
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Coordinated tellurenium(II) and telluronium(IV) cations: synthesis, structure and hydrolysis†
Martin
Hejda
 a,
Lukáš
Doležal
a,
Jan
Blahut
b,
Emanuel
Hupf
c,
Jiří
Tydlitát
d,
Roman
Jambor
a,
Aleš
Růžička
a,
Jens
Beckmann
*c and
Libor
Dostál
*a
a,
Lukáš
Doležal
a,
Jan
Blahut
b,
Emanuel
Hupf
c,
Jiří
Tydlitát
d,
Roman
Jambor
a,
Aleš
Růžička
a,
Jens
Beckmann
*c and
Libor
Dostál
*a
aDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, CZ-532 10 Pardubice, Czech Republic. E-mail: libor.dostal@upce.cz; Tel: +420466037163
bInstitute of Organic Chemistry and Biochemistry, Czech Academy of Science, Flemingovo nám. 2, 16610, Prague, Czech Republic
cInstitut für Anorganische Chemie und Kristallographie, Universität Bremen, Leobener Straße 7, 28359 Bremen, Germany. E-mail: j.beckmann@uni-bremen.de
dInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, CZ-532 10 Pardubice, Czech Republic
First published on 28th September 2023
Abstract
A set of N-coordinated tellurium(II) compounds containing either C,N-chelating ligands CNR (where CN = 2-(RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)C6H4, R = tBu or Dipp; Dipp = 2,6-iPr2C6H3) or N,C,N pincer ligands NCNR (where NCN = 2,6-(RNCH)2C6H4, R = tBu or Dipp) were synthesized. In the case of C,N-chelated compounds, the reaction of CNDippLi with Te(dtc)2 (where dtc = Et2NCS2) in a 1
CH)C6H4, R = tBu or Dipp; Dipp = 2,6-iPr2C6H3) or N,C,N pincer ligands NCNR (where NCN = 2,6-(RNCH)2C6H4, R = tBu or Dipp) were synthesized. In the case of C,N-chelated compounds, the reaction of CNDippLi with Te(dtc)2 (where dtc = Et2NCS2) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio smoothly provided the carbamate CNDippTe(dtc) which upon treatment with 2 eq. of HCl provided the chloride CNDippTeCl. In contrast, the analogous conversion of NCNRLi with Te(dtc)2 surprisingly furnished ionic bromides [NCNRTe]Br as a result of the exchange of dtc by Br coming from nBuBr present in the reaction mixture. Furthermore, the reaction of CNDippTeCl or [NCNRTe]Br with silver salts AgX (X = OTf or SbF6) provided the expected tellurenium cations [CNDippTe]SbF6 and [NCNRTe]X. To further increase the Lewis acidity of the central atom, the oxidation of selected compounds with 1 eq. of SO2Cl2 was examined yielding stable compounds [CNtBuTeCl2]X and [NCNtBuTeCl2]X. The oxidation of the Dipp substituted compounds proved to be more challenging and an excess of SO2Cl2 was necessary to obtain the oxidized products [CNDippTeCl2]SbF6 and [NCNDippTeCl2]SbF6, which could solely be characterized in solution. Compounds [CNtBuTeCl2]OTf and [NCNtBuTeCl2]OTf were shown to undergo a controlled hydrolysis to the corresponding telluroxanes. All compounds were studied by multinuclear NMR spectroscopy in solution and for selected compounds solid state 125Te NMR spectroscopy and single-crystal X-ray diffraction analysis were performed. The Lewis acidity of the studied cations was examined by the Gutmann–Beckett method using Et3PO as the probing agent. The Te–N chalcogen bonding situation of selected compounds has also been examined computationally by a set of real-space bonding indicators.
:1 molar ratio smoothly provided the carbamate CNDippTe(dtc) which upon treatment with 2 eq. of HCl provided the chloride CNDippTeCl. In contrast, the analogous conversion of NCNRLi with Te(dtc)2 surprisingly furnished ionic bromides [NCNRTe]Br as a result of the exchange of dtc by Br coming from nBuBr present in the reaction mixture. Furthermore, the reaction of CNDippTeCl or [NCNRTe]Br with silver salts AgX (X = OTf or SbF6) provided the expected tellurenium cations [CNDippTe]SbF6 and [NCNRTe]X. To further increase the Lewis acidity of the central atom, the oxidation of selected compounds with 1 eq. of SO2Cl2 was examined yielding stable compounds [CNtBuTeCl2]X and [NCNtBuTeCl2]X. The oxidation of the Dipp substituted compounds proved to be more challenging and an excess of SO2Cl2 was necessary to obtain the oxidized products [CNDippTeCl2]SbF6 and [NCNDippTeCl2]SbF6, which could solely be characterized in solution. Compounds [CNtBuTeCl2]OTf and [NCNtBuTeCl2]OTf were shown to undergo a controlled hydrolysis to the corresponding telluroxanes. All compounds were studied by multinuclear NMR spectroscopy in solution and for selected compounds solid state 125Te NMR spectroscopy and single-crystal X-ray diffraction analysis were performed. The Lewis acidity of the studied cations was examined by the Gutmann–Beckett method using Et3PO as the probing agent. The Te–N chalcogen bonding situation of selected compounds has also been examined computationally by a set of real-space bonding indicators.
Introduction
Tellurium can be called the maverick of the chalcogen group,1 being an element with very rich organometallic and coordination chemistry that provides various bonding possibilities as well as remarkable flexibility among its oxidation states.1 Not surprisingly, organotellurium compounds have found widespread applications as precursors for materials, in organic synthesis and catalysis.2 Among these compounds, organotellurium cations certainly represent a prominent compound class that have recently gained considerable attention.3 In fact, two classes of these species are recognized based on the oxidation state of the tellurium, i.e. divalent organotellurenium(II) [RTe]+ and tetravalent organotelluronium(IV) [R3Te]+ cations. The uncoordinated naked tellurenium cations (Fig. 1A) are unknown due to very high reactivity caused mainly by the inherent electron deficiency of the tellurium atom; therefore, the presence of an external Lewis base (LB) is crucial for their stabilization.3 The utilization of inter-molecularly coordinated LBs has proved to be highly useful in this regard (Fig. 1B).4 Amongst the intramolecularly coordinating ligands, N,C,N-pincer type ligands have shown to be versatile in the stabilization of the Te center (Fig. 1C).5 | ||
| Fig. 1 Relevant examples of organotellurium cations for this study. | ||
In contrast, the organotelluronium(IV) cations provide more possibilities regarding the number of carbon bonded ligands, thus [R3Te]+, [R2TeX]+ and [RTeX2]+ species can be distinguished.6–8 A structurally fully characterized dihaloorganotelluronium cation [RTeX2]+ was unknown for a long time and only recently the first example has been synthesized by using an N,C,N-pincer ligand (Fig. 1D).9 The S,C,S-pincer ligands10 (Fig. 1E) and carbenes11 even allowed the isolation of diorganotellurium(II) dications. Noteworthily, the tellurium cations of both types have gained increasing attention with regard to their utilization in catalysis or ion recognition.12
Organotelluroxanes, bearing at least one Te–C bond, constitute another interesting class of tellurium compounds that are usually synthesized by a hydrolysis of respective organotellurium halides.13 For example, a controlled base hydrolysis in the case of divalent RTeX results in the formation of oxygen bridged (RTe)2(μ-O) telluroxanes.13 The chemistry of tetravalent telluroxanes is more diverse. The hydrolysis of R2TeX2 should lead to diorganotelluroxide R2TeO, but these compounds mostly exist in their aggregated – dimeric or oligomeric forms [R2TeO]n.13,14 The monomeric species with a terminal Te–O bond could be isolated either using the support of a C,N-chelating ligand15 or by the protonation of the terminal oxygen by a strong Brønsted acid.7a,16 The oxidation of diorganotellurium compounds R2Te also provided interesting dicationic oligotelluroxanes [R2Te+–[R2TeO]n–O–Te+R2][X]2 (where X = OTf, n = 1–4).17 The hydrolysis of monoorganotellurium(IV) compounds RTeX3 is even more complicated, furnishing various products depending on the condensation and aggregation steps,18 but even monomeric organotellurinic acid RTe(O)(OH) could be isolated.19 By analogy, a unique dimer of a organotelluronic acid [RTe(O)(OH)3]2 could be isolated first using sufficient steric shielding,20 but later on related tellurium(IV) compounds could be obtained also with C,N-chelating ligands.19,21 Finally, diorganotellurones R2Te(O)2 with two terminal Te–O bonds were isolated after oxidation of the starting R2Te with NaIO4.22
We have recently reported the synthesis of a set of tellurenium cations stabilized by a C,N-chelating ligand CNtBu with weakly coordinating anions.23 We have also shown that in the case of the [CB11H12]− carborane counter-anion, a remarkable B–H bond activation by the tellurium centre is accessible.24
The aim of this study is to further develop this family of promising N-coordinated organotellurenium(II) cations [RTe]+ and to examine the possibility of the isolation of organotelluronium(IV) cations [RTeCl2]+. For this purpose, either C,N-chelating ligands CNR (where CN = 2-(RNCH)C6H4, R = tBu or Dipp; Dipp = 2,6-iPr2C6H3) or N,C,N-pincer ligands NCNR (where NCN = 2,6-(RNCH)2C6H3, R = tBu or Dipp) were selected. These ligands enable us to follow the influence of the R group attached to the imino-function and the number of these donor moieties. The description of a controlled hydrolysis of selected dichloroorganotelluronium cations is also included.
