEO (anti-CF3CHOO) ≈ kTHEO (CH2OO) ≪ kTHEO (anti-CF3CFOO) < kTHEO (syn-CF3CFOO). In general syn & anti-CF3 substituents reduce overall sCI reactivity compared to similar non-halogenated sCI species, whereas both syn & anti-F substituents significantly increase HFO-sCI reactivity. While HFO-sCI reactivity is largely dictated by the identity and location of the sCI substituent groups, there are co-reactants that alter these observed trends in reactivity, for example HCl reacts more rapidly with CH2OO than it does with syn- & anti-CF3CFOO.
Environmental significanceEmissions of new short-lived hydrofluoroolefin coolants have become increasingly widespread in the last decade and the breakdown of these hydrofluoroolefins will generate many secondary products, including a range of hydrofluoroolefin-derived stabilised Criegee intermediates (HFO-sCIs). Criegee intermediates are important OH radical sources and strong tropospheric sinks for many pollutants, but HFO-sCI reactivity has not yet been fully characterised. This computation chemistry study provides a greater understanding of the implications of the increased tropospheric HFO-sCI abundance. The enhanced bimolecular reactivity found for HFO-sCIs in this study indicates that several of the HFO-sCIs have heightened capacities to: react with toxins (e.g. methanol & trifluoroacetic acid); produce hydroperoxides implicated with forestry damage; and convert SO2 to SO3, a process implicated with generating H2SO4 and aerosols. |
1 Introduction
The recent commercialisation of highly volatile hydrochlorofluoroolefins and hydrofluoroolefins (referred to collectively herein as HFOs) for use as coolants in refrigerator units have been developed to reduce greenhouse gas emissions and reduce stratospheric ozone loss.1–6 Furthermore, as well as being used as blowing foam and as propellants, HFOs are very important working fluids for waste heat recovery applications in heat pumps, as they are stable, have favourable toxicity profiles and are compatible with many plastics and elastomers used in such set-ups.7,8 When haloalkenes breakdown it is the Cl & Br radicals that are primarily implicated in ozone destruction in the upper atmosphere and therefore only HFOs containing Cl & Br atoms (e.g. HCFO-1233zd(E)) appear to marginally affect stratospheric ozone levels.9–12 But their compact lifetime means HFOs are often depleted in the troposphere and contribute to only minimal ozone depletion.9–12 Additionally, they are relatively non-toxic, have minimal impact on global warming due to their short lifespans, and have high ignition energies compared to the first generation of refrigerants used in the 19th century (e.g. NH3, SO2 & CH3CHO).1–3,13–17 Moreover, whereas CO2 dominates the lower IR wave numbers (600–750 cm−1), HFCs/HCFCs are also strongly IR active in the higher wavenumbers (1000–1400 cm−1), increasing their heat trapping ability.9,18 In contrast, the longevity of the second-generation chlorofluorocarbon (CFC) refrigerants leads to depletion of stratospheric O3, which results in more cases of skin cancer from the greater ground-level penetration of the carcinogenic high-frequency UV light.19–21The growing interest in alternative refrigerants stems from the fact that, while the hydrofluorocarbon (HFC) and hydrochlorofluorocarbon (HCFC) replacements that have been phased in since the 1970s do not replicate this ozone depletion, the lengthy lifetimes and high levels of IR activity of HFCs & HCFCs give them very high global warming potentials (GWP100).2,9,19,22–26 In areas as widespread as China, the USA and the EU, the phasing in of HFOs has been increasingly integrated into regional regulations, to try and eliminate HCFC & HFC emissions, and this has led to HFOs seeing increasing use in refrigerators, insulation and vehicle cooling units.1,27,28 For example, a UK government 2022 report on the “F gas regulation in Great Britain” documents that HFO-1234yf has dominated total refrigerant usage in new small vehicles since 2017 and HFO–HFC blends use could eliminate high GWP HFCs from transport refrigeration by 2050.29 Modest concerns about both the tropospheric implications of HFO breakdown (e.g. TFA formation) and about the carbon-intensity of HFO production has meant that both the UK and the EU are currently assessing the role of HFOs in future climate regulations.30 Many refrigerant experts and industrial leaders continue to strongly support HFO use and are consulting on such regulations with governments currently, especially as the alternatives to HFOs also have trade-offs in the areas of toxicity, flammability and/or high-pressure requirements.31,32 Examples of CFCs, HCFCs, HFCs and HFOs with their GWP100 and ODP values and lifespan are displayed in Table 1.
| Name | Structure | GWP100 | ODP | Lifetime (year) | HFO-sCIs | Ref. |
|---|---|---|---|---|---|---|
| HFO-1234yf | CF3CF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
4 | 0 | 10.5 days | CH2OO | 2, 4 and 9 |
| Syn-/anti-CF3CFOO | ||||||
| HCFO-1233zd(E) | E-CF3CHCHCl |
14 | 0.0005 | 26 days | Syn-/anti-ClCHOO | 2, 5 and 33 |
| Syn-/anti-CF3CHOO | ||||||
| HFO-1234ze(E) | E-CF3CHCHF |
<1 | 0 | 16.4 days | Syn-/anti-FCHOO | 2, 9 and 34 |
| Syn-/anti-CF3CHOO | ||||||
| CFC-11 | CFCl3 | 4660 | 1 | 45 | — | 2 and 9 |
| CFC-12 | CCl2F2 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
0.82 | 100 | — | 2 |
| HFC-134a | CF3CH2F | 3710 | 0.0013 | 13.4 | — | 2 and 26 |
| HFC-245fa | CF3CH2CHF2 | 858 | 0 | 7.7 | — | 2 and 9 |
One of the reasons for the short-lived nature of HFOs listed in Table 1, HFO-1234yf, HCFO-1233zd(E) and HFO-1234ze(E), all of which are now widely used as refrigerants or insulation foam, is that the unsaturated 〉CC〈 bond makes HFOs more susceptible to these chemical loss mechanisms.1,3–5,27,35–39 The gas-phase reaction of HFOs with radicals, such as OH & Cl, and other reactive species, like O3, are important atmospheric loss mechanisms for many such tropospheric alkenes, for example isoprene reaction with O3 is responsible for ∼10% of the loss of this non-halogenated alkene globally.4–6,34,40–44 The importance of this HFO ‘ozonolysis’ loss process is increasing year-on-year because the rise of HFO emissions particularly in O3-rich urban areas is likely to result in sizable increases tropospheric populations of the Criegee intermediate (CI) and aldehyde/ketone products of this process (see Fig. 1).4,5,45–48 A recent rise in tropospheric trifluoracetic acid (TFA) abundance in Beijing has already been partially attributed to the reaction of OH radicals with HFOs and, as a similar increase in other HFO-derived products is likely in similar circumstances, determining the tropospheric impact these of new HFO-derived stabilised Criegee intermediates (HFO-sCIs) is very necessary.3,28,36,49–51
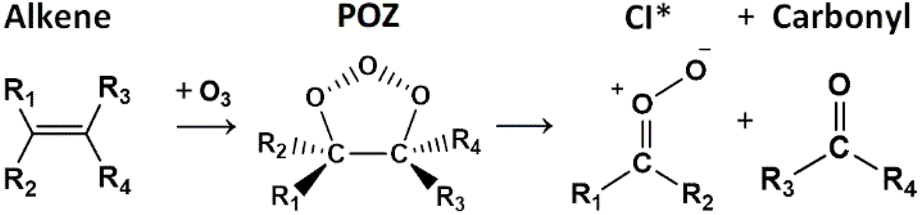 | ||
| Fig. 1 The general schematic for the ozonolysis of a standard alkene via a short-lived primary ozonide (POZ) intermediate products, which produces in a pair of final products: a Criegee intermediate (CI) and a carbonyl product (either aldehyde or ketone). A significant portion of the CI population is formed with a high degree of energy indicated by asterisk. | ||
The efficiency of ozonolysis as a tropospheric depletion mechanism for HFOs is disputed because while the presence of halogenated groups in the HFO reduces the overall effectiveness of most major alkene depletion pathways, such as via reaction with OH or Cl, this varies considerably depending on the HFO.45–47,52,53 The maximum ozonolysis yield for most HFOs reviewed are around 1%, but a few studies indicate that up to 10% of the removal of some haloalkenes occur via reaction with O3, a significant enough pathway that HFO ozonolysis is being investigated thoroughly at present.47,52 Furthermore, while direct CI branching fraction measurements for HFO ozonolysis are not found in the literature, indirect measurements of their aldehyde/ketone co-products show strong yields for CF3CHOO species and other similar CIs (>40%).52,54 This indicates that ozonolysis of haloalkenes in general are a major pathway for producing HFO-sCIs, even if these halogenated sCIs are low in abundance and ozonolysis is not the dominant pathway for removal of HFOs. This is a highly important topic and of relevance to the study of Criegee intermediates. Relative HFO ozonolysis percentages have been avoided here due to this need for a much more thorough re-investigation.
CIs are produced from the ozonolysis of a large variety of both halo-alkenes and non-halogenated alkenes and the atmospheric role of these CIs have been the subject of increased scientific study in the literature in recent years.5,6,34,48 This is due to the fact that a large portion of the CIs produced by alkene ozonolysis (35–50%) are formed with such excess of internal energy that they undergo a rapid unimolecular fragmentation process, which is an important non-photolytic source of the atmospheric ‘detergent’ HO˙ species. The remaining fraction are stabilised by collision, generating stabilised Criegee intermediates (sCIs), that can participate in bimolecular reactions with other atmospheric species. These sCIs deplete pollutants to such an extent that the simplest sCI, formaldehyde oxide (CH2OO), can compete with the atmospheric “detergent” HO˙ species as a sink for polar pollutants, such as HNO3 and SO2, especially in CH2OO-rich boreal forest environments ([CH2OO] ∼ 1–5 × 104 molec. per cm3).42,55 To examine the capacity of sCIs to deplete pollutants is the purpose of this study, as well as much of the increased scientific examination of sCIs overall.
The theoretical literature (see Table 1) shows that the ozonolysis of prominent HFOs produces several particular HFO-derived stabilised Criegee intermediates (HFO-sCIs): CH2OO, syn- & anti-FCHOO, syn- & anti-ClCHOO, syn- & anti-CF3CHOO and syn- & anti-CF3CFOO.4,5,45,46,53,56 Only the bimolecular chemistry of CH2OO species has been explored in the experimental literature, although syn- & anti-CF3CHOO and syn- & anti-ClCHOO have been experimentally identified.57–66 In a small number of studies, the bimolecular chemistry of HFO-sCIs has been interrogated using computational chemistry techniques: CH2OO somewhat exhaustively;67–71syn- & anti-FCHOO and syn- & anti-ClCHOO in reactions with CO2, H2, MeOH and CH4;72–76 and syn- & anti-CF3CHOO in reactions with CO2, CF3CHCH2 and H2.72,73,77 The literature has very little analysis of syn- or anti-CF3CFOO. As much of the bimolecular chemistry of syn- & anti-FCHOO and syn- & anti-ClCHOO has already been previously studied in the computational literature, the HFO-sCIs explored herein are instead CH2OO, syn-CF3CHOO, anti-CF3CHOO, syn-CF3CFOO and anti-CF3CFOO, which are labelled as sCIs 1–5 and are displayed in Fig. 2.72–75
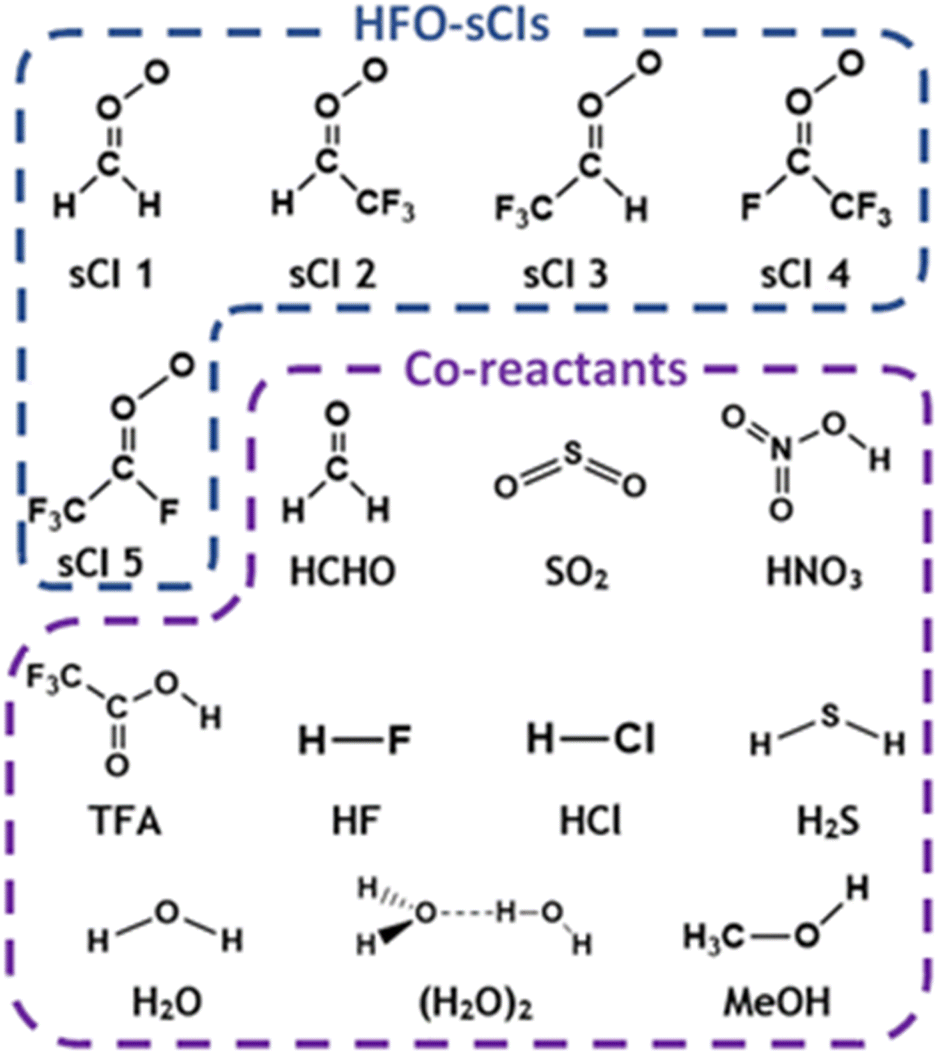 | ||
| Fig. 2 Geometric structures of hydrofluoroolefin-derived stabilised Criegee intermediates, HFO-sCIs, labelled as sCIs 1–5, and co-reactants examined in this in this study. In each case, rate constants for each HFO-sCI + co-reactant reaction are evaluated. | ||
Also displayed in Fig. 2 is the targeted range of co-reactants selected for this computational study, including both H2O & (H2O)2 because the global abundance of water vapour (∼0–40000 ppm) means that these molecules are responsible for reacting with up to 95% of the total global flux of some atmospheric sCIs.67,68,78–83 As shown previously with the high abundance of TFA in Beijing, HFO breakdown produces a variety of secondary products, including HCHO, and so both TFA & HCHO are studied here as they like to have significant overlapping local concentrations with HFO-sCIs.3,4,28,36,49–51
Some co-reactants (HNO3, HCl & H2S) are selected for study here because there are some environments in which CH2OO competes with OH radicals to facilitate the depletion of these co-reactants, as noted by Khan et al.42 Furthermore, some co-reactants studied here, such as SO2, are of interest because their reactions with sCIs are a competitive source for tropospherically relevant species such as SO3, which is considered vital in aerosol nucleation pathways.84,85 Another example is that sCI + MeOH & H2O reactions produce a large proportion of the global flux of the α-alkoxyalkylhydroperoxides (AAAH) and α-hydroxy-hydroperoxides (HHP). These are oxidising species that cause both forest damage and SOA formation.67,68,72,78–80,83,86
One last area of consideration in co-reactant targeting is that Kumar et al. propose that sCI reactivity with H2X (X = O, S, Se, and Te) increases with the size of the X atom, referred to as heteroatom tuning of the co-reactants.87 In this study, the co-reactants with low mass heteroatoms, H2O & HF, are compared with species which have the same structures but larger heteroatoms of the same periodic group, H2S & HCl, to see if all the HFO-sCIs in this chapter replicate the co-reactant heteroatom tuning patterns observed in the Kumar et al. study.87 The last reactions featured are examined because the HCHO + sCIs 2 & 3 and sCI 1 + CF3CHO reactions are part of the same multistep potential energy surface (PES) and so the theoretical rate constants (kTHEO) and product branching ratios (ΓTHEO) of sCI 1 + CF3CHO can be determined with little additional computational cost. This is also the case for HCHO + sCIs 4 & 5 and sCI 1 + CF3CFO reactions which are all part of the same multistep potential energy surface.
By using computational chemistry methods to review reactions with this variety of common tropospheric gas species, this study aims to evaluate to what degree the bimolecular chemistry of HFO-sCI (syn- & anti-CF3CHOO and syn- & anti-CF3CFOO) differs from that of CH2OO, as well as the other non-halogenated sCIs.
2 Methodologies
This work employs a density functional theory (DFT) approach, using the hybrid B3LYP functional, and the Dunning correlation-consistent aug-cc-pVTZ basis set (B3LYP/aug-cc-pVTZ), is used to optimise the geometric structures of all minimum and transition state (TS) geometries, referred to collectively as stationary points.88–99 Each minimum or TS structure is verified through evaluation of the number of imaginary frequencies from subsequent harmonic frequency calculations, performed at the same level of theory as the optimisation. TSs identified in this manner are mapped to the geometric minima that they bisect using intrinsic reaction coordinate (IRC) calculations. For very shallow TSs and barrierless reactions, IRC calculations become computationally challenging, therefore in these cases relaxed potential energy scans are performed using incremental separation of a molecular bond (which forms a key component of the reaction coordinate) until the local minima is reached. The relax surface scans for non-barrierless channels each have a local energy maximum, which denote the location of the TS structure on the potential energy surface, whereas the energy diagram for barrierless channels only display asymptotic Morse-like behaviour. Optimisations, harmonic frequencies, IRCs and potential energy scans are all performed using the Gaussian09 computational chemistry programme.100 All optimisations in this study were performed on singlet surfaces.Rate constants and PESs are constructed by using DFT optimised structures with energies refined using a higher level, ab initio approach. Single point molecular energy calculations, for all stationary points, are calculated using a localised, density fitted coupled cluster method with the same Dunning basis set (DF-HF/DF-LCCSD(T)-F12a/aug-cc-pVTZ).91,94–96,101–111 These molecular energies are calculated in the MOLPRO computational chemistry package.112 All zero-point energies or Gibbs free energies in this study are calculated using the zero-point and free energy corrections acquired from the DFT harmonic frequency calculations. All relative energies (ΔE) referenced herein in the main body of this manuscript and in all PESs are zero-point corrected. This overall computational approach, when applied to the variety of sizes of sCIs and co-reactants in this study, provides both accurate energies and manageable computational cost. This computational approach is used in other studies in the literature too and it effectively describes the chemistry of sCI reactions with TFA & alcohols.61,74 For further verification of the veracity of this computational approach, the theoretical rate constants (kTHEO) for the bimolecular sCI 1 reactions obtained in this study are benchmarked against experimental rate constants (kEXP) from the literature.
The open source Master Equation Solver for Multi-Energy Well Reactions (MESMER) software is used to calculate the microcanonical kinetic reaction rates by deriving the individual energy-grained master equation (EGME) rate coefficients, which are then subsequently used to generate the overall master equation rate constant (kTHEO) and product branching ratios (ΓTHEO) of the system over a range of temperatures.113 Computational chemistry rate constants from other studies are also referred to as kTHEO constants, even if generated using differing methods. Master equation rate constants are referred to as kME values only in the ESI† to distinguish them from other theoretical rate constants. This software has been used extensively to solve analogous chemical problems, and has been utilised previously to evaluate Criegee intermediate reaction kinetics.76,84,114–119
These MESMER calculations include a rigid rotor harmonic oscillator approximation, with structures and vibrational frequencies again taken from the DFT calculations, as shown by the example MESMER input in ESI Section S9.†113 An inverse Laplace transform (ILT) capture rate coefficient of 1 × 10−10 cm3 s−1 and an excess reactant concentration of 1 × 1016 molecule cm−3 are assumed.76,113 The collisional energy transfer factor, 〈ΔEdown〉, was assigned as 300 cm−1 for all minima. This has been shown to be a good approximation for systems involving nitrogen as a gas bath.76,113,120 A 10 cm−1 grain size was routinely used for the EGME calculations, however, for the larger systems, grain sizes are adjusted to reduce computational expense (ESI Sections 1.1–1.4†).
