
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photolytic aging of organic aerosol from pyrolyzed urban materials†
Katherine S.
Hopstock
 a,
Alexandra L.
Klodt
a,
Qiaorong
Xie
b,
Michael A.
Alvarado
a,
Alexander
Laskin
bc and
Sergey A.
Nizkorodov
*a
a,
Alexandra L.
Klodt
a,
Qiaorong
Xie
b,
Michael A.
Alvarado
a,
Alexander
Laskin
bc and
Sergey A.
Nizkorodov
*a
aDepartment of Chemistry, University of California, Irvine, Irvine, CA 92697, USA. E-mail: nizkorod@uci.edu
bDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
cDepartment of Earth, Atmospheric and Planetary Sciences, Purdue University, West Lafayette, IN 47907, USA
First published on 20th July 2023
Abstract
Emissions from large-scale fires significantly contribute to the atmospheric burden of primary organic aerosol (OA). The frequency of fires occurring at the wildland–urban interface (WUI) is increasing, with biomass and a wide range of human-engineered materials serving as fuels. The chemical composition and optical properties of OA from WUI fires are poorly characterized, and this work seeks to understand how direct photolytic aging alters the light absorbing properties of particulate matter generated from WUI fires. Ten flammable urban materials were selected to represent structural and furnishing components commonly combusted in urban fires: electrical 12 AWG wire (white PVC coating), ceiling tile, synthetic fabric, electrical 23 AWG wire (purple PVC coating), lumber, drywall, fiberboard, vinyl flooring, plywood, and carpet. Each material was pyrolyzed at 600 °C under N2 in a tube furnace and resulting smoke particles were collected on Teflon filters or deposited on fused silica optical windows. OA samples were aged by exposing them to simulated solar radiation (near-UV radiation, 280–400 nm) directly on the collection substrates. Mass absorption coefficients (MAC) and Absorption Angström Exponents (AAE) of the unaged and aged OA were calculated from spectrophotometry measurements taken after extracting OA in suitable organic solvents. We observed variable trends after short-term exposure to UV radiation: six out of ten types of urban OA exhibited photoenhancement, two exhibited photobleaching, and the remaining two experienced negligible change after 2 h of photolytic aging. The average AAE value for unaged OA was 8.8 ± 1.3 and decreased to 7.3 ± 0.9 after aging, reflecting increased absorption in the visible range of the spectrum after the UV exposure, and an evolution from very weakly to weakly absorbing brown carbon. Long-term UV exposure results indicate that most photochemical change occurs within the first few hours of irradiation. These results suggest that WUI fires efficiently produce brown carbon, which becomes increasingly light-absorbing in sunlight.
Environmental significanceThe frequency of fires occurring at the wildland–urban interface is increasing, with biomass and a wide range of human-engineered materials serving as fuels. Urban material combustion is an important source of atmospheric particulate matter emissions. Smoke particles that can absorb visible solar radiation are especially important because they reduce visibility and lead to atmospheric warming. This work determines the extent to which particulate matter from the burning of different urban materials can absorb sunlight. Additionally, it shows that light-absorbing properties of this particulate matter change upon exposure to solar UV radiation by (1) decomposing light-absorbing compounds (photobleaching) or (2) producing stronger light-absorbing compounds from photooxidation of smaller molecules (photoenhancement). On average, photoenhancement is a more important effect resulting in increasing light absorption by the particulate matter after it travels through the air. This means that smoke particles emitted during wildland–urban interface fires may have a strong effect on visibility and climate. |
1. Introduction
Aerosols affect the propagation of radiation through Earth's atmosphere by directly absorbing or scattering incoming solar radiation. Black carbon (BC) and brown carbon (BrC) are components of carbonaceous aerosol particles that absorb solar radiation and contribute to direct forcing on climate by aerosols. BC absorbs strongly over a broad wavelength range and is commonly attributed to soot particles emitted from biomass burning and incomplete combustion of fossil fuels.1 BrC is characterized by a negligible absorption coefficient at the red edge of the visible spectrum (600–700 nm) and has a much stronger absorption coefficient in the near-UV and blue range of the spectrum (300–500 nm), attributing to its brown color.2–4 BrC can be emitted from the burning of biomass materials (wildfires,5–8 domestic wood burning,9 and cookstove emissions10) and its optical properties are highly variable and dependent upon their biofuel source and combustion conditions.11–13 Atmospheric BrC can also result from the oxidation of aromatic hydrocarbons producing light-absorbing secondary organic aerosol (SOA).The high chemical reactivity of OA alters its chemical composition and optical properties on atmospherically relevant timescales. Solar radiation drives many of these aging processes by directly photolyzing light-absorbing species, and also through indirect photochemical processes, such as secondary reactions of OA compounds with photochemically produced free radicals and photosensitized reactions.10,14 Many studies have been conducted to examine the aqueous photochemistry of OA extracts to simulate the photochemical processing of organics dissolved in fog and cloud water.15–23 Less attention has been given to simulating photochemical processing that occurs inside the organic phase of particles. This organic phase represents a unique environment for photochemistry because of its higher viscosity and reduced polarity compared to an aqueous solution resulting in different photochemical mechanisms in organic particles and in aqueous solutions. Aerosol particles have been estimated to spend 85% of their respective life cycles under non-cloud-activated conditions, thus photochemistry will often take place in the predominantly organic phase.24 Even under cloud-activated conditions, photochemistry in the organic phase is also important as only a fraction of OA is water soluble.
Previous studies of condensed-phase photochemistry in OA have primarily focused on the photodegradation of SOA particles generated from terpene ozonolysis.25–27 Mass loss was observed upon UV irradiation of α-pinene and D-limonene ozonolysis SOA,28–30 which was driven by the direct photolysis of peroxide and carbonyl functionalities as well as secondary reactions of the resulting free radicals.28,31–34 A few studies have characterized the mass loss and absorption changes of SOA-containing nitrophenols during UV irradiation.30,35–37 Additionally, there have been a few studies observing the optical properties of suspended OA produced from burning or pyrolysis, and these generally observe an increase in absorption at long wavelengths with UV irradiation, sometimes followed by photobleaching.38–40 However, it was difficult to cleanly separate the effects of gas-phase and condensed-phase photochemistry in these experiments.
Even less attention has been given to the photolytic aging of biomass burning organic aerosol (BBOA). Fleming et al. (2020) identified major chromophores in BBOA from the combustion of several biofuel types and observed the loss of individual chromophores by utilizing chromatographic separation followed by photodiode array spectrophotometry and high-resolution mass spectrometry.21 They found that the majority of chromophores were lost after approximately 33 hours of UV irradiation, with the exception of a few photorecalcitrant compounds.21 To the best of our knowledge, this has been the only study to investigate changes in the optical properties of BBOA particles induced by photolysis in the organic particle phase. It is still unclear how photolytic aging impacts the chemical composition and optical properties of OA generated from biomass combustion. Further, even less is known about the optical properties and photochemical aging of OA generated by fires at the wildland–urban interface (WUI).
The frequency of fires occurring at the WUI is increasing, indicating that the composition and properties of the wildfire related OA are dictated not only by burning biomass but also by burning human-engineered materials.41 The WUI is defined as the area where urban development encroaches upon borders of private and public wildlands.42 In 2000, 39% of all housing units in the continental United States occupied WUI land.43 Burke et al. (2021) estimated that 50 million homes (and counting) are located in WUI areas in the United States.44 Climate change has led to more frequent occurrences of large scale forest wildfires that devastatingly destroy urban homes and buildings located at the WUI.45,46 The effect of urban fire emissions (and its subsequent change) on Earth's radiative forcing is unknown. Blomqvist et al. (2007) reported that wood, particleboard, paper, textiles, polyvinyl chloride (PVC), polyurethane, polyethylene, polystyrene, acrylonitrile butadiene styrene, tarred roofing, linoleum, and rubber products were present during building fires in Sweden (1999) that lead to the emissions of volatile organic compounds (VOCs), polycyclic aromatic hydrocarbons (PAHs), and dioxins.47 Prominent VOCs related to the burning of urban buildings, farms, and household materials have been previously characterized,47–49 however, neither the chemical composition nor the aging of this WUI particulate matter have been characterized. This study is the first to conduct photolytic aging experiments using laboratory generated OA representing emissions from WUI fires. Materials representative of engineered wood, plastics, construction materials, textiles, and furnishings were selected to cover the major categories of urban materials combusted in WUI fires.
2. Experimental
Flowcharts of experimental procedures can be found in the ESI in Schemes S1 and S2.†2.1 Ten urban materials
Ten samples of flammable urban materials were selected in this study to represent structural and furnishing components present in urban fires: electrical 12 AWG wire (white PVC coating), ceiling tile, fabric, electrical 23 AWG wire (purple PVC coating), lumber, drywall, fiberboard, vinyl flooring, plywood, and industrial carpet. More specific details on the materials can be found in Table S1.† The 12 AWG wire (white PVC coating) and 23 AWG wire (purple PVC coating) will be referred to as “thick PVC wire” and “thin PVC wire”, respectively. All urban materials melt or thermally degrade at temperatures less than that used in our pyrolysis experiments (600 °C).2.2 Organic aerosol (OA) generation
Experiments were conducted similarly to our previous work on biomass pyrolysis.50 Pyrolysis is the thermal decomposition of biomass under oxygen limited conditions with temperatures between 300 and 800 °C.51 Fires often transition between smoldering (450–700 °C) and flaming (1500–1800 °C) combustion with their genesis process being pyrolysis.52 Therefore, 600 °C is suitable to mimic realistic burning conditions. For each pyrolysis experiment, approximately 200 mg of one tested flammable material (Table S1†) was placed in a ceramic pyrolysis boat and then inserted into a 2.1 cm I.D./2.5 cm O.D. fused silica tube, upstream from the Thermolyne F21135 Tube Furnace (Scheme S1b†). The tube was sealed and continuously purged with N2 gas (∼0.7 standard liters per minute). A 40 cm section of the tube, downstream from the pyrolysis boat, was heated to 600 °C, and once at this temperature, the tube was pushed inside to bring the urban material into the heated zone to start the pyrolysis. The starting pressure was ∼760 Torr and did not exceed 770 Torr during the experiment. A flow of N2 gas was constant during pyrolysis and was utilized to push the generated aerosol and fumes through the fused silica tube for collection. The setup was not reopened until collection had ceased. OA sample from each urban material was collected for 10 min starting from the moment the smoke appeared in the flow (Fig. S1†). Samples of tested carpet material were also pyrolyzed at 800 °C and 1000 °C to explore the effects of higher pyrolysis temperatures (Fig. S2†). Pyrolysis experiments at 600 °C produced urban OA that favored BrC production, and all subsequent experiments were done at this temperature.Samples were collected on two different substrates approximately 1 m downstream from the heated zone, at which point the flow cooled to ∼40 °C. For most experiments, the collection was done on PTFE 47 mm Teflon filters, which are stable at temperatures less than 350 °C (Millipore Sigma, Fluoropore Membrane Filter, FGLP04700, 47 mm, 0.22 μm pore size) (Fig. S1†). A subset of samples were collected on optical windows (ISP Optics, Fused Silica IR Grade Windows, QI-W-12-1, 12.7 mm diameter, 1 mm thick) using a single-stage electrostatic precipitator (ESP) (TSI 3089 Nanometer Aerosol Sampler, −5 kV) equipped with a Kr-85 neutralizer (TSI Model 3077 Aerosol Neutralizer). Samples (both Teflon filters and fused silica windows) were weighed after collection using a Sartorius ME5-F microbalance (±1 μg precision) and the OA mass was calculated. For the samples collected on Teflon filters, the emission factors (EF), defined as the mass of OA collected per urban material mass used in pyrolysis (g kg−1), were calculated from the measured mass collected, assuming that the filters had 100% collection efficiency.