Results and discussion
Synthesis
The recently presented synthetic protocol developed for CNtBuTeCl,25 including the conversion of CNRLi with Te(dtc)2 (where dtc = Et2NCS2) in a 1:1 molar ratio followed by the treatment with 2 eq. of HCl, was successfully applied for the synthesis of CNDippTeCl (Scheme 1A). In contrast, the treatment of NCNRLi with Te(dtc)2 surprisingly led to the isolation of ionic bromides [NCNRTe]Br. Importantly, both compounds precipitated directly from the reaction mixtures as the least soluble compound pointing to the fact that their ionic nature plays a crucial role (vide infra). The auto-ionization of both compounds is obviously caused by the presence of the second donor group in the pincer ligand. Although the dithiocarbamates NCNRTe(dtc) are formed first, due to the presence of nBuBr in the reaction mixture (as a result of in situ lithiation of the ligand) an exchange of dtc with Br occurs giving [NCNRTe]Br as isolable products. Low solubility helps in promoting this procedure (Scheme 1B), although the isolated yields are still rather moderate. This hypothesis has been clearly proven by the inspection of the mother liquors after the reactions, where the presence of nBu(dtc) was established by NMR spectroscopy (Fig. S89–S91†) and the obtained data also agree with those published elsewhere.26
 | ||
| Scheme 1 Synthesis of starting compounds along with the proposed mechanism for the formation of [NCNRTe]Br. | ||
The chloride or bromide in CNDippTeCl or [NCNRTe]Br can subsequently be substituted by less nucleophilic anions using silver salts AgX (X = OTf or SbF6, Scheme 2) and the corresponding tellurenium cations [CNDippTe]X and [NCNRTe]X can be isolated in quantitative yields as crystalline solids.
 | ||
| Scheme 2 Synthesis of tellurium(II) cations. | ||
Finally, selected tellurenium cations were oxidized by 1 eq. of SO2Cl2 producing rare examples of monoorganotelluronium(IV) cations [CNtBuTeCl2]X and [NCNtBuTeCl2]X (X = OTf or SbF6) that were obtained as yellow solids in high yields (Scheme 3). In contrast, the oxidation of the Dipp substituted compounds [CNDippTe]SbF6 and [NCNDippTe]SbF6 was more challenging and an excess (5 to 10 eq.) of SO2Cl2 was necessary for complete oxidation. Furthermore, telluronium cations are highly sensitive towards even traces of moisture, which hampered the isolation of Dipp substituted compounds as pure solid samples. In the case of [CNDippTeCl2]SbF6 and [NCNDippTeCl2]SbF6, this hydrolysis always led to complicated mixtures of products. Interestingly, the tBu species [CNtBuTeCl2]OTf and [NCNtBuTeCl2]OTf can be hydrolyzed in a controlled manner, e.g. in wet acetonitrile, giving [COTeCl2]2O and [OCNtBuTeCl2]2O in very high yields (Scheme 3), where CO = 2-(OCH)C6H4, OCNtBu = 2-(–O–CH)-6-(tBuNCH)C6H3.
 | ||
| Scheme 3 Oxidation of tellurium(II) cations and subsequent hydrolysis. | ||
Close inspection of the molecular structures of both hydrolyzed species revealed substantial structural differences (vide infra for sc-XRD analysis). In the case of [COTeCl2]2O, the oxygen bridges two tellurium atoms and its formation can be, thus, explained by a conventional hydrolysis of the pendant imino group to an aldehyde with a subsequent condensation of two tellurium hydroxides to give the final telluroxane [COTeCl2]2O (Scheme 4A). Similar condensation reactions are well documented in the literature.18a,19
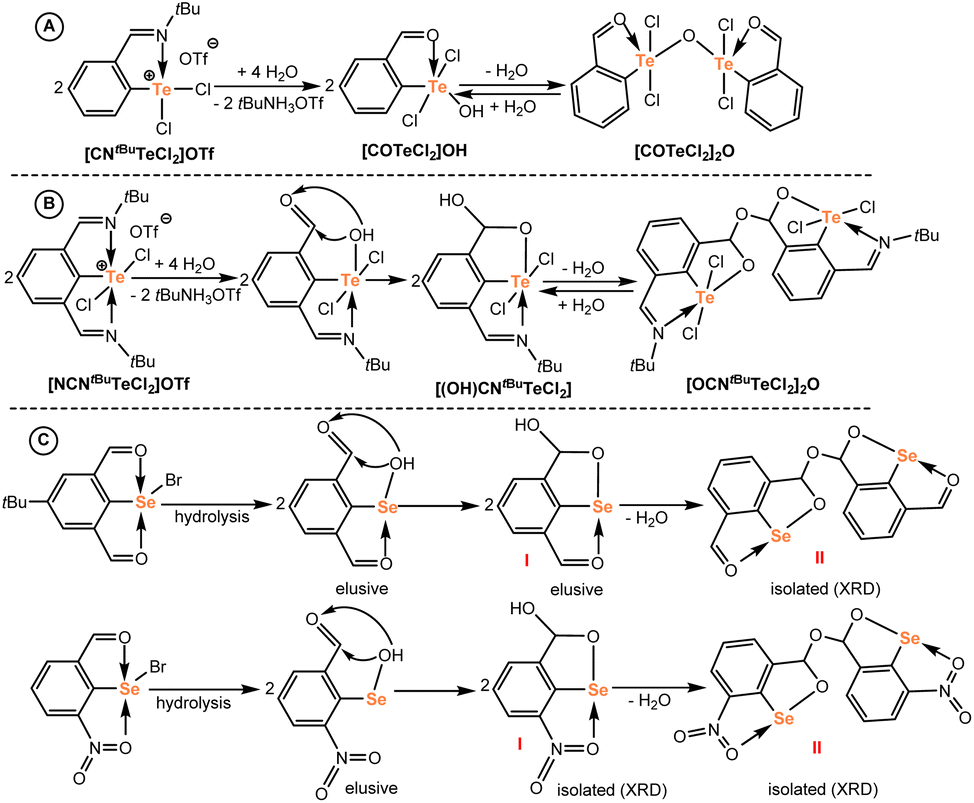 | ||
| Scheme 4 Proposed mechanisms for the hydrolysis of telluronium(VI) cations (A and B) and comparison with the literature (C, ref. 27 and 28). | ||
In contrast, the oxygen atom in [OCNtBuTeCl2]2O bridges the tellurium fragments via two CH groups and no Te–O–Te linkage is formed. Considering the hydrolysis of the nitrogen function in the first step, the resulting Te–OH species does not undergo a classical condensation, but an attack of the Te–OH moiety to the CHO function seems to be feasible (Scheme 4B). Importantly, similar reactions have been recently well documented during an accidental hydrolysis of selenium(II) bromides (Scheme 4C).27,28 Attack of the Se–OH group leads to intermediates I, which dimerize to II with concomitant water elimination. In both the reported cases, the second ortho-position next to the selenium is occupied by a group containing formally an oxygen donor atom and the corresponding molecular structures revealed non-negligible Se⋯O intramolecular interactions in all cases. Entirely the same situation is found in our case, when the intact imino-group may coordinate the central tellurium, thereby probably facilitating the Te–OH attack on the aldehyde function instead of a simple condensation toward the Te–O–Te dimer (Scheme 4B). Thus, the intermediate [(OH)CNtBuTeCl2] (where (OH)CNtBu = 2-(O–C(OH)H)-6-(tBuN = CH)C6H3), vide infra then condenses to the final product [OCNtBuTeCl2]2O as the least soluble compound similarly to the selenium analogues mentioned above.27,28
Multinuclear NMR spectroscopy
All tellurium(II) compounds were characterized using high-resolution MALDI MS spectra (see the Experimental section and Fig. S99–S105†), but it was not possible for corresponding tellurium(IV) compounds due to their very high sensitivity to moisture; a similar problem was encountered with combustion analysis of the studied compounds. Nevertheless, all compounds were in detail investigated by 1H, 13C{1H}, 125Te{1H} and 1H–15N HMBC NMR spectroscopy. The presence of the appropriate ligand was established mainly by diagnostic signals for the CHN group in the range of 8.83–10.31 ppm for δ(1H) and 159.4–171.1 ppm for δ(13C) (see Table 1). One signal was also obtained in 2D 1H–15N HMBC experiments. Compounds substituted with tBuN groups showed signals with δ(15N) = −86.7 to −130.2 ppm, while for DippN substituted compounds values between −102.8 to −174.9 ppm were detected. Selected examples were also characterized by solid state 125Te NMR vide infra, where they are discussed together with solution 125Te NMR data.
| Compound |
CHN group |
δ(125Te) | δ(15N)a | |
|---|---|---|---|---|
| δ(1H) | δ(13C) | |||
| a Determined by 1H, 15N HMBC experiments. b From ref. 23. c From ref. 25. d Not measured. | ||||
| Covalent CN-chelated tellurium(II) compounds | ||||
| CNtBuTeCl | 8.58 | 159.6 | −93.4 | 1259 |
| CNDippTeCl | 9.16 | 164.7 | −121.4 | 1392 |
| Ionic CN-chelated tellurium(II) compounds | ||||
| [CNtBuTe]OTf | 9.53 | 163.8 | −123.0 | 1753 |
| [CNtBuTe]SbF6 | 9.54 | 165.5 | −130.2 | 1897 |
| [CNDippTe]SbF6 | 9.37 | 171.1 | −174.9 | 2113 |
| Ionic NCN-chelated tellurium(II) compounds | ||||
| [NCNtBuTe]Br | 10.27 | 160.7 | −91.2 | 1394 |
| [NCNtBuTe]OTf | 9.75 | 160.0 | −88.8 | 1400 |
| [NCNtBuTe]SbF6 | 9.57 | 159.4 | −89.5 | 1409 |
| [NCNDippTe]Br | 10.31 | 166.5 | −119.8 | 1522 |
| [NCNDippTe]SbF6 | 9.60 | 165.9 | −117.4 | 1557 |
| CN-chelated tellurium(IV) compounds | ||||
| CNtBuTeCl3 | 8.86 | 163.4 | 1177 | |
| [CNtBuTeCl2]OTf | 9.27 | 168.2 | −86.8 | 1373 |
| [CNtBuTeCl2]SbF6 | 9.27 | 168.6 | −87.1 | 1399 |
| [CNDippTeCl2]SbF6 | 8.83 | 170.3 | −102.8 | 1428 |
| NCN-chelated tellurium(IV) compounds | ||||
| [NCNtBuTeCl2]OTf | 9.45 | 163.2 | −86.7 | 1180 |
| [NCNtBuTeCl2]SbF6 | 9.40 | 163.2 | −87.0 | 1179 |
| [NCNDippTeCl2]SbF6 | 9.22 | 168.2 | −112.9 | 1308 |
The hydrolysed products [COTeCl2]2O and [OCNtBuTeCl2]2O exhibited only limited solubility in most organic deuterated solvents. [COTeCl2]2O was reasonably soluble in thf-d8 that allowed us to obtain 1H and 13C{1H} NMR spectra. Surprisingly, if non-dried thf-d8 is used, the spectra contained two sets of chemical shifts pointing to the presence of two compounds (i.e.[COTeCl2]2O and [COTeCl2]OHvide infra) that both display shifts at 10.22 and 10.38 ppm (198.3 and 200.5 ppm) for the CHO group (Fig. S52 and 53†), respectively. Furthermore, the 1H NMR spectrum also contained a singlet at 7.74 ppm that lacks any cross-peak in the 2D-1H–13C HSQC NMR spectrum corresponding to the OH group in [COTeCl2]OH. Furthermore, the 1H–1H EXSY NMR spectrum unambiguously proved a dynamic mutual exchange between both species and traces of present water (Fig. S55†). 125Te{1H} NMR spectra also showed two signals at 1342 ([COTeCl2]OH) and 1405 ppm ([COTeCl2]2O). Based on these findings, we propose that upon dissolving of [COTeCl2]2O in wet thf-D8 a partial hydrolysis of the Te–O–Te bridge occurred during the formation of [COTeCl2]OH (Scheme 4A). This assumption was verified by the addition of 10 μL of water to this sample that led to a complete disappearance of the signals attributable to the telluroxane [COTeCl2]2O and only one set of signals for [COTeCl2]OH remained present (Fig. S56–58†). The use of freshly dried thf-d8 almost suppressed the formation of [COTeCl2]OH, but traces remain detectable in the NMR spectra most probably as a result of the moisture present in the original sample after hydrolytic synthesis of [COTeCl2]2O (Fig. S59–S62†).
