
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and computational study of the exchange interaction between the V(III) centers in the vanadium-cyclal dimer†
Andreas
Reiß
a,
Maximilian Kai
Reimann
 b,
Chengyu
Jin
c,
Martha
Wachter-Lehn
d,
Reinhard K.
Kremer
e,
Rainer
Pöttgen
b,
Karin
Fink
c,
Wim
Klopper
*cd and
Claus
Feldmann
*a
b,
Chengyu
Jin
c,
Martha
Wachter-Lehn
d,
Reinhard K.
Kremer
e,
Rainer
Pöttgen
b,
Karin
Fink
c,
Wim
Klopper
*cd and
Claus
Feldmann
*a
aInstitut für Anorganische Chemie, Karlsruhe Institute of Technology (KIT), Engesserstraße 15, 76131 Karlsruhe, Germany. E-mail: claus.feldmann@kit.edu
bInstitut für Anorganische und Analytische Chemie, Universität Münster, Corrensstraße 30, 48149 Münster, Germany
cInstitut für Nanotechnologie, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInstitut für Physikalische Chemie, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany
eMax-Planck-Institut für Festkörperforschung, Heisenbergstraße 1, 70569 Stuttgart, Germany
First published on 3rd November 2023
Abstract
[V2(HCyclal)2] is prepared by controlled oxidation of vanadium nanoparticles at 50 °C in toluene. The V(0) nanoparticles are synthesized in THF by reduction of VCl3 with lithium naphthalenide. They exhibit very small particle sizes of 1.2 ± 0.2 nm and a high reactivity (e.g. with air or water). By reaction of V(0) nanoparticles with the azacrown ether H4Cyclal, [V2(HCyclal)2] is obtained with deep green crystals and high yield. The title compound exhibits a V(III) dimer (V⋯V: 304.1(1) pm) with two deprotonated [HCyclal]3− ligands as anions. V(0) nanoparticles as well as the sole coordination of V(III) by a crown ether as the ligand and nitrogen as sole coordinating atom are shown for the first time. Magnetic measurements and computational results point to antiferromagnetic coupling within the V(III) couple, establishing an antiferromagnetic spin S = 1 dimer with the magnetic susceptibility determined by the thermal population of the total spin ranging from ST = 0 to ST = 2.
Introduction
Binuclear coordination compounds with M⋯M dimers (M: metal) and certain binding between the metal atoms are well-known and have been intensely studied.1 Prominent examples comprise Mg–Mg, Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Re, W
Re, W![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) W, or Cr
W, or Cr![[quadruple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e003.gif) Cr dimers with the respective single to fourfold bonding.2 Typical methods to prove the type of metal–metal interaction are crystal-structure analysis (e.g. evaluation of M⋯M distance), magnetism (e.g. presence/absence of non-paired electrons), or computational results (e.g. analysis of bonding situation).1,2 In the case of vanadium, a wide variety of binuclear compounds ranging from V(I),3 V(II)4via V(III)5 to V(IV)6 with different interaction of the vanadium dimer was reported. Most often chalcogenide-based ligands were used to coordinate and to stabilize the V2 dimer. As a synthesis approach, the reduction of vanadium species with a higher oxidation state (most often V(IV), V(V)) is the most widely applied.3–6
Cr dimers with the respective single to fourfold bonding.2 Typical methods to prove the type of metal–metal interaction are crystal-structure analysis (e.g. evaluation of M⋯M distance), magnetism (e.g. presence/absence of non-paired electrons), or computational results (e.g. analysis of bonding situation).1,2 In the case of vanadium, a wide variety of binuclear compounds ranging from V(I),3 V(II)4via V(III)5 to V(IV)6 with different interaction of the vanadium dimer was reported. Most often chalcogenide-based ligands were used to coordinate and to stabilize the V2 dimer. As a synthesis approach, the reduction of vanadium species with a higher oxidation state (most often V(IV), V(V)) is the most widely applied.3–6
As a part of our studies on the synthesis of base-metal nanoparticles and their reactivity,7 we have realized zerovalent vanadium (V(0)) nanoparticles, which we present here for the first time. The synthesis of V(0) nanoparticles follows our general approach of reducing metal halides after dissolution in tetrahydrofuran (THF) with a solution of lithium or sodium naphthalenide ([LiNaph], [NaNaph]) in THF. This liquid-phase synthesis typically results in 1–5 nm-sized metal nanoparticles and is suitable for a large group of base-metal nanoparticles (i.e. Mg, Al, all group IIIb to group VIIb transition metals, all rare-earth metals from Sc to Lu).7,8 At present, the knowledge on V(0) nanoparticles is low and limited to gas-phase methods.9 Moreover, a stabilization of the metal core against oxidation by a vanadium carbide shell as passivating layer was described.10 However, a reliable synthesis strategy for V(0) nanoparticles <10 nm is unknown, so far. This situation can be also related to the absence of chemically stable vanadium carbonyls as starting materials, which, therefore, cannot be thermally decomposed in the liquid phase as in the case of other transition metals (e.g., Cr, Mn, Fe, Co).11
With this study, we present a reliable liquid-phase synthesis of V(0) nanoparticles with a size <10 nm. To evaluate their reactivity in the liquid phase near room temperature (≤100 °C),12 we exemplarily show the reaction and reactivity of the V(0) nanoparticles with an azacrown ether to obtain single crystals of [V2(HCyclal)2]. [V2(HCyclal)2] contains a dimer of V(III) cations, which show antiferromagnetic coupling and a rare coordination of vanadium, only with the azacrown ether as a ligand and nitrogen as sole donor atom. The exchange interaction between the V(III) centers is analysed based on magnetic measurements and computational studies.