2.3 UV-visible spectroscopy measurements and effects of solvents
After collection, Teflon filters were cut into four equal pieces and each quarter was extracted, separately in 10 mL, using four different solvents: “orgmix” (acetonitrile, dichloromethane, hexane 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 by volume), methanol, water, and n-octanol. A 2 mL aliquot of each OA extract was transferred into a 1 cm fused silica cuvette and a dual-beam spectrophotometer (Shimadzu UV-2450) was used to take UV/Vis absorption spectra, with the corresponding solvent used as a reference. The best-suited extraction solvent for each OA sample was the one that gave the highest absorbance of the extracted material (Table S1 and Fig. S3†). Wavelength-dependent mass absorption coefficients (MAC), in the units of cm2 g−1, were calculated from the base-10 absorbance (A10), the estimated concentration of OA extracted in solution, Cmass (g cm−3), and path length of the cuvette, b (cm) (eqn (1)).
:2:1 by volume), methanol, water, and n-octanol. A 2 mL aliquot of each OA extract was transferred into a 1 cm fused silica cuvette and a dual-beam spectrophotometer (Shimadzu UV-2450) was used to take UV/Vis absorption spectra, with the corresponding solvent used as a reference. The best-suited extraction solvent for each OA sample was the one that gave the highest absorbance of the extracted material (Table S1 and Fig. S3†). Wavelength-dependent mass absorption coefficients (MAC), in the units of cm2 g−1, were calculated from the base-10 absorbance (A10), the estimated concentration of OA extracted in solution, Cmass (g cm−3), and path length of the cuvette, b (cm) (eqn (1)).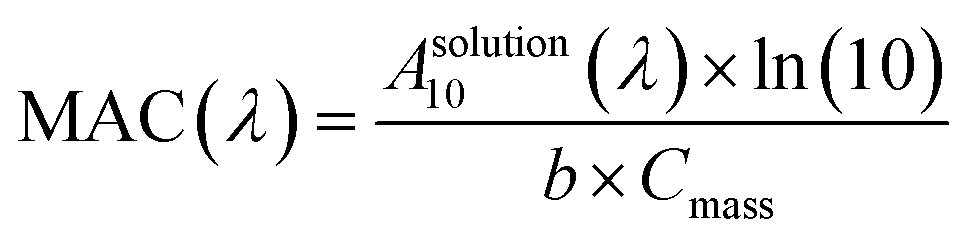 | (1) |
To minimize the effects of small baseline drifts during the measurement, the calculated MAC values were corrected by subtracting the average MAC between 680 to 700 nm (the absorbance in this wavelength range was generally negligible). Absorption Angström Exponents (AAE) were calculated from the slopes of the linear fits applied to the log–log plot of MAC(λ) values between 280 and 480 nm.
2.4 Photolysis experiments
On-filter photolysis experiments were performed as described previously.37 Briefly, samples were irradiated with near-UV radiation from a xenon arc lamp (Newport Model 66902) with UVC and visible wavelengths removed using a dichroic mirror, a 295 nm long-pass filter (Schott WG295), and a UV bandpass filter (Schott BG1), leaving the majority of the radiation between 280 and 400 nm. A comparison between our lamp spectrum and ambient solar flux is provided in Fig. S4 and Table S2.† One hour in our photolysis setup is approximately equivalent to 0.7 h under the 2 h average Los Angeles solar flux (average taken for 11 AM to 1 PM on November 9th) as calculated using the “Quick TUV” calculator (Madronich, S. ACOM: Quick TUV, 2019),53 assuming a photolysis wavelength range of 280 to 400 nm (matching our lamp's radiation). This irradiation time (utilizing 2 h average flux) was chosen to match the rooftop photolysis experiments described below. We note that for photochemical processes that take place inside the particles, photolysis of particles immobilized on an inert substrate (such as fibers of a Teflon filter) should give the same products as photolysis of particles suspended in air as long as particles have access to ambient air. There may be a small enhancement of absorption by particles on the filter due to multiple scattering by the filter fibers and changes in particle shapes,54,55 which is neglected in our estimation of equivalent UV exposure times.Filter samples were cut into quarters, and each quarter was weighed to estimate the mass of OA on each quarter (as the OA distribution on the filter was not uniform). For irradiation, one quarter was placed vertically in the path of the near-UV beam, such that the filter surface was uncovered and open to laboratory air (photographs of filters before and after irradiation are provided in Table S3†). The quarters not being photolyzed were re-sealed until irradiation. After photolysis, the filter quarter was extracted with methanol, n-octanol, or orgmix (using the previously best determined solvent for that urban material) to take a UV-Vis spectrum (Fig. S5–S7†). Since the UV-Vis sample preparation for the filter samples was destructive, each time point required an entire filter quarter and only four time points were taken per filter. The samples corresponding to 0 h, (unaged), 0.5 h, 1 h, and 2 h of irradiation were chosen as reference time points. MAC values were then calculated with eqn (1). All MAC calculations were performed using the mass of the unaged sample, i.e., assuming no evaporation or other mass loss occurred. Based on previous SOA photolysis experiments, we do expect some mass loss to occur with photolysis,29,56 so all MAC values reported in this work should be considered as lower limits.
For most samples, the extracted filter quarters exhibited the original white color of the Teflon filter, suggesting that all the light-absorbing material was extracted. However, residual OA material was visible on the Teflon filters after photolysis and extraction for three materials: lumber, fabric, and vinyl flooring (Fig. S8†). To remedy this, we collected lumber, fabric, and vinyl flooring OA on fused silica windows to facilitate extraction after photolysis (Scheme S2†). Similarly to the Teflon filter experiments, samples on fused silica windows were photolyzed for 0.5 h, 1 h, and 2 h. Each photolyzed sample was extracted from the window in the previously determined best solvent, absorption spectra were taken, and spectra from the fused silica window experiments were compared with those of the Teflon experiments (Fig. S9–S12†). Each sample extraction from a fused silica window was destructive, meaning that each time point required the preparation of a new pyrolysis sample.
2.5 High-resolution mass spectrometry experiments
One sample, namely thin PVC wire OA, was selected for bulk compositional analysis using high resolution mass spectrometry (HRMS) to identify the chromophore responsible for the distinct absorption bands in its absorption spectrum. As this was the only material to showcase distinctive absorption band features, only thin PVC wire OA was examined with HRMS in this study. Mass spectra were recorded with a Thermo Scientific Vanquish Horizon ultrahigh pressure liquid chromatograph coupled to a Vanquish Horizon photodiode array spectrophotometer and to a Q Exactive Plus high-resolution mass spectrometer (UHPLC-PDA-HRMS) using heated electrospray ionization in the negative mode (Fig. S13†). More information can be found in Appendix A of the ESI.†In additional experiments, portions of selected OA filter samples (carpet, vinyl tile, and fiberboard OA) were loaded individually onto copper pot stubs used in the temperature-programmed desorption – direct analysis in real time – high resolution mass spectrometry (TPD-DART-HRMS) experiments57 using the Q-Exactive™ HF-X Orbitrap mass spectrometer. Mass spectra were recorded in negative mode, at mass resolving power of m/Δm ∼240000 at m/z 200 and acquisition frequency of 1.5 Hz in the full MS scan mode in the mass range of 100–1000 Da (Fig. S14 and S15†). HRMS peak lists were inferred from total ion chromatograms recorded in TPD experiments, integrated over elution times corresponding to selected temperature ranges, and then were background subtracted using the Xcalibur software. Peak lists were extracted with the Decon2LS software and processed using Excel macros developed for m/z peak alignment, blank subtraction, and 13C isotope clustering and filtering.58 The following constraints were applied for all formula assignments: C1–40, H1–100, O1–25, and N0–3, mass tolerance of ±2.0 ppm. Further details of these experiments, associated data interpretation and data processing can be found elsewhere.57
2.6 Additional photolysis experiments
Carpet, vinyl flooring, and fiberboard OA were selected to evaluate how urban OA absorption properties change over longer time scales. Following the same procedures described previously, carpet, vinyl flooring, and fiberboard OA were collected on Teflon filters, cut into four equal fractions, and aged under the xenon arc lamp. Subsequent extraction and absorption measurements were taken (Fig. S16†) at 0 (unaged), 1, 6, and 18 h time points.Drywall OA was selected to compare photolytic exposure from the laboratory photolysis setup (described previously) versus exposure from ambient sunlight. The sample was placed outside on the rooftop of the UC Irvine chemistry building (Rowland Hall) on November 9, 2022 (Fig. S4†). Spectral flux density calculations (Table S2†) concluded that one hour of exposure under the xenon arc lamp equated to 0.7 h of sunlight between the hours of greatest solar flux in Irvine, CA (11 AM to 1 PM) on November 9th. Subsequent extraction and absorption measurements were taken and compared with an unaged drywall OA sample (Fig. S17†).
3. Results and discussion
3.1 Solubility of tested OA
Ten urban flammable materials (Table S1†) were pyrolyzed (under N2) in a tube furnace and resulting OA particles were collected on substrates. Fig. S1† shows representative photographs of resulting filter-collected OA during pyrolysis at 600 °C. The visual appearance of the collected OA varied amongst materials, ranging from gray to light and dark yellow. In addition, carpet OA appearance varied with increasing pyrolysis temperature (Fig. S2†). OA had a brown color resembling that of biomass burning BrC at 600 °C and transformed to OA resembling elemental, black carbon at temperatures exceeding 800 °C. Hence, 600 °C was chosen for all subsequent experiments because samples prepared at >800 °C could not be efficiently extracted in organic solvents.Corresponding EFs (Table S1†) ranged from 1 g kg−1 (drywall) to 47 g kg−1 (lumber), indicating a wide range in the capacity for producing particulate matter from the selected urban materials. Our EF values are likely underestimated due to particle wall loss in the pyrolysis setup. The generated smoke moved from the heated zone, through the fused silica and Teflon tubing before collection on the filter (Scheme S1b†), providing plenty of surfaces for the wall loss. Under-ventilated conditions (i.e., smoldering combustion) tend to produce incomplete combustion with enhanced particulate matter production.59 Because real fires undergo various stages of flaming (well-ventilated conditions) and smoldering (under-ventilated) conditions, our EF values cannot be directly compared with those reported for combustion experiments. With that being said, our wood-based urban materials (lumber and plywood) are well within reported EF ranges for flaming and smoldering combustion.48
Methanol extraction, typically the solvent system of choice, is not as effective in BrC extraction from BBOA as methanol/dichloromethane (1:1 and 1:2, v/v), tetrahydrofuran, and non-dimethylformamide.60 Therefore, solvent extractions efficiencies were assessed for all OA samples. The best extraction solvent was determined by the highest overall MAC values in Fig. S3† and are listed in Table S1.† Consistent with prior BBOA extraction studies,60,61 for all OA samples tested here, water extracted the least OA, as indicated by lower MAC values compared to the other solvent systems (Fig. S3†). The samples had comparable solubility in orgmix, methanol, and octanol. With the notable exception of the thin PVC wire, the MAC spectra in all organic solvents had the same shape, suggesting near complete extraction of all material from the filter.
