
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiply engaged molecular gears composed of a cerium(IV) double-decker of a triptycene-functionalized porphyrin†
Toshio
Nishino
 *a,
Masafumi
Fukumura
a,
Shohei
Katao
a,
Kazuma
Yasuhara
ab and
Gwénaël
Rapenne
*ac
*a,
Masafumi
Fukumura
a,
Shohei
Katao
a,
Kazuma
Yasuhara
ab and
Gwénaël
Rapenne
*ac
aDivision of Materials Science, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara, Japan
bCentre of Digital Green-Inovation, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara, Japan
cCEMES, Université de Toulouse, CNRS, 29, rue Jeanne Marvig, 31055 Toulouse, France. E-mail: rapenne@cemes.fr
First published on 9th August 2023
Abstract
Intramolecular gearing motions are studied in a cerium(IV) double-decker of triptycene-functionalised porphyrins using single crystal X-ray analysis and variable temperature 1H-NMR.
A molecular machine1 is a compound or an assembly of molecular sub-units designed to perform a precise function in response to a specific stimulus. Building machines at the molecular scale has been recognised by the Nobel Prize in 2016.2 In the case of a molecular motor,3 a controlled unidirectional rotary motion has been achieved but only a few studies have seen their work being used for practical applications, for instance the rotation of a glass cylinder 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times bigger than the motor which induces such rotation,4 the transport of a macroscopic droplet,5 the light-induced contraction of a gel6 or the opening of a cell membrane.7
000 times bigger than the motor which induces such rotation,4 the transport of a macroscopic droplet,5 the light-induced contraction of a gel6 or the opening of a cell membrane.7
Among the rotating machines, molecular gears8 appear as essential elementary units allowing the mechanical transmission of the energy induced by this rotating motion to other molecules through a train of gears and consecutively transferring the corresponding energy to another region of the nanospace. In the last few years, many cogwheels have been synthesised9 to be anchored and studied on metallic surfaces and intermolecular rotation transfer has been achieved up to three consecutive molecules.10 At the intramolecular level, the pioneering works of Iwamura and Mislow11 used oxygen or methylene-bridged triptycenes as an archetype of a bevel gear (i.e. with the rotation axis of each rotating sub-unit not being parallel). Many bevel gears with up to six triptycenyl rotating sub-units12 have been described, showing correlated motion in an intermeshed framework. Triptycene moieties have been extensively used as prototypical molecular cogwheels with intrinsic-C3 symmetry. Gears with the rotation axis of each rotating sub-unit being parallel (i.e. spur gears) have also been synthesised and studied.13 Double-decker architectures are very suitable to access a wide variety of 3D arrangements of rotating cogwheels. As a seminal example, Shinkai, Takeuchi et al. synthesised a bis(porphyrinate)lanthanum heteroleptic double-decker used as a suitable central platform for cogwheels and multiple rotating parts.14 In this impressive system, the rotation of the upper porphyrin of the double-decker (in red) induced the rotation of two peripheral porphyrins perpendicularly connected to the lower porphyrin of the double-decker (Fig. 1). This modular platform displayed correlated motion of the four red toothed cogwheels with the two lateral blue cogwheels.
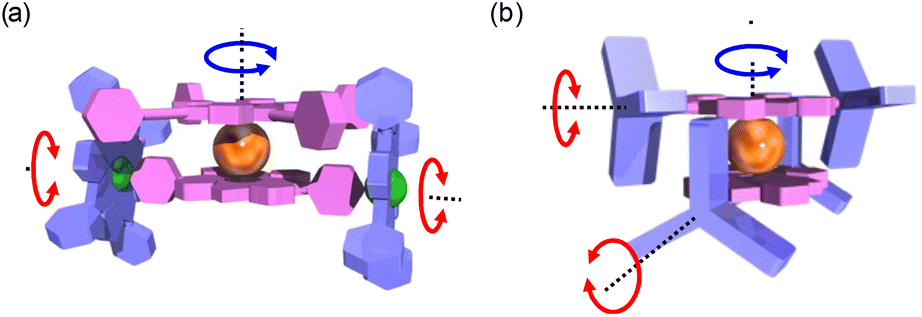 | ||
| Fig. 1 (a) Illustration of the molecular rotating motions in the heteroleptic bis(porphyrinate)lanthanum double-decker system of Shinkai and Takeuchi and (b) in this work. | ||
However, it is still challenging to design and synthesise molecules in which plural motions are linked and synchronised. Recently, we reported the construction of nanocars with ethynyltriptycene wheels at two opposite meso positions.15 Herein we thus report the use of this triptycene-functionalised porphyrin as a sub-unit in the formation of a multiply engaged molecular gearing system composed of a cerium(IV) double-decker. Assembling two of these porphyrins around a lanthanide ion led to the construction of a molecular gear in which the triptycenes are meshed with each other.
