
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insight into cobalt-mediated [2 + 2 + 2]-cycloaddition reactions with γ-alkylidenebutenolide and γ-alkylidenebuterolactam as 2π partners†
Léo
Chaussy‡
 ,
Marion
Delorme‡
,
Alexander
Punter
,
Yannick
Carissan
,
Jean-Luc
Parrain
,
Muriel
Amatore
*,
Paola
Nava
* and
Laurent
Commeiras
*
,
Marion
Delorme‡
,
Alexander
Punter
,
Yannick
Carissan
,
Jean-Luc
Parrain
,
Muriel
Amatore
*,
Paola
Nava
* and
Laurent
Commeiras
*
Aix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: laurent.commeiras@univ-amu.f; paola.nava@univ-amu.fr; muriel.amatore@univ-amu.fr
First published on 21st September 2023
Abstract
The molecular complexity of recently reported cobalt(III) polycyclic complexes, resulting from an intramolecular formal (2 + 2 + 3) cycloaddition reaction on an enediyne containing a lactone moiety, has prompted us to computationally review the mechanisms of cobalt cycloaddition reactions with γ-alkylidenebutenolide or γ-alkylidenebuterolactam as 2π partners. Computed mechanisms are compared, leading to either cobalt(III)- or cobalt(I)-spiro complexes depending of both the nature of the reaction (intra- vs. intermolecular pathway) and the nature of the 2π partner (γ-alkylidenebutenolide vs. γ-alkylidenebuterolactam). These proposed mechanisms are supported by experiments, allowing us to report the synthesis and characterization of the predicted compounds.
Introduction
Among synthetic strategies, metal-catalysed cyclotrimerisations of alkynes represent elegant approaches to functionalised aromatic and heteroaromatic polycyclic systems.1 [2 + 2 + 2] cycloaddition reactions are also convenient solutions for the synthesis of cyclohexadiene cores when two alkynes and one alkene are engaged as 2π partners in the process.2 Among active species for metal-mediated or metal-catalysed [2 + 2 + 2] cycloaddition reactions, CpCoL2 (L = CO, PR3, alkenes) complexes have proven their efficiency both for the synthesis of arenes and cyclohexadienes. In this context, the general mechanism of the cobalt-catalysed process involving two alkynes and one alkene has been proposed based on both experimental and computational studies.3–5 Recently, targeting a standard cobalt-catalysed [2 + 2 + 2] cycloaddition to γ-spiro-γ-lactones, we have eventually reported the intriguing reactivity of enediynes featuring γ-alkylidenebutenolide compounds (Scheme 1). Due to the electronic specificities of the employed enediynes, a serendipitous Co(I)-mediated formal (2 + 2 + 3) cycloaddition reaction occurs allowing the highly regio- and diastereoselective formation of original cobalt(III) complexes. Preliminary computational studies have revealed an unusual mechanistic pathway relying on a fragmentation of the butenolide moiety (Scheme 1).6 More precisely, after the oxidative coupling from IO, coordination of the alkene moiety would readily occur to give IIO, due to its proximity to the metal centre, followed by the insertion step leading to IIIO. At this point, the mechanism differs from that of the classic [2 + 2 + 2] cycloaddition reaction. Instead of a reductive elimination step leading to the cobalt-complex-γ-spiro-γ-lactones P2O, a 7-membered ring would be generated through the formation of the C2–C14 bond. This would cause the spontaneous fragmentation of the lactone, resulting into the formation of the crucial β-oxo-ketene intermediate IVO. From here, after a conformational change, a final step involving Co–C15 and C8–C16 bond formations would lead to the Co(III) complex P1O.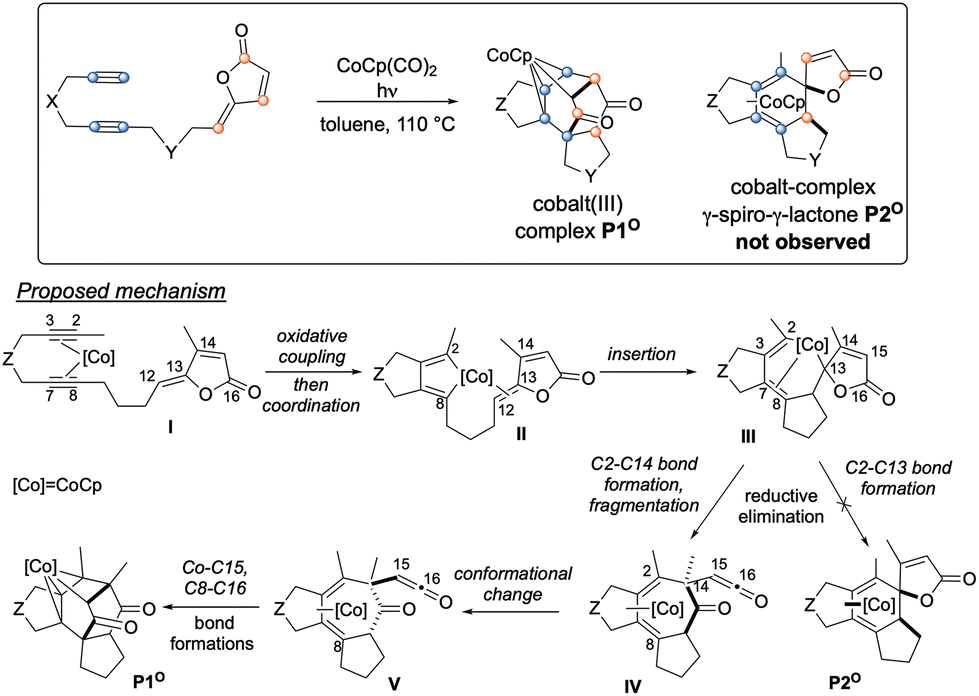 | ||
| Scheme 1 Proposed mechanism for the cobalt-mediated formal (2 + 2 + 3) reaction. | ||
The molecular complexity of these unprecedented cobalt(III) complexes pushed us to perform further theoretical calculations by comparing intra- versus intermolecular approaches (Fig. 1a), and by computing the outcomes of a related Co(I)-mediated reaction employing γ-alkylidenebuterolactam as 2π partner, instead of γ-alkylidenebutenolide (Fig. 1b). The computational studies are systematically supported by experiments, which enable us to report the synthesis of new compounds.