3.2 Unaged urban OA absorption spectra characteristics
Because BrC is a complex mixture of individual chromophores, the absorption spectra of ambient BrC are typically featureless, with the absorption coefficient smoothly increasing from visible to UV wavelengths.2 The absorption coefficient of BrC often scales with wavelength and a power law, ∝λ−AAE, where AAE stands for Absorption Angström Exponent. For ambient BrC, AAE varies from 2 to 11, depending on the OA composition.2,62,63Fig. 1a shows the MAC(λ) values of all unaged OA samples extracted in the best-suited solvent. All ten OA absorb strongly at shorter wavelengths (UV) and have weak absorption over the visible range, characteristic of BrC.2 Most OA have a slight absorption band at ∼250 nm, attributed to a π to π* transition of conjugated/aromatic organic compounds.39,64,65 The resulting AAE values ranged from 6.0 to 11.6 with the average being 8.8 ± 1.3 (value ± standard deviation). These values fall within expected AAE values for BrC previously reported for SOA BrC and BBOA BrC.2,62,63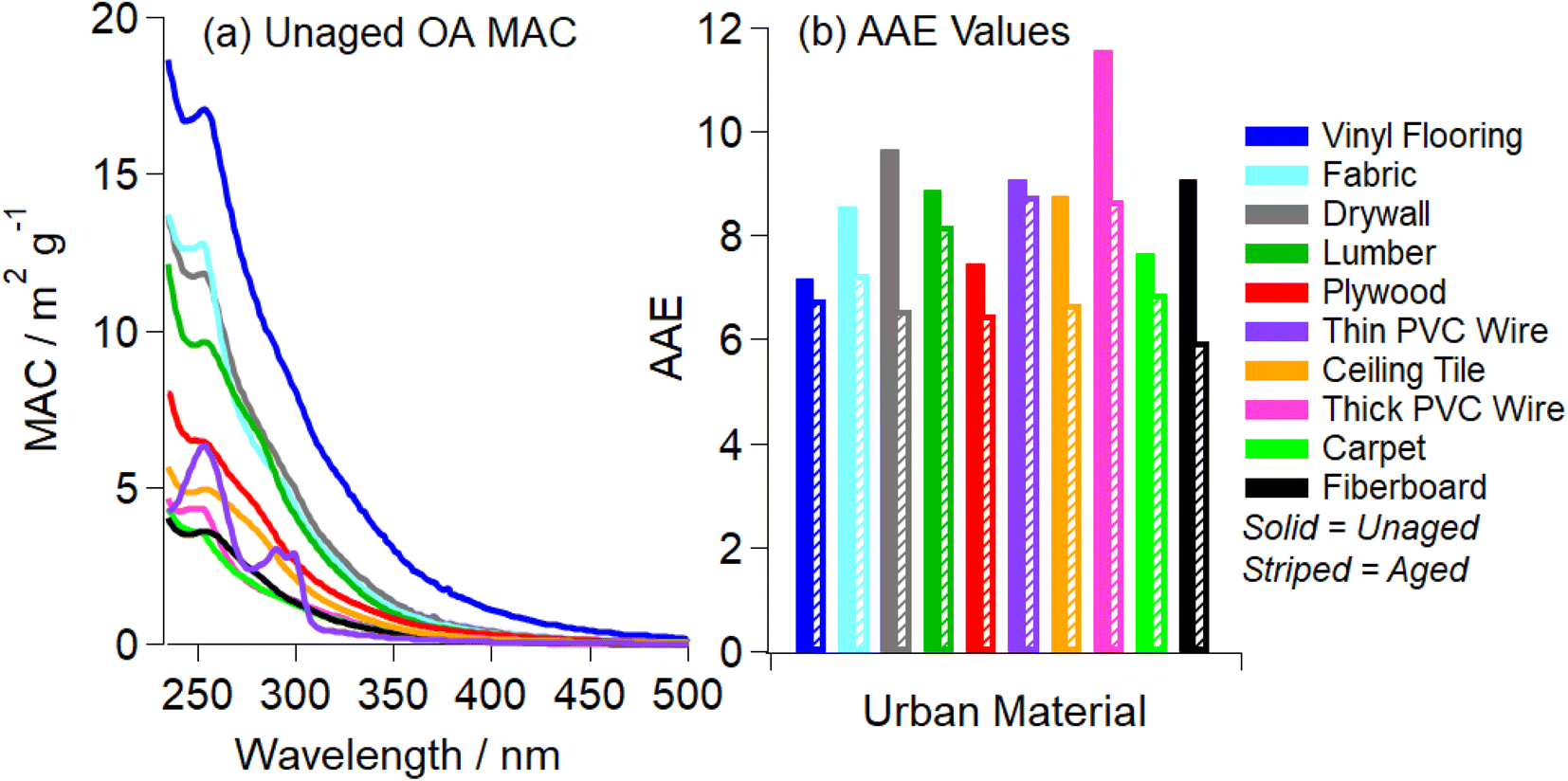 | ||
| Fig. 1 Panel (a) shows the wavelength-dependent mass absorption coefficient (MAC) values recorded after pyrolysis of urban materials and extraction in the best-suited organic solvent (Table S1†). MAC values were calculated assuming 100% extraction efficiency from the Teflon filter. Boxcar averaging of the MAC data (over a 5 nm range) was utilized to improve signal-to-noise ratio. Panel (b) shows the AAE values for unaged OA (solid bars) and 2 h aged OA (striped bars). The effective AAE values were obtained from the linear fits of log(MAC) vs. log(λ) data in Fig. S5† in the 280–480 nm range. | ||
3.3 Transformations of OA absorption caused by photolysis
Three distinct color changes of the OA samples were observed: photoenhancement (samples became visibly darker in color), photobleaching (samples became lighter in color), and unchanged (no perceptible color change was observed). These qualitative visual observations were confirmed by the UV-Vis spectroscopy measurements that were taken after extracting OA at each irradiation time point.Fig. 2 shows the MAC values for two OA samples generated from the pyrolysis of drywall (Fig. 2a and b) and thin PVC wire (Fig. 2c and d). The subplots in each panel show the MAC values integrated over their respective wavelength range (280 to 400 nm for the near-UV and 400 to 700 nm for the visible range), normalized to their corresponding unaged MAC values. Both drywall OA and thin PVC wire OA show a similar decrease in the UV range integrated MAC (Fig. 2a and c), but different trends were observed in the visible range. There was an increase by a factor of 2.75 (relative to the unaged value) for the drywall OA visible integrated MAC (Fig. 2b), whereas thin PVC wire OA experienced a decrease by a factor of 1.21 compared to the unaged sample (Fig. 2d). These results confirmed visual observations that photoenhancement in the absorption coefficient occurred for drywall OA and photobleaching occurred for thin PVC wire OA (Table S3†). Similar to the results in Fig. 2, Fig. S6† shows that fiberboard OA exhibited photoenhancement (increase by a factor of 3) and carpet OA exhibited photobleaching (2 h aged sample was factor of 1.5 lower than unaged sample). In addition, three other OA samples (thick PVC wire, ceiling tile, and plywood) exhibited photoenhancement effects in the visible range (increase by factors of 1.8, 1.6, and 1.5, respectively, compared to the unaged value) upon 2 h of direct photolytic aging (Fig. S7†).
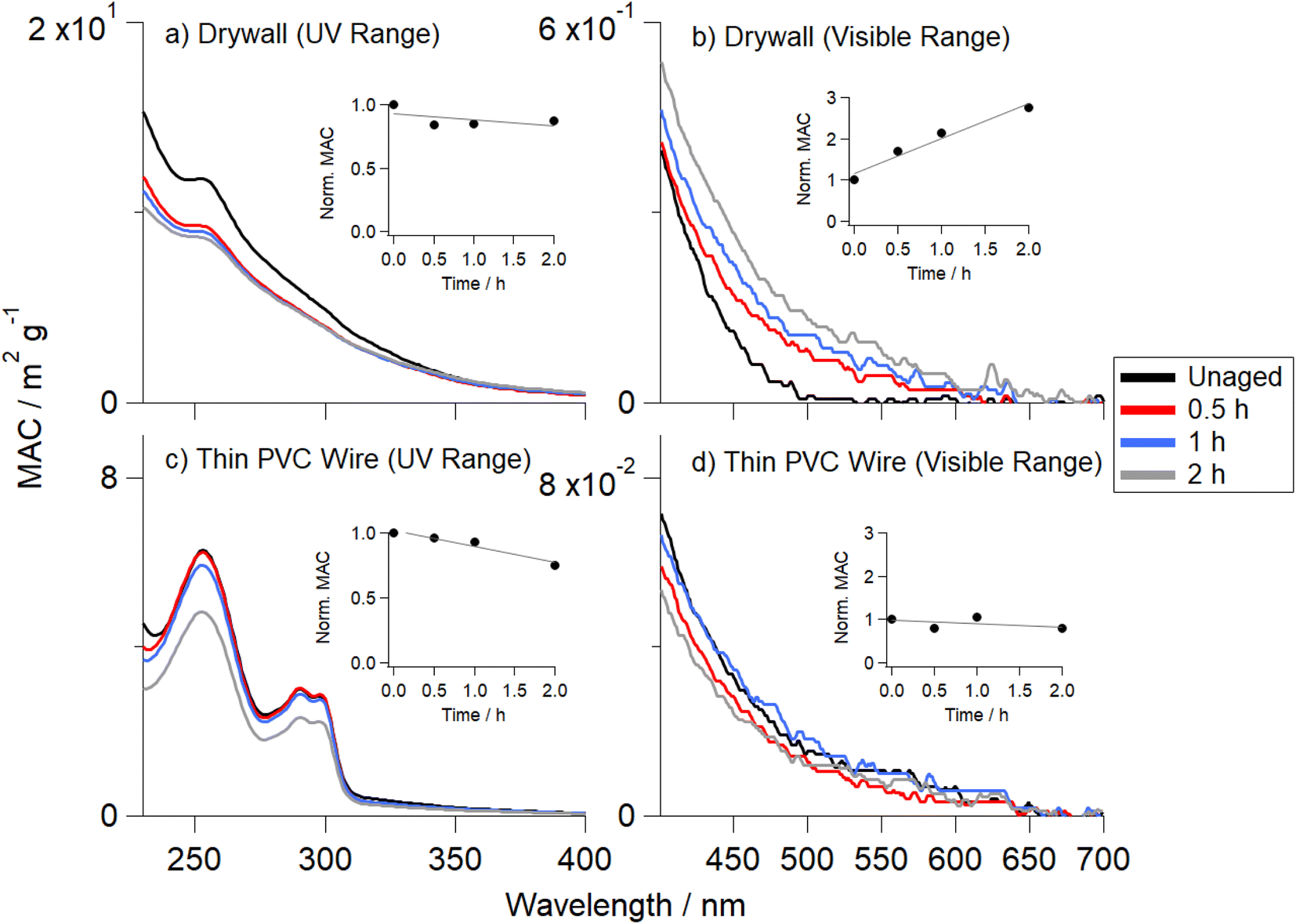 | ||
| Fig. 2 Wavelength-dependent MAC values recorded after photolysis on Teflon filters and the subsequent extractions of drywall OA (a) and (b) and thin PVC wire OA (c) and (d). Each quarter of the filter was aged separately for 0.5 h (red), 1 h (blue), or 2 h (gray) and is compared with the unaged (0 h) fraction (black). The integrated MAC values (normalized to the unaged value) are shown as a subplot in each panel, with the UV integrated over 280–400 nm and the visible integrated over 400–700 nm. Boxcar averaging of the MAC data (over a 5 nm range) was utilized to improve signal-to-noise ratio. | ||
The UV exposure not only changed the color of the filters but, in some cases, appeared to affect the solubility of the residual material. Specifically, lumber, fabric, and vinyl flooring OA could not be fully extracted from the Teflon filters after the irradiation (Fig. S8†). This complicated classification of the irradiation effects into categories of photoenhancement or photobleaching. For example, lumber OA results show a decrease in integrated MAC value by a factor of 1.02 in the visible range Teflon filter experiments (Fig. S9b†), but this result contradicted the obvious filter darkening observed visually (Table S3†). To resolve this issue, photolysis experiments for these materials were repeated using OA collected on fused silica windows after passing through an ESP (−5 kV). This collection method allowed for a more complete extraction of lumber, fabric, and vinyl flooring OA post-photolysis (Scheme S2†). While the Teflon filters collected particles >0.22 μm, the collection by ESP favored smaller particle sizes.66,67 To this end, composition of smaller particles may have enhanced the solubility of photolyzed OA. ESP experiments for lumber OA exhibited an increase in the UV by a factor of 4 and an increase in the visible by a factor of 5.5 (Fig. S9c and d†) indicating that the residual OA prevented the full representation of light-absorptive properties of the sample after photolytic aging. Fabric and vinyl flooring OA exhibited no net change (0.9 > ratio (2 h aged to unaged) < 1.1) in both the Teflon and fused silica window experiments (Fig. S10 and S11†).