In the expected double-decker complex, the porphyrin will be able to rotate around the central metal like a ball bearing. The convergent synthetic strategy to obtain the molecular gear Ce(L)2 and its less sterically demanding analogue Ce(3)2 is shown in Scheme 1. The common precursor Zn2 was synthesised in three steps from 3,5-dibutoxybenzaldehyde following a similar methodology developed to synthesise a family of dipolar nanocars.15 Its condensation with dipyrromethane, followed by oxidation by DDQ gave H21 which was then quantitatively converted to the zinc complex Zn1 and its meso-positions brominated to give Zn2 in a 40% overall yield (for 3 steps). After the introduction of 9-ethynyltriptycene16 or ethynylphenyl subunits at the two brominated meso positions of Zn2 through Sonogashira coupling reaction,17 the central zinc ion was removed using TFA to afford the corresponding free-base porphyrins H2L and H23. In general, prolonged heating in a high boiling solvent (e.g. 1,2,4-trichlorobenzene or 1-chloronaphthalene) is required for the formation of multi-decker complexes of porphyrinoid ligands, such as porphyrin and phthalocyanine, with lanthanoid ions. Kadish et al. reported that the microwave-mediated method is effective for the synthesis of multi-decker complexes with bulky substituents.18 Following their synthetic methodology, the formation of the homoleptic cerium(IV) double-decker complexes was achieved by complexation of free-base porphyrins with Ce(acac)3·nH2O under microwave irradiation in o-dichlorobenzene. The double-decker-type complex of the triptycene-functionalised porphyrin Ce(L)2 was obtained in 58% yield, whereas its less sterically demanding analogue Ce(3)2 was obtained in 31% yield.
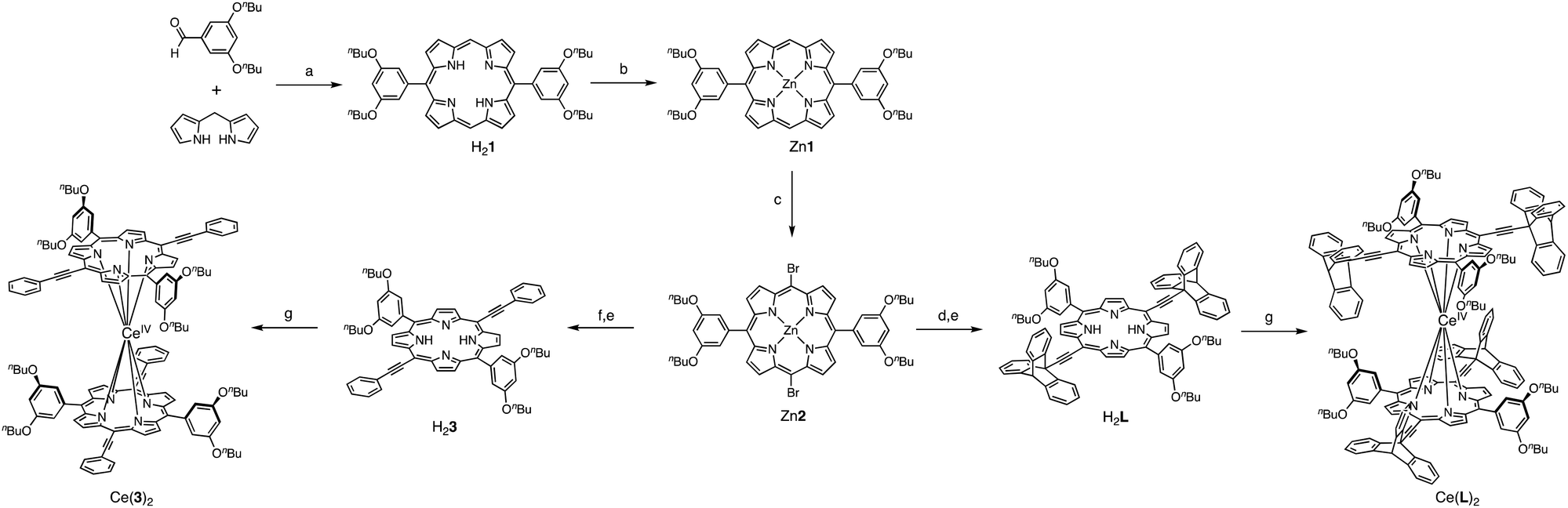 | ||
| Scheme 1 Synthesis of the molecular gear Ce(L)2 and its less sterically demanding analogue Ce(3)2. Reaction conditions: (a) (i)TFA, CH2Cl2, RT; (ii) DDQ, Et3N 41%; (b) Zn(Oac)2·2H2O, CHCl3, MeOH 100%; (c) NBS, CH2Cl2, pyridine, −20 °C, 12 h, 96%; (d) 9-ethynyltriptycene, Pd(PPh3)2Cl2, CuI, Et3N, THF, N2, reflux, 3 h, 96%; (e) TFA, CH2Cl2, RT, 1 h, 96–100%; (f) phenylacetylene, Pd(PPh3)2Cl2, CuI, Et3N, THF, N2, reflux, 3 h, 83%; and (g) Ce(acac)3·nH2O, o-dichlorobenzene, MW heating, 220–250 °C, 31% for Ce(3)2 58% for Ce(L)2. For full description refer to the ESI.† | ||