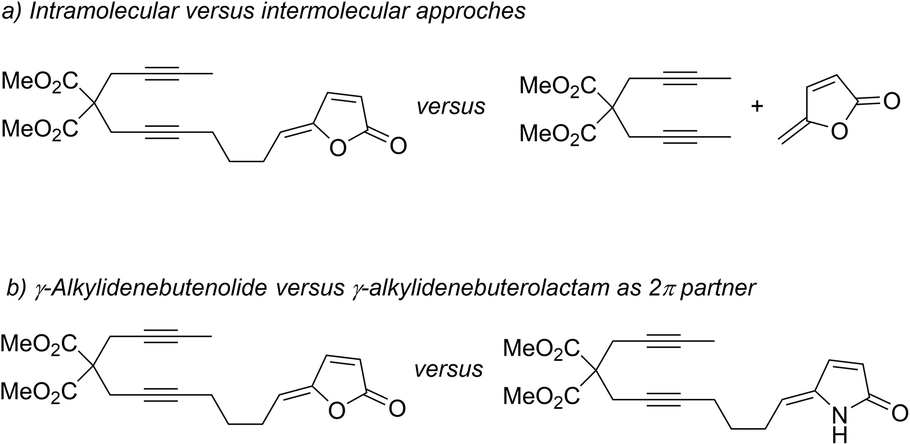 | ||
| Fig. 1 Computational and experimental case studies. | ||
Results and discussion
Intermolecular vs. intramolecular approaches
We first computationally studied the [2 + 2 + 2] cycloaddition reaction in an intermolecular fashion between the diyne derivative (2π + 2π partner) and the γ-alkylidenebutenolide (2π partner). The initial steps, which lead to the coordination of the alkene moiety to the cobaltacyclopentadiene, have been previously investigated for similar systems.3,4,7 Notably, it has been already shown that the initial oxidative coupling between the two alkyne moieties leading to an intermediate of type II is preferred over a process implicating an oxidative coupling between a triple and a double bond.3b Therefore, the ΔG profiles (at 383.15 K) have been computed from intermediates of type II, obtained after the oxidative coupling and the coordination of the exo-cyclic double bond (Scheme 1). On the basis of Density Functional Theory calculations (DFT), it has been suggested that the final reductive elimination step of the [2 + 2 + 2] cycloaddition reaction would occur on a triplet potential energy surface. However, recent ab initio calculations reveal that the hopping on the triplet state surface is not necessary: the whole reaction can take place on the singlet potential energy surface.5 Thus, no inter system crossing is to be invoked.In the intermolecular reactivity, four coordination approaches are possible between the cobaltacyclopentadiene and the two double bonds of the γ-alkylidenebutenolide (Fig. 2): either the exo- and endo-approaches with the exo-cyclic double bond, or the exo- and endo-approaches with the endo-cylic double bond. The energy of IIexo was chosen as reference.
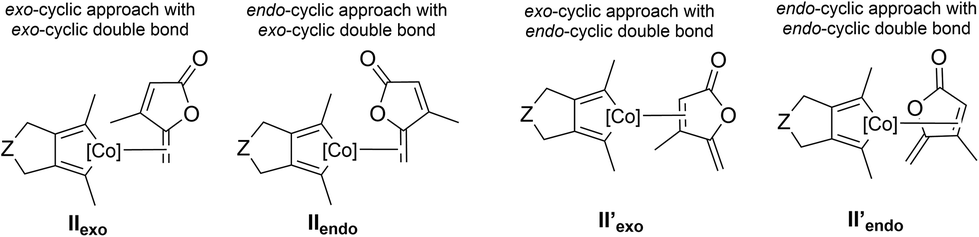 | ||
| Fig. 2 Different coordination modes for the intermolecular approach. | ||
From IIexo (Fig. 3 left), two regioselectivities can be envisioned for the insertion step of the exo-cyclic double bond: either the C2–C10 bond is formed to provide IIIexo1 (orange path), preventing the formation of the diketone product, or the C8–C9 bond is created to obtain IIIexo2 (blue path). By following the orange pathway, intermediate IIIexo1 evolves to the spirolactone P2exo product through a transition state located at only 13.7 kcal mol−1. The transition state leading to IIIexo2 is sterically more accessible and thus lower in energy (7.8 kcal mol−1) than the one leading to IIIexo1 (10.5 kcal mol−1). This accessibility results in intermediates that are significantly different in energy, with IIIexo2 more stable. From IIIexo2 two pathways can be considered: a reductive elimination step implicating the formation of the C2–C10 bond, would lead to the spirolactone P2exo product, while the creation of the C2–C11 bond would cause the spontaneous fragmentation of the lactone. Two further steps are necessary to obtain the Co(III) complex P1exo. These two transition states are similar in energy, however barriers from IIIexo2 are quite high. Therefore, overall, even if the insertion step apparently would favour the process through IIIexo2, the approach through IIIexo1 is preferred, thus excluding the possibility to form P1exo.
