
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced luminescence properties through heavy ancillary ligands in [Pt(C^N^C)(L)] complexes, L = AsPh3 and SbPh3†
Rose
Jordan
 a,
Iván
Maisuls
b,
Shruthi S.
Nair
cd,
Benjamin
Dietzek-Ivanšić
*cd,
Cristian A.
Strassert
*b and
Axel
Klein
*a
a,
Iván
Maisuls
b,
Shruthi S.
Nair
cd,
Benjamin
Dietzek-Ivanšić
*cd,
Cristian A.
Strassert
*b and
Axel
Klein
*a
aUniversity of Cologne, Faculty for Mathematics and Natural Sciences, Department of Chemistry, Institute for Inorganic Chemistry, Greinstrasse 6, D-50939 Köln, Germany. E-mail: rjordan1@uni-koeln.de; axel.klein@uni-koeln.de
bUniversität Münster, Institut für Anorganische und Analytische Chemie, CiMIC, CeNTech, Heisenbergstraße 11, D-48149 Münster, Germany. E-mail: maisuls@uni-muenster.de; cstra_01@uni-muenster.de
cFriedrich Schiller University Jena, Institute for Physical Chemistry (IPC), Helmholtzweg 4, 07743 Jena, Germany. E-mail: benjamin.dietzek@uni-jena.de
dLeibniz Institute for Photonic Technologies Jena (IPHT), Research Department Functional Interfaces, Albert-Einstein-Str. 9, 07745 Jena, Germany. E-mail: shruthi.santoshnair@leibniz-ipht.de; benjamin.dietzek@leibniz-ipht.de
First published on 21st November 2023
Abstract
In the frame of our research aiming to develop efficient triplet-emitting materials, we are exploring the concept of introducing additional heavy atoms into cyclometalated transition metal complexes to enhance intersystem-crossing (ISC) and thus triplet emission through increased spin–orbit coupling (SOC). In an in-depth proof-of-principle study we investigated the double cyclometalated Pt(II) complexes [Pt(C^N^C)(PnPh3)] (HC^N^CH = 2,6-diphenyl-pyridine (H2dpp) or dibenzoacridine (H2dba); Pn = pnictogen atoms P, As, Sb, or Bi) through a combined experimental and theoretical approach. The derivatives containing Pn = P, As, and Sb were synthesised and characterised comprehensively using single crystal X-ray diffraction (scXRD), UV-vis absorption and emission spectroscopy, transient absorption (TA) spectroscopy and cyclic voltammetry (CV). Across the series P < As < Sb, a red-shift is observed concerning absorption and emission maxima as well as optical and electrochemical HOMO–LUMO gaps. Increased photoluminescence quantum yields ΦL and radiative rates kr from mixed metal-to-ligand charge transfer (MLCT)/ligand centred (LC) triplet states are observed for the heavier homologues. Transient absorption spectroscopy showed processes in the ps range that were assigned to the population of the T1 state by ISC. The heavy PnPh3 ancillary ligands are found to enhance the emission efficiency due to both higher Pt–Pn bond strength and stronger SOC related to increased MLCT character of the excited states. The experimental findings are mirrored in hybrid (TD-)DFT calculations. This allowed for extrapolation to the rather elusive Bi derivatives, which were synthetically not accessible. This shortcoming is attributed to the transmetalation of phenyl groups from BiPh3 to Pt, as supported by experimental NMR/MS as well as DFT studies.
Introduction
Luminescent organometallic transition metal complexes with efficient phosphorescence at ambient temperature are the subject of academic and technological interest with promising applications in illumination, display technology, sensors, therapeutic agents, and biological probes.1–15 High quantum efficiency of such materials is due to strong spin–orbit coupling (SOC), which allows for triplet (electro)luminescence.5,7 However, many transition metal complexes are scarcely emissive at room temperature, often because their excited metal-centred (d–d*) states are subject to efficient non-radiative deactivation.5,8,9,11 Cyclometalated heteroaromatic ligands appear to be a good strategy towards effective emitters in many regards. The very strong ligand field of the cyclometalated carbanions raises the energy of the dissociative d–d* states impeding the population of these non-emissive, so-called dark states.8,11 The rigidity introduced by these ligands prohibits large distortions of the excited state compared with the electronic ground state. High rigidity of both the organic ligand and the coordination polyhedron around the metal are considered to be important to generate long-lived and efficient triplet emitting complexes.5,9,16–42 Such a strategy has led to many cyclometalated Pt(II) and a number of related Pd(II) complexes, which are emissive either from triplet states with primarily ligand-centred 3LC (3π–π*) character, from metal-to-ligand charge transfer 3MLCT states, or mixtures thereof.5,8,16–29On the way to introducing Ni(II) to the field, replacing Pt or Pd, the intrinsically much smaller spin–orbit coupling (SOC) and ligand field splitting of Ni(II) pose an obstacle to the realisation of efficient phosphorescence.29,34,42–46 Considering the question of how to facilitate spin-forbidden transitions and to also reach significant phosphorescence rates in Ni(II) complexes has led us to the idea of introducing additional heavy main group elements into the compounds, specifically in the form of PnPh3 (Pn = P, As, Sb, Bi) ancillary ligands for tridentate ligand systems. The ancillary ligand or coligand represents an important point of access for tuning the properties of square planar complexes with tridentate ligands.17,26,47–49 In order to evaluate whether our concept of improving phosphorescence using heavy ancillary ligands for tridentate ligand systems is viable, we initially resorted to Pt(II) complexes as a synthetically well-accessible model system. After a first study on a series of Pt(II) complexes [Pt(N^N^N)(PnPh3)] (Pn = P, As, Sb) containing the tridentate N^N^N = 2,6-bis(3-(trifluoromethyl)-1H-1,2,4-triazol-5-yl)pyridine and 2,6-bis(3-(tert-butyl)-1H-1,2,4-triazol-5-yl)pyridine ligands found little systematic variation between the PnPh3 homologues due to a saturation effect from Pt on the emission efficiency in this N^N^N system,30 we moved on to cyclometalated C^N^C ligand systems.31
Herein, we report two series of double cyclometalated Pt(II) complexes [Pt(C^N^C)(PnPh3)] (Pn = P, As, Sb) based on the two established C^N^C ligand systems dpp (H2dpp = 2,6-diphenyl-pyridine)31,50 and dba (H2dba = dibenzo[c,h]acridine)31,51 which showed an enhancement of the phosphorescence quantum yields for their heavier congeners. The role of radiative and radiationless deactivation rates was studied by combining extensive photoluminescence (PL) and transient absorption spectroscopy (TAS) with density functional theory (DFT) calculations.
Results and discussion
Synthesis and structural characterisation
The new complexes [Pt(C^N^C)(AsPh3)] and [Pt(C^N^C)(SbPh3)] were synthesised in the same way as the previously reported lighter congeners [Pt(C^N^C)(PPh3)]50,51via a facile ligand exchange reaction from the corresponding [Pt(C^N^C)(dmso)] precursors as shown in Scheme 1 in good yields ranging from 64% to 84% (more information in the ESI†). The new complexes were characterised by nuclear magnetic resonance (NMR) spectroscopy (see Fig. S1–S20 in the ESI†) as well as high-resolution electrospray ionisation mass spectrometry (HR-ESI-MS) and single-crystal X-ray diffraction (scXRD). | ||
| Scheme 1 General synthetic procedure for the complexes. | ||
Similar to our previous report on a series of Pt(II) complexes [Pt(N^N^N)(PnPh3)] (N^N^N = 2,6-bis(3-(trifluoromethyl)-1H-1,2,4-triazol-5-yl)pyridine and 2,6-bis(3-(tert-butyl)-1H-1,2,4-triazol-5-yl)pyridine; Pn = P, As, Sb)30 the BiPh3 derivatives of the dpp and dba complexes were not isolated, but MS and NMR evidence suggests their transient formation. Based on our observation of progressively de-phenylated fragments of BiPh3 and progressively phenylated derivatives of the dpp ligand in ESI-MS reaction control samples as well as the relative depletion of BiPh3 next to [Pt(dpp)(dmso)] during in situ1H NMR studies (Fig. S21, ESI†), we suspect that the reason for our failure to isolate [Pt(dpp)(BiPh3)] lies in the catalytic decomposition of BiPh3 in the presence of [Pt(dpp)(dmso)]. A free DFT geometry optimisation of the postulated structure of [Pt(dpp)(BiPh3)] yielded a pentacoordinate geometry [Pt(C^N^C)(Ph)(BiPh2)] where a phenyl group from BiPh3 moved to the Pt centre in the C^N^C coordination plane while the remaining BiPh2 fragment is displaced into a distal position with a Pt⋯Bi distance of 2.72 Å (Fig. S26 and Table S5, ESI†). This further supports the assumption that the decomposition of BiPh3 under the reaction conditions is triggered by the transmetalation of phenyl groups to Pt as initial reaction, which is then followed by a cascade of decomposition reactions. Detailed information on our experimental studies leading to this conclusion as well as computational details can be found in the ESI.† For further DFT calculations on the elusive [Pt(dpp)(BiPh3)] and [Pt(dba)(BiPh3)] complexes, the predicted transmetalation of a phenyl group was suppressed during the geometry optimisation by excluding the N-Pt-Bi angle from the optimisation process (Fig. S26 and Table S5, ESI†).