The structures of both complexes Ce(L)2 and Ce(3)2 were confirmed by MALDI-TOF MS, 1H-NMR, 13C NMR, and elemental analyses. MALDI-TOF MS showed molecular peaks with the expected isotopic distribution pattern (Fig. S1†). Crystals of Ce(L)2 suitable for single crystal X-ray analysis were obtained by the diffusion of methanol into a chloroform solution of the sample. The central Ce4+ in Ce(L)2 is coordinated by the eight pyrrolic nitrogen atoms of two porphyrinato ligands L, resulting in a square antiprismatic coordination geometry for the Ce4+ ion with an average twist angle of 47° between the two ligands (Fig. 2). The average distances between two sets of bridgehead carbon atoms, namely the 9 and 10 positions of triptycene (refer to Fig. 2 for the numbering), were 7.2 and 9.0 Å, respectively. In the molecular gear of Shionoya et al.,12 in which six triptycenes are arranged circularly around a hexasubstituted phenyl ring, the distances between the bridgehead positions are 5.5 and 8.1 Å, respectively. These shorter distances compared to the similar distances in Ce(L)2 are due to the difference in the size of the core structure with the porphyrin being larger than a phenyl ring.
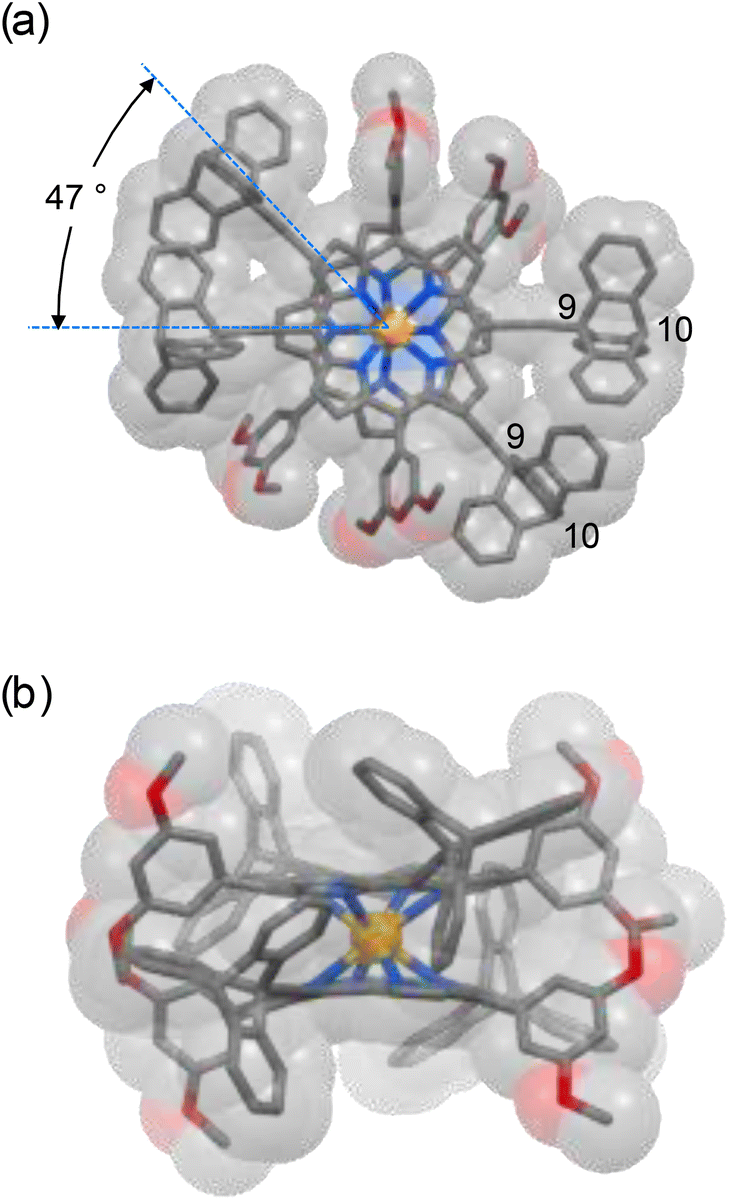 | ||
| Fig. 2 Top view (a) and side view (b) of the X-ray crystal structure of the double decker complex Ce(L)2. Hydrogen atoms, solvent molecules and disordered atoms are omitted for clarity. Only the first carbon atom of the butyl chain is shown for each Bu group. Color code: Ce, orange; C, gray; N, blue; and O; red. | ||
The NMR spectra of L and Ce(L)2 are compared in Fig. 3 (for the full 1H-NMR spectrum with the attribution of all signals, see Fig. S2†). The peaks corresponding to the triptycene moieties appeared at 8.39, 7.56, 7.23 and 7.18 ppm before complexation. After complexation, these peaks shifted to 8.58, 7.58, 7.08 and 6.91 ppm (Fig. 3 red lines). This change in their chemical shift clearly illustrates the influence of the neighbouring phenyl sub-units at the meso-positions