
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Different chlorine and hydroxyl radical environments impact m-xylene oxidation products†
Nirvan
Bhattacharyya
 ,
Mrinali
Modi
,
Leif G.
Jahn
and
Lea
Hildebrandt Ruiz
*
,
Mrinali
Modi
,
Leif G.
Jahn
and
Lea
Hildebrandt Ruiz
*
McKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: lhr@che.utexas.edu
First published on 30th May 2023
Abstract
Airborne emissions of aromatic hydrocarbons including benzene, ethylbenzene, toluene, and xylenes are associated with anthropogenic activities (e.g. transportation) and form secondary organic aerosol (SOA) when oxidized. While hydroxyl radicals (OH) dominate oxidation, chlorine radicals (Cl) react with alkyl substituted aromatics more rapidly and favor a different oxidative pathway. High concentrations of reactive chlorine species have been observed in continental and coastal regions, where mixed Cl/OH chemistry is expected to influence regional SOA formation and composition. This study uses environmental chamber experiments to assess SOA formation and composition from the oxidation of m-xylene in mixed Cl/OH oxidation environments. Experiments were conducted with hydrogen peroxide (H2O2), chlorine (Cl2), and nitryl chloride (ClNO2) radical precursors under high and low NOx conditions. Data was collected with an Aerosol Chemical Speciation Monitor (ACSM), Scanning Electrical Mobility System (SEMS) and time of flight chemical ionization mass spectrometer with filter desorption (FIGAERO-CIMS) utilizing H3O+ and I− reagent ions. Different oxidative pathways in H2O2 and Cl2 experiments resulted in bicyclic peroxide and methylbenzoquinone species, respectively. When Cl2 was the sole radical precursor, SOA was more highly oxidized and less fragmented. ClNO2 experiments formed substantial amounts of bicyclic peroxide and minimal methylbenzoquinone in the gas phase and less oxidized SOA with a lower fraction of organochlorides. These differences are related to secondary OH formation and slower ClNO2 photolysis driving lower Cl radical concentrations. This study provides evidence that gas and particle-phase products vary depending on the oxidative environment and underlines the importance of studying novel oxidants and oxidative pathways.
Environmental significanceAromatic hydrocarbon oxidation forms atmospheric aerosols which impact health and climate. Previous environmental chamber studies focus on OH radical oxidation, but Cl radicals react faster with alkyl substituted aromatics along an alternate reaction pathway. Experiments with H2O2, Cl2, and ClNO2 examine gas phase chemistry, particle formation, and product composition from oxidation of m-xylene. We demonstrate OH and Cl oxidation results in different products. More highly oxidized organic aerosol is formed in experiments when only Cl2 is a radical precursor. Using ClNO2 as a radical precursor results in different chemistry and product distributions than Cl2 + NOx, indicating ClNO2-laden air reacts differently than Cl2 emissions in high NOx environments. Exploration of radical precursors is required to constrain Cl oxidation impacts. |
1 Introduction
Aromatic hydrocarbons are a substantial fraction of volatile organic compound (VOC) emissions globally and regionally.1–3 In urban environments, small aromatics including benzene, toluene, ethylbenzene, and xylenes (BTEX) often dominate observed VOCs, and their main source is thought to be transportation.4,5 Atmospheric oxidation of these species leads to substantial formation of ozone, oxidized volatile organic compounds (OVOCs),6,7 and secondary organic aerosol (SOA).8–10 SOA impacts climate forcing11 and human health,12 and a complete understanding of formation mechanisms is necessary to evaluate policy options to reduce SOA. Environmental chamber experiments on hydroxyl radical (OH) oxidation of BTEX compounds show SOA yields ranging from 5–40% dependent on seed aerosol, NOx concentration, and relative humidity (RH).10,13–15 Estimated SOA production from these aromatics ranges from 2–12 Tg annually.9While hydroxyl radicals (OH) are the most abundant radical in the atmosphere and are thought to dominate aromatic hydrocarbon oxidation,10,16 chlorine radicals (Cl) may also have a substantial impact due to their more rapid reaction rate with substituted aromatic hydrocarbons.17 However, atmospheric Cl radical concentrations have been historically underestimated and consequently understudied.18 Rapid oxidation by Cl may enhance SOA formation due to rapid formation of semivolatile organic compounds (SVOCs).10,19 In addition, while OH radicals add to the aromatic ring in over 90% of initial oxidations,7,20 Cl radicals perform hydrogen abstraction from alkyl substituents on the aromatic ring.17,21 Previous work on toluene + Cl oxidation has illustrated differences in gas phase products compared to OH radical oxidation including formation of aldehyde, peroxy acid, and quinone species.22 Particles were more highly oxidized from Cl-initiated chemistry than from OH-initiated reactions, and chlorinated species were observed.22,23
Prior work on chlorine oxidation typically utilized molecular chlorine (Cl2) as the chlorine radical precursor.22–27 While Cl2 has been observed far inland of marine and coastal sources including in urban environments,28,29 nitryl chloride (ClNO2) is often more abundant than Cl2 in these environments, particularly in early morning air masses.30 Near oil and gas production well sites, Cl2 concentrations were observed to be higher than ClNO2.31 Atmospheric sources of Cl2 are largely associated with industrial emissions and biocides, though some heterogenous reactions on chloride aerosols have been shown to produce Cl2.32,33 ClNO2 is primarily produced from heterogeneous reaction of dinitrogen pentoxide (N2O5) with particulate or aqueous chloride from sources including sea salt aerosol, biomass burning aerosol, saline snowpack, playa dusts, and aerosol in polluted megacities.34–37 Beyond being an atmospherically relevant Cl source, ClNO2 photolysis generates NO2, which photolyzes to NO. Under these high NOx conditions, Cl radical oxidation leads to formation of secondary OH radicals as NO reacts with HO2.38 Secondary hydroxyl radical production under high NOx conditions has been shown to obscure the impact of Cl radicals from traditional VOC decay techniques for assessing Cl radical concentrations.39 Furthermore, the presence of NOx suppresses SOA formation from aromatics due to a shift in peroxy radical fate and subsequent fragmentation.10,14,40 Nitryl chloride generates a complex mixture of radicals and oxidants with substantial impacts on gas phase chemistry, SOA formation, and SOA composition.
Here, we evaluate the impacts of different oxidative environments on the formation of gas and particle phase products from the oxidation of m-xylene in an environmental chamber. We utilize chlorine gas, nitryl chloride, and hydrogen peroxide as radical precursors under high and low NOx conditions, generating environments in which only OH, only Cl or a mixture of both radicals are initiating oxidation of hydrocarbons. These different oxidative environments have substantial impacts on gas and particle phase products and composition. We observe and discuss the formation of several gas phase products, and evaluate chemically-speciated particle composition using a thermal desorption technique. We also assess SOA mass yield and bulk composition.
2 Methods
2.1 Chamber photo-oxidation experiments
Oxidation experiments were conducted in a 10 m3 Teflon environmental chamber at 298 K at 30% relative humidity (RH) using UV black lights with a peak emission at 354 nm to generate radicals. Clean dry air was supplied to the chamber using a clean-air generator (model 727R, AADCO). The chamber was flushed overnight with humidified clean air to reach the target 30% RH. Dried ammonium sulfate seed particles were generated from 0.05 M aqueous ammonium sulfate injected with an aerosol generation system (AGS 2002, Brechtel). Chlorine gas (100.3 ppm in N2, Airgas) was directly injected into the chamber. Multipoint calibrations of the I− CIMS for Cl2 were performed based on repeated injections into the chamber. Nitryl chloride was generated reactively by passing Cl2 (100.3 ppm in N2) at a low flow rate (0.25 L min−1) over a solution of sodium nitrite as previously described.41 Based on data from the iodide mode chemical ionization mass spectrometer (I− CIMS), >99% of Cl2 is consumed at this flow rate (<1% of initial Cl2 signal remains) and for calibration purposes, it is assumed that 100% of Cl2 injected is converted to ClNO2 during ClNO2 injection to the chamber. Formation of the common NO2 byproduct during ClNO2 synthesis is not observed in this study. Multipoint I− CIMS calibration for ClNO2 was performed based on repeated injection into the chamber based on this 100% conversion. Hydrogen peroxide was injected as an OH radical precursor by bubbling clean dry air at 2 L min−1 through a 30% hydrogen peroxide solution (Sigma Aldrich) for 1 hour. Hydrogen peroxide concentration was estimated at 1 ppm based on interference in the 254 nm photometric ozone monitor and the relative ratios of the absorption cross sections of ozone and hydrogen peroxide at 254 nm. I− CIMS measurements of hydrogen peroxide injection indicated that injections were repeatable, and response was linear with respect to injection volume. m-Xylene (>99% purity, Sigma Aldrich) was injected into a glass gas sampling tube which was flushed into the chamber with heated clean air at 2 L min−1 for 30 minutes. Complete vaporization and injection of m-xylene into the chamber is assumed in calculating initial concentrations. Nitric acid (NO) (50.3 ppm in N2, Airgas) was directly injected into the chamber for high NOx experiments. Initial VOC and oxidant precursor concentrations are listed in Table 1.| # | Radical precursors | [VOC]0 (μg m−3) | [Cl2]0 (ppb) | [ClNO2]0 (ppb) | [H2O2]0 (ppm) | [NOx]0 (ppb) | Max SOAa (μg m−3) | Yieldb (%) |
|---|---|---|---|---|---|---|---|---|
| a Peak SOA is calculated based on wall loss corrected ACSM organic loading following the UV illumination period. b Yield (organic aerosol mass yield) is calculated for experiments where H3O+ CIMS data are available to measure xylene consumption. c Aerosol data unavailable for this experiment. | ||||||||
| 1 | ClNO2 | 340 | <0.1 | 50 | <0.1 | <2 | —c | — |
| 2 | ClNO2 | 340 | <0.1 | 50 | <0.1 | <2 | 12.7 | 4.8 |
| 3 | Cl2 + NOx | 340 | 25 | <0.1 | <0.1 | 20 | 12.0 | — |
| 4 | Cl2 | 340 | 25 | <0.1 | <0.1 | <2 | 24.3 | 8.8 |
| 5 | Cl2 | 340 | 25 | <0.1 | <0.1 | <2 | —c | — |
| 6 | H2O2 + NOx | 340 | <0.1 | <0.1 | 1 | 20 | 10.1 | 4.4 |
| 7 | H2O2 | 340 | <0.1 | <0.1 | 1 | <2 | 11.2 | — |
| 8 | Cl2 + ClNO2 | 340 | 10 | 30 | <0.1 | <2 | 11.3 | — |
| 9 | Cl2 + H2O2 | 340 | 10 | <0.1 | 1 | <2 | —c | — |
| 10 | Cl2 + ClNO2 + H2O2 | 340 | 5 | 15 | 1 | <2 | 10.1 | — |
An example experimental time series is shown in Fig. 1. Following injection, VOCs, oxidant precursors, and seeds were allowed to mix and then UV lights were turned on (time = 0 in Fig. 1) to initiate photo-oxidation. Cl2 (or other radical precursors) are photolyzed to form radicals which oxidize m-xylene to form oxidation products including SOA and OVOCs. Photo-oxidation continued for 1 to 2 hours until organic aerosol concentrations stabilized. Organic aerosol concentrations stabilized earlier for Cl2 experiments due to its rapid photolysis rate compared to ClNO2 and H2O2. m-Xylene was typically not completely consumed. “Blank” experiments were conducted the day before and after photo-oxidation experiments where only oxidant precursors and ammonium sulfate seed were present. Minimal SOA formation (<1 μg m−3) was observed in “blank” experiments.