Low mass atom motion within the reaction mechanism is sometimes facilitated by quantum tunnelling, often accelerating the reaction. The effect of tunneling on the rate constant is determined herein using a non-ab initio tunneling correction called the asymmetrical Eckart constant (kECKART).121 This kECKART is calculated and then integrated directly into the kTHEO and ΓTHEO values through the MESMER software package, unless the reaction barriers are too low to support this methodology (these exceptions are stated in the ESI Section S1.1†).76,113,121 In calculating the kECKART function, the probability of transmission through the one-dimensional energy barrier, p(E), needs to be calculated using eqn (1)–(7):
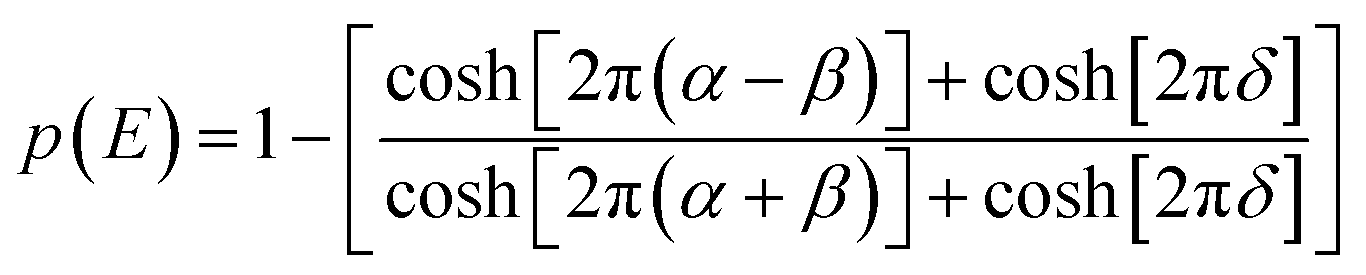 | (1) |
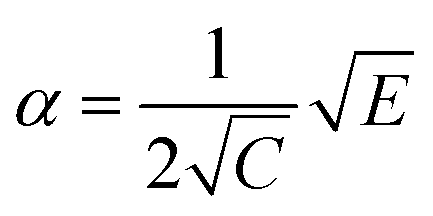 | (2) |
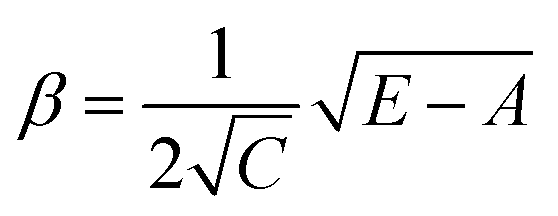 | (3) |
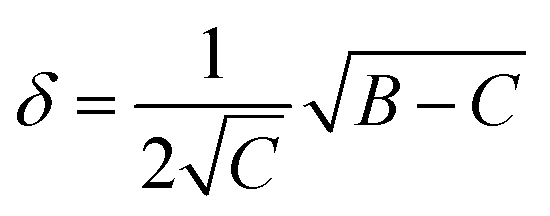 | (4) |
| A = ΔH‡,0Kf − ΔH‡r | (5) |
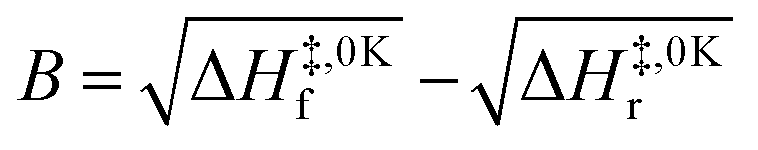 | (6) |
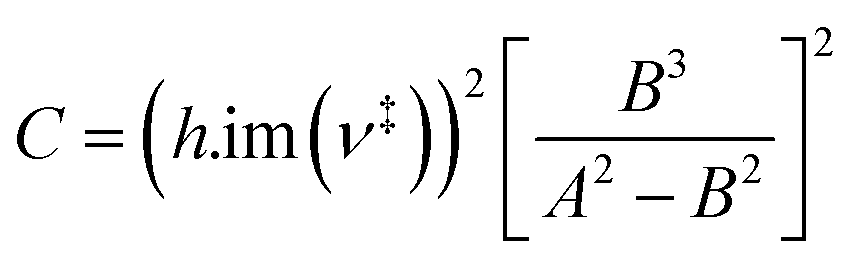 | (7) |
Calculating the probability of transmission involves estimating the barrier width between the reactants and products using: the forward and reverse zero-point corrected energy barriers, ΔH‡,0Kf and ΔH‡,0Kr, and the imaginary frequency of the associated TS describing the reaction coordinate, v‡. The kECKART value is then determined using p(E) in eqn (8):
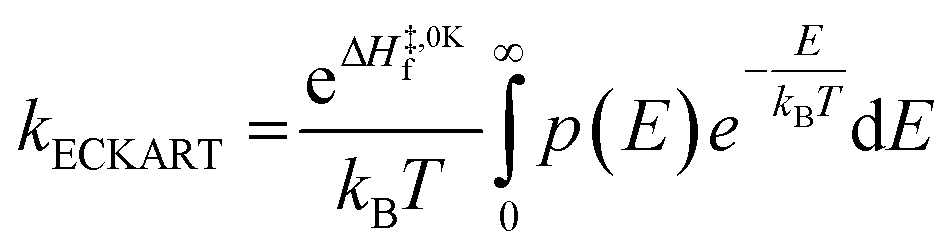 | (8) |
The irreversible final product formation processes are accommodated by the MESMER software by means of using an infinite “sink” approximation. In a system where a particular reaction channel that produces multiple products via a post-reaction complex (i.e. reactant → TS → post-reaction complex → product 1 + product 2) herein the formation of the post-reaction complex is treated as irreversible by MESMER when determining the kECKART & ΓTHEO values and it is assumed that ∼100% of this post-reaction complex separates into the final individual bimolecular products. This approximation reduces the computational intensity of MESMER calculations and introduces an error of less than 1%.122,123 This use of the post-reaction complex can be seen in the MESMER example file in the ESI.†
In some multi-step reactions, a small number of the intermediate product → final products fragmentations involve progressing over an asymptotic Morse-like energy curve until the final separated products are formed, rather than fragmenting via a transition state. When Peltola et al. examined CH2OO decomposing into HCO + OH and Blitz et al. studied CH3NH breaking down into CH3 + NH, both used a reverse inverse Laplace transformation (reverse ILT) method in MESMER to interpret these chemical pathways,117,124 and both use the reverse ILT method with the experimental rate constant (kEXP) of the equivalent forward reaction as the pre-exponential factor.117,124 However, as the reaction channels in this study where the reverse ILT method is used have no literature kEXP values instead the dipole–dipole capture limit rate constant, kd–d, (discussed in the next paragraph) are used as the pre-exponential factors instead. Furthermore, this reverse ILT method requires using the term reverse = true in the activation energy component and assigning both final products as separate sink structures.
In a few previous studies, sCI reactions with other trace atmospheric species (e.g.sCI 1 + HCOOH, CH3COOH or CF3COOH) have produced such large kTHEO & kEXP values that each sCI + co-reactant collision leads to a reaction.59–61,74,125 Under such conditions, it is proposed that kTHEO values can be more accurately modelled using a collision frequency model, such as the dipole–dipole capture limit, kd–d. The kd–d value is calculated using eqn (9) and (10):
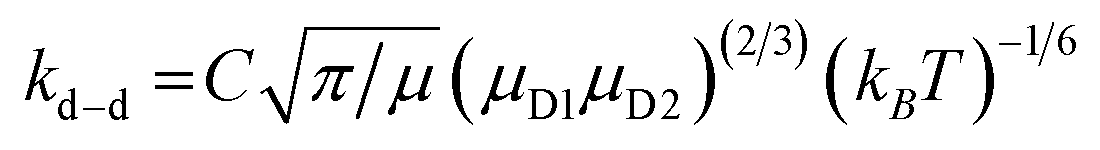 | (9) |
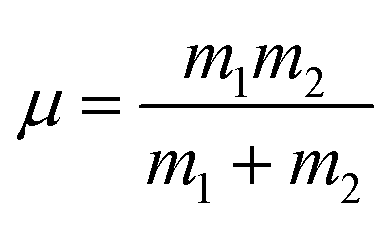 | (10) |
C is a constant dependent on the anisotropy of the capture potential; μD1 and μD2 are the dipole moments of reactants 1 & 2; and μ is the total reduced mass of the system calculated using the mass of reactants 1 & 2 (m1 & m2).126 The C value adopted can be either isotropic (4.08), adiabatic anisotropic (2.68) and non-adiabatic anisotropic (1.953).126 The isotropic kd–d constant is used in this study, as it is much used in the literature for bimolecular sCI reactions.61,125
Frequently in this study, the rate determining step is barrierless or kTHEO > kd–d (usually where kTHEO ≥ ∼10−10) and so the kd–d is used as the overall rate constant, as it is a more true reflection of the tropospheric rate constant. As sCIs are polar in character, the isotropic kd–d value is used over the more traditional collision limit (kCOLL) too, as kd–d is better able to model the effect this polarity has on sCI — co-reactant collision frequency. However, the kCOLL values for each reaction, along with the adiabatic anisotropic and non-adiabatic anisotropic kd–d values, are all listed in the ESI Section S3† for comparison. The rate constants generated in this work are analysed using the effective first-order rate constant, kEFF, which accounts for the concentration of the co-reactant:35
| kEFF = kTHEO[co-reactant] | (11) |
These kEFF values in this work are calculated using previously reported co-reactant concentrations in example ‘important’ local environments. This data is displayed in Table 2 in the next section, alongside any known experimental rate constants for the same reactions. Further examples of other local co-reactant abundances in other environments and the effect they have on the kEFF values are listed in ESI Section S4.†
3 Results & discussion
The bimolecular master equation rate constant (kTHEO), the dipole–dipole capture limit (kd–d), and the effective rate constant (kEFF), for the HFO-sCI reactions calculated in this study are found in Table 2. The co-reactant abundances featured in Table 2 are selected from locations where either the pollutant has a high enough concentration to be a potentially significant sCI sink and/or an area where humans may face a significant degree exposure of either the pollutant or the potential product of this reaction. These values are then used to generate the kEFF values seen in Table 2 (to view the extended literature survey of the tropospheric abundances of the co-reactants, see ESI Section S4.2†). As most of these values are averages from single locations across a protracted period of time or a generalised upper-limit in certain urban environments from a variety of locations, most measurements occur over a range of temperatures that include 298 K. Either the kTHEO value or the kd–d value are in bold to highlight which the rate constant is used to calculate each kEFF value. Any ΓTHEO branching fractions of the two-step HFO-sCI reactions with aldehydes & SO2 are not displayed in Table 2 and can be instead found in Sections 3.1 & 3.2.| [Co-reactant] (molec. per cm3) | sCI | k THEO (cm3 s−1) | k d–d (cm3 s−1) | k EXP (cm3 s−1) | k EFF (s−1) |
|---|---|---|---|---|---|
| a “≫kd–d” refers to barrierless reactions that yield no kTHEO and that the kd–d value should be used instead. b In bold is the overall theoretical rate constant, which is the lowest between the kTHEO & kd–d values. | |||||
| HCHO, 2.0 × 1012 (Urban heavy traffic)128 | 1 | 2.79 × 10−12 | 1.06 × 10−9 | 4.1 × 10−12 (ref. 127) | 5.60 |
| 2 | 2.69 × 10−13 | 8.04 × 10−10 | — | 0.54 | |
| 3 | 2.49 × 10−12 | 6.46 × 10−10 | — | 5.0 | |
| 4 | ≫kd–d | 7.20 × 10−10 | — | 1447 | |
| 5 | 1.66 × 10−11 | 7.07 × 10−10 | — | 33.4 | |
| CF3CHO | 1 | ≫kd–d | 6.50 × 10−10 | — | — |
| CF3CFO | 1 | 1.98 × 10−12 | 3.34 × 10−10 | — | — |
| SO2, 4.1 × 1011 (Beijing urban area)129 | 1 | ≫kd–d | 7.25 × 10−10 | 3.9 × 10−11 (ref. 63) | 300 |
| 2 | 1.90 × 10−12 | 5.08 × 10−10 | — | 0.79 | |
| 3 | ≫kd–d | 4.08 × 10−10 | — | 169 | |
| 4 | ≫kd–d | 4.51 × 10−10 | — | 186 | |
| 5 | ≫kd–d | 4.42 × 10−10 | — | 183 | |
| HNO3, 1.1 × 1011 (Houston, Texas)130 | 1 | 8.22 × 10−9 | 8.45 × 10−10 | 5.1 × 10−10 (ref. 59) | 94 |
| 2 | 2.06 × 10−11 | 5.93 × 10−10 | — | 2.3 | |
| 3 | 8.87 × 10−11 | 4.76 × 10−10 | — | 9.8 | |
| 4 | 9.25 × 10−8 | 5.26 × 10−10 | — | 58 | |
| 5 | 7.53 × 10−11 | 5.16 × 10−10 | — | 8.4 | |
| TFA, 3.3 × 107 (Beijing higher emission areas)28 | 1 | ≫kd–d | 7.54 × 10−10 | 3.5 × 10−10 (ref. 61) | 2.5 × 10−2 |
| 2 | ≫kd–d | 4.96 × 10−10 | — | 1.7 × 10−2 | |
| 3 | ≫kd–d | 3.98 × 10−10 | — | 1.3 × 10−2 | |
| 4 | ≫kd–d | 4.35 × 10−10 | — | 1.5 × 10−2 | |
| 5 | ≫kd–d | 4.27 × 10−10 | — | 1.4 × 10−2 | |
| H2O, 6.2 × 1017 (∼80% humidity Sao Paulo)132,133 | 1 | 1.18 × 10−16 | 1.06 × 10−9 | 2.4 × 10−16 (ref. 131) | 73 |
| 2 | 1.35 × 10−18 | 8.38 × 10−10 | — | 0.83 | |
| 3 | 1.12 × 10−16 | 6.73 × 10−10 | — | 70 | |
| 4 | 5.13 × 10−12 | 7.54 × 10−10 | — | 3.2 × 106 | |
| 5 | 2.27 × 10−13 | 7.40 × 10−10 | — | 1.4 × 105 | |
| (H2O)2, 8.7 × 1014 (∼80% humidity Sao Paulo)132,133 | 1 | 3.28 × 10−12 | 1.07 × 10−9 | 7.5 × 10−12 (ref. 131) | 2.9 × 103 |
| 2 | 2.71 × 10−12 | 7.99 × 10−10 | — | 2.4 × 103 | |
| 3 | 3.74 × 10−12 | 6.42 × 10−10 | — | 3.3 × 103 | |
| 4 | 1.14 × 10−6 | 7.14 × 10−10 | — | 6.2 × 105 | |
| 5 | 1.46 × 10−9 | 7.01 × 10−10 | — | 6.1 × 105 | |
| MeOH, 8.4 × 1011 (Sao Paulo, Brazil)135 | 1 | 1.20 × 10−14 | 8.15 × 10−10 | 1.1 × 10−13 (ref. 134) | 0.010 |
| 2 | 9.43 × 10−17 | 6.14 × 10−10 | — | 7.9 × 10−5 | |
| 3 | 3.30 × 10−14 | 4.93 × 10−10 | — | 0.028 | |
| 4 | ≫kd–d | 5.50 × 10−10 | — | 462 | |
| 5 | 3.31 × 10−11 | 5.39 × 10−10 | — | 28 | |
| H2S, 1.2 × 1013 (industrial landfill)137 | 1 | 7.06 × 10−15 | 5.68 × 10−10 | 1.7 × 10−13 (ref. 136) | 0.052 |
| 2 | 1.96 × 10−16 | 4.26 × 10−10 | — | 1.4 × 10−3 | |
| 3 | 5.72 × 10−15 | 3.42 × 10−10 | — | 0.042 | |
| 4 | 6.08 × 10−13 | 3.81 × 10−10 | — | 4.5 | |
| 5 | 1.23 × 10−14 | 3.74 × 10−10 | — | 0.091 | |
| HCl, 1.2 × 1011 (daytime Los Angeles)138 | 1 | 4.70 × 10−10 | 6.06 × 10−10 | 4.1 × 10−11 (ref. 59) | 58 |
| 2 | 4.04 × 10−15 | 4.51 × 10−10 | — | 5.0 × 10−3 | |
| 3 | 7.28 × 10−11 | 3.62 × 10−10 | — | 8.96 | |
| 4 | 1.64 × 10−10 | 4.03 × 10−10 | — | 20 | |
| 5 | 1.13 × 10−10 | 3.96 × 10−10 | — | 14 | |
| HF, 3.7 × 1011 (near volcano vicinity ambient T)139 | 1 | 3.32 × 10−13 | 8.15 × 10−10 | — | 0.12 |
| 2 | 6.67 × 10−18 | 6.14 × 10−10 | — | 2.5 × 10−6 | |
| 3 | 2.86 × 10−15 | 4.93 × 10−10 | — | 1.1 × 10−3 | |
| 4 | 1.19 × 10−11 | 5.50 × 10−10 | — | 4.4 | |
| 5 | 3.66 × 10−13 | 5.39 × 10−10 | — | 0.14 | |
3.1 HFO-sCI reaction with formaldehyde (HCHO)
During an ozonolysis event that generates a Criegee intermediate, a carbonyl containing species (aldehydes and ketones) are very common co-products and as such these carbonyl moieties are co-located with sCIs during formation such that a further reaction between these two proximate species is probable. Formaldehyde (HCHO) is produced from the ozonolysis of a variety of alkenes, notably for this work that includes HFO-1234yf (see Fig. 3), and therefore examining any possible secondary reaction with HCHO is likely vital to understanding the overall tropospheric chemistry of these HFO-sCIs.4,48,140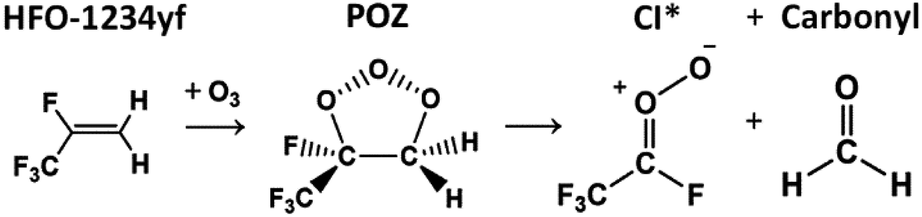 | ||
| Fig. 3 The general schematic for the ozonolysis of HFO-1234yf producing a short-lived primary ozonide (POZ) followed by one of the pairs of final products, a Criegee intermediate (sCI 5 or anti-CF3CFOO) and a carbonyl product (formaldehyde). | ||
Also, as HCHO emissions are strong in areas of heavy urban traffic (up to 170 μg m−3) and HFOs are used in vehicle coolant units, there is a high probability of emissions overlap between HCHO and HFO-sCIs.36,128,141,142 Additionally as HCHO is carcinogenic and toxic if inhaled, sCIs depletion of tropospheric HCHO should be included in many atmospheric models.143–145
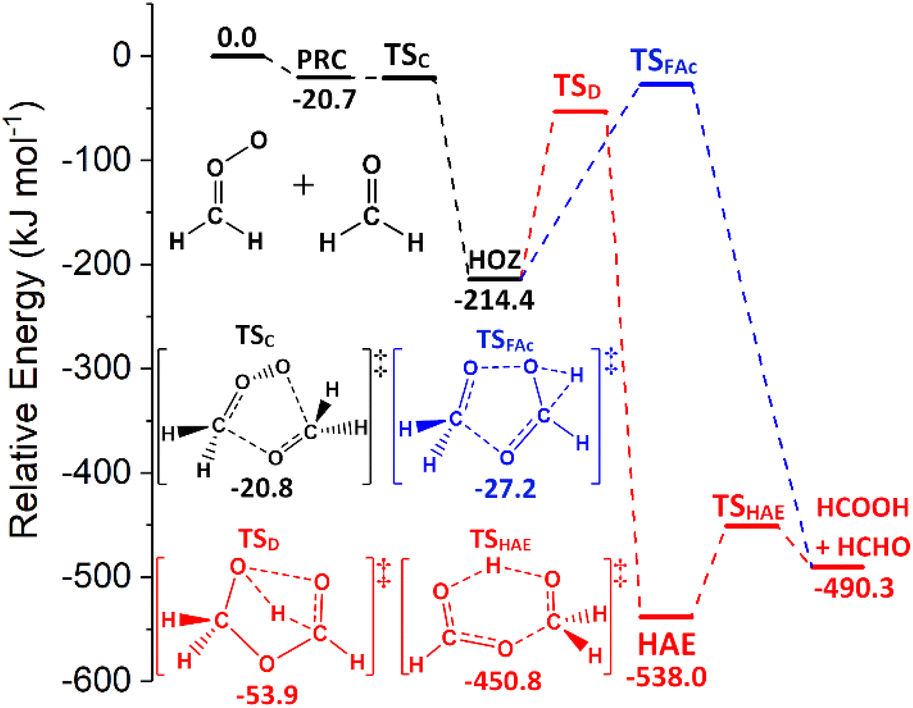 | ||
| Fig. 4 The potential energy surface for the sCI 1 + HCHO reaction. The lowest energy transition state for each HOZ fragmentation pathway is displayed. The 1-step direct transfer channel via the TSFAc barrier is in blue and a 2-step hydroxyalkyl ester (HAE) channel, via the TSD & TSHAE barriers, is red. Energies are relative to the raw reactants. | ||
Computational assessments of the sCI 1 + HCHO reaction (Fig. 4) are characterised by a low energy TSC barrier (∼−25 to −20 kJ mol−1), which leads to high theoretical rate constants both here (kTHEO ∼ 2.79 × 10−11 cm3 s−1) and in the literature (kTHEO ∼ 4.03 × 10−10 cm3 s−1).71,146 The kTHEO value determined here is also similar to the kEXP values measured for sCI 1 + HCHO by Luo et al., (4.11 ± 0.25) × 10−12, as well as those determined for studies of the analogous sCI 1 + CH3CHO reaction (3.0 × 10−13 to 1.7 × 10−12 cm3 s−1).64,65,127,147
The sCI 1 + HCHO reaction produces only one final product set, HCOOH + HCHO, and of the two HOZ fragmentation mechanism, the TSD barrier of the two-step HAE channel has a lower energy than the TSFAc barrier to the one-step HOZ fragmentation channel (Fig. 4). Both findings are authenticated by similar results in prior studies.71,146 Obtaining reliable TSD & TSFAc barriers is important because whereas the absolute height of the energy barrier(s) of the first reaction step dictates the kTHEO value, the crucial factor in determining the ΓTHEO branching fractions is the energy difference between the HOZ fragmentation barriers. This study also agrees with other computational studies in that that the second step of the two-step HOZ fragmentation, the breakdown of the HAE, involves a very low TSHAE barrier (Fig. 4).71,146
Given that the HAE is formed with considerably higher energy than the HAE → TSHAE barrier height, the MESMER results discussed here are calculated on the assumption that formation of the HAE is the final step of that reaction channel and that all the HAE population reacts further to produce their acid + aldehyde products, with negligible populations of HAE remaining. These MESMER simulations yield a very similar result (ΓTHEO ∼ 0.973) to those in which the HCOOH + HCHO are set as the final products (ΓTHEO ∼ 0.978). As this methodology has been shown to have little effect on the accuracy, this ∼100% HAE → acid + aldehyde conversion principle is applied to the HCHO reactions with sCIs 2–5, for the purpose of reducing computational cost.