Crystal structures and DFT-calculated molecular structures
The structures of the four new complexes [Pt(C^N^C)(AsPh3)] and [Pt(C^N^C)(SbPh3)] (C^N^C = dpp, dba) were determined by scXRD (Fig. 1 and Figs. 22–24, ESI†). Details on data collection, structure solution and refinement as well as selected structural parameters are listed in Tables S1–S3 (ESI†) alongside with the data for the previously reported [Pt(dpp)(PPh3)] and [Pt(dba)(PPh3)].50,51 The crystal structures feature long Pt⋯Pt distances ranging from about 7 to 10 Å (far beyond a significant interaction between the Pt atoms, typically below 3.5 Å); thus, no significant Pt⋯Pt interactions nor π-stacking interactions were found.52,53 In contrast, the dba containing structures show pronounced π-stacking of the acridine moieties with centroid distances between 3.5 and 3.6 Å. However, due to the antiparallel alignment of the [Pt(dba)] fragments as dictated by the bulky PnPh3 ancillary ligands, significant Pt⋯Pt interactions can still be ruled out from large Pt⋯Pt distances ranging from 6.9 to 7.7 Å (Fig. 1). | ||
| Fig. 1 Molecular structures of the complexes [Pt(dpp)(AsPh3)] (left) and [Pt(dba)(AsPh3)] (right) with numbering, as obtained from scXRD. Displacement ellipsoids are shown at 50% probability, H atoms omitted for clarity. | ||
As expected from the increasing radii of the progressively heavier pnictogens, the Pt–Pn bond lengths increase by about 0.11 Å from the P to the As derivatives and by another 0.15 to 0.18 Å from the As to the Sb derivatives for both C^N^C ligands. This aligns with our findings for the previously reported [Pt(N^N^N)(PnPh3)] series. However, the Pt–Pn distances are overall slightly shorter in the herein reported C^N^C complexes by about 0.07 to 0.02 Å.30 This indicates a slightly higher bond strength, possibly due to an increased covalent character of the Pt–Pn bond in the herein studied system. The differences between Pt–Pn bond lengths (if comparing the C^N^C versus N^N^N complexes) are largest for the PPh3 complexes and smallest for the SbPh3 derivatives. This may be related to the Pt–Sb bond being generally more covalent than the Pt–P or Pt–As bond due to better orbital overlap between atoms of more similar atomic or ionic radii and polarisability. The averaged C–Pn–C angles decrease slightly along P > As > Sb for both series of complexes, from P (∼104°) to As (∼103°) and more markedly to Sb (99° for dpp, 101° for dba) (data in Tables S2 and S3, ESI†), exceeding the C–Pn–C angles of the free ligands by around 3 to 4°. We have previously described this widening of the angles upon binding to Pt as compensation of the hybridisation defect at the Pn atom.30
Notably for the complexes with the more flexible dpp ligand, a significant deviation from a coplanar orientation between the pyridine moiety and one of the phenyl moieties is observed in the solid state, with dihedral angles reaching up to 12° in the PPh3 derivative.50 This pronounced unilateral twist is not predicted by DFT geometry optimisations using BP86/def2-TZVP/CPCM(CH2Cl2)54–58 (Tables S2 and S3, ESI†) and the underlying reason are likely packing effects in the solid state. Apart from this deviation, the experimentally observed structural trends in the solid state are well reproduced by DFT. The optimised structures were used as the basis for all further calculations using the hybrid functional TPSSh, which was found to provide qualitatively good to excellent results for organometallic transition metal complexes in previous studies.34,59–62
Electrochemistry and DFT-calculated frontier orbitals
Cyclic voltammograms showed irreversible oxidations at around +0.6 V (Fig. 2, Fig. S27–S32 and Table S6, ESI†) for all complexes with slightly higher values for the dba derivatives and almost no variation within the dpp and dba series. They are assigned to essentially metal centred Pt(II)/Pt(III) couples. The DFT-calculated frontier molecular orbital (FMO) landscapes (Fig. S33 and S34, ESI†), which show energetically virtually invariant highest occupied molecular orbitals (HOMOs) with strong metal centred character for the complexes within each series, support this assignment. | ||
| Fig. 2 Electrochemical data from cyclic voltammetry in 0.1 M n-Bu4NPF6 THF solutions at a scan rate of 50 mV s−1 (dots); DFT-calculated frontier orbitals, and energies for [Pt(dpp)(PnPh3)] (left) and [Pt(dba)(PnPh3)] (right; Pn = P, As, Sb). Values for the redox potentials are half half-wave potentials E1/2 for the reversible first reduction and potentials Ep for the irreversible oxidations and second and third reductions (Table S6, ESI†). | ||
Two reduction processes were observed for all complexes (Fig. 2). Interestingly, their potentials are virtually identical for [Pt(dpp)(PPh3)] and [Pt(dpp)(AsPh3)] and appear at −2.36 and −2.96 V (vs. ferrocene/ferrocenium). For the dba complexes three reduction waves were observed, the first ones at markedly less negative potentials. In both series the SbPh3 complexes exhibit the least negative values. The DFT-calculated lowest unoccupied molecular orbitals (LUMO) represent exclusively the π* levels of the C^N^C ligands. This fits well to the general separation of the reduction potential of the two systems into dpp and dba complexes. The observed PnPh3 ancillary ligand-dependent differences (with the Sb derivatives having the least negative potentials) are fully in line with the DFT-calculated LUMO+1 showing contributions from the PnPh3 ligands. These assignments are further substantiated through spectroelectrochemical (UV-vis-SEC) studies (Fig. S39–S42, ESI†). Upon reduction, the complexes show several bands indicative of reduced phenyl-pyridine or acridine moieties, and pointing to ligand-centred LUMOs mirroring what has been reported for related complexes.25,34,51,63,64 Oxidation of the dpp complexes leads to a general bleaching of the long-wavelength bands in line with a depopulation of the metal-dominated HOMO. For the dba derivatives, the long-wavelength bands also bleach upon oxidation, but also a broad absorption band ranging from 500 to 350 nm appears. This slightly resembles the acridinyl radical (also called “C radical”), which absorbs in this range65,66 and agrees with the more pronounced contribution of the dba ligand to the HOMO of the complex, particularly when compared with dpp.
The electrochemical HOMO–LUMO gaps range from about 3.0 to 2.6 eV narrowing along the series P > As > Sb for both the dpp and the dba complexes. The smaller gaps for the dba complexes (2.72 to 2.62 eV) compared with the dpp derivatives (3.01 to 2.87 eV) by about 0.2 to 0.3 eV can be rationalised by the more extended π-system in the dba complexes and are reproduced by the DFT-calculations (Fig. 2, Fig. S33 and S34, ESI†). Parallel trends are predicted by DFT, which shows a decrease in the HOMO–LUMO gaps between the PPh3 and SbPh3 derivatives by 0.07 eV for the dpp complexes and 0.06 eV for the dba complexes. The DFT calculated values are generally slightly smaller than the experimental values. It should be noted that the predictive power of DFT reproduces trends much better than absolute numbers, as specifically the HOMO–LUMO gap depends strongly on how much Hartree–Fock exchange is included in the functional.67,68 Energetically above the strongly ligand-centred LUMOs (LUMO and LUMO+1 for dpp; LUMO to LUMO+2 for dba), a metal centred MO of strong dx2–y2 character is found in the DFT calculated FMO landscapes. This orbital is significantly destabilised across both series from P to Sb, indicating that the ligand-field splitting increases down the group 15 elements due to their increasing polarisability and atomic radii, as discussed earlier. The described trends for the PPh3, AsPh3, and SbPh3 derivatives of both the dpp and dba complexes are continued in the calculated FMO landscapes for the hypothetical BiPh3 derivatives.