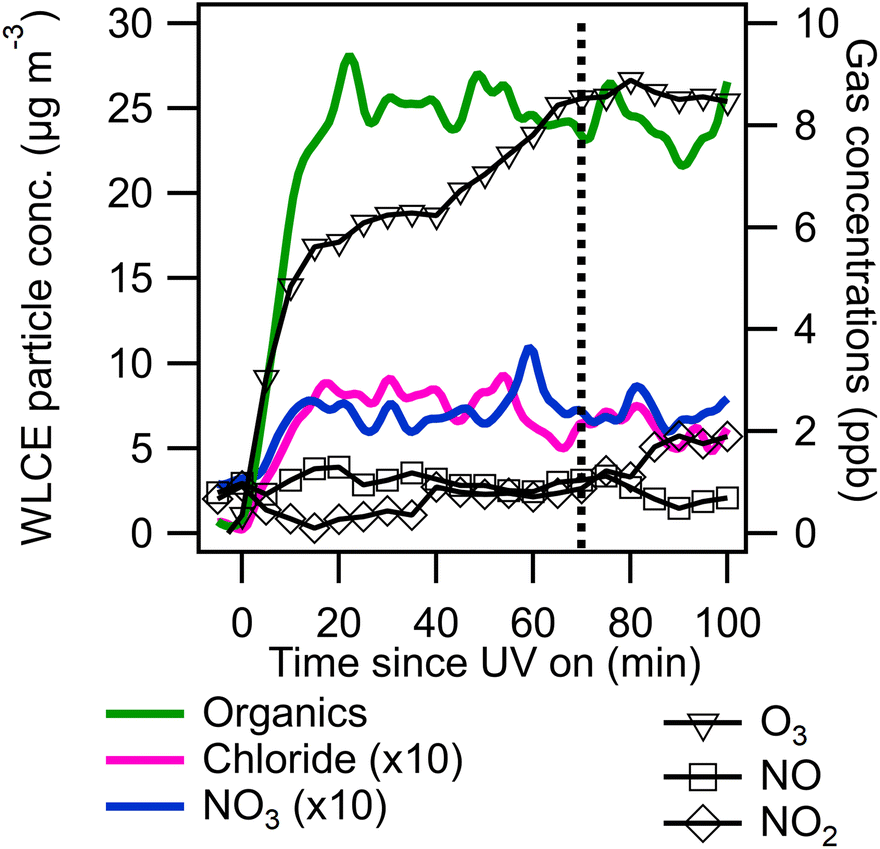 | ||
| Fig. 1 Experiment 4 (Cl2) wall loss and collection efficiency corrected (WLCE) ACSM data (left axis) and trace gas monitor data (right axis). Chloride and nitrate traces are multiplied by 10 for legibility. Vertical dashed line indicates end of UV light on period. | ||
2.2 Instrumentation
NO and total NOx concentrations were monitored with a chemiluminescent monitor (200E, Teledyne); limit of detection (LOD) 0.4 ppb and ±0.2 ppb accuracy. NO2 concentrations were measured directly using a monitor from Environnement (Model AS32M) using a cavity attenuated phase shift (CAPS) method; LOD 0.1 ppb and ±0.05 ppb accuracy. Ozone concentrations were monitored with a photometric ozone analyzer (400E, Teledyne); LOD 0.6 ppb and ±0.3 ppb accuracy. Particle size distributions in the 10–1000 nm size range were measured using a scanning electrical mobility system (SEMS, Brechtel model 2002). Bulk particle phase composition was assessed using an aerosol chemical speciation monitor (ACSM, Aerodyne),42 calibrated with 300 nm size-selected ammonium nitrate and ammonium sulfate particles. Particle phase concentrations from the ACSM were corrected for chamber wall losses and instrument collection efficiency by taking the ratio of organic to sulfate signal and multiplying by the initial ammonium sulfate seed concentration measured by the SEMS data as previously described.15 Wall loss and collection efficiency corrected (WLCE) ACSM data were used to calculate SOA yield for experiments 2, 4, and 6 where m-xylene consumption was measured by switching the CIMS reagent ion to the positive hydronium ion (H3O+ CIMS). A single CIMS system was utilized in this study and was switched between negative (I−) and positive (H3O+) modes for some experiments. SOA yield was calculated as the change in wall loss corrected ACSM organic mass from before and after the UV photo-oxidation period divided by the mass of m-xylene consumed.Gas and particle phase composition was assessed using a Filter Inlet for Gases and Aerosols coupled to an iodide mode chemical ionization mass spectrometer (FIGAERO-CIMS, Aerodyne). Iodide mode FIGAERO-CIMS has been described in detail elsewhere.43,44 In this application, real time gas phase chemistry was observed using I− CIMS including the decay of oxidant precursors and the formation of oxidized VOCs. In addition, H3O+ CIMS was utilized to examine the decay of m-xylene during some oxidation experiments. Following UV photooxidation, lights were turned off and particles were collected on the FIGAERO-CIMS PTFE filter (1 μm pore Zefon filters) at 3 SLPM for 45 minutes. Filters were desorbed using heated N2 (Airgas) which began at 25 °C and was heated to 300 °C over 30 minutes and held at 300 °C for 10 minutes. This particle desorption method allows for detailed speciation of a wide range of particle phase organic species.
For both gas and particle phase analysis of CIMS, high resolution peak fitting and identification was performed using the Tofware data analysis package. For I− CIMS, all identified species are iodide adducts (elemental formula contains I−) and it is assumed that I− is from ionization in the instrument and was not present on the analyte. Similarly, H+ or H3O+ adducts are formed in positive mode CIMS. CIMS can only provide elemental compositions without information about molecular structure. When species are discussed, their likely structures are inferred based on expected reaction mechanisms and products. I− CIMS calibration was performed as described above for radical precursor species (H2O2, Cl2, and ClNO2). Such calibrations were performed within 6 weeks of the experiment to which they were applied. All other species detected in the I− CIMS are presented normalized to I− reagent ion signal where analyte signal is divided at each time point by I− signal and multiplied by 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. This normalization procedure adjusts for variability in reagent ion concentration. I− reagent ion signal varied by 10–30% during experiments, particularly during Cl2 experiments as Cl2 depletes I− reagent ion. I− CIMS sensitivity is also highly dependent on sample humidity.43 To stabilize instrument response, reagent ion flow is humidified to above 70% RH by bubbling the reagent ion source N2 through purified water (MilliQ water purification system, ThermoScientific). Based on prior literature,43 I− CIMS limits of detection for compounds discussed here are in the 10 s of ppt or lower and accuracy is within ±10 ppt.
000. This normalization procedure adjusts for variability in reagent ion concentration. I− reagent ion signal varied by 10–30% during experiments, particularly during Cl2 experiments as Cl2 depletes I− reagent ion. I− CIMS sensitivity is also highly dependent on sample humidity.43 To stabilize instrument response, reagent ion flow is humidified to above 70% RH by bubbling the reagent ion source N2 through purified water (MilliQ water purification system, ThermoScientific). Based on prior literature,43 I− CIMS limits of detection for compounds discussed here are in the 10 s of ppt or lower and accuracy is within ±10 ppt.
Particle phase data from the FIGAERO is background subtracted and then integrated across the full thermal desorption period. In several cases, species are discussed and grouped based on their elemental composition. For example, organochlorine species with the composition C2–10HyOzCl1–3I− are discussed and consist of the total summed signal of all species which contain 2 to 10 carbon atoms, 1 or more hydrogen, 1 or more oxygen, and 1 to 3 chlorine atoms.