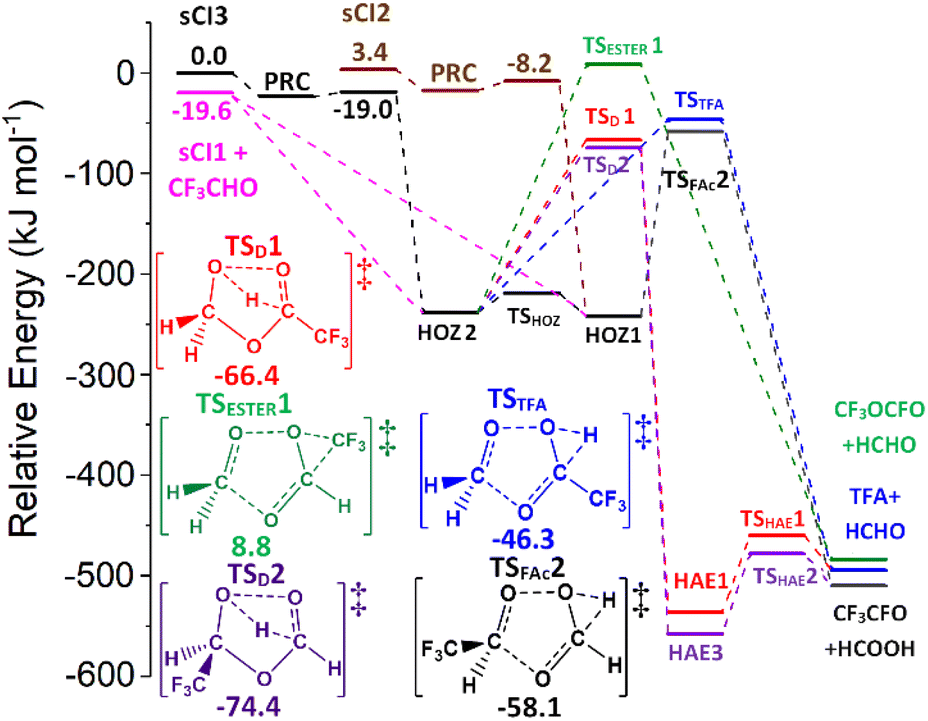 | ||
| Fig. 5 The collective potential energy surface for sCIs 2 & 3 + HCHO and sCI 1 + CF3CHO reactions with minima energy structures of each HOZ fragmentation displayed. Energies are relative to the raw sCI 3 + HCHO reactants. | ||
The relatively low TSC barriers seen for the sCIs 2 & 3 reactions with HCHO in Fig. 5, lead to large kTHEO values (2.69 & 24.9 × 10−13 cm3 s−1), although both reactions are less reactive than sCI 1 + HCHO (see Table 3). Studies of the bimolecular HFO-sCI chemistry are limited, but computational work on sCIs 1–3 reactions with H2, CO2 & CF3CHCH2 are present in the literature and show similar trends to those identified here (Table 3).72,73,77 For instance, the barriers to the HFO-sCIs + CO2 reaction follow the same reactivity order (ETS (sCI 2) > ETS (sCI 1) > ETS (sCI 3)) seen for these HCHO reactions.72 Furthermore, the anti-orientated sCI 3 is more reactive with CF3CHCH2 than the syn-orientated sCI 2, the same result observed for sCIs 2 & 3 + HCHO.77
While the inclusion of a single –CF3 group does not significantly enhance the reactivity of sCIs 2 & 3, the presence of a single –CF3 substituent in the aldehyde co-reactant increases its reactivity to such a degree that the cycloaddition of sCI 1 + CF3CHO is barrierless and the kd–d value (∼6.50 × 10−10 cm3 s−1) is used as the rate constant. This could be due to a hypothetical inductive effect caused simply by adding any alkyl substituent to the aldehyde. But, despite the increased number of alkyl groups on CH3CHO & (CH3)2CO the experimental literature suggests that their reactions with sCI 1 (kEXP ∼ 10−1–10−12 cm3 s−1) do not show the enhanced reactivity seen for the sCI 1 + CF3CHO reaction.65,71,147 However, the role of fluorine in –CF3 substituents is explored in an experimental study by Taatjes and co-workers and they show that sCI 1 + (CF3)2COO has a larger kEXP (∼3.0 ± 0.3 × 10−11 cm3 s−1) compared to the kEXP of sCI 1 + (CH3)2COO (∼2.3 ± 0.3 × 10−13 cm3 s−1).65 This confirms the high reactivity seen for sCI 1 + CF3CHO must emerge from the inductive impact that the fluorenes in the –CF3 substituents have on the aldehyde reactivity.
Both HCHO + sCI 2 and HCHO + sCI 3 (Fig. 6) produce high yields of the sCI 1 + CF3CHO (∼0.91–0.94), due to the barrierless nature of their cycloreversion mechanisms, with small contributions from ΓTFA+HCHO (∼0.012–0.016) and ΓHCOOH+CF3CHO (∼0.052–0.068). In contrast, during the sCI 1 + CF3CHO reaction, the production of HCHO + sCIs 2 & 3 gives very low branching ratios (<0.001) because the energy barriers for producing HCHO + sCIs 2 & 3 are much larger than other HOZ fragmentation pathways. Consequently, the overall ΓTHEO of sCI 1 + CF3CHO is dominated by ΓHCOOH+CF3CHO ∼ 0.823 with most of this contribution coming from the two-step HAE pathways (ΓTSD1 ∼ 0.746) with a smaller contribution from the one-step channel (ΓTSFAc ∼ 0.077). The secondary product yield, ΓTFA+HCHO ∼ 0.177, also has most of its contribution emerge from the two-step HAE pathways, ΓTSD2&TSD3 ∼ 0.166. The TSESTER barrier is too high for the ester formation to have anything but a negligible yield for all three reactions.
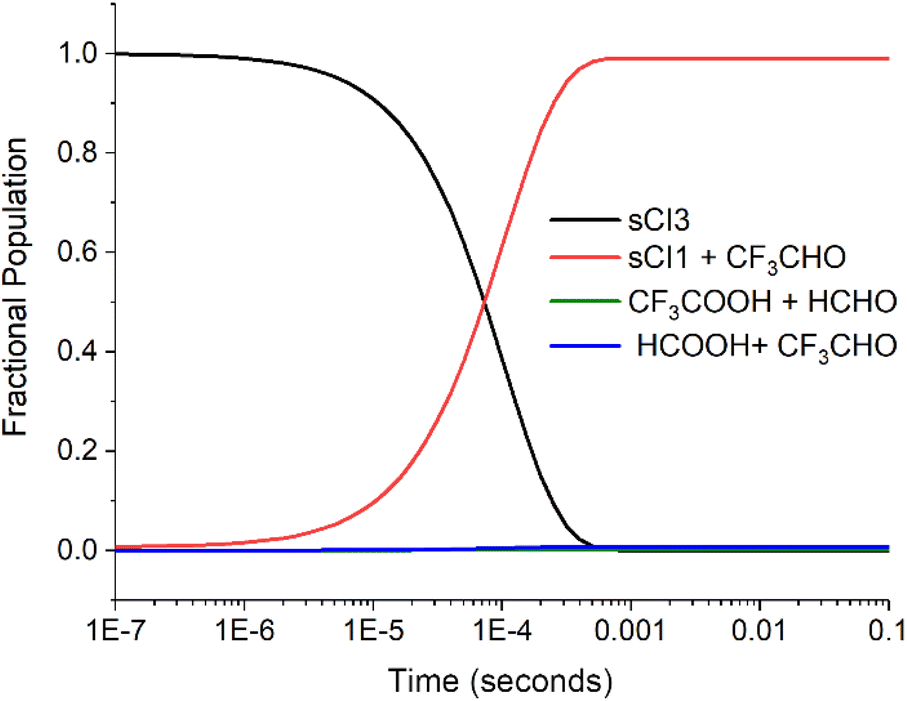 | ||
| Fig. 6 The fractional populations of reactants and products for the sCI 3 + HCHO reaction over time (the product branching fractions for the barrierless cycloreversion channels to sCI 1 + CF3CHO are calculated using the same reverse ILT method described in the Methods section.117,124 Excess HCHO reagent concentration is ∼1.0 × 1016 molec. per cm3. For other details on the conditions of the reaction see ESI Section S11†). | ||
While no experimental or theoretical product ratios for sCI 1 + CF3CHO are available in the literature, some of the analysis of the similar sCI 1 + CH3CHO & PhCHO reactions does include computational calculations of HOZ fragmentation energy barriers. Examination of both the sCI 1 + CH3CHO & PhCHO systems show patterns consistent with this work, including: that the formation of smaller acid final products was preferred over larger acid products, that the 2-step HAE channel was the favoured formation mechanism for these acids, and that ester formation was negligible.
sCI 4 & sCI 5 reactions with HCHO are both integrated into the same reaction system as sCI 1 + CF3CFO and therefore all sCI + aldehyde reactant pairs can also be produced during HOZ fragmentation in this overall reaction system. The ΓTHEO for both the sCIs 4 & 5 + HCHO reactions are dominated by yields of sCI 1 + CF3CFO (∼0.84) with the remainder being the ΓHCOOH+CF3CFO contribution (∼0.16). In contrast, the sCI 1 + CF3CFO reaction has only negligible yields for sCIs 4 & 5 + HCHO (<0.0001) because their cycloreversion TSC barriers are >100 kJ mol−1 higher than the lower energy HOZ fragmentation barriers.
The primary product of the sCI 1 + CF3CFO reaction is HCOOH + CF3CFO (ΓHCOOH+CF3CFO > 0.99), with the direct H-transfer TSFAc channel producing almost all the formic acid yield. The higher barriers to the HAE pathway compared to the direct H-transfer channel seen here for the sCI 1 + CF3CFO reaction system (see ESI Section S5†) is uncommon both in this study and in the literature.71,146 However, in a literature analysis of anti-PhCHOO + PhCHO, the direct H-transfer mechanism also has a higher barrier (−19.2 kJ mol−1) to phenylacetic acid formation than the two step HAE channel (−17.6 kJ mol−1).146 As noted in Table 4, ester formation continues to be negligible for all sCI + aldehyde reactions.
| Reaction sCI + HCHO | Final product branching ratio (ΓTHEO) | ||||
|---|---|---|---|---|---|
| Γ sCI1+R1CR2O | Γ HCHO+R1CR2OO | Γ HCOOH+R1CR2O | Γ TFA+HCHO | Γ ester+HCHO | |
| a The product branching fractions of the barrierless cycloreversion channels that leads to either sCI 1 + CF3CHO or sCI 4 + HCHO are calculated using the same reverse ILT method described in Method section.117,124 | |||||
| sCI 2 + HCHO | 0.993a | 7.40 × 10−5 | 0.006 | 0.001 | 1.78 × 10−7 |
| sCI 3 + HCHO | 0.991a | 3.92 × 10−6 | 0.007 | 0.002 | 1.02 × 10−7 |
| sCI 4 + HCHO | 0.843 | 9.40 × 10−9 | 0.156 | — | 3.06 × 10−5 |
| sCI 5 + HCHO | 0.842 | 8.13 × 10−9a | 0.158 | — | 4.98 × 10−5 |
| sCI 1 + R1CR2O | Γ HCHO+anti-R1CR2OO | Γ HCHO+syn-R1CR2OO | Γ HCOOH+R1CR2O | Γ TFA+HCHO | Γ ester+HCHO |
| sCI 1 + CF3CHO | 1.47 × 10−6 | 2.49 × 10−5 | 0.823 | 0.177 | 2.14 × 10−8 |
| sCI 1 + CF3CFO | 3.10 × 10−14 | 3.51 × 10−12a | >0.999 | — | 1.74 × 10−7 |
3.2 HFO-sCIs reactions with sulphur dioxide (SO2)
Toxic sulphur dioxide (SO2) emitted from various sources, including the burning of fossil fuels and volcanic eruptions, has a major role as an effective gaseous sCI sink and the sCI + SO2 reaction is therefore of significant prominence in the literature.63,78,81,82,84,85,131,149,152–158 This reaction is considered responsible for between ∼1% of the loss of syn-CH3CHOO in rural environments to ∼22% of the loss of “syn-CH3-anti-(trans-CH2CH)–COO” in boreal forests.159 Furthermore, the importance of this reaction is increased because the dominant final product of this reaction is SO3, which is a source of organosulphates and is involved in the formation and growth of secondary organic aerosol (SOA) nuclei.84,85,160 The promotion of SOA nucleation, caused by increased SO3 yields, is vital to understand because the prevalence of aerosols in urban environments can cause serious respiratory problems and premature death.161–165 Also, depending on the aerosol type, they can absorb and scatter solar radiation and therefore influence climate temperatures.161,164,165
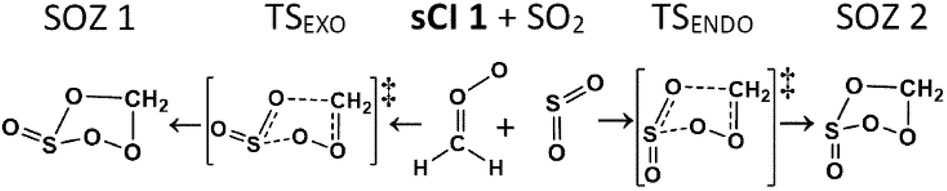 | ||
| Fig. 7 A Schematic of the cycloaddition process for sCI 1 + SO2: featuring the endo- & exo-cycloaddition transition states (TSEXO & TSENDO) and the endo- & exo-SOZ intermediate products (SOZs 1 & 2). | ||
The SOZ interconversion via the inversion of the S atom is known to require >180 kJ mol−1 excess energy, which exceeds barriers to both cycloreversion (via TSEXO & TSENDO) and SOZ fragmentation (Fig. 8).84 This would impede SOZ interconversion but, as the R1 and R2 substituents in sCI 1 are identical, SOZs 1 & 2 can freely interconvert over low energy pseudorotation TSSOZ barriers (see Fig. 8), which this means that SOZs 1 & 2 behaves as a single SOZ species.
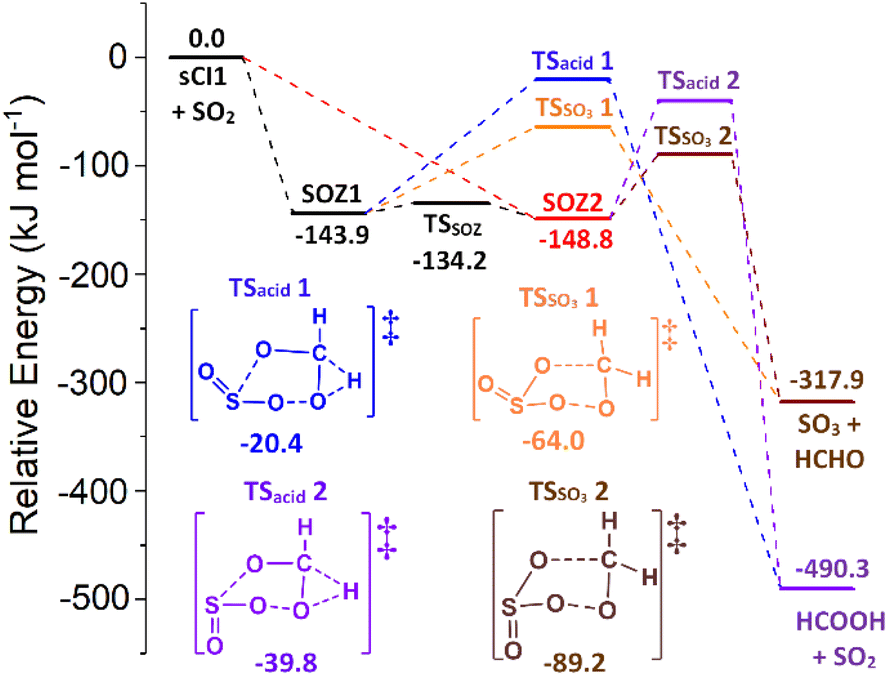 | ||
| Fig. 8 The potential energy surface of sCI 1 + SO2 with minima energy structures of each SOZ fragmentation displayed. Energies are relative to the raw reactants. | ||
The next stage of the sCI 1 + SO2 reaction is the fragmentation of the SOZ to produce either SO3 + HCHO via the TSSO3 channel or SO2 + HCOOH via the TSacid channel. All SOZ fragmentation channels studied here are single-step mechanisms and the acid-producing mechanisms from each HFO-sCI + SO2 reaction system exclusively produces a single acid species. Kuwett et al. have performed a comprehensive analysis of both SO2 reactions with sCIs 1 & (CH3)2COO comprising a numerous range of major and minor SOZ fragmentation pathways, including a homolytic O–O bond fission to produce a 1,5-(bis)oxy diradical intermediate product.84 While the self-reaction of this diradical intermediate produces important products, such as SOCO rings, sulphurous anhydrides or 1,3-(bis)oxy diradicals, its yield is so minor that 1,5-(bis)oxy diradical pathways are not explored for sCIs 1–5 + SO2 here.84
The final product branching fraction for sCI 1 + SO2 is dominated by SO3 + HCHO (ΓSO3 ∼ 0.966) with a subordinate yield HCOOH + SO2 (Γacid ∼ 0.034) because the TSSO3 1 & 2 barriers (Fig. 8) are significantly lower in energy than the TSacid 1 & 2 structures. This dominance of SO3 + CF3CHO in the ΓTHEO value is in agreement with that calculated in the literature (ΓSO3 ∼ 0.973–0.984).84
:0.31. Even though all other SO2 reactions with sCIs 1–5 are barrierless, sCI 2 + SO2 still has a substantially large theoretical rate constant (kTHEO = 1.90 × 10−12 cm3 s−1). As both the TSEXO & TSENDO channels of sCI 3 + SO2 are barrierless, the kd–d value (4.08 × 10−10 cm3 s−1) is assigned as the rate constant and SOZs 3 & 4 are produced in equal fractions.