UV-vis absorption spectroscopy and TD-DFT calculations
The UV-vis absorption spectra of the dba complexes [Pt(dba)(PnPh3)] (Pn = P, As, Sb) feature absorption bands down to almost 600 nm, which are structured into four double bands around 290, 340, 400 and 520 nm (Fig. 3, Table 1, Fig. S35 and S37, ESI†). The dpp complexes [Pt(dpp)(PnPh3)] (Pn = P, As, Sb) show only two intense, structured absorption bands, one at about 250 to 300 nm and a second at around 350 nm. Additionally, very weak absorption bands trail down to about 500 nm. UV-bands in both systems have been previously assigned to transitions into LC (π–π*) states, whereas mixed LC/metal-to-ligand charge transfer (MLCT) states were discussed for increasingly long-wavelength bands.31,51,69,70 | ||
| Fig. 3 Left: experimental UV-vis absorption spectra (molar absorption coefficient ε as a function of wavelength) of [Pt(C^N^C)(SbPh3)] in CH2Cl2 solution at 298 K. Inset: magnification of the 350–630 nm region. Right: experimental UV-vis absorption spectrum in CH2Cl2 solution at 298 K and TD-DFT-calculated spectrum with selected transition difference densities for [Pt(dba)(SbPh3)]. | ||
| λ 1 | λ 2 | λ 3 | λ 4 | λ 5 | λ 6 | λ 7 | λ 8 | ||
|---|---|---|---|---|---|---|---|---|---|
| a In CH2Cl2; absorption maxima λ in nm. b TPSSh/def2-TZVP/CPCM(CH2Cl2), maxima from convoluted spectrum. | |||||||||
| [Pt(dba)(PPh3)] | Exp.a | 286 | 296 | 333 | 348 | 387 | 409 | 499 | 533 |
| Calc.b | 306 | 363 | 430 | 552 | |||||
| [Pt(dba)(AsPh3)] | Exp.a | 285 | 296 | 333 | 354 | 390 | 412 | 505 | 539 |
| Calc.b | 307 | 364 | 432 | 560 | |||||
| [Pt(dba)(SbPh3)] | Exp.a | 286 | 296 | 335 | 351 | 392 | 416 | 511 | 546 |
| Calc.b | 308 | 365 | 434 | 567 | |||||
TD-DFT calculated absorption spectra excellently match the experimental results and support these assignments (Fig. 3, Table 1, Fig. S35–S38, Tables S6 and S7, ESI†). Fig. 3 (right) shows selected transition difference densities for the TD-DFT calculated transitions for [Pt(dba)(SbPh3)] as a representative example, while illustrating their mixed yet predominantly 1LC and 1MLCT character, in agreement with previously reported data for related compounds.25,28–31,34,35,51,52 Across the series P > As > Sb, a slight red-shift of the UV-vis absorption energies is both observed experimentally and predicted theoretically, matching the electrochemical data and the electronic structure modelled by DFT.
The absorption cut-off extrapolated from the low-energy slope of the lowest energy absorption band, representing the optical HOMO–LUMO gap, is about 565 nm (2.19 eV) for [Pt(dba)(PPh3)] to 580 nm (2.14 eV) for [Pt(dba)(SbPh3)]. While the optical HOMO–LUMO gap is expected to exceed the electrochemical HOMO–LUMO gap (which probes the molecule in its geometric ground state), the opposite is the case here, as the electrochemical HOMO–LUMO gaps determined from CVs are larger by about 0.5 eV than the optical gaps obtained from UV-vis absorption spectroscopy. Since negative reorganisation energy (i.e., reorganisation into a higher energy conformation upon oxidation/reduction) is physically not reasonable, we have to consider the contribution of direct absorption from the S0 ground state into lowest 3MLCT states producing the low-energy absorption bands and thus the low optical gaps. This is discussed for Pt(II) systems in the literature,71–74 with a lower energy and weakly intense band related to a 3MLCT state mirroring the absorption into the 1MLCT state. Remarkably, our TD-DFT calculations also predict these very low energy transitions, even if they do not include such triplet admixtures. DFT calculated T1–S0 energy gaps of 1.97 eV for [Pt(dba)(PPh3)] and 1.95 eV for the As and Sb derivatives (Table 2) are roughly consistent with this assumption and reinforcing the assumed correlation between optical and actual HOMO–LUMO gaps.
| T | λ max (nm) | Φ L ± 0.02 | τ (μs)e | k r (10−4 s−1) | k nr (10−4 s−1) | 〈Sn|HSO|T1〉2 (cm−2)f | ΔE(T1–S0) (eV / nm)f | |
|---|---|---|---|---|---|---|---|---|
a Excitation wavelength λexc = 350 nm.
b Measured in a frozen glassy matrix of CH2Cl2/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 77 K.
c Measured in fluid CH2Cl2 solution upon purging with Ar.
d Shoulder.
e For bi-exponential photoluminescence decays, amplitude-weighted average lifetimes are shown. Raw time-resolved photoluminescence decays along with the fitting parameters are shown in the ESI.†
f (TD-)DFT calculated, computational details in the ESI.† :1) at 77 K.
c Measured in fluid CH2Cl2 solution upon purging with Ar.
d Shoulder.
e For bi-exponential photoluminescence decays, amplitude-weighted average lifetimes are shown. Raw time-resolved photoluminescence decays along with the fitting parameters are shown in the ESI.†
f (TD-)DFT calculated, computational details in the ESI.†
|
||||||||
| [Pt(dpp)(PPh3)] | 77 Kb | 511, 550, 590d | 0.48 | 12.70 ± 0.01 | 3.8 ± 0.2 | 4.1 ± 0.2 | 9.18 × 105 (n = 0) | 2.06 / 602 |
| 1.23 × 104 (n = 2) | ||||||||
| [Pt(dpp)(AsPh3)] | 77 Kb | 517, 557, 588d | 0.70 | 11.80 ± 0.01 | 5.9 ± 0.2 | 2.5 ± 0.2 | 9.06 × 105 (n = 0) | 2.04 / 608 |
| 1.41 × 104 (n = 2) | ||||||||
| [Pt(dpp)(SbPh3)] | 77 Kb | 521, 562, 592d | 0.68 | 11.60 ± 0.01 | 5.6 ± 0.2 | 3.0 ± 0.2 | 8.14 × 105 (n = 0) | 2.05 / 605 |
| 1.11 × 104 (n = 2) | ||||||||
| [Pt(dba)(PPh3)] | 77 Kb | 576, 625, 684 | 0.90 | 27.90 ± 0.06 | 3.2 ± 0.1 | 0.5 ± 0.1 | 2.99 × 105 (n = 0) | 1.97 / 629 |
| 298 Kc | 590, 635d, 695d | 0.03 | 0.81 ± 0.01 | 0.4 ± 0.2 | 12.0 ± 0.3 | 1.20 × 105 (n = 2) | ||
| [Pt(dba)(AsPh3)] | 77 Kb | 580, 632, 692 | 0.90 | 23.70 ± 0.07 | 3.8 ± 0.1 | 0.4 ± 0.1 | 2.94 × 105 (n = 0) | 1.95 / 636 |
| 298 Kc | 594, 636d, 700d | 0.04 | 0.270 ± 0.001 | 1.5 ± 0.7 | 35.4 ± 0.8 | 1.38 × 105 (n = 2) | ||
| [Pt(dba)(SbPh3)] | 77 Kb | 584, 635, 694 | 0.94 | 21.90 ± 0.02 | 4.3 ± 0.1 | 0.3 ± 0.1 | 2.22 × 105 (n = 0) | 1.95 / 636 |
| 298 Kc | 598, 640d, 702d | 0.07 | 0.470 ± 0.001 | 1.5 ± 0.4 | 19.8 ± 0.4 | 1.41 × 105 (n = 2) | ||
Time-resolved and steady-state photoluminescence spectroscopy
Photoluminescence spectroscopy showed the dpp complexes [Pt(dpp)(PnPh3)] (Pn = P, As, Sb) to be emissive at 77 K in frozen CH2Cl2/MeOH (1:1) glassy matrices, but not in solution at 298 K. The spectra (Fig. S43–S45, ESI†) show two sharp emission bands around 515 and 560 nm and a shoulder around 590 nm. This corresponds to an approximate energy of around 2.4 eV for the 0–0 transitions, which is markedly larger than the DFT calculated ΔE(T1–S0) values (around 2.05 eV, see Table 2). The dba derivatives [Pt(dba)(PnPh3)] (Pn = P, As, Sb) are emissive both at 77 K in frozen glassy matrices and, albeit weakly, at 298 K in solution (spectra in Fig. 4 and Fig. S46–S48, ESI†). At 77 K, three sharp bands with emission maxima around 580, 630 and 690 nm are found. At 298 K, these maxima are shifted to around 595, 640 and 700 nm, where the latter two are not sharp bands but shoulders. Also in this case, the energy of the 0–0 transitions according to spectroscopy (around 2.1 eV) exceeds the DFT calculated ΔE(T1–S0) values discussed above, but not as markedly as for the dpp series. Like for the absorption spectra, a red-shift is observed for the emission spectra across the series P < As < Sb for both systems, whereas the calculated ΔE(T1–S0) values show a marginal (and in the case of dpp imperfect) downwards trend on the second decimal place. All data is compiled in Table 2.
 | ||
| Fig. 4 Photoluminescence spectra of the complexes [Pt(dba)(PnPh3)] (Pn = P, As, Sb) at 77 K in a frozen glassy matrix of MeOH/CH2Cl2 (1:1) (solid lines) and in CH2Cl2 solution at 298 K (dashed lines). | ||
For the dpp series, photoluminescence quantum yields (ΦL) of 0.70 and 0.68 were found for the AsPh3 and SbPh3 derivatives, significantly exceeding the value of 0.48 for [Pt(dpp)(PPh3)]. The ΦL are in concordance with literature reported values for similar Pt(II) complexes with flexible double cyclometalated C^N^C ligands.49,51,64,69,75,76 For the dba series at 77 K, values of ΦL near unity (0.90 for PPh3 and AsPh3, 0.94 for SbPh3) were determined. For similar complexes bearing several different ancillary ligands on the [Pt(dba)] fragment, we recently reported ΦL at 77 K between 0.80 and 0.90.51 At 298 K, the dba complexes exhibit much lower ΦL with values (below 0.1, see Table 2). At room temperature (298 K), all three dba complexes are practically non-emissive, with their ΦL values approaching the experimental uncertainty (except for the SbPh3 complex, which exhibits a modest yet measurable ΦL). The excited state lifetimes (τ) determined for both series at 77 K are within the range typically reported for similar C^N^C Pt(II) complexes at 77 K (ref. 47–49, 51, 64, 69, 75 and 76) and decrease across the series P > As > Sb.