Experimental section
General
All sample handling and reactions were performed under argon atmosphere using standard Schlenk techniques and gloveboxes (MBraun Unilab, O2/H2O < 0.1 ppm). Reactions were carried out in Schlenk flasks or glass ampoules. Prior to use, all glassware was evacuated (p ≤ 10−3 mbar), heated, and flushed with argon thrice to remove all moisture.Chemicals
Tetrahydrofurane (THF, Seulberger, 99%) and toluene (Seulberger, 99%) were refluxed over sodium with benzophenone and distilled off prior to use. Lithium metal (Alfa Aesar, 99%) was freshly cut under argon atmosphere prior to use. 1,4,8,12-Tetraazacyclopentadecane (H4Cyclal, Sigma-Aldrich, 97%), naphthalene (Alfa Aesar, ≥99%), and VCl3 (Alfa Aesar, 99%) were used as purchased.V(0) nanoparticles
16.7 mg of lithium (2.40 mmol), 323.0 mg of naphthalene (2.52 mmol), and 125.8 mg of VCl3 (0.80 mmol) were dissolved in 15 mL of THF and stirred for 12 hours, resulting in a deep black suspension. The as-prepared V(0) nanoparticles were separated by centrifugation (45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g) and purified by washing twice with 15 mL of THF. Finally, the V(0) nanoparticles were redispersed in THF to obtain long-term stable suspensions or dried in vacuum to obtain powder samples with a yield of about 70%.
000g) and purified by washing twice with 15 mL of THF. Finally, the V(0) nanoparticles were redispersed in THF to obtain long-term stable suspensions or dried in vacuum to obtain powder samples with a yield of about 70%.
V2(HCyclal)2 (1)
25.7 mg of dried V(0) nanoparticles (0.51 mmol), 108.4 mg of H4Cyclal (0.51 mmol), and 0.4 mL of toluene were filled into a Schlenk tube. The Schlenk tube was heated to 50 °C for 7 days. After natural cooling to room temperature, deep green crystals of 1 were obtained with a yield of about 75% in relation to the employed amount of H4Cyclal. The title compound is sensitive to air and moisture and readily oxidized, generating a brown, amorphous crust covering the crystal surfaces of 1.Analytical equipment
Details related to analytical techniques, crystallographic data, and computational methods are described in the ESI.†Results and discussion
Synthesis and characterization of V(0) nanoparticles
Zerovalent vanadium nanoparticles were prepared utilizing a general synthesis strategy to realize zerovalent base-metal nanoparticles via naphthalenide-driven reduction of simple metal halides, which we established recently.7,8 To obtain zerovalent vanadium (V(0)) nanoparticles, VCl3 was used as the starting material, lithium naphthalenide ([LiNaph]) as the reducing agent, and tetrahydrofuran (THF) as the solvent (Fig. 1). Due to the low solubility of VCl3 in THF, VCl3, lithium metal and naphthalene were added to THF in a one-pot approach (Fig. 1a). Thus, VCl3 largely remains as an insoluble solid at the bottom of the Schlenk tube, and lithium metal is located on top of the liquid phase due to its low density. Upon intense stirring over 12 hours, [LiNaph] is formed resulting in a greenish solution. VCl3 is slowly dissolved, but – after dissolution – instantaneously reacts with [LiNaph] to form V(0) nanoparticles. Despite these – from a general perspective – non-optimal conditions for nanoparticle nucleation (i.e., lithium and VCl3 as solid phases instead of a homogeneous solution), two essential aspects nevertheless promote the nucleation of very small nanoparticles. First, the reduction of VCl3 is very fast. Second, the solubility of zerovalent vanadium in THF is extremely low, so that a high supersaturation is reached. In sum, VCl3 and lithium metal have reacted completely after 12 hours of vigorous stirring, resulting in a deep black suspension of V(0) nanoparticles (Fig. 1b). | ||
| Fig. 1 Scheme illustrating the synthesis of V(0) nanoparticles with (a) starting materials, (b) the as-prepared V(0) nanoparticles in THF, (c) single crystal and unit cell of [V2(HCyclal)2] after liquid-phase oxidation of the V(0) nanoparticles. | ||
The V(0) nanoparticles were purified by centrifugation and repeated redispersion/centrifugation in/from THF to remove LiCl and naphthalene. Thereafter, the nanoparticles can be redispersed either in THF or in toluene to obtain colloidally stable suspensions, or they can be dried in vacuum to obtain powder samples. For their handling, caution needs to be paid since the V(0) nanoparticle suspensions and powder samples are highly reactive when in contact to O2, H2O or other oxidizing agents. Specifically, the reaction of powder samples with oxygen and/or water can result in violent reactions and explosions. Consequently, the V(0) nanoparticles need to be handled and stored under inert conditions (argon, nitrogen).