Due to the high energy barrier for interconversion via inversion of the S atom (>180 kJ mol−1), the SOZ 1 & 2 conformers, produced from the SO2 reaction with the monosubstituted sCI 2, cannot interconvert with each other without reverting to the initial reactants. This likewise applies to the mono-substituted sCI 3 + SO2, which also produces endo and exo conformers SOZs 3 & 4. During some sCI + SO2 reactions, the SOZs produced can interconvert via a pseudorotation TSSOZ mechanism and circumvent the higher barrier via S atom inversion, but this can only transpire during reactions with sCIs with the same R1 & R2 substituents (e.g.sCI 1 or (CH3)2COO). Instead, when SOZ 1 surmounts the pseudorotation TSSOZ 1 barrier it isomerises into the SOZ 3 conformer and the TSSOZ 2 pseudorotation makes possible the SOZ 2 ↔ SOZ 4 interconversion (Fig. 9). This also means that SO2 reactions with sCIs 2 & 3 are part of the same integrated PES, through the TSSOZ 1 & 2 isomerisation channels (Fig. 9 and 10). This phenomena of 2 × non-interchangeable sets of SOZs also applies to SO2 reactions with sCIs 4 & 5 and, according to the literature, SO2 reactions with syn- & anti-CH3CHOO.84
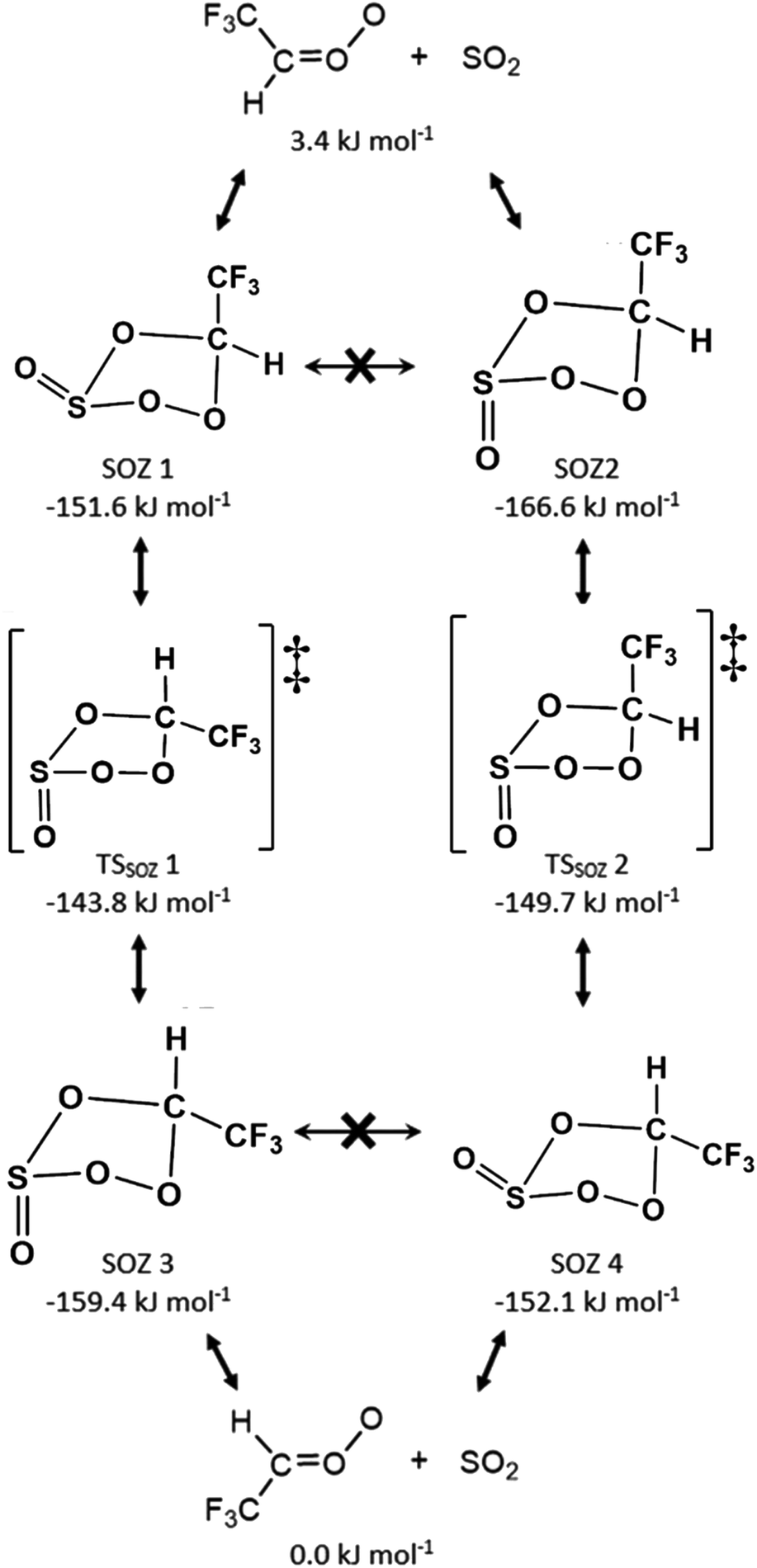 | ||
| Fig. 9 Collective diagram of all SOZs produced by sCIs 2 & 3 + SO2 reactions and the pseudorotation TSSOZ barriers that facilitate SOZ 1 ↔ SOZ 3 and SOZ 2 ↔ SOZ 4 interconversions. Structures included and energies relative to sCI 3 + SO2. | ||
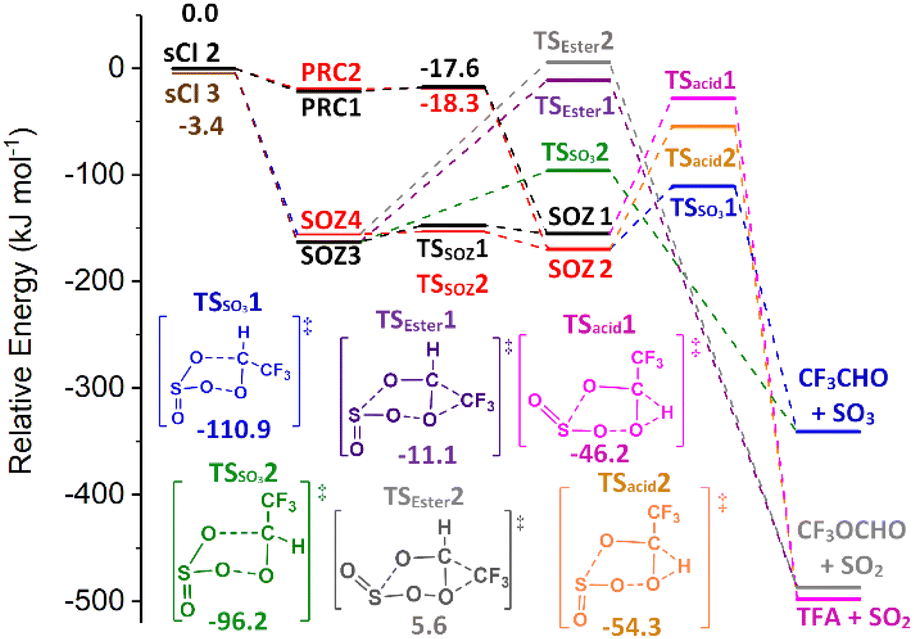 | ||
| Fig. 10 The collective potential energy surface for sCIs 2 & 3 + SO2 reactions with minima energy structures of each SOZ fragmentation displayed. Energies are relative to the raw sCI 2 + SO2 reactants. | ||
During the SOZ fragmentation of either sCI 2 + SO2 & sCI 3 + SO2 reactions, the reverse sCI + SO2 reactant pairs can be produced through cycloreversion, as both reactions are part of the same potential energy surface. The SOZ fragmentation can also proceed through: TSSO3 into SO3 + CF3CHO, TSacid into SO2 + TFA, or TSEster into SO2 + CF3OCHO. It was noted above that the cycloaddition step of the SO2 + sCI 2 reaction produced a large yield of the SOZ 2 & 4 conformer set and a small yield of the SOZ 1 & 3 conformer set, whereas the SO2 + sCI 3 produced fairly even yields of each SOZ set. Furthermore, these stereochemically distinct SOZ sets each have their own fragmentation pathways, as SOZ 1 & 3 breakdown via TSSO3 2, TSacid 1 & TSEster 1 and SOZ 2 & 4 fragments via TSSO3 1, TSacid 2 & TSEster 2. The overall product branching ratio for sCI 2 + SO2 is very similar to that of sCI 3 + SO2, so it appears that the differing SOZ yields seen in these two reactions has little overall effect on the product branching fraction.
sCI 2 + SO2 does produce a small yield of sCI 3 + SO2 from the cycloreversion channel, whereas the sCI 2 + SO2 yield from sCI 3 + SO2 reaction is negligible. This is because cycloreversion to sCI 2 + SO2 has to overcome TS barriers, whereas cycloreversion to sCI 3 + SO2 only has to overcome the energy of required to reform the raw reactants. The product branching fraction for ester formation is slight. The dominant final products for both the sCIs 2 & 3 + SO2 reactions are CF3CHO + SO3 (ΓTHEO ∼ 0.995) with only a very minor yield of TFA + SO2 (ΓTHEO ∼ 0.003).
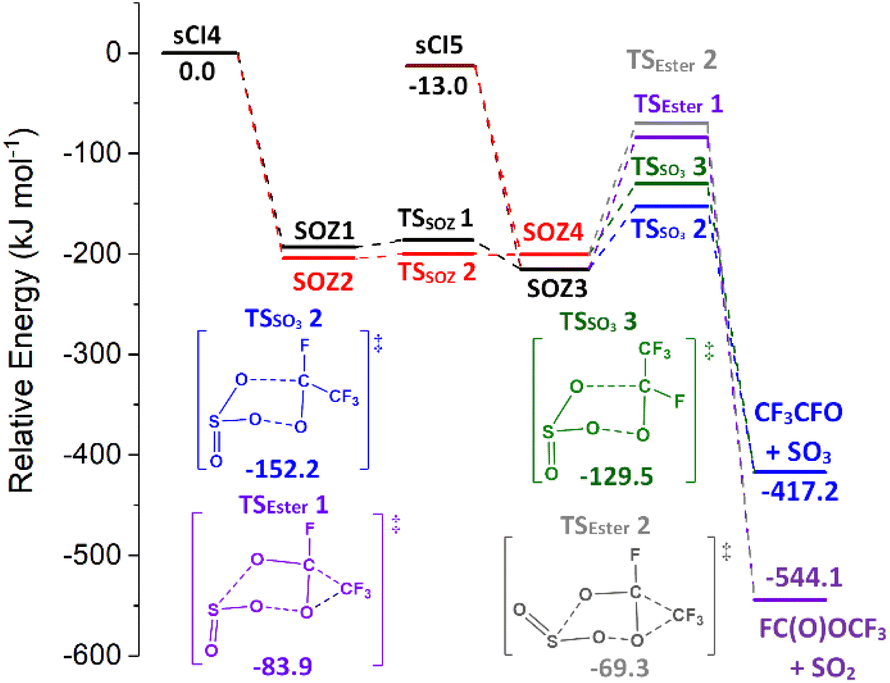 | ||
| Fig. 11 The collective potential energy surface for sCIs 4 & 5 + SO2 reactions with minima energy structures of each SOZ fragmentation displayed. Energies are relative to the raw sCI 4 + SO2 reactants. | ||
| Reaction | sCI# | Product branching ratio (ΓTHEO) | |||
|---|---|---|---|---|---|
| Γ SO3+aldehyde | Γ SO2+acid | Γ SO2+ester | Γ SO2+other CI | ||
| a The product branching fractions of the barrierless cycloreversion channels that lead to SO2 + sCIs 3, 4 or 5 are calculated using the same reverse ILT method described in Method section.117,124 | |||||
| CH2OO + SO2 | 1 | 0.968 | 0.032 | N/A | N/A |
| Syn-CF3CHOO + SO2 | 2 | 0.9922 | 0.0019 | <0.0001 | SO2 + sCI3: 0.0059a |
| Anti-CF3CHOO + SO2 | 3 | 0.9973 | 0.0027 | <0.0001 | SO2 + sCI2: <0.0001 |
| Syn-CF3CFOO + SO2 | 4 | 0.9860 | N/A | 0.0090 | SO2 + sCI5: 0.0050a |
| Anti-CF3CFOO + SO2 | 5 | 0.9955 | N/A | 0.0045 | SO2 + sCI4: <0.0001a |
The only other SOZ fragmentation calculated here are TSSO3 & TSESTER channels, as an SOZ → acid channel cannot transpire from SO2 reactions with sCIs 4 & 5 (Fig. 11). The cycloreversion channel has a negligible yield in the sCI 5 + SO2 reaction and a minor role in sCI 4 + SO2 reaction (Table 5). While TSESTER 1 & 2 are not extremely high in energy, the ester yield is still small (ΓSO2+ CF3OCHO ∼ 0.01). The low TSSO3 barriers and the minor yields of other products means that the predominant products from SOZ fragmentation are SO3 + CF3CHO (∼0.986 & 0.996).
3.3 HFO-sCIs reactions with organic and inorganic acids
Both organic acids such as TFA & carboxylic acids, and inorganic acids including HNO3 & HCl, are prevalent in various tropospheric environments (see ESI Section S4.2† for more details) and sCIs is their capacity to deplete these acids.49,59,77,130,138,139,167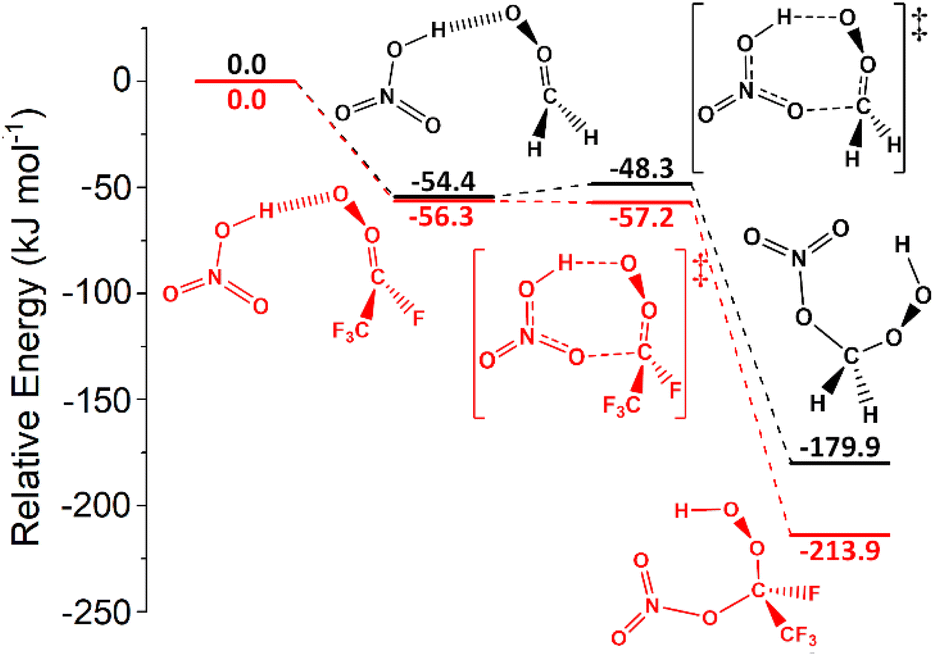 | ||
| Fig. 12 The potential energy surfaces for the sCI 1 + HNO3 (black) and sCI 4 + HNO3 (red) reactions. Energies are relative to raw reactants. | ||
None of the 1,4-insertion processes for HFO-sCI + HNO3 reactions studied here are barrierless, but the high reactivity of sCI 1 + HNO3 means that the kTHEO value (8.21 × 10−9 cm3 s−1) is so great that the kd–d limit (8.45 × 10−10 cm3 s−1) is used as the rate constant instead. This kd–d limit is also similar to the kEXP [295 K] value (5.4 × 10−10 cm3 s−1) from the experimental Foreman et al. study.59
The trend in reactivity indicated by these HFO-sCI + HNO3 relative barrier heights, ΔETS (sCI 2) > ΔETS (sCI 3) > ΔETS (sCI 1) > ΔETS (sCI 5) > ΔETS (sCI 4), further verifies the HFO-sCI reactivity series seen in previous sections. However, the overall reactivity trend, kTHEO (sCI 2) < kTHEO (sCI 3) < kTHEO (sCI 5) < kd–d (sCI 4) < kd–d (sCI 1), is altered by the rate constants of HNO3 reactions with sCIs 1 & 4, which reaching the kd–d limit, and an surprisingly low kTHEO value for sCI 5 + HNO3 (7.57 × 10−11 cm3 s−1).
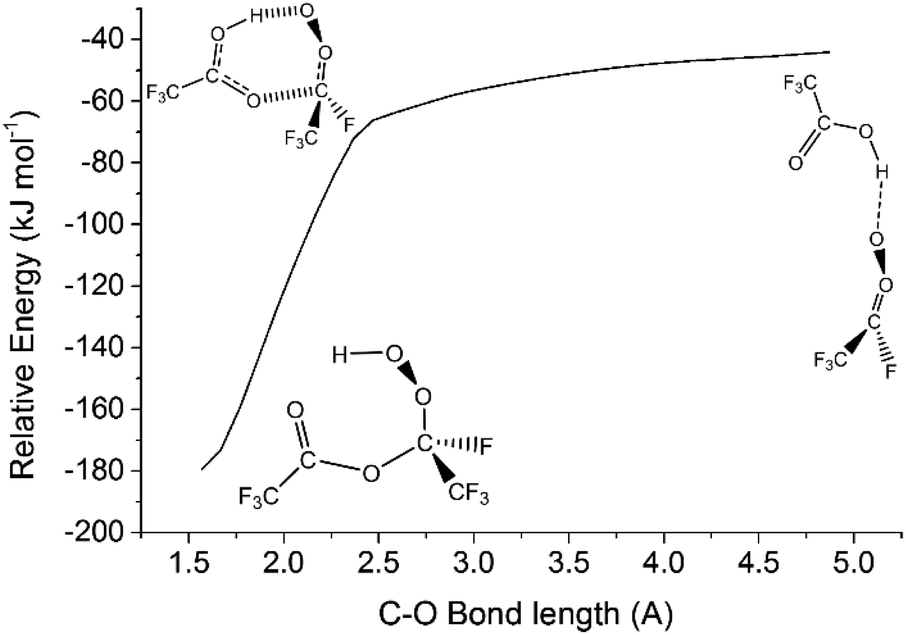 | ||
| Fig. 13 The barrierless minimum energy pathway for the sCI 4 + TFA reaction. Energies displayed here are derived from using the B3LYP/aug-cc-pVTZ approach. Energies displayed here are relative to raw reactants. | ||
Altering the sCI substituents does not alter the barrierless nature of the HFO-sCI + TFA reaction profiles studied here (see ESI Section S8.5†). For this reason, all the sCIs 1–5 + TFA reactions use the kd–d values as rate constants (3.98–7.54 × 10−10 cm3 s−1).
3.4 HFO-sCI reactions with water monomer and dimer
Due to the high tropospheric concentration of water vapour, water acts as the most prevalent bimolecular sink for sCIs, and therefore examining the chemistry of sCIs 1–5 reactions with H2O & (H2O)2 is essential. sCIs 1–5 reactions with H2O proceed through only two reaction channels (TSH2O 1 and TSH2O 2), whereas sCIs 1–5 reactions with (H2O)2 proceed via four pathways: TS(H2O)2 1, TS(H2O)2 2, TS(H2O)2 3 and TS(H2O)2 4 (see ESI Section S8.9†). One commonality between these reactions is that the primary product is an α-hydroxy-hydroperoxide (HHP), which is implicated in both forest damage and generating crucial secondary products (inc. H2O2, organic acids & aldehydes).86,170–176As the yields of the HHP products are sizable, the atmospheric fate of these HHPs is the subject of many scientific studies.67,68,131,150,157,177–179 Under standard conditions, photolysis, deposition, and reaction with hydroxyl radicals are usually the main sinks for HHPs, but the excess internal energy within the HHP at the point of formation increases the significance of thermal unimolecular HHP decay.131,177 The primary unimolecular decay mechanism for hydroxymethyl hydroperoxide (HMHP), the product of CH2OO + H2O, is the elimination of H2O2 to produce formaldehyde, with some yields from minor channels that produce either H2O + HCOOH or OH + OCH2OH.180,181 Sheps et al. showed in a study of CH2OO + (H2O)2 that while ∼55% of the HMHP remained stable, this excess energy led to ∼40% of the HMHP fragmenting to produce H2O2 and formaldehyde, both of which are implicated in biogenic degradation.131 Halogenated HHPs produced from sCIs 2–5 are also likely to be produced with excess energy, and therefore exploring the breakdown of these products may be useful in the future.
