
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The preservation of sarin and O,O′-diisopropyl fluorophosphate inside coordination cage hosts†
Jack C.
Dorrat
a,
Rosemary J.
Young
a,
Christopher G. P.
Taylor
d,
Max B.
Tipping
d,
Andrew J.
Blok
 b,
David R.
Turner
a,
Alasdair I.
McKay
a,
Simon
Ovenden
b,
Michael D.
Ward
*d,
Genevieve H.
Dennison
*bc and
Kellie L.
Tuck
*a
b,
David R.
Turner
a,
Alasdair I.
McKay
a,
Simon
Ovenden
b,
Michael D.
Ward
*d,
Genevieve H.
Dennison
*bc and
Kellie L.
Tuck
*a
aSchool of Chemistry, Monash University, Melbourne, VIC 3800, Australia. E-mail: Kellie.Tuck@monash.edu
bCBRN Defence Branch, Sensors and Effectors Division, Defence Science and Technology Group, Fishermans Bend, VIC 3207, Australia
cWeapon Seekers and Tactical Sensors Branch, Sensors and Effectors Division, Defence Science and Technology Group, Edinburgh, SA 5111, Australia. E-mail: Genevieve.Dennison@defence.gov.au
dDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: M.D.Ward@warwick.ac.uk
First published on 30th May 2023
Abstract
The host–guest chemistry of O,O′-diisopropyl fluorophosphate (DFP), a phosphonofluoridate G-series chemical warfare agent simulant, was investigated in the presence of a number of octanuclear cubic coordination cage hosts. The aim was to demonstrate cage-catalysed hydrolysis of DFP at near neutral pH: however, two octanuclear coordination cages, HPEG (containing water-solubilising PEG groups) and HW (containing water-solubilising hydroxymethyl groups), were actually found to increase the lifetime of DFP in aqueous buffer solution (pH 8.7). Crystallographic analysis of DFP with a structurally related host cage revealed that DFP binds to windows in the cage surface, not in the internal cavity. The phosphorus–fluorine bond is directed into the cavity rather than towards the external environment, with the cage/DFP association protecting DFP from hydrolysis. Initial studies with the chemical warfare agent (CWA) sarin (GB) with HPEG cage in a buffered solution also showed a drastically reduced rate of hydrolysis for sarin when bound in the host cage. The ability of these cages to inhibit hydrolysis of these P–F bond containing organophosphorus guests, by encapsulation, may have applications in forensic sample preservation and analysis.
Introduction
The organophosphorus chemical warfare agent (OP CWA) sarin (O-isopropyl methylphosphonofluoridate, or GB; Scheme 1), a G-series nerve agent, is a volatile, highly toxic and potent acetylcholinesterase inhibitor and, in its pure form, is colourless and odourless.1 Understandably, the management and remediation of OP CWA contamination to safe levels is of critical importance. The P–F bond of GB can be hydrolysed in aqueous solutions, to produce O-isopropyl methylphosphonic acid (IMPA), which is significantly less toxic (Scheme 1). In the field, decontamination of G-series agents is typically achieved by using mixtures of sodium hypochlorite (bleach), ethanol and water. Use of this corrosive mixture however, can result in significant damage to expensive and sensitive equipment.2 The development of any species which can catalyse the decomposition of GB at near neutral pH, in a localised and controlled environment, would allow for a less destructive decontamination process for such equipment.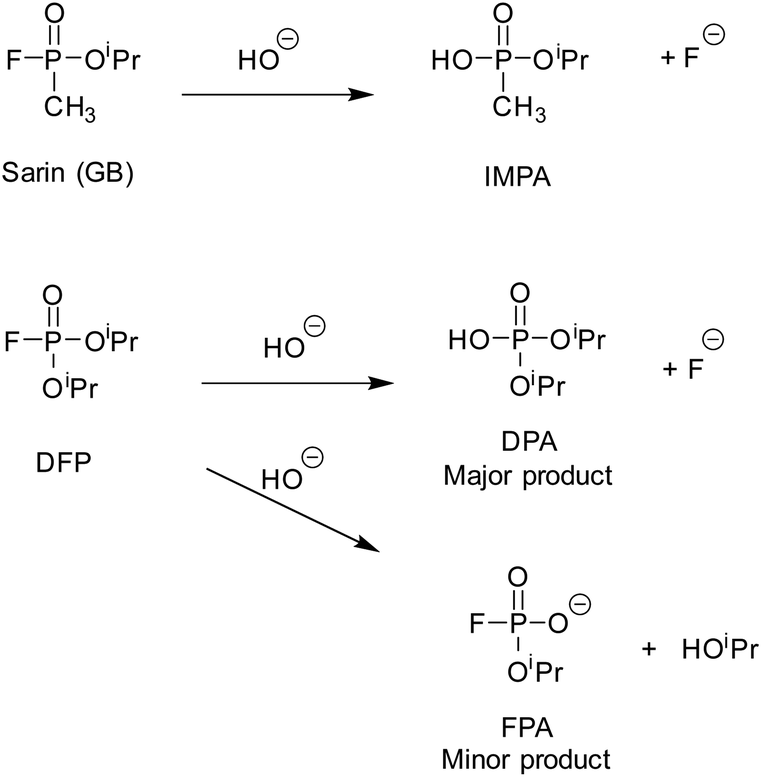 | ||
| Scheme 1 Hydrolysis of GB (sarin) and the simulant O,O′-diisopropyl fluorophosphate (DFP) under basic aqueous conditions; IMPA – O-isopropyl methylphosphonic acid, DPA – O,O′-diisopropyl phosphoric acid, FPA – isopropyl fluorophosphonate. | ||
Coordination cages have been shown to display a range of host–guest chemistries with varying functions, including catalysis,3–7 guest absorption and subsequent separation,8–10 and molecular sensing.11–15 In recent years we have reported a series of octanuclear cubic cages with the general structure [M8L12]16+ (Fig. 1a and b) which differ in the external substituents attached to the pyridine rings: these cages are abbreviated as either H (R = H),16HW (R = CH2OH),17 or HPEG (R = (CH2OCH2)3CH2OCH3),18 see Fig. 1a for ligand structures. The ligands have a 1,5-naphthalene-diyl core linking two bidentate pyrazolylpyridine chelating termini. When coordinated to Co2+ or Cd2+ ions the resulting cage complexes have been demonstrated to be effective hydrophobic hosts, encapsulating hydrophobic organic guests in water.19–24 The interior cavity has a volume of approximately 400 Å3 meaning that (following the “55% rule” proposed by Rebek)25 the ideal volume of a guest molecule is just over 200 Å3, which is sufficient for a wide range of organic substrates to be viable guests,19–24 though the highest binding constants (of up to 106 M−1 in water) are associated with guests close to this optimal size.20
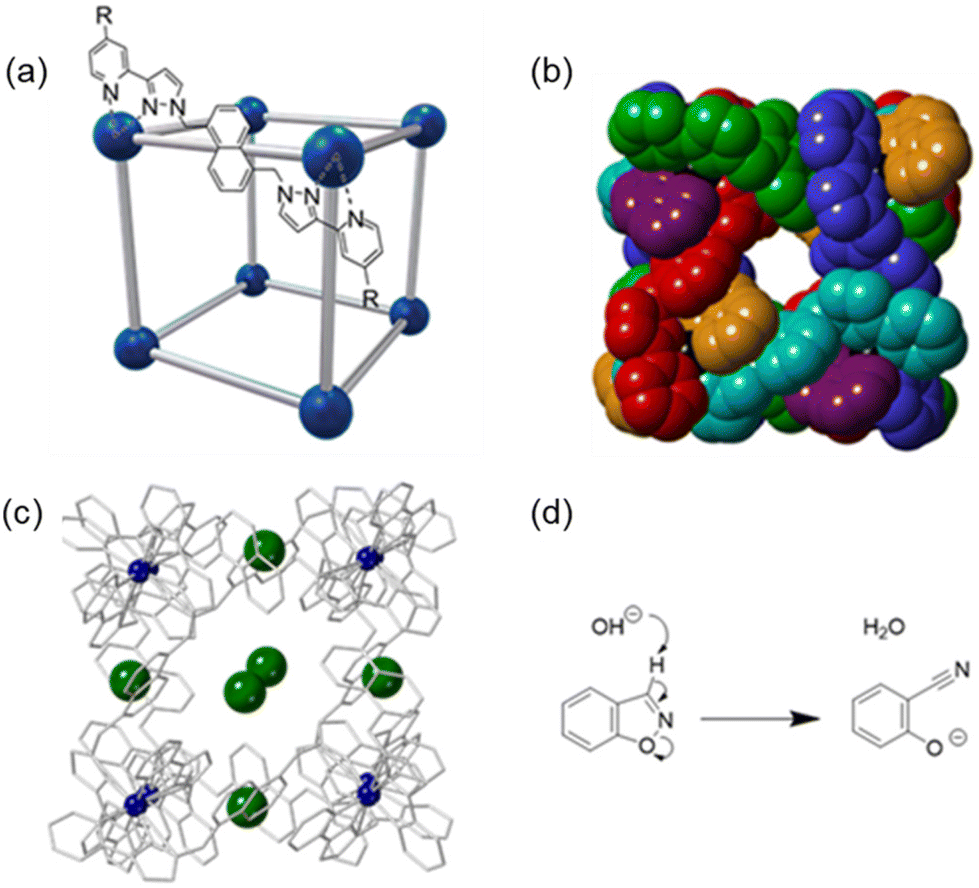 | ||
Fig. 1 The cages [M8L12]16+ (M defines the cationic metal ion, L the ligand) are abbreviated as either H (R = H), HW (R = CH2OH), or HPEG (R = (CH2OCH2)3CH2OCH3). (a) A representation of the cage emphasising the cubic array of metal (M2+) ions as blue spheres connected by the bridging ligands (L) as grey rods; (b) space-filling model showing a view of the hollow cavity of the cubic cage (R = H), with each ligand coloured differently for clarity;30 (c) crystal structure (wireframe) of H·Cl16 showing the position of the cage anions (chloride, green) in the windows and Co2+ metal ions (blue, CCDC #1581566![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30), disordered cage anions are not shown; (d) the Kemp elimination reaction of benzisoxazole, which has a faster rate in the presence of cage HW.31 30), disordered cage anions are not shown; (d) the Kemp elimination reaction of benzisoxazole, which has a faster rate in the presence of cage HW.31 | ||
The nature of the counter-ion in the chemistry of these cages (which carry a 16+ charge) has turned out to be as important as the substrate binding in the central cavity.26–28 The counter-ion of the as-synthesised cages is often BF4− or Cl− for solution studies, but we have also investigated a variety of other counter ions for crystallographic studies (for example NO3−, ClO4−, BPh4−, PF6−, SO42− and CF3SO3−).26–30 We observe that the anions always bind to the surface of the cage in the windows in the face centres: these windows seem to be an ideal size to present a convergent array of C–H groups to any anion located there. Judicious selection of counter ions, coupled with the modification of the ligand R substituent, has been shown to improve the aqueous solubility of the cages, as well as modify the reactivity behaviours of the cage/guest complexes.26
In some cases we have observed catalysed reactions of bound guests with a range of reaction types in aqueous environments.30–33 For example, HW (water-solubilised by the external hydroxymethyl substituents) substantially enhances the rate of the Kemp elimination reaction of cavity-bound benzisoxazole when compared to the control buffer solution (Fig. 1d, kcat/kuncat of 2 × 105, pD 8.5).31 This catalysis occurs because the accumulation of hydroxide ions in the portals around the 16+ cage surface, even under very weakly basic conditions, results in a much higher local pH (>5 pH units) around the cavity-bound hydrophobic guest compared to the bulk solution. The product of this reaction, 2-cyanophenolate, is negatively charged at the prevailing pH and more hydrophilic, and thus has reduced binding affinity in the cage cavity and moves into the external aqueous solution where it is solvated, allowing the host to bind another molecule of guest and exhibit catalytic turnover.31 The basis of the catalysis is therefore that the hydrophobic interaction of these guests with the cage brings the substrates into close proximity with the high local concentration of hydroxide ions which surround the surface, attracted by the positive charge of the cage: these two interactions (hydrophobic and electrostatic) are essentially orthogonal.28