[OCNtBuTeCl2]2O exhibited good solubility in dmso-d6 (anhydrous) only and the obtained 1H and 13C{1H} NMR spectra agreed well with the proposed structure. The presence of the intact CHNtBu group is reflected by the observation of the signal at 9.14 ppm (157.3 ppm), while the chemical shift at 7.3 ppm (100.7 ppm) corresponds to the CH group involved in the bridge between both tellurium fragments. The 125Te{1H} NMR spectrum showed one signal at 1381 ppm (Fig. S64–S66†). The utilization of undried dmso-d6 resulted in the appearance of the second set of signals that was tentatively assigned to [(OH)CNtBuTeCl2] pointing to a reversible hydrolysis of the present bridge (Scheme 4B); but in contrast to [COTeCl2]2O, the full conversion to [(OH)CNtBuTeCl2] was not obtained even after 8 days, with about 15% of intact [OCNtBuTeCl2]2O still present (see Fig. S67–72†).29 The 1H NMR spectrum of [(OH)CNtBuTeCl2] contained two singlets at 6.88 and 9.06 ppm corresponding to CHO and CHN groups (δ(13C) = 99.9 and 156.7 ppm) respectively, along with a broad one at 7.30 ppm for a new OH group (no cross-peak in 2D-1H, 13C HSQC NMR). The 125Te{1H} NMR spectrum also revealed a new signal at 1374 ppm that is, however, only very slightly shifted in comparison to the starting [OCNtBuTeCl2]2O (cf. 1381 ppm), indicating a rather similar bonding situation around the tellurium atom (Fig. S68†). Note that the cleavage of the Te–O–Te bridge ongoing from [COTeCl2]2O to [COTeCl2]OH was accompanied by more pronounced downfield shift by 63 ppm. Unfortunately, all attempts to crystallize either [COTeCl2]OH or [(OH)CNtBuTeCl2] remained unsuccessful. Nevertheless, their existence in solution supported the proposed mechanism for the hydrolysis of telluronium cations shown in Scheme 4.
Solid state structures
Molecular structures determined by sc-X-Ray diffraction analysis are shown in Fig. 2–5 and the relevant geometrical parameters are compiled in Table 2. | ||
| Fig. 2 ORTEP drawings of molecular structures of CNDippTeCl. The thermal ellipsoids are given with 30% probability and hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 3 ORTEP drawings of molecular structures of tellurenium(II) cations. The thermal ellipsoids are given with 30% probability and hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 4 ORTEP drawings of molecular structures of telluronium(IV) cations. The thermal ellipsoids are given with 30% probability and hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 5 ORTEP drawings of molecular structures of compounds [COTeCl2]2O and [OCNtBuTeCl2]2O. The thermal ellipsoids are given with 30% probability and hydrogen atoms except for CH group in the latter are omitted for clarity. Selected bond lengths [Å] for [COTeCl2]2O: Te(1)–C(1) 2.124(3), Te(1)–O(1) 1.9526(17), Te(1)–O(2) 2.5474(18), Te(1)–Cl(1) 2.5607(7), Te(1)–Cl(2) 2.4411(8), Te(2)–C(8) 2.117(2), Te(2)–O(1) 1.9432(18), Te(2)–O(3) 2.569(2), Te(2)–Cl(3) 2.4487(7), Te(2)–Cl(4) 2.5440(8). For [OCNtBuTeCl2]2O: Te(1)–C(1) 2.070(3), Te(1)–N(1) 2.483(2), Te(1)–O(1) 1.997(2), Te(1)–Cl(1) 2.4861(14), Te(1)–Cl(2) 2.5375(14), Te(2)–C(13) 2.060(3), Te(2)–N(2) 2.456(2), Te(2)–O(2) 2.006(2), Te(2)–Cl(3) 2.4928(14), Te(2)–Cl(4) 2.5445(14), O(3)–C(12) 1.418(3), O(3)–C(24) 1.424(3), O(1)–C(12) 1.424(4), O(2)–C(24) 1.417(3). | ||
| Compound | N1–Te1 | N2–Te1 | Other important contactsa |
|---|---|---|---|
| a Contacts include either intermolecular contacts of the type Te–X⋯Te or contacts between the cation and the anion. b From ref. 23. c From ref. 25. d Two closely related molecules present and data for only one are displayed. | |||
| Covalent CN-chelated tellurium( II ) compounds | |||
| CNtBuTeCl | 2.203(2) | — | Te–Cl⋯Te 3.579 |
| CNDippTeCl | 2.249(4) | — | — |
| Ionic CN-chelated tellurium( II ) compounds | |||
| [CNtBuTe]OTf | 2.113(1) | — | Te⋯O(OTf) 2.500 |
| [CNtBuTe]SbF6 | 2.073(2) | — | Te⋯F(SbF6) 2.687 |
| [CNDippTe]SbF6 | 2.0645(16) | — | Te⋯F(SbF6) 2.666 |
| Ionic NCN-chelated tellurium( II ) compounds | |||
| [NCNtBuTe]Br | 2.286(4) | 2.303(4) | Te⋯Br 3.609 |
| [NCNtBuTe]SbF6 | 2.259(2) | 2.259(2) | — |
| [NCNDippTe]SbF6 | 2.259(2) | 2.2596(19) | — |
| CN-chelated tellurium(IV) compounds | |||
| CNtBuTeCl3 | 2.286(1) | — | Te–Cl⋯Te 3.506(3) |
| [CNtBuTeCl2]OTf | 2.308(3) | — | Te⋯O(intra-OTf) 2.523 Te⋯O(inter-OTf) 3.042 |
| NCN-chelated tellurium(IV) compounds | |||
| [NCNtBuTeCl2]OTf | 2.281(4) | 2.311(4) | Te–Cl⋯Te 3.699 |
| [NCNtBuTeCl2]SbF6 | 2.300(5) | 2.294(5) | Te–Cl⋯Te 3.620 |
The Te(1)–N(1) distance in CNDippTeCl of 2.249(4) Å (cf. Σcov(Te–N) = 2.07 Å,30Fig. 1) is slightly elongated in comparison to that in CNtBuTeCl (2.203(2) Å),25 but the former remains exclusively monomeric, while the latter forms a dimer via very weak Te–Cl⋯Te contacts (3.579 Å, cf. Σcov(Te–Cl) = 2.35 Å (ref. 30)) that can be probably ascribed to the steric effects of the Dipp group. The Te(1)–N(1) distance becomes significantly shorter (2.0645(16) Å) upon abstraction of the chlorine atom in [CNDippTe]SbF6 (Fig. 3) and is comparable to its tBu counterpart [CNtBuTe]SbF6 (2.073(2) Å).23 The Te(1) atom is di-coordinated with a weak chalcogen Te⋯F bond with the counter-anion. As mentioned above, the utilization of the pincer ligands resulted in the autoionization even in the case of the bromide [NCNtBuTe]Br, where the Br(1) atom again forms a weak chalcogen bond with Te(1) atom (3.609 Å, cf. Σcov(Te–Br) = 2.50 Å (ref. 30)). In the case of hexafluoroantimonates [NCNtBuTe]SbF6 and [NCNDippTe]SbF6, the tellurium cation remains without a similar contact. The Te(1) atom is three-coordinated with a T-shaped geometry and the Te–N distances span over a narrow interval of 2.259(2)–2.303(4) Å, which are elongated in comparison with C,N chelated cations (Table 2) but are well comparable to other N,C,N-pincer ligand stabilized tellurenium cations (2.225–2.392 Å).5b–d
The molecular structures of the telluronium(IV) cations differ significantly based on the used ligand. In the case of [CNtBuTeCl2]OTf, the tellurium atom adopts an octahedral array (Fig. 4), where C(1)/N(1) and Cl(1)/Cl(2) atoms are mutually cis coordinated. The Te(1)–N(1) distance 2.308(3) Å is similar to that in CNtBuTeCl3 2.286(1) Å,25 while the Te(1)–Cl(1/2) bonds 2.4528(8)/2.3350(9) Å, respectively, correspond to single bonds albeit the first one is a bit elongated in comparison to Σcov(Te–Cl) = 2.35 Å.30 The triflate coordinates to the Te(1) atom with the bond length Te(1)–O(1) 2.523(2) Å that is significantly elongated in comparison to Σcov(Te–O) = 2.35 Å (ref. 30) and the octahedron is completed by the intermolecular contact with a second triflate (Te(1)–O(3a) 3.042(3) Å), thereby forming a dimer. In the case of [NCNtBuTeCl2]OTf and [NCNtBuTeCl2]SbF6, the presence of the pincer ligand again supported the formation of ionic species and both OTf and SbF6 are situated outside the coordination sphere of the central atom. The Te(1) atom is captured by the pincer ligand with the Te–N distances ranging from 2.281(4) to 2.311(4) Å and these values are shorter than those established for the only structurally characterized analogue, i.e. [2,6-(Me2NCH2)2C6H4TeBr2][Br3] 2.372(2) and 2.435(2) Å.9 The chlorine atoms are coordinated mutually in trans positions, which is in contrast to [CNtBuTeCl2]OTf and the Te–Cl bond lengths are in the interval of 2.4532(16)–2.5147(17) Å, again a bit elongated in comparison with Σcov(Te–Cl) = 2.35 Å.30 The shape of the coordination polyhedron can be described as a distorted square-pyramid with the pincer ipso-carbon atom in the apical position. The closest intermolecular, but negligible, contact with the chlorine atom (3.699 and 3.620 Å, respectively) from the adjacent molecule is not considered.