2.3 Chamber box modeling
A chamber box model utilizing the Carbon Bond Mechanism (version 6, revision 4) from the Comprehensive Air Quality Model with Extensions (CAMx)45 with previously described modifications for chlorine radical chemistry46 was used to model these experiments. Radical precursor, m-xylene, NOx, temperature, and humidity concentrations were initialized in the model. UV light intensity in the model is based on previously measured blacklight spectra (P-200 spectroradiometer, Apogee Instruments). Chamber characterization experiments47 provided measured NO2 photolysis rates, chamber leak rate (Cl2 utilized as a leak tracer), and wall effects. Modeling was used to capture OH and Cl radical concentrations. Fig. 2a–c shows measured and modeled consumption of radical precursors alongside modeled m-xylene consumption for low NOx Cl2, ClNO2, and H2O2 experiments. Model results indicate that Cl2 is rapidly consumed, while ClNO2 and H2O2 have much slower modeled rates of consumption due to a lower photolysis rate under chamber lighting conditions. While chlorine consumption shows good model agreement, nitryl chloride consumption deviates slightly and hydrogen peroxide consumption deviates substantially from model predictions. This difference in modeled and measured radical precursor decay may be due to uncertainties associated with the measured UV spectrum. The absorption cross sections of H2O2 and ClNO2 overlap only slightly with the measured emission spectrum of the UV lights and small errors in spectrum or absorption cross section measurements may substantially alter modeled photolysis rates. Absorption cross sections for radical precursors and the measured light spectrum used in the chamber model are shown in Fig. S1.†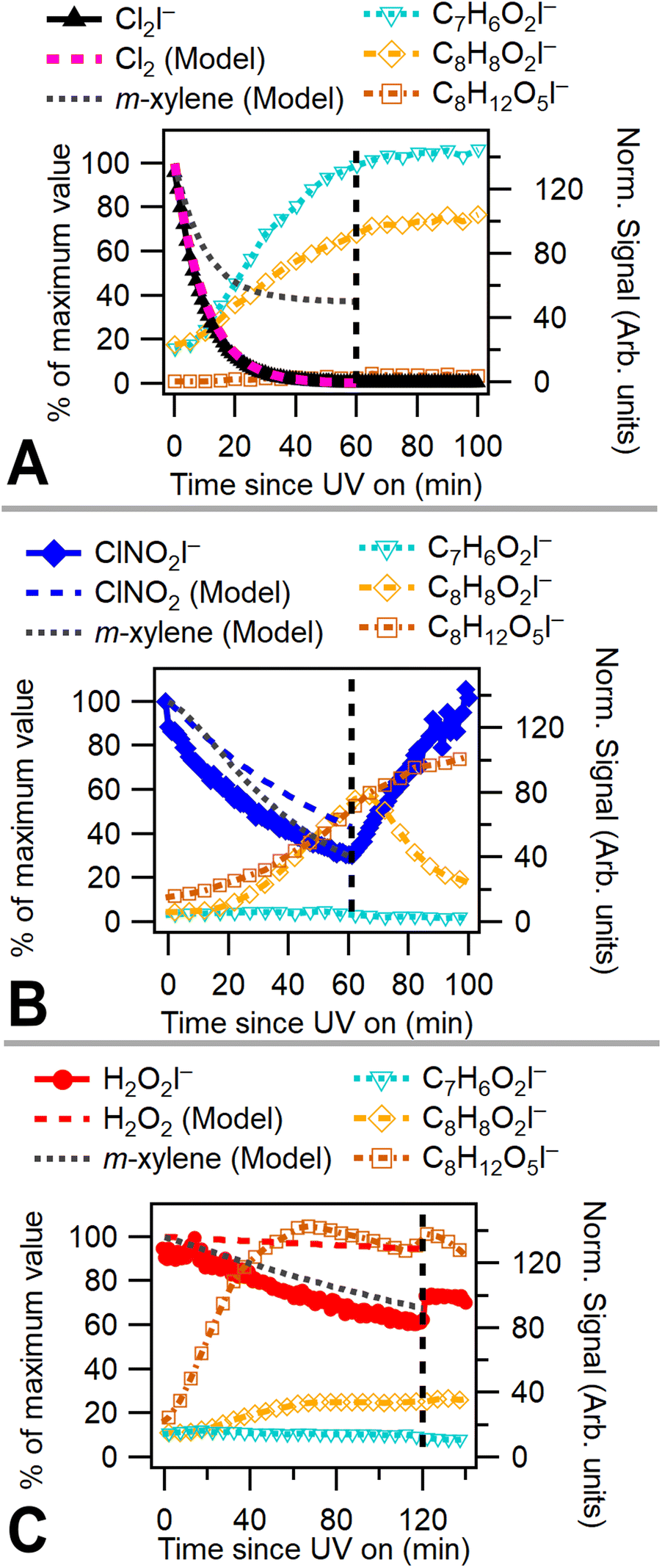 | ||
| Fig. 2 (A)–(C) Normalized I− CIMS data and chamber model results from (A) experiment 5 – Cl2 (B) experiment 1 – ClNO2 (C) experiment 7 – H2O2. Model and radical precursor time series are normalized to maximum value. Vertical dashes show UV lights were turned off. C7H6O2I− is consistent with 2-methyl-1,4-benzoquinone, C8H8O2I− with m-toluic acid, and C8H12O5I− with bicyclic peroxide. | ||
3 Results and discussion
3.1 Radical precursor identity alters gas phase products and degree of oxidation
Chlorine and hydroxyl radicals utilize different primary oxidation pathways as illustrated in Schemes 1 (Cl) and 2 (OH). Three selected products illustrate these reaction pathway differences: 2-methyl-1,4-benzoquinone is formed primarily though hydrogen abstraction by the Cl radical, the bicyclic peroxide is formed from OH radical addition, and m-toluic acid can be formed by both radicals. Formation of these products is described briefly here.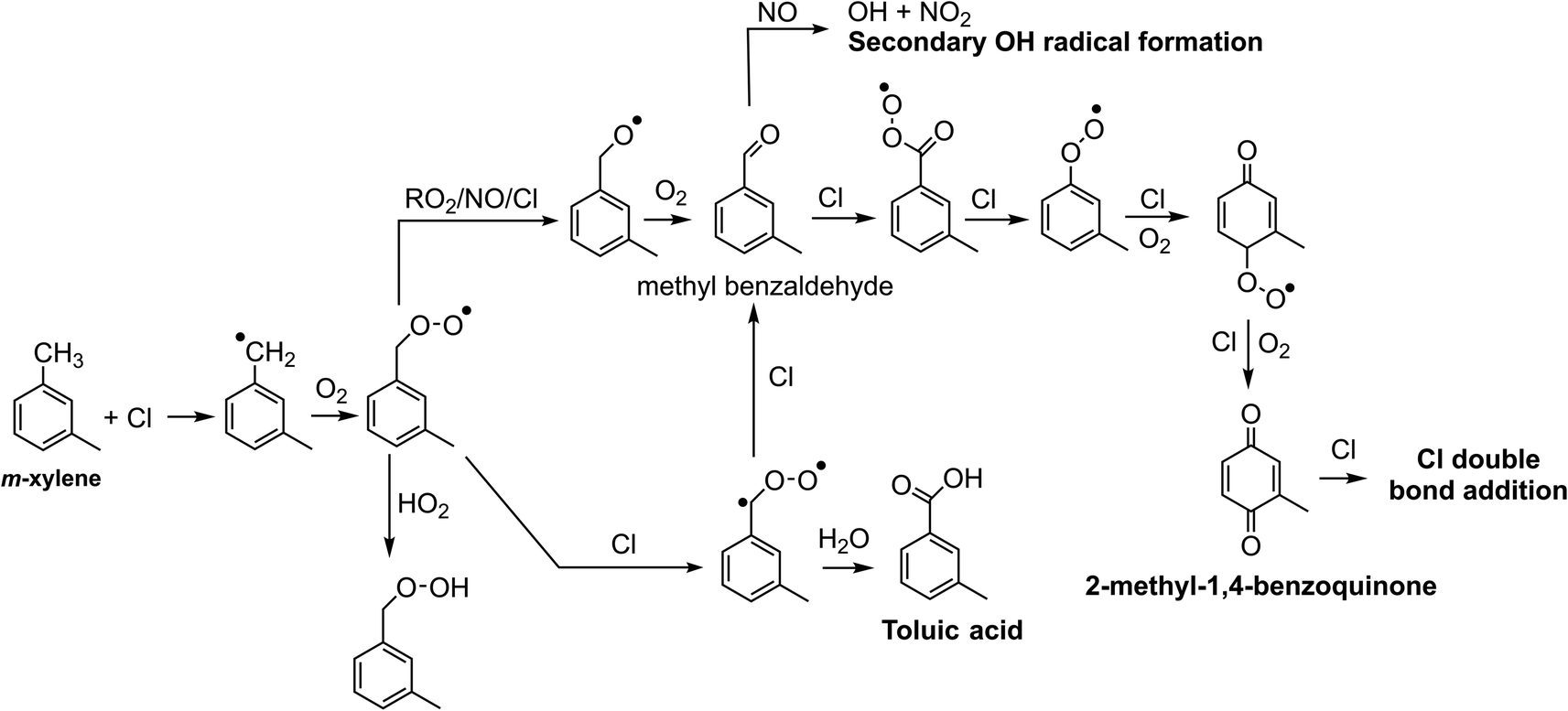 | ||
| Scheme 1 Selected oxidation pathways for m-xylene + Cl. | ||
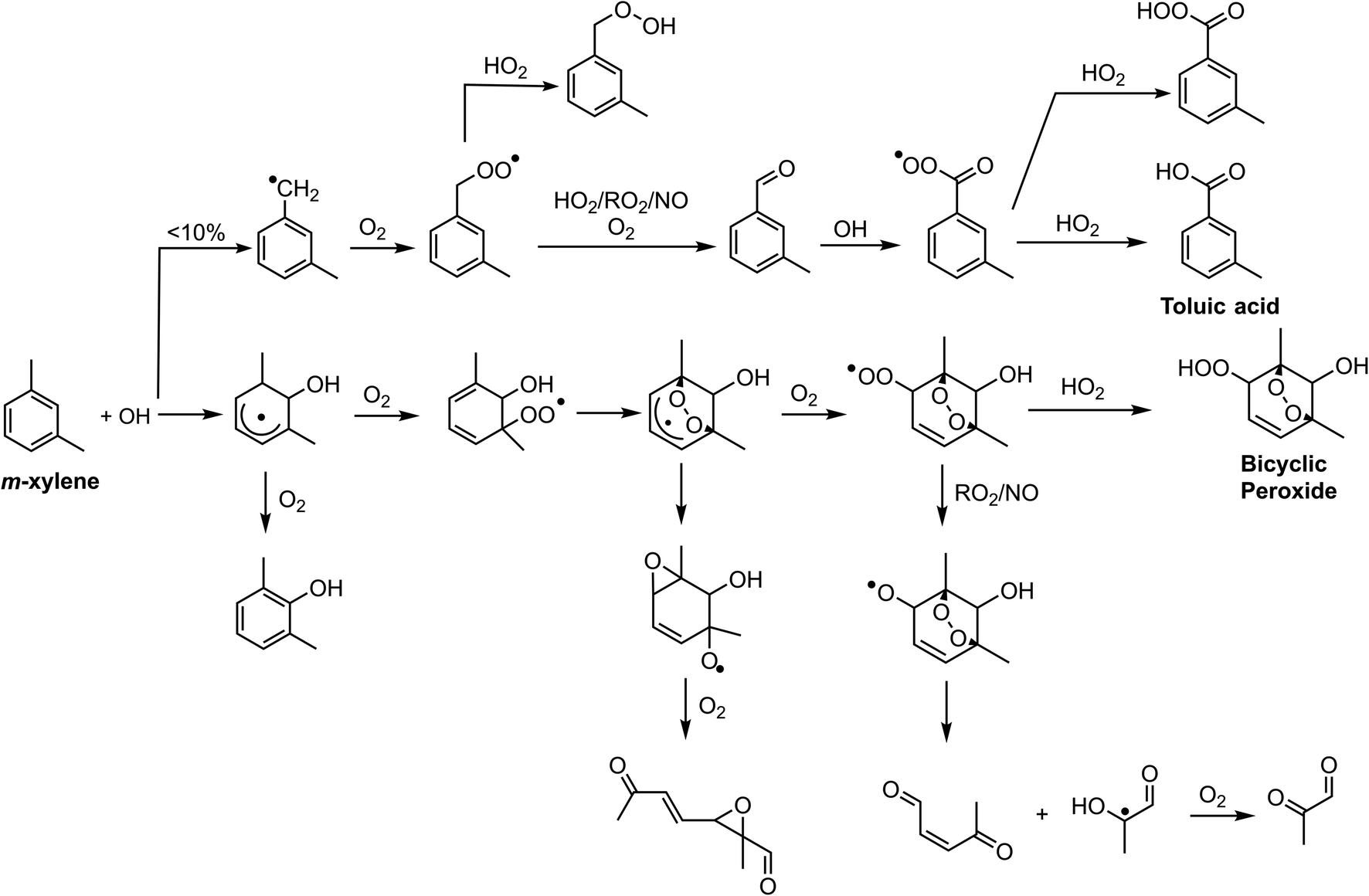 | ||
| Scheme 2 Selected oxidation pathways for m-xylene + OH. | ||
Chlorine initiated oxidation of toluene forms 1,4-benzoquinone due to hydrogen abstraction of aldehydic hydrogen and additional Cl radical attack on peroxy radicals to form ClO.22,23,48 Applying this reaction pathway to m-xylene predicts the analogous species, 2-methyl-1,4-benzoquinone (C7H6O2) measured here. Hydroxyl radicals can form quinone species through an OH ring addition pathway resulting in methyl phenol products, though this pathway is less common.49,50
The OH ring addition pathway generates a bicyclic radical species with a wide variety of fates, including epoxidation, fragmentation, and the bicyclic peroxide product (C8H12O5)20,51 discussed here. Chlorine oxidation is not observed to cause ring addition and bicyclic radical species are not formed.17
Toluic acid is the result of hydrogen abstraction at the methyl group resulting in the carboxylic acid species. OH and Cl radicals form this product following attack on an aldehydic hydrogen. In addition, Cl oxidation generates toluic acid after forming a Criegee intermediate. Criegee intermediates form following the attack of chlorine on the peroxy radical52 and have been previously discussed for toluene + Cl systems.53
Fig. 2a–c plots 2-methyl-1,4-benzoquinone, the bicyclic peroxide and m-toluic acid for Cl2, ClNO2, and H2O2 experiments. Growth of 2-methyl-1,4-benzoquinone is only substantial when Cl2 is present, including under high NOx conditions (Fig. S2†), indicating that high Cl radical concentrations initiate this multigenerational chemistry. Greater relative formation of 2-methyl-1,4-benzoquinone over m-toluic acid and bicyclic peroxide occurred in both low NOx Cl2 experiments (Exp. 4 and 5). According to our chamber box model (Fig. S10†), integrated Cl radical exposures during the full UV exposure period are 8 × 109 molecules per cm3 per s and 14 × 109 molecules per cm3 per s under high and low NOx conditions, respectively. In contrast, little growth of the quinone product is observed in a ClNO2 experiment with modeled Cl radical exposure of 5 × 109 molecules per cm3 per s. In addition, model results (Fig. S10†) indicate Cl2 generates peak Cl radical concentrations nearly 1 order of magnitude higher than ClNO2 as a result of the high Cl2 photolysis rate. Increased Cl radical concentrations may offer more opportunity for multiple radical oxidation events which drive formation of the quinone product. This mirrors previously observed differences in SOA formation from high NOxm-xylene + OH systems.10 In this prior work, use of photolabile HONO produced a rapid plume of OH radicals which generated SOA not observed in classical high NOx photooxidation experiments which rely on NOx and HOx cycling to generate OH radicals more slowly. Higher radical concentrations reduce the timescale of multigenerational chemistry and lower SVOCs wall losses in addition to potentially altering radical fate branching pathways.10