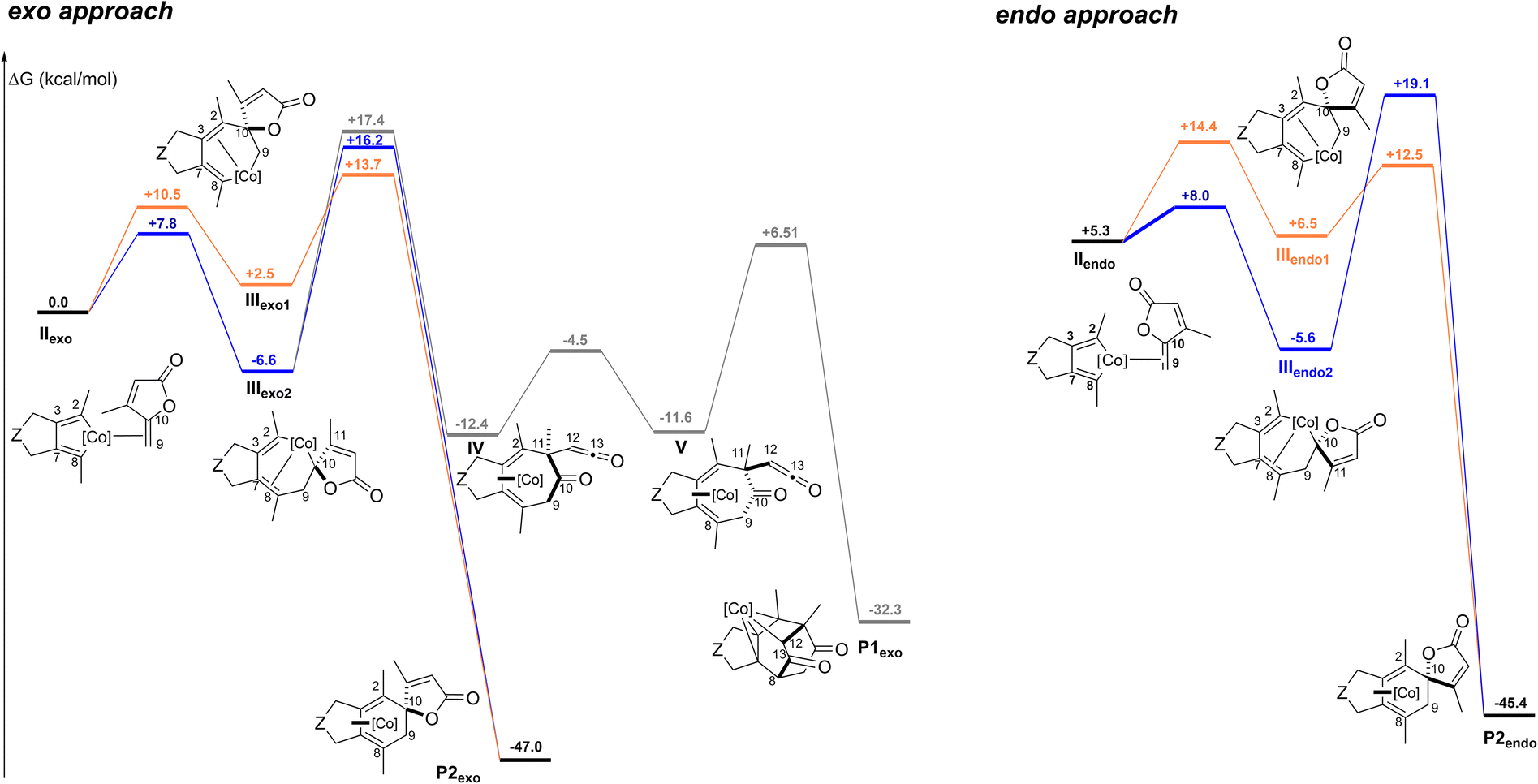 | ||
| Fig. 3 Computed exo- and endo-pathways. Z = C(CO2Me)2, [Co] = CpCo. ΔG are computed at the TPSS-D3/def2-TZVP level (T = 383.15 K). Solvent effects are added as single point calculations with the COSMO implicit model (toluene). | ||
In the pathway resulting from an endo approach (Fig. 3 right), two different insertion positions are again formally possible, with the creation of either the C2–C10 bond to IIIendo1 (orange path), or the C8–C9 bond to IIIendo2 (blue path). In both cases, only the spirolactone P2endo product could be obtained. The obtention of the Co(III) complex would require the formation of the C2–C11 bond. However, in IIIendo2 the C11 is not correctly positioned face to C2. As for the exo approach, even if the insertion step is less accessible from the lactone side (orange path), the pathway through this approach is overall favoured, as the barrier for the reductive elimination is significantly lower.
We also computationally studied the coordination of the cobaltacyclopentadiene with the endo-cyclic double bond of the γ-alkylidenebutenolide (II′exo and II′endo, Fig. 2). The system evolves towards the coordination of the cobaltacyclopentadiene with the exo-cyclic double bond of the 2π partner.
Our calculations demonstrate that the Co(III) complex is not accessible in the intermolecular version of the reaction. The overall costs to the γ-spiro-γ-lactone products P2exo and P2endo are similar: the highest transition states are located at 13.7 and 14.4 kcal mol−1 (less than 1 kcal mol−1 difference) for the exo and endo approaches, respectively, suggesting that the two products could be obtained. In both cases the insertion step through the lactone side (orange path) leads to an intermediate with an accessible transition state for the reductive elimination step, preventing the possibility to form the β-oxo-ketene intermediate. This is not the case in the intramolecular approach. The presence of the tether between the diyne and the γ-alkylidenebutenolide facilitates the coordination of the alkene moiety by imposing an exo-like approach (Scheme 1). The resulting intermediate III has a connectivity similar to that of IIIexo2, which differs from that of IIIexo1. Importantly, only a connectivity as in IIIexo2 allows to access to the Co(III) complex (see ESI† for details on the structures).