which are close to the triptycenes after complexation. It should be mentioned that the presence of four signals for the triptycene sub-units reflects the chemical equivalence of the three phenyl rings, indicating that the rotation of the two triptycenes, which formed a gear-like engaged structure in the crystal, was fast at room temperature, compared to the NMR timescale. As for the porphyrin core, the signals corresponding to protons at the β-positions appeared at 9.99 and 9.11 ppm as two sets of doublets before complexation reflecting the C2v symmetry of the precursor. After complexation, these signals shifted to a higher field and each of them split into two sets of doublets at 9.85, 9.56 and 8.90, 8.58 ppm (Fig. 3 blue lines). This splitting is due to a lower symmetry in the complex (D2) with a non-equivalent environment of the four protons at the β-position because of the staggered configuration of the porphyrin ligands, meaning that the rotation of the two porphyrins around the Ce(IV) ion was slow compared to the NMR timescale. The double-decker complex in which two trans-A2B2-type porphyrins coordinate to a central metal ion in a square antiprismatic fashion has D2 symmetry and in consequence it is chiral. Two enantiomeric complexes, a left handed (Λ) and a right handed (Δ) Ce(IV) complexes, co-exist as shown in Fig. 4. The signals corresponding to the 3,5-dibutoxyphenyl rings are also strongly modified, especially the two ortho protons to the meso position are split from one doublet at 7.58 ppm to two signals at 5.80 and 9.10 ppm. This strong effect illustrates the very different chemical environment felt by the ortho protons after complexation, one being close to the cerium centre while the other one being outside of its influence. This splitting also clearly indicates that the 3,5-dibutoxyphenyl ring is not freely rotating. On the other hand, the proton at the para position is almost unchanged at around 6.9 pm. In addition, since the signals corresponding to the protons at the β-position of the porphyrins present four doublets, there is no rotation of the porphyrin subunits relative to each other around the porphyrin–cerium–porphyrin axis.