However, we observe continued formation of 2-methyl-1,4-benzoquinone in Cl2 + NO experiments even after Cl radical concentrations fall below those observed in ClNO2 experiments (Fig. S2 and S10†). This is likely related to differences in initial RO2 radical fate which drive precursor availability. High initial Cl radical concentrations combined with the presence of NO bias Cl2 + NO oxidation environments toward RO2 + NO radical terminations or shift the fate of the Criegee intermediate toward reaction with Cl to form methyl benzaldehyde. Formation of these key precursors is suppressed in the ClNO2 case due to slower formation of NO and higher OH to Cl ratio which drives HO2 formation and efficient formation of the bicyclic peroxide radical. The presence of benzaldehyde and other precursors in Cl2 + NO experiments enables formation of the benzoquinone products even as the OH to Cl ratio increases.
Bicyclic peroxide product formation is observed for all experiments except the low NOx Cl2 case. Unsurprisingly, hydrogen peroxide experiments rapidly generated this OH oxidation product. Chlorine gas in the absence of NOx formed minimal secondary OH radicals with an OH radical exposure of 1.5 × 109 molecules per cm3 per s. In contrast, Cl2 + NO and ClNO2 experiments have OH radical exposures of 43 and 35 × 109 molecules per cm3 per s, respectively. This NOx enhancement of hydroxyl radicals is driven both by the effect of OH regeneration through NOx cycling and the presence of secondary OH formation pathways which are enhanced by NOx reaction with peroxy radicals.39 The formation of bicyclic peroxides in the ClNO2 case was more rapid than in the Cl2 + NO case (Fig. S2†) and was observed in both ClNO2 experiments (Exp. 1 and 2). Under both conditions, similar OH radical concentrations/exposures are achieved (Fig. S10†); however, slower formation of Cl radicals from ClNO2 likely biases initial oxidation toward OH radical addition pathways compared to initial Cl oxidations in the Cl2 + NO case. As discussed, peak Cl radical concentrations in the Cl2 + NO case result in multigenerational Cl chemistry. The reduced Cl concentration in the nitryl chloride system generated an approximate 1:10 Cl:OH radical ratio, similar to ambient measurements of a 1 to 2 order of magnitude difference in Cl and OH radical concentrations in inland Texas.28 In the case of Cl2 + NO, we observe a shifting Cl:OH radical ratio of 1:2 initially to nearly 1:50 during hour-long photo-oxidation. In addition, dark chemistry is observed following photooxidation in the ClNO2 experiment, as evidenced by growth in nitryl chloride and a decline in m-toluic acid. This may be related to heterogeneous chemistry which forms nitryl chloride from N2O5 interactions with chloride containing particles. We do see growth of N2O5 in the dark for ClNO2 experiments (Fig. S6†), possibly from reaction of NO2 and NO3 in the gas phase which goes on to react with chloride and form nocturnal ClNO2. This behavior is not observed when ClNO2 is used in combination with Cl2 and/or H2O2 (Exp. 8 and 10, Fig. S3 and S4†), likely due to lower availability of N2O5 precursors. Taken together, these results indicate that oxidative environments from fresh chlorine emissions in high NOx areas differ from ClNO2 rich air masses typically observed in the early morning.
m-Toluic acid is observed to form during photo-oxidation in all experiments. Formation of toluic acid is substantially lower in H2O2 experiments, consistent with the OH oxidation pathway which favors ring addition over hydrogen abstraction. Under high NOx Cl2 conditions (Fig. S2†), formation of toluic acid is more rapid than 2-methyl-1,4-benzoquinone. This may indicate termination of radical propagation is more rapid under high NOx conditions or may be a result of additional competition for the Cl radical as it forms ClNO, ClONO, and ClNO2 in the presence of NOx.
In addition to these species, organochlorine species are formed in chlorine radical systems. Gas phase organochlorine fraction was calculated by dividing the total C2–10HyOzCl1–3I− signal by the total C1–10I− signal (i.e. total organic species signal) averaged for 10 minutes following the end of UV photolysis. We assume a uniform sensitivity for all organic species. Under low NOx conditions, we find a gas phase organochlorine fraction of 0.22 which falls to 0.18 under Cl2 + NO and falls further to 0.07 when ClNO2 is used. This is consistent with the growth in OH radical concentrations observed from Cl2 to Cl2 + NO to ClNO2 experiments. These gas phase chlorinated species are a result of chlorine addition reactions, likely to available double bonds following aromatic ring breaking.