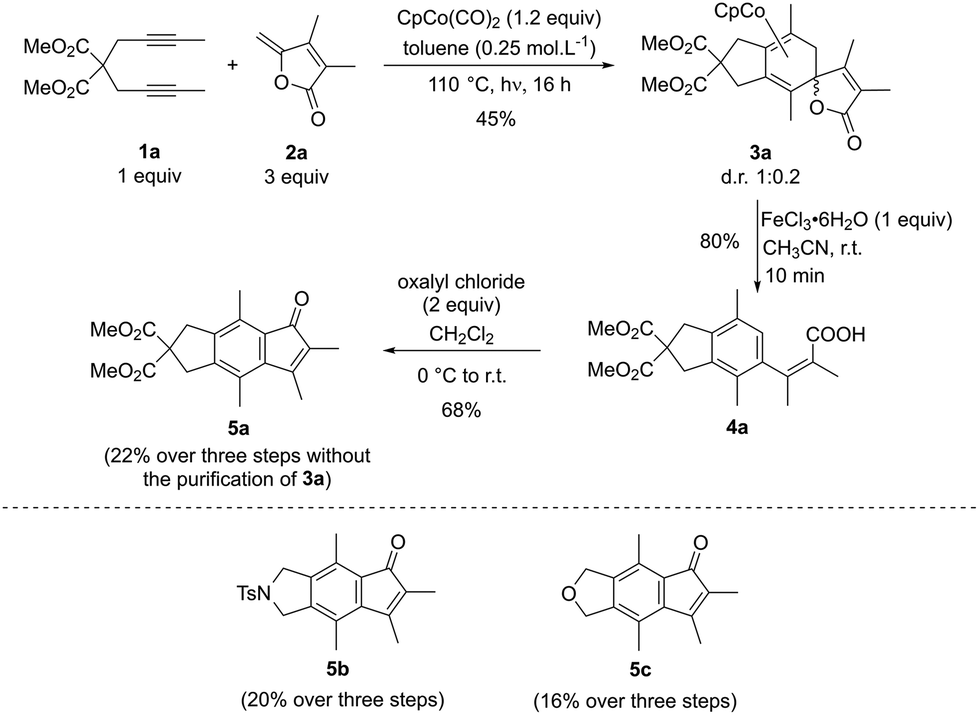
Experiments were then performed to corroborate the computational studies. Slightly reoptimized conditions developed for the intramolecular formation of Co(III) complex were applied for the intermolecular strategy (Scheme 2). The diyne partner 1a (1 equiv.) and the γ-alkylidenebutenolide 2a (3 equiv.) were exposed to a stoichiometric amount of CpCo(CO)2 (1.2 equiv.) in toluene (0.25 mol L−1) at 110 °C under irradiation (halogen 400 W) for one night. In these conditions, the formation of the predicted tricyclic spirolactone cobalt(I) complex 3a was observed and isolated in 45% yield as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 mixture of two diastereomers. As the γ-alkylidenebutenolide employed displays two methyl substituents, it slightly differs from the lactone in Fig. 3. Calculations were therefore performed with the doubly substituted substrate, as well. The computed profiles (in the ESI†) are sensibly the same as those reported in Fig. 3, allowing to draw the same conclusion. The formation of the γ-spiro-γ-lactone cobalt complex is thus favoured over that of the Co(III) complex and the experimental result validates the conclusions of the theoretical study.
:0.2 mixture of two diastereomers. As the γ-alkylidenebutenolide employed displays two methyl substituents, it slightly differs from the lactone in Fig. 3. Calculations were therefore performed with the doubly substituted substrate, as well. The computed profiles (in the ESI†) are sensibly the same as those reported in Fig. 3, allowing to draw the same conclusion. The formation of the γ-spiro-γ-lactone cobalt complex is thus favoured over that of the Co(III) complex and the experimental result validates the conclusions of the theoretical study.
 | ||
| Scheme 2 Experimental evidence and post-functionalisation. | ||
To enhance the synthetic potential of this tricyclic spirolactone cobalt complex 3a, we then investigated its oxidative demetallation. When 3a is submitted to 1 equivalent of FeCl3·6H2O in acetonitrile, carboxylic acid 4a was isolated in 80%, instead of the expected γ-spiro-γ-lactone compound. The formation of 4a could be explained by a β-elimination step, after the decomplexation of the cobalt, which is favoured by the aromatisation of the 6-membered ring. It is worth noting that 4a could be obtained in similar yield (33%), without the purification of the spirolactone cobalt complex 3a. Finally, the indanone 5a could be obtained in 68% yield when 4a is treated with an excess of oxalyl chloride.
This sequential three-step synthesis was applied with two other diynes (Z = NTs and Z = O), providing the corresponding indenones 5b–5c in 20 and 16% yields respectively.