The kTHEO value produced for sCI 1 + H2O (1.18 × 10−16 cm3 s−1) is within the range of literature kEXP values (0.25–13 × 10−16 cm3 s−1), and is particularly close to the rate constant found by Sheps et al. (kEXP [293 K, 50 Torr] ∼ 2.4 × 10−16 cm3 s−1).64,78,131,182,183 Furthermore, both the kTHEO value in this study and the kEXP value found in the literature agree that sCI 1 + H2O reactions slower than sCI 1 reactions with HNO3, SO2 or TFA.59,61,63,64,85,153,154,166
The computational rate constants of the sCIs 4 & 5 + H2O reactions were found to have a negative temperature dependence, where 200 K < T < 400 K, whereas sCIs 1, 2 & 3 + H2O all have a positive temperature dependence within the same range (see ESI Section 1.2†). The general reactivity trend for these sCI + H2O reactions continues the trend observed throughout most of this study kTHEO (sCI 2) < kTHEO (sCI 3) ≤ kTHEO (sCI 1) < kTHEO (sCI 5) < kTHEO (sCI 4). The theoretical trend of sCI 4 being more reactive than sCI 5 is partially obscured by rate constants being near to or exceeding the kd–d limit, however as neither the kTHEO values for sCI 4 + H2O (5.13 × 10−12 cm3 s−1) or sCI 5 + H2O (2.27 × 10−13 cm3 s−1) reach the kd–d limit, sCI + H2O would provide an experimental case study to verify if kTHEO (sCI 4) does exceed kTHEO (sCI 5) in bimolecular reactions.
In contrast, HFO-sCI + (H2O)2 reactions are faster as a result of sCI 5 + (H2O)2 having a lower reaction barrier than sCI 5 + H2O (Fig. 14). The greater reactivity of sCI + (H2O)2 reactions is evidenced by kEXP values found for sCI 1 + (H2O)2 (4.0–8.2 × 10−12 cm3 s−1), which are in close proximity to the large kTHEO value calculated here (3.28 × 10−12 cm3 s−1).78,131,156–158,184 In line with previous HFO-sCI reactions, sCI 2 + (H2O)2 has the lowest kTHEO value (2.71 × 10−12 cm3 s−1) but unusually the sCI 3 reaction with (H2O)2 has a greater kTHEO value (3.74 × 10−12 cm3 s−1) than sCI 1. None of the 4 × TS(H2O)2 channels for H2O reactions with either sCI 4 or sCI 5 are barrierless, but the very high reactivity of these reactions leads to very large kTHEO values (1.14 × 10−6 & 1.46 × 10−9 cm3 s−1). This means that the kd–d constants of sCIs 4 & 5 + H2O (∼7 × 10−10 cm3 s−1) are used as the rate constants instead. Overall the reactivity trend for HFO-sCI + (H2O)2 reactions: kTHEO (sCI 2) < kTHEO (sCI 1) ≈ kTHEO (sCI 3) < kd–d (sCI 5) ≈ kd–d (sCI 4).
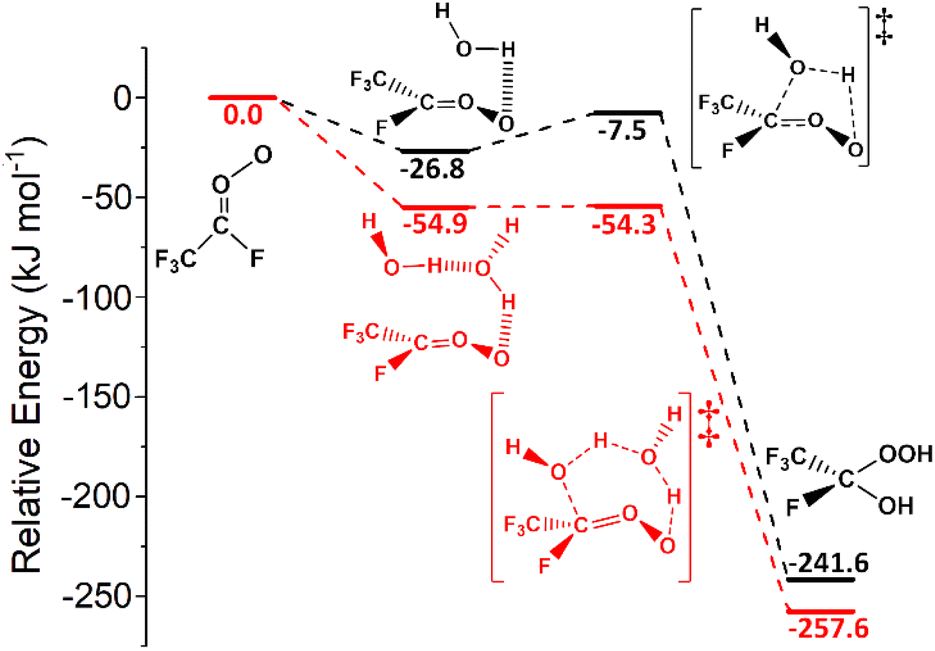 | ||
| Fig. 14 The potential energy surfaces for the sCI 5 + H2O (in black) and sCI 5 + (H2O)2 (in red) reactions. Energies are relative to raw reactants. | ||
Criegee intermediates exhibit a mixed biradical and zwitterionic character and it has been shown that the more reactive zwitterionic dominated COO moieties have the larger ratio of OO and CO bond lengths, ROO/RCO or q.68,74 Given these observations, sCIs 2 & 3 (q = 1.064 & 1.071) appear to have a more biradical COO moiety than sCI 1 (q = 1.078), whereas sCIs 5 & 4 (q = 1.101 & 1.114) are more zwitterionic. Anglada et al. noted in a computational study of sCI + H2O reactions that the degree of this zwitterionic nature of the carbonyl oxide moiety, and consequentially it's reactivity, can be tuned by modification of the substituents.68 Furthermore, in a computational study of sCI + alcohol reactions it is noted that the electron-withdrawing nature of F substituents in syn- & anti-FCHOO is likely to increase the unsaturated nature of the zwitterionic carbonyl oxide. This then generates a more electropositive central C atom, which is more vulnerable to nucleophilic attack by the alcohol's electronegative O atom. This study shows the same positive relationship between electron-withdrawing groups, zwitterionic COO moiety and high reactivity seen in the literature, as shown by the fact that increases in q values in the HFO-sCI series mostly correlates with the growth in rate constant: kTHEO (sCI 2) < kTHEO (sCI 3) ≈ kTHEO (sCI 1) < kTHEO (sCI 5) < kTHEO (sCI 4).
3.5 Impact of heteroatom tuning on HFO-sCI reactions
It has been suggested that for sCI + H2X (X = O, Si, Se & Te) reactions, as the central X atom in the H2X co-reactant is substituted with larger group XVI elements, the rate constant increases.87 In this section, the computational analysis of the sCIs 1–5 + H2S reactions is compared with sCIs 1–5 reactions with H2O (& MeOH) to determine if these heteroatom tuning trends apply to HFO-sCIs too. To see if the effects observed from heteroatom tuning group XVI-centred co-reactants also apply to group XVII-centred co-reactants, analysis of HFO-sCI + HF & HCl reactions is also included in this section.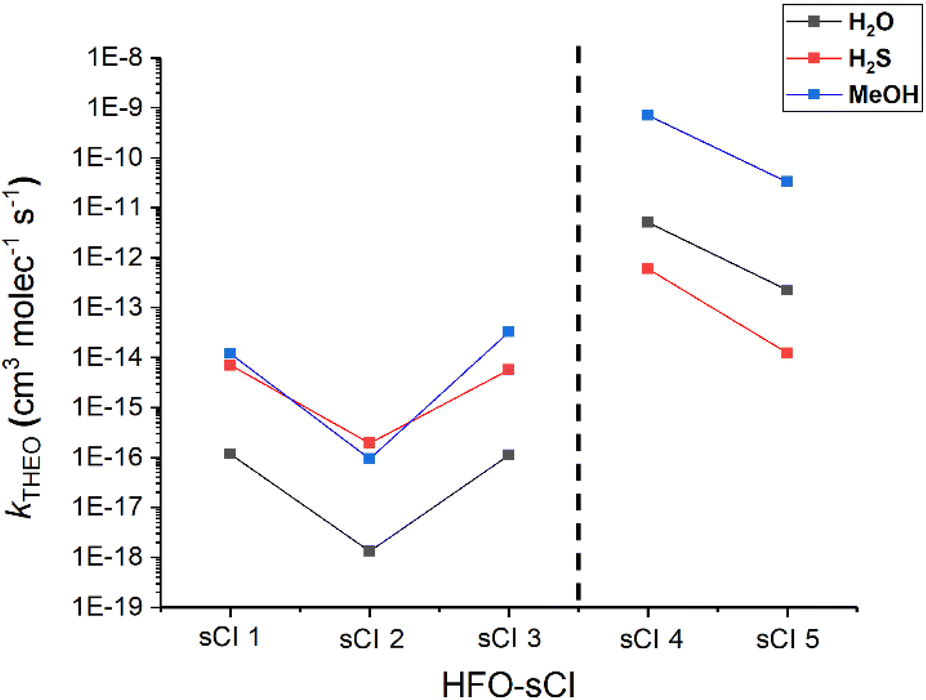 | ||
| Fig. 15 A comparison of the impact that heteroatom tuning of the group XVI-centred co-reactant (identity in the legend) has on the bimolecular rate constant (kTHEO) of different HFO-sCI reaction. Of note in this figure is the large increase in reactivity for sCIs 4 & 5 + H2O and MeOH that is not observed for H2S reactions. | ||
One other point that may be noteworthy is that sCI 3 & sCI 1 exhibit similar rate constants throughout all three reactions featured in Fig. 15, as well as throughout much of this study. In much of the literature the non-fluorinated equivalent to sCI 3, anti-CH3CHOO, exhibits a higher bimolecular rate constant to sCI 1 in reactions with H2O, MeOH & SO2.61,74,79,148,151 This may well be due to the presence of hyperconjugative α-H atoms in the anti-CH3 substituent and that α-F atoms on the anti-CF3 group rather than hyperconjugative α-H atoms reduces the inductive impact the substituent has on sCI 3 reactivity.56
In contrast to the patterns seen for the sCIs 1–3 + H2S reactions, the rate constants observed for the H2S reactions with sCIs 4 & 5 are both smaller than those of the equivalent H2O reactions (Fig. 15). To determine whether this stagnation of the sCI + H2S rate constant was due to increased overall steric bulk, the kTHEO values were for the HFO-sCI reactions with the bulkier MeOH (which also proceeds via a hydrogen abstraction mechanism) were determined. However, no such stagnation in the rate constants of sCI 4 & 5 + MeOH reactions emerges (Fig. 15).
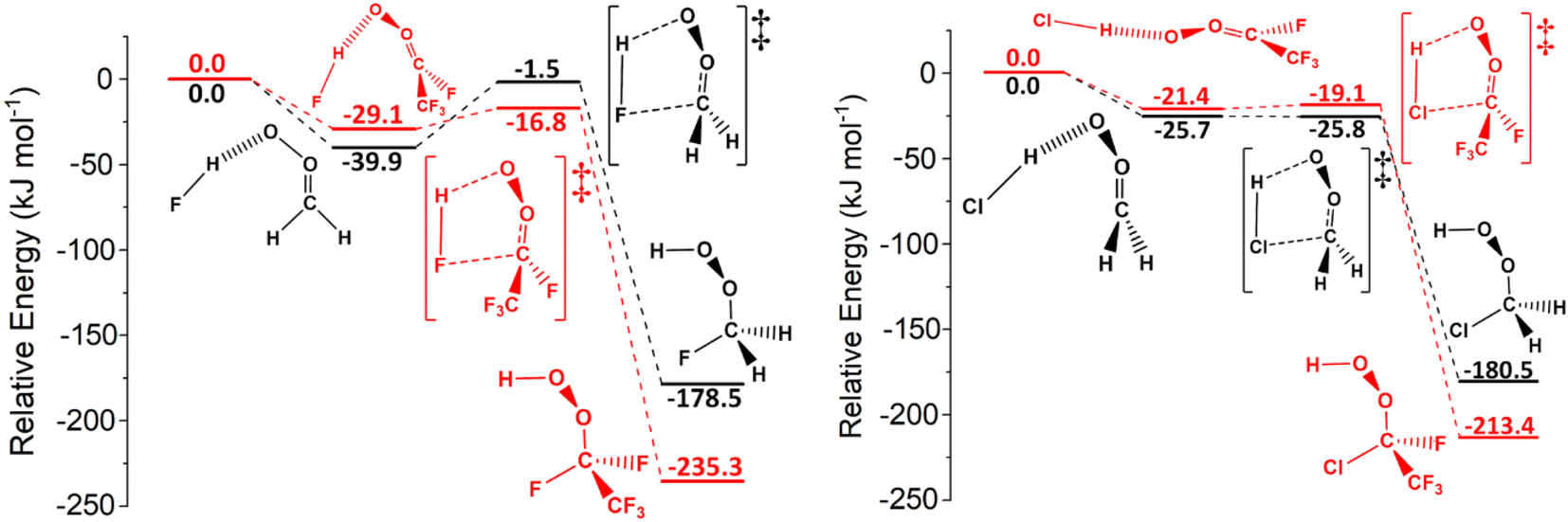 | ||
| Fig. 16 The potential energy surfaces for selected sCI reactions with HF and HCl. On the potential energy surfaces on the left, feature sCI 1 + HF, in black, and sCI 4 + HF, in red. On the potential energy surfaces on the right, feature sCI 1 + HCl, in black, and sCI 4 + HCl, in red. Energies are relative to raw reactants. | ||
Fig. 16 displays that the fluorinated sCI 4 has much lower reaction barrier with HF than the unsubstituted sCI 1 has, demonstrating the continued activating effect that the –F substituent has on the sCI reactivity. The inductive impact of the –F substituent results in sCI 4 and sCI 5 having substantially larger rate constants (1.19 × 10−11 & 3.66 × 10−13 cm3 s−1) compared to the sCI 1 + HF reaction. In contrast, the energy barrier of the sCI 4 + HCl reaction does not decrease compared to that of sCI 1 + HCl (Fig. 16), replicating the observation that heteroatom tuning does not have an inductive effect on reactions involving sCIs 4 & 5. In fact, –F substituents in sCIs 4 & 5 in combination with the heteroatom tuning of the co-reactant leads not just to the same stagnation effect seen for sCIs 4 & 5 + H2S reactions, but instead to a reductive effect on reactivity for the sCIs 4 & 5 + HCl reactions, compared to sCIs 4 & 5 + HF. This reductive effect is so clear that the rate constants for HCl reactions with sCI 4 (1.64 × 10−10 cm3 s−1) and sCI 5 (1.13 × 10−10 cm3 s−1) are both smaller than that of the equivalent sCI 1 reaction for the only time in this study. The cause of these stagnation/reductive effects can potentially be explained in terms of the repulsion between the sCI substituent groups and increased steric interactions, although a deeper exploration into such factors would probably require experimental analysis and verification.56
4. Atmospheric implications of HFO-sCIs bimolecular sinks
HFO emissions, and subsequently HFO-sCIs, are projected to become much more abundant as the use of HFO become increasingly widespread in refrigerators, insulation and vehicle cooling units, as is being seen in China, the USA and the EU.1,27,28 The chemistry of sCIs and other atmospheric species are typically included in atmospheric models in sets of like chemistries or taxonomic groups, where their reactivity can be estimated on the basis of their structure, and this work suggest sCIs 2–5 can be grouped into any such wider sCI classification system.40,70,187,188 Similar taxonomic groupings are already used in atmospheric models to estimate the chemistry of large groups of molecules and therefore reduce the computational processing power required to run simulations.189 However, integrating HFO-sCIs into a wider taxonomic sCI framework faces particular difficulties, principally because of the differing effects non-halogenated groups (–CH3 and –H) and halogenated groups (–CF3 and –F) have on the conjugation, hyperconjugation and steric interactions the sCI.56,72–75 One example of this is the difficulty of including the stagnation/reduction in reactivity of sCIs 4 & 5 seen during reactions with H2S & HCl, a phenomenon not exhibited by other sCIs studied here or in the prior literature.59,64,78,87,131,136,182,183 This shows that system-by-system computational analysis, or indeed ultimately experimental analysis is still vital for understanding the chemistry of many bimolecular sCI reactions.The effective rate constants, kEFF, a product of the rate constant and the atmospheric abundance of the co-reactant, is used here as a primitive system of measuring the potential tropospheric impact of each bimolecular HFO-sCI reaction studied (Table 1).
The sCI reactions with water are the dominant bimolecular sink for the HFO-sCIs due to the abundance water vapour.42,67,68,70,150,179 However, whether the sCI reaction with H2O or (H2O)2 is more prevalent depends not only on the relative abundance of the H2O or (H2O)2, but also on the identity of the sCI. For instance, the kEFF ((H2O)2) for sCI 2 is around 103 s−1 larger than the equivalent kEFF (H2O), because the natural abundance of H2O (6.2 × 1017 molec. per cm3) over that of (H2O)2 (8.7 × 1014 molec. per cm3) is overcome by the fact that the sCI 2 has a 106 cm3 s−1 larger kTHEO ((H2O)2) than kTHEO (H2O). sCIs 1 & 3 also exhibit larger kEFF ((H2O)2) than kEFF (H2O) by between 101–103 s−1, meaning that tropospheric removal of the sCI occurs more readily via reaction with the water dimer than the monomer. But, whereas the rate constants for sCIs 4 & 5 + H2O reactions are much enhanced, the sCIs 4 & 5 + (H2O)2 systems are so reactive that the kd–d capture limits are used as the rate constants, and the gap between these kd–d ((H2O)2) and kTHEO (H2O) values is much reduced. Consequently, the kEFF (H2O) for sCI 5 is similar to the equivalent kEFF ((H2O)2) value and the kEFF (H2O) for sCI 4 is larger than kEFF ((H2O)2), as seen in Table 6.