The cage H (water solubilised by use of chloride as the counter-ion)30 enhances the rate of the hydrolysis of the organophosphate (OP) pesticides dichlorvos (2,2-dichlorovinyl dimethyl phosphate, kcat/kuncat of 14, pD 7.7) and paraoxon-methyl (4-nitrophenyl dimethyl phosphate, kcat/kuncat of 11, pD 7.8): structures shown in Fig. 2a.32 In this case, unexpectedly, the hydrolysis reaction of dichlorvos and paraoxon-methyl in solution was determined to occur at the exterior surface of the cage H rather than in the cavity.32 However in the crystal structure, obtained under forcing non-equilibrium conditions and with high guest concentration, dichlorvos was observed to bind inside the cavity as well as at the exterior surface (Fig. 2b). More recently, we have observed that diacetyl fluorescein (ca. 300 Å3), which is too long to bind in the cage cavity, interacts strongly with the exterior surface of HW (K = 1.5 × 104 M−1) and is brought into proximity to the shell of hydroxide ions close to the cage, catalysing hydrolysis of the ester groups, with a kcat/kuncat ratio of ca. 50. This study also demonstrated that 4-nitrophenyl acetate binds inside the cage cavity but, in this case, the hydrolysis reaction was actually inhibited.33 Also of note to the present study are the previously reported ‘crystal sponge’ experiments, where under forcing conditions two CWA simulants, O,O′-dimethyl methylphosphonate (DMMP) and O,O′-diisopropyl methylphosphonate (DIMP), demonstrated binding to the interior cavity of the H·(BF4)16 cage (Fig. 2c and d).341H paramagnetic NMR spectroscopic studies with HW·(BF4)16 in water revealed that DMMP was in fast exchange (K = 7 M−1) and DIMP, the larger guest, was in slow exchange with the host (K = 390 M−1).34
 | ||
| Fig. 2 (a) The chemical structures of dichlorvos, paraoxon-methyl, DMMP and DIMP; (b) crystal structure of H·(BF4)16 with dichlorvos (CCDC #1959406),32 (c) crystal structure of H·(BF4)16 with one DIMP guest molecule (CCDC #1458106);34 (d) crystal structure of H·(BF4)16 with two guest molecules of DMMP (CCDC #1458107).34 H atoms and BF4 anions are omitted for clarity: Co – dark blue, C – black, N – blue, O – red, P – purple, F – light green, Cl – dark green. | ||
Due to the findings in our previous research outlined above, it was of interest to investigate the host–guest properties of OP CWAs such as GB with these coordination cages and determine if the rate of hydrolysis of GB and O,O′-diisopropyl fluorophosphate (DFP), a simulant of GB (see Scheme 1 for chemical structures),35 increased in the presence of cage due to catalysis of the reaction with hydroxide; or alternatively, if the rate of hydrolysis was inhibited because the cavity-bound substrate is protected from the surrounding environment – we have seen examples of both types of behaviour. In this paper, we present our findings involving the host–guest chemistry of the substrate DFP in the presence of the coordination cages H, HW, and HPEG (Fig. 1a). Interestingly, for several cages a substantial improvement in stability for DFP in buffered aqueous solution was observed; this observation was also reproduced in an experiment using GB with HPEG as the host. NMR spectroscopy experiments demonstrated that DFP is cavity-bound with HW and HPEG in solution; and crystallographic experiments with H revealed that, in the solid state, DFP is bound to the coordination cage in the windows in a way which protects the reactive P–F bond from the aqueous environment, thereby providing a rationale for the increased stability of both DFP and GB in aqueous solution.
Results and discussion
Caution, the CWA GB (sarin) and its simulant DFP are both acetylcholinesterase inhibitors, and can cause incapacitation and death at low concentrations. All of the work reported herein was conducted by trained professionals in specialised facilities, accredited for the safe handling and experimentation of GB for protective purposes. GB is a Schedule 1 chemical under the Chemical Weapons Convention, and its synthesis and experimentation is highly regulated under national laws and with international oversight from the Organisation for the Prohibition of Chemical Weapons (OPCW) in The Hague, Netherlands.NMR spectroscopic analysis of DFP in the presence of [M8L12]16+ cages
Under near neutral aqueous conditions (50 mM borate buffer, pH 8.7), the CWA simulant DFP was observed to hydrolyse into two major products, O,O′-diisopropyl phosphoric acid (DPA) and fluoride, and two minor products, O-isopropyl fluorophosphonate (FPA) and isopropanol (Scheme 1). As noted above, it was expected that DFP would bind to the cages via the hydrophobic effect, in a way similar to that observed for DMMP and DIMP.34 It was also expected that the rate of aqueous hydrolysis would increase due to the high local hydroxide concentration at the exterior of the cage surface, following other examples of cage-based catalysis of reactions with hydroxide ions.31–33 We have recently observed that catalytic reactions with all cages in the presence of chloride ions are slower than in their absence.26,31,33 When chloride ions are present they accumulate around the cage more than hydroxide ions as they are more readily desolvated: the local hydroxide ion concentration is therefore lowered and the chloride ions bind to the cage windows, not only displacing hydroxide ions but also blocking access of potential substrates to the cavity.31 Therefore, we investigated the cages HW·OH (with M = Co2+) and HPEG·OH (with M = Cd2+), for their hydrolytic properties with DFP; and compared our findings to cages with the chloride anion (H·Cl16 and HW·Cl16, M = Co2+) present following anion metathesis. Previous studies by our group have shown, in the cases where the same guests were examined, that the guest binding constants were generally comparable for cages with M = Co2+ or Cd2+. This is not unexpected as the cages are isostructural, and have the same charge. HW and HPEG were prepared as their tetrafluoroborate and nitrate salts respectively. For solution use they were digested in buffer until dissolved. As the catalytic activity of the coordination cages is due to HO− anions, for the purposes of this research we have termed the soluble form as HW·OH and HPEG·OH respectively. As H·Cl16 and HW·Cl16 were obtained following anion exchange we have chosen to note their empirical formula when referenced.The rate of DFP hydrolysis can be determined by monitoring either the 31P NMR spectra for the disappearance of resonances related to DFP (d, −10.64 ppm, 1JP–F = 974 Hz), and the concurrent appearance of resonances due to either DPA (s, −0.91 ppm) or FPA (d, 5.97 ppm, 1JP–F = 925 Hz), or via19F NMR spectroscopy monitoring the disappearance of resonances related to DFP (d, −77.8 ppm, 1JF–P = 974 Hz), and the appearance of resonances related to FPA (d, −76.8 ppm, 1JF–P = 925 Hz) or fluoride ions (s, −120.1 ppm). The typical spectral changes, observed over time, of DFP in the presence of HPEG·OH are reproduced in Fig. 3. The NMR spectra of DFP in the presence of the HPEG·OH cage, which contained diamagnetic Cd2+ ions, resulted in narrow resonance lines, whereas for cages containing paramagnetic Co2+ ions (H·Cl16, HW·Cl16 and HW·OH) resonances were both broadened and substantially shifted. Within the research described, the reported rate constants and half-lives were calculated, unless otherwise stated, via analysis of the 19F NMR spectra. This was due to improved signal to noise (S/N) that is a result of the greater receptivity of the 19F nuclei than the 31P nuclei.36 Whilst not analysed in detail, the rates determined from analysis of the 31P NMR spectra were consistent with values obtained via analysis of the corresponding 19F NMR spectra.