The structures of the hydrolysis products [COTeCl2]2O and [OCNtBuTeCl2]2O are shown in Fig. 5. The molecular structure of the former proved the hydrolysis of the imino-tBuNCH function to the benzaldehyde framework while the condensation of the Te–OH groups produced a central –Cl2Te(μ-OTe)Cl2– linkage. Similar hydrolytic products were obtained upon basic hydrolysis of neutral tellurium(IV) complexes ArTeCl3 (Ar = 2-pyC6H419 or 8-Me2NC10H618a). The coordination polyhedron of the central atoms is best described as a distorted square-pyramidal. The Te1/2-O2/3 distances 2.5474(18)/2.569(2) Å, respectively, are elongated in comparison with Σcov(Te–O) = 1.99 Å,30 but still indicate intramolecular Te⋯O chalcogen interactions. Both Te(1) and Te(2) atoms are bridged by the O(1) atom and the bond lengths of 1.9526(18) and 1.9432(18) Å correspond well to the single-bond30 and are also comparable to [ArTe(Cl2)]2(μ-O) (Ar = 2-pyC6H419 1.969(3) and 1.963(3) Å or 8-Me2NC10H618a 1.97(1) and 1.96(1) Å). The Te(1)–O(1)–Te(2) bonding angle of 123.96(13)° also approaches reported values for the abovementioned analogues 124.9(2)19 and 125.4(5)°,18a respectively. Each of the two tellurium atoms is further coordinated by two chlorine atoms and in each case one of the Te–Cl bonds is a bit longer than the other (cf. 2.4411(8) vs. 2.5607(7) for Te(1) and 2.4487(8) vs. 2.5440(8) Å for Te(2)).
The structure of [OCNtBuTeCl2]2O differs significantly from that of its counterpart [COTeCl2]2O as mentioned above. Two tellurium fragments are connected via the C(12)–O(3)–C(24) bridge and both bond lengths correspond to single bonds 1.418(3) and 1.424(3) Å (ref. 30) (bonding angle 113.5(2)°), while the geometry around carbon atoms C12 and C24 is tetrahedral corresponding to the sp3 hybridization. Both tellurium atoms Te(1) and Te(2) form single bonds with O(1) and O(2) atoms (1.997(2) and 2.006(2) Å, respectively) and each is further coordinated by one nitrogen atom N(1) or N(2) (2.483(2) and 2.456(2) Å). The distorted square pyramidal array around the central atoms is completed by two chlorine atoms (the range of bond lengths 2.4861(14)–2.5445(14) Å) and ipso-carbon atoms of the pincer ligands in the axial position. The geometrical framework in [OCNtBuTeCl2]2O is only rarely found for heavier p-block elements. With the exception of two selenium compounds27,28 depicted in Scheme 4, only pure organic compounds were reported.31
125Te NMR spectroscopy and the Gutmann–Beckett method
All 125Te NMR chemical shifts of the studied compounds along with those of relevant earlier published compounds are compiled in Table 1. It is to be noted that the values of the 125Te NMR chemical shifts of compounds containing the CNDipp ligand are always downfield shifted in comparison with that of CNtBu compounds, which is evidently a consequence of the electron withdrawing aromatic Dipp group in the former. For selected tellurium(II) compounds, solid state 125Te NMR spectroscopy has also been examined to gain insight into the relationship between solution and solid state structure.Regarding the C,N-chelated compounds, it becomes obvious that the abstraction of the chloride ion from the neutral precursors CNtBuTeCl (1259 ppm) and CNDippTeCl (1392 ppm) by weakly coordinating anions resulted in a significant downfield shift of the signals in [CNtBuTe]X (X = OTf 1753 and SbF6 1897 ppm) and [CNDippTe]SbF6 (2113 ppm). The latter represents the highest obtained value among reported compounds pointing to its high Lewis acidity (vide infra further discussion). In the case of pincer complexes, the 125Te NMR chemical shifts are found in a narrow interval i.e. 1394–1409 ppm for [NCNtBuTe]X (X = Br, OTf and SbF6) and 1522–1557 ppm for [NCNDippTe]X (X = Br or SbF6) pointing to the fact that all compounds form practically identical ionic pairs in solution even in the case of bromides. These values also suggest a significant shielding of the central atom in comparison with the C,N-chelated analogues due to the presence of the second donor functionality.
In the solid state, the δiso(125Te) NMR chemical shifts for the starting CNDippTeCl (1403 and 1349 ppm, two independent molecules in the unit cell) are close to the value found in solution (1392 ppm). In the case of CNtBuTeCl, δiso(125Te) = 1340 ppm is shifted in comparison with solution (1259 ppm) most probably indicating the absence of the weak intermolecular Te⋯Cl contact in solution.25 In contrast, δiso(125Te) values of tellurenium cations [CNtBuTe]X (X = OTf 1742 and SbF6 1895 and 1842 ppm, two independent molecules in the unit cell) and [NCNtBuTe]SbF6 (1388 ppm) are all close to the values found in solution proving an analogous structure in both phases.
Looking to solution δ(125Te) of tellurium(IV) compounds, the formal abstraction of one of the chlorides in neutral CNtBuTeCl3 (1177 ppm)25 resulted in a downfield shift, but it is significantly less pronounced in comparison to the abovementioned tellurenium cations, cf.[CNtBuTeCl2]X (X = OTf 1373 and SbF6 1399 ppm) and [CNDippTeCl2]SbF6 (1428 ppm). Again, the presence of the second ligand arm in pincer compounds helps to shield the tellurium atom more efficiently based on the obtained δ(125Te), cf.[NCNtBuTeCl2]X (X = OTf 1180 and SbF6 1179 ppm) and [NCNDippTeCl2]SbF6 (1308 ppm).
The Gutmann–Beckett method,32 even with its shortcomings regarding steric effects and(or) “Pearson hardness”,33 is still used as a gauge of Lewis acidity that enables a reasonable and straightforward scaling within one class of compounds, being often applied to p-block elements as well.34 We used this method to shed more light on the Lewis acidity of the studied compounds using Et3PO as the probing agent (Table 3). As we are aware that 31P NMR chemical shifts of both Et3PO and its complexes with Lewis acids are solvent dependent, we refrain from calculating the exact values of Gutmann–Beckett acceptor numbers and rather compare Δδ(31P), defined as Δδ(31P) = δ(31P)obs − δ(31P of Et3PO), within the set of compounds and with relevant examples known from the literature. All samples were analysed in dichloromethane-d2 as the mostly used solvent, except for [NCNtBuTeCl2]OTf and [NCNtBuTeCl2]SbF6 that are completely insoluble and acetonitrile had to be used instead. The Dipp substituted telluronium cations are also not included as they are not obtainable as pure compounds (vide supra).
:5 molar ratio, dichloromethane-d2); Δδ(31P) = δ(31P)obs − δ(31P, Et3PO) [ppm]
| Entry | Compound | δ(31P)obs | Δδ(31P) |
|---|---|---|---|
| a Measured in acetonitrile-d3 due to the solubility problems. | |||
| 1 | Pure Et3PO | 50.7 | 0 |
| 2 | CNtBuTeCl | 50.8 | 0.1 |
| 3 | CNDippTeCl | 50.7 | 0 |
| 4 | [CNtBuTe]OTf | 69.7 | 19.0 |
| 5 | [CNtBuTe]SbF6 | 70.3 | 19.6 |
| 6 | [CNDippTe]SbF6 | 72.7 | 22.0 |
| 7 | [NCNtBuTe]Br | 51.4 | 0.8 |
| 8 | [NCNtBuTe]OTf | 54.4 | 3.7 |
| 9 | [NCNtBuTe]SbF6 | 51.1 | 0.4 |
| 10 | [NCNDippTe]Br | 51.5 | 0.8 |
| 11 | [NCNDippTe]SbF6 | 54.6 | 3.9 |
| 12 | [CNtBuTeCl2]OTf | 88.6 | 37.9 |
| 13 | [CNtBuTeCl2]SbF6 | 96.2 | 45.5 |
| 14 | Pure Et3POa | 50.4 | 0 |
| 15 | [NCNtBuTeCl2]OTf | 66.2 | 15.8 |
| 16 | [NCNtBuTeCl2]SbF6 | 54.6 | 4.2 |
Inspection of the Δδ(31P) values summarized in Table 3 revealed that neutral tellurium(II) compounds, not surprisingly, exhibited limited shift difference (entries 2 and 3); similarly the N,C,N-chelated tellurenium cations (entries 7–11) showed only negligible influence as the Lewis acidity of the central atom is effectively compensated by the pincer ligands. In contrast, in the case of C,N-coordinated tellurenium cations (entries 4–6), Δδ(31P) in dichloromethane-d2 between 19.0 and 22.0 ppm indicate remarkable Lewis acidity as these values approach that of the well-established borane B(C6F5)3 with Δδ(31P) ∼ 26 ppm.34i,j Regarding the telluronium cations bearing pincer ligands (entries 15 and 16), the Δδ(31P) is surprisingly quite different depending on the counter-anion. It could be speculated that the forced utilization of acetonitrile-d3 may play a role in this phenomenon. In contrast, the C,N-chelated telluronium cations (entries 12 and 13) showed impressive Δδ(31P) values of 37.9 and 45.5 ppm, for [CNtBuTeCl2]OTf and [CNtBuTeCl2]SbF6 respectively. These values are well comparable e.g. to those reported for silylium ions (39–45 ppm),34a,c neutral super Lewis acidic silanes34e,f,m (∼35 ppm) or organofluorophosphonium salts (∼35 ppm).35 This fact also well corresponds to their extremely high affinity to hydrolysis and make it challenging to utilize them as Lewis acids in catalysis.