| Co-reactant | k EFF (s−1) | ||||
|---|---|---|---|---|---|
| CH2OO | Syn-CF3CHOO | Anti-CF3CHOO | Syn-CF3CFOO | Anti-CF3CFOO | |
| sCI 1 | sCI 2 | sCI 3 | sCI 4 | sCI 5 | |
| HCHO | 5.6 | 0.54 | 5.0 | 1447 | 33.4 |
| SO2 | 300 | 0.79 | 169 | 186 | 183 |
| HNO3 | 94 | 2.3 | 9.8 | 58 | 8.4 |
| TFA | 2.5 × 10−2 | 1.7 × 10−2 | 1.3 × 10−2 | 1.2 × 10−2 | 1.2 × 10−2 |
| H2O | 73 | 0.83 | 70 | 3.2 × 106 | 1.4 × 105 |
| (H2O)2 | 2.9 × 103 | 2.4 × 103 | 3.3 × 103 | 6.2 × 105 | 6.1 × 105 |
| MeOH | 0.010 | 7.9 × 10−5 | 0.028 | 462 | 28 |
| H2S | 0.085 | 1.4 × 10−3 | 0.042 | 4.5 | 0.091 |
| HCl | 58 | 5.0 × 10−3 | 8.96 | 20 | 14 |
| HF | 0.12 | 2.5 × 10−6 | 1.1 × 10−3 | 4.4 | 0.14 |
This great preference for HFO-sCI + H2O & (H2O)2 reactions leads to the significant production of fluorinated α-hydroxy-hydroperoxides, which have been implicated in forest damage and associated with generating important secondary products like H2O2, organic acids and aldehydes, albeit that the primary emission locations for HFO are likely to be in urban environments.86,170–176 HFO-sCI + MeOH reactions are not as competitive as the equivalent H2O & (H2O)2 reactions, but because, MeOH is emitted from vehicles that run off biofuel and HFOs are used in vehicle coolant units, the HFO-sCI & MeOH species have greater concentration overlap in many urban environments.135,190,191 Under such circumstances, emissions of α-alkyoxyalkyl hydroperoxides (AAAHs), a significant functionalised peroxide, are increased and these AAAHs have also been implicated in forest damage and SOA formation.67,68,78–80,83,86,170
HFO-sCI reactions with aldehydes are quite uncompetitive compared to the reactions with H2O & (H2O)2 but the products of their reactions include organofluoride aldehydes and TFA, both known to contribute to respiratory problems.192 Furthermore the sCIs 2–5 reactions with HCHO lead to high yields of sCI 1 sometimes with a large excess of energy, that easily leads to CI fragmentation and therefore contributes to the tropospheric population of the ‘atmospheric detergent’ OH radical.70,82
The sCI + SO2 reaction is known to act as a sink for ∼20% of some sCIs in boreal forest, however the high uptake of the reactions with H2O & (H2O)2 means that they do not contribute hugely to the overall depletion of sCIs 2–5.159 However the SO2 reactions with sCIs 2–5 also seem to produce SO3 at a higher efficiency than reactions with sCI 1 and (CH3)2COO, and this tropospheric SO3 will then contribute to atmospheric H2SO4 and aerosol formation.84,193–196
TFA may have a disproportionate impact on HFO-sCI bimolecular chemistry because both HFO-sCIs and TFA emerge from the breakdown of HFOs and so they may see a large population overlap with HFO-sCIs. The large rate constants from sCI reactions with TFA, HNO3 and HCl found here indicate a high reactivity between sCIs and other organic and inorganic acids. However, even in seaside areas, where HCl concentrations are elevated, the contribution that sCI 1 could make the minor and the role of sCIs 4 & 5 would be even more reduced, due to the high kEFF values of the sCI + H2O & (H2O)2 reactions. The non-water reactions (e.g. sCI reactions with HF, SO2 & H2S) would contribute more to the depletion of HFO-sCI population in dry environments, such as near active volcanoes.82,139,197–199
After collisional stabilisation, sCIs are less energetic so fewer unimolecular fragmentation mechanisms have energy barriers low enough to compete with bimolecular reactions as sinks for sCIs under tropospheric temperature and pressure conditions.42 According to Guidry et al., the central mechanism for the fragmentation of both syn- & anti-CF3CHOO is via a 1,3-ring closure to produce a dioxirane ring, both of which produce energy barriers (∼80 kJ mol−1) higher than anti-CH3CHOO (∼70 kJ mol−1) and more similar to those of CH2OO (70–100 kJ mol−1).200 The unimolecular rate constant (kUNI) of a reaction path and kEFF are both equivalent and effective tools to show the efficiency of the respective reaction path as an sCI sink but, while Wang et al. have identified syn- & anti-CF3CHOO experimentally, no kUNI values were identified in either the Wang et al. or Guidry et al. studies.66,200 Therefore, the kUNI for CH2OO (∼0.3 s−1) is used here instead as it may be the closest analogue to that of syn- & anti-CF3CHOO.42,70 On the basis of these premises, while the kUNI value used here is greater than many of the kEFF value for syn- & anti-CF3CHOO found in Table 6, the kEFF value for other reactions, particularly syn- & anti-CF3CHOO + (H2O)2, are greater still. This indicates that bimolecular reactions can compete with unimolecular decomposition.
No equivalent kUNI analogue could be found for syn- & anti-CF3CFOO especially considering the role of the –F substituent in CI fragmentation in isolation is also challenging. One study by Ljubić and Sabljić calculated that the anti-F substituent induced a lower anti-FCHOO fragmentation barrier (TSUNI ∼ 47.3 kJ mol−1), whereas syn-FCHOO saw no such effect (TSUNI ∼ 83.5 kJ mol−1).201 It is quite possible that the anti-F substituent could therefore theoretically induce a higher kUNI for syn-CF3CFOO, but this might be offset by the lack of any tunnelling effect and the steric hinderance of the syn-CF3 substituent. Nevertheless, no computational or experimental literature exists of the syn- & anti-CF3CFOO unimolecular fragmentation reactions, and an assessment of these reactions has been not been deliberated here, due to the need for a more in-depth analysis of these reactions than the kind of provisional evaluation that would be covered in this study.
5. Conclusion
It has been shown that the individual steric and electronic features of HFO-sCIs impact upon their bimolecular chemistry in a way that transcends the choice of tropospheric co-reactant species. The chemistry of sCI 2 (syn-CF3CHOO) & sCI 3 (anti-CF3CHOO) have consistency with existing reactivity trends in the literature, particularly that sCIs with a syn-orientated sterically bulky group are significantly less reactive than anti-orientated sCIs. However, sCI 3 is distinctive because it exhibits similar rate constants to the unsubstituted sCI 1 (CH2OO), whereas sCIs with similar orientations like anti-CH3CHOO yields larger rate constants. The literature indicates that the presence of hyperconjugative α-H atoms in the anti-CH3 group has an inductive impact on the sCI, but here the presence of α-F atoms in the anti-CF3 group appears to reverse this activating effect.The especially large bimolecular rate constants for sCI 4 (syn-CF3CFOO) & sCI 5 (anti-CF3CFOO) emerge from the inductive –F substituents they possess. Moreover, the inductive impact of the –F substituent is greater in the case of sCI 4 because of the anti-position of the –F substituent, and so the kTHEO (sCI 4) exceeds the kTHEO (sCI 5). However, with some notable exceptions, the HFO-sCIs studied here follow the reactivity series: kTHEO (sCI 2) < kTHEO (sCI 3) ∼ kTHEO (sCI 1) < kTHEO (sCI 5) < kTHEO (sCI 4).
One exceptionally distinct observation in this study is that the overall sCI reactivity series is influenced by the choice of co-reactant, as shown when contrasting the reactivity of sCIs 1–5 reactions with different group-XVI and group-XVII centred co-reactants. When sCI + H2O & HF are compared with sCI + H2S & HCl respectively, sCIs 1–3 reactions with H2S & HCl produced comparatively larger rate constants, in line with previous literature observations.87 However, this does not always apply as the –F substituent appear to obstruct the large S and Cl atoms from reaching the reaction site during sCIs 4 & 5 reactions with H2S and HCl and this leads to a smaller rate constant.
This work demonstrates that for all sCIs considered, reaction with H2O or (H2O)2 is the biggest tropospheric sink of HFO-sCIs, due primarily to the natural abundance of water vapour. Furthermore, these sCI reactions with water produced functionalised hydroxide products called α-hydroxy-hydroperoxide (HHP) that have been implicated with biogenic damage.42,67,68,78–80,83,86,134,170 While bimolecular reaction with other trace pollutants are less likely to be sinks for HFO-sCIs, sCIs 2–5 all have a significant capacity to react with tropospheric species such as MeOH, TFA & HNO3 and yield other functionalised hydroperoxides.
The uncommon nature of these HFO-sCIs is also demonstrated in the multi-step reactions with HCHO, where sCIs 2–5 react (via a short-lived HOZ) to produce another sCI, a highly energised sCI 1, a product not commonly found in other studies of sCI + aldehyde reactions.65,71,147 A similar full mechanistic analysis of the reactions using a two-step HFO-sCI + SO2 systems show that these reactions have very high rate constants and all produce high yields of SO3 + R1R2CO (0.90–0.99). This production of SO3 would also lead to H2SO4 formation and aerosol nucleation. Furthermore, HFO-sCI reactions with both HCHO & SO2 systems are found to produce CF3CHO or CF3CFO, which are known to contribute to respiratory problems.
In summary, HFO-sCIs are shown to have very unique chemistry, because the –F substituent has a large activation affect on the COO group, whereas the α-F atoms in the CF3 group are mildly deactivating. If any sCI taxonomic classification is established, sCIs 2 & 3 could be categorised as part of syn-sCI and anti-sCI groups respectively, whereas sCIs 4 & 5 could be categorised in a separate “electronegative substituent” taxonomic group.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Abbreviations
| sCI | Stabilized Criegee intermediate |
| AAAHs | Alkoxyalkyl hydroperoxides |
| HHP | Hydroxy-hydroperoxides |
| HFO-sCIs | Hydrofluoroolefin-derived stabilized Criegee intermediate |
| TFA | Trifluoracetic acid |
| HOZ | Heteroozonide |
| SOZ | Secondary ozonide |
| NAHP | Nitrooxyalkyl hydroperoxide |
| HAE | Hydroxyalkyl formate |
| CH2OO | sCI 1 |
| syn-CF3CHOO | sCI 2 |
| anti-CF3CHOO | sCI 3 |
| syn-CF3CFOO | sCI 4 |
| anti-CF3CFOO | sCI 5 |
Acknowledgements
This work was partially supported by Advanced Research Computing at Cardiff and High Performance Computing (HPC) Wales, a company formed between the Universities and the private sector in Wales which provides the UK's largest distributed supercomputing network. Dr J. M. Beames was supported in initiating this research through Marie Skłodowska Curie Individual Fellowship NPTC (701593). This work was partially supported by the Natural Environment Research Council (NERC) through the Integrated Research Observation System for Clean Air (OSCA) grant (NE/T001984/1). Information on the data underpinning the results presented here is shown in the ESI.†References
- O. US EPA, Refrigerant Transition & Environmental Impacts, https://www.epa.gov/mvac/refrigerant-transition-environmental-impacts, accessed 9 January 2019.
- Fifth Assessment Report - Climate Change 2013, http://www.ipcc.ch/report/ar5/wg1/, accessed 26 August 2018.
- M. K. Vollmer, S. Reimann, M. Hill and D. Brunner, First Observations of the Fourth Generation Synthetic Halocarbons HFC-1234yf, HFC-1234ze(E), and HCFC-1233zd(E) in the Atmosphere, Environ. Sci. Technol., 2015, 49, 2703–2708 CrossRef CAS PubMed.
- S. Paul, R. C. Deka and N. K. Gour, Kinetics, mechanism, and global warming potentials of HFO-1234yf initiated by O3 molecules and NO3 radicals: insights from quantum study, Environ. Sci. Pollut. Res., 2018, 25, 26144–26156 CrossRef CAS PubMed.
- P. K. Rao and S. P. Gejji, Atmospheric degradation of HCFO-1233zd(E) initiated by OH radical, Cl atom and O3 molecule: Kinetics, reaction mechanisms and implications, J. Fluorine Chem., 2018, 211, 180–193 CrossRef CAS.
- P. K. Rao and S. P. Gejji, Molecular insights for the HFO-1345fz +X (X = Cl, O3 or NO3) reaction and fate of alkoxy radicals initiated by Cl: DFT investigations, J. Fluorine Chem., 2017, 204, 65–75 CrossRef CAS.
- C. Arpagaus, F. Bless, M. Uhlmann, E. Büchel, S. Frei, J. Schiffmann and S. Bertsch, High Temperature Heat Pump Using HFO and HCFO Refrigerants - System Design, Simulation, and First Experimental Results, International Refrigeration and Air Conditioning Conference, 2018, p. 1875 Search PubMed.
- J. Navarro-Esbrí, A. Fernández-Moreno and A. Mota-Babiloni, Modelling and evaluation of a high-temperature heat pump two-stage cascade with refrigerant mixtures as a fossil fuel boiler alternative for industry decarbonization, Energy, 2022, 254, 124308 CrossRef.
- T. J. Wallington, M. P. Sulbaek Andersen and O. J. Nielsen, Atmospheric chemistry of short-chain haloolefins: Photochemical ozone creation potentials (POCPs), global warming potentials (GWPs), and ozone depletion potentials (ODPs), Chemosphere, 2015, 129, 135–141 CrossRef CAS PubMed.
- T. J. Wallington, W. F. Schneider, J. Sehested and O. J. Nielsen, Hydrofluorocarbons and stratospheric ozone, Faraday Discuss., 1995, 100, 55–64 RSC.
- A. R. Ravishankara, A. A. Turnipseed, N. R. Jensen, S. Barone, M. Mills, C. J. Howard and S. Solomon, Do Hydrofluorocarbons Destroy Stratospheric Ozone?, Science, 1994, 263, 71–75 CrossRef CAS PubMed.
- K. O. Patten and D. J. Wuebbles, Atmospheric lifetimes and Ozone Depletion Potentials of trans-1-chloro-3,3,3-trifluoropropylene and trans-1,2-dichloroethylene in a three-dimensional model, Atmos. Chem. Phys., 2010, 10, 10867–10874 CrossRef CAS.
- F. P. E. Carré, Improvement in apparatus for freezing liquids, US Pat., US30201A, 1860 Search PubMed.
- F. P. E. Carré, Improvement in ice-machines, US Pat., USRE5287E, 1873 Search PubMed.
- E. C. Smith, Pioneers of Refrigeration, Nature, 1943, 151, 412–413 CrossRef.
- J. M. Calm, The next generation of refrigerants – Historical review, considerations, and outlook, Int. J. Refrig., 2008, 31, 1123–1133 CrossRef CAS.
- E. Askar, V. Schröder, T. Schmid and M. Schwarze, Explosion characteristics of mildly flammable refrigerants ignited with high-energy ignition sources in closed systems, Int. J. Refrig., 2018, 90, 249–256 CrossRef CAS.
- Ø. Hodnebrog, M. Etminan, J. S. Fuglestvedt, G. Marston, G. Myhre, C. J. Nielsen, K. P. Shine and T. J. Wallington, Global warming potentials and radiative efficiencies of halocarbons and related compounds: A comprehensive review, Rev. Geophys., 2013, 51, 300–378 CrossRef.
- M. J. Molina and F. S. Rowland, Stratospheric sink for chlorofluoromethanes: chlorine atom-catalysed destruction of ozone, Nature, 1974, 249, 810–812 CrossRef CAS.
- D. L. Narayanan, R. N. Saladi and J. L. Fox, Ultraviolet radiation and skin cancer, Int. J. Dermatol., 2010, 49, 978–986 CrossRef PubMed.
- Chlorofluorocarbons and Ozone Depletion, https://www.acs.org/content/acs/en/education/whatischemistry/landmarks/cfcs-ozone.html, accessed 9 January 2019.
- The Montreal Protocol, https://web.archive.org/web/20130602153542/http://ozone.unep.org/new_site/en/montreal_protocol.php, accessed 4 May 2020.
- G. J. M. Velders, S. O. Andersen, J. S. Daniel, D. W. Fahey and M. McFarland, The importance of the Montreal Protocol in protecting climate, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 4814–4819 CrossRef CAS PubMed.
- R. McKenzie, G. Bernhard, B. Liley, P. Disterhoft, S. Rhodes, A. Bais, O. Morgenstern, P. Newman, L. Oman, C. Brogniez and S. Simic, Success of Montreal Protocol Demonstrated by Comparing High-Quality UV Measurements with “World Avoided” Calculations from Two Chemistry-Climate Models, Sci. Rep., 2019, 9, 1–13 CrossRef CAS PubMed.
- O. US EPA, Updating Ozone Calculations and Emissions Profiles for Use in the Atmospheric and Health Effects Framework Model, https://www.epa.gov/ozone-layer-protection/updating-ozone-calculations-and-emissions-profiles-use-atmospheric-and-health, accessed 7 August 2020.
- M. M. Hurwitz, E. L. Fleming, P. A. Newman, F. Li, E. Mlawer, K. Cady-Pereira and R. Bailey, Ozone depletion by hydrofluorocarbons, Geophys. Res. Lett., 2015, 42, 8686–8692 CrossRef CAS.
- European Commission - PRESS RELEASES - Press release - Refrigerants used in mobile air condition systems (MAC) - State of play, http://europa.eu/rapid/press-release_MEMO-14-50_en.htm, accessed 9 January 2019.
- B. Zhang, Z. Zhai and J. Zhang, Distribution of trifluoroacetic acid in gas and particulate phases in Beijing from 2013 to 2016, Sci. Total Environ., 2018, 634, 471–477 CrossRef CAS PubMed.
- Department for Environment, Food & Rural Affairs, Assessment of the F Gas Regulation in Great Britain, 2022 Search PubMed.
- J. Soulet, F-Gas Regulation Could Have Chilling Effect on EU Businesses and Their Customers, EURACTIV Media network, 2023 Search PubMed.
- N. Everitt, Industry Urged to Engage with UK F-Gas Survey', Cooling Post, 2023 Search PubMed.
- D. Fleet, J. Hanlon, K. Osborne, M. La Vedrine and P. Ashford, Study on Environmental and Health Effects of HFO Refrigerants, Norwegian Environment Agency, 2017 Search PubMed.
- V. L. Orkin, L. E. Martynova and M. J. Kurylo, Photochemical Properties of trans-1-Chloro-3,3,3-trifluoropropene (trans-CHClCHCF3): OH Reaction Rate Constant, UV and IR Absorption Spectra, Global Warming Potential, and Ozone Depletion Potential, J. Phys. Chem. A, 2014, 118, 5263–5271 CrossRef CAS PubMed.
- M. Antiñolo, I. Bravo, E. Jiménez, B. Ballesteros and J. Albaladejo, Atmospheric Chemistry of E- and Z-CF3CHCHF (HFO-1234ze): OH Reaction Kinetics as a Function of Temperature and UV and IR Absorption Cross Sections, J. Phys. Chem. A, 2017, 121, 8322–8331 CrossRef PubMed.
- J. Navarro-Esbrí, M. Amat-Albuixech, F. Molés, C. Mateu-Royo, A. Mota-Babiloni and R. Collado, HCFO-1224yd(Z) as HFC-245fa drop-in alternative in low temperature ORC systems: Experimental analysis in a waste heat recovery real facility, Energy, 2020, 193, 116701 CrossRef.
- D. J. Luecken, R. L. Waterland, S. Papasavva, K. N. Taddonio, W. T. Hutzell, J. P. Rugh and S. O. Andersen, Ozone and TFA Impacts in North America from Degradation of 2,3,3,3-Tetrafluoropropene (HFO-1234yf), A Potential Greenhouse Gas Replacement, Environ. Sci. Technol., 2010, 44, 343–348 CrossRef CAS PubMed.
- Honeywell starts up HFO refrigerants plant in Louisiana - Chemical Engineering, https://www.chemengonline.com/honeywell-starts-up-hfo-refrigerants-plant-in-louisiana/?printmode=1, accessed 24 October 2018.
- R. R. Singh, H. T. Pham, D. P. Wilson and R. H.Thomas, Azeotrope-like compositions of tetrafluoropropene and trifluoroiodomethane, US Pat., US6969701B2, 2005 Search PubMed.
- M. Nayak, U.S. companies brace for climate-friendly alternatives in cooling systems, Reuters, 2016 Search PubMed.
- M. R. McGillen, T. J. Carey, A. T. Archibald, J. C. Wenger, D. E. Shallcross and C. J. Percival, Structure–activity relationship (SAR) for the gas-phase ozonolysis of aliphatic alkenes and dialkenes, Phys. Chem. Chem. Phys., 2008, 10, 1757–1768 RSC.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Milller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. Wild, S. S. Brown, A. H. Goldstein, J. A. de Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Atmospheric fates of Criegee intermediates in the ozonolysis of isoprene, Phys. Chem. Chem. Phys., 2016, 18, 10241–10254 RSC.
- M. A. H. Khan, C. J. Percival, R. L. Caravan, C. A. Taatjes and D. E. Shallcross, Criegee intermediates and their impacts on the troposphere, Environ. Sci.: Processes Impacts, 2018, 20, 437–453 RSC.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I - gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 2004, 4, 1461–1738 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, J. A. Kerr, M. J. Rossi and J. Troe, Evaluated Kinetic, Photochemical and Heterogeneous Data for Atmospheric Chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 1997, 26, 521–1011 CrossRef CAS.
- O. J. Nielsen, M. S. Javadi, M. P. Sulbaek Andersen, M. D. Hurley, T. J. Wallington and R. Singh, Atmospheric chemistry of CF3CFCH2: Kinetics and mechanisms of gas-phase reactions with Cl atoms, OH radicals, and O3, Chem. Phys. Lett., 2007, 439, 18–22 CrossRef CAS.
- M. P. S. Andersen, E. J. K. Nilsson, O. J. Nielsen, M. S. Johnson, M. D. Hurley and T. J. Wallington, Atmospheric chemistry of trans-CF3CHCHCl: Kinetics of the gas-phase reactions with Cl atoms, OH radicals, and O3, J. Photochem. Photobiol., A, 2008, 199, 92–97 CrossRef CAS.
- M. P. S. Andersen, O. J. Nielsen, A. Toft, T. Nakayama, Y. Matsumi, R. L. Waterland, R. C. Buck, M. D. Hurley and T. J. Wallington, Atmospheric chemistry of CxF2x+1CHCH2 (x=1, 2, 4, 6, and 8): Kinetics of gas-phase reactions with Cl atoms, OH radicals, and O3, J. Photochem. Photobiol., A, 2005, 176, 124–128 CrossRef CAS.