Particle size and shape of the V(0) nanoparticles were examined by transmission electron microscopy (TEM). Accordingly, spherical nanoparticles with uniform size were obtained with a size range of 1–2 nm and a low degree of agglomeration (Fig. 2a). Statistical evaluation of >200 nanoparticles on TEM images results in a mean diameter of 1.2 ± 0.2 nm (Fig. 2c). High-resolution (HR)TEM images prove the as-prepared V(0) nanoparticles to be monocrystalline with lattice fringes through the whole particle (Fig. 2b). The lattice plane distance of 2.1 ± 0.1 Å is in good agreement with cubic bulk vanadium (V(0): d110 with 2.1 Å).13 Composition and surface functionalization of the as-prepared V(0) nanoparticles were further examined by Fourier-transform infrared (FT-IR) spectroscopy and element analysis (EA). FT-IR spectra, as expected, indicate the solvent THF to be adsorbed on the particle surface (Fig. 2d). The observed weak vibrations relate to THF (ν(C–H): 3000–2800 cm−1, ν(C–O): 1100–900 cm−1, fingerprint area: 1500–500 cm−1). EA indicates C and H contents of 53.4 and 4.1 wt% with a C:H ratio of 13, which is higher as expected for THF (C:H = 6). This indicates certain amount of naphthalene also to be adhered on the particle surface. The comparably high C/H content is to be expected in view of the small particle size (1.2 ± 0.2 nm) and a monomolecular adsorption layer. Finally, it must be noticed that the synthesis of V(0) nanoparticles with [LiNaph] is the first liquid-phase synthesis of vanadium nanoparticles with a diameter <10 nm until now.
 | ||
| Fig. 2 Characterization of the as-prepared V(0) nanoparticles: (a) TEM overview image, (b) single V(0) nanoparticle with lattice fringes, (c) size distribution (based on statistical evaluation of >200 nanoparticles on TEM images), (d) FT-IR spectrum (with THF and naphthalene as references). | ||
Synthesis and structure of [V2(HCyclal)2]
The reaction of, for instance, oxygen or water already points to the high reactivity of the as-prepared V(0) nanoparticles. As expected, the reactivity of the V(0) nanoparticles is much higher as compared to bulk vanadium. This high reactivity already indicates the absence of passivation layers (e.g. vanadium oxides/hydroxides). Violent reactions such as in air or in water, however, are not controlled in terms of reproducible conditions or the formation of a specific compound. To probe the reactivity of the V(0) nanoparticles and to perform a reaction in a controlled manner, we have reacted them in the liquid phase near room temperature (≤100 °C) with the azacrown ether 1,4,8,12-tetraazacyclopentadecane (H4Cyclal) as a mild oxidizing agent and sterically demanding ligand. This seemed specifically promising in regard of the different oxidation states of vanadium between ±0 and +V as well as in regard of a potential metal–metal interaction. For this purpose, the V(0) nanoparticles were reacted with H4Cyclal at 50 °C in toluene for 7 days, which results in the formation of deep green, moisture- and air-sensitive single crystals of V2(HCyclal)2 with visible evolution of hydrogen (Fig. 1c). The title compound was obtained with a yield of about 75% (in relation the applied amount of V(0) nanoparticles). The synthesis can be ascribed to the following reaction:| 2V(0) + 2H4Cyclal → [V2(HCyclal)2] + 3H2 |