 | ||
| Fig. 3 (a) 19F NMR spectra of DFP (7.5 mM) with HPEG·OH (0.50 mM) after (i) 48 min, (ii) 6 days, and (iii) 16.5 days. (b) 31P NMR spectra of DFP (7.5 mM) with HPEG·OH (0.50 mM) after (i) 21 min, (ii) 6 days, and (iii) 16.5 days; in H2O/D2O (90:10 v/v) with 50 mM borate buffer, pH 8.7. DFP (*), FPA (#), fluoride ions (^) and DPA (x). | ||
In the absence of any cage, in buffer solution alone, the hydrolysis rate of DFP (7.5 mM DFP, 50 mM borate buffer, 90% H2O:10% D2O, pH 8.7) was first-order with respect to DFP and had an apparent initial first-order rate constant of 1.6 × 10−5 s−1 and t1/2 = 11.8 h (Table 1, entry 1). This was determined by fitting the concentration change of DFP over time to a one-phase decay model (Fig. S1†). As expected, the rate of formation of fluoride ions was also first-order with the same apparent initial first-order rate constant within error, 1.7 × 10−5 s−1. After 5 days (ca. 10 half-lives) the resonances due to DFP could no longer be observed. FPA, the minor product from the hydrolysis reaction (Scheme 1), was also observed (6%) alongside the major product (fluoride ions). Under identical conditions, except with the addition of 0.5 mM HPEG·OH, a substantial decrease in the rate of hydrolysis of DFP was observed (Fig. 4). Surprisingly, the resonance for DFP remained observable in the 19F NMR spectrum after 35 days, with 1% of DFP remaining after this time. The preservation of pesticides by supramolecular hosts has been previously documented, as demonstrated by the preservation of chloroamidophos in the presence of β-cyclodextrin.37 However, to the best of our knowledge, the preservation of DFP, a CWA simulant – and CWAs like sarin (see below) – has not been reported previously. It was noted that after 7 days, for the sample with the addition of 0.5 mM HPEG·OH, that the resonance due to the fluoride ion shifted downfield slightly and the resonance broadened to such an extent that the data was no longer suitable for inclusion in the analysis. Correlation of the 19F integration value with the concentration of the species being analysed prior to this 7 day timepoint was confirmed in a separate experiment. Fluoride ions from NaF (7.5 mM), representing the fluoride ions generated by hydrolysis of DFP, were added to an aqueous buffered solution of 0.2 mM HPEG·OH (Fig. S2†). Under these conditions identical signal broadening was observed, indicating that the fluoride ion was interacting with the dimagnetic cage. Importantly, integration of this resonance immediately after addition and at 7 days post addition, was identical to that obtained for a 7.5 mM solution of NaF in buffer alone and as a result, it was concluded that monitoring the formation of fluoride ions over the initial 7 day period is representative of the rate of formation of DPA. Plotting the disappearance of DFP over time, and the appearance of fluoride ions (major product) and FPA (minor product), demonstrated that the hydrolysis of DFP has two distinctly different rate profiles (Fig. 4a and b). No additional fluoride formation was observed after 5 days, whereas progressive formation of FPA occurred throughout the 35 day period. The differences in rates between the control experiment (buffer solution only), and in the presence of HPEG·OH, are further evident when the combined integration products of the reaction (fluoride ion and FPA) are plotted versus time (Fig. 4c).
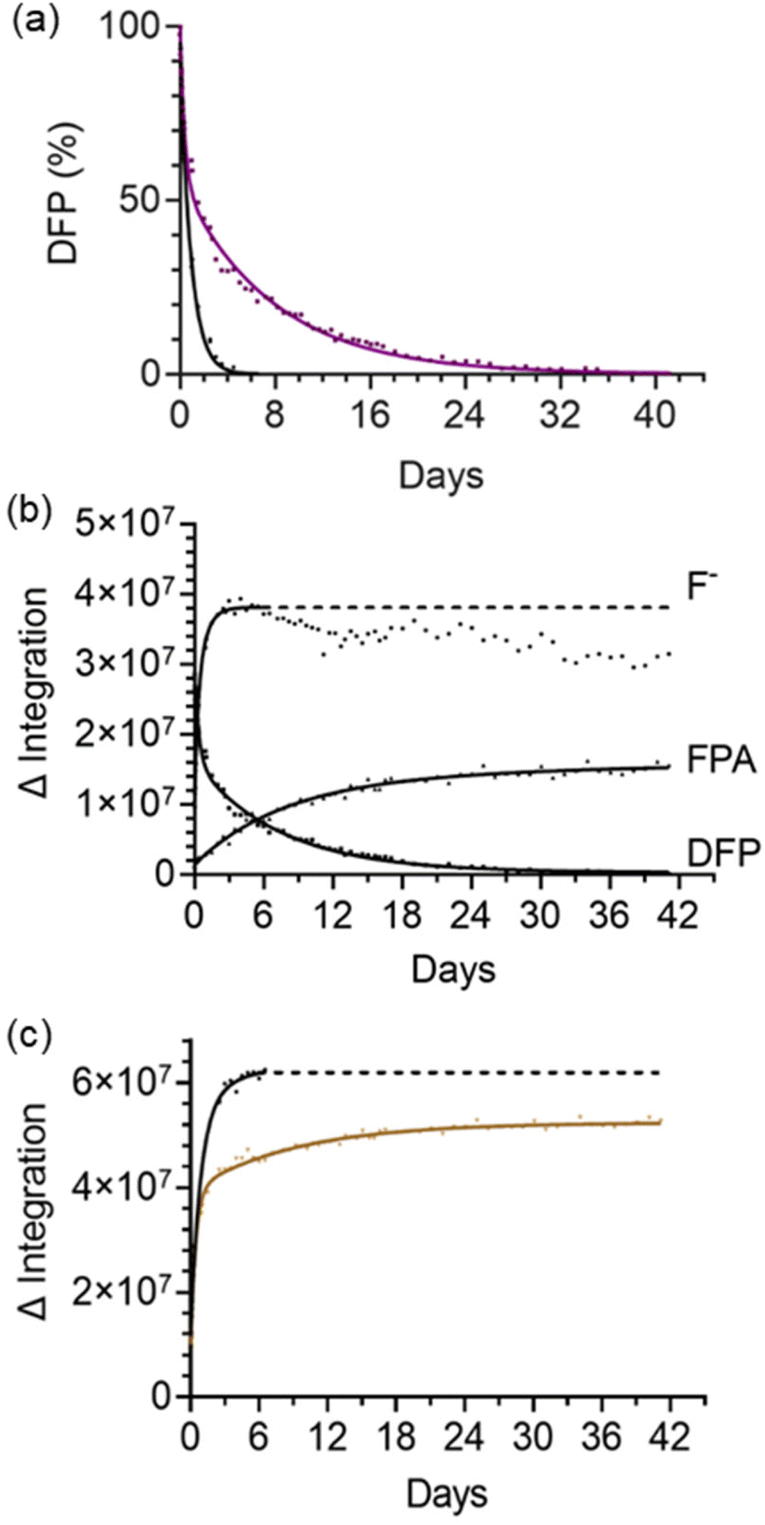 | ||
| Fig. 4 Reaction progress profiles of the hydrolysis of DFP; (a) percentage of DFP remaining over time with buffer only (black) and with HPEG·OH (0.50 mM, purple). (b) Change in the concentration of fluoride ions, FPA and DFP over time with HPEG·OH (0.5 mM), as determined by 19F NMR integration (arbitrary units). Due to broadness observed for the fluoride resonance after 7 days this data was not included in the analysis, the concentration remaining is inferred by the dotted line. (c) The combined concentration of fluoride ions and FPA, determined by NMR integration (arbitrary units), buffer only (black) and with HPEG·OH (0.5 mM, brown). Determined by 19F NMR spectroscopic analysis; 7.5 mM DFP, H2O/D2O (90:10 v/v), 50 mM borate buffer, pH 8.7. | ||
| Entry | Cage | [cage]/mM | [DFP]/mM | k 1 × 10−5 s−1 | t 1/2/h |
|---|---|---|---|---|---|
| a Control (i.e. DFP in buffer). b Rate determined over the first 9 hours. c Rate determined after 5 days. d Goodness-of-fit score < 0.95. ND – not determined. | |||||
| 1 | —a | — | 7.5 | 1.6 ± 0.07 | 11.8 ± 0.5 |
| 2a | HPEG·OH | 0.5 | 7.5 | 1.2 ± 0.06b | 16.0 ± 0.8 |
| 2b | HPEG·OH | 0.5 | 7.5 | 0.13 ± 0.04c | 145 ± 5 |
| 3a | HW·OH | 0.5 | 7.5 uncomplexed DFP | 1.7 ± 0.06 | 11.5 ± 0.4 |
| 3b | HW·OH | 0.5 | 7.5 complexed DFP | 0.69 ± 0.06 | 27.9 ± 2.6 |
| 4 | HW·Cl16 | 0.5 | 7.5 | 2.6 ± 0.20 | 7.5 ± 0.6 |
| 5 | H·Cl16 | 0.5 | 7.5 | 2.5 ± 0.10 | 7.6 ± 0.3 |
| 6a | HW·OH | 0.7 | 3.0 uncomplexed DFP | 1.2 ± 0.14 | 16.2 ± 1.9 |
| 6b | HW·OH | 0.7 | 3.0 complexed DPF | ND | >21d |
| 7 | H·Cl16 | 0.7 | 3.0 | 1.7 ± 0.09 | 11.3 ± 0.6 |
| 8 | HW·Cl16 | 0.7 | 3.0 | 3.3 ± 0.12 | 5.8 ± 0.2 |
Over the first 9 h, the hydrolysis rate of reaction of DFP in the presence of HPEG·OH was observed to be first-order with respect to DFP, with an observed rate constant of 1.2 × 10−5 s−1 and an observed half-life of t1/2 = 16.0 h (Table 1, entry 2a). After formation of fluoride ions ceases (5 days) the rate of reaction, whilst still first-order, decreases significantly (k1 = 0.13 × 10−5 s−1, t1/2 = 145 h, Table 1 entry 2b). The rate for formation of FPA after 5 days is identical to the rate constant observed for hydrolysis of DFP in buffer alone (k1 = 0.13 × 10−5 s−1) indicating that after this time period FPA is the major product. In the absence of cage, FPA – the hydrolysis product formed via loss of the isopropoxide – is formed in 6% yield, whereas in the presence of HPEG·OH the conversion to this product increases to 29%.