3.2 Chlorine oxidation yields more highly oxidized SOA
Differences in gas phase oxidation discussed above lead to differences in particle phase composition. Fig. 3 shows integrated particle composition data from the FIGAERO-CIMS from experiments 1, 4, 6, and 7. Particles were collected for 45 minutes following the end of UV exposure and desorbed on a 45 minutes heating cycle. Integration of the thermal desorption signal is performed over the full desorption temperature range (25–180 °C). Experiments using Cl2 as radical precursor resulted in more highly oxidized organic aerosol; for example, the experiments formed a higher fraction of C7–8O4–8 species compared to experiments using ClNO2 which formed a much smaller fraction of species with more than four oxygen atoms. This additional oxygen content may be related to the more rapid formation of Cl radicals under Cl2 oxidation conditions, driving multigenerational oxidation chemistry. In addition, the fraction of ring retaining C8 species is higher in SOA formed when Cl2 was used as the radical precursor, consistent with functionalization of the methyl group after hydrogen abstraction. SOA formed in experiments where ClNO2 or H2O2 serves as the radical precursor has a higher fraction of C7 species. Even greater fragmentation to C4 and C5 species is observed in hydrogen peroxide experiments, consistent with the ring breaking fragmentation products resulting from the bicyclic radicals formed by OH ring addition chemistry.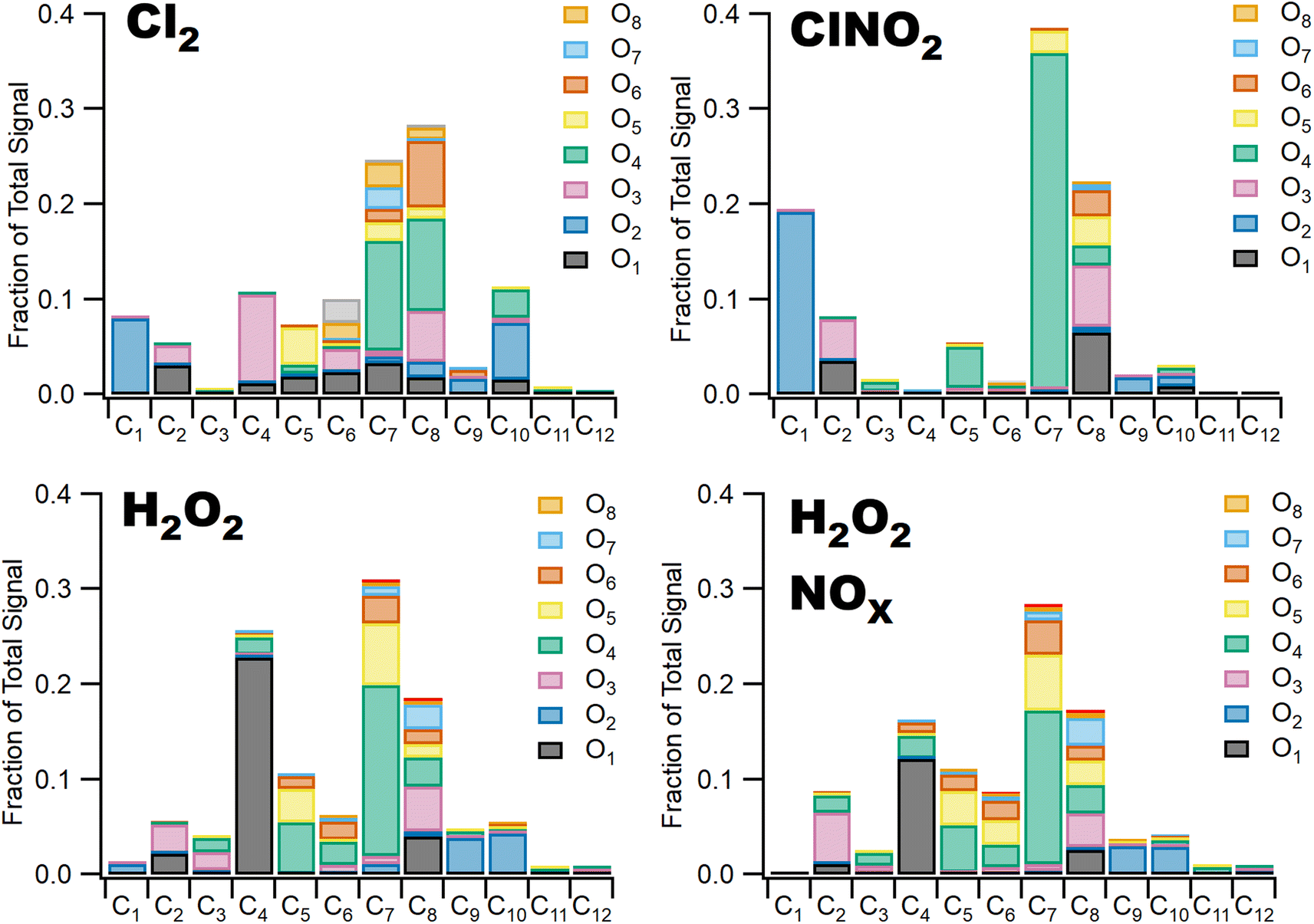 | ||
| Fig. 3 I− FIGAERO-CIMS desorption data from experiment 1 (ClNO2), 4 (Cl2), 6 (H2O2 + NOx) and 7 (H2O2). Particle phase species are categorized by carbon and oxygen number, integrated over their desorption curves, summed and divided by total integrated desorption signal of represented species. | ||
These observations are consistent with bulk particle composition from the ACSM. Triangle plot analysis54 (Fig. 4) shows that the fraction of the total organic signal due to organic mass at m/z 44 (f44) was elevated and f43 was reduced in Cl2 experiments. This indicated use of pure Cl2 as a radical precursor resulted in more highly oxidized SOA with increased formation of acid groups. Use of ClNO2 resulted in similar f44 fractions to H2O2 experiments. In addition, f43 fractions were higher in H2O2 experiments, indicating that less oxidized SOA was the norm when primary OH radicals are present. This is consistent with observations of toluene + Cl and toluene + OH chemistry from Dhulipala et al. (2019)22 where toluene + Cl SOA had a lower f43 and higher f44 measured in the ACSM than toluene + OH SOA. This was attributed to formation of ring opened products from OH oxidation with unoxidized methyl groups related to higher f43 values. SOA from m-xylene + H2O2 experiments in Loza et al. (2011)55 resulted in higher f43 values measured in the aerosol mass spectrometer (AMS) than reported here, though f44 values are similar.
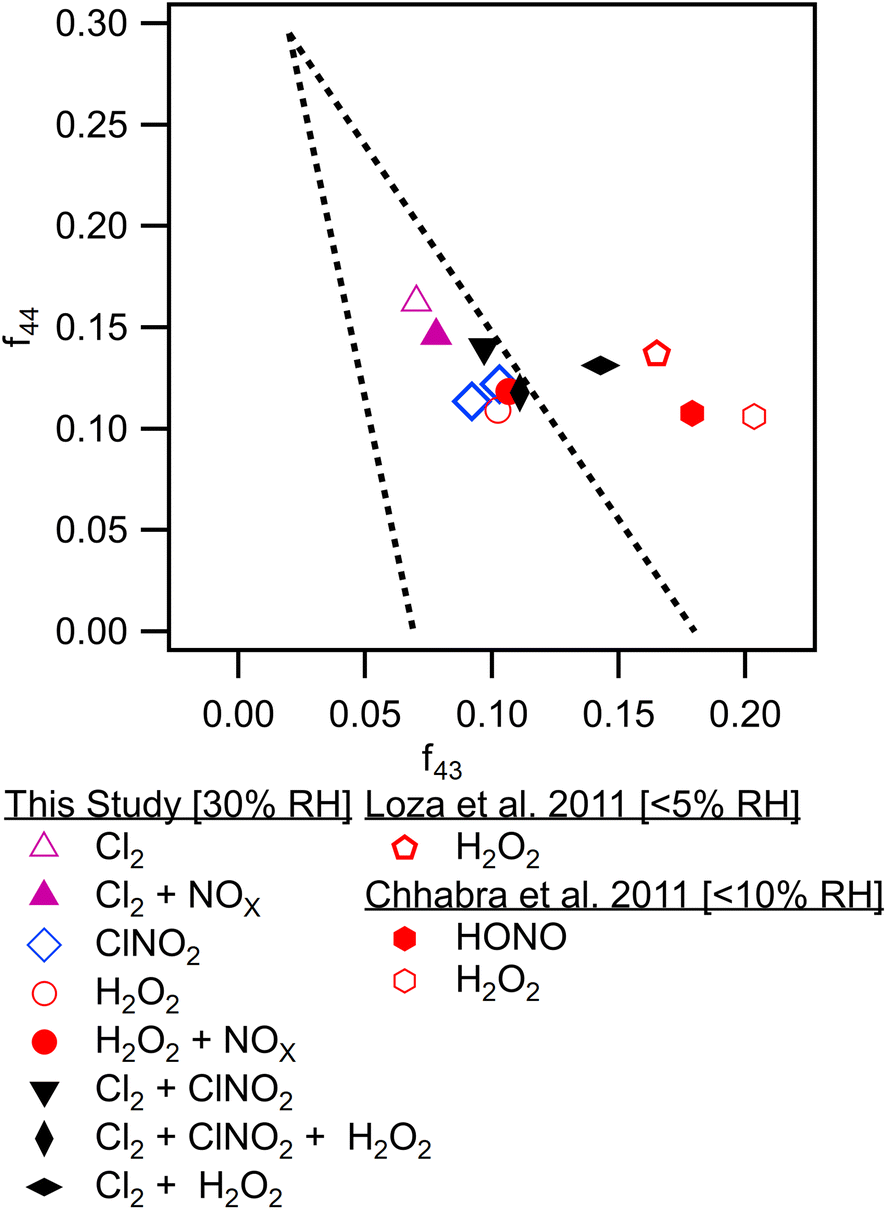 | ||
| Fig. 4 f 44 to f43 triangle plot for all chamber experiments from final average ACSM bulk particle composition. Data from literature sources is shown.46,47 | ||
Particle phase organochlorine content varied with radical precursor as shown by the ratio of HCl+ (m/z 36) to organics in the ACSM. ACSM HCl+ to organics ratio has been suggested as an improved semi-quantitative measure of particulate organochlorine compared to the detection utilizing Cl+ (m/z 35).24 Here, we found that the HCl+ to organics ratio decreased from 0.037 in a Cl2 experiment to 0.025 and 0.011 in Cl2 + NO and ClNO2 experiments, respectively. FIGAERO-CIMS particle desorption signals show that in Cl2 experiments organochlorine species make up 0.27 of total identified desorption signal. This falls to 0.07 for ClNO2 experiments. This behavior is qualitatively consistent with gas phase decreases in organochlorine content discussed earlier. The behavior is also qualitatively consistent with the ACSM HCl/Organics ratio.