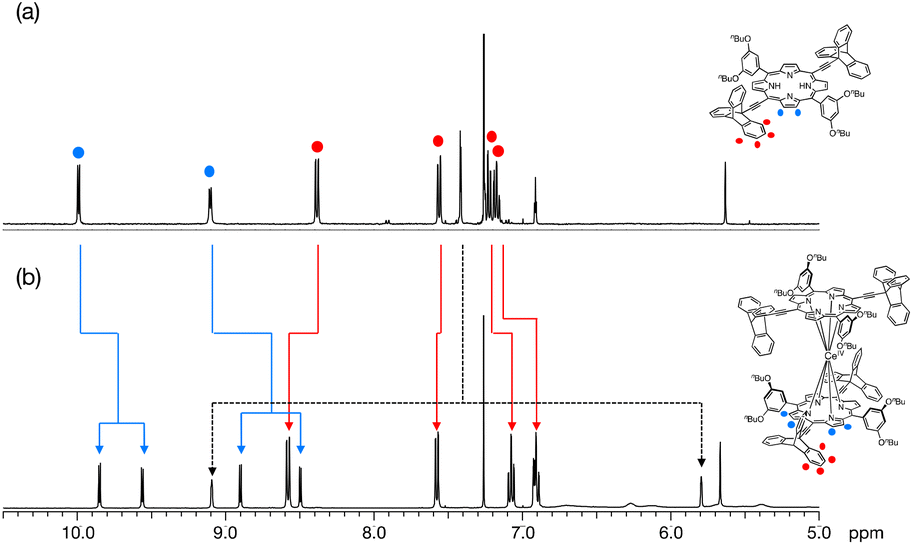 | ||
| Fig. 3 Aromatic region of the 1H-NMR spectra of (a) H2L and (b) Ce(L)2 in CDCl3 (400 MHz, 293 K). The blue lines illustrate the shifting and splitting of the porphyrinic signals, whereas the red lines indicate the shifting of the triptycene signals. In addition, the large splitting of the two ortho protons of the 3,5-dibutoxyphenyl rings is shown with black dotted lines. | ||
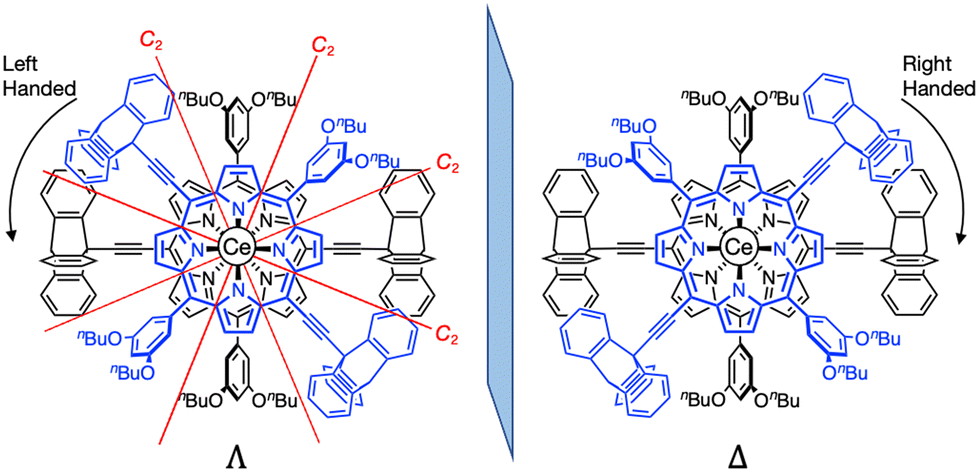 | ||
| Fig. 4 The two enantiomers of Ce(L)2: left handed (Λ) and right handed (Δ) with the position of the C2 axis of symmetry. | ||
The dynamic behaviour of Ce(L)2 in solution was clarified by variable temperature 1H-NMR (Fig. S3†). At room temperature, we know from the 1H-NMR data that the triptycenes are freely rotating, whereas the 3,5-dibutoxyphenyl and the porphyrin rings are not. Low-temperature NMR measurements were carried out to elucidate the effect of temperature on the gearing rotation of the triptycenes. Upon cooling down the sample solution to 213 K, a broadening of peaks was observed, implying that the rotational speed of triptycenes became slower and close to the NMR timescale. However, the coalescence and splitting of peaks attributed to triptycene units were not observed until 213 K but attempts to go below this temperature failed due to the limitation of the solubility of Ce(L)2 in toluene-d8. Rotation between porphyrin ligands was also investigated at higher temperature. If the intra-ligand rotation becomes faster than the NMR timescale, the signals corresponding to the protons at the β-position are expected to coalesce and be finally observed as two sets of doublets upon the increase of the temperature. Surprisingly, the peak splitting pattern did not change up to 373 K (Fig. S3†). This result indicated that the relative angle of two porphyrins coordinating to the Ce ions was maintained even at high temperature and Ce(L)2 has a high activation energy for intra-ligand rotation. This result is very different from the observation of Aida and coworkers19 who showed that a Ce(IV) bis(ditolyl) trans A2B2 porphyrin complex had coalescence at 13 °C. Intrigued by this observation, we wanted to clarify if this restriction in the rotation came from a large steric hindrance caused by the presence of the four triptycenes. We then studied the less sterically demanding complex Ce(3)2. This analogue compound functionalised with ethynyl phenyl groups at its meso-position instead of ethynyltriptycenes (Scheme 1) also did not show the coalescence behaviour reflecting the rotation of porphyrin around the cerium(IV) ion in a similar temperature range (Fig. S4†). This result may be due to the steric hindrance between the four phenyl groups at the meso-position or the characteristics of the central cerium(IV) ion, rather than to the steric hindrance between neighbouring triptycenes in Ce(L)2. The result that no coalescence of the porphyrin β-position signals was observed from NMR measurements in Ce(L)2 over a wide temperature range indicated that the activation energy of Ce(L)2 for racemisation was higher than that of the double-decker complexes reported by Aida and coworkers.