Lactone vs. lactam
Next, we investigated the outcome of the intramolecular cycloaddition reaction in the case where the lactone moiety is replaced by a lactam one. In the reactivity to the Co(III) complexes (Scheme 1), the insertion of the exo-cyclic double bond, the C12–C14 bond formation and the fragmentation of the lactone to the β-oxo-ketene are accompanied by the creation of two C–C σ-bonds, thus implying that two sp2 carbon atoms, (C12 and C14, Scheme 1) are converted into sp3 carbon atoms. To mimic this transformation, from sp2 to sp3 carbon atoms, two hypothetical structures were constructed by the addition of CH4 (an H atom onto C12 and a CH3 group onto C14) to both γ-alkylidenebutenolide and γ-alkylidenebuterolactam (Fig. 4a and b).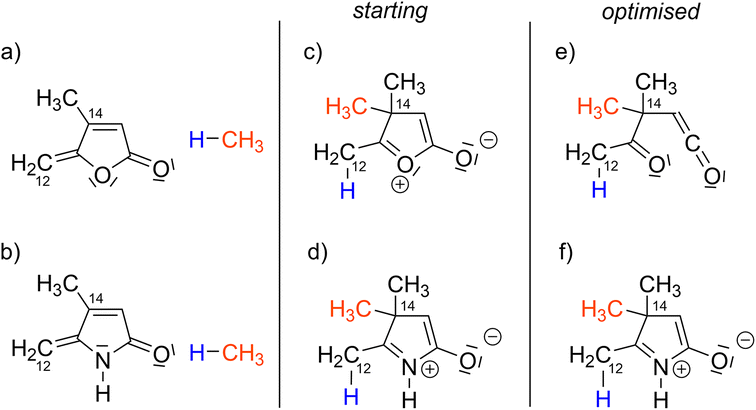 | ||
| Fig. 4 Model reactivity. ‘Starting’ denotes the initial structures and ‘Optimised’ denotes the structure obtained after the geometry optimisations (PBE0/def2-TZVP). | ||
One the one hand, the Lewis structure of the lactone-derived system (Fig. 4c) displays an oxygen atom carrying a positive charge and the other oxygen atom carrying a negative charge. This charge separation suggests that it would not be stable. Indeed, a geometry optimisation was performed, and a spontaneous cleavage of the C–O bond is observed (Fig. 4e). On the other hand, the Lewis structure for the lactam-derived system (Fig. 4d) presents a charge separation, which is compatible with the electronegativity of the atoms, the nitrogen atom being formally charged plus and the oxygen minus. The geometry optimisation did not show the cleavage of the C–N bond (Fig. 4f). In this case, the fragmentation of the lactam ring is not spontaneous, which suggests that its transformation to β-imino-ketene moiety is less accessible than for the lactone moiety.
To verify this hypothesis, the mechanisms of the [2 + 2 + 2] cycloaddition reactions with a lactam moiety have been compared to those obtained with the lactone (Fig. 5). For clarity reason, we report on top of Fig. 5 the computed mechanism for the lactone case. The overall picture of this mechanism is the same as the one presented in Scheme 1 and previously reported, although ΔG values differ slightly, since we employ here a different level of calculations (see the computational details).6 Moreover, we have detailed in the present work the coordination step from IIaO to IIO through TsOII that was not reported in our previous contribution.
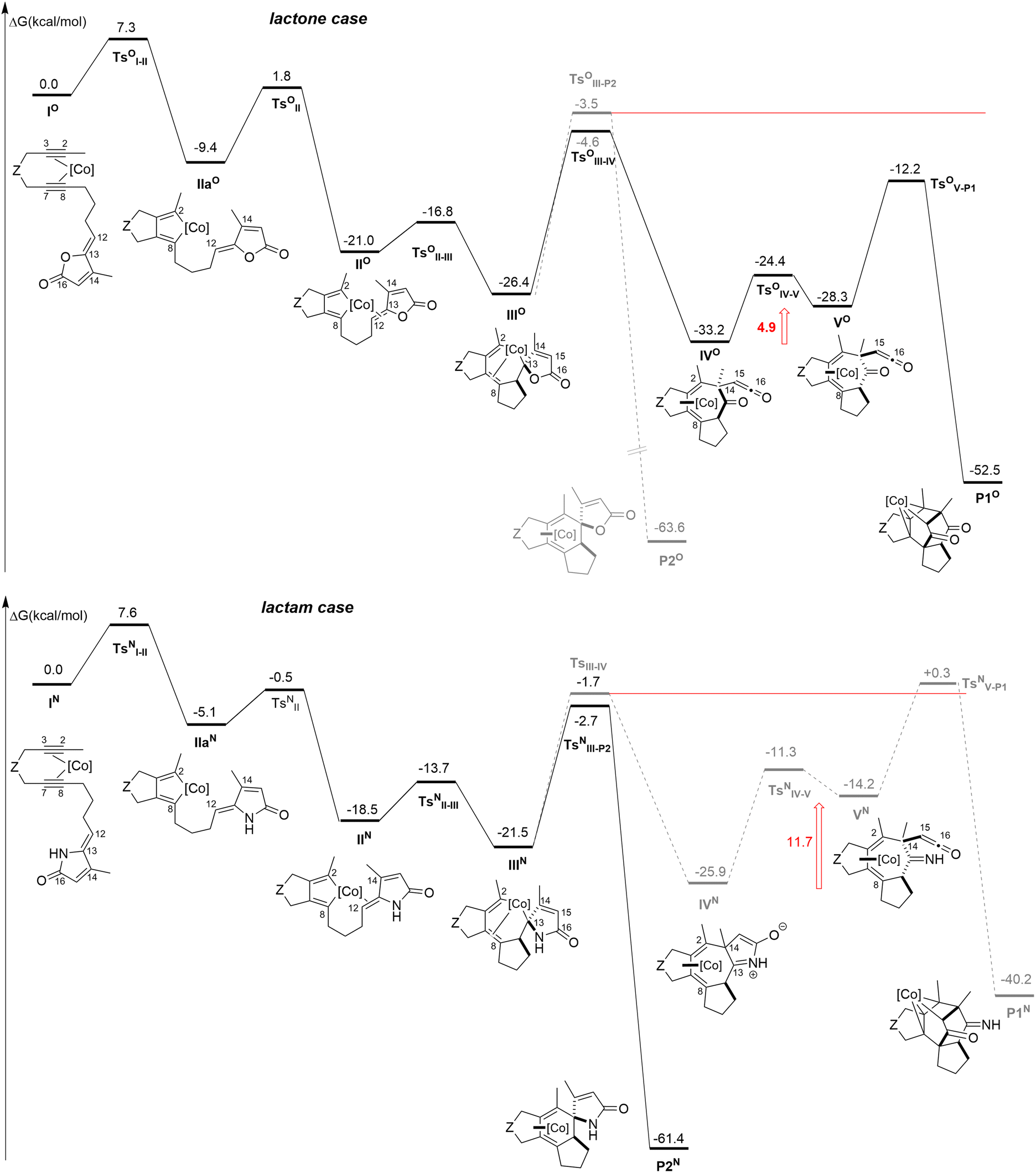 | ||
| Fig. 5 Computed pathways for the possible intramolecular reactions. On top, lactone case, and lactam case at the bottom, Z = C(C(O)OMe)2, [Co] = CpCo. ΔG are computed at the TPSS-D3/def2-TZVP level (T = 383.15 K). Solvent effects are added as single point calculations with the COSMO implicit model (toluene). | ||
Let us now describe the computed pathways for the lactam case. From IN, the pathway would be the following: the oxidative coupling takes place leading to IIaN in a singlet state. The triplet state of IIaN is lower in energy (ΔΔG = ΔG(1IIaN) − ΔG(3IIaN) = 5.2 kcal mol−1), however the coordination of the alkene moiety to the metal is not possible on the triplet potential energy surface, while this coordination is readily accessible on the singlet state to IIN. The insertion step would give IIIN.