Based on the obtained ΦL and excited-state lifetimes, we estimated the average radiative (kr) and non-radiative (knr) rate constants, as detailed in Table 2 and Fig. 5.
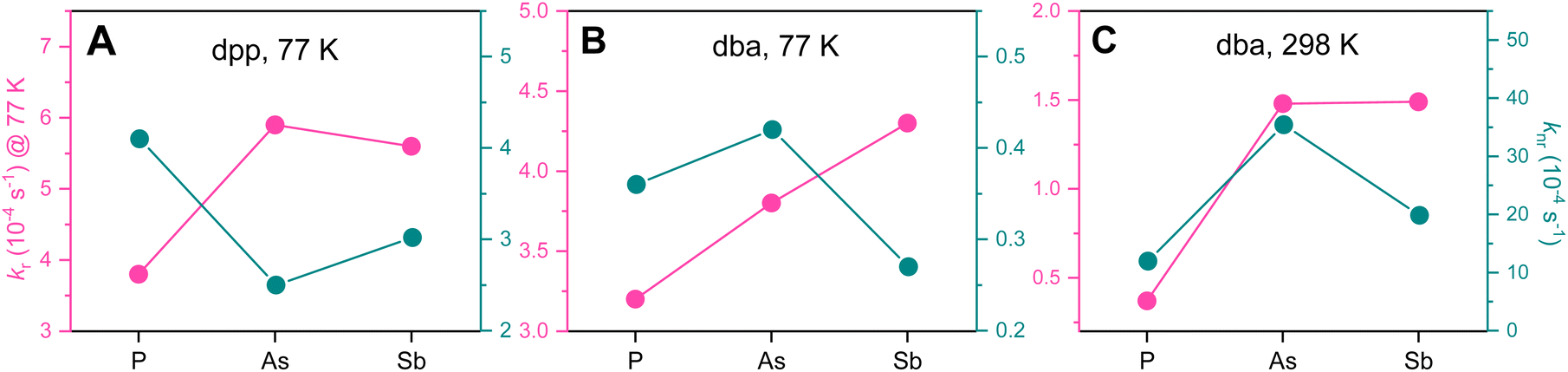 | ||
| Fig. 5 Visualisation of kr (pink) and knr (green) for the complexes [Pt(dpp)(PnPh3)] (A) and [Pt(dba)(PnPh3)] at 77 K (B) and 298 K (C) (Pn = P, As, Sb). | ||
At 77 K, we see a trend of increasing kr values and decreasing knr values from P to Sb in both the dpp and the dba series (Fig. 5A and B). We assume that the observed deviations from the linear trend (especially for knr) are due to 3d-contraction anomaly related to the first filling of a d-shell before the element As.77,78 At 298 K, kr for the dba complexes also increases from P to Sb, but no clear trend can be deduced from the knr values, which we attribute to more complex roto-vibronic relaxation pathways between T1 and S0 or possibly via the population of dissociative 3MC states at ambient temperature (Fig. 5C).
To further our understanding of the interplay between SOC and roto-vibronic aspects in the photophysics of the two series of complexes, we expanded our computational work to include TD-DFT spin–orbit coupling calculations at the DFT optimised T1 geometries (Table S4, ESI†).79–81 The results indicate significant mixing of the T1 states with the S0 and S2 states. Surprisingly, the S1 states only marginally couple with the T1 states. The corresponding SOC matrix elements 〈Sn|HSO|T1〉2 are listed in Table 2. In general, the values of 〈S2|HSO|T1〉2 are larger for the dba series than in the dpp series by a factor of 10. This may be related to the rigidity of the dba framework, thus allowing for a stronger coupling with higher excited states as more geometric overlap is preserved.
For the dba complexes, an increase in 〈S2|HSO|T1〉2 across the P < As < Sb series is predicted alongside a decrease in 〈S0|HSO|T1〉2. For the dpp derivatives, the 〈S0|HSO|T1〉2 values again decrease when moving down the pnictogen group. Interestingly, an unexpected trend in 〈S2|HSO|T1〉2 is observed for the dpp series, namely Sb < P < As. Thus, in the particular case of [Pt(dpp)(SbPh3)], our calculations predict a counterintuitively diminished SOC.
A possible explanation for these differences between the dpp and dba complexes lies in the character of the emissive states. The MLCT character of the emissive state is expected to decrease in frozen matrix at 77 K. Furthermore, the extended π-system of the dba ligand leads to smaller MLCT and larger LC contributions compared with the dpp system. We assume that for the dpp complexes at 77 K and for the dba derivatives at 298 K we have a larger MLCT contribution compared with dba at 77 K. With a higher MLCT character in the excited state, the heavier SbPh3 ligand is not able to develop its potential for higher SOC as the effect is already strong due to the participation of the Pt centre.30 Only in case of the LC-dominated dba systems at 77 K, SbPh3 reveals its superior SOC capacity over the lighter homologues.
To confirm this, we assessed the character of the emissive T1 states for both series of complexes as well as for the hypothetical BiPh3 theoretically via exciton analysis using the open-source software TheoDORE (Fig. 6, see ESI† for details).82 On first view, they confirm that for the dpp series that MLCT character of the T1 excited state is higher than for the dba complexes (∼22% vs. ∼17%) and the LC character dominates the latter (∼65% vs. ∼75%).
 | ||
| Fig. 6 Decomposition of the emissive T1 states into MLCT, L′MCT, LL′CT, LC and MC contributions for [Pt(dpp)(PnPh3)] (left) and [Pt(dba)(PnPh3)] (right; Pn = P, As, Sb, Bi) based on TD-DFT calculations (TPSSh/def2-TZVP/CPCM(CH2Cl2)).82 | ||
In both series, the LC character decreases slightly down the series from P to Sb, while the MLCT contribution increases. This is in line with the above discussed higher polarisability of the PnPh3 ligands leading to the observed red-shifts in absorption and emission as well as to the reduced electrochemical gaps.
Femtosecond transient absorption spectroscopy
The excited state dynamics of the two series of complexes [Pt(dpp)(PnPh3)] and [Pt(dba)(PnPh3)] (Pn = P, As, Sb) were studied further using femtosecond transient absorption (fsTA) spectroscopy under ambient conditions. A particular focus was on observing the ISC from the singlet to the triplet manifold and obtaining time constants for this process in the different PnPh3 derivatives. The ISC from singlet excited states into the triplet manifold is an ultrafast process for Pt(II) complexes, with reported ISC time constants for organometallic Pt(II) complexes being typically <5 ps and often even <1 ps.83–86When exciting the dpp complexes at 340 nm we observed intense excited state absorption (ESA) with broad features around 430 and 550 nm and no ground state bleach (spectra in Fig. 7a and Fig. S49, ESI†). Global analysis of the TA data using an exponential kinetic scheme reveals two characteristic time constants, one very fast (<10 ps) and one slower process beyond 10 ns after excitation. The first process is fastest in the PPh3 derivative and significantly slower in the SbPh3 complex. We assign this to an ISC in accordance with literature values for similar complexes.83–86 The counterintuitive trend in the <10 ps time constants (P ≈ As < Sb) roughly parallel the trend in decreasing SOC calculated by TD-DFT for the dpp series. Another concurring factor is probably the increasing Pt–Pn bond strength slowing down all radiationless processes (including ISC). The second process with time constants around 1.7 to 2.9 ns can be assigned to the relaxation from the T1 state to the ground state. This is consistent with absence of phosphorescence in the μs time range at 298 K. For [Pt(dpp)(dmso)], a relaxation pathway via T2 is described in the literature based on detailed theoretical analysis.72 It is therefore in principle also possible that the observed ISC occurs between S1 and T2 followed by a rapid decay from T2 to T1, even though we did not make any experimental observations specifically pointing in that direction.