While NOx resulted in less oxidized SOA in Cl2 experiments, it had little effect within peroxide experiments. This may be driven by H2O2 photolysis generating substantial HO2 and favoring HO2 + RO2 and RO2 + RO2 chemistry over NOx + RO2 pathways. Use of HONO as a high NOx source of OH radicals promoted a more substantial shift in SOA oxidation state compared to H2O2 for m-xylene oxidation in Chhabra et al. (2011).56 This may be because the photolysis of HONO produces a lower HO2 to RO2 ratio as well as higher initial OH radical concentrations. The f43/f44 differences in this study compared to literature may be related to instrument variability as noted in Ng et al. (2010).54 Differences in f43/f44 may also be driven by increased humidity as discussed in Zhang et al. (2019),13 where a shift in O:C ratio was observed for m-xylene + OH experiments. The higher RH in these experiments may reduce the degree of oxidation observed in the particle phase by reducing the formation of low-volatility oligomers. This effect has also been reported in toluene57 and isoprene58 studies.
3.3 Oxidant environment shifts SOA formation
In addition to SOA composition, SOA formation and yield is also dependent on radical precursor choice. Table 1 lists peak wall loss corrected SOA concentrations. Peak SOA formation is greatest for the low NOx Cl2 condition (experiment 4) compared to SOA formation for ClNO2 (experiment 2) and high NOx Cl2 (experiment 3). This reduction in SOA formation due to NOx has been previously reported for some aromatic and hydroxyl radical systems.10 Peroxy radical fate drives these differences, with NO + RO2 pathways resulting in more volatile products compared to HO2 + RO2 reaction products.9Secondary organic aerosol yields are determined as the mass concentration of SOA formed divided by the mass concentration of precursor consumed. Yields are reported for experiments 2, 4, and 6, the three experiments were measurements of m-xylene consumption were available based on H3O+ CIMS measurements. Measured and modeled m-xylene consumption is shown in Fig. S7–S9.†Fig. 5 compares yields from this study to literature. The highest yield (8.8%) is observed for the low NOx Cl2 condition. Yield for the m-xylene + ClNO2 system was found to be 4.8% here. While yields for the m-xylene + Cl reaction has not been previously reported, toluene + Cl2 experiments report yields as low as 5% (ref. 23) and as high at 82%.22 Toluene and m-xylene are similar molecules, though the additional methyl group on m-xylene may alter the volatility of expected product by providing an additional site for Cl hydrogen abstraction. In addition, previous studies differ substantially in use of seed aerosol, relative humidity, and Cl2/VOC ratio from this one. Prior studies have observed a substantial dependence of SOA formation from Cl radicals on Cl2/VOC ratio.23,53
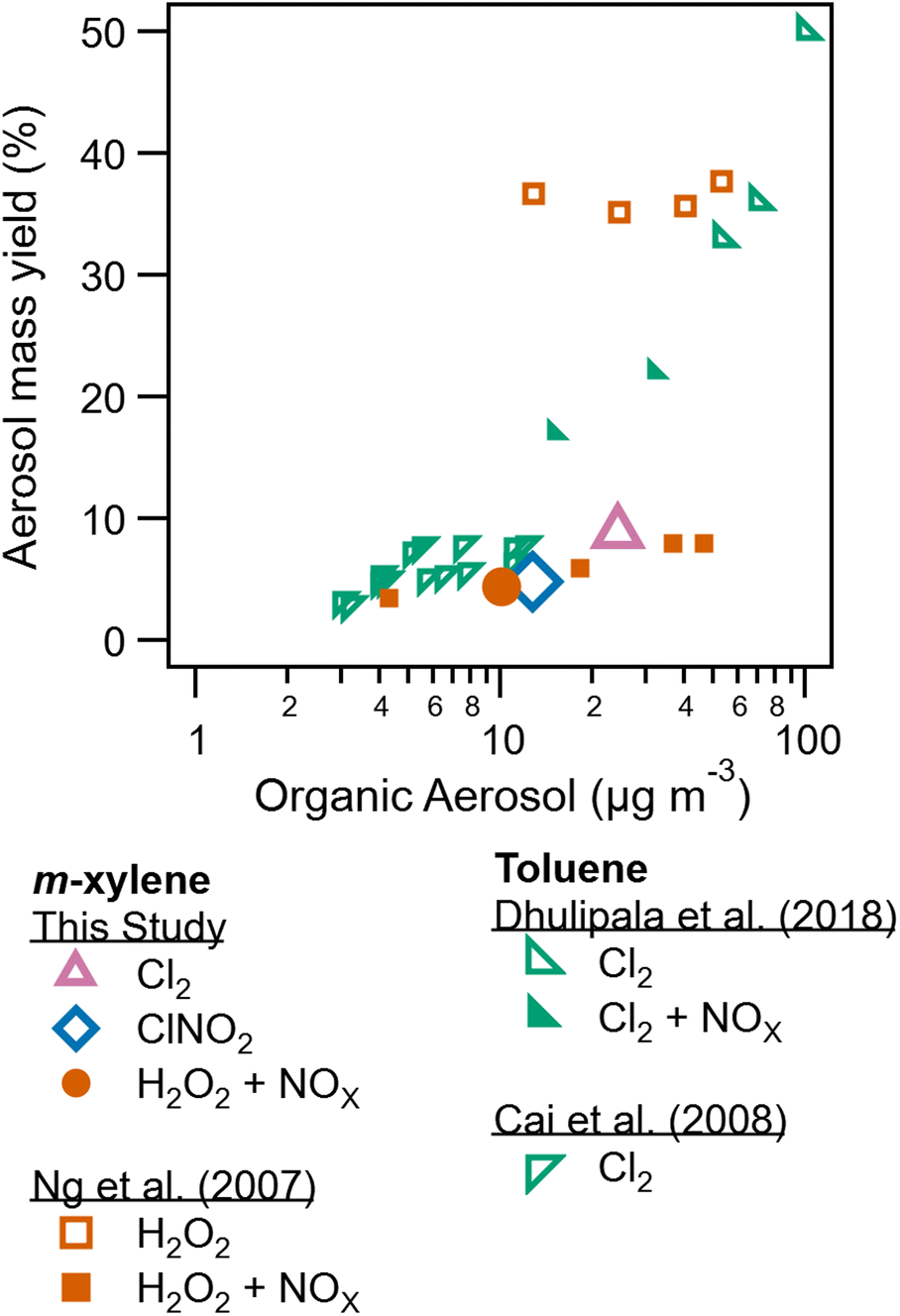 | ||
| Fig. 5 Aerosol mass yield plotted against organic aerosol concentration from this study, Ng et al. (2007),10 Dhulipala et al. (2018),33 and Cai et al. (2008).23 This study and Ng et al. (2007)10 utilize m-xylene while the other studies utilize toluene. Data from this study are shown in larger symbols for emphasis. | ||
For the high NOx H2O2 system, yields agree with those presented in Ng et al. (2007).10 Substantially higher formation and yield is observed in Ng et al. (2007) for low NOx conditions, though differences in humidity between these experiments and the one described there may play a role. Zhang et al. (2019)13 show a sharp drop in SOA yield with increasing RH, related to increased wall losses of semi volatile species and reduced oligomerization as previously discussed.
4 Conclusions
This environmental chamber study examined the impact of radical precursor identity on Cl and OH oxidation of m-xylene. Hydrogen peroxide experiments resulted in well studied hydroxyl radical chemistry which primarily leads to bicyclic radical formation and subsequent fragmentation due to OH ring addition. Chlorine gas used as a Cl precursor drove rapid oxidation by hydrogen abstraction from methyl substituents and addition of oxygenated functional groups. In addition, chlorine gas drove the formation of a multigenerational product (2-methyl-1,4-benzoquinone) under both high and low NOx conditions. In contrast, nitryl chloride generated a different high NOx Cl radical environment which did not form 2-methyl-1,4-benzoquinone. Furthermore, the formation of secondary hydroxyl radicals is substantial and results in formation of bicyclic radical species in high NOx Cl radical environments. In ambient air masses with high concentrations of chlorine gas, including marine regions or certain industrial plumes, functionalized aromatics and toxic compounds including quinones and organochlorine species will be rapidly formed. In those ambient early morning air masses with high nitryl chloride concentrations, greater fragmentation of aromatic hydrocarbons is anticipated with reduced formation of organochlorine species as secondary OH radicals dominate oxidation. Broadly, differences in chlorine radical exposure, peak chlorine radical concentrations, and formation of secondary hydroxyl radicals all drive different chemical outcomes for environments dominated by fresh chlorine gas in the presence of NOx compared to those dominated by nitryl chloride.In the particle phase, Cl2 drove less fragmentation and greater functionalization of organic species than H2O2 consistent with previous observations of toluene + Cl/OH chemistry. Reduced fragmentation was also observed when ClNO2 was used, though chlorine gas generated more highly oxidized aerosol. This is consistent with gas phase oxidation pathways described above as OH ring addition chemistry leads to the fragmentation of the bicyclic radical intermediate. Consistent with rates and degrees of precursor consumption, SOA mass formed under low NOx Cl2 conditions was greatest, while the presence of NOx suppressed formation and yield as seen in other studies. Despite the high secondary hydroxyl radical formation observed in Cl radical experiments, the composition of organic aerosol from Cl2 and ClNO2 differs substantially from OH oxidation alone. Atmospherically, the reduced fragmentation and increased functionalization of particle phase aromatics following Cl oxidation may substantially alter SOA properties. Functionalized aromatics may act as chromophores which alter aerosol light absorption, while organochlorine species may be toxic. Furthermore, organochlorine species in the aerosol phase may contribute to heterogenous reactions that regenerate Cl2 and ClNO2. Accounting for the particle phase differences resulting from Cl oxidation compared to OH oxidation is necessary to fully capture climate and health impacts.