This change in the balance between the two reaction pathways, in addition to the varied rates observed over time mentioned above, provides an insight to the mechanism of action of HPEG·OH. We hypothesise that the increase in conversion to FPA is the direct result of the orientation in which the DFP guest binds to the coordination cage. The increased occurrence of –OiPr as the leaving group, rather than –F, could be for two reasons. Firstly, the orientation of the P–F bond inside the cage cavity is in such a way that it is protected from and unable to react with the surrounding shell of hydroxide ions. This contrasts with the position of the P–OiPr moiety that appears to be accessible by the shell of hydroxide ions. Secondly, it can be hypothesised that in this binding mode the position of the fluorine leaving group is in a more hydrophobic environment than the –OiPr moiety which is directed outside the cage. If the fluorine atom is directed to the more hydrophobic environment of the cage interior rather than the bulk aqueous phase, it will be less well solvated and less able to tolerate a growing negative charge than when it is free in aqueous solution; the outcome of this would be that the pKa of the fluoride ion increases in a hydrophobic environment, such that it is a less labile leaving group than in a normal aqueous phase. Whilst it is recognised that nucleophilic attack on DPA by fluoride can also lead to the formation of FPA, we do not believe that this mechanism is applicable in this case, since DPA is not a strongly binding guest (see later).
Using the experimental conditions above with HW·OH (0.5 mM, 50 mM borate buffer, 90% H2O:10% D2O, pH 8.7) and 7.5 mM DFP, it was observed that the decrease in the integration of DFP and increase in the cumulative integration of the products FPA and fluoride ions did not correlate. This was ascribed to the presence of BF4− anions that were used in the initial synthesis of HW·(BF4)16 hydrolysing to hydroxyfluoroborates and fluoride ions during the experiment. As a result, the hydrolysis rate of DFP in the presence of this cage was determined by analysis of DFP consumption alone, as that will not be affected by the presence of any fluoride ions arising from decomposition of the BF4− counter-ions. For this experiment an additional resonance in the 19F NMR spectrum (d, −88.4 ppm, 1JF–P = 977 Hz), that was not observed with the diamagnetic HPEG·OH cage (Cd2+), and was upfield to the resonance of DFP, was noted (Fig. S3†); this was reasoned to be due to the HW·OH·DFP complex. The addition of cycloundecanone (CUD, 22 mM), a high affinity guest (K = 1.2 × 106 M−1) that is known to block the cage cavity,20 to the NMR sample resulted in disappearance of the resonance at −88.4 ppm with the intensity of the resonance at −77.8 ppm, attributed to non-complexed DFP, increasing in equal measure. Thus, we conclude that DFP was cavity bound and the signal at −88.4 ppm was indeed due to the HW·OH·DFP complex. Due to the complexed and non-complexed DFP having different NMR environments the number of molecules of DFP associated to each HW·OH cage could be determined directly from integration of the resonances. At this concentration of host and guest, ca. 1.5 molecules of DFP are bound per cage according to the 19F NMR signal integrations. NMR binding experiments, discussed later, confirmed that DFP was in slow exchange with the cage, which is consistent with the presence of the two resonances observed in the 19F NMR spectrum.
The preservation effect of DFP (7.5 mM) in the presence of HW·OH (0.5 mM) was only observed after 9 h (Fig. 5 and S4†). It is hypothesised that for DFP to be preserved it needs to be cavity-bound, and at earlier timepoints the hydrolysis of the non-complexed fraction of DFP dominates the calculated rate constant. The use of a higher number of equivalents of DFP to cage (3.0 mM DFP, 0.7 mM HW·OH) did show more clearly preservation of DFP in the first 9 h, and this is discussed further below. Experiments with a higher concentration of cage could not be conducted as 0.7 mM was the upper limit of cage solubility. The observed hydrolysis rates of both complexed and non-complexed DFP could be independently measured using the separate 19F NMR resonances, and both were noted to be first-order with respect to DFP over the initial 5 days. The hydrolysis rate of the non-complexed DFP was 1.7 × 10−5 s−1 (t1/2 = 11.5 h, Table 1 entry 3a), and was comparable to the control (t1/2 = 11.8 h, Table 1 entry 1). For the complexed DFP the rate of hydrolysis, as expected, was significantly reduced, with a reaction rate of 0.69 × 10−5 s−1 (t1/2 = 27.9 h, Table 1 entry 3b) and bound, preserved, DFP was still detected after 15 days.
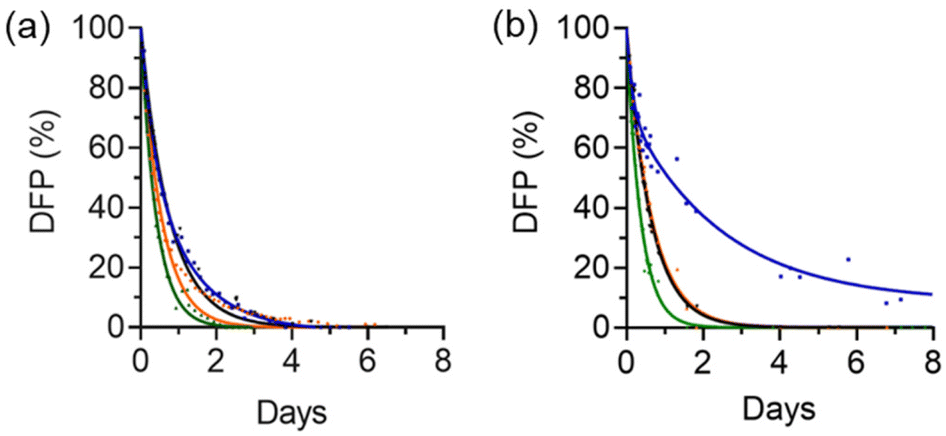 | ||
| Fig. 5 Reaction progress profiles of the hydrolysis of DFP; (a) DFP (7.5 mM), buffer only (black), 0.5 mM HW·OH (blue), 0.5 mM HW·Cl16 (green) and 0.5 mM H·Cl16 (orange); and (b) DFP (3.0 mM), buffer only (black), 0.7 mM HW·OH (blue), 0.7 mM HW·Cl16 (green) and 0.7 mM H·Cl16 (orange). Determined by 19F NMR spectroscopic analysis, H2O/D2O (90:10 v/v), 50 mM borate buffer, pH 8.7. For HW·OH the percentage of DFP remaining is the sum of the bound and unbound DFP. | ||
The hydrolysis rate of DFP (7.5 mM) was also investigated in the presence of HW·Cl16 and H·Cl16 (Fig. 5). As mentioned above, chloride ions are known to inhibit cage catalytic activity by effectively blocking the windows of the cage.26,31 As a result, it was expected that guest binding and subsequent preservation would be negatively affected by the presence of chloride anions. No preservation effect was observed with these cages and there was no evidence of formation of the cage·DFP complex by 19F NMR spectroscopic analysis. With the HW·Cl16 or H·Cl16 cages weak catalysis was observed (HW·Cl16k1 = 2.6 × 10−5 s−1, t1/2 = 7.5 h, Table 1 entry 4; and H·Cl16k1 = 2.5 × 10−5 s−1, t1/2 = 7.6 h, Table 1 entry 5) when compared to the control (Table 1, entry 1). This is consistent with a weak catalytic (rather than a protective) effect, most likely due to chloride ions preventing binding of DFP to the cage and catalysis occurring at the external surface.32,33
The above experiments indicate that cavity binding of DFP to HW·OH and HPEG·OH has a preservation effect, and it was reasoned that increasing the ratio of cage to DFP would allow the preservation effect to be more readily observed by ensuring that a higher fraction of DFP is bound. This was investigated initially with HW·OH, as the reaction rates of both the complexed and non-complexed DFP could be determined from the 19F NMR data. For HW·OH (0.7 mM, 3.0 mM DFP, 50 mM borate buffer, 90% H2O:10% D2O, pH 8.7) using the integration of HW·OH·DFP complex and the non-complexed DFP, it was calculated that 1.4 equiv. of DFP was bound per cage (i.e. 1 mM of the 3 mM present), consistent with the number of guest molecules per cage calculated previously. Cavity binding of DFP was again confirmed by addition of CUD and disappearance of the resonance at −88.4 ppm (Fig. S5†). At these relative concentrations, DFP preservation was clearly observed (Fig. 5b) which can be ascribed to the larger proportion of the DFP present bound in the cage (33%) compared to unbound (67%). The results obtained were comparable with those observed above for 7.5 mM DFP and 0.5 mM HW·OH; the hydrolysis rate for non-complexed DFP was 1.2 × 10−5 s−1, t1/2 = 16.2 h (Table 1 entry 6a), and for the bound fraction of DFP a half-life of greater than 21 h was observed (Table 1, entry 6b). Due to the low S/N ratio in the 19F NMR spectrum, for the HW·OH·DFP complex, an acceptable fit to pseudo-first order decay could not be achieved and thus a rate constant was not determined.