Fig. 3 presents the ratios of 2 h aged to unaged MAC values integrated over the visible range for all 10 urban materials. As mentioned above, three types of trends were observed over the 2 h aging time: photobleached, unchanged, and photoenhanced. In addition to these results in the visible range, Fig. S12† shows that all but one OA sample (lumber) decreased in UV range MAC values after 2 h of photo-aging, as indicated by ratio greater than 1.1. Carpet OA and thin PVC wire OA (red region in Fig. 3) photobleached upon UV exposure, indicating chromophore fragmentation occurred more efficiently than chromophore formation. Similar results were observed by Ye et al. (2019) as they studied the aqueous-phase photochemical oxidation of a model BBOA compound, 4-ethylphenol.68 They observed degradation and fragmentation upon initial exposure to hydroxyl radical oxidation under UV light.68 Since we conducted photolytic aging experiments without intentionally added OH radical sources, we hypothesize that the observed photobleaching effects were a result of UV light induced OH radical production, leading to the degradation of chromophoric species in carpet OA and thin PVC wire OA.
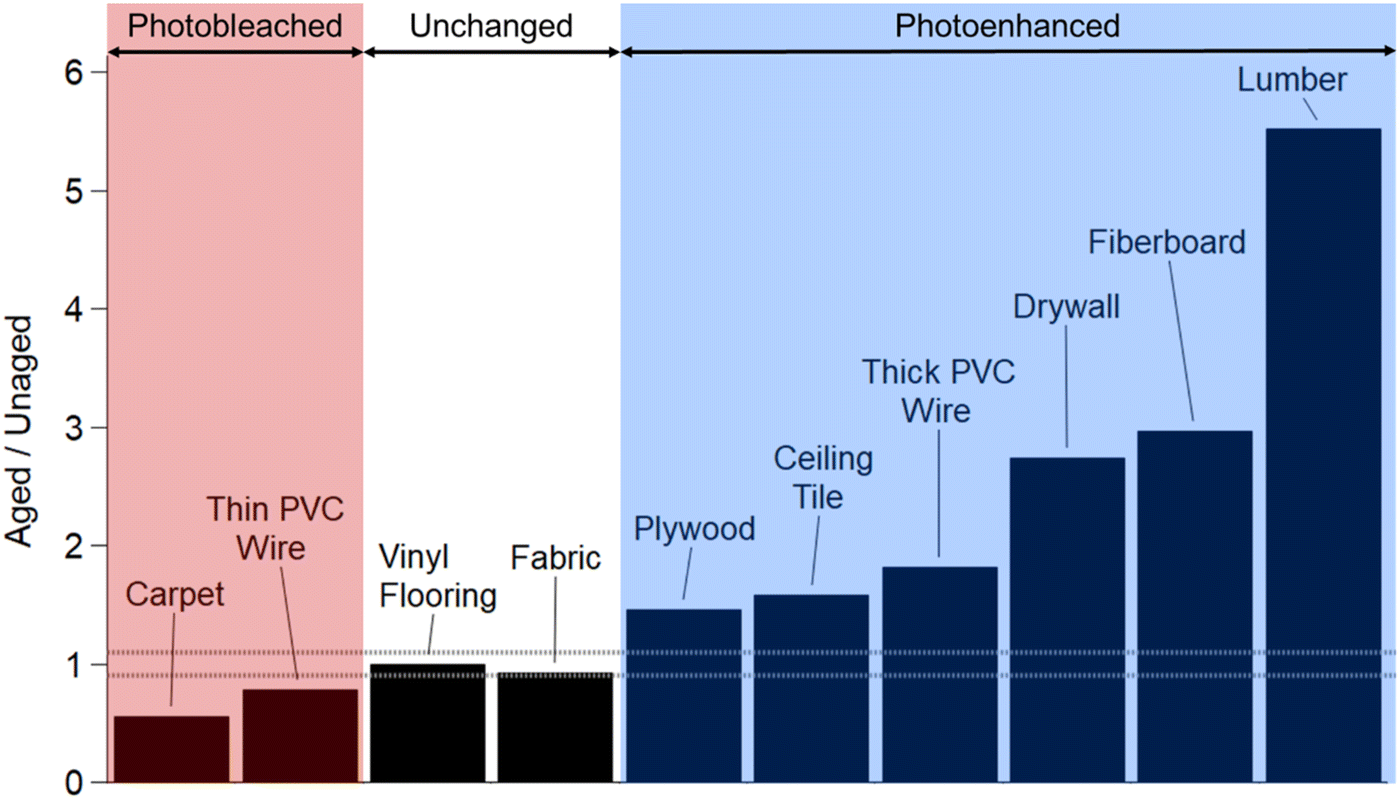 | ||
| Fig. 3 Ratios of the 2 h aged to unaged integrated, visible range MAC values. The reported ratios were calculated by integrating the MAC values in the visible range (400–700 nm) then referencing the 2 h aged, integrated MAC value to that of the unaged, integrated MAC value. The shading color corresponds to classification of OA into three categories: photobleached (red), unchanged (white) and photoenhanced (blue). The dashed line (0.9 to 1.1) indicates the threshold for the unchanged category. | ||
Six urban materials (blue region in Fig. 3) exhibited photoenhancement. We hypothesize that photochemically-driven formation of stronger chromophores occurred by free radical (OH) and triplet-excited state reactions occurring in the OA material. Such reactions have been shown to lead to the formation of chromophores in aqueous photochemistry,69–72 but they have also been reported to occur in model OA material.35,36 More specifically for model BBOA chromophores, Hems and Abbatt (2018) observed absorbance enhancement in the visible wavelength range for nitrocatechol, nitroguaiacol, and dinitrophenol due to the formation of functionalized products after exposure to a steady state concentration of OH radical.73 These studies proved that OH radical oxidation of dissolved BBOA compounds can lead to functionalized and oligomerized products with enhanced absorption in visible wavelengths.
Fabric OA and vinyl flooring OA MAC values (white region in Fig. 3) exhibited ratios greater than 0.9 but less than 1.1 when 2 h aged measurements were referenced to the unaged (Fig. S10 and S11,† respectively). Noticeably for these two materials, photoenhancement occurred in the first hour of aging then photodegradation occurred between 1 and 2 h of aging, thus returning the MAC back to the initial unaged value. Previous studies investigating BrC photolytic aging have observed similar trends. Secondary BrC has been observed to rapidly photobleach,74 whereas primary BrC from biomass burning has been observed to undergo both photoenhancement and photobleaching during the course of UV exposure, with initial photoenhancement followed by photobleaching.22,38,39 Aqueous-phase UV/OH radical exposure studies showed that product formation occurred during oxidation of organic compounds and initial increases in absorption are attributed to functionalization of chromophores.16,22,73 Over time, loss of absorption occurs when further reactivity leads to fragmentation.16,22,73,75 We observe this “dynamic” nature in the fabric OA and vinyl flooring OA cases. Further mass spectrometry analysis (at each irradiation time point) is necessary to determine the mechanism(s) of evolution.
All MAC and AAE values for the OA samples unaged (0 h) and after 2 h of photolysis (Fig. 1 and S5†) are incorporated in Fig. 4. The MAC405 of unaged OA (black markers) are compared to OA aged for 2 h (blue markers), placed into the context of the optically based BrC absorption strength classification introduced by Saleh (2020) and modified by Hettiyadura et al. (2021).62,76 Note that these MAC values were calculated using the mass of the unaged sample, and some mass loss is expected with photolysis. Therefore, reported MAC values are likely to be underestimations of the true MAC values post-photolysis. Upon photolysis, there is a downward shift in AAE values from Very Weak → Weak BrC. These results are similar to those reported in Hettiyadura et al. (2021) for fresh biomass burning BrC and biomass burning BrC that darkened with NO3 chemistry.76 The average AAE value for all ten urban OA after photolyzing for 2 h reduced from 8.8 ± 1.3 to 7.3 ± 0.9. All materials experienced a reduction in AAE value upon short term aging, as indicated by striped bars in Fig. 1b. As we are examining components commonly combusted in fires at the WUI, the average AAE shift shown in Fig. 4 reflects that WUI OA exhibits an overall absorption enhancement effect with UV aging.