According to single-crystal structure analysis, [V2(HCyclal)2] crystallizes in the monoclinic space group P21/n (ESI: Table S1 and Fig. S1†). The title compound is composed of a binuclear vanadium-cyclal complex, in which each V center is trigonal-bipyramidally coordinated by four nitrogen atoms of one azacrown ether and one nitrogen atom of the second azacrown ether (Fig. 3; ESI: Fig. S2†). Furthermore, one nitrogen atom of both azacrown ethers shows μ2-bridging of the V2 couple. Here, it needs to be noticed that the azacrown ether is disordered in the crystal structure, so that the propyl and ethyl groups of the azacrown ether cannot be localized in a specific position. In the structure refinement, this was taken care of by assuming split-atom positions with 50% occupancy for each atom (ESI: Fig. S3†). The V–N distances amount to 209.9(9) pm (N1′) and 215.0(10) pm (N1) within the V2N2 ring as well as to 191.8(7) (N2), 200.9(13) (N3), and 224.3(5) (N4) to the non-bridging N atoms. The comparably high standard deviations observed for the V–N distances can be attributed to the aforementioned disorder of the azacrown-ether ligands. The angles in the V2N2 quadrangle (N1–V1–N1′: 88.6(4), V1–N1–V1′: 91.4(4)°) are in accordance with the different distances and the deviation from an ideal squared arrangement. As expected, the bridging N atoms (N1, N1′) show larger V–N distances than N2 and N3. The longest V–N distance, however, is observed for N4 and – together with the V–N–C angles of 109.4(6)–116.0(6)° – indicates a tetrahedral arrangement with a non-protonated NH group. In contrast, N1 to N3 are deprotonated as indicated by V–N–C angles of 115.3(5)–134.2(7)°, corresponding to a distorted trigonal planar coordination. In sum, this points to the presence of an (HCyclal)3− ligand and, vice versa, an oxidation state of +III of vanadium. This oxidation state is also in accordance with the deep green color of the title compound (Fig. 1c) and further validated by magnetic measurements and computational analysis (see below). A coordination of vanadium by crown ethers as a ligand is rare, even with the most conventional crown ethers 15-crown-5 or 18-crown-6.14 Most often thiol crown ethers were used as ligands.15 A coordination with azacrown ethers was only reported for [V(IV)O(cyclam)Cl]Cl16 and [(cyclen)V(III)(CN)3],17 which both represent mononuclear coordination compounds with additional ligands coordinated to the vanadium center. Moreover, [V(IV)O(cyclam)Cl]Cl and [(cyclen)V(III)(CN)3] were prepared via Lewis-acid–base reactions with VO(SO4) or VCl3 as starting materials, and the azacrown ether serves as neutral ligand without any deprotonation.16,17
 | ||
| Fig. 3 Molecular structure of [V2(HCyclal)2] (disorder of azacrown ether and H atoms not shown for clarity). | ||
Beside single-crystal structure analysis, structure and purity of the title compound were validated by X-ray powder diffraction (ESI: Fig. S4†). To verify the presence of a remaining NH group of the azacrown ether, Fourier-transform infrared (FT-IR) spectroscopy was performed and compared to the pure ligand H4Cyclal (Fig. 4). In general, all vibrations of the ligand are visible for the title compound. Intensity, splitting, and position of the vibrations are of course slightly different to the free, non-bound ligand due to the different point symmetry and binding situation. In regard of the N–H vibrations at 3300–3000 cm−1, the intensity is significantly lower for [V2(HCyclal)2] as for free H4Cyclal. However, the N–H vibrations are still visible, which is in accordance with a partial deprotonation of the azacrown ether (Fig. 4). Together with the tetrahedral arrangement of a sole nitrogen atom, FT-IR spectra confirms the presence of a remaining NH-group of the (HCyclal)3− anion.
 | ||
| Fig. 4 FT-IR spectra of V2(HCyclal)2 and H4Cyclal as a reference. | ||
The V–V distance in [V2(HCyclal)2] is 304.1(1) pm, which indicates an interaction of the V(III) dimer. In comparison to other dinuclear vanadium units, however, this distance is at the upper end of observed separation (Table 1). Most often dimers of V(II) were reported that exhibit a significantly shorter threefold VV bond. V(III) dimers, typically with chalcogenide-type ligands, also exhibit distances below about 290 pm (Table 1). In this regard, it needs to be noticed that [V2(HCyclal)2] is the only V(III) dimer with sole coordination by nitrogen.