Investigation of the hydrolysis of DFP (3.0 mM) in the presence of H·Cl16 (0.5 mM) was consistent with that observed for the control (k1 = 1.7 × 10−5 s−1, t1/2 = 11.3 h, Table 1 entry 7), and with HW·Cl16 the rate of DFP hydrolysis was shown to increase slightly (kcat = 3.3 × 10−5 s−1, t1/2 = 5.8 h, Table 1 entry 8). A second-order rate constant was calculated after subtraction of the observed background rate and taking the concentration of cage into consideration. This rate constant, k2 = 0.03 M−1 s−1, is comparable to what we have observed for surface-based cage-catalysed hydrolysis of other organophosphates.32
The association constants for DFP binding to HW·OH were determined by a conventional 1H NMR spectroscopic titration study. As noted in our previous research, the resonances of the paramagnetic cages (M = Co2+) in the 1H NMR spectrum occur over a range of 200 ppm, and spectral changes due to guest binding can be easily observed whether the guest is in fast or slow exchange.17 The addition of DFP (0.25–24.9 equiv.) to HW·OH resulted in new resonances being observed for the complex HW·OH·DFP, allowing us to conclude that DFP was in slow exchange with this cage on the NMR timescale (Fig. 6). Notably, and supporting the cavity binding that was demonstrated by displacement of DFP following addition of CUD, new resonances at ∼−5 ppm result from the altered chemical environment of the guest bound inside the paramagnetic cavity of the cage in slow exchange.22,23,29 Using the knowledge from the 19F NMR spectroscopy experiments that the number of molecules of DFP complexed to each HW·OH (cage 0.7 mM, DFP 3.0 mM) was ca. 1.5, indicating that both 1:1 (H·G) and 1:2 (H·G2) complexes are present in the equilibrium, it was concluded that both give similar changes in the 1H NMR spectra of the bound guest. With this knowledge, for a host:guest ratio of 1:0.74, the association constant – assuming that only H·G was present – was K11 = 900 M−1. At a higher host:guest ratio (1:10.8), and with the assumption at this concentration that H·G2 is the predominant species, the association constant was calculated to be K12 = 140 M−2. The association constant for the second stepwise binding event is accordingly calculated as ca. 0.16 M−1, entirely consistent with guest cavity binding31 and supporting the findings above. In a similar experiment with O,O′-diisopropyl phosphoric acid (DPA) (see Scheme 1 for chemical structure), the hydrolysis product of DFP, we observed no spectral changes after the addition of 3 equiv. of DFP to HW·OH, confirming that it is not a strongly binding guest. With H·Cl16 and HW·Cl16, no host–guest interaction was observed via1H NMR analysis with up to 20 equiv. of DFP added. This further confirms that chloride ions inhibit cavity-binding of guests, as we have noted in other studies.28,30
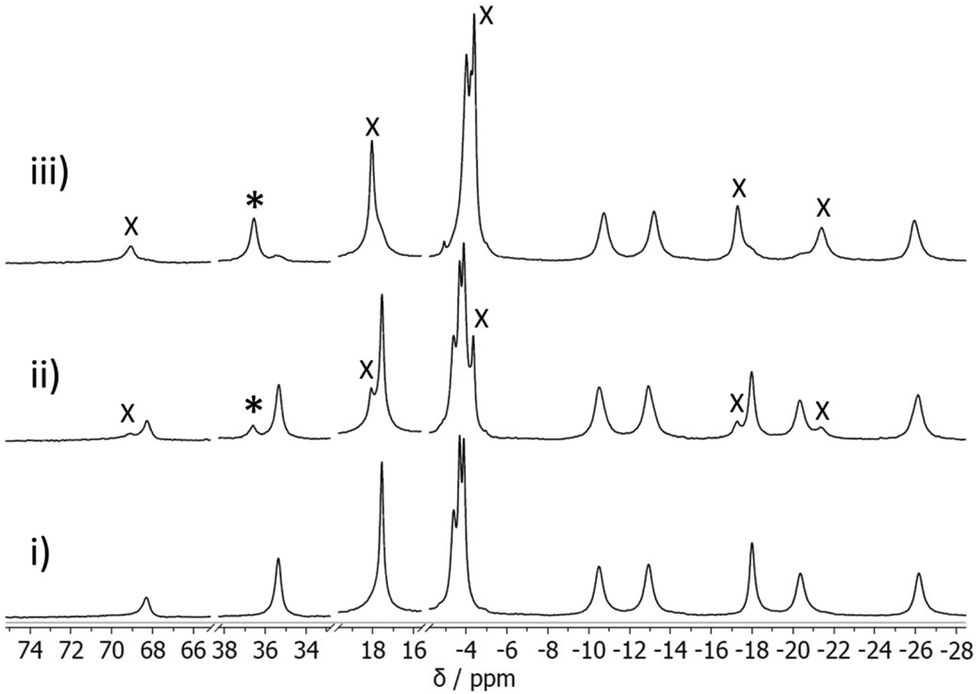 | ||
| Fig. 6 1H NMR titration study of DFP into a buffered solution of HW·OH (0.59 mM); representative spectral changes as guest binding occurs, with (i) 0, (ii) 0.74, (iii) 6.01 equiv. of DFP respectively in D2O, with 50 mM borate buffer, pH 8.7. Signals due to the host–guest complex (HW·OH·DFP) are denoted with either an asterisk (*) or cross (X), those marked with an asterisk (*) were used to determine the association constant. | ||
From the above studies it can be concluded that DFP guest binding to HPEG·OH or HW·OH inhibits the hydrolysis rate significantly: and if DFP is not complexed then the rate of hydrolysis is essentially the same as in the control. For cages with chloride as the counter anion (H·Cl16 and HW·Cl16), guest binding did not occur, as evidenced by NMR spectroscopic titration experiments, although generally a slight enhancement of hydrolysis rate was detected when compared to buffer solution alone. For these cages it can be deduced that the presence of chloride ions inhibits guest binding in the cavity and also that some surface binding must be possible (as observed with some other organophosphates)32 resulting in weak catalysis. This adds further evidence to the fact that cavity binding with these cages is not needed for catalysis to occur.
Crystallographic studies
To understand the location and orientation of DFP binding to the cages, crystallographic studies on the host–guest complexes were attempted. Unfortunately, host–guest crystal structures with DFP and HW·OH or HPEG·OH could not be not obtained, due to high levels of disorder and/or poor crystallinity. However we could obtain good-quality crystals of H·Cl16 (having the same cubic cage structure without external solubilising substituents) by slow vapour diffusion of THF into an aqueous solution of the cage. These crystals were then soaked in concentrated solutions of guest under forcing, non-equilibrium conditions to obtain H·Cl16·DFP, utilising the ‘crystal sponge’ method38,39 that we have successfully used.40 After treatment with high concentrations of guest (400 mmol for 6 h), crystals were carefully washed (THF/H2O, 85:15 v/v) to remove the excess guest molecules in solution and to ensure safe handling prior to single-crystal X-ray diffraction analysis.
Analysis of the crystal structure of H·Cl16·DFP revealed that two guest molecules are bound to the cage, located in a pair of opposite windows, with the other four windows occupied by chloride anions (Fig. 7a). Chloride anions were also observed in the crystalline lattice between the cage structures, and there were disordered water molecules in the interior of the cage.24‡ Solvent-accessible channels were observed in the crystal packing lattice though which guests could diffuse (Fig. S6†). At each of the two binding sites the DFP guests are disordered over two positions in the cage windows, with the major position refined to a crystallographic occupancy of 58% (Fig. 7) and the minor position to 16% (Fig. S7†). Despite the low crystallographic occupancies of these guests, their identity is unmistakable due to the tetrahedral geometry around the heavy phosphorus atom, which is unlike any solvents or anions present. This result was repeatable, with several crystal structures of H·Cl16 obtained from different crystalline sponge experiments on multiple occasions. The total chemical occupancies of 74% at each of the two guest binding sites indicated that the H·Cl16 cage binds to, on average, 1.5 equiv. of DFP (cf. the solution measurements).