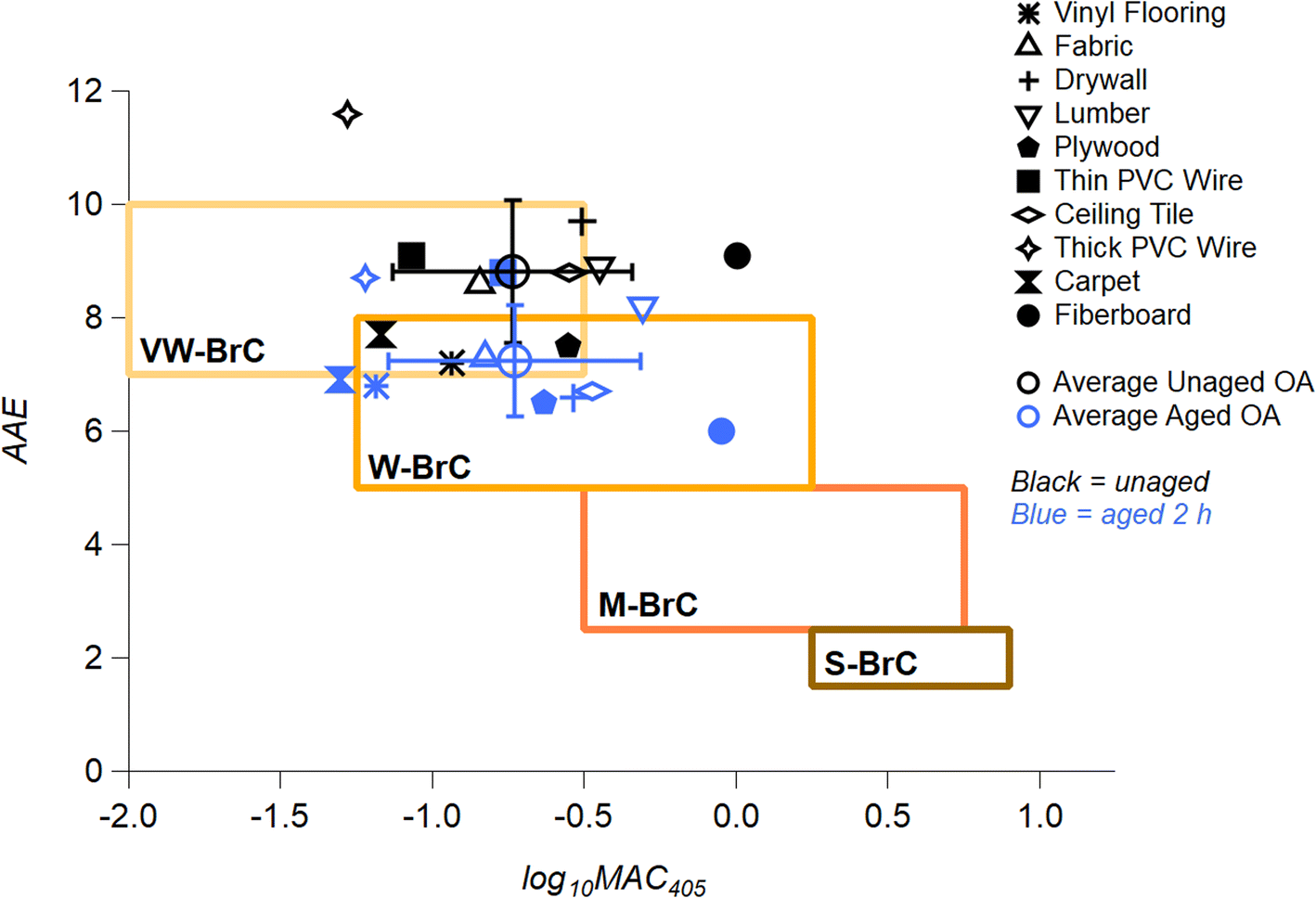 | ||
| Fig. 4 Urban OA BrC optical properties mapped in AAE vs. log10(MAC405) space following the classification introduced by Saleh (2020) and further adapted by Hettiyadura et al. (2021).62,76 Boxed areas represent very weakly (VW), weakly (W), moderately (M), and strongly (S) absorbing BrC classes. The markers correspond to the values of AAE calculated for a wavelength range of 280–480 nm for unaged urban OA (black markers) and the 2 h aged urban OA (blue markers). The global average, unaged OA (black open circle) and average, 2 h aged OA (blue open circle) are shown to illustrate a change upon photolytic aging. | ||
3.4 Compositional results of unaged and aged OA
Thin PVC wire OA is an anomaly in this data set in two ways. First, the two types of PVC wire OA (thin and thick) exhibited opposite trends: OA from thin PVC wire (purple) photobleached, whereas OA from thick PVC wire (white) photoenhanced after UV exposure. Second, thin PVC wire OA was the only material to exhibit distinct absorption bands, one at 253 nm and a doublet at 290 nm and 300 nm (Fig. 1). Through UHPLC-PDA-HRMS analysis (Fig. S13 and Appendix A†), we sought to identify the chromophore responsible for these spectral features. While we could not identify the specific chromophore whose absorption would match the distinct doublet at 290 + 300 nm, we were able to identify phthalic acid as a major contributor to the sample's overall light absorption. Phthalic acid is used in the production of PVC plasticizer, polyester resin, and dyes and is a derivative of phthalic anhydride, a common chemical intermediate in plastic production.77 Since the thin PVC wire material tested in this study was purple, it is reasonable to suspect that phthalic acid was added alongside a purple dye into the PVC coating. It is possible that the photobleaching effect exhibited by thin PVC wire OA is due to degradation of the phthalic acid chromophore by continued OH radical generation and aging.75 This (and other trends) will need to be systematically explored in future works with mass spectrometry analysis of all urban OA samples.To further assess the corresponding changes in chemical composition of OA samples after direct photolysis, we conducted TPD-DART-HRMS experiments to detect chemical transformations in three selected OA samples representative of photobleached (carpet), photoenhanced (fiberboard) and unchanged (vinyl tile) cases observed in our study (Fig. 3). The reconstructed HRMS plots of all detected organic compounds in both unaged and aged aerosols are exhibited in Fig. S14 and S15.† There were 1698/1479 (carpet), 633/379 (vinyl tile), and 956/988 (fiberboard) compounds detected in each of the unaged/aged pairs of OA samples, respectively (Fig. S14†). In each of the pairs, compounds that were affected by the photolysis experiments were selected based on the following criteria: (1) components unique to unaged or aged OA samples and (2) components that decreased or increased by >10% in the aged OA samples. There were 1036/871 (carpet), 470/206 (vinyl tile) and 185/205 (fiberboard) components that satisfied these criteria. The reconstructed HRMS plots summarizing these photolysis-affected compounds are shown in Fig. S15† and the corresponding DBE (double bond equivalent) values versus C (number of carbon atoms) plots are shown in Fig. 5.
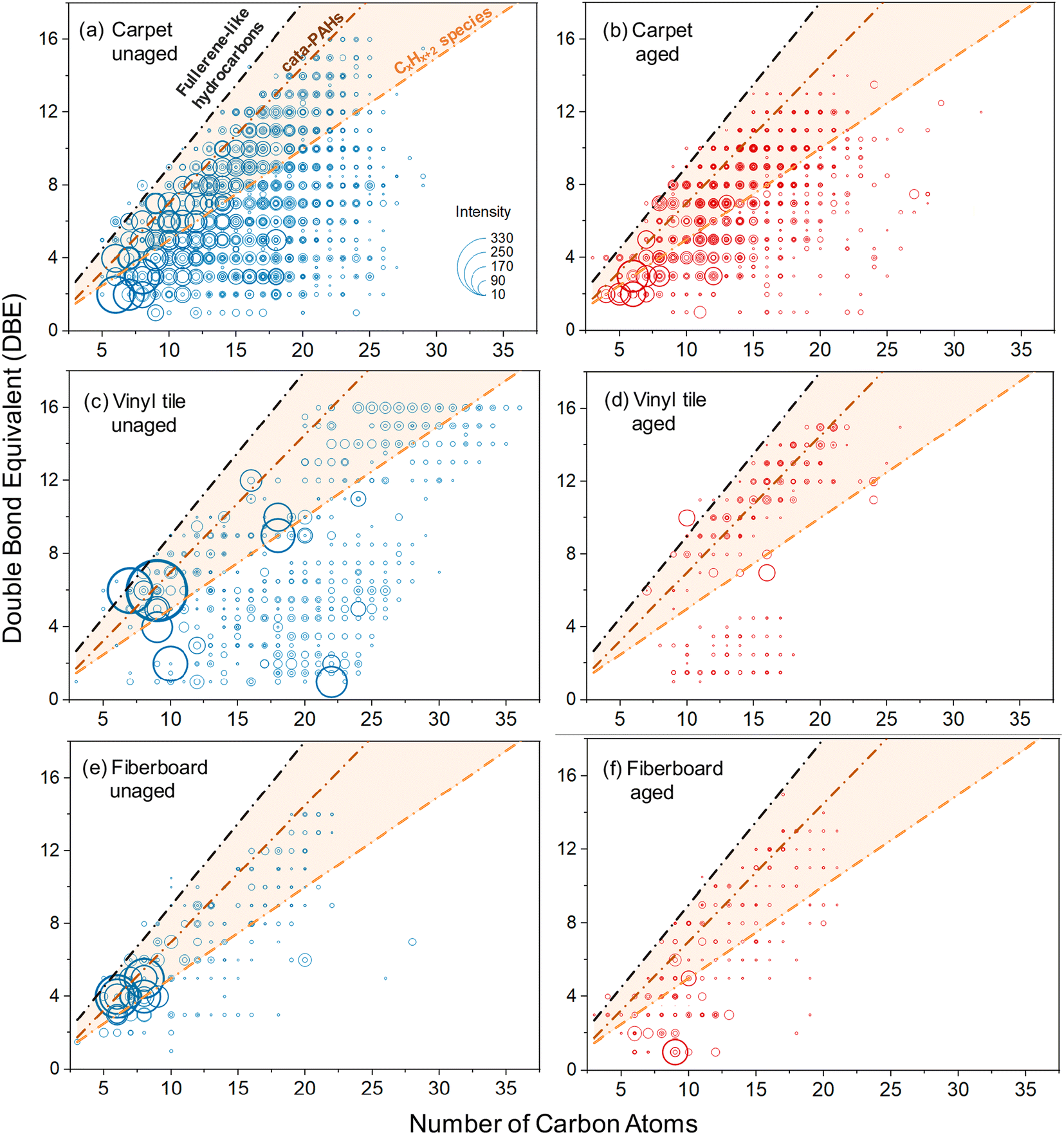 | ||
| Fig. 5 The double bond equivalent (DBE) values vs. number of carbon atoms corresponding to the OA components affected by the photolysis reactions (i.e., those that exhibited a >10% change in relative abundance) detected in unaged (left panels) and aged (right panels) OA samples collected from pyrolysis of carpet (a) and (b), vinyl tile (c) and (d), and fiberboard (e) and (f) samples. Lines indicate DBE reference values of linear conjugated polyenes CxHx+2 (orange line), cata-condensed PAHs (brown line), and fullerene-like hydrocarbons (black line).78 Data points inside the orange shaded area are potential BrC chromophores. Sizes of the individual points are arbitrarily scaled to the cubic root of the corresponding MS peaks intensities. Occasional datapoints located above the fullerene-like limiting line correspond to highly aromatic N-containing species. | ||
Because efficient absorption of visible light by organic molecules requires uninterrupted conjugation of π-bonds across significant parts of their carbon skeleton, compounds with DBE/C ratio greater than that of polyenes are potential BrC chromophores,79 which are located in the plots within shaded areas (Fig. 5). Qualitative assessment of the BrC chromophore transformations after photolysis can be inferred from visual comparison of Fig. 5 and S15† featuring differences between aged and unaged OA samples. Aged carpet, vinyl flooring, and fiberboard peaks changed by 59%, 54%, and 21%, respectively, compared to the unaged peaks. For the photobleached OA sample (carpet), both the number and intensity of BrC chromophores decreased significantly after photolysis, which is consistent with the observed and commonly expected photobleaching effect. However, more complex compositional changes are observed for the photoenhanced (fiberboard) and unchanged (vinyl tile) OA samples. In both cases, there are relatively small C6–9 BrC compounds with high intensity present in unaged samples, which largely disappear from the aged samples. On the other hand, formation of highly aromatic BrC chromophores of larger C12–18 molecular sizes is clearly observed in the corresponding aged samples. This trend can be plausibly explained by the photochemically induced oligomerization reactions that consume C6–9 monomers and form C12–18 dimer BrC products with stronger absorption properties. In both cases (fiberboard and vinyl tile OA), the overall transformation of the BrC light absorption by the aged OA samples is a combined effect of the degradation of small chromophores and the buildup of the larger chromophores. Depending on the chromophore-specific light absorbing properties, the integrated BrC absorbance of the decayed and the newly formed chromophores may show either null (vinyl tile OA) or photoenhanced (fiberboard OA) effects. These observations demonstrate that urban material BrC pyrolysis products undergo complex reaction chemistry which modifies composition of various BrC chromophores but, simultaneously, their integrated optical properties show no detectable change.