| Compound | V–V distance/pm | Oxidation state of V |
|---|---|---|
| DPhF: N,N′-diphenylformamidine; hpp: 1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidin; DFM: N,N′-di-(p-tolyl)formamidinate. | ||
| K(THF)3[V+IV+II(DPhF)4]3 | 192.95(8) | +I/+II |
| [V+II2(hpp)4]4a | 193.2(1) | +II |
| [V+II2(DFM)4]4b | 197.5(4) | |
| [V+II2(2,6-dimethoxyphenyl)4]4c | 220.0(2) | |
| [V+III2(salophen)2Na2(THF)6]5a | 240.6(3) | +III |
| [V+III6Se8O(PMe3)6]5b | 279.4–283.9 | |
| [(π-MeC5H4)4V+III4(μ3-S)4]5c | 286.5–287.0 | |
| [V+III2(HCyclal)2] (title compound) | 304.1(1) | |
| [(π-MeC5H4)5V+III4V+IV(μ3-S)6]5d | 305.9(2)–321.7(2) | +III/+IV |
| [V+IV2(μ-S2)2{(C4H9)2NCS2}4]6a | 285.1 | +IV |
| [V+IV2(S2)2(S2CNEt2)4]6b | 288.4 | |
Magnetic properties of [V2(HCyclal)2]
To verify the interaction within the V2 dimer in [V2(HCyclal)2], magnetic measurements were performed with the molar susceptibility determined in an external magnetic field of 10 kOe (Fig. 5a). Apart from a strong divergence of the susceptibility towards lowest temperatures, which can be ascribed to Curie-like susceptibility contributions from free V(III) entities, the increase of the magnetic susceptibility with increasing temperature is characteristic and indicates a gradual thermal population of excited states with a magnetic moment. | ||
| Fig. 5 Temperature-dependent molar susceptibility of [V2(HCyclal)2]: (a) experimental molar susceptibility per two V(III) centers (filled black circles) and calculation (red solid line, based on eqn (1) (solid blue line) to which a Curie–Weiss term (black dashed line) of the form c/(T − Tc) was added (c = 0.02 cm3 mol−1 K, Tc = 33 K) with antiferromagnetic spin exchange of J = −299 K, a g-factor of 2.06, and a temperature-independent contribution (TIC) of χ0 = 1.04 × 10−4 cm3 mol−1. The turquoise solid line shows the result from fitting only J = −289 K with the program PHI, without TIC but with a paramagnetic triplet impurity of 6%; (b) energy diagram for a S = 1 dinuclear unit with an antiferromagnetic spin exchange of J = −299 K. (c) Magnetization isotherms of [V2(HCyclal)2] recorded at 3.1, 10 and 50 K. The red lines correspond to fits according to an S = 1 Brillouin function. The refined concentration of free V(III) is indicated and consistent with the data in (a). | ||
To describe the susceptibility of the title compound, each V(III) cation is assumed to have a S = 1 state, i.e., an orbital singlet of the 3F d2 ground state and an antiferromagnetic spin–spin coupling of the nearby V(III) cations, 302 pm in distance, to a dinuclear unit. The Hamiltonian of such dinuclear unit is given by:

The magnetic susceptibility of such a spin S = 1 dinuclear unit (for two V(III) centers) is given by:18
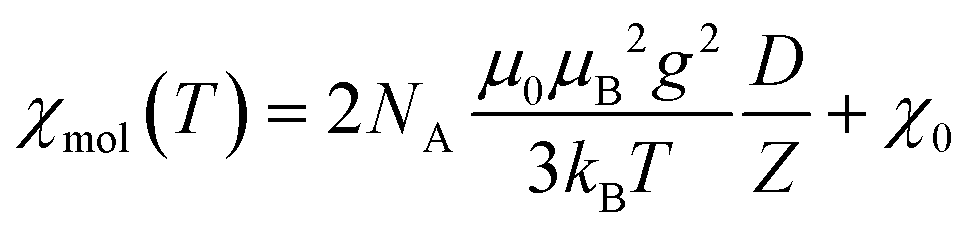 | (1a) |
| D = {3exp(2x) + 15exp(6x)} | (1b) |
| Z = {1 + 3exp(2x) + 5exp(6x)} | (1c) |
 | (1d) |
Computation of exchange interactions
The experimental findings are confirmed by ab initio quantum-chemical calculations, both by density-functional-theory (DFT) calculations and configuration-interaction (CI) calculations (see ESI† for details). The calculations reveal that the dinuclear V(III) complex [V2(HCyclal)2] can be described perfectly well in terms of two antiferromagnetically coupled 3d2 centers with spins S1 = S2 = 1.DFT calculations were performed at the B3LYP-D3(BJ)-abc level using the def2-TZVPP basis set for vanadium and nitrogen, the def2-TZVP basis for carbon, and the def2-SV(P) basis for hydrogen (ESI: Table S2†). Since the open-shell singlet ground state of the complex cannot be described by a single Kohn–Sham determinant, we have approximated the energy of the ground state (GS) as a linear combination of the energies of the high-spin (HS) determinant (with MS1 = 1 and MS2 = 1; 〈Ŝ2〉 ≈ 6ħ2) and a broken-symmetry (BS, Fig. 6) determinant with MS1 = 1, MS2 = −1, and 〈Ŝ2〉 ≈ 2ħ2:
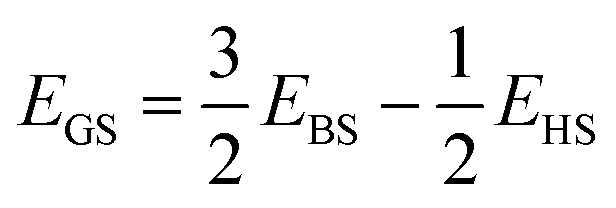 | (2) |
 | ||
| Fig. 6 Spin density (plotted at an isovalue of ±0.02a0−3) of the broken-symmetry Kohn–Sham determinant as obtained at the B3LYP-D3(BJ)-abc level (H atoms omitted for clarity). | ||
This equation follows from assuming that the relevant 〈Ŝ2〉 expectation values are equal to 6ħ2 and 2ħ2, thereby neglecting the dependence of the geometry on these expectation values.21–23
After optimizing the ground-state geometry, which displays Ci point-group symmetry, the exchange coupling parameter was computed according to the formula:
 | (3) |
At the B3LYP-D3(BJ)-abc level, we obtained J = −251 K at the optimized ground-state geometry. We have repeated the DFT calculations using the TPSSh functional (ESI: Table S3†), but the geometry optimization with this functional yielded a much too short V–V distance (Table 2). Therefore, we decided to adopt the B3LYP optimized structure for our investigations at the CI level. Note that also the N1–V–N1′ and V1–N1–V1′ angles (being decisive for the strength of the exchange coupling) of the B3LYP geometry agree much better with those of the experimental X-ray structure than the TPSSh ones.