- J. G. Calvert, J. J. Orlando, W. R. Stockwell and T. J. Wallington, The Mechanisms of Reactions Influencing Atmospheric Ozone, Oxford University Press, 2015 Search PubMed.
- Z. Wang, Y. Wang, J. Li, S. Henne, B. Zhang, J. Hu and J. Zhang, Impacts of the Degradation of 2,3,3,3-Tetrafluoropropene into Trifluoroacetic Acid from Its Application in Automobile Air Conditioners in China, the United States, and Europe, Environ. Sci. Technol., 2018, 52, 2819–2826 CrossRef CAS PubMed.
- S. Henne, D. E. Shallcross, S. Reimann, P. Xiao, D. Brunner, S. O'Doherty and B. Buchmann, Future Emissions and Atmospheric Fate of HFC-1234yf from Mobile Air Conditioners in Europe, Environ. Sci. Technol., 2012, 46, 1650–1658 CrossRef CAS PubMed.
- M. P. Sulbaek Andersen, J. A. Schmidt, A. Volkova and D. J. Wuebbles, A three-dimensional model of the atmospheric chemistry of E and Z-CF3CH=CHCl (HCFO-1233(zd) (E/Z)), Atmos. Environ., 2018, 179, 250–259 CrossRef CAS.
- A. Soto, B. Ballesteros, E. Jiménez, M. Antiñolo, E. Martínez and J. Albaladejo, Kinetic and mechanistic study of the gas-phase reaction of CxF2x+1CH=CH2 (x=1, 2, 3, 4 and 6) with O3 under atmospheric conditions, Chemosphere, 2018, 201, 318–327 CrossRef CAS PubMed.
- R. Søndergaard, O. J. Nielsen, M. D. Hurley, T. J. Wallington and R. Singh, Atmospheric chemistry of trans-CF3CHCHCl: Kinetics of the gas-phase reactions with Cl atoms, OH radicals, and O3, Chem. Phys. Lett., 2007, 443, 199–204 CrossRef.
- M. P. S. Andersen, T. I. Sølling, L. L. Andersen, A. Volkova, D. Hovanessian, C. Britzman, O. J. Nielsen and T. J. Wallington, Atmospheric chemistry of (Z)-CF3CHCHCl: products and mechanisms of the Cl atom, OH radical and O3 reactions, and role of (E)–(Z) isomerization, Phys. Chem. Chem. Phys., 2018, 20, 27949–27958 RSC.
- A. Novelli, K. Hens, C. T. Ernest, M. Martinez, A. Nolscher, V. Sinha, P. Paasonen, T. Petaja, M. Sipila, T. Elste and D. Kubistin, Estimating the Atmospheric Concentration of Criegee Intermediates and Their Possible Interference in a FAGE-LIF Instrument, Faculty of Science, Medicine and Health - Papers, 2017, pp. 7807–7826 Search PubMed.
- N. Watson, An Analysis of the Sources and Sinks for Criegee Intermediates: an Extended Computational Study, Cardiff University, 2021 Search PubMed.
- C. Cabezas, J.-C. Guillemin and Y. Endo, Fourier-transform microwave spectroscopy of a halogen substituted Criegee intermediate ClCHOO, J. Chem. Phys., 2016, 145, 184304 CrossRef PubMed.
- K. M. Kapnas, B. W. Toulson, E. S. Foreman, S. A. Block, J. G. Hill and C. Murray, UV photodissociation dynamics of CHI2Cl and its role as a photolytic precursor for a chlorinated Criegee intermediate, Phys. Chem. Chem. Phys., 2017, 19, 31039–31053 RSC.
- E. S. Foreman, K. M. Kapnas and C. Murray, Reactions between Criegee Intermediates and the Inorganic Acids HCl and HNO3: Kinetics and Atmospheric Implications, Angew. Chem., Int. Ed., 2016, 55, 10419–10422 CrossRef CAS PubMed.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, M. A. H. Khan, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Rate Coefficients of C1 and C2 Criegee Intermediate Reactions with Formic and Acetic Acid Near the Collision Limit: Direct Kinetics Measurements and Atmospheric Implications, Angew. Chem., Int. Ed., 2014, 53, 4547–4550 CrossRef CAS PubMed.
- R. Chhantyal-Pun, M. R. McGillen, J. M. Beames, M. A. H. Khan, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Temperature-Dependence of the Rates of Reaction of Trifluoroacetic Acid with Criegee Intermediates, Angew. Chem., Int. Ed., 2017, 56, 9044–9047 CrossRef CAS PubMed.
- M. R. McGillen, B. F. E. Curchod, R. Chhantyal-Pun, J. M. Beames, N. Watson, M. A. H. Khan, L. McMahon, D. E. Shallcross and A. J. Orr-Ewing, Criegee Intermediate–Alcohol Reactions, A Potential Source of Functionalized Hydroperoxides in the Atmosphere, ACS Earth Space Chem., 2017, 1, 664–672 CrossRef CAS.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Direct Kinetic Measurements of Criegee Intermediate (CH2OO) Formed by Reaction of CH2I with O2, Science, 2012, 335, 204–207 CrossRef CAS PubMed.
- D. Stone, M. Blitz, L. Daubney, N. U. M. Howes and P. Seakins, Kinetics of CH2OO reactions with SO2, NO2, NO, H2O and CH3CHO as a function of pressure, Phys. Chem. Chem. Phys., 2014, 16, 1139–1149 RSC.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, E. P. F. Lee, J. M. Dyke, D. W. K. Mok, D. E. Shallcross and C. J. Percival, Direct measurement of Criegee intermediate (CH2OO) reactions with acetone, acetaldehyde, and hexafluoroacetone, Phys. Chem. Chem. Phys., 2012, 14, 10391–10400 RSC.
- L. Wang, Z. Wu, B. Lu and X. Zeng, Spectroscopic characterization and photochemistry of the Criegee intermediate CF3C(H)OO, J. Environ. Sci., 2022, 114, 160–169 CrossRef CAS PubMed.
- J. M. Anglada and A. Solé, Impact of the water dimer on the atmospheric reactivity of carbonyl oxides, Phys. Chem. Chem. Phys., 2016, 18, 17698–17712 RSC.
- J. M. Anglada, J. González and M. Torrent-Sucarrat, Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water, Phys. Chem. Chem. Phys., 2011, 13, 13034–13045 RSC.
- M. Kumar, D. H. Busch, B. Subramaniam and W. H. Thompson, Criegee Intermediate Reaction with CO: Mechanism, Barriers, Conformer-Dependence, and Implications for Ozonolysis Chemistry, J. Phys. Chem. A, 2014, 118, 1887–1894 CrossRef CAS PubMed.
- L. Vereecken, A. Novelli and D. Taraborrelli, Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- A. Jalan, J. W. Allen and W. H. Green, Chemically activated formation of organic acids in reactions of the Criegee intermediate with aldehydes and ketones, Phys. Chem. Chem. Phys., 2013, 15, 16841–16852 RSC.
- M. Kumar and J. S. Francisco, Reactions of Criegee Intermediates with Non-Water Greenhouse Gases: Implications for Metal Free Chemical Fixation of Carbon Dioxide, J. Phys. Chem. Lett., 2017, 8, 4206–4213 CrossRef CAS PubMed.
- M. Kumar and J. S. Francisco, H–X. (X = H, CH3, CH2F, CHF2, CF3, and SiH3) Bond Activation by Criegee Intermediates: A Theoretical Perspective, J. Phys. Chem. A, 2017, 121, 9421–9428 CrossRef CAS PubMed.
- N. A. I. Watson, J. A. Black, T. M. Stonelake, P. J. Knowles and J. M. Beames, An Extended Computational Study of Criegee Intermediate–Alcohol Reactions, J. Phys. Chem. A, 2019, 123, 218–229 CrossRef CAS PubMed.
- K. Xu, W. Wang, W. Wei, W. Feng, Q. Sun and P. Li, Insights into the Reaction Mechanism of Criegee Intermediate CH2OO with Methane and Implications for the Formation of Methanol, J. Phys. Chem. A, 2017, 121, 7236–7245 CrossRef CAS PubMed.
- R. Chhantyal-Pun, R. Shannon, D. P. Tew, R. L. Caravan, M. Duchi, C. Wong, A. Ingham, C. Feldman, M. R. McGillen, M. A. H. H. Khan, I. O. Antonov, B. Rotavera, K. Ramasesha, D. L. Osborn, C. A. Taatjes, C. Percival, D. E. Shallcross and A. Orr-Ewing, Experimental and Computational Studies of Criegee Intermediate Reactions with NH3 and CH3NH2, Phys. Chem. Chem. Phys., 2019, 21, 14042–14052 RSC.
- L. Vereecken, H. Harder and A. Novelli, The reactions of Criegee intermediates with alkenes, ozone, and carbonyl oxides, Phys. Chem. Chem. Phys., 2014, 16, 4039 RSC.
- W. Chao, J.-T. Hsieh, C.-H. Chang and J. J.-M. Lin, Direct kinetic measurement of the reaction of the simplest Criegee intermediate with water vapor, Science, 2015, 347, 751–754 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Direct Measurements of Conformer-Dependent Reactivity of the Criegee Intermediate CH3CHOO, Science, 2013, 340, 177–180 CrossRef CAS PubMed.
- M. J. Newland, A. R. Rickard, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Atmospheric isoprene ozonolysis: impacts of stabilised Criegee intermediate reactions with SO2, H2O and dimethyl sulfide, Atmos. Chem. Phys., 2015, 15, 9521–9536 CrossRef CAS.
- G. Ritchie, Atmospheric Chemistry: from the Surface to the Stratosphere, World Scientific Publishing Company, 2017 Search PubMed.
- A. M. Holloway and R. P. Wayne, Atmospheric Chemistry, Royal Society of Chemistry, 2010 Search PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, Atmospheric Chemistry of Criegee Intermediates: Unimolecular Reactions and Reactions with Water, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS PubMed.
- K. T. Kuwata, E. J. Guinn, M. R. Hermes, J. A. Fernandez, J. M. Mathison and K. Huang, A Computational Re-examination of the Criegee Intermediate–Sulfur Dioxide Reaction, J. Phys. Chem. A, 2015, 119, 10316–10335 CrossRef CAS PubMed.
- R. Chhantyal-Pun, A. Davey, D. E. Shallcross, C. J. Percival and A. J. Orr-Ewing, A kinetic study of the CH2OO Criegee intermediate self-reaction, reaction with SO2 and unimolecular reaction using cavity ring-down spectroscopy, Phys. Chem. Chem. Phys., 2015, 17, 3617–3626 RSC.
- S. Gäb, E. Hellpointner, W. V. Turner and F. Koŕte, Hydroxymethyl hydroperoxide and bis(hydroxymethyl) peroxide from gas-phase ozonolysis of naturally occurring alkenes, Nature, 1985, 316, 535–536 CrossRef.
- M. Kumar and J. S. Francisco, Heteroatom Tuning of Bimolecular Criegee Reactions and Its Implications, Angew. Chem., Int. Ed., 2016, 55, 13432–13435 CrossRef CAS PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. F. Sousa, P. A. Fernandes and M. J. Ramos, General Performance of Density Functionals, J. Phys. Chem. A, 2007, 111, 10439–10452 CrossRef CAS PubMed.
- null Car and null Parrinello, Unified approach for molecular dynamics and density-functional theory, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef PubMed.
- J. Harris and R. O. Jones, The surface energy of a bounded electron gas, J. Phys. F: Met. Phys., 1974, 4, 1170–1186 CrossRef.
- J. Poater, M. Duran and M. Solà, Parametrization of the Becke3-LYP hybrid functional for a series of small molecules using quantum molecular similarity techniques, J. Comput. Chem., 2001, 22, 1666–1678 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, Adiabatic Connection for Kinetics, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS.
- E. G. Lewars, Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics, Springer Science & Business Media, 2010 Search PubMed.
- T. Kupka and C. Lim, Polarization-Consistent versus Correlation-Consistent Basis Sets in Predicting Molecular and Spectroscopic Properties, J. Phys. Chem. A, 2007, 111, 1927–1932 CrossRef CAS PubMed.
- K. A. Peterson and T. H. Dunning, Accurate correlation consistent basis sets for molecular core–valence correlation effects: The second row atoms Al–Ar, and the first row atoms B–Ne revisited, J. Chem. Phys., 2002, 117, 10548–10560 CrossRef CAS.
- D. Feller, K. A. Peterson and T. D. Crawford, Sources of error in electronic structure calculations on small chemical systems, J. Chem. Phys., 2006, 124, 054107 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc, Wallingford CT, 2016 Search PubMed.
- P. Atkins, J. de Paula and R. Friedman, Physical Chemistry: Quanta, Matter, and Change, OUP, Oxford, 2013 Search PubMed.
- D. Spellmeyer, Annual Reports in Computational Chemistry, Elsevier, 2006 Search PubMed.
- P. Pulay, Localizability of dynamic electron correlation, Chem. Phys. Lett., 1983, 100, 151–154 CrossRef CAS.
- P. Pulay and S. Saebø, Orbital-invariant formulation and second-order gradient evaluation in Møller-Plesset perturbation theory, Theor. Chim. Acta, 1986, 69, 357–368 CrossRef CAS.
- R. Polly, H.-J. Werner, F. R. Manby and P. J. Knowles, Fast Hartree–Fock theory using local density fitting approximations, Mol. Phys., 2004, 102, 2311–2321 CrossRef CAS.
- H.-J. Werner and K. Pflüger, in Annual Reports in Computational Chemistry, ed. D. C. Spellmeyer, Elsevier, 2006, vol. 2, pp. 53–80 Search PubMed.
- S. Loibl, F. R. Manby and M. Schütz, Density fitted, local Hartree–Fock treatment of NMR chemical shifts using London atomic orbitals, Mol. Phys., 2010, 108, 477–485 CrossRef CAS.
- T. B. Adler and H.-J. Werner, An explicitly correlated local coupled cluster method for calculations of large molecules close to the basis set limit, J. Chem. Phys., 2011, 135, 144117 CrossRef PubMed.
- T. B. Adler, G. Knizia and H.-J. Werner, A simple and efficient CCSD(T)-F12 approximation, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, Simplified CCSD(T)-F12 methods: Theory and benchmarks, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- K. Wang, W. Li and S. Li, Generalized Energy-Based Fragmentation CCSD(T)-F12a Method and Application to the Relative Energies of Water Clusters (H2O)20, J. Chem. Theory Comput., 2014, 10, 1546–1553 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: a general-purpose quantum chemistry program package: Molpro, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, MESMER: An Open-Source Master Equation Solver for Multi-Energy Well Reactions, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- S. J. Klippenstein and J. A. Miller, From the Time-Dependent, Multiple-Well Master Equation to Phenomenological Rate Coefficients, J. Phys. Chem. A, 2002, 106, 9267–9277 CrossRef CAS.
- M. F. Vansco, R. L. Caravan, K. Zuraski, F. A. F. Winiberg, K. Au, N. Trongsiriwat, P. J. Walsh, D. L. Osborn, C. J. Percival, M. A. H. Khan, D. E. Shallcross, C. A. Taatjes and M. I. Lester, Experimental Evidence of Dioxole Unimolecular Decay Pathway for Isoprene-Derived Criegee Intermediates, J. Phys. Chem. A, 2020, 124, 3542–3554 CrossRef CAS PubMed.
- S. Sarkar and B. Bandyopadhyay, Singlet (1Δg) O2 as an Efficient Tropospheric Oxidizing Agent: Gas Phase Reaction with Simplest Criegee Intermediate, Phys. Chem. Chem. Phys., 2020, 22, 19870–19876 RSC.
- J. Peltola, P. Seal, A. Inkilä and A. Eskola, Time-resolved, broadband UV-absorption spectrometry measurements of Criegee intermediate kinetics using a new photolytic precursor: unimolecular decomposition of CH2OO and its reaction with formic acid, Phys. Chem. Chem. Phys., 2020, 22, 11797–11808 RSC.
- B. Long, J. L. Bao and D. G. Truhlar, Rapid unimolecular reaction of stabilized Criegee intermediates and implications for atmospheric chemistry, Nat. Commun., 2019, 10, 2003 CrossRef PubMed.
- D. Stone, K. Au, S. Sime, D. J. Medeiros, M. Blitz, P. W. Seakins, Z. Decker and L. Sheps, Unimolecular decomposition kinetics of the stabilised Criegee intermediates CH2OO and CD2OO, Phys. Chem. Chem. Phys., 2018, 20, 24940–24954 RSC.
- J. M. Gomez and J. M. C. Plane, Reaction Kinetics of CaOH with H and O2 and O2CaOH with O: Implications for the Atmospheric Chemistry of Meteoric Calcium, ACS Earth Space Chem., 2017, 1, 431–441 CrossRef PubMed.
- S. Canneaux, F. Bohr and E. Henon, KiSThelP: a program to predict thermodynamic properties and rate constants from quantum chemistry results, J. Comput. Chem., 2014, 35, 82–93 CrossRef CAS PubMed.
- S. A. Carr, T. J. Still, M. A. Blitz, A. J. Eskola, M. J. Pilling, P. W. Seakins, R. J. Shannon, B. Wang and S. H. Robertson, Experimental and Theoretical Study of the Kinetics and Mechanism of the Reaction of OH Radicals with Dimethyl Ether, J. Phys. Chem. A, 2013, 117, 11142–11154 CrossRef CAS PubMed.
- R. J. Shannon, R. L. Caravan, M. A. Blitz and D. E. Heard, A combined experimental and theoretical study of reactions between the hydroxyl radical and oxygenated hydrocarbons relevant to astrochemical environments, Phys. Chem. Chem. Phys., 2014, 16, 3466–3478 RSC.
- M. A. Blitz, D. Talbi, P. W. Seakins and I. W. M. Smith, Rate Constants and Branching Ratios for the Reaction of CH Radicals with NH3: A Combined Experimental and Theoretical Study, J. Phys. Chem. A, 2012, 116, 5877–5885 CrossRef CAS PubMed.
- R. Chhantyal-Pun, B. Rotavera, M. R. McGillen, M. A. H. Khan, A. J. Eskola, R. L. Caravan, L. Blacker, D. P. Tew, D. L. Osborn, C. J. Percival, C. A. Taatjes, D. E. Shallcross and A. J. Orr-Ewing, Criegee Intermediate Reactions with Carboxylic Acids: A Potential Source of Secondary Organic Aerosol in the Atmosphere, ACS Earth Space Chem., 2018, 2, 833–842 CrossRef CAS.
- A. I. Maergoiz, E. E. Nikitin, J. Troe and V. G. Ushakov, Classical trajectory and adiabatic channel study of the transition from adiabatic to sudden capture dynamics. III. Dipole–dipole capture, J. Chem. Phys., 1996, 105, 6277–6284 CrossRef CAS.
- P.-L. Luo, I.-Y. Chen, M. A. H. Khan and D. E. Shallcross, Direct gas-phase formation of formic acid through reaction of Criegee intermediates with formaldehyde, Commun. Chem., 2023, 6, 1–10 CrossRef PubMed.
- Air quality guidelines for Europe, https://www.euro.who.int/en/publications/abstracts/air-quality-guidelines-for-europe, accessed 22 January 2021.
- W. Lin, X. Xu, Z. Ma, H. Zhao, X. Liu and Y. Wang, Characteristics and recent trends of sulfur dioxide at urban, rural, and background sites in North China: Effectiveness of control measures, J. Environ. Sci., 2012, 24, 34–49 CrossRef CAS PubMed.
- Y. J. Leong, A. P. Rutter, H. Y. Wong, C. V. Gutierrez, M. Junaid, E. Scheuer, L. Gong, R. Lewicki, J. E. Dibb, F. K. Tittel and R. J. Griffin, Impact of environmental variables on the reduction of nitric acid by proxies for volatile organic compounds emitted by motor vehicles, Atmos. Pollut. Res., 2016, 7, 221–227 CrossRef.
- L. Sheps, B. Rotavera, A. J. Eskola, D. L. Osborn, C. A. Taatjes, K. Au, D. E. Shallcross, M. A. H. Khan and C. J. Percival, The reaction of Criegee intermediate CH2OO with water dimer: primary products and atmospheric impact, Phys. Chem. Chem. Phys., 2017, 19, 21970–21979 RSC.
- J. M. Anglada, G. J. Hoffman, L. V. Slipchenko, M. M. Costa, M. F. Ruiz-López and J. S. Francisco, Atmospheric Significance of Water Clusters and Ozone–Water Complexes, J. Phys. Chem. A, 2013, 117, 10381–10396 CrossRef CAS PubMed.