3.5 Additional direct photolysis experiments
Considering the lifetime of an aerosol in the lower atmosphere is not limited to a few hours, we performed additional experiments at longer time scales to see if the trends observed over our 2 h period would continue over longer times. To this end, one OA from each result category (photobleached, unchanged, and photoenhanced) was photolyzed for up to 18 hours (Fig. S16†). For our photobleaching material (carpet) and our unchanged material (vinyl flooring) the trends over the 18 h time scale matched those in the 2 h time scale. The visible region of the carpet OA continued to photobleach between 1 and 6 h of irradiation but the visible region of the vinyl flooring OA exhibited negligible change over the 18 h of photolysis. For both OA, the most change happened in the first 6 hours of irradiation, producing products that were no longer photochemically active.In the case of the fiberboard OA (Fig. S16e and f†), which photoenhanced during short-term irradiation, photobleaching was observed over longer time scales. It appears that this type of OA first increases in visible absorbance and then ultimately bleaches with continued photolysis. This trend mirrors work done on heterogeneous OH oxidation of particulate BrC,80 aqueous phase photo-oxidation of biomass burning BrC,16,23 and photolysis of aqueous biomass burning BrC.15,16 In the case of primary BrC generated from smoldering pine wood, heterogeneous OH oxidation resulted in absorption enhancement over the first 2.5 h (followed by photobleaching),80 which is similar to the timeframe observed here. Once again, chromophore build up may be attributed to increased oxidation through OH reactions or oligomerization through either OH or triplet excited state reactions.3,81 Future work will implement mass spectrometry to identify whether the optical changes seen in this work are attributed to OH functionalization reactions or oligomerization processes.
The experiments described above relied on the filtered Xe-lamp source for driving photochemistry in the OA material. To demonstrate that actual sunlight produces similar results, we selected drywall OA, a material that exhibited significant change with photolysis, and exposed a portion of the filter to direct sunlight and another portion to flux from our photolysis set-up (Fig. S17†). As 1 h under our lamp was estimated to be equivalent to 0.7 h in direct sunlight, the filter portion was exposed to the lamp for 2 h and the other portion was exposed to outside sunlight for 1.4 h. Integration of the UV portion of the MAC (280 to 400 nm) shows that the two filter portions exhibited similar degrees of change, within 5% (Fig. S17†).
4. Conclusions and future work
This work explored the optical properties of OA produced from the smoldering pyrolysis of flammable urban materials and their transformations during exposure to solar UV radiation. Atmospheric aging of OA (via direct photolysis) altered the optical properties of this BrC by (1) decomposing BrC chromophores (photobleaching) or (2) producing stronger chromophores from photooxidation of smaller molecules (photoenhancement).15,22,23 Plywood, ceiling tile, thick PVC coated wire, drywall, fiberboard, and lumber OA samples exhibited photoenhancement effects, whereas carpet and thin PVC coated wire OA exhibited photobleaching effects with exposure to UV light. Vinyl flooring and fabric OA remained unchanged after photolytic aging. We have observed that, on average, OA from the WUI exhibits increased absorption after the UV exposure. This means that BrC from WUI fires may have stronger effect on visibility and radiative forcing as it undergoes aging in the atmosphere.Previous work investigated the photolysis of BBOA between 0 to 16 h of irradiation and determined that photobleaching is slow due to competing aging mechanisms by heterogeneous OH oxidation.21 Photolytic aging of BrC from BBOA has also been seen to have significant OH concentrations in OA extracts when no additional OH source was added.16 In the present study, we observed significant absorption enhancement of OA from the WUI over the first few hours of photolysis, indirectly suggesting that OH (and likely other free radicals) are produced in the organic phase of the particles during irradiation. To understand the chemical changes responsible for our optical observations, identification and structural characterization of WUI BrC chromophores (before and after photolysis) are needed to determine the mechanism in which this OA transforms and identify if it evolves similarly to BrC from BBOA.
Author contributions
K. S. H., A. L. K., and M. A. A. conducted pyrolysis and photolysis experiments. Q. X. conducted experiments and write up of TPD-DART-HRMS results. K. S. H. conducted UHPLC-PDA-HRMS experiments, data analysis, and wrote the manuscript. A. L. K. assisted in manuscript writing. S. A. N. and A. L. designed the experiments, assisted in interpretation of results, and manuscript co-editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Oceanic and Atmospheric Administration grants NA22OAR4310196 (University of California, Irvine) and NA22OAR4310195 (Purdue University). The photolysis experiments were done with support from National Science Foundation grant NSF AGS-1853639 (University of California, Irvine).References
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. DeAngelo, M. G. Flanner, S. Ghan, B. Kärcher, D. Koch, S. Kinne, Y. Kondo, P. K. Quinn, M. C. Sarofim, M. G. Schultz, M. Schulz, C. Venkataraman, H. Zhang, S. Zhang, N. Bellouin, S. K. Guttikunda, P. K. Hopke, M. Z. Jacobson, J. W. Kaiser, Z. Klimont, U. Lohmann, J. P. Schwarz, D. Shindell, T. Storelvmo, S. G. Warren and C. S. Zender, Bounding the Role of Black Carbon in the Climate System: A Scientific Assessment, J. Geophys. Res.: Atmos., 2013, 118(11), 5380–5552, DOI:10.1002/jgrd.50171.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Chemistry of Atmospheric Brown Carbon, Chem. Rev., 2015, 115(10), 4335–4382, DOI:10.1021/cr5006167.
- R. F. Hems, E. G. Schnitzler, C. Liu-Kang, C. D. Cappa and J. P. D. Abbatt, Aging of Atmospheric Brown Carbon Aerosol, ACS Earth Space Chem., 2021, 5(4), 722–748, DOI:10.1021/acsearthspacechem.0c00346.
- M. O. Andreae and A. Gelencser, Black Carbon or Brown Carbon? The Nature of Light-Absorbing Carbonaceous Aerosols, Atmos. Chem. Phys., 2006, 18 Search PubMed.
- R. A. Washenfelder, A. R. Attwood, C. A. Brock, H. Guo, L. Xu, R. J. Weber, N. L. Ng, H. M. Allen, B. R. Ayres, K. Baumann, R. C. Cohen, D. C. Draper, K. C. Duffey, E. Edgerton, J. L. Fry, W. W. Hu, J. L. Jimenez, B. B. Palm, P. Romer, E. A. Stone, P. J. Wooldridge and S. S. Brown, Biomass Burning Dominates Brown Carbon Absorption in the Rural Southeastern United States, Geophys. Res. Lett., 2015, 42(2), 653–664, DOI:10.1002/2014GL062444.
- V. Ramanathan, F. Li, M. V. Ramana, P. S. Praveen, D. Kim, C. E. Corrigan, H. Nguyen, E. A. Stone, J. J. Schauer, G. R. Carmichael, B. Adhikary and S. C. Yoon, Atmospheric Brown Clouds: Hemispherical and Regional Variations in Long-Range Transport, Absorption, and Radiative Forcing, J. Geophys. Res.: Atmos., 2007, 112, D22S21, DOI:10.1029/2006JD008124.
- P. Lin, N. Bluvshtein, Y. Rudich, S. A. Nizkorodov, J. Laskin and A. Laskin, Molecular Chemistry of Atmospheric Brown Carbon Inferred from a Nationwide Biomass Burning Event, Environ. Sci. Technol., 2017, 51(20), 11561–11570, DOI:10.1021/acs.est.7b02276.
- Y. Chen and T. C. Bond, Light Absorption by Organic Carbon from Wood Combustion, Atmos. Chem. Phys., 2010, 10(4), 1773–1787, DOI:10.5194/acp-10-1773-2010.
- S. H. Budisulistiorini, M. Riva, M. Williams, J. Chen, M. Itoh, J. D. Surratt and M. Kuwata, Light-Absorbing Brown Carbon Aerosol Constituents from Combustion of Indonesian Peat and Biomass, Environ. Sci. Technol., 2017, 51(8), 4415–4423, DOI:10.1021/acs.est.7b00397.
- L. T. Fleming, P. Lin, A. Laskin, J. Laskin, R. Weltman, R. D. Edwards, N. K. Arora, A. Yadav, S. Meinardi, D. R. Blake, A. Pillarisetti, K. R. Smith and S. A. Nizkorodov, Molecular Composition of Particulate Matter Emissions from Dung and Brushwood Burning Household Cookstoves in Haryana, India, Atmos. Chem. Phys., 2018, 18(4), 2461–2480, DOI:10.5194/acp-18-2461-2018.
- T. W. Kirchstetter, T. Novakov and P. V. Hobbs, Evidence That the Spectral Dependence of Light Absorption by Aerosols Is Affected by Organic Carbon, J. Geophys. Res.: Atmos., 2004, 109, D21208, DOI:10.1029/2004JD004999.
- L. V. Rizzo, A. L. Correia, P. Artaxo, A. S. Procópio and M. O. Andreae, Spectral Dependence of Aerosol Light Absorption over the Amazon Basin, Atmos. Chem. Phys., 2011, 11(17), 8899–8912, DOI:10.5194/acp-11-8899-2011.
- X. Zhang, Y.-H. Lin, J. D. Surratt and R. J. Weber, Sources, Composition and Absorption Ångström Exponent of Light-Absorbing Organic Components in Aerosol Extracts from the Los Angeles Basin, Environ. Sci. Technol., 2013, 47(8), 3685–3693, DOI:10.1021/es305047b.