| Parameter | TPSSh | B3LYP | Exptl. |
|---|---|---|---|
| V1–V1 distance (pm) | 288.6 | 301.4 | 304.1(1) |
| V1–N1 distance (pm) | 211.9 | 214.7 | 215.8(10) |
| V1–N1′ distance (pm) | 207.1 | 208.6 | 209.9(9) |
| V1–N2 distance (pm) | 194.9 | 195.1 | 191.8(7) |
| V1–N3 distance (pm) | 192.6 | 193.3 | 200.9(13) |
| V1–N4 distance (pm) | 225.2 | 227.1 | 224.3(5) |
| N1–V–N1′ angle (°) | 92.9 | 89.2 | 88.6(4) and 89.0(4) |
| V1–N1–V1′ angle (°) | 87.1 | 90.8 | 91.0(4) and 91.4(4) |
Finally, we show localized (majority-spin) spin–orbitals of the [V2(HCyclal)2] complex as obtained for the high-spin state (MS = 2). Fig. 7 shows the coordination of one of the two equivalent V(III) centers. The same orbitals are also found on the other V(III) center. Each V(III) center has two singly occupied 3d orbitals and is coordinated by five N atoms, which all contribute one σ-donor orbital. The atoms N1, N1′ and N4 are sp3-hybridized and only contribute a single σ-donor orbital. The atoms N2 and N3, however, are sp2-hybridized and their lone-pair 2p-orbital shows π-donating character towards to V(III) center, leading to V1–N2 and V1–N3 distances that are about 23–25 pm shorter than the other three V1–N bond distances.
 | ||
| Fig. 7 Localized spin orbitals that describe the bonding situation in the [V2(HCyclal)2] complex, plotted at an isovalue of ±0.05a0−3/2: (a) first singly occupied 3d orbital on V1. (b) Second singly occupied 3d orbital on V1. (c) Two 2p orbitals on N2 and N3 with π-donating character. (d) Five σ-donor orbitals (of which four are visible). | ||
Based on the B3LYP-D3(BJ)-abc optimized structure of the singlet state, modified complete-active-space configuration-interaction (modified CASCI) calculations24 were performed in a (4,4) active space of the partly occupied 3d orbitals with the complete-active-space spin–orbit configuration interaction (CASOCI)25 program (ESI: Table S4†). The charge-transfer states were shifted down by 11.48 eV. The calculated susceptibilities are compared to the measured data (ESI: Fig. S5†). This approach yielded a magnetic exchange coupling constant of J = 247 K. Assuming a paramagnetic impurity of 6% by a potential V(III) monomer in a triplet state, J was fitted with the program PHI26 to the experimental data with J = −289 K as best value. Though the calculated and fitted values of the magnetic exchange constants show some differences in the exact value, all of them show a strong antiferromagnetic coupling of the two vanadium centers with an overall singlet ground state. At temperatures below 50 K, the occupation of higher states is neglectable (ESI: Table S5†). The strong increase below 50 K must be connected to a paramagnetic impurity. Even at higher temperatures, the absolut value of the magnetic susceptibility remains rather small due to the strong coupling. At 300 K, 69% of the [V2(HCyclal)2] molecules are in the singlet ground state, 30% in the triplet state, and 1% in the quintet state (ESI: Table S5†). This corresponds to χT = 0.34 cm3 K mol−1. Combining with 6% of a paramagnetic impurity and 94% of V2(HCyclal)2, χT = 0.38 cm3 K mol−1, is in good agreement with the experimental value.