From IIIN, the two pathways are considered. Notably, the transition state leading to the γ-spiro-γ-lactam cobalt complex (TsNIII-P2 to P2N) is slightly lower than the one leading to the formation of the C2–C14 bond (TsNIII-IV to IVN). Interestingly and as predicted by the model (Fig. 4), the lactam ring does not open after TsNIII-IV to IVN. The following transformation, implying the fragmentation of the lactam moiety, would give an intermediate that is 11.7 kcal mol−1 higher than IVN and the transition state leading to the hypothetical Co(III) product (TsNV-P1 to P1N) is even higher than TsNIII-IV.
Finally, in the case of the lactam, the mechanisms of the [2 + 2 + 2] cycloaddition reaction is preferred over that leading to the P1N product. The overall ΔG profiles differ from those computed for the lactone, where the 5-membered ring fragments immediately after the formation of the C2–C14 bond. Notably VO would be only 4.9 kcal mol−1 higher in energy than IVO and the last transition state (TsOIII-IV) is lower than TsOIII-IV (Fig. 5).
The intramolecular [2 + 2 + 2] cycloaddition reaction of the ene-1,6-diyne containing a lactam scaffold, was then explored to validate the computational study (Scheme 3). The benchmark substrate 8 was prepared as a partially separable mixture of E/Z isomers.8 After separation, each enriched diastereomer was submitted to the same reaction conditions as those for the intramolecular [2 + 2 + 2] cycloaddition reaction with a lactone scaffold (1.2 equiv. of CpCo(CO)2 in toluene at 110 °C during 16 hours under irradiation (halogen 400 W)). Pleasingly, as predicted by the theoretical study, the cycloaddition reaction proceeded stereospecifically to give the desired γ-spiro-γ-lactam cobalt complexes 9-cis and 9-trans, even with no full conversion. It is interesting to note an erosion of the stereospecificity of the cycloaddition reaction, which could be explained by the partial isomerization of the exo-cyclic double bond of the lactam moiety under the reaction conditions.
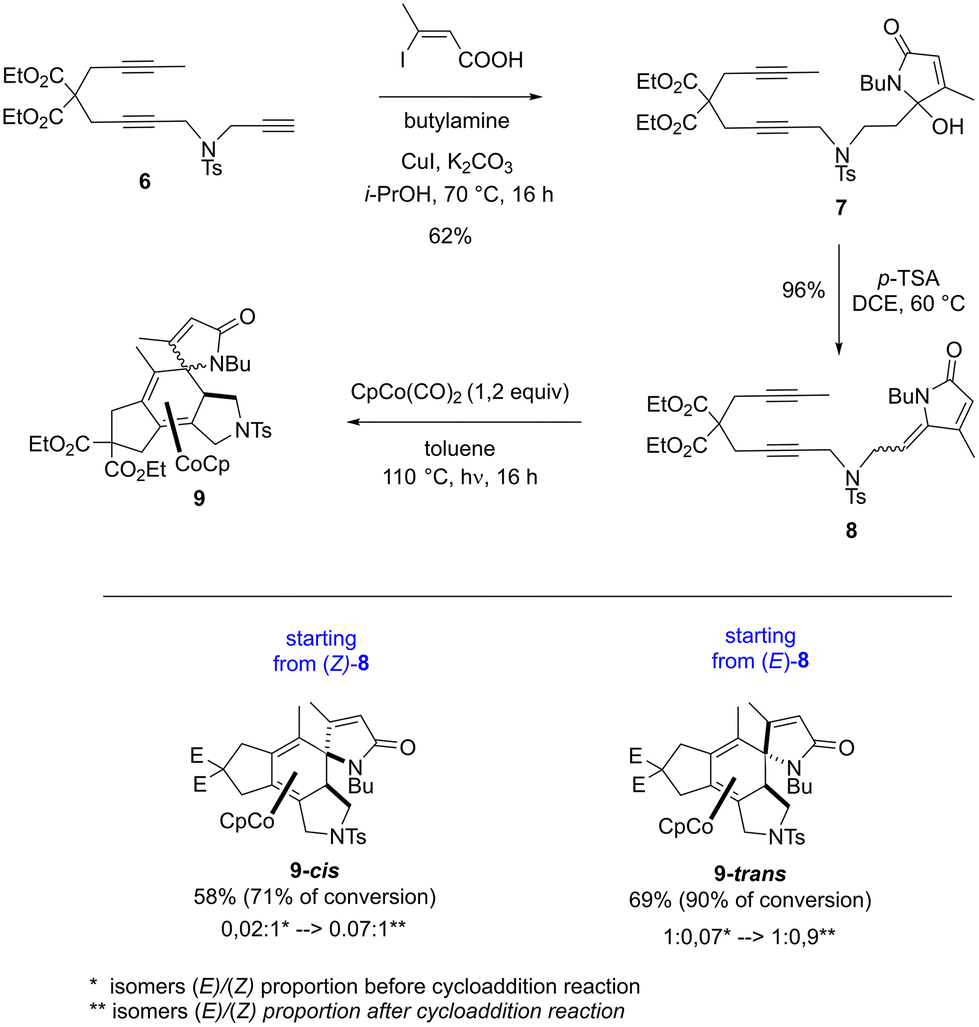 | ||
| Scheme 3 Experimental evidence for the synthesis of γ-spiro-γ-lactam cobalt complexes 9. | ||
As the benchmark substrate 8 exists in the Z and E form, calculations were also performed on the latter. The computed mechanism shows that only a spiro product can be obtained from the E isomer, as detailed in the ESI.†
Conclusions
Two typologies of mechanisms are envisioned for cobalt cycloaddition reactions with γ-alkylidenebutenolide or γ-alkylidenebuterolactam as 2π partners, leading to either cobalt(III)- or cobalt(I)-spiro-complexes, the latter issued from standard [2 + 2 + 2] cycloadditions. The reductive elimination step, occurring on the singlet potential energy surface, is crucial for the selectivity: in one case the C–C bond formation leads to a cycloheptadiene that can further evolve to the cobalt(III) complex; in the other case, the generated C–C bond leads to the spiro product containing a cyclohexadiene. Calculations suggest that two ingredients are essential to obtain the cobalt(III) complex: the reaction needs to be performed in an intramolecular fashion, with a substrate carrying a lactone. Indeed, several coordination modes of the 2π partner to the metal are possible in the intermolecular case, while the molecular link joining the lactone to the diyne moiety imposes the structural motif favourable to the formation of the cycloheptadiene in the intramolecular approach. Furthermore, the comparison between the lactam and lactone reveals that the lactam does not fragment after the reductive elimination, while the lactone breaks spontaneously to generate the ketene intermediate, which reacts further. Finally, experimental studies have confirmed the theoretical predictions: spiro compounds were obtained through [2 + 2 + 2] cycloaddition reactions starting from lactone substrates in an intermolecular fashion and from lactam derivatives in an intramolecular way.Experimental