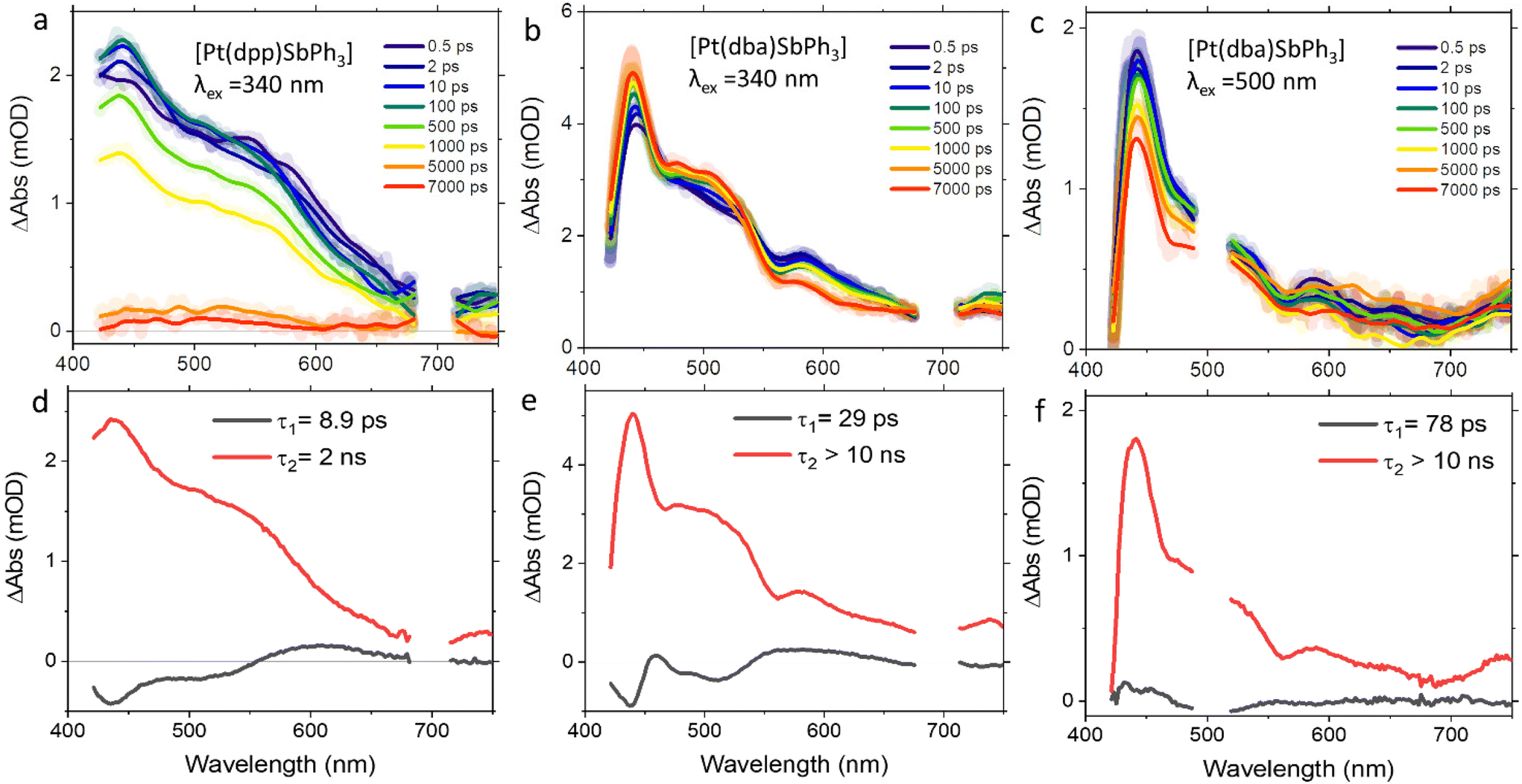 | ||
| Fig. 7 Transient absorption spectra (TAS) at selected delay times (a–c) and decay-associated spectra (d–f) for [Pt(dpp)(SbPh3)] following excitation at 340 nm (a and d), [Pt(dba)(SbPh3)] upon excitation at 340 nm (b and e) and 500 nm (c and f) in THF. The thicker lines in TAS shows smoothed data. | ||
For the dba complexes, we performed transient absorption experiments upon excitation at 340 and 500 nm. In all cases, we also observed intense ESA with maxima at 430 nm and shoulders at 500 and 580 nm. The evolution of signals could be fitted to a model taking into account two distinct processes (see Fig. 7 and Fig. S50 and S51, ESI†), with a slower process that is assigned to the decay of the emissive T1 state exceeding the 10 ns window.
Upon 340 nm excitation, we observed a fast process characterised by time constants around 30 ps for all complexes Pn = P, As, and Sb. We again assign this sub-100 ps kinetics to ISC, based on the characteristic time constants and the similar shapes regarding the decay-associated spectra for the fast process in the dpp derivatives (i.e. decrease of ESA at 580 nm and increase of ESA at 430 and 500 nm). For the dba derivatives, this process is overall slower than in the dpp complexes due to the larger LC contributions to the excited states.
Upon excitation at 500 nm, we see less pronounced spectral changes in the process represented by the first component, if compared to that observed at 340 nm excitation, leading us to assume the population of a different state (with significant triplet admixtures already upon initial excitation). It is possible that intermediate steps (like partial ISC) are not resolved from the relatively slow effective τ1. The slower ISC rate could be attributed to increased LC character. The second component is longer-lived and its spectral profile resembles that observed upon 340 nm excitation.
Generally, the lifetimes of the S1 states for the more rigid dba complexes are significantly longer than for the dpp derivatives, pointing to a slower ISC. For both series, the ISC under ambient conditions is not significantly faster for the heavy SbPh3 derivatives compared to their lighter PPh3 and AsPh3 congeners as originally expected. As discussed above, the effect of the PnPh3 variation is multifaceted. The initially predicted enhancement of SOC for the heavier derivatives is observed only in the dba series, due to the predominant LC character of their emissive triplet states. With increasing MLCT character of the emissive triplet state, the heavy atom effect is progressively masked by the dominating effect of the 5d metal, as seen in the dpp complexes. Furthermore, the variation of the Pn atom induces not only changes in SOC, but also in the Pt–Pn bond strength, influencing the photophysical properties by affecting the vibronic properties.
Conclusions
The effects of progressively heavy pnictogen-based ancillary ligands PnPh3 on the electronic and photophysical properties two series of Pt(II) C^N^C cyclometalated complexes [Pt(dpp)PnPh3] and [Pt(dba)PnPh3] (Pn = P, As, Sb; H2dpp = 2,6-diphenyl-pyridine, H2dba = dibenzoacridine) were explored. The electrochemical HOMO–LUMO gaps and the long-wavelength UV-vis absorption and emission maxima are red-shifted along the series P < As < Sb for both the dpp and the dba framework, in line with increasing electron density at the Pt centre, which we interpret as stronger σ-donation of the heavier PnPh3 ligands to Pt. The latter correlates with an increasing ligand-field splitting along P < As < Sb. The photoluminescence quantum yields ΦL are increased for the heavy congeners reaching almost unity for the dba complex. The radiative rate constants kr are increased by introduction of the heavier homologues while the non-radiative rate constants knr are reduced. As an important factor for this behaviour we identify increased Pt–Pn bond strength in the heavier derivatives, especially with SbPh3, due to better orbital overlap resulting from more similar atomic/ionic radii and polarisability. The initially sought-after heavy atom effect with increased spin orbit coupling (SOC) for the heavier PnPh3 ancillary ligands is modulated by the character of the excited states. In our two series of complexes the increased SOC is most evident for the dba complexes at 77 K, where the LC character of the excited state is dominant over the MLCT character. Thus, the enhancement of phosphorescence via introduction of heavy ligands is less prominent in systems with emissive states of strong MLCT character.Based on this analysis, we are confident that the present design principle of combining a cyclometalating, rigid tridentate ligand with a heavy element containing ancillary ligand can be applied to further systems, especially with LC-dominated excited states to enhance triplet emission. For the present series of complexes, we were unable to obtain the Bi derivative, which therefore remains an open synthetic challenge. In the future, the isolation of this or related Bi-containing complexes will show whether its heavy atom effect can further enhance kr in complexes with weak SOC. Further promising candidates for heavy ancillary ligands may also include the anionic tetrelate derivatives GeR3−, SnR3−, and PbR3−.
Author contributions
R.J. carried out syntheses, cyclic voltammetry, UV-vis absorption spectroscopy and spectroelectrochemistry measurements as well as the computational work. I.M. carried out the emission spectroscopy. S.S.N and R.J. carried out the transient absorption spectroscopy measurements. A.K., B.D.-I., and C.A.S. supervised the project. R.J. and A.K. wrote the original draft, all authors revised the manuscript. All authors agreed with the final version.Data availability
All relevant data are available from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft [DFG Priority Programme 2102 “Light-controlled Reactivity of Metal Complexes” STR 1186/6-1 and 6-2 (C. A. S.), KL1194/16-1 and 16-2 (A. K.), and DI1517/19-1 (B. D.-I.)] and the Studienstiftung des Deutschen Volkes e.V. (R. J.) for funding of this project. We would also like to thank the Regional Computing Center of the University of Cologne (RRZK) for providing computing time on the DFG-funded High Performance Computing (HPC) system CHEOPS as well as for technical support.References
- K. Mori and H. Yamashita, Metal Complexes Supported on Solid Matrices for Visible–Light–Driven Molecular Transformations, Chem. – Eur. J., 2016, 22, 11122–11137, DOI:10.1002/chem.201600441.