In conclusion, we synthesised double-decker type molecular gears through the complexation of triptycene-functionalised porphyrins with a cerium(IV) ion. Single crystal X-ray analysis revealed that the two triptycenes introduced onto the porphyrin ring were doubly engaged. Variable temperature 1H-NMR measurements revealed that the two kinds of rotations, namely engaged triptycenes’ rotation, and porphyrins’ rotation, were independent. The rotation speed between two porphyrins around the metal ion was slower than the NMR timescale at room temperature and the two engaged triptycenes rotate faster than the NMR timescale.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI Grant-in-Aid for Challenging Research (20K21131; GR), the JSPS KAKENHI Grant-in-Aid for Basic Research A (22H00325; GR) and the JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (21K14406; TN). The University Paul Sabatier (Toulouse) and NAIST are also warmly thanked for providing a crossed position to GR. We thank Mr Fumio Asanoma for his help with NMR measurements.References
- V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines. Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- (a) J.-P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080 CrossRef CAS PubMed; (b) J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094 CrossRef CAS PubMed; (c) B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060 CrossRef CAS PubMed.
- (a) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592 RSC; (b) M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200 CrossRef CAS PubMed; (c) D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310 CrossRef CAS PubMed; (d) V. García-López, D. Liu and J. M. Tour, Chem. Rev., 2020, 120, 79 CrossRef PubMed; (e) U. G. E. Perera, F. Ample, H. Kersell, Y. Zhang, G. Vives, J. Echeverria, M. Grisolia, G. Rapenne, C. Joachim and S.-W. Hla, Nat. Nanotechnol., 2013, 8, 46 CrossRef CAS PubMed; (f) Y. Zhang, H. Kersell, R. Stefak, J. Echeverria, V. Iancu, U. G. E. Perera, Y. Li, A. Deshpande, K.-F. Braun, C. Joachim, G. Rapenne and S.-W. Hla, Nat. Nanotechnol., 2016, 11, 706 CrossRef CAS PubMed; (g) H. P. Jacquot de Rouville, R. Garbage, F. Ample, A. Nickel, J. Meyer, F. Moresco, C. Joachim and G. Rapenne, Chem. – Eur. J., 2012, 18, 8925 CrossRef CAS; (h) Y. Zhang, J. P. Calupitan, T. Rojas, R. Tumbleson, G. Erbland, C. Kammerer, T. M. Ajayi, S. Wang, L. C. Curtiss, A. T. Ngo, S. E. Ulloa, G. Rapenne and S. W. Hla, Nat. Commun., 2019, 10, 3742 CrossRef.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 128, 14397 CAS.