Conclusion
[V2(HCyclal)2] contains a novel V(III) dimer with the vanadium centers coordinated and bridged by deprotonated [HCyclal]3− azacrown ligands. Such coordination of vanadium only by crown ethers as ligands and with nitrogen as sole donor is reported here for the first time. The title compound was prepared by controlled oxidation of vanadium nanoparticles with the azacrown ether H4Cyclal in the liquid phase (toluene) near room temperature (50 °C). Such a redox approach was rarely reported and allows irreversible reactions due to the removal of hydrogen. Whereas bulk vanadium is more-or-less non-reactive at low temperature in the liquid phase, the as-used V(0) nanoparticles are highly suitable as starting material. They were synthesized in THF by reduction of VCl3 with lithium naphthalenide and are characterised by a very small particle size of 1.2 ± 0.2 nm. Such V(0) nanoparticles and their use as a starting material open new options for reactions in the liquid phase. The V(III) dimer (V⋯V: 304.1(1) pm) in [V2(HCyclal)2] exhibits a very strong antiferromagnetic coupling of the two S = 1 centres to a spin-ladder ranging from ST = 0 to ST = 2. Because of the strong coupling, the ST = 2 state remains unoccupied while about 30% of the molecules are in the ST = 1 state at room temperature. The experimental data of magnetic measurements were confirmed by quantum chemical calculations, which both point to an antiferromagnetic coupling within the V(III) dimer.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. R., C. J., K. F., W. K. and C. F. acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through the Collaborative Research Centre “4f for Future” (CRC 1573, project number 471424360), projects A4 and Q. Moreover, the authors thank Prof. Dr Peter Roesky, Dr Michael Gamer and Dr Adrian Hauser for data collection on a Stoe IPDS II diffractometer.References
- J. A. Chipman and J. F. Berry, Chem. Rev., 2020, 120, 2409–2447 CrossRef CAS PubMed.
- (a) S. G. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed; (b) M. Bochmann, G. Wilkinson, A. M. R. Galas, M. B. Hursthouse and K. M. A. Malik, J. Chem. Soc., Dalton Trans., 1980, 1797–1799 RSC; (c) M. L. Green, J. D. Hubert and P. Mountford, J. Chem. Soc., Dalton Trans., 1990, 3793–3800 RSC; (d) J. N. Van Niekerk and F. R. L. Schoening, Nature, 1953, 171, 36–37 CrossRef CAS.
- F. A. Cotton, E. A. Hillard and C. A. Murillo, J. Am. Chem. Soc., 2003, 125, 2026–2027 CrossRef CAS PubMed.
- (a) F. A. Cotton and D. J. Timmons, Polyhedron, 1998, 17, 179–184 CrossRef CAS; (b) F. A. Cotton, L. M. Daniels and C. A. Murillo, Inorg. Chem., 1993, 32, 2881–2885 CrossRef CAS; (c) F. A. Cotton and M. Millar, J. Am. Chem. Soc., 1977, 99, 7886–7891 CrossRef CAS.
- (a) S. Gambarotta, M. Mazzanti, C. Floriani and M. Zehnder, J. Chem. Soc., Chem. Commun., 1984, 1116–1118 RSC; (b) D. Fenske, A. Grissinger, M. Loos and J. Magull, Z. Anorg. Allg. Chem., 1991, 598, 121–128 CrossRef; (c) C. M. Bolinger, J. Darkwa, G. Gammle, S. D. Gammon, J. W. Lyding, T. B. Rauchfuss and S. R. Wilson, Organometallics, 1986, 5, 2386–2388 CrossRef CAS; (d) I. L. Eremenko, A. S. Katugin, A. A. Pasynskii, Y. T. Struchkov and V. E. Shklover, J. Organomet. Chem., 1988, 345, 79–86 CrossRef CAS.
- (a) I. L. Eremenko, A. A. Pasynskii, A. S. Katugin, O. G. Éllert, V. E. Shklover and Y. T. Struchkov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1984, 33, 1531–1532 CrossRef; (b) T. R. Halbert, L. L. Hutchings, R. Rhodes and E. I. Stiefel, J. Am. Chem. Soc., 1986, 108, 6437–6438 CrossRef CAS; (c) S. C. Sendlinger, J. R. Nicholson, E. B. Lobkovsky, J. C. Huffman, D. Rehder and G. Christou, Inorg. Chem., 1993, 32, 204–210 CrossRef CAS.
- C. Schöttle, P. Bockstaller, R. Popescu, D. Gerthsen and C. Feldmann, Angew. Chem., Int. Ed., 2015, 54, 9866–9870 CrossRef PubMed.
- (a) S. Riegsinger, R. Popescu, D. Gerthsen and C. Feldmann, Chem. Commun., 2022, 58, 7499–7502 RSC; (b) D. Bartenbach, O. Wenzel, R. Popescu, L.-P. Faden, A. Reiß, M. Kaiser, A. Zimina, J.-D. Grunwaldt, D. Gerthsen and C. Feldmann, Angew. Chem., Int. Ed., 2021, 60, 17373–17377 CrossRef CAS PubMed; (c) A. Egeberg, L.-P. Faden, A. Zimina, J.-D. Grunwaldt, D. Gerthsen and C. Feldmann, Chem. Commun., 2021, 57, 3648–3651 RSC; (d) A. Egeberg, T. Block, O. Janka, O. Wenzel, D. Gerthsen, R. Pöttgen and C. Feldmann, Small, 2019, 15, 1902321 CrossRef PubMed.