- M. Parasram and V. Gevorgyan, Visible light-induced transition metal-catalyzed transformations: beyond conventional photosensitizers, Chem. Soc. Rev., 2017, 46, 6227–6240, 10.1039/C7CS00226B.
- Q. Zhao, F. Li and C. Huang, Phosphorescent chemosensors based on heavy-metal complexes, Chem. Soc. Rev., 2010, 39, 3007–3030, 10.1039/B915340C.
- A. S.-Y. Law, L. C.-C. Lee, K. K.-W. Lo and V. W.-W. Yam, Aggregation and Supramolecular Self-Assembly of Low-Energy Red Luminescent Alkynylplatinum(II) Complexes for RNA Detection, Nucleolus Imaging, and RNA Synthesis Inhibitor Screening, J. Am. Chem. Soc., 2021, 143, 5396–5405, DOI:10.1021/jacs.0c13327.
- K. Li, G. S. M. Tong, Q. Wang, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Highly phosphorescent platinum(II) emitters: photophysics, materials and biological applications, Chem. Sci., 2016, 7, 1653–1673, 10.1039/C5SC03766B.
- S. Fantacci and F. De Angelis, A computational approach to the electronic and optical properties of Ru(II) and Ir(III) polypyridyl complexes: Applications to DSC, OLED and NLO, Coord. Chem. Rev., 2011, 255, 2704–2726, DOI:10.1016/j.ccr.2011.03.008.
- C. E. Housecroft and E. C. Constable, Over the LEC rainbow: Colour and stability tuning of cyclometallated iridium(III) complexes in light-emitting electrochemical cells, Coord. Chem. Rev., 2017, 350, 155–177, DOI:10.1016/j.ccr.2017.06.016.
- H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH, Weinheim, Germany, 2008. DOI:10.1002/9783527621309.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Sustainable metal complexes for organic light-emitting diodes (OLEDs), Coord. Chem. Rev., 2018, 373, 49–82, DOI:10.1016/j.ccr.2017.09.011.
- L. Ravotto and P. Ceroni, Aggregation induced phosphorescence of metal complexes: From principles to applications, Coord. Chem. Rev., 2017, 346, 62–76, DOI:10.1016/j.ccr.2017.01.006.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, The triplet state of organo-transition metal compounds. Triplet harvesting and singlet harvesting for efficient OLEDs, Coord. Chem. Rev., 2011, 255, 2622–2652, DOI:10.1016/j.ccr.2011.01.042.
- X. Zhen, R. Qu, W. Chen, W. Wu and X. Jiang, The development of phosphorescent probes for in vitro and in vivo bioimaging, Biomater. Sci., 2021, 9, 285–300, 10.1039/d0bm00819b.
- Y. Chen, R. Guan, C. Zhang, J. Huang, L. Ji and H. Chao, Two-photon luminescent metal complexes for bioimaging and cancer phototherapy, Coord. Chem. Rev., 2016, 310, 16–40, DOI:10.1016/j.ccr.2015.09.010.
- C. Balachandran, K. Yokoi, K. Naito, J. Haribabu, Y. Tamura, M. Umezawa, K. Tsuchiya, T. Yoshihara, S. Tobita and S. Aoki, Cyclometalated Iridium(III) Complex–Cationic Peptide Hybrids Trigger Paraptosis in Cancer Cells via an Intracellular Ca2+ Overload from the Endoplasmic Reticulum and a Decrease in Mitochondrial Membrane Potential, Molecules, 2021, 26, 7028, DOI:10.3390/molecules26227028.
- J. Haribabu, Y. Tamura, K. Yokoi, C. Balachandran, M. Umezawa, K. Tsuchiya, Y. Yamada, R. Karvembu and S. Aoki, Synthesis and Anticancer Properties of Bis- and Mono(cationic peptide) Hybrids of Cyclometalated Iridium(III) Complexes: Effect of the Number of Peptide Units on Anticancer Activity, Eur. J. Inorg. Chem., 2021, 2021, 1796–1814, DOI:10.1002/ejic.202100154.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Light-emitting devices based on organometallic platinum complexes as emitters, Coord. Chem. Rev., 2011, 255, 2401–2425, DOI:10.1016/j.ccr.2011.01.049.
- I. O. Koshevoy, M. Krause and A. Klein, Non-covalent intramolecular interactions through ligand-design promoting efficient photoluminescence from transition metal complexes, Coord. Chem. Rev., 2020, 405, 213094, DOI:10.1016/j.ccr.2019.213094.
- L. M. Cinninger, L. D. Bastatas, Y. Shen, B. J. Holliday and J. D. Slinker, Luminescent properties of a 3, 5-diphenylpyrazole bridged Pt(II) dimer, Dalton Trans., 2019, 48, 9684–9691, 10.1039/C9DT00795D.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Light-Emitting Self-Assembled Materials Based on d8 and d10 Transition Metal Complexes, Chem. Rev., 2015, 115, 7589–7728, DOI:10.1021/acs.chemrev.5b00074.
- M. Cnudde, D. Brünink, N. L. Doltsinis and C. A. Strassert, Tetradentate N^N°N^N-type luminophores for Pt(II) complexes: Synthesis, photophysical and quantum-chemical investigation, Inorg. Chim. Acta, 2021, 518, 120090, DOI:10.1016/j.ica.2020.120090.
- G. Li, L. Ameri, T. Fleetham, Z.-Q. Zhu and J. Li, Stable and efficient blue and green organic light emitting diodes employing tetradentate Pt(II) complexes, Appl. Phys. Lett., 2020, 117, 253301, DOI:10.1063/5.0033023.
- F. Yu, Y. Sheng, D. Wu, K. Qin, H. Li, G. Xie, Q. Xue, Z. Sun, Z. Lu, H. Ma and X.-C. Hang, Blue-Phosphorescent Pt(II) Complexes of Tetradentate Pyridyl–Carbolinyl Ligands: Synthesis, Structure, Photophysics, and Electroluminescence, Inorg. Chem., 2020, 59, 14493–14500, DOI:10.1021/acs.inorgchem.0c02244.
- G. Cheng, Y. Kwak, W.-P. To, T.-L. Lam, G. S. M. Tong, M.-K. Sit, S. Gong, B. Choi, W. Choi, C. Yang and C.-M. Che, High-efficiency solution-processed organic light-emitting diodes with tetradentate platinum(II) emitters, ACS Appl. Mater. Interfaces, 2019, 11, 45161–45170, DOI:10.1021/acsami.9b11715.
- T. Fleetham, G. Li and J. Li, Phosphorescent Pt(II) and Pd(II) complexes for efficient, high–color–quality, and stable OLEDs, Adv. Mater., 2016, 29, 1601861, DOI:10.1002/adma.201601861.
- M. Krause, R. von der Stück, D. Brünink, S. Buss, N. L. Doltsinis, C. A. Strassert and A. Klein, Platinum and palladium complexes of tridentate C^N^N (phen-ide)-pyridine-thiazol ligands – A case study involving spectroelectrochemistry, photoluminescence spectroscopy and TD-DFT calculations, Inorg. Chim. Acta, 2021, 518, 120093, DOI:10.1016/j.ica.2020.120093.
- M. Hebenbrock, D. González-Abradelo, A. Hepp, J. Meadowcroft, N. Lefringhausen, C. A. Strassert and J. Müller, Influence of the ancillary ligands on the luminescence of platinum(II) complexes with a triazole-based tridentate C^N^N luminophore, Inorg. Chim. Acta, 2021, 516, 119988, DOI:10.1016/j.ica.2020.119988.
- V. Sivchik, A. Kochetov, T. Eskelinen, K. S. Kisel, A. I. Solomatina, E. V. Grachova, S. P. Tunik, P. Hirva and I. O. Koshevoy, Modulation of metallophilic and π–π interactions in platinum cyclometalated luminophores with halogen bonding, Chem. – Eur. J., 2021, 27, 1787–1794, DOI:10.1002/chem.202003952.
- E. V. Puttock, J. Sturala, J. C. M. Kistemaker and J. A. G. Williams, Platinum(II) Complexes of Tridentate–Coordinating Ligands Based on Imides, Amides, and Hydrazides: Synthesis and Luminescence Properties, Eur. J. Inorg. Chem., 2021, 2021, 335–347, DOI:10.1002/ejic.202000879.
- T. Eskelinen, S. Buss, S. K. Petrovskii, E. V. Grachova, M. Krause, L. Kletsch, A. Klein, C. A. Strassert, I. O. Koshevoy and P. Hirva, Photophysics and Excited State Dynamics of Cyclometalated [M (Phbpy)(CN)](M = Ni, Pd, Pt) Complexes: A Theoretical and Experimental Study, Inorg. Chem., 2021, 60, 8777–8789, DOI:10.1021/acs.inorgchem.1c00680.
- S. C. Gangadharappa, I. Maisuls, D. A. Schwab, J. Kösters, N. L. Doltsinis and C. A. Strassert, Compensation of Hybridization Defects in Phosphorescent Complexes with Pnictogen-Based Ligands—A Structural, Photophysical, and Theoretical Case-Study with Predictive Character, J. Am. Chem. Soc., 2020, 142, 21353–21367, DOI:10.1021/jacs.0c09467.