- M. A. Bravo, J. Son, C. U. de Freitas, N. Gouveia and M. L. Bell, Air pollution and mortality in São Paulo, Brazil: Effects of multiple pollutants and analysis of susceptible populations, J. Exposure Sci. Environ. Epidemiol., 2016, 26, 150–161 CrossRef CAS PubMed.
- S. V. Tadayon, E. S. Foreman and C. Murray, Kinetics of the Reactions between the Criegee Intermediate CH2OO and Alcohol, J. Phys. Chem. A, 2018, 122, 258–268 CrossRef CAS PubMed.
- H. T.-H. Nguyen, N. Takenaka, H. Bandow, Y. Maeda, S. T. de Oliva, M. M. f. Botelho and T. M. Tavares, Atmospheric alcohols and aldehydes concentrations measured in Osaka, Japan and in Sao Paulo, Brazil, Atmos. Environ., 2001, 35, 3075–3083 CrossRef CAS.
- M. C. Smith, W. Chao, M. Kumar, J. S. Francisco, K. Takahashi and J. J.-M. Lin, Temperature-Dependent Rate Coefficients for the Reaction of CH2OO with Hydrogen Sulfide, J. Phys. Chem. A, 2017, 121, 938–945 CrossRef CAS PubMed.
- National Research Council (US) Committee, Acute Exposure Guideline Levels, Hydrogen Sulfide Acute Exposure Guideline Levels, National Academies Press (US), 2010 Search PubMed.
- T. A. Crisp, B. M. Lerner, E. J. Williams, P. K. Quinn, T. S. Bates and T. H. Bertram, Observations of gas phase hydrochloric acid in the polluted marine boundary layer, J. Geophys. Res.: Atmos., 2014, 119, 6897–6915 CrossRef CAS.
- M.-D. Cheng, Atmospheric chemistry of hydrogen fluoride, J. Atmos. Chem., 2018, 75, 1–16 CrossRef CAS.
- E. V. Avzianova and P. A. Ariya, Temperature-dependent kinetic study for ozonolysis of selected tropospheric alkenes, Int. J. Chem. Kinet., 2002, 34, 678–684 CrossRef CAS.
- G. J. Raw, S. K. D. Coward, V. M. Brown and D. R. Crump, Exposure to air pollutants in English homes, J. Exposure Sci. Environ. Epidemiol., 2004, 14, S85–S94 CrossRef CAS PubMed.
- J. Kazil, S. McKeen, S.-W. Kim, R. Ahmadov, G. A. Grell, R. K. Talukdar and A. R. Ravishankara, Deposition and rainwater concentrations of trifluoroacetic acid in the United States from the use of HFO-1234yf, J. Geophys. Res.: Atmos., 2014, 119, 14059–14079 CrossRef CAS.
- A. Tiwary and I. Williams, Air Pollution: Measurement, Modelling and Mitigation, CRC Press, 4h edn, 2018 Search PubMed.
- L. Zhu, D. J. Jacob, F. N. Keutsch, L. J. Mickley, R. Scheffe, M. Strum, G. González Abad, K. Chance, K. Yang, B. Rappenglück, D. B. Millet, M. Baasandorj, L. Jaeglé and V. Shah, Formaldehyde (HCHO) As a Hazardous Air Pollutant: Mapping Surface Air Concentrations from Satellite and Inferring Cancer Risks in the United States, Environ. Sci. Technol., 2017, 51, 5650–5657 CrossRef CAS PubMed.
- J. C. Salamanca, J. Meehan-Atrash, S. Vreeke, J. O. Escobedo, D. H. Peyton and R. M. Strongin, E-cigarettes can emit formaldehyde at high levels under conditions that have been reported to be non-averse to users, Sci. Rep., 2018, 8, 7559 CrossRef PubMed.
- Q. Ma, X. Lin, C. Yang, B. Long, Y. Gai and W. Zhang, The influences of ammonia on aerosol formation in the ozonolysis of styrene: roles of Criegee intermediate reactions, R. Soc. Open Sci., 2018, 5, 172171 CrossRef PubMed.
- R. M. I. Elsamra, A. Jalan, Z. J. Buras, J. E. Middaugh and W. H. Green, Temperature- and Pressure-Dependent Kinetics of CH2OO + CH3COCH3 and CH2OO + CH3CHO: Direct Measurements and Theoretical Analysis, Int. J. Chem. Kinet., 2016, 48, 474–488 CrossRef CAS.
- Y.-H. Lin, K. Takahashi and J. J.-M. Lin, Reactivity of Criegee Intermediates toward Carbon Dioxide, J. Phys. Chem. Lett., 2018, 9, 184–188 CrossRef CAS PubMed.
- A. B. Ryzhkov and P. A. Ariya, A theoretical study of the reactions of parent and substituted Criegee intermediates with water and the water dimer, Phys. Chem. Chem. Phys., 2004, 6, 5042–5050 RSC.
- L.-C. Lin, H.-T. Chang, C.-H. Chang, W. Chao, M. C. Smith, C.-H. Chang, J. J.-M. Lin and K. Takahashi, Competition between H2O and (H2O)2 reactions with CH2OO/CH3CHOO, Phys. Chem. Chem. Phys., 2016, 18, 4557–4568 RSC.
- L. Sheps, A. M. Scully and K. Au, UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO, Phys. Chem. Chem. Phys., 2014, 16, 26701–26706 RSC.
- T. Berndt, J. Voigtländer, F. Stratmann, H. Junninen, R. L. M. Iii, M. Sipilä, M. Kulmala and H. Herrmann, Competing atmospheric reactions of CH2OO with SO2 and water vapour, Phys. Chem. Chem. Phys., 2014, 16, 19130–19136 RSC.
- L. Sheps, Absolute Ultraviolet Absorption Spectrum of a Criegee Intermediate CH2OO, J. Phys. Chem. Lett., 2013, 4, 4201–4205 CrossRef CAS PubMed.
- Y. Liu, K. D. Bayes and S. P. Sander, Measuring Rate Constants for Reactions of the Simplest Criegee Intermediate (CH2OO) by Monitoring the OH Radical, J. Phys. Chem. A, 2014, 118, 741–747 CrossRef CAS PubMed.
- T. Kurtén, J. R. Lane, S. Jørgensen and H. G. Kjaergaard, A Computational Study of the Oxidation of SO2 to SO3 by Gas-Phase Organic Oxidants, J. Phys. Chem. A, 2011, 115, 8669–8681 CrossRef PubMed.
- Y. Liu, F. Liu, S. Liu, D. Dai, W. Dong and X. Yang, A kinetic study of the CH2OO Criegee intermediate reaction with SO2, (H2O)2, CH2I2 and I atoms using OH laser induced fluorescence, Phys. Chem. Chem. Phys., 2017, 19, 20786–20794 RSC.
- M. C. Smith, C.-H. Chang, W. Chao, L.-C. Lin, K. Takahashi, K. A. Boering and J. J.-M. Lin, Strong Negative Temperature Dependence of the Simplest Criegee Intermediate CH2OO Reaction with Water Dimer, J. Phys. Chem. Lett., 2015, 6, 2708–2713 CrossRef CAS PubMed.
- R. Yajima, Y. Sakamoto, S. Inomata and J. Hirokawa, Relative Reactivity Measurements of Stabilized CH2OO, Produced by Ethene Ozonolysis, Toward Acetic Acid and Water Vapor Using Chemical Ionization Mass Spectrometry, J. Phys. Chem. A, 2017, 121, 6440–6449 CrossRef CAS PubMed.
- L. Vereecken, H. Harder and A. Novelli, The reaction of Criegee intermediates with NO, RO2, and SO2, and their fate in the atmosphere, Phys. Chem. Chem. Phys., 2012, 14, 14682–14695 RSC.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- O. US EPA, Benefits and Costs of the Clean Air Act 1990-2020, Report Documents and Graphics, https://www.epa.gov/clean-air-act-overview/benefits-and-costs-clean-air-act-1990-2020-report-documents-and-graphics, accessed 20 July 2020.
- D. Huang, J. Xu and S. Zhang, Valuing the health risks of particulate air pollution in the Pearl River Delta, China, Environ. Sci. Policy, 2012, 15, 38–47 CrossRef CAS.
- J. Wang, S. Wang, A. S. Voorhees, B. Zhao, C. Jang, J. Jiang, J. S. Fu, D. Ding, Y. Zhu and J. Hao, Assessment of short-term PM2.5-related mortality due to different emission sources in the Yangtze River Delta, China, Atmos. Environ., 2015, 123, 440–448 CrossRef CAS.
- J. Baek, Y. Hu, M. T. Odman and A. G. Russell, Modeling secondary organic aerosol in CMAQ using multigenerational oxidation of semi-volatile organic compounds, J. Geophys. Res.: Atmos., 2015, 116, 22204–22216 Search PubMed.
- Z. Ren-Jian, H. Kin-Fai and S. Zhen-Xing, The Role of Aerosol in Climate Change, the Environment, and Human Health, Atmos. Ocean. Sci. Lett., 2015, 5(2), 156–161 CrossRef.
- T. Berndt, H. Herrmann and T. Kurtén, Direct Probing of Criegee Intermediates from Gas-Phase Ozonolysis Using Chemical Ionization Mass Spectrometry, J. Am. Chem. Soc., 2017, 139, 13387–13392 CrossRef CAS PubMed.
- E. Sanhueza, Hydrochloric acid from chlorocarbons: a significant global source of background rain acidity, Tellus B, 2001, 53, 122–132 CrossRef.
- P. Raghunath, Y.-P. Lee and M. C. Lin, Computational Chemical Kinetics for the Reaction of Criegee Intermediate CH2OO with HNO3 and Its Catalytic Conversion to OH, J. Phys. Chem. A, 2017, 121, 3871–3878 CrossRef CAS PubMed.
- L. Vereecken, The reaction of Criegee intermediates with acids and enols, Phys. Chem. Chem. Phys., 2017, 19, 28630–28640 RSC.
- K. H. Becker, K. J. Brockmann and J. Bechara, Production of hydrogen peroxide in forest air by reaction of ozone with terpenes, Nature, 1990, 346, 256–258 CrossRef CAS.
- P. Neeb, F. Sauer, O. Horie and G. K. Moortgat, Formation of hydroxymethyl hydroperoxide and formic acid in alkene ozonolysis in the presence of water vapour, Atmos. Environ., 1997, 31, 1417–1423 CrossRef.
- F. Sauer, C. Schäfer, P. Neeb, O. Horie and G. K. Moortgat, Formation of hydrogen peroxide in the ozonolysis of isoprene and simple alkenes under humid conditions, Atmos. Environ., 1999, 33, 229–241 CrossRef CAS.
- O. Horie and G. K. Moortgat, Gas-Phase Ozonolysis of Alkenes. Recent Advances in Mechanistic Investigations, Acc. Chem. Res., 1998, 31, 387–396 CrossRef CAS.
- R. Crehuet, J. M. Anglada and J. M. Bofill, Tropospheric Formation of Hydroxymethyl Hydroperoxide, Formic Acid, H2O2, and OH from Carbonyl Oxide in the Presence of Water Vapor: A Theoretical Study of the Reaction Mechanism, Chem.–Eur. J., 2001, 7, 2227–2235 CrossRef CAS PubMed.
- E. Jiménez, S. González, M. Cazaunau, H. Chen, B. Ballesteros, V. Daële, J. Albaladejo and A. Mellouki, Atmospheric Degradation Initiated by OH Radicals of the Potential Foam Expansion Agent, CF3(CF2)2CH=CH2 (HFC-1447fz): Kinetics and Formation of Gaseous Products and Secondary Organic Aerosols, Environ. Sci. Technol., 2016, 50, 1234–1242 CrossRef PubMed.
- P. Aplincourt and J. M. Anglada, Theoretical Studies on Isoprene Ozonolysis under Tropospheric Conditions. 1. Reaction of Substituted Carbonyl Oxides with Water, J. Phys. Chem. A, 2003, 107, 5798–5811 CrossRef CAS.
- L. Chen, Y. Huang, Y. Xue, Z. Shen, J. Cao and W. Wang, Mechanistic and kinetics investigations of oligomer formation from Criegee intermediate reactions with hydroxyalkyl hydroperoxides, Atmos. Chem. Phys., 2019, 19, 4075–4091 CrossRef CAS.
- Z. J. Buras, R. M. I. Elsamra, A. Jalan, J. E. Middaugh and W. H. Green, Direct Kinetic Measurements of Reactions between the Simplest Criegee Intermediate CH2OO and Alkenes, J. Phys. Chem. A, 2014, 118, 1997–2006 CrossRef CAS PubMed.
- L.-C. Lin, W. Chao, C.-H. Chang, K. Takahashi and J. J.-M. Lin, Temperature dependence of the reaction of anti-CH3CHOO with water vapor, Phys. Chem. Chem. Phys., 2016, 18, 28189–28197 RSC.
- S. Bauerle and G. K. Moortgat, Absorption cross-sections of HOCH2OOH vapor between 205 and 360 nm at 298 K, Chem. Phys. Lett., 1999, 309, 43–48 CrossRef CAS.
- H. M. Allen, J. D. Crounse, K. H. Bates, A. P. Teng, M. P. Krawiec-Thayer, J. C. Rivera-Rios, F. N. Keutsch, J. M. St. Clair, T. F. Hanisco, K. H. Møller, H. G. Kjaergaard and P. O. Wennberg, Kinetics and Product Yields of the OH Initiated Oxidation of Hydroxymethyl Hydroperoxide, J. Phys. Chem. A, 2018, 122, 6292–6302 CrossRef CAS PubMed.
- B. Ouyang, M. W. McLeod, R. L. Jones and W. J. Bloss, NO3 radical production from the reaction between the Criegee intermediate CH2OO and NO2, Phys. Chem. Chem. Phys., 2013, 15, 17070–17075 RSC.
- T. Berndt, R. Kaethner, J. Voigtländer, F. Stratmann, M. Pfeifle, P. Reichle, M. Sipilä, M. Kulmala and M. Olzmann, Kinetics of the unimolecular reaction of CH2OO and the bimolecular reactions with the water monomer, acetaldehyde and acetone under atmospheric conditions, Phys. Chem. Chem. Phys., 2015, 17, 19862–19873 RSC.
- T. R. Lewis, M. A. Blitz, D. E. Heard and P. W. Seakins, Direct evidence for a substantive reaction between the Criegee intermediate, CH2OO, and the water vapour dimer, Phys. Chem. Chem. Phys., 2015, 17, 4859–4863 RSC.
- M. Kumar, J. Zhong, J. S. Francisco and X. C. Zeng, Criegee intermediate-hydrogen sulfide chemistry at the air/water interface, Chem. Sci., 2017, 8, 5385–5391 RSC.
- S. Liu, X. Zhou, Y. Chen, Y. Liu, S. Yu, K. Takahashi, H. Ding, Z. Ding, X. Yang and W. Dong, Experimental and Computational Studies of Criegee Intermediate syn-CH3CHOO Reaction with Hydrogen Chloride, J. Phys. Chem. A, 2021, 125, 8587–8594 CrossRef CAS PubMed.
- M. R. McGillen, A. T. Archibald, T. Carey, K. E. Leather, D. E. Shallcross, J. C. Wenger and C. J. Percival, Structure–activity relationship (SAR) for the prediction of gas-phase ozonolysis rate coefficients: an extension towards heteroatomic unsaturated species, Phys. Chem. Chem. Phys., 2011, 13, 2842–2849 RSC.
- K. E. Leather, M. R. McGillen and C. J. Percival, Temperature-dependent ozonolysis kinetics of selected alkenes in the gas phase: an experimental and structure–activity relationship (SAR) study, Phys. Chem. Chem. Phys., 2010, 12, 2935–2943 RSC.
- R. Sommariva, S. Cox, C. Martin, K. Borońska, J. Young, P. Jimack, M. J. Pilling, V. N. Matthaios, M. J. Newland, M. Panagi, W. J. Bloss, P. S. Monks and A. R. Rickard, AtChem, an open source box-model for the Master Chemical Mechanism, Geosci. Model Dev., 2019, 1–27 Search PubMed.
- J. Pu, H. Xu, B. Yao, Y. Yu, Y. Jiang, Q. Ma and L. Chen, Estimate of Hydrofluorocarbon Emissions for 2012–16 in the Yangtze River Delta, China, Adv. Atmos. Sci., 2020, 37, 576–585 CrossRef CAS.
- É. C. M. de Lanes, P. M. de A. Costa and S. Y. Motoike, Alternative fuels, https://www.nature.com/articles/511031a, accessed 27 March 2018.
- Committee on Acute Exposure Guideline Levels, Committee on Toxicology, Board on Environmental Studies and Toxicology; Division on Earth and Life Studies and National Research Council, Acute Exposure Guideline Levels for Selected Airborne Chemicals, National Academies Press (US), 2012, vol. 13 Search PubMed.
- R. L. Mauldin III, T. Berndt, M. Sipilä, P. Paasonen, T. Petäjä, S. Kim, T. Kurtén, F. Stratmann, V.-M. Kerminen and M. Kulmala, A new atmospherically relevant oxidant of sulphur dioxide, Nature, 2012, 488, 193–196 CrossRef PubMed.
- R. A. Cox and S. A. Penkett, Oxidation of Atmospheric SO2 by Products of the Ozone–Olefin Reaction, Nature, 1971, 230, 321–322 CrossRef CAS PubMed.
- M. Jang, N. M. Czoschke, S. Lee and R. M. Kamens, Heterogeneous Atmospheric Aerosol Production by Acid-Catalyzed Particle-Phase Reactions, Science, 2002, 298, 814–817 CrossRef CAS PubMed.
- M. Boy, D. Mogensen, S. Smolander, L. Zhou, T. Nieminen, P. Paasonen, C. Plass-Dülmer, M. Sipilä, T. Petäjä, L. Mauldin, H. Berresheim and M. Kulmala, Oxidation of SO2 by stabilized Criegee intermediate (sCI) radicals as a crucial source for atmospheric sulfuric acid concentrations, Atmos. Chem. Phys., 2013, 13, 3865–3879 CrossRef.
- T. A. Mather, Volcanoes and the environment: Lessons for understanding Earth's past and future from studies of present-day volcanic emissions, J. Volcanol. Geotherm. Res., 2015, 304, 160–179 CrossRef CAS.
- A. Carfora, C. P. Campobasso, P. Cassandro, F. La Sala, A. Maiellaro, A. Perna, R. Petrella and R. Borriello, Fatal inhalation of volcanic gases in three tourists of a geothermal area, Forensic Sci. Int., 2019, 297, e1–e7 CrossRef CAS PubMed.
- A. Aiuppa, A. Franco, R. von Glasow, A. G. Allen, W. D'Alessandro, T. A. Mather, D. M. Pyle and M. Valenza, The tropospheric processing of acidic gases and hydrogen sulphide in volcanic gas plumes as inferred from field and model investigations, Atmos. Chem. Phys., 2007, 7, 1441–1450 CrossRef CAS.
- L. M. Guidry, C. A. Poirier, J. M. Ratliff, E. Antwi, B. Marchetti and T. N. V. Karsili, Modeling the Unimolecular Decay Dynamics of the Fluorinated Criegee Intermediate, CF3CHOO, Photochem, 2023, 3, 327–335 CrossRef.
- I. Ljubić and A. Sabljić, Fluorocarbonyl oxide: CASSCF/CASPT2 study of structure, cis effect and unimolecular decomposition paths, Chem. Phys., 2005, 309, 157–165 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: A full breakdown of the master equation rate constants (kME) and canonical rate constants (kCAN) through temperature range of 200 K < T < 500 K at p = 760 Torr for sCIs 1–5 reactions with the following co-reactants: HCHO, SO2, HNO3, TFA, HF, HCl, H2S, H2O, (H2O)2, MeOH; calculated product yields for sCIs 1–5 reactions with HCHO, SO2; calculated values for sCI 1 + co-reactant rate constants at selected temperatures and pressures for direct comparison with experiment; calculated values for the effective rate constants for sCI + co-reactant reactions under various conditions; relative energies [kJ mol−1] of stationary point for sCI + co-reactant reactions; computed dipole–dipole and gas collision limits; discussion on the partially barrierless sCI 4 + MeOH reaction; Cartesian coordinates and vibrational frequencies of all stationary points, and IRCs of all transition states in this study; an example MEMSER input file for the sCI 1 + HNO3 (PDF). See DOI: https://doi.org/10.1039/d3ea00102d |
| This journal is © The Royal Society of Chemistry 2023 |