Oxidation of aromatic hydrocarbons and subsequent SOA formation may have substantial impacts on tropospheric pollution. As shown here, Cl2 and ClNO2 generate substantially different radical environments and subsequently form different product mixtures. Radical precursor identity and the resulting oxidative environment demands future study in both laboratory and field measurement settings. With growing evidence of high particulate chloride in polluted megacities and the evolution of chlorine radical precursors from industrial and mineral sources, there is a premium on better understanding and constraining the fate and transport of chlorine in the environment.
Data availability
Experimental data are available in the ICARUS database. Data published in the main manuscript's figures are available via the Texas Data Repository https://doi.org/10.18738/T8/6XWHN9. Underlying research data are also available by request to Lea Hildebrandt Ruiz (E-mail: lhr@che.utexas.edu).Author contributions
Nirvan Bhattacharyya: conceptualization, methodology, investigation, formal analysis, writing – original draft. Mrinali Modi: software, formal analysis, writing – review & editing. Leif G. Jahn: writing – review & editing. Lea Hildebrandt Ruiz: conceptualization, supervision, funding acquisition, writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to acknowledge the support of staff at the Center for Engineering and Environmental Resources including Denzil Smith, Susan McCoy and Nohemi Cazares in building management, ordering, and other logistics. The authors are also grateful for funding support from the National Science Foundation (Grant number 1653625) and the Welch Foundation (Grant numbers F-1925-20170325 and F-1925-20200401).References
- G. Huang, R. Brook, M. Crippa, G. Janssens-Maenhout, C. Schieberle, C. Dore, D. Guizzardi, M. Muntean, E. Schaaf and R. Friedrich, Speciation of anthropogenic emissions of non-methane volatile organic compounds: a global gridded data set for 1970-2012, Atmos. Chem. Phys., 2017, 17, 7683–7701 CrossRef CAS.
- T. Salameh, S. Sauvage, C. Afif, A. Borbon and N. Locoge, Source apportionment vs. emission inventories of non-methane hydrocarbons (NMHC) in an urban area of the Middle East: local and global perspectives, Atmos. Chem. Phys., 2016, 16, 3595–3607 CrossRef CAS.
- J. An, Y. Huang, C. Huang, X. Wang, R. Yan, Q. Wang, H. Wang, S. Jing, Y. Zhang, Y. Liu, Y. Chen, C. Xu, L. Qiao, M. Zhou, S. Zhu, Q. Hu, J. Lu and C. Chen, Emission inventory of air pollutants and chemical speciation for specific anthropogenic sources based on local measurements in the Yangtze River Delta region, China, Atmos. Chem. Phys., 2021, 21, 2003–2025 CrossRef CAS.
- R. R. Hoque, P. S. Khillare, T. Agarwal, V. Shridhar and S. Balachandran, Spatial and temporal variation of BTEX in the urban atmosphere of Delhi, India, Sci. Total Environ., 2008, 392, 30–40 CrossRef CAS PubMed.
- S. C. Lee, M. Y. Chiu, K. F. Ho, S. C. Zou and X. Wang, Volatile organic compounds (VOCs) in urban atmosphere of Hong Kong, Chemosphere, 2002, 48, 375–382 CrossRef CAS PubMed.
- J. Zhao, R. Zhang, K. Misawa and K. Shibuya, Experimental product study of the OH-initiated oxidation of m-xylene, J. Photochem. Photobiol., A, 2005, 176, 199–207 CrossRef CAS.
- R. Atkinson and J. Arey, Mechanisms of the gas-phase reactions of aromatic hydrocarbons and PAHS with OH and NO3 radicals, Polycyclic Aromat. Compd., 2007, 27, 15–40 CrossRef CAS.
- M. Wang, D. Chen, M. Xiao, Q. Ye, D. Stolzenburg, V. Hofbauer, P. Ye, A. L. Vogel, R. L. Mauldin, A. Amorim, A. Baccarini, B. Baumgartner, S. Brilke, L. Dada, A. Dias, J. Duplissy, H. Finkenzeller, O. Garmash, X. C. He, C. R. Hoyle, C. Kim, A. Kvashnin, K. Lehtipalo, L. Fischer, U. Molteni, T. Petäjä, V. Pospisilova, L. L. J. Quéléver, M. Rissanen, M. Simon, C. Tauber, A. Tomé, A. C. Wagner, L. Weitz, R. Volkamer, P. M. Winkler, J. Kirkby, D. R. Worsnop, M. Kulmala, U. Baltensperger, J. Dommen, I. El-Haddad and N. M. Donahue, Photo-oxidation of Aromatic Hydrocarbons Produces Low-Volatility Organic Compounds, Environ. Sci. Technol., 2020, 54, 7911–7921 CrossRef CAS PubMed.
- D. K. Henze, J. H. Seinfeld, N. L. Ng, J. H. Kroll, T. M. Fu, D. J. Jacob and C. L. Heald, Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: high- vs. low-yield pathways, Atmos. Chem. Phys., 2008, 8, 2405–2421 CrossRef CAS.
- N. L. Ng, J. H. Kroll, A. W. H. Chan, P. S. Chhabra, R. C. Flagan and J. H. Seinfeld, Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 2007, 7, 3909–3922 CrossRef CAS.
- C. G. Fletcher, B. Kravitz and B. Badawy, Quantifying uncertainty from aerosol and atmospheric parameters and their impact on climate sensitivity, Atmos. Chem. Phys., 2018, 18, 17529–17543 CrossRef CAS.
- M. Brauer, G. Freedman, J. Frostad, A. van Donkelaar, R. V. Martin, F. Dentener, R. van Dingenen, K. Estep, H. Amini, J. S. Apte, K. Balakrishnan, L. Barregard, D. Broday, V. Feigin, S. Ghosh, P. K. Hopke, L. D. Knibbs, Y. Kokubo, Y. Liu, S. Ma, L. Morawska, J. L. T. Sangrador, G. Shaddick, H. R. Anderson, T. Vos, M. H. Forouzanfar, R. T. Burnett and A. Cohen, Ambient Air Pollution Exposure Estimation for the Global Burden of Disease 2013, Environ. Sci. Technol., 2016, 50, 79–88 CrossRef CAS PubMed.
- Q. Zhang, Y. Xu and L. Jia, Secondary organic aerosol formation from OH-initiated oxidation of m-xylene: effects of relative humidity on yield and chemical composition, Atmos. Chem. Phys., 2019, 19, 15007–15021 CrossRef CAS.
- C. Song, K. Na, B. Warren, Q. Malloy and D. R. Cocker, Secondary organic aerosol formation from m-xylene in the absence of NOx, Environ. Sci. Technol., 2007, 41, 7409–7416 CrossRef CAS PubMed.
- L. Hildebrandt, N. M. Donahue and S. N. Pandis, High formation of secondary organic aerosol from the photo-oxidation of toluene, Atmos. Chem. Phys., 2009, 9, 2973–2986 CrossRef CAS.
- X. Wang, D. J. Jacob, S. D. Eastham, M. P. Sulprizio, L. Zhu, Q. Chen, B. Alexander, T. Sherwen, M. J. Evans, B. H. Lee, J. D. Haskins, F. D. Lopez-Hilfiker, J. A. Thornton, G. L. Huey and H. Liao, The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 2019, 19, 3981–4003 CrossRef CAS.
- L. Wang, J. Arey and R. Atkinson, Reactions of chlorine atoms with a series of aromatic hydrocarbons, Environ. Sci. Technol., 2005, 39, 5302–5310 CrossRef CAS PubMed.
- T. P. Riedel, T. H. Bertram, T. A. Crisp, E. J. Williams, B. M. Lerner, A. Vlasenko, S.-M. Li, J. Gilman, J. de Gouw, D. M. Bon, N. L. Wagner, S. S. Brown and J. A. Thornton, Nitryl Chloride and Molecular Chlorine in the Coastal Marine Boundary Layer, Environ. Sci. Technol., 2012, 46, 10463–10470 CrossRef CAS PubMed.
- A. W. H. Chan, J. H. Kroll, N. L. Ng and J. H. Seinfeld, Kinetic modeling of secondary organic aerosol formation: effects of particle-and gas-phase reactions of semivolatile products, Atmos. Chem. Phys., 2007, 7, 4135–4147 CrossRef CAS.
- J. Fan and R. Zhang, Density functional theory study on OH-initiated atmospheric oxidation of m-xylene, J. Phys. Chem. A, 2008, 112, 4314–4323 CrossRef CAS PubMed.
- P. J. Ziemann and R. Atkinson, Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 2012, 41, 6582–6605 RSC.
- S. V. Dhulipala, S. Bhandari and L. Hildebrandt Ruiz, Formation of oxidized organic compounds from Cl-initiated oxidation of toluene, Atmos. Environ., 2019, 199, 265–273 CrossRef CAS.
- X. Cai, L. D. Ziemba and R. J. Griffin, Secondary aerosol formation from the oxidation of toluene by chlorine atoms, Atmos. Environ., 2008, 42, 7348–7359 CrossRef CAS.
- D. S. Wang and L. Hildebrandt Ruiz, Secondary organic aerosol from chlorine-initiated oxidation of isoprene, Atmos. Chem. Phys., 2017, 17, 13491–13508 CrossRef CAS.
- D. S. Wang and L. Hildebrandt Ruiz, Chlorine-initiated oxidation of n-alkanes under high-NOx conditions: insights into secondary organic aerosol composition and volatility using a FIGAERO-CIMS, Atmos. Chem. Phys., 2018, 18, 15535–15553 CrossRef CAS.