DFT computations
Density functional theory (DFT) computations were conducted on the compounds [CNtBuTeCl2]SbF6 and [CNDippTe]SbF6 as well as on the cations [NCNtBuTe]+, [NCNtBuTeCl2]+ and [NCNDippTe]+. In order to gain insights into the N–Te bonding, a set of real-space bonding descriptors have been studied. According to the atoms-in-molecules36 analysis, N–Te bond critical points (bcp) have been observed in all studied cases, showing characteristics of polar covalent bonding contributions. The effect of the Dipp-substituent on the nature of the N–Te bond was studied by comparing the compound pairs [CNRTe]SbF6 (R = t-Bu23 and Dipp) and [NCNRTe]+ (R = t-Bu and Dipp) and revealed no substantial differences in the N–Te bonding (Table S2†). The additional steric effects imposed by the Dipp group are also discernible in the AIM picture, as additional CH⋯Te bcps are observed, which are absent in the respective t-Bu species (Fig. S93–S98†).In contrast, single (CN) or double (NCN) N-coordination to the Te-atom has a significant effect on the (individual) N–Te chalcogen bonds. Key parameters of the AIM analysis gave rise to smaller electron densities (ρbcp(r)) and Laplacians (∇2(ρbcp(r))) at the bcp for [NCNRTe]+ (0.51–0.56 e Å−3/3.0–3.3 e Å−5) compared to [CNRTe]SbF6 (0.73–0.77 e Å−3/4.7–5.0 e Å−5), indicating a stronger coordination of the single N-ligand in the latter (Table S1†). This is also revealed in the kinetics (G/ρbcp(r)) and total energies (H/ρbcp(r)) over ρ values. Both parameters are closer to zero in [NCNRTe]+ than in [CNRTe]SbF6, suggesting weaker ionic and covalent bonding contributions in the pincer type compounds (Table S1†). However, it should be noted that these pincer ligands lead to a more balanced coordination in [NCNRTe]+, whereas in [CNRTe]SbF6 there is a stronger (N–Te) and weaker (F–Te) coordination. This is also reflected upon inspection of the Wiberg bond indices, NLMO/NPA bond orders and the delocalization indices for the N–Te interaction (Table S6†), which show smaller values for [NCNRTe]+ compared to [CNDippTe]SbF6. Interestingly, the electron population of the disynaptic ELI-D37 V2(N,Te) bonding basin is less affected by the single or double coordination, showing populations of 2.48 e ([CNtBuTe]SbF623), 2.58 e ([CNDippTe]SbF6), 2.49/2.58 e ([NCNtBuTe]+) and 2.48/2.49 e ([NCNDippTe]+). The non-covalent interaction (NCI) index38 is a powerful tool in unravelling weak, non-covalent bonding contributions and for the N–Te bonds in [NCNDippTe]+ clear red-colored shaped rings are observed, which is barely visible in the single N–Te bond of [CNDippTe]SbF6, indicating the stronger covalent bonding contributions in the latter (see Fig. S95†). Interestingly, based on the AIM analysis there is only a little effect on the doubly coordinating N–Te interaction upon chlorination from [NCNtBuTe]+ to [NCNtBuTeCl2]+. The ρbcp(r) and H/ρbcp(r) values are very similar in both compounds and the slight decrease of the Laplacians (∇2(ρbcp(r))) and G/ρbcp(r) values in [NCNtBuTeCl2]+ points to a minimal reduction of ionic bonding contributions.
NBO39 analysis reveals no distinct Te–N bonding orbitals, but second order perturbation theory gives rise to LP(N) → LV(Te) or LP(N) → RY(Te) donation of a total of E2 = 103.73–119.73 kcal mol−1 for the pincer ligands [NCNRTe]+ (Table S4†), which is in agreement with similar NCN-stabilized Te(II) cations.5d,9
For [CNtBuTeCl2]SbF6, we investigated two possible structural isomers: one with a linear Cl–Te–Cl linkage and another with a nearly rectangular Cl–Te–Cl linkage. The gas-phase structure of the latter form is energetically favoured by −51.24 kJ mol−1 and resembles also the experimentally obtained geometry (Fig. 4, although with OTf− as anion). Interestingly, the bonding situation in both isomers differs significantly. With a rectangular Cl–Te–Cl linkage the N–Te bonding can be compared to the N–Te interaction in the pincer compounds of the [NCNtBuTe]+ type with ρbcp(r) values and respective Laplacians of 0.51 e Å−3/2.2 e Å−5. Also the G/ρbcp(r) and H/ρbcp(r) values are very similar to the ones in [NCNtBuTe]+. The isomer with a linear Cl–Te–Cl linkage shows stronger N–Te coordination as observed e.g. by higher ρbcp(r) and Laplacian values (0.64 e Å−3/2.8 e Å−5), but are smaller compared to the Te(II) species [CNRTe]SbF6. The F–Te interaction in the rectangular Cl–Te–Cl form is also weakened in comparison to the form with a linear Cl–Te–Cl linkage. The effects in both isomers on the F–Te and N–Te interaction can also be observed in the NCI. The incomplete red-colored ring around the N–Te in the linear Cl–Te–Cl form (Fig. 6, right) points towards stronger covalent interactions compared to the clearly visible ring-shape in the rectangular form (Fig. 6, left). Furthermore, the considerably weaker F–Te interaction in the rectangular form is reflected by the blue disk-shaped area on the F–Te axis in the NCI (Fig. 6, left).
 | ||
| Fig. 6 AIM molecular graph of [CNtBuTeCl2]SbF6 with rectangular Cl–Te–Cl linkage (left) and linear Cl–Te–Cl linkage (right) with bond critical points as red spheres and bond paths in orange as well as NCI iso-surfaces at s(r) = 0.5 colour coded with sign(λ2)ρ in a. u. Blue surfaces refer to attractive forces and red to repulsive forces. Green indicates weak interactions. | ||
Interestingly, the NBO analysis of the rectangular form shows a 3-center, 4-electron Cl–Te–N bond, with Cl–Te/Te–N contributions of 65.0/35.0% (Table S5†).
Conclusions
We have demonstrated the potential of both C,N- and N,C,N-chelating ligands, containing imino-donor function(s), for the stabilization of tellurenium and rare examples of dichlorotelluronium cations. The combined solution and solid-state 125Te NMR study showed that the structure revealed by sc-X-ray diffraction analysis is retained in solution in the case of tellurenium cations. Two dichlorotelluronium cations underwent a controlled hydrolysis with a concomitant condensation producing either a telluroxane with the –Te(Cl)2–O–Te(Cl)2– bridge or, in the case of the pincer compound, the condensation occurred at the ligand arm.Furthermore, the Gutmann–Beckett method was applied for a straightforward assessment of the Lewis acidity of the reported cations. Based on the obtained results, it became obvious that pincer ligands significantly suppress the acidity of the tellurium atom due to the presence of the second nitrogen donor atom. In contrast, the C,N-chelated tellurenium cations showed values comparable to B(C6F5)3, thereby indicating their potential in Lewis acid induced activation of various substrates. This is consistent with recently reported B–H bond activation of a carborane cage24 and the field will be developed in the future. More importantly, the C,N-chelated telluronium cations possess remarkable Lewis acidity as one can deduce from the very high Δδ(31P) values obtained by the Gutmann–Beckett method. Unfortunately, the extremely high propensity of these compounds to hydrolysis may hamper their further reasonable application.
Our next steps will be directed towards the extension of the applicability of C,N-chelated tellurenium cations in the activation of E–H bonds (E = B or Si) and building of more robust C,N-chelated telluronium cations. In particular, the latter as Lewis acids hold remarkable potential for further investigation of various bond activations or catalysis.
Experimental
General consideration
All syntheses were performed under an argon atmosphere using standard Schlenk techniques except for the hydrolysis of [NCNtBuTeCl2]OTf and [CNtBuTeCl2]OTf. All used solvents were dried using MD7 Pure Solv instrument (Innovative Technology, MA, USA), degassed and stored in Young-valve containers over potassium mirror or molecular sieves (3 Å). Deuterated solvents were dried by standard procedures and stored over molecular sieves. Procedures for the synthesis of all already published compounds are referenced in the main text. All other chemicals were purchased from commercial companies and used as delivered. 1H, 13C{1H}, and 125Te{1H} NMR spectra were recorded on a Bruker 400 or Bruker 500 spectrometers, using a 5 mm tunable broadband probe or the cryoprobe Prodigy. Chemical shifts in the 1H and 13C{1H} NMR spectra were referenced to the residual solvent signals (CDCl3: δ(1H) = 7.27 ppm, δ(13C) = 77.23 ppm, CD2Cl2: δ(1H) = 5.32 ppm, δ(13C) = 54.00 ppm, acetonitrile-d3: δ(1H) = 1.94 ppm, δ(13C) = 1.39 ppm, dmso-d6: δ(1H) = 2.50 ppm, and δ(13C) = 39.51 ppm). 15N NMR spectra are referenced to external neat nitromethane [δ(15N) = 0.0 ppm]. All 15N chemical shifts were obtained via 2D 15N, 1H HMBC experiment. The 125Te NMR chemical shifts are referenced to an external CDCl3 solution of Ph2Te2 [δ(125Te) = 422 ppm relative to Me2Te]. Solid-state NMR spectra of 125Te (Fig. S89†) were acquired on a JEOL 600 MHz spectrometer under magic-angle-spinning conditions at 18 kHz MAS frequency without active temperature regulation. Samples were packed into 3.2 mm zirconia rotors under an inert atmosphere and spun using N2. Spectra were acquired using direct excitation experiment (90° pulse of 3.5 us) with direct FID detection. Typical pre-acquisition delay was set 120 s. The isotropic shift was identified by comparison with spectra acquired at a slower MAS rate and referenced using an external sample of solid Te(OH)6 with δ(125Te) = 692.2 and 685.6 ppm (two independent molecules in the unit cell).40 High-resolution MALDI MS spectra were measured with a MALDI mass spectrometer LTQ Orbitrap XL (Thermo Fisher Scientific, Bremen, Germany) equipped with a nitrogen UV laser (337 nm, 60 Hz). The LTQ Orbitrap instrument was operated in positive-ion mode over a normal mass range (m/z 50–2000) with 100000 resolution at m/z = 400. The survey crystal positioning system (survey CPS) was set for the random choice of shot position by automatic crystal recognition. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as a matrix. Mass spectra were averaged over the whole MS record for all measured samples.