Computational details
Density functional theory calculations were performed using the TURBOMOLE program package.9 The TPSS functional was employed to obtain the mechanistic profiles, together with the empirical dispersion corrections D3 (TPSS-D3).10 The basis set is of def2-TZVP quality for all atoms and the corresponding auxiliary basis sets for the RI treatment were selected.11 Solvent effects are added as single point calculations with the COSMO implicit model (toluene).12The mechanism for the lactone substrate in Fig. 5 was newly calculated to provide comparable results with those proposed in this contribution. Results between calculations performed at the BP86-D3 level (the method used in our preliminary work)6 and those obtained at the TPSS-D3 level here reported are almost identical. However, we decided to systematically employ the TPSS-D3 functional because it belongs to the meta-GGA functional family, which represents an improvement over the GGA functionals (as BP86). The TPSS functional is a general purpose functional that works well for transition metal complexes (notably 3d metals)10f and provides, as well, good results on the energetics of cobalt complexes, when comparing singlet and triplet states. Singlet and triplet potential energy surfaces get close for some intermediates of the cobalt-catalysed or mediated cycloaddition reactions, although we focused in this work on steps occurring on the singlet potential energy surface. We employ here the TPSS-D3 functional because it provides the triplet vs. singlet energy ordering predicted by ab initio calculations, whereas methods as B3LYP or PBE0 tend to over-stabilise triplets in some cases, in particular for the insertion intermediate.5
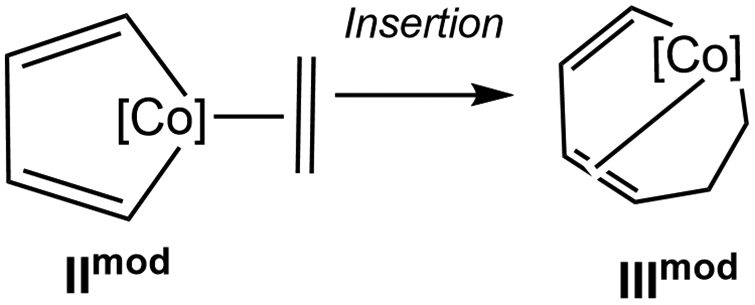
The lactam reaction pathway was computed by three different DFT methods (BP86-D3,10a,b,e,13 TPSS-D3, PBE0-D3 functionals,10a,b,e,14 def2-TZVP basis set, see ESI†). Importantly, all computations deliver the same qualitative picture, predicting that the pathway to the Co(III) complex is less favoured than the one to the spirocyclic lactam Co(I) complex. However, relative energies are somewhat sensitive to the method used, notably the difference in energy between IIIN and IIN is of −3.0 kcal mol−1 at the TPSS-D3 level and decreases to −10.3 kcal mol−1 at the PBE0-D3 level. We have then checked that the TPSS-D3 method would provide reasonable results: we considered the model systems for the cobaltacyclopentadiene coordinated by an alkene, IImod, and for the intermediate following the insertion, IIImod. The energy differences (ΔE = EIIImod − EIImod, without ZPE, gas phase) between these complexes, computed at DFT (BP86-D3, TPSS-D3, PBE0-D3 functionals, def2-TZVP basis set) and CCSD(T) levels, are reported in Table 1. CCSD(T) calculations, performed with the MOLPRO program package, are single point calculations on TPSS structures and employ the aug-cc-pwCVTZ-DK, cc-pVTZ-DK and cc-pVDZ-DK basis sets for Co, C and H, respectively. Results from TPSS-D3 and BP86-D3 are close to the CCSD(T) value (within 1.6 kcal mol−1), whereas the PBE0 method predicts the insertion intermediate somewhat too stable than IImod. We concluded that the TPSS-D3 calculations would give a more accurate global picture of the treated mechanisms than the hybrid GGA (Generalized Gradient Approximation) PBE0 functional in these cases.