- P. Corral Arroyo, T. Bartels-Rausch, P. A. Alpert, S. Dumas, S. Perrier, C. George and M. Ammann, Particle-Phase Photosensitized Radical Production and Aerosol Aging, Environ. Sci. Technol., 2018, 52(14), 7680–7688, DOI:10.1021/acs.est.8b00329.
- J. P. S. Wong, A. Nenes and R. J. Weber, Changes in Light Absorptivity of Molecular Weight Separated Brown Carbon Due to Photolytic Aging, Environ. Sci. Technol., 2017, 51(15), 8414–8421, DOI:10.1021/acs.est.7b01739.
- J. P. S. Wong, M. Tsagkaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes and R. J. Weber, Atmospheric Evolution of Molecular-Weight-Separated Brown Carbon from Biomass Burning, Atmos. Chem. Phys., 2019, 19(11), 7319–7334, DOI:10.5194/acp-19-7319-2019.
- P. Lin, P. K. Aiona, Y. Li, M. Shiraiwa, J. Laskin, S. A. Nizkorodov and A. Laskin, Molecular Characterization of Brown Carbon in Biomass Burning Aerosol Particles, Environ. Sci. Technol., 2016, 50(21), 11815–11824, DOI:10.1021/acs.est.6b03024.
- P. K. Aiona, J. L. Luek, S. A. Timko, L. C. Powers, M. Gonsior and S. A. Nizkorodov, Effect of Photolysis on Absorption and Fluorescence Spectra of Light-Absorbing Secondary Organic Aerosols, ACS Earth Space Chem., 2018, 2(3), 235–245, DOI:10.1021/acsearthspacechem.7b00153.
- J. H. Kroll, J. D. Smith, D. L. Che, S. H. Kessler, D. R. Worsnop and K. R. Wilson, Measurement of Fragmentation and Functionalization Pathways in the Heterogeneous Oxidation of Oxidized Organic Aerosol, Phys. Chem. Chem. Phys., 2009, 11(36), 8005, 10.1039/b905289e.
- H. J. J. Lee, P. K. Aiona, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of Solar Radiation on the Optical Properties and Molecular Composition of Laboratory Proxies of Atmospheric Brown Carbon, Environ. Sci. Technol., 2014, 48(17), 10217–10226, DOI:10.1021/es502515r.
- L. T. Fleming, P. Lin, J. M. Roberts, V. Selimovic, R. Yokelson, J. Laskin, A. Laskin and S. A. Nizkorodov, Molecular Composition and Photochemical Lifetimes of Brown Carbon Chromophores in Biomass Burning Organic Aerosol, Atmos. Chem. Phys., 2020, 20(2), 1105–1129, DOI:10.5194/acp-20-1105-2020.
- R. Zhao, A. K. Y. Lee, L. Huang, X. Li, F. Yang and J. P. D. Abbatt, Photochemical Processing of Aqueous Atmospheric Brown Carbon, Atmos. Chem. Phys., 2015, 15(11), 6087–6100, DOI:10.5194/acp-15-6087-2015.
- R. F. Hems, E. G. Schnitzler, M. Bastawrous, R. Soong, A. J. Simpson and J. P. D. Abbatt, Aqueous Photoreactions of Wood Smoke Brown Carbon, ACS Earth Space Chem., 2020, 4(7), 1149–1160, DOI:10.1021/acsearthspacechem.0c00117.
- H. R. Pruppacher and R. Jaenicke, The Processing of Water Vapor and Aerosols by Atmospheric Clouds, a Global Estimate, Atmos. Res., 1995, 38(1), 283–295, DOI:10.1016/0169-8095(94)00098-X.
- M. L. Walser, J. Park, A. L. Gomez, A. R. Russell and S. A. Nizkorodov, Photochemical Aging of Secondary Organic Aerosol Particles Generated from the Oxidation of D-Limonene, J. Phys. Chem. A, 2007, 111(10), 1907–1913, DOI:10.1021/jp066293l.
- S. A. Mang, D. K. Henricksen, A. P. Bateman, M. P. S. Andersen, D. R. Blake and S. A. Nizkorodov, Contribution of Carbonyl Photochemistry to Aging of Atmospheric Secondary Organic Aerosol, J. Phys. Chem. A, 2008, 112(36), 8337–8344, DOI:10.1021/jp804376c.
- X. Pan, J. S. Underwood, J.-H. Xing, S. A. Mang and S. A. Nizkorodov, Photodegradation of Secondary Organic Aerosol Generated from Limonene Oxidation by Ozone Studied with Chemical Ionization Mass Spectrometry, Atmos. Chem. Phys., 2009, 9(12), 3851–3865, DOI:10.5194/acp-9-3851-2009.
- K. M. Henry and N. M. Donahue, Photochemical Aging of α-Pinene Secondary Organic Aerosol: Effects of OH Radical Sources and Photolysis, J. Phys. Chem. A, 2012, 116(24), 5932–5940, DOI:10.1021/jp210288s.
- K. T. Malecha, Z. Cai and S. A. Nizkorodov, Photodegradation of Secondary Organic Aerosol Material Quantified with a Quartz Crystal Microbalance, Environ. Sci. Technol. Lett., 2018, 5(6), 366–371, DOI:10.1021/acs.estlett.8b00231.
- V. J. Baboomian, Y. Gu and S. A. Nizkorodov, Photodegradation of Secondary Organic Aerosols by Long-Term Exposure to Solar Actinic Radiation, ACS Earth Space Chem., 2020, 4(7), 1078–1089, DOI:10.1021/acsearthspacechem.0c00088.
- H.-M. Hung, Y.-Q. Chen and S. T. Martin, Reactive Aging of Films of Secondary Organic Material Studied by Infrared Spectroscopy, J. Phys. Chem. A, 2013, 117(1), 108–116, DOI:10.1021/jp309470z.
- S. A. Epstein, S. L. Blair and S. A. Nizkorodov, Direct Photolysis of α-Pinene Ozonolysis Secondary Organic Aerosol: Effect on Particle Mass and Peroxide Content, Environ. Sci. Technol., 2014, 48(19), 11251–11258, DOI:10.1021/es502350u.
- K. M. Badali, S. Zhou, D. Aljawhary, M. Antiñolo, W. J. Chen, A. Lok, E. Mungall, J. P. S. Wong, R. Zhao and J. P. D. Abbatt, Formation of Hydroxyl Radicals from Photolysis of Secondary Organic Aerosol Material, Atmos. Chem. Phys., 2015, 15(14), 7831–7840, DOI:10.5194/acp-15-7831-2015.
- M. Krapf, I. El Haddad, E. A. Bruns, U. Molteni, K. R. Daellenbach, A. S. H. Prévôt, U. Baltensperger and J. Dommen, Labile Peroxides in Secondary Organic Aerosol, Chem, 2016, 1(4), 603–616, DOI:10.1016/j.chempr.2016.09.007.
- M. L. Hinks, M. V. Brady, H. Lignell, M. Song, J. W. Grayson, A. K. Bertram, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of Viscosity on Photodegradation Rates in Complex Secondary Organic Aerosol Materials, Phys. Chem. Chem. Phys., 2016, 18(13), 8785–8793, 10.1039/C5CP05226B.
- A. B. Dalton and S. A. Nizkorodov, Photochemical Degradation of 4-Nitrocatechol and 2,4-Dinitrophenol in a Sugar-Glass Secondary Organic Aerosol Surrogate, Environ. Sci. Technol., 2021, 55(21), 14586–14594, DOI:10.1021/acs.est.1c04975.
- A. L. Klodt, M. Adamek, M. Dibley, S. A. Nizkorodov and R. E. O'Brien, Effects of the Sample Matrix on the Photobleaching and Photodegradation of Toluene-Derived Secondary Organic Aerosol Compounds, Atmos. Chem. Phys., 2022, 22(15), 10155–10171, DOI:10.5194/acp-22-10155-2022.
- R. Saleh, C. J. Hennigan, G. R. McMeeking, W. K. Chuang, E. S. Robinson, H. Coe, N. M. Donahue and A. L. Robinson, Absorptivity of Brown Carbon in Fresh and Photo-Chemically Aged Biomass-Burning Emissions, Atmos. Chem. Phys., 2013, 13(15), 7683–7693, DOI:10.5194/acp-13-7683-2013.
- M. Zhong and M. Jang, Dynamic Light Absorption of Biomass-Burning Organic Carbon Photochemically Aged under Natural Sunlight, Atmos. Chem. Phys., 2014, 14(3), 1517–1525, DOI:10.5194/acp-14-1517-2014.
- B. J. Sumlin, A. Pandey, M. J. Walker, R. S. Pattison, B. J. Williams and R. K. Chakrabarty, Atmospheric Photooxidation Diminishes Light Absorption by Primary Brown Carbon Aerosol from Biomass Burning, Environ. Sci. Technol. Lett., 2017, 4(12), 540–545, DOI:10.1021/acs.estlett.7b00393.
- D. E. Calkin, J. D. Cohen, M. A. Finney and M. P. Thompson, How Risk Management Can Prevent Future Wildfire Disasters in the Wildland-Urban Interface, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(2), 746–751, DOI:10.1073/pnas.1315088111.
- J. B. Davis, The Wildland-Urban Interface: Paradise or Battleground?, J. For., 1990, 88(1), 26–31 Search PubMed.
- V. C. Radeloff, R. B. Hammer, S. I. Stewart, J. S. Fried, S. S. Holcomb and J. F. McKeefry, The Wildland–Urban Interface in the United States, Ecological Applications, 2005, 15(3), 799–805, DOI:10.1890/04-1413.
- M. Burke, A. Driscoll, S. Heft-Neal, J. Xue, J. Burney and M. Wara, The Changing Risk and Burden of Wildfire in the United States, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(2), e2011048118, DOI:10.1073/pnas.2011048118.
- R. Barbero, J. T. Abatzoglou, N. K. Larkin, C. A. Kolden, B. Stocks, R. Barbero, J. T. Abatzoglou, N. K. Larkin, C. A. Kolden and B. Stocks, Climate Change Presents Increased Potential for Very Large Fires in the Contiguous United States, Int. J. Wildland Fire, 2015, 24(7), 892–899, DOI:10.1071/WF15083.
- M. Goss, D. L. Swain, J. T. Abatzoglou, A. Sarhadi, C. A. Kolden, A. P. Williams and N. S. Diffenbaugh, Climate Change Is Increasing the Likelihood of Extreme Autumn Wildfire Conditions across California, Environ. Res. Lett., 2020, 15(9), 094016, DOI:10.1088/1748-9326/ab83a7.
- P. Blomqvist, B. Persson and M. McNamee, Fire Emissions of Organics into the Atmosphere, Fire Technol., 2007, 43, 213–231, DOI:10.1007/s10694-007-0011-y.
- F. Reisen, M. Bhujel and J. Leonard, Particle and Volatile Organic Emissions from the Combustion of a Range of Building and Furnishing Materials Using a Cone Calorimeter, Fire Saf. J., 2014, 69, 76–88, DOI:10.1016/j.firesaf.2014.08.008.