The bulk purity of tellurium(II) compounds was established by high-resolution MALDI MS spectra in combination with multinuclear NMR spectroscopy (see the ESI†). Unfortunately, telluronium cations could be characterized by multinuclear NMR spectroscopy only (see the ESI†) because their high sensitivity toward hydrolysis hampered our attempts to get satisfactory high-resolution MS spectra or combustion analysis.
Syntheses of starting compounds
N] ppm. 13C{1H} NMR (100.61 MHz, CDCl3): δ 24.4 [iPr–CH3]; 25.1 [iPr–CH3]; 28.6 [iPr–CH]; 124.2 [Ar–C]; 126.9 [Ar–C]; 129.0 [Ar–C]; 132.3 [Ar–C]; 133.1 [Ar–C]; 133.8 [Ar–C]; 135.1 [Ar–C], 138.9 [Ar–C]; 142.3 [Ar–C]; 144.1 [Ar–C]; 164.7 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CDCl3): δ −121.4 ppm. 125Te{1H} NMR (158 MHz, CDCl3): δ 1392 ppm. 125Te NMR (solid): δiso 1349 and 1403 ppm (two independent molecules in the unit cell). HRMS (MALDI) m/z calc. for C19H22N130Te: 394.0809 [M − Cl]+, found 394.0805.
N] ppm. 13C{1H} NMR (125.61 MHz, CD2Cl2): δ 32.4 [tBu–CH3]; 63.3 [tBu–C]; 129.3 [Ar–C]; 135.5 [Ar–C]; 136.6 [Ar–C], 136.8 [Ar–C]; 160.7 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD2Cl2): δ −91.2 ppm. 125Te{1H} NMR (158 MHz, CD2Cl2): δ 1394 ppm. HRMS (MALDI) m/z calc. for C19H22N130Te: 373.0918 [M − Br]+, found 394.0913.
N] ppm. 13C{1H} NMR (125.6 MHz, CDCl3): δ 24.3 [iPr–CH3]; 24.8 [iPr–CH3]; 28.8 [iPr–CH]; 124.5 [Ar–C]; 129.6 [Ar–C]; 130.4 [Ar–C]; 134.5 [Ar–C]; 138.6 [Ar–C]; 140.2 [Ar–C]; 141.3 [Ar–C], 141.6 [Ar–C]; 166.5 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CDCl3) δ −119.8 ppm. 125Te{1H} NMR (158 MHz, CDCl3) δ 2113 ppm. HRMS (MALDI) m/z calc. for C32H39N2130Te: 581.2170 [M − Br]+, found 581.2161.
General procedure for the synthesis of tellurium(II) cations
The particular tellurium precursor was mixed with 1 eq. of the corresponding silver salt AgX (X = OTf or SbF6) and dichloromethane was added to this mixture. The reaction mixture was stirred for 1 h while the reaction flasks were protected from ambient light by an aluminum foil. Then, the precipitated AgX (X = Cl or Br) was filtered and the filtrate was evaporated and the resulting solid was washed with hexane to give the target tellurium(II) cations. For particular loadings, yields and experimental data, see below.N] ppm. 13C{1H} NMR (100.61 MHz, CD2Cl2): δ 24.3 [iPr–CH3]; 25.0 [iPr–CH3]; 29.2 [iPr–CH]; 125.5 [Ar–C]; 129.4 [Ar–C]; 132.0 [Ar–C]; 132.5 [Ar–C]; 135.3 [overlap of two signals based on HSQC, Ar–C]; 136.7 [Ar–C]; 137.1 [Ar–C], 143.8 [Ar–C]; 150.7 [Ar–C]; 171.1 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD2Cl2) δ −174.9 ppm. 125Te{1H} NMR (158 MHz, CD2Cl2) δ 1392 ppm. HRMS (MALDI) m/z calc. for C19H22N130Te: 394.0809 [M − SbF6]+, found 394.0803.
N] ppm. 13C{1H} NMR (125.61 MHz, CD2Cl2): δ 32.3 [tBu–CH3]; 63.4 [tBu–C]; 121.6 [q, 1JF,C = 322 Hz, CF3]; 129.6 [Ar–C]; 135.3 [Ar–C]; 136.5 [Ar–C], 136.8 [Ar–C]; 160.0 [CHN] ppm. 19F{1H} NMR (376.5 MHz, CD2Cl2) δ −78.8 ppm. 15N NMR (via15N, 1H HMBC, CD2Cl2) δ −88.8 ppm. 125Te{1H} NMR (158 MHz, CD2Cl2) δ 1400 ppm. HRMS (MALDI) m/z calc. for C19H22N130Te: 373.0918 [M − CF3O3S]+, found 373.091.
N] ppm. 13C{1H} NMR (125.61 MHz, CD2Cl2): δ 32.3 [tBu–CH3]; 63.5 [tBu–C]; 129.7 [Ar–C]; 135.2 [Ar–C]; 136.3 [Ar–C], 137.1 [Ar–C]; 159.4 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD2Cl2) δ −89.5 ppm. 125Te{1H} NMR (158 MHz, CD2Cl2) δ 1409 ppm. 125Te NMR (solid): δiso 1388 ppm. HRMS (MALDI) m/z calc. for C19H22N130Te: 373.0918 [M − SbF6]+, found 373.0912.
N] ppm. 13C{1H} NMR (125.6 MHz, CD2Cl2): δ 24.5 [iPr–CH3]; 24.6 [iPr–CH3]; 29.2 [iPr–CH]; 125.0 [Ar–C]; 130.1 [Ar–C]; 130.7 [Ar–C]; 134.7 [Ar–C]; 138.8 [Ar–C]; 139.0 [Ar–C]; 142.0 [Ar–C], 142.8 [Ar–C]; 166.9 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD2Cl2) δ −117.4 ppm. 125Te{1H} NMR (158 MHz, CD2Cl2): δ 1557 ppm. HRMS (MALDI) m/z calc. for C32H39N2130Te: 581.2170 [M − SbF6]+, found 581.2156.
Oxidation of tellurium(II) cations by SO2Cl2
N] ppm. 13C{1H} NMR (125.61 MHz, CD3CN): δ 30.5 [tBu–CH3]; 65.2 [tBu–C]; 120.9 [q, 1JF,C = 319 Hz, CF3]; 132.0 [Ar–C]; 135.1 [Ar–C]; 135.5 [Ar–C], 136.5 [Ar–C]; 137.1 [Ar–C]; 143.8 [Ar–C]; 168.3 [CHN] ppm. 19F{1H} NMR (376.5 MHz, CD2Cl2) δ −79.2 ppm. 15N NMR (via15N, 1H HMBC, CD3CN) δ −86.8 ppm. 125Te{1H} NMR (158 MHz, CD3CN) δ 1373 ppm.
N] ppm. 13C{1H} NMR (125.61 MHz, CD3CN): δ 30.4 [tBu–CH3]; 65.5 [tBu–C]; 132.7 [Ar–C]; 135.8 [Ar–C]; 136.0 [Ar–C], 136.8 [Ar–C]; 137.5 [Ar–C]; 141.7 [Ar–C]; 168.6 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD3CN) δ −87.1 ppm. 125Te{1H} NMR (158 MHz, CD3CN) δ 1399 ppm.
N] ppm. 13C{1H} NMR (125.61 MHz, CD3CN): δ 30.6 [tBu–CH3]; 66.8 [tBu–C]; 121.5 [q, 1JF,C = 320 Hz, CF3]; 134.5 [Ar–C]; 136.0 [Ar–C]; 138.5 [Ar–C], 145.8 [Ar–C]; 163.2 [CHN] ppm. 19F{1H} NMR (376.5 MHz, CD3CN) δ −79.2 ppm. 15N NMR (via15N, 1H HMBC, CD3CN) δ −86.7 ppm. 125Te{1H} NMR (158 MHz, CD3CN) δ 1180 ppm.
N] ppm. 13C{1H} NMR (125.61 MHz, CD3CN): δ 30.7 [tBu–CH3]; 66.9 [tBu–C]; 134.5 [Ar–C]; 136.1 [Ar–C]; 138.6 [Ar–C], 146.0 [Ar–C]; 163.2 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD3CN) δ −87.0 ppm. 125Te{1H} NMR (158 MHz, CD3CN) δ 1179 ppm.
1H NMR (500 MHz, CD2Cl2): δ 1.20 [6H, d, 3JH,H = 6.7 Hz, iPr–CH3]; 1.30 [6H, d, 3JH,H = 6.7 Hz, iPr–CH3]; 2.84 [2H, sept, 3JH,H = 6.7 Hz, iPr–CH]; 7.37 [2H, d, 3JH,H = 7.8 Hz, Ar–H]; 7.47 [1H, t, 3JH,H = 7.8 Hz, Ar–H]; 8.11 [1H, t, 3JH,H = 7.8 Hz, Ar–H]; 8.20 [1H, t, 3JH,H = 8.0 Hz, Ar–H]; 8.25 [1H, d, 3JH,H = 7.5 Hz, Ar–H]; 8.78 [1H, d, 3JH,H = 8.0 Hz, Ar–H]; 8.83 [1H, s, CHN] ppm. 13C{1H} NMR (125.6 MHz, CD2Cl2): δ 24.2 [iPr–CH3]; 25.9 [iPr–CH3]; 30.0 [iPr–CH]; 125.7 [Ar–C]; 130.6 [Ar–C]; 134.5 [Ar–C]; 134.8 [Ar–C]; 136.2 [overlap of two signals based on HSQC, Ar–C]; 137.2 [Ar–C]; 138.1 [Ar–C]; 138.9 [Ar–C], 142.9 [Ar–C]; 170.3 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD2Cl2) δ −102.8 ppm. 125Te{1H} NMR (158 MHz, CD2Cl2) δ 1428 ppm.