- C. G. Masoud and L. Hildebrandt Ruiz, Chlorine-Initiated Oxidation of α-Pinene: Formation of Secondary Organic Aerosol and Highly Oxygenated Organic Molecules, ACS Earth Space Chem., 2021, 5, 2307–2319 CrossRef CAS.
- P. L. Tanaka, S. Oldfield, J. D. Neece, C. B. Mullins and D. T. Allen, Anthropogenic sources of chlorine and ozone formation in urban atmospheres, Environ. Sci. Technol., 2000, 34, 4470–4473 CrossRef CAS.
- C. B. Faxon, J. Bean and L. Hildebrandt Ruiz, Inland Concentrations of Cl2 and ClNO2 in Southeast Texas Suggest Chlorine Chemistry Significantly Contributes to Atmospheric Reactivity, Atmosphere, 2015, 6, 1487–1506 CrossRef CAS.
- H. D. Osthoff, J. M. Roberts, A. R. Ravishankara, E. J. Williams, B. M. Lerner, R. Sommariva, T. S. Bates, D. Coffman, P. K. Quinn, J. E. Dibb, H. Stark, J. B. Burkholder, R. K. Talukdar, J. Meagher, F. C. Fehsenfeld and S. S. Brown, High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 2008, 1, 324–328 CrossRef CAS.
- G. J. Phillips, M. J. Tang, J. Thieser, B. Brickwedde, G. Schuster, B. Bohn, J. Lelieveld and J. N. Crowley, Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 2012, 9(10), L10811 Search PubMed.
- C. Masoud, M. Modi, N. Bhattacharyya, L. Jahn, K. McPherson, P. Abue, K. Patel, D. Allen and L. Hildebrandt Ruiz, High chlorine concentrations in an unconventional oil and gas development region and impacts on atmospheric chemistry, Environ. Sci. Technol. Lett., 2023,(Submitted) Search PubMed.
- C. B. Faxon and D. T. Allen, Chlorine chemistry in urban atmospheres: a review, Environ. Chem., 2013, 10, 221–233 CrossRef CAS.
- C. B. Faxon, S. V. Dhulipala, D. T. Allen and L. Hildebrandt Ruiz, Heterogeneous production of Cl2 from particulate chloride: effects of composition and relative humidity, AIChE J., 2018, 64, 3151–3158 CrossRef CAS.
- A. T. Ahern, L. Goldberger, L. Jahl, J. Thornton and R. C. Sullivan, Production of N2O5 and ClNO2 through Nocturnal Processing of Biomass-Burning Aerosol, Environ. Sci. Technol., 2018, 52, 550–559 CrossRef CAS PubMed.
- S. M. McNamara, Q. Chen, J. Edebeli, K. D. Kulju, J. Mumpfield, J. D. Fuentes, S. B. Bertman and K. A. Pratt, Observation of N2O5 Deposition and ClNO2 Production on the Saline Snowpack, ACS Earth Space Chem., 2021, 5, 1020–1031 CrossRef CAS.
- D. Mitroo, T. E. Gill, S. Haas, K. A. Pratt and C. J. Gaston, ClNO2 Production from N2O5 Uptake on Saline Playa Dusts: New Insights into Potential Inland Sources of ClNO2, Environ. Sci. Technol., 2019, 53, 7442–7452 CrossRef CAS PubMed.
- S. Gani, S. Bhandari, S. Seraj, D. S. Wang, K. Patel, P. Soni, Z. Arub, G. Habib, L. Hildebrandt Ruiz and J. S. Apte, Submicron aerosol composition in the world's most polluted megacity: the Delhi Aerosol Supersite study, Atmos. Chem. Phys., 2019, 19, 6843–6859 CrossRef CAS.
- R. Atkinson, Gas-phase tropospheric chemistry of organic compounds: a review, Atmos. Environ., 2007, 41, 200–240 CrossRef.
- C. J. Young, R. A. Washenfelder, P. M. Edwards, D. D. Parrish, J. B. Gilman, W. C. Kuster, L. H. Mielke, H. D. Osthoff, C. Tsai, O. Pikelnaya, J. Stutz, P. R. Veres, J. M. Roberts, S. Griffith, S. Dusanter, P. S. Stevens, J. Flynn, N. Grossberg, B. Lefer, J. S. Holloway, J. Peischl, T. B. Ryerson, E. L. Atlas, D. R. Blake and S. S. Brown, Chlorine as a primary radical: evaluation of methods to understand its role in initiation of oxidative cycles, Atmos. Chem. Phys., 2014, 14, 3427–3440 CrossRef.
- C. Song, K. Na and D. R. Cocker, Impact of the hydrocarbon to NOx ratio on secondary organic aerosol formation, Environ. Sci. Technol., 2005, 39, 3143–3149 CrossRef CAS PubMed.
- R. D. Thaler, L. H. Mielke and H. D. Osthoff, Quantification of Nitryl Chloride at Part Per Trillion Mixing Ratios by Thermal Dissociation Cavity Ring-Down Spectroscopy, Anal. Chem., 2011, 83, 2761–2766 CrossRef CAS PubMed.
- N. L. Ng, S. C. Herndon, A. Trimborn, M. R. Canagaratna, P. L. Croteau, T. B. Onasch, D. Sueper, D. R. Worsnop, Q. Zhang, Y. L. Sun and J. T. Jayne, An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol, Aerosol Sci. Technol., 2011, 45, 780–794 CrossRef CAS.
- B. H. Lee, F. D. Lopez-Hilfiker, C. Mohr, T. K. Kurtén, D. R. Worsnop and J. A. Thornton, An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 2014, 48, 6309–6317 CrossRef CAS PubMed.
- F. D. Lopez-Hilfiker, C. Mohr, M. Ehn, F. Rubach, E. Kleist, J. Wildt, T. F. Mentel, A. Lutz, M. Hallquist, D. Worsnop and J. A. Thornton, A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 2014, 7, 983–1001 CrossRef.
- W. P. L. Carter, Development of a condensed SAPRC-07 chemical mechanism, Atmos. Environ., 2010, 44, 5336–5345 CrossRef CAS.
- P. L. Tanaka, D. T. Allen, E. C. McDonald-Buller, S. Chang, Y. Kimura, C. B. Mullins, G. Yarwood and J. D. Neece, Development of a chlorine mechanism for use in the carbon bond IV chemistry model, J. Geophys. Res.: Atmos., 2003, 108(D4) DOI:10.1029/2002jd002432.
- W. P. L. Carter, D. R. Cocker, D. R. Fitz, I. L. Malkina, K. Bumiller, C. G. Sauer, J. T. Pisano, C. Bufalino and C. Song, A new environmental chamber for evaluation of gas-phase chemical mechanisms and secondary aerosol formation, Atmos. Environ., 2005, 39, 7768–7788 CrossRef CAS.
- M. Huang, W. Zhang, X. Gu, C. Hu, W. Zhao, Z. Wang and L. Fang, Size distribution and chemical composition of secondary organic aerosol formed from Cl-initiated oxidation of toluene, J. Environ. Sci., 2012, 24, 860–864 CrossRef CAS PubMed.
- C. Bloss, V. Wagner, M. E. Jenkin, R. Volkamer, W. J. Bloss, J. D. Lee, D. E. Heard, K. Wirtz, M. Martin-Reviejo, G. Rea, J. C. Wenger and M. J. Pilling, Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 2005, 5, 641–664 CrossRef CAS.
- R. H. Schwantes, K. A. Schilling, R. C. McVay, H. Lignell, M. M. Coggon, X. Zhang, P. O. Wennberg and J. H. Seinfeld, Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 2017, 17, 3453–3474 CrossRef CAS.
- A. W. Birdsall, J. F. Andreoni and M. J. Elrod, Investigation of the Role of Bicyclic Peroxy Radicals in the Oxidation Mechanism of Toluene, J. Phys. Chem. A, 2010, 114, 10655–10663 CrossRef CAS PubMed.
- M. M. Maricq, J. J. Szente, E. W. Kaiser and J. Shi, Reaction of Chlorine Atoms with Methylperoxy and Ethylperoxy Radicals, J. Phys. Chem., 1994, 98, 2083 CrossRef CAS.
- R. S. Karlsson, J. J. Szente, J. C. Ball and M. M. Maricq, Homogeneous aerosol formation by the chlorine atom initiated oxidation of toluene, J. Phys. Chem. A, 2001, 105, 82–96 CrossRef CAS.
- N. L. Ng, M. R. Canagaratna, Q. Zhang, J. L. Jimenez, J. Tian, I. M. Ulbrich, J. H. Kroll, K. S. Docherty, P. S. Chhabra, R. Bahreini, S. M. Murphy, J. H. Seinfeld, L. Hildebrandt, N. M. Donahue, P. F. Decarlo, V. A. Lanz, A. S. H. Prévôt, E. Dinar, Y. Rudich and D. R. Worsnop, Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 2010, 10, 4625–4641 CrossRef CAS.
- C. L. Loza, P. S. Chhabra, L. D. Yee, J. S. Craven, R. C. Flagan and J. H. Seinfeld, Chemical aging of m-xylene secondary organic aerosol: laboratory chamber study, Atmos. Chem. Phys., 2012, 12, 151–167 CrossRef CAS.
- P. S. Chhabra, N. L. Ng, M. R. Canagaratna, A. L. Corrigan, L. M. Russell, D. R. Worsnop, R. C. Flagan and J. H. Seinfeld, Elemental composition and oxidation of chamber organic aerosol, Atmos. Chem. Phys., 2011, 11, 8827–8845 CrossRef CAS.
- M. L. Hinks, J. Montoya-Aguilera, L. Ellison, P. Lin, A. Laskin, J. Laskin, M. Shiraiwa, D. Dabdub and S. A. Nizkorodov, Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene, Atmos. Chem. Phys., 2018, 18, 1643–1652 CrossRef CAS.
- T. B. Nguyen, P. J. Roach, J. Laskin, A. Laskin and S. A. Nizkorodov, Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 2011, 11, 6931–6944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ea00024a |
| This journal is © The Royal Society of Chemistry 2023 |