- (a) C.-C. Yang, W.-L. Huang, Y.-H. Lin, C.-Y. Wenig, Z.-Y. Mo and Y.-Y. Chen, IEEE Trans. Magn., 2011, 47, 3535–3537 CAS; (b) A. B. Phillips, G. Myeni and B. S. Shivaram, AIP Conf. Proc., 2006, 837, 250–254 CrossRef CAS.
- R. Zacharia, K. Y. Kim, A. K. M. Fazle Kibria and K. S. Nahm, Chem. Phys. Lett., 2005, 412, 369–375 CrossRef CAS.
- (a) C. Janiak, Z. Naturforsch., B: Chem. Sci., 2013, 68, 1059–1089 CrossRef CAS; (b) E. Redel, R. Thomann and C. Janiak, Chem. Commun., 2008, 1789–1791 RSC.
- (a) E. Ahmed and M. Ruck, Coord. Chem. Rev., 2011, 255, 2892–2903 CrossRef CAS; (b) M. C. Buzzeo, R. G. Evans and R. G. Compton, ChemPhysChem, 2004, 5, 1106–1120 CrossRef CAS PubMed.
- H. E. Swanson, H. F. McMurdie, M. C. Morris, E. H. Evans, B. Paretzkin, J. H. DeGroot and S. J. Carmel, Natl. Bur. Stand. (U.S.) Monogr., 1971, 9, 58 Search PubMed.
- (a) G. Frenzen, W. Massa, T. Ernst and K. Dehnicke, Z. Naturforsch., B: Chem. Sci., 1990, 45, 1393–1397 CrossRef CAS; (b) M. Plate, G. Frenzen and K. Dehnicke, Z. Naturforsch., B: Chem. Sci., 1993, 48, 149–155 CrossRef CAS; (c) U. Kynast, S. G. Bott and J. L. Atwood, J. Coord. Chem., 1988, 17, 53–61 CrossRef CAS.
- (a) C. D. Beard, L. Carr, M. F. Davis, J. Evans, W. Levason, L. D. Norman, G. Reid and M. Webster, Eur. J. Inorg. Chem., 2006, 4399–4406 CrossRef CAS; (b) M. C. Durrant, S. C. Davies, D. L. Hughes, C. Le Floc'h, R. L. Richards, H. R. Sanders, N. R. Champness, S. J. Pope and G. Reid, Inorg. Chim. Acta, 1996, 251, 13–14 CrossRef CAS.
- A. Ross, D. C. Soares, D. Covelli, C. Pannecouque, L. Budd, A. Collins, N. Robertson, S. Parsons, E. De Clercq, P. Kennepohl and P. J. Sadler, Inorg. Chem., 2010, 49, 1122–1132 CrossRef CAS PubMed.
- I. S. Lee and J. R. Long, Dalton Trans., 2004, 3434–3436 RSC.
- H. Lueken, in Magnetochemie, ed. B. G. Teubner, Stuttgart, Leipzig, 1999, p. 319 Search PubMed.
- A. Abragam and B. Bleaney, Electron Paramagnetic Resonance of Transition Ions, Clarendon Press, Oxford, 1970 Search PubMed.
- (a) K. Kanamori, Coord. Chem. Rev., 2003, 237, 147–161 CrossRef CAS; (b) J. R. Rambo, S. L. Castro, K. Folting, S. L. Bartley, R. t A. Heintz and G. Christou, Inorg. Chem., 1996, 35, 6844–6852 CrossRef CAS PubMed.
- N. N. Nair, E. Schreiner, R. Pollet, V. Staemmler and D. Marx, J. Chem. Theory Comput., 2008, 4, 1174–1188 CrossRef CAS PubMed.
- Y. Kitagawa, T. Saito, Y. Nakanishi, Y. Kataoka, T. Matsui, T. Kawakami, M. Okumura and K. Yamaguchi, J. Phys. Chem. A, 2009, 113, 15041–15046 CrossRef CAS PubMed.
- P. Hou, S. Peschtrich, N. Huber, W. Feuerstein, A. Bihlmeier, I. Krummenacher, R. Schoch, W. Klopper, F. Breher and J. Paradies, Chem. Europ. J., 2022, 28, e202200478 CrossRef CAS PubMed.
- K. Fink and V. Staemmler, Mol. Phys., 2013, 111, 2594–2605 CrossRef CAS.
- T. Bodenstein, A. Heimermann, K. Fink and C. van Wüllen, ChemPhysChem, 2022, 23, e202100648 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical techniques, crystallographic data, and computational methods. CCDC 2289310. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03243d |
| This journal is © The Royal Society of Chemistry 2023 |