- J. Berna, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Perez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704 CrossRef CAS PubMed.
- Q. Li, G. Fuks, E. Moulin, M. Maaloum, M. Rawiso, I. Kulic, J. T. Foy and N. Giuseppone, Nat. Nanotechnol., 2015, 10, 161 CrossRef CAS PubMed.
- V. García-López, F. Chen, L. G. Nilewski, G. Duret, A. Aliyan, A. B. Kolomeisky, J. T. Robinson, G. Wang, R. Pal and J. M. Tour, Nature, 2017, 548, 567 CrossRef PubMed.
- Y. Gisbert, S. Abid, C. Kammerer and G. Rapenne, Chem. – Eur. J., 2021, 27, 12019 CrossRef CAS PubMed.
- (a) G. Erbland, S. Abid, Y. Gisbert, N. Saffon-Merceron, Y. Hashimoto, L. Andreoni, T. Guérin, C. Kammerer and G. Rapenne, Chem. – Eur. J., 2019, 25, 16328 CrossRef CAS PubMed; (b) Y. Gisbert, S. Abid, G. Bertrand, N. Saffon-Merceron, C. Kammerer and G. Rapenne, Chem. Commun., 2019, 55, 14689 RSC; (c) S. Abid, Y. Gisbert, M. Kojima, N. Saffon-Merceron, J. Cuny, C. Kammerer and G. Rapenne, Chem. Sci., 2021, 12, 4709 RSC; (d) K. Omoto, M. Shi, K. Yasuhara, C. Kammerer and G. Rapenne, Chem. – Eur. J., 2023, 29, e202203483 CrossRef CAS PubMed.
- K. H. Au Yeung, T. Kühne, F. Eisenhut, M. Kleinwächter, Y. Gisbert, R. Robles, N. Lorente, G. Cuniberti, C. Joachim, G. Rapenne, C. Kammerer and F. Moresco, J. Phys. Chem. Lett., 2020, 11, 6892 CrossRef CAS PubMed.
- H. Iwamura and K. Mislow, Acc. Chem. Res., 1988, 21, 175 CrossRef CAS.
- (a) H. Ube, R. Yamada, J. Ishida, H. Sato, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2017, 139, 16470 CrossRef CAS PubMed; (b) H. Ube, Y. Yasuda, H. Sato and M. Shionoya, Nat. Commun., 2017, 8, 14296 CrossRef CAS PubMed.
- (a) J. C. Bryan, R. A. Sachleben, A. A. Gakh and G. J. Bunick, J. Chem. Crystallogr., 1999, 29, 513 CrossRef CAS; (b) S. Toyota, T. Shimizu, T. Iwanaga and K. Wakamatsu, Chem. Lett., 2011, 40, 312 CrossRef CAS; (c) D. K. Frantz, A. Linden, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 2012, 134, 1528 CrossRef CAS PubMed.
- S. Ogi, T. Ikeda, R. Wakabayashi, S. Shinkai and M. Takeuchi, Chem. – Eur. J., 2010, 16, 8285 CrossRef CAS PubMed.
- T. Nishino, C. J. Martin, H. Takeuchi, F. Lim, K. Yasuhara, Y. Gisbert, S. Abid, N. Saffon-Merceron, C. Kammerer and G. Rapenne, Chem. – Eur. J., 2020, 26, 12010 CrossRef CAS PubMed.
- G. Rapenne and G. Jimenez-Bueno, Tetrahedron, 2007, 63, 7018 CrossRef CAS.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- H.-G. Jin, X. Jiang, I. A. Kühne, S. Clair, V. Monnier, C. Chendo, G. Novitchi, A. K. Powell, K. M. Kadish and T. S. Balaban, Inorg. Chem., 2017, 56, 4864 CrossRef CAS PubMed.
- K. Tashiro, K. Konishi and T. Aida, Angew. Chem., Int. Ed. Engl., 1997, 36, 856 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation of compounds, and spectra of new compounds. CCDC 2258829. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02443a |
| This journal is © The Royal Society of Chemistry 2023 |