1H NMR (500 MHz, CD2Cl2): δ 1.28 [12H, d, 3JH,H = 6.7 Hz, iPr–CH3]; 1.31 [12H, d, 3JH,H = 6.7 Hz, iPr–CH3]; 3.20 [4H, sept, 3JH,H = 6.7 Hz, iPr–CH]; 7.42 [4H, d, 3JH,H = 7.8 Hz, Ar–H]; 7.54 [2H, t, 3JH,H = 7.8 Hz, Ar–H]; 8.33 [1H, t, 3JH,H = 7.6 Hz, Ar–H]; 8.79 [2H, d, 3JH,H = 7.6 Hz, Ar–H]; 9.22 [2H, s, CHN] ppm. 13C{1H} NMR (125.6 MHz, CD2Cl2): δ 25.0 [iPr–CH3]; 25.6 [iPr–CH3]; 30.1 [iPr–CH]; 126.1 [Ar–C]; 131.5 [Ar–C]; 132.8 [Ar–C]; 136.1 [Ar–C]; 139.0 [Ar–C]; 140.6 [Ar–C]; 144.0 [Ar–C], 150.0 [Ar–C]; 168.2 [CHN] ppm. 15N NMR (via15N, 1H HMBC, CD2Cl2) δ −112.9 ppm. 125Te{1H} NMR (158 MHz, CD2Cl2) δ 1308 ppm.
Preparation of crystals of [COTeCl2]2O and [OCNtBuTeCl2]2O
O] ppm. 13C{1H} NMR (125.61 MHz, thf-d8): δ 130.4 [Ar–C]; 132.8 [Ar–C]; 134.9 [Ar–C], 136.6 [Ar–C]; 139.6 [Ar–C]; 151.5 [Ar–C]; 200.5 [CHO] ppm. 125Te{1H} NMR (158 MHz, thf-d8) δ 1405 ppm.
The NMR data for [COTeCl2]OH: 1H NMR (500 MHz, thf-d8/water): δ 7.75 [1H, t, 3JH,H = 7.4 Hz, Ar–H]; 8.00 [1H, t, 3JH,H = 7.4 Hz, Ar–H]; 8.17 [1H, d, 3JH,H = 7.3 Hz, Ar–H]; 8.38 [1H, d, 3JH,H = 7.5 Hz, Ar–H]; 10.22 [1H, s, CHO] ppm. 13C{1H} NMR (125.61 MHz, thf-d8/water): δ 127.2 [Ar–C]; 132.3 [Ar–C]; 134.3 [Ar–C], 136.3 [Ar–C]; 138.1 [Ar–C]; 150.2 [Ar–C]; 198.3 [CHO] ppm. 125Te{1H} NMR (158 MHz, d8/water) δ 1342 ppm.
N] ppm. 13C{1H} NMR (125.61 MHz, dmso-d6): δ 30.1 [tBu–CH3]; 61.3 [tBu–C]; 100.7 [CH]; 130.5 [Ar–C]; 132.0 [Ar–C]; 132.8 [Ar–C], 132.9 [Ar–C]; 142.6 [Ar–C]; 143.3 [Ar–C]; 157.3 [CHN] ppm. 125Te{1H} NMR (158 MHz, dmso-d6) δ 1381 ppm.
Computational methodology
Geometry optimizations of the isolated molecular structures were carried out using density functional theory (DFT) at the B3PW91/6-311+G(2df,p)41,42 level of theory using the Gaussian1643 software package. For the Sb and Te atoms, effective core potentials (ECP28MDF) and corresponding cc-pVTZ basis sets44 were used. Dispersion effects were modelled using Grimme's GD3BJ parameters.45 Wavefunction files were used for a topological analysis of the electron density according to the Atoms-In-Molecules partitioning scheme36 using AIMAll,46 whereas DGRID47 was used to generate and analyze the Electron-Localizability-Indicator (ELI-D)37 related real-space bonding descriptors applying a grid step size of 0.05 a.u. NBO39 analysis was carried out using the NBO6 software.48 The NCI grids were computed with NCIplot.49 AIM, ELI-D and NCI figures are displayed using Multiwfn 3.850 and VMD.51Crystallography
The X-ray data for colourless crystals of studied compounds were obtained at 150 K using an Oxford Cryostream low-temperature device with a Bruker D8-Venture diffractometer equipped with a Mo (Mo/Kα radiation; λ = 0.71073 Å) microfocus X-ray (IμS) source, and a photon CMOS detector was used for data collection. The obtained data were treated with XT-version 2019/1 and SHELXL-2019/1 software implemented in APEX3 v2016.9-0 (Bruker AXS) system.52The frames for all complexes were integrated with the Bruker SAINT software package using a narrow-frame algorithm. Data were corrected for absorption effects using the Multi-Scan method (SADABS). The structures were solved and refined using the Bruker SHELXTL software package. Hydrogen atoms were mostly localized on a difference Fourier map; however, to ensure uniformity of treatment of crystals, most of the hydrogen atoms were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2Ueq (pivot atom) or 1.5Ueq (methyl). H atoms in methyl, methine moieties and C–H of imine or located in aromatic rings were placed with C–H distances of 0.96, 0.97, 0.98 and 0.93 Å. In [NCNtBuTeCl2]OTf, there is an electron density hole between the tellurium atom and ipso carbon of the aromatic ring of ∼2.7 e− Å−3, which has no chemical sense.
Crystallographic data for all structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC 2254918 (CNtBuTeCl2OTf), 2254919 (CNDippTeCl), 2254920 ([COTeCl2]2O), 2254921 ([NCNtBuTeCl2]SbF6), 2254922 ([NCNtBuTeCl2]OTf), 2254923 ([NCNtBuTe]SbF6), 2254924 ([NCNtBuTe]Br), 2254925 ([CNDippTe]SbF6), 2254926 ([OCNtBuTeCl2]2O) and 2254927 ([NCNDippTe]SbF6).†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Kateřina Drábková and Thi Dieu Linh Do for their experimental contribution. L. D. is thankful to the Czech Science Foundation project no. 22-17230S for financial support.References
- T. Chivers and R. S. Laitinen, Chem. Soc. Rev., 2015, 44, 1725–1739 RSC.
- For recent examples see: (a) A. Arora, P. Oswal, D. Sharma, S. Purohit, A. Tyagi, P. Sharma and A. Kumar, Dalton Trans., 2022, 51, 17114–17144 RSC; (b) A. Arora, P. Oswal, A. Datta and A. Kumar, Coord. Chem. Rev., 2022, 459, 214406 CrossRef; (c) A. Pop, C. Silvestru and A. Silvestru, Phys. Sci. Rev., 2019, 4 Search PubMed, UNSP 20180061; (d) V. K. Jain, Dalton Trans., 2020, 49, 8817–8835 RSC.
- A. Gupta, S. Kumar and H. B. Singh, Proc. Natl. Acad. Sci., India, Sect. A, 2016, 86, 465–498 CrossRef.
- (a) J. Jenske, W. W. du Mont, F. Ruthe, P. G. Jones, L. M. Mercuri and P. Deplano, Eur. J. Inorg. Chem., 2000, 1591–1599 CrossRef; (b) J. Jenske, W. W. du Mont and P. G. Jones, Angew. Chem., Int. Ed. Engl., 1997, 36, 2219–2221 CrossRef; (c) K. Sugamata, T. Sasamori and N. Tokitoh, Eur. J. Inorg. Chem., 2012, 755–778 Search PubMed; (d) C. Kollemann and F. Sladky, J. Organomet. Chem., 1990, 396, C1–C3 CrossRef; (e) J. Beckmann, J. Bolsinger, A. Duthie, P. Finke, E. Lork, C. Ludtke, O. Mallow and S. Mebs, Inorg. Chem., 2012, 51, 12395–12406 CrossRef PubMed; (f) J. Beckmann, P. Finke, S. Heitz and M. Hesse, Eur. J. Inorg. Chem., 2008, 1921–1925 CrossRef; (g) S. Yadav, S. Raju, H. B. Singh and R. J. Butcher, Dalton Trans., 2016, 45, 8458–8467 RSC.
- (a) H. Fujihara, H. Mima and N. Furukawa, J. Am. Chem. Soc., 1995, 117, 10153–10154 CrossRef; (b) A. Beleaga, V. A. Bojan, A. Pollnitz, C. I. Rat and C. Silvestru, Dalton Trans., 2011, 40, 8830–8838 RSC; (c) A. Gupta, R. Deka, H. B. Singh and R. J. Butcher, New J. Chem., 2019, 43, 13225–18838 RSC; (d) V. Rani, M. Boda, S. Raju, G. Naresh Patwari, H. B. Singh and R. J. Butcher, Dalton Trans., 2018, 47, 9114–9127 RSC.
- Examples of [R3Te]+: (a) J. Jeske, W. W. du Mont and P. G. Jones, Angew. Chem., Int. Ed. Engl., 1996, 35, 2653–2655 CrossRef; (b) F. R. Knight, R. A. M. Randall, K. S. A. Arachchige, L. Wakefield, J. M. Griffin, S. E. Ashbrook, M. Bühl, A. M. Slawin and J. D. Woollins, Inorg. Chem., 2012, 51, 11087–11097 CrossRef PubMed; (c) T. P. Lin and F. P. Gabbaï, Angew. Chem., Int. Ed., 2013, 52, 3864–3868 CrossRef PubMed.
- Examples of [R2TeX]+: (a) J. Beckmann, J. Bolsinger, A. Duthie and P. Finke, Organometallics, 2011, 31, 238–245 CrossRef; (b) J. Beckmann, J. Bolsinger, A. Duthie and P. Finke, Dalton Trans., 2013, 42, 12193–12202 RSC; (c) J. Beckmann, D. Dakternieks, A. Duthie, N. A. Lewcenko, C. Mitchell and M. Schürmann, Z. Anorg. Allg. Chem., 2005, 631, 1856–1862 CrossRef; (d) M. R. Detty, A. J. Williams, J. M. Hewitt and M. McMillan, Organometallics, 1995, 14, 5258–5262 CrossRef.