- S. Garbe, M. Krause, A. Klimpel, I. Neundorf, P. Lippmann, I. Ott, D. Brünink, C. A. Strassert, N. L. Doltsinis and A. Klein, Cyclometalated Pt Complexes of CNC Pincer Ligands: Luminescence and Cytotoxic Evaluation, Organometallics, 2020, 39, 746–756, DOI:10.1021/acs.organomet.0c00015.
- A. Iwakiri, Y. Konno and K. Shinozaki, Determination of excimer emission quantum yield of Pt(dpb)Cl (dpbH = 1,3-di(2-pyridyl)benzene and its analogues in solution, J. Lumin., 2019, 207, 482–490, DOI:10.1016/j.jlumin.2018.11.042.
- E. Garoni, J. Boixel, V. Dorcet, T. Roisnel, D. Roberto, D. Jacquemin and V. Guerchais, Controlling the emission in flexibly-linked (N^C^N) platinum dyads, Dalton Trans., 2018, 47, 224–232, 10.1039/C7DT03695G.
- L. Kletsch, R. Jordan, A. S. Köcher, S. Buss, C. A. Strassert and A. Klein, Photoluminescence of Ni(II), Pd(II), and Pt(II) Complexes [M(Me2dpb)Cl] Obtained from C-H Activation of 1,5-Di(2-pyridyl)-2,4-dimethylbenzene (Me2dpbH), Molecules, 2021, 26, 5051, DOI:10.3390/molecules26165051.
- B. Schulze, C. Friebe, M. Jäger, H. Görls, E. Birckner, A. Winter and U. S. Schubert, PtII Phosphors with Click-Derived 1,2,3-Triazole-Containing Tridentate Chelates, Organometallics, 2018, 37, 145–155, DOI:10.1021/acs.organomet.7b00777.
- M. Hebenbrock, L. Stegemann, J. Kösters, N. L. Doltsinis, J. Müller and C. A. Strassert, Phosphorescent Pt(II) complexes bearing a monoanionic C^N^N luminophore and tunable ancillary ligands, Dalton Trans., 2017, 46, 3160–3169, 10.1039/C7DT00393E.
- A. Rodrigue-Witchel, D. L. Rochester, S.-B. Zhao, K. B. Lavelle, J. A. G. Williams, S. Wang, W. B. Connick and C. Reber, Pressure-induced variations of MLCT and ligand-centered luminescence spectra in square-planar platinum(II) complexes, Polyhedron, 2016, 108, 151–155, DOI:10.1016/j.poly.2015.12.011.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Improving the performance of Pt(II) complexes for blue light emission by enhancing the molecular rigidity, Inorg. Chem., 2012, 51, 312–319, DOI:10.1021/ic201664v.
- P. Pander, A. V. Zaytsev, A. Sil, J. A. G. Williams, V. N. Kozhevnikov and F. B. Dias, Enhancement of thermally activated delayed fluorescence properties by substitution of ancillary halogen in a multiple resonance-like diplatinum(II) complex, J. Mater. Chem. C, 2022, 10, 4851–4860, 10.1039/d1tc05026e.
- G. Li, J. Wen, F. Zhan, W. Lou, Y.-F. Yang, Y. Hu and Y. She, Fused 6/5/6 metallocycle-based tetradentate Pt(II) emitters for efficient green phosphorescent OLEDs, Inorg. Chem., 2022, 61, 11218–11231, DOI:10.1021/acs.inorgchem.2c01202.
- L. Zhu, C. Sha, A. Lv, W. Xie, K. Shen, Y. Chen, G. Xie, H. Ma, H. Li and X.-C. Hang, Tetradentate Pt(II) complexes with peripheral hindrances for highly efficient solution-processed blue phosphorescent OLEDs, Inorg. Chem., 2022, 61, 10402–10409, DOI:10.1021/acs.inorgchem.2c01063.
- K. A. Van Houte, D. C. Heath, C. A. Barringer, A. L. Rheingold and R. S. Pilato, Functionalized 2-Pyridyl-Substituted Metallo-1,2-enedithiolates. Synthesis, Characterization, and Photophysical Properties of (dppe)M{S2C2(2-pyridine(ium))(CH2CH2OR′′)} and (dppe)M[{S2C2(CH2CH2-N-2-pyridinium)}]+ (R′′ = H, Acetyl, Lauroyl; M = Pd, Pt; dppe = 1,2-Bis(diphenylphosphino)ethane), Inorg. Chem., 1998, 37, 4647–4653, DOI:10.1021/ic980273j.
- N. Vogt, A. Sandleben, L. Kletsch, S. Schäfer, M. T. Chin, D. A. Vicic, G. Hörner and A. Klein, Role of the X Coligands in Cyclometalated [Ni(Phbpy)X] Complexes (HPhbpy = 6-Phenyl-2,2′-bipyridine), Organometallics, 2021, 40, 1776–1785, DOI:10.1021/acs.organomet.1c00237.
- C. Hamacher, N. Hurkes, A. Kaiser, A. Klein and A. Schüren, Electrochemistry and spectroscopy of organometallic terpyridine nickel complexes, Inorg. Chem., 2009, 48, 9947–9951, DOI:10.1021/ic900753r.
- H. Houjou, Y. Hoga, M. Yi-Lan, H. Achira, I. Yoshikawa, T. Mutai and K. Matsumura, Dinuclear fused salen complexes of group-10 metals: Peculiarity of the crystal structure and near-infrared luminescence of a bis (Pt-salen) complex, Inorg. Chim. Acta, 2017, 461, 27–34, DOI:10.1016/j.ica.2017.01.031.
- Y.-S. Wong, M.-C. Tang, M. Ng and V. W.-W. Yam, Toward the design of phosphorescent emitters of cyclometalated earth-abundant nickel(II) and their supramolecular study, J. Am. Chem. Soc., 2020, 142(16), 7638–7646, DOI:10.1021/jacs.0c02172.
- W. Lu, M. C. W. Chan, K.-K. Cheung and C.-M. Che, π–π Interactions in Organometallic Systems. Crystal Structures and Spectroscopic Properties of Luminescent Mono-, Bi-, and Trinuclear Trans-cyclometalated Platinum(II) Complexes Derived from 2,6-Diphenylpyridine, Organometallics, 2001, 20, 2477–2486, DOI:10.1021/om0009839.
- V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong, X.-X. Lu, K.-K. Cheung and N. Zhu, Syntheses, Electronic Absorption, Emission, and Ion-Binding Studies of Platinum(II) C^N^C and Terpyridyl Complexes Containing Crown Ether Pendants, Chem. – Eur. J., 2002, 8, 4066–4076, DOI:10.1002/1521-3765(20020902)8:17<4066::AID-CHEM4066>3.0.CO;2-O.
- S. C. F. Kui, F.-F. Hung, S.-L. Lai, M.-Y. Yuen, C.-C. Kwok, K.-H. Low, S. S.-Y. Chui and C.-M. Che, Luminescent organoplatinum(II) complexes with functionalized cyclometalated C^N^C ligands: structures, photophysical properties, and material applications, Chem. – Eur. J., 2012, 18, 96–109, DOI:10.1002/chem.201101880.
- A. Kergreis, R. M. Lord and S. J. Pike, Influence of ligand and nuclearity on the cytotoxicity of cyclometallated C^N^C platinum(II) complexes, Chem. – Eur. J., 2020, 26, 14938–14946, DOI:10.1002/chem.202002517.
- J. N. Friedel, M. Krause, R. Jordan, I. Maisuls, D. Bruenink, D. Schwab, N. L. Doltsinis, C. A. Strassert and A. Klein, Triplet Emitting C^N^C Cyclometalated Dibenzo[c,h]Acridine Pt(II) Complexes, Molecules, 2022, 27, 8054, DOI:10.3390/molecules27228054.
- V. W.-W. Yam and A. S. Y. Law, Luminescent d8 metal complexes of platinum(II) and gold(III): From photophysics to photofunctional materials and probes, Coord. Chem. Rev., 2020, 414, 213298, DOI:10.1016/j.ccr.2020.213298.
- C. Janiak, A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands, J. Chem. Soc., Dalton Trans., 2000, 21, 3885–3896, 10.1039/B003010O.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297, 10.1039/b508541a.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100, DOI:10.1103/PhysRevA.38.3098.
- J. P. Perdew and W. Yue, Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802, DOI:10.1103/PhysRevB.33.8800.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102, 1995–2001, DOI:10.1021/jp9716997.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energiesstructures, and electronic properties of molecules in solution with the C–PCM solvation model, J. Comput. Chem., 2003, 24, 669–681, DOI:10.1002/jcc.10189.
- J. Tao, J. P. Perdew, V. N. Stavoverov and G. E. Scuseria, Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids, Phys. Rev. Lett., 2003, 91, 146401, DOI:10.1103/PhysRevLett.91.146401.
- R. Jordan, M. Niazi, S. Schäfer, W. Kaim and A. Klein, Rhenium Tricarbonyl Complexes of Azodicarboxylate Ligands, Molecules, 2022, 27, 8159, DOI:10.3390/molecules27238159.