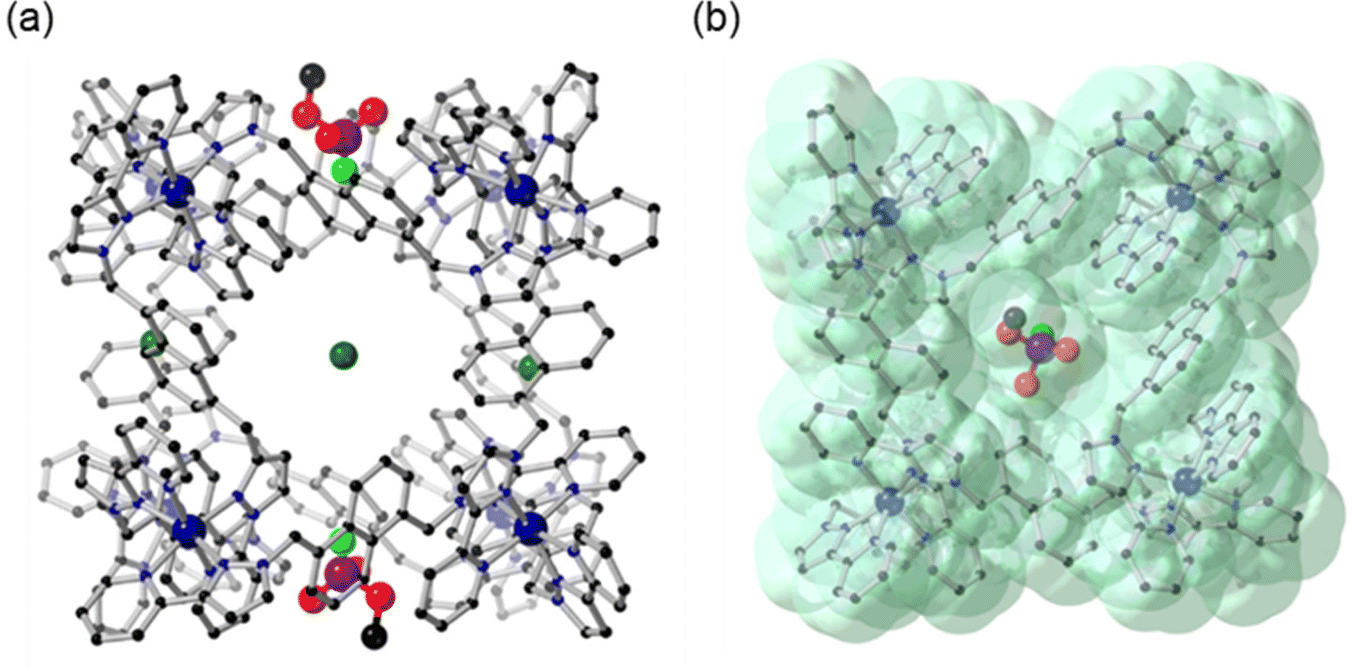 | ||
| Fig. 7 Crystal structures of H·Cl16·DFP (CCDC #2237215); (a) rotated so that the Cl− anions and DFP in the windows are visible; and (b) showing the van der Waals surface, shown in green. H atoms are omitted for clarity, Co – dark blue, C – black, N – blue, O – red, P – purple, F – light green, Cl – dark green. | ||
While it is recognised that the crystal site occupancy cannot be directly compared to that observed in solution via NMR spectroscopic studies with HW·OH, it is clear that binding of DFP to the cage occurs in both solid and solution states. In the crystal structure of H·Cl16·DFP, the lower-occupancy position of DFP (16%) was observed to involve binding 2.88 ± 0.02 Å further inside the window of the cage compared to the DFP guest in the major position (shown in Fig. 7, distance based on the position of the phosphorus centre at each of the two DFP positions). In all cases, the P–F bond of DFP was oriented into the cavity of the cage (Fig. 7). Due to the similar electron densities of fluorine and oxygen atoms, the modelling of the P–F bond was based on the internuclear distance and analysis of the residual electron density map. This model does support the reactivity observed in the solution studies as it can be seen that the P–F bond is protected from the aqueous medium. The isopropyl carbon chains on DFP, whilst disordered and mainly unresolvable, are clearly directed outside the cavity of the cage. As evident in the van der Waals surface (Fig. 7b), the shape of the DFP guest complements the cage window, indicating its affinity for this site. This results in the F atom being located in a more hydrophobic, and potentially sterically hindered, environment compared to the bulk aqueous solution,‡ which would slow the rate of hydrolysis for reasons discussed above.
Whilst these crystal structures show DFP guest binding to the (unsubstituted) H cage, it should be noted that for all three cages the core structures and the cavity shape/size are the same, and we thus expect guest binding properties to be similar. We have shown via crystallographic analysis that the shape of the cage core, its cavity, and the locations of anions binding to various salts of H·Cl16, HW·Cl16 and HW·OH are structurally similar between cages, with the exception of the R groups at the cage vertices (for solubilisation) that do not influence guest binding.17,18 The binding location of the DFP guests in the windows (Fig. 8) in the crystals is the same as that of anions which generally also occupy these pockets in both crystal structures and in solution, implying competition for binding.28 This is substantiated firstly via1H paramagnetic NMR spectroscopy studies, showing that the host·guest·DFP complex was formed in the presence of HW·OH (Fig. 6), but not HW·Cl16. Secondly, DFP was not protected from hydrolysis in solution in the presence of the cages when chloride anions were also present following anion metathesis of HW·OH to HW·Cl16 (Fig. 5), again implying that chloride ions competitively prevent DFP from binding such that any protective effect from the cage is lost. We know from previous work that poorly-solvated anions bind strongly to the cage, generally to the surface26,27 but also sometimes in the cavity.28
 | ||
| Fig. 8 The superimposed crystal structures of H·Cl16 (CCDC #1581566) and H·Cl16·DFP (CCDC #2237215) showing the binding location of DFP and Cl− guests inside the cage window. Hydrogen bonds are noted with green dashed lines; Co – dark blue, C – black, N – blue, H – white, O – red, P –purple, F – light green, Cl – dark green. | ||
NMR spectroscopic analysis of GB in the presence of HPEG·OH
Initial studies were conducted to investigate the rate of hydrolysis of the CWA GB with HPEG·OH, as this cage exhibited the greatest preservation effect on the simulant DFP. Due to regulations surrounding the use of Schedule 1 materials and its high toxicity, crystallographic studies with GB were not possible to determine guest binding. The hydrolysis rate of GB was monitored via31P rather than 19F NMR spectroscopic analysis (Fig. 9a) due to instrumental limitations, by either the disappearance of the resonance due to GB itself (d, 34 ppm, 1JP–F = 1048 Hz) or the appearance of the 31P resonance corresponding to isopropyl methylphosphonic acid (IMPA, s, 25.4 ppm), the hydrolysis product of GB (Scheme 1). In aqueous buffer alone (control, pH 8.7, 50 mM borate buffer) less than 7% of GB (initial concentration 7.5 mM) remained after 77 minutes (k1 = 41.5 × 10−5 s−1, t1/2 = 27.9 min). The hydrolysis rate of GB in the control experiment (no cage present) was noted to be substantially faster than for the CWA simulant DFP (t1/2 = 11.8 h), and this rate difference should be considered where DFP is used as a simulant of GB in laboratory studies. Significantly, the hydrolysis rate of GB in the presence of HPEG·OH was substantially reduced (0.5 mM, 7.5 mM GB, 50 mM borate buffer, 90% H2O:10% D2O, pH 8.7) and analysis at 77 minutes post GB addition revealed that 58% of GB remained (Fig. 9b and S8†). No cleavage of the OiPr group, which would result in the product methyl fluorophosphonate (MFPA), was observed. The results of this study, which allows the comparison of GB hydrolysis rates to those obtained using DFP in the absence and presence of HPEG·OH, are encouraging. This finding also supports the current literature that DFP,35 whilst sterically more demanding, is an appropriate complexation simulant for GB.
 | ||
| Fig. 9 (a) 31P NMR spectra of GB (7.5 mM) 69 minutes post addition; (i) without cage and (ii) with HPEG·OH (0.5 mM), showing substantial retention of unreacted GB in the presence of HPEG·OH. (b) Reaction progress profiles of the hydrolysis of GB, determined by NMR integration; without cage (black) and with HPEG·OH (0.50 mM, purple), measurements in D2O/H2O/MeCN (10:85:5 v/v/v) with 50 mM borate buffer, pH 8.7 determined via31P NMR spectroscopic analysis, a.u. – arbitrary units. | ||
In our experiments we have noted that the presence of 0.5 mM HPEG·OH with 7.5 mM DFP increases the half-life by ca. 1.4 times in the first 9 hours and 12.3 times after 5 days; the two different environments that the simulant experiences, bound and free, result in two different reaction rates, and the greater protection effect later in the reaction is associated with a higher fraction of residual DFP being cavity-bound. With GB, whilst the hydrolysis rate is substantially faster in the control (GB t1/2 = 27.9 min; vs. DFP t1/2 = 11.8 h, monitored for 67 min), the addition of HPEG·OH increases the half-life by ca. 2.8 times in a similar timeframe (77 min). These findings highlight the potential utility of these cages to capture and increase the stability of P–F bond containing OP CWAs, which may be of interest for sample collection, preservation and subsequent analysis.