- Note that also [TeX3]+ were reported for example see: T. A. Engesser, P. Hrobárik, N. Trapp, P. Eiden, H. Scherer, M. Kaupp and I. Krossing, ChemPlusChem, 2012, 77, 643–651 CrossRef and references cited therein.
- R. Deka, A. Gupta, A. Sarkar, R. J. Butcher and H. B. Singh, Eur. J. Inorg. Chem., 2020, 47, 4170–4179 CrossRef.
- A. B. Bergholdt, K. Koayashi, E. Horn, O. Takahashi, S. Sato, N. Furukawa, M. Yokoyama and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 1230–1236 CrossRef.
- A. Kozma, J. Petuškova, C. W. Lehmann and M. Alcarazo, Chem. Commun., 2013, 49, 4145–4147 RSC.
- For recent examples see: (a) B. Zhou and F. P. Gabbaï, J. Am. Chem. Soc., 2021, 143, 8625–8630 CrossRef PubMed; (b) B. Zhou and F. P. Gabbaï, Organometallics, 2021, 40, 2371–2374 CrossRef; (c) B. Zhou and F. P. Gabbaï, Chem. Sci., 2020, 11, 7495–7500 RSC; (d) S. Benz, A. I. Poblador-Bahamonde, N. Low-Ders and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 5408–5412 CrossRef PubMed; (e) P. Wonner, A. Dreger, L. Vogel, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2019, 58, 16923–16927 CrossRef PubMed; (f) R. Weiss, E. Aubert, P. Pale and V. Mamane, Angew. Chem., Int. Ed., 2021, 60, 19281–19286 CrossRef PubMed; (g) P. Wonner, T. Steinke, L. Vogel and S. M. Huber, Chem. – Eur. J., 2020, 26, 1258–1262 CrossRef PubMed; (h) L. Groslambert, A. Padilla-Hernandez, R. Weiss, P. Pale and V. Mamane, Chem. – Eur. J., 2023, e202203372 CrossRef PubMed; (i) T. Steinke, P. Wonner, E. Engelage and S. M. Huber, Synthesis, 2021, 53, 2043–2050 CrossRef.
- K. Srivastava, P. Shah, H. B. Singh and R. J. Butcher, Organometallics, 2011, 30, 534–546 CrossRef and references cited therein.
- For example see: (a) N. W. Alcock and W. D. Harrison, J. Chem. Soc., Dalton Trans., 1982, 709–712 RSC; (b) J. Beckmann, D. Dakternieks, A. Duthie, F. Ribot, M. Schürmann and N. A. Lewcenko, Organometallics, 2003, 22, 3257–3261 CrossRef; (c) M. Oba, Y. Okada, M. Endo, K. Tanaka, K. Nishiyama, S. Shimada and W. Ando, Inorg. Chem., 2010, 49, 10680–10686 CrossRef PubMed.
- T. M. Klapötke, B. Krumm and M. Scherr, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 1347–1354 CrossRef.
- A. Gupta, R. Deka, S. Raju, H. B. Singh and R. J. Butcher, J. Organomet. Chem., 2019, 894, 10–17 CrossRef.
- (a) K. Kobayashi, N. Deguchi, O. Takahashi, K. Tanaka, E. Horn, O. Kituchi and N. Furukawa, Angew. Chem., Int. Ed., 1999, 38, 1638–1640 CrossRef; (b) K. Takagi, N. Sakakibara, S. Kikkawa and S. Tsuzuki, Chem. Commun., 2021, 51, 13736–13739 RSC.
- (a) J. Beckmann, J. Bolsinger and A. Duthie, Chem. – Eur. J., 2011, 17, 930–940 CrossRef PubMed and references cited therein; (b) J. Beckmann, A. Duthie, T. M. Gesing, T. Koehne and E. Lork, Organometallics, 2012, 31, 3451–3454 CrossRef; (c) J. Beckmann, P. Finke, M. Hesse and B. Wettig, Angew. Chem., Int. Ed., 2008, 47, 9982–9984 CrossRef PubMed; (d) M. Oba, K. Nishiyama, S. Koguchi, S. Shimada and W. Ando, Organometallics, 2013, 32, 6620–6623 CrossRef; (e) K. Srivastava, S. Sharma, H. B. Singh, U. P. Singh and R. J. Butcher, Chem. Commun., 2010, 46, 1130–1132 RSC.
- R. Deka, A. Sarkar, R. J. Butcher, P. C. Junk, D. R. Turner, G. B. Deacon and H. B. Singh, Dalton Trans., 2020, 49, 1173–1180 RSC.
- J. Beckmann, J. Bolsinger, P. Finke and M. Hesse, Angew. Chem., Int. Ed., 2010, 49, 8030–8032 CrossRef PubMed.
- (a) J. Beckmann, J. Bolsinger, A. Duthie and P. Finke, Organometallics, 2012, 31, 289–293 CrossRef; (b) A. Gupta, R. Deka, A. Sarkar, H. B. Singh and R. J. Butcher, Dalton Trans., 2019, 48, 10979–10985 RSC.
- M. Oba, Y. Okada, K. Nishiyama, S. Shimada and W. Ando, Chem. Commun., 2008, 5378–5380 RSC.
- M. Hejda, D. Duvinage, E. Lork, A. Lyčka, S. Mebs, L. Dostál and J. Beckmann, Organometallics, 2020, 39, 1202–1212 CrossRef.
- M. Hejda, D. Duvinage, E. Lork, A. Lyčka, Z. Černošek, J. Macháček, S. Makarov, S. Ketkov, S. Mebs, L. Dostál and J. Beckmann, Chem. – Eur. J., 2021, 27, 14577–14581 CrossRef PubMed.
- M. Hejda, E. Lork, S. Mebs, L. Dostál and J. Beckmann, Eur. J. Inorg. Chem., 2017, 3435–3445 CrossRef.
- N. Azizi, F. Aryanasab and M. R. Siadi, Org. Lett., 2006, 8, 5272–5277 Search PubMed.
- S. S. Zade, H. B. Singh and R. J. Butcher, Angew. Chem., Int. Ed., 2004, 43, 4513–4515 CrossRef PubMed.
- V. P. Singh, H. B. Singh and R. J. Butcher, Chem. - Asian J., 2011, 6, 1431–1442 CrossRef PubMed.
- Unfortunately, an attempt to add additional water to this mixture of [OCNtBuTeCl2]2O and [(OH)CNtBuTeCl2] (as in the case of [COTeCl2]2O to give [COTeCl2](OH) resulted in an uncontrolled further hydrolysis of the sample only.
- For comparison with the Σcov see: P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- (a) M. Si, J. Yan, Y. Ding and H. Huang, Org. Biomol. Chem., 2022, 20, 3917–3921 RSC; (b) E. Oloughlin and E. J. Valente, J. Chem. Crystallogr., 2019, 49, 193–205 CrossRef.
- (a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef; (b) M. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef.
- (a) L. Greb, Chem. – Eur. J., 2018, 24, 17881–17896 CrossRef PubMed; (b) P. Erdmann and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef PubMed.
- For recent examples see: (a) A. L. Liberman-Martin, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 5328–5331 CrossRef PubMed; (b) B. Pan and F. P. Gabbaï, J. Am. Chem. Soc., 2014, 136, 9564–9567 CrossRef PubMed; (c) H. Grosskappenberg, M. Reissmann, M. Schmidtmann and T. Müller, Organometallics, 2015, 34, 4952–4958 CrossRef; (d) J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC and references cited therein; (e) D. Hartmann, M. Schädler and L. Greb, Chem. Sci., 2019, 10, 7379–7388 RSC; (f) F. S. Tschernuth, T. Thorwart, L. Greb, F. Hanusch and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 25799–25803 CrossRef PubMed; (g) A. T. Henry, T. P. L. Cosby, P. D. Boyle and K. M. Baines, Dalton Trans., 2021, 50, 15906–15913 RSC; (h) R. Kannan, S. Kumar, A. P. Andrews, E. D. Jemmins and A. Venugopal, Inorg. Chem., 2017, 56, 9391–9395 CrossRef PubMed; (i) M. O. Akram, J. R. Tidwell, J. L. Dutton and C. D. Martin, Angew. Chem., Int. Ed., 2022, 61, e202212073 CrossRef PubMed; (j) L. A. Körte, J. Schwabedissen, M. Soffner, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy, B. Neumann, H. G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2017, 56, 8578–8582 CrossRef PubMed; (k) D. Roth, J. Stirn, D. W. Stephan and L. Greb, J. Am. Chem. Soc., 2021, 143, 15845–15851 CrossRef PubMed; (l) A. Hanft, K. Radacki and C. Lichtenberg, Chem. – Eur. J., 2021, 27, 6230–6239 CrossRef PubMed; (m) C. P. Hsu, C. A. Liu, C. C. Wen, Y. H. Liu, Y. F. Lin and C. W. Chiu, ChemCatChem, 2022, 14, e202101715 CrossRef; (n) R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717–1720 CrossRef PubMed.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2023, 341, 1374–1377 CrossRef PubMed.
- R. W. F. Bader, Atoms in Molecules. A Quantum Theory, Cambridge University Press, Oxford U.K., 1991 Search PubMed.
- (a) M. Kohout, Int. J. Quantum Chem., 2004, 97, 651–658 CrossRef; (b) M. Kohout, F. R. Wagner and Y. Grin, Theor. Chem. Acc., 2008, 119, 413–420 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 Search PubMed.
- M. J. Collins, A. J. Ripmeester and J. F. Sawyer, J. Am. Chem. Soc., 1987, 109, 4113–4115 CrossRef.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef; (b) J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef PubMed.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef; (b) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 (RevisionC.01), Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- (a) B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef; (b) K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef; (c) K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- A. Todd, AIMAll (Version 15.09.27), Keith, TK Gristmill Software, Overland Park KS, USA, 2015 (aim.tkgristmill.com).
- M. Kohout, DGrid, version 5.1, Dresden, 2019 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013 Search PubMed.
- J. Contreras-García, E. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR, Raman and mass spectra, crystallographic data and details for theoretical studies. CCDC 2254918–2254927. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02404k |
| This journal is © The Royal Society of Chemistry 2023 |