- L. Kletsch, G. Hörner and A. Klein, Cyclometalated Ni(II) Complexes [Ni(N^C^N)X] of the Tridentate 2,6-di(2-pyridyl)phen-ide Ligand, Organometallics, 2020, 39, 2820–2829, DOI:10.1021/acs.organomet.0c00355.
- R. Alrefai, G. Hörner, H. Schubert and A. Berkefeld, Broadly versus Barely Variable Complex Chromophores of Planar Nickel(II) from κ3-N,N′,C and κ3-N,N′,O Donor Platforms, Organometallics, 2021, 40, 1163–1177, DOI:10.1021/acs.organomet.1c00121.
- R. von der Stück, M. Krause, D. Brünink, S. Buss, N. L. Doltsinis, C. A. Strassert and A. Klein, Luminescent Pd(II) Complexes with Tridentate−C^N^N Arylpyridine-(benzo)thiazole Ligands, Z. Anorg. Allg. Chem., 2022, 648, e202100278, DOI:10.1002/zaac.202100278.
- M. Krause, D. Kourkoulos, D. González-Abradelo, K. Meerholz, C. A. Strassert and A. Klein, Luminescent PtII Complexes of Tridentate Cyclometalating 2,5-Bis(aryl)-pyridine Ligands, Eur. J. Inorg. Chem., 2017, 2017, 5215–5223, DOI:10.1002/ejic.201700792.
- A. Kellmann and F. Tfibel, Radicals Produced from the Laser-Induced Photoionization of Acridine in Solution, J. Photochem., 1982, 18, 81–88, DOI:10.1016/0047-2670(82)80009-9.
- X. Liu, T. N. V. Karsili, A. L. Sobolewski and W. Domcke, Photocatalytic Water Splitting with the Acridine Chromophore: A Computational Study, J. Phys. Chem. B, 2015, 119, 10664–10672, DOI:10.1021/acs.jpcb.5b04833.
- G. Zhang and C. B. Musgrave, Comparison of DFT methods for molecular orbital eigenvalue calculations, J. Phys. Chem. A, 2007, 111, 1554–1561, DOI:10.1021/jp061633o.
- E. J. Baerends, O. V. Gritsenko and R. van Meer, The Kohn–Sham gap, the fundamental gap and the optical gap: the physical meaning of occupied and virtual Kohn–Sham orbital energies, Phys. Chem. Chem. Phys., 2013, 15, 16408–16425, 10.1039/C3CP52547C.
- Q. Zhang, S. Wang, Y. Zhu, C. Zhang, W. M. Cao, X. Tian, J. Wu, H. Zhou and Y. Tian, Functional Platinum(II) complexes with four-photon absorption activity, lysosome specificity, and precise cancer therapy, Inorg. Chem., 2021, 60, 2362–2371, DOI:10.1021/acs.inorgchem.0c03245.
- R. Zhu, X. Chen, N. Shu, Y. Shang, Y. Wang, P. Yang, Y. Tang, F. Wang and J. Xu, Computational study of photochemical relaxation pathways of platinum(II) complexes, J. Phys. Chem. A, 2021, 125, 10144–10154, DOI:10.1021/acs.jpca.1c07017.
- M. Wang and K. Ishii, Photochemical properties of phthalocyanines with transition metal ions, Coord. Chem. Rev., 2022, 468, 214626, DOI:10.1016/j.ccr.2022.214626.
- S. Záliš, Y.-C. Lam, H. B. Gray and A. Vlček, Spin–Orbit TDDFT Electronic Structure of Diplatinum(II, II) Complexes, Inorg. Chem., 2015, 54, 3491–3500, DOI:10.1021/acs.inorgchem.5b00063.
- T. J. McKay, J. A. Bolger, J. Staromlynska and J. R. Davy, Linear and nonlinear optical properties of platinum-ethynyl, J. Chem. Phys., 1998, 108, 5537–5541, DOI:10.1063/1.475943.
- V. M. Miskowski and V. H. Houlding, Linear and nonlinear optical properties of platinum-ethynyl, Inorg. Chem., 1989, 28, 1529–1533, DOI:10.1021/ic00307a021.
- J. S. Wilson, N. Chawdhury, M. R. A. Al-Mandhary, M. Younus, M. S. Khan, P. R. Raithby, A. Köhler and R. H. Friend, The energy gap law for triplet states in Pt-containing conjugated polymers and monomers, J. Am. Chem. Soc., 2001, 123, 9412–9417, DOI:10.1021/ja010986s.
- G. S.-M. Tong and C.-M. Che, Emissive or nonemissive? A theoretical analysis of the phosphorescence efficiencies of cyclometalated platinum(II) complexes, Chem. – Eur. J., 2009, 15, 7225–7237, DOI:10.1002/chem.200802485.
- J. E. Huheey and C. L. Huheey, Anomalous Properties of Elements that Follow “Long Periods” of Elements, J. Chem. Educ., 1972, 49, 227–230, DOI:10.1021/ed049p227.
- S. Haupt and K. Seppelt, Solid State Structures of AsCl5 and SbCl5, Z. Anorg. Allg. Chem., 2002, 628, 729–734, DOI:10.1002/1521-3749(200205)628:4<729::AID-ZAAC729>3.0.CO;2-E.
- D. Hernández-Castillo, R. E. P. Nau, M.-A. Schmid, S. Tschierlei, S. Rau and L. González, Multiple Triplet Metal-Centered Jahn-Teller Isomers Determine Temperature-Dependent Luminescence Lifetimes in [Ru(bpy)3]2+, Angew. Chem., Int. Ed., 2023, e202308803, DOI:10.1002/anie.202308803.
- K. Li, G. S. M. Tong, J. Yuan, C. Ma, L. Du, C. Yang, W.-M. Kwok, D. L. Phillips and C.-M. Che, Excitation-Wavelength-Dependent and Auxiliary-Ligand-Tuned Intersystem-Crossing Efficiency in Cyclometalated Platinum(II) Complexes: Spectroscopic and Theoretical Studies, Inorg. Chem., 2020, 59, 14654–14665, DOI:10.1021/acs.inorgchem.0c01192.
- W. H. Lam, E. S.-H. Lam and V. W.-W. Yam, Computational Studies on the Excited States of Luminescent Platinum(II) Alkynyl Systems of Tridentate Pincer Ligands in Radiative and Nonradiative Processes, J. Am. Chem. Soc., 2013, 135, 15135–15143, DOI:10.1021/ja406810a.
- F. Plasser, TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations, J. Chem. Phys., 2020, 152, 084108, DOI:10.1063/1.5143076.
- A. Lázaro, C. Cunha, R. Bosque, J. Pina, J. S. Ward, K.-N. Truong, K. Rissanen, J. C. Lima, M. Crespo, J. S. S. de Melo and L. Rodríguez, Room-temperature phosphorescence and efficient singlet oxygen production by cyclometalated Pt(II) complexes with aromatic alkynyl ligands, Inorg. Chem., 2020, 59, 8220–8230, DOI:10.1021/acs.inorgchem.0c00577.
- K. Li, G. S. M. Tong, J. Yuan, C. Ma, L. Du, C. Yang, W.-M. Kwok, D. L. Phillips and C.-M. Che, Excitation-Wavelength-Dependent and Auxiliary-Ligand-Tuned Intersystem-Crossing Efficiency in Cyclometalated Platinum(II) Complexes: Spectroscopic and Theoretical Studies, Inorg. Chem., 2020, 59, 14654–14665, DOI:10.1021/acs.inorgchem.0c01192.
- P. K. Chow, C. Ma, W.-P. To, G. S. M. Tong, S. L. Lai, S. C. F. Kui, W.-M. Kwok and C.-M. Che, Strongly Phosphorescent Palladium(II) Complexes of Tetradentate Ligands with Mixed Oxygen, Carbon, and Nitrogen Donor Atoms: Photophysics, Photochemistry, and Applications, Angew. Chem., Int. Ed., 2013, 52, 11775–11779, DOI:10.1002/anie.201305590.
- T. Theiss, S. Buss, I. Maisuls, R. López-Arteaga, D. Brünink, J. Kösters, A. Hepp, N. L. Doltsinis, E. A. Weiss and C. A. Strassert, Room-Temperature Phosphorescence from Pd(II) and Pt(II) Complexes as Supramolecular Luminophores: The Role of Self-Assembly, Metal–Metal Interactions, Spin–Orbit Coupling, and Ligand-Field Splitting, J. Am. Chem. Soc., 2023, 145, 3937–3951, DOI:10.1021/jacs.2c09775.
Footnote |
| † Electronic supplementary information (ESI) available: All other data including experimental procedures and characterisation data for all new compounds. CCDC 2149899, 2257280, 2194357 and 2208288 for [Pt(dpp)(AsPh3)], [Pt(dpp)(SbPh3)], [Pt(dba)(AsPh3)] and [Pt(dba)(SbPh3)]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03225f |
| This journal is © The Royal Society of Chemistry 2023 |