Conclusions
In conclusion, we have shown that the presence of the cubic coordination cages HPEG·OH and HW·OH significantly increases the hydrolysis half-life of DFP, and HPEG·OH significantly increases the hydrolysis half-life of the OP CWA GB, both in borate buffer (50 mM, pH 8.7). An understanding of the host–guest chemistry, by NMR spectroscopic analysis and structural analysis of crystals formed by the ‘crystal sponge’ method, allowed us to propose that DFP binds to the cage: in the solid state it occupies windows around the cage surface, but the fact that in solution it is prevented from binding when the cavity-blocking inhibitor CUD is present suggests that DFP occupies the central cavity in the solution phase. The increase in stability of the OP guests when bound is postulated to be due to a reduction in reactivity of the P–F bond, as this bond is sterically protected from the hydroxide ions surrounding the cage exterior and in bulk solution; and/or it could be due to the pKa of the leaving group increasing in the more hydrophobic environment of the cage interior, making the P–F bond less susceptible to cleavage, as the F atom is surrounded by C–H groups of the cage rather than aqueous solvent. When using cages with chloride as the counter anion (H·Cl16 and HW·Cl16), guest binding did not occur as chloride ions competitively inhibit guest binding. For HW·Cl16 some catalysis of DFP hydrolysis was observed which we ascribe to interaction with the cage exterior surface. We have also shown that GB in the presence of HPEG·OH displayed a substantially reduced hydrolysis rate, with 58% of GB remaining un-hydrolysed in aqueous solution after 77 minutes compared to ca. 7% in the control (no HPEG·OH cage) under the same conditions. These findings highlight the ability of our cubic coordination cages HW·OH and HPEG·OH to encapsulate P–F containing guests and, when complexed, protect the P–F bond from aqueous hydrolysis. A focus of our future studies is further investigation of the binding of GB to cage complexes and application of this protection effect to the preservation of forensic samples and analytical applications.Experimental
The preparation of cages for solution studies was as previously described and further outlined in the ESI.†17–19O,O′-Diisopropyl fluorophosphate (DFP) (99%) was purchased from Advanced Molecular Technologies and used without purification. The absence of O,O′-diisopropyl phosphoric acid (DPA) was confirmed by 31P NMR analysis prior to each use. GB (sarin) and IMPA were synthesised using in-house methods at Defence Science and Technology Group, Fishermans Bend, Victoria, Australia.
For the DFP experiments, NMR spectra were recorded on either a Bruker Avance III or Bruker Avance Neo NMR spectrometer equipped with a 5 mm BBFO probe, operating at 400 MHz (1H), 377 MHz (19F), or 162 MHz (31P). Paramagnetic proton nuclear magnetic resonance (1H paramagnetic NMR) were recorded using a spin-echo pulse sequence, over a spectral range of −130 ppm to +150 ppm. For GB, NMR experiments were recorded on a Bruker Avance III NMR spectrometer equipped with a 5 mm BBO probe, operating at 202 MHz (31P).
All NMR data was collected using standard Bruker pulse sequences. The probe temperature was set to 298 K, and standard processing parameters were used for 1H, 19F and 31P spectra. Line broadening was set to 3 Hz for 19F processing. Chemical shifts (δ) are reported in ppm and were referenced to the residual solvent signals (1H) or to external standards (19F and 31P).
NMR spectral analysis of DFP with cages
The analysis of DFP in the presence of each of the cages (H·Cl16, HW·Cl16, HW·OH and HPEG·OH) is as described in the main text and corresponding figure captions. The 31P NMR and 19F NMR spectra of DFP, in the presence or absence of cage, were monitored at specified time intervals. Only data from the 19F NMR spectra was used to determine rate constants. In all cases the guest and host were dissolved in H2O/D2O (90:10 v/v) with borate buffer (50 mM, pH 8.7).
NMR spectral analysis of GB in the presence of cages
A stock solution of GB (150 mM) in MeCN was diluted to a concentration of 7.5 mM (5% v/v) by addition into the D2O/H2O (10:90) NMR samples containing borate buffer (final buffer concentration of 50 mM, pH 8.7) with or without HPEG·OH. GB was added as a MeCN solution to ensure safe handling and to improve the accuracy of addition. The 31P NMR spectrum of GB (7.5 mM) with and without HPEG·OH (0.5 mM) in D2O/H2O/MeCN (10:85:5) in borate buffer (50 mM, pH 8.7) was monitored every 7 minutes for 66 minutes without cage and 77 minutes with cage.
Determination of rate constants
First-order rate constants (k1) were calculated from the NMR spectral integration of DFP or GB with the equation A = A0 × exp(−k1t) (GraphPad Prism software version 9.3.1). 95% confidence intervals are presented as the error (±CI/2). Representative fits are shown in Fig. S1 and S4.† The fits used the constraints; k > 0, A0 = initial concentration of DFP/GB and plateau = 0, unless specified otherwise. Rate constants are noted only for fits with goodness-of-fit score >0.94. Half-lives were calculated using the equation ln2/k1.
For HPEG·OH, rate constants for DFP hydrolysis were calculated with data from 0–9 h and 5–41 days, and the rate constant for formation of FPA was calculated with data from 5–41 days. For FPA the fit was constrained to go through the initial datapoint in this time period. For 0.5 mM HW·OH and 7.5 mM DFP, resonances for bound and unbound DFP were used to separately calculate rate constants for DFP hydrolysis (0–5 days). For 0.7 mM HW·OH and 3.0 mM DFP, due to a lower S/N, the fit for the unbound DFP was constrained to go through the initial datapoint, and a satisfactory fit for the bound DFP could not be obtained. For all other cages the fit was over the time period 0–10 h. Rate constants for GB hydrolysis in the presence of HPEG·OH were calculated with data obtained from 0–69 min.
Determination of association constants
DFP was separately titrated into solutions of either H·Cl16 (0.27 mM), HW·Cl16 (0.59 mM) or HW·OH (0.59 mM) in D2O (pH 8.7, 50 mM borate buffer) and the resulting 1H paramagnetic NMR spectrum was acquired. The equivalents of DFP in these experiments were; H·Cl16 – 0, 0.35, 0.70, 1.00, 3.62, 39.7 and 221; HW·Cl16 – 0, 0.31, 0.62, 1.25, 2.24, 3.74, 6.24, 12.5, 18.7, 25.0; and HW·OH – 0, 0.25, 0.74, 0.98, 1.23, 2.20, 3.16, 4.12, 5.07, 6.01, 10.2, 15.5, 24.9.For HW·OH, in all cases slow binding of DFP was observed. It was assumed that at low guest concentration, 0.43 mM DFP (host:guest ratio of 1:0.74), that H·G is the predominant species The association constant (K11) was calculated using the following equation:34
| K11 = [H·G]/[H] × [G] |
At high guest concentration, 6.04 mM DFP (host:guest ratio of 1:10.2), it is assumed that H·G2 is the predominant species. The association constant (K12) was accordingly calculated using the following equation:
| K12 = [H1·G2]/[H] × [G]2 |
The ratio of the host and host:guest complex (H·G or H·G2) was calculated using integration of the resonances at 36.5 and 35.5 ppm, corresponding to the free and bound cage respectively, at the above guest concentrations. The concentration of unbound guest (G) was calculated from the initial concentration of guest added minus the concentration of host:guest complex.
Crystallisation studies of cages with guests
Crystals of H·Cl16 were prepared via recrystallisation from H2O with slow vapour diffusion of THF to give large, pale orange block crystals suitable for single-crystal X-ray diffraction. The crystals were washed with 85% THF(aq) three times. To the resulting solvated crystals, neat DFP was added (29 μmol) to the crystals submerged in 85% THF(aq), and the vials were sealed and the crystals left soaking for a period of 6 h. Afterwards, excess DFP was removed via three additional washes with 85% THF(aq). Data collection was performed at the MX2 beamline of the Australian Synchrotron.41,42 Using Olex2, the structure was solved with the SHELXT structure solution program using Intrinsic Phasing and refined with the SHELXL refinement package using Least Squares minimisation.43–46 Excess solvent molecules which could not be modelled were removed from the refinement using the ‘SQUEEZE’ function in the PLATON software.47 CrystalMaker Software Ltd was utilised to construct the figures. Information on crystal properties, data collection and refinement details associated with the structures of H·Cl16 and DFP are in ESI† CIF file.Author contributions
Cage synthesis: CGPT and MBT. Highly toxic chemical handling: GHD. NMR spectroscopy measurements: JCD, GHD, AJB, SO, AIM. Binding constant measurements: JCD. X-ray crystallography: JCD, RJY, DRT. Data analysis: JCD, KLT, GHD. Manuscript preparation: JCD, KLT, GHD, MDW. Project conception and supervision: KLT, GHD, MDW.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the School of Chemistry, Monash University; the Australian Department of Defence and Defence Science and Technology Group; and the Department of Chemistry, University of Warwick. We thank the Australian Department of Defence for research funding and highly toxic chemical testing, the Universities of Warwick and Monash University for a ‘Monash/Warwick Alliance’ Accelerator Grant to MDW, KLT, GHD and DRT, the Royal Society for an International Exchange Grant IES/R3/170177 to MDW and KLT, and Monash University, School of Chemistry for a Dean's Scholarship to JCD. Part of this work was conducted using the MX2 beamline at the Australian synchrotron, part of ANSTO, using the Australian Cancer Research Foundation (ACRF). We thank Dr Roger Mulder (CSIRO Clayton) for his assistance with 19F and 31P proton decoupled NMR experiments.References
- S. W. Wiener and R. S. Hoffman, Nerve agents: a comprehensive review, J. Intensive Care Med., 2004, 19, 22–37 CrossRef PubMed.