- F. Hewitt, A. Christou, K. Dickens, R. Walker and A. A. Stec, Release of Volatile and Semi-Volatile Toxicants during House Fires, Chemosphere, 2017, 173, 580–593, DOI:10.1016/j.chemosphere.2016.12.079.
- K. S. Hopstock, B. P. Carpenter, J. P. Patterson, H. A. Al-Abadleh and S. A. Nizkorodov, Formation of Insoluble Brown Carbon through Iron-Catalyzed Reaction of Biomass Burning Organics, Environ. Sci.: Atmos., 2023, 3, 207–220, 10.1039/D2EA00141A.
- M. Devi, S. Rawat and S. Sharma, A Comprehensive Review of the Pyrolysis Process: From Carbon Nanomaterial Synthesis to Waste Treatment, Oxford Open Materials Science, 2021, 1(1), itab014, DOI:10.1093/oxfmat/itab014.
- M. A. Santoso, E. G. Christensen, J. Yang and G. Rein, Review of the Transition from Smouldering to Flaming Combustion in Wildfires, Front. Mech. Eng., 2019, 5, 49, DOI:10.3389/fmech.2019.00049.
- S. Maldronich, ACOM: Quick TUV Calculator, National Center for Atmospheric Research Atmospheric Chemistry Observations and Modeling, https://www.acom.ucar.edu/Models/TUV/Interactive_TUV/, accessed 2022-11-03 Search PubMed.
- T. C. Bond, T. L. Anderson and D. Campbell, Calibration and Intercomparison of Filter-Based Measurements of Visible Light Absorption by Aerosols, Aerosol Sci. Technol., 1999, 30(6), 582–600, DOI:10.1080/027868299304435.
- D. A. Lack, C. D. Cappa, D. S. Covert, T. Baynard, P. Massoli, B. Sierau, T. S. Bates, P. K. Quinn, E. R. Lovejoy and A. R. Ravishankara, Bias in Filter-Based Aerosol Light Absorption Measurements Due to Organic Aerosol Loading: Evidence from Ambient Measurements, Aerosol Sci. Technol., 2008, 42(12), 1033–1041, DOI:10.1080/02786820802389277.
- R. E. O'Brien and J. H. Kroll, Photolytic Aging of Secondary Organic Aerosol: Evidence for a Substantial Photo-Recalcitrant Fraction, J. Phys. Chem. Lett., 2019, 10(14), 4003–4009, DOI:10.1021/acs.jpclett.9b01417.
- C. P. West, Y.-J. Hsu, K. T. MacFeely, S. M. Huston, B. P. Aridjis-Olivos, A. C. Morales and A. Laskin, Volatility Measurements of Individual Components in Organic Aerosol Mixtures Using Temperature-Programmed Desorption–Direct Analysis in Real Time–High Resolution Mass Spectrometry, Anal. Chem., 2023, 95(19), 7403–7408, DOI:10.1021/acs.analchem.3c00923.
- P. J. Roach, J. Laskin and A. Laskin, Higher-Order Mass Defect Analysis for Mass Spectra of Complex Organic Mixtures, Anal. Chem., 2011, 83(12), 4924–4929, DOI:10.1021/ac200654j.
- P. Blomqvist, M. S. McNamee, A. A. Stec, D. Gylestam and D. Karlsson, Detailed Study of Distribution Patterns of Polycyclic Aromatic Hydrocarbons and Isocyanates under Different Fire Conditions, Fire Mater., 2014, 38(1), 125–144, DOI:10.1002/fam.2173.
- Z. Xu, W. Feng, Y. Wang, H. Ye, Y. Wang, H. Liao and M. Xie, Potential Underestimation of Ambient Brown Carbon Absorption Based on the Methanol Extraction Method and Its Impacts on Source Analysis, Atmos. Chem. Phys., 2022, 22(20), 13739–13752, DOI:10.5194/acp-22-13739-2022.
- D. Sengupta, V. Samburova, C. Bhattarai, E. Kirillova, L. Mazzoleni, M. Iaukea-Lum, A. Watts, H. Moosmüller and A. Khlystov, Light Absorption by Polar and Non-Polar Aerosol Compounds from Laboratory Biomass Combustion, Atmos. Chem. Phys., 2018, 18(15), 10849–10867, DOI:10.5194/acp-18-10849-2018.
- R. Saleh, From Measurements to Models: Toward Accurate Representation of Brown Carbon in Climate Calculations, Curr. Pollut. Rep., 2020, 6(2), 90–104, DOI:10.1007/s40726-020-00139-3.
- T. Moise, J. M. Flores and Y. Rudich, Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes, Chem. Rev., 2015, 115(10), 4400–4439, DOI:10.1021/cr5005259.
- H. Chin, K. S. Hopstock, L. T. Fleming, S. A. Nizkorodov and H. A. Al-Abadleh, Effect of Aromatic Ring Substituents on the Ability of Catechol to Produce Brown Carbon in Iron(III)-Catalyzed Reactions, Environ. Sci.: Atmos., 2021, 1(2), 64–78, 10.1039/D0EA00007H.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, Wiley-Interscience; Wiley, New York, 2nd edn, 2003 Search PubMed.
- J. Leskinen, J. Tissari, O. Uski, A. Virén, T. Torvela, T. Kaivosoja, H. Lamberg, I. Nuutinen, T. Kettunen, J. Joutsensaari, P. I. Jalava, O. Sippula, M.-R. Hirvonen and J. Jokiniemi, Fine Particle Emissions in Three Different Combustion Conditions of a Wood Chip-Fired Appliance – Particulate Physico-Chemical Properties and Induced Cell Death, Atmos. Environ., 2014, 86, 129–139, DOI:10.1016/j.atmosenv.2013.12.012.
- O. Molchanov, K. Krpec, J. Horák, L. Kuboňová and F. Hopan, Comparison of Methods for Evaluating Particle Charges in the Electrostatic Precipitation of Fly-Ash from Small-Scale Solid Fuel Combustion, Sep. Purif. Technol., 2020, 248, 117057, DOI:10.1016/j.seppur.2020.117057.
- Z. Ye, Z. Qu, S. Ma, S. Luo, Y. Chen, H. Chen, Y. Chen, Z. Zhao, M. Chen and X. Ge, A Comprehensive Investigation of Aqueous-Phase Photochemical Oxidation of 4-Ethylphenol, Sci. Total Environ., 2019, 685, 976–985, DOI:10.1016/j.scitotenv.2019.06.276.
- J. Cai, X. Zeng, G. Zhi, S. Gligorovski, G. Sheng, Z. Yu, X. Wang and P. Peng, Molecular Composition and Photochemical Evolution of Water-Soluble Organic Carbon (WSOC) Extracted from Field Biomass Burning Aerosols Using High-Resolution Mass Spectrometry, Atmos. Chem. Phys., 2020, 20(10), 6115–6128, DOI:10.5194/acp-20-6115-2020.
- M. Mekic, J. Liu, W. Zhou, G. Loisel, J. Cai, T. He, B. Jiang, Z. Yu, Y. G. Lazarou, X. Li, M. Brigante, D. Vione and S. Gligorovski, Formation of Highly Oxygenated Multifunctional Compounds from Cross-Reactions of Carbonyl Compounds in the Atmospheric Aqueous Phase, Atmos. Environ., 2019, 219, 117046, DOI:10.1016/j.atmosenv.2019.117046.
- Z. Mu, Q. Chen, L. Zhang, D. Guan and H. Li, Photodegradation of Atmospheric Chromophores: Changes in Oxidation State and Photochemical Reactivity, Atmos. Chem. Phys., 2021, 21(15), 11581–11591, DOI:10.5194/acp-21-11581-2021.
- W. Jiang, M. V. Misovich, A. P. S. Hettiyadura, A. Laskin, A. S. McFall, C. Anastasio and Q. Zhang, Photosensitized Reactions of a Phenolic Carbonyl from Wood Combustion in the Aqueous Phase—Chemical Evolution and Light Absorption Properties of AqSOA, Environ. Sci. Technol., 2021, 55(8), 5199–5211, DOI:10.1021/acs.est.0c07581.
- R. F. Hems and J. P. D. Abbatt, Aqueous Phase Photo-Oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Space Chem., 2018, 2(3), 225–234, DOI:10.1021/acsearthspacechem.7b00123.
- N. Sareen, S. G. Moussa and V. F. McNeill, Photochemical Aging of Light-Absorbing Secondary Organic Aerosol Material, J. Phys. Chem. A, 2013, 117(14), 2987–2996, DOI:10.1021/jp309413j.
- Z. Ye, Y. Zhuang, Y. Chen, Z. Zhao, S. Ma, H. Huang, Y. Chen and X. Ge, Aqueous-Phase Oxidation of Three Phenolic Compounds by Hydroxyl Radical: Insight into Secondary Organic Aerosol Formation Yields, Mechanisms, Products and Optical Properties, Atmos. Environ., 2020, 223, 117240, DOI:10.1016/j.atmosenv.2019.117240.
- A. P. S. Hettiyadura, V. Garcia, C. Li, C. P. West, J. Tomlin, Q. He, Y. Rudich and A. Laskin, Chemical Composition and Molecular-Specific Optical Properties of Atmospheric Brown Carbon Associated with Biomass Burning, Environ. Sci. Technol., 2021, 55(4), 2511–2521, DOI:10.1021/acs.est.0c05883.
- B.-Z. Ren, C.-H. Hou, H.-G. Chong, W.-R. Li and H.-J. Song, Solubility of O-Phthalic Acid in Methanol + Water and Methanol + Butyl Acetate from (295.87 to 359.75) K, J. Chem. Eng. Data, 2006, 51(6), 2022–2025, DOI:10.1021/je0600505.
- J. Cain, A. Laskin, M. R. Kholghy, M. J. Thomson and H. Wang, Molecular Characterization of Organic Content of Soot along the Centerline of a Coflow Diffusion Flame, Phys. Chem. Chem. Phys., 2014, 16(47), 25862–25875, 10.1039/C4CP03330B.
- P. Lin, L. T. Fleming, S. A. Nizkorodov, J. Laskin and A. Laskin, Comprehensive Molecular Characterization of Atmospheric Brown Carbon by High Resolution Mass Spectrometry with Electrospray and Atmospheric Pressure Photoionization, Anal. Chem., 2018, 90(21), 12493–12502, DOI:10.1021/acs.analchem.8b02177.
- E. G. Schnitzler, T. Liu, R. F. Hems and J. P. D. Abbatt, Emerging Investigator Series: Heterogeneous OH Oxidation of Primary Brown Carbon Aerosol: Effects of Relative Humidity and Volatility, Environ. Sci.: Processes Impacts, 2020, 22(11), 2162–2171, 10.1039/D0EM00311E.
- L. Yu, J. Smith, A. Laskin, C. Anastasio, J. Laskin and Q. Zhang, Chemical Characterization of SOA Formed from Aqueous-Phase Reactions of Phenols with the Triplet Excited State of Carbonyl and Hydroxyl Radical, Atmos. Chem. Phys., 2014, 14(24), 13801–13816, DOI:10.5194/acp-14-13801-2014.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00078h |
| This journal is © The Royal Society of Chemistry 2023 |