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Destruction and detection of chemical warfare agents, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- I. Sinha and P. S. Mukherjee, Chemical Transformations in Confined Space of Coordination Architectures, Inorg. Chem., 2018, 57, 4205–4221 CrossRef CAS PubMed.
- T. Murase, S. Horiuchi and M. Fujita, Naphthalene Diels−Alder in a Self-Assembled Molecular Flask, J. Am. Chem. Soc., 2010, 132, 2866–2867 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Supramolecular Catalysis in Metal–Ligand Cluster Hosts, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Supramolecular Coordination Cages for Artificial Photosynthesis and Synthetic Photocatalysis, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS PubMed.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Catalytic reactions within the cavity of coordination cages, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- D. F. Sava, V. C. Kravtsov, J. Eckert, J. F. Eubank, F. Nouar and M. Eddaoudi, Exceptional Stability and High Hydrogen Uptake in Hydrogen-Bonded Metal−Organic Cubes Possessing ACO and AST Zeolite-like Topologies, J. Am. Chem. Soc., 2009, 131, 10394–10396 CrossRef CAS PubMed.
- S. Mukherjee, A. V. Desai and S. K. Ghosh, Potential of metal–organic frameworks for adsorptive separation of industrially and environmentally relevant liquid mixtures, Coord. Chem. Rev., 2018, 367, 82–126 CrossRef CAS.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Metal-organic cages for molecular separations, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- K. Ono, M. Yoshizawa, M. Akita, T. Kato, Y. Tsunobuchi, S.-i. Ohkoshi and M. Fujita, Spin Crossover by Encapsulation, J. Am. Chem. Soc., 2009, 131, 2782–2783 CrossRef CAS PubMed.
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Metal–organic frameworks: functional luminescent and photonic materials for sensing applications, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC.
- M.-C. Daniel and D. Astruc, Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Fluorescent sensors: A bright future for cages, Coord. Chem. Rev., 2021, 434, 213820 CrossRef.
- E. C. Guo, A. C. Sedgwick, T. Hirao and J. L. Sessler, Supramolecular Fluorescent sensors: An historical overview and update, Coord. Chem. Rev., 2021, 427, 213560 CrossRef PubMed.
- I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, Octanuclear Cubic Coordination Cages, J. Am. Chem. Soc., 2008, 130, 15167–15175 CrossRef CAS PubMed.
- M. Whitehead, S. Turega, A. Stephenson, C. A. Hunter and M. D. Ward, Quantification of solvent effects on molecular recognition in polyhedral coordination cage hosts, Chem. Sci., 2013, 4, 2744–2751 RSC.
- G. D. Jackson, M. B. Tipping, C. G. P. Taylor, J. R. Piper, C. Pritchard, C. Mozaceanu and M. D. Ward, A Family of Externally-Functionalised Coordination Cages, Chemistry, 2021, 3, 1203–1214 CrossRef CAS.
- M. D. Ward, C. A. Hunter and N. H. Williams, Coordination Cages Based on Bis(pyrazolylpyridine) Ligands: Structures, Dynamic Behavior, Guest Binding, and Catalysis, Acc. Chem. Res., 2018, 51, 2073–2082 CrossRef CAS PubMed.
- S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, Mapping the Internal Recognition Surface of an Octanuclear Coordination Cage Using Guest Libraries, J. Am. Chem. Soc., 2014, 136, 8475–8483 CrossRef CAS PubMed.
- J. R. Piper, L. Cletheroe, C. G. P. Taylor, A. J. Metherell, J. A. Weinstein, I. V. Sazanovich and M. D. Ward, Photoinduced energy- and electron-transfer from a photoactive coordination cage to bound guests, Chem. Commun., 2017, 53, 408–411 RSC.
- W. Cullen, K. A. Thomas, C. A. Hunter and M. D. Ward, pH-Controlled selection between one of three guests from a mixture using a coordination cage host, Chem. Sci., 2015, 6, 4025–4028 RSC.
- W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, pH-dependent binding of guests in the cavity of a polyhedral coordination cage: reversible uptake and release of drug molecules, Chem. Sci., 2015, 6, 625–631 RSC.
- A. J. Metherell, W. Cullen, N. H. Williams and M. D. Ward, Binding of Hydrophobic Guests in a Coordination Cage Cavity is Driven by Liberation of “High-Energy” Water, Chem. – Eur. J., 2018, 24, 1554–1560 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek, The 55% Solution: A Formula for Molecular Recognition in the Liquid State, Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- M. D. Ludden, C. G. Taylor, M. B. Tipping, J. S. Train, N. H. Williams, J. C. Dorrat, K. L. Tuck and M. D. Ward, Interaction of anions with the surface of a coordination cage in aqueous solution probed by their effect on a cage-catalysed Kemp elimination, Chem. Sci., 2021, 12, 14781–14791 RSC.
- M. D. Ludden and M. D. Ward, Outside the box: quantifying interactions of anions with the exterior surface of a cationic coordination cage, Dalton Trans., 2021, 50, 2782–2791 RSC.
- M. D. Ludden, C. G. P. Taylor and M. D. Ward, Orthogonal binding and displacement of different guest types using a coordination cage host with cavity-based and surface-based binding sites, Chem. Sci., 2021, 12, 12640–12650 RSC.
- S. Turega, M. Whitehead, B. R. Hall, A. J. Meijer, C. A. Hunter and M. D. Ward, Shape-, size-, and functional group-selective binding of small organic guests in a paramagnetic coordination cage, Inorg. Chem., 2013, 52, 1122–1132 CrossRef CAS PubMed.
- W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, Catalysis in a Cationic Coordination Cage Using a Cavity-Bound Guest and Surface-Bound Anions: Inhibition, Activation, and Autocatalysis, J. Am. Chem. Soc., 2018, 140, 2821–2828 CrossRef CAS PubMed.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Highly efficient catalysis of the Kemp elimination in the cavity of a cubic coordination cage, Nat. Chem., 2016, 8, 231 CrossRef CAS PubMed.
- C. G. P. Taylor, A. J. Metherell, S. P. Argent, F. M. Ashour, N. H. Williams and M. D. Ward, Coordination-Cage-Catalysed Hydrolysis of Organophosphates: Cavity- or Surface-Based?, Chem. – Eur. J., 2020, 26, 3065–3073 CrossRef CAS PubMed.
- A. B. Solea, B. Sudittapong, C. G. P. Taylor and M. D. Ward, Inside or outside the box? Effect of substrate location on coordination-cage based catalysis, Dalton Trans., 2022, 51, 11277–11285 RSC.
- C. G. Taylor, J. R. Piper and M. D. Ward, Binding of chemical warfare agent simulants as guests in a coordination cage: contributions to binding and a fluorescence-based response, Chem. Commun., 2016, 52, 6225–6228 RSC.
- S. L. Bartelt-Hunt, D. R. Knappe and M. A. Barlaz, A review of chemical warfare agent simulants for the study of environmental behavior, Crit. Rev. Environ. Sci. Technol., 2008, 38, 112–136 CrossRef CAS.
- Properties of Common Nuclear Spins, in High Resolution NMR (Third Edition), ed. E. D. Becker, Academic Press, San Diego, 2000, pp. 381–384 Search PubMed.
- S. Zhou, L. Wang, A. Zhang, K. Lin and W. Liu, Preparation, stabilization, and bioefficacy of beta-cyclodextrin inclusion compounds of chloramidophos, J. Agric. Food Chem., 2008, 56, 2708–2713 CrossRef CAS PubMed.
- M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, The crystalline sponge method updated, IUCrJ, 2016, 3, 139–151 CrossRef CAS PubMed.
- K. Rissanen, Crystallography of encapsulated molecules, Chem. Soc. Rev., 2017, 46, 2638–2648 RSC.
- C. G. P. Taylor, S. P. Argent, M. D. Ludden, J. R. Piper, C. Mozaceanu, S. A. Barnett and M. D. Ward, One Guest or Two? A Crystallographic and Solution Study of Guest Binding in a Cubic Coordination Cage, Chem. – Eur. J., 2020, 26, 3054–3064 CrossRef CAS PubMed.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price and A. Riboldi-Tunnicliffe, MX1: a bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo and N. Mudie, MX2: a high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. Spek, PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cage preparation and supplemental figures. CCDC 2237215. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01378b |
| ‡ Note that the presence of a network of water molecules inside the cage cavity in the solid state crystal structure does not change the fact that the cage interior is more hydrophobic than the bulk solution phase. If this were not the case, hydrophobic guests would have no reason to bind inside the cage cavity in aqueous solution,19 and displace those ‘high-energy’ or ‘frustrated’ water molecules24 into an environment where they can form a better network of hydrogen-bonds. The well-established guest-binding ability of the cage cavity accordingly confirms its hydrophobicity relative to the bulk aqueous phase in solution. |
| This journal is © The Royal Society of Chemistry 2023 |