|
|
|
|---|---|
| Method | ΔE (kcal mol−1) |
| CCSD(T) | −8.0 |
| BP86-D3 | −6.6 |
| TPSS-D3 | −6.4 |
| PBE0-D3 | −13.1 |
General
1H Nuclear Magnetic Resonance (NMR) spectra were recorded using an internal deuterium lock at ambient temperatures on the following instruments: Bruker AC400 (400 MHz) and Bruker AC300 (300 MHz). The internal references of 1H 7.26 was used for the residual protons in CDCl3. Data are presented as follows: chemical shift (in ppm), integration, interpretation, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quintet = quin, m = multiplet, dd = doublet of doublet, dt = doublet of triplet, br = broad) and coupling constant (J in Hz). 13C NMR spectra were recorded on a Bruker AC400 (101 MHz) and a Bruker AC300 (75 MHz) spectrometers with complete proton decoupling. Chemical shifts were reported in ppm from the internal solvent signal (peak at 77.16 ppm in the case of CDCl3).Infra-red spectra were recorded on a Bruker VERTEX70 Fourier transform infrared spectrometer fitted with a single reflection diamond ATR Bruker A222 accessory. The measurements were done for pure samples. For each individual spectrum, about 30 scans were averaged at 4 cm−1 resolution. The diamond crystal without sample served as reference. All the system was purged with dry air. The identification of peaks was done with the standard method proposed in OPUS 6.0 software. Wavelengths of maximum absorbance (max) are quoted in cm−1.
High resolution MS experiments were performed with a QSTAR Elite mass spectrometer (Applied Biosystems SCIEX) or a SYNAPT G2 HDMS mass spectrometer (Waters) equipped with an electrospray ionization source operated in the positive ion mode. In this hybrid instrument, ions were measured using an orthogonal acceleration time-of-flight (oa-TOF) mass analyzer.
Analytical thin layer chromatography (TLC) was carried out on Merck® Kieselgel 60 F254 plates and achieved under a 254 nM UV light, visualized with a KMnO4 solution or anisaldehyde solution.
Synthesis
:1) to give 3a as 1/0.2 mixture of diastereomers (28 mg, 45% yield). Major diastereomer: mp 159 °C; 1H NMR (300 MHz, chloroform-d) δ 0.89 (s, 3H), 1.11 (br d, J = 15.8 Hz, 1H), 1.29 (br s, 3H), 1.36 (br q, J = 1.0 Hz, 3H), 1.69 (br q, J = 1.0 Hz, 3H), 1.92 (d, J = 15.8 Hz, 1H), 3.13 (d, J = 16.4 Hz, 1H), 3.17 (d, J = 16.4 Hz, 1H), 3.67 (d, J = 16.4 Hz, 1H), 3.69 (d, J = 16.4 Hz, 1H), 3.80 (s, 3H), 3.89 (s, 3H), 4.48 (s, 5H); 13C NMR (75 MHz, chloroform-d) δ 8.5, 10.3, 16.4, 23.5, 39.0, 39.4, 44.2, 53.2, 53.4, 55.9, 56.2, 60.2, 82.8 (5C), 94.5, 94.7, 94.9, 122.5, 161.1, 172.4, 173.0, 174.5; HRMS (ESI-MS) calcd for C25H30O6Co+ [M + H]+ 485.1369, found 485.1364; IR νmax/cm−1 2956, 1735, 1436, 1257, 1199, 116.
:1 to 8:2) to give 4a (12 mg, 80% yield). Mp 171 °C; 1H NMR (300 MHz, chloroform-d) δ 1.99 (br q, J = 1.1 Hz, 3H), 2.02 (br q, J = 1.1 Hz, 3H), 2.03 (s, 3H) 2.17 (s, 3H), 3.54 (s, 4H), 3.75 (s, 3H), 3.77 (s, 3H), 6.61 (s, 1H), OH not observed; 13C NMR (75 MHz, chloroform-d) δ 15.8, 15.9, 18.7, 23.3, 39.9, 40.3, 53.1 (2C), 59.6, 125.0, 126.6, 127.1, 130.9, 137.6, 139.2, 142.3, 147.8, 171.2, 172.4, 172.7; HRMS (ESI-MS) calcd for C20H28NO6+ [M + NH4]+ 378.1911, found 378.1914; IR νmax/cm−1 2010, 1726, 1658, 1433, 1280, 1255, 1190, 1155, 1076, 1057.
:2) to give 5a (6.5 mg, 68% yield). Mp 198 °C; 1H NMR (400 MHz, chloroform-d) δ 1.74 (br q, J = 1.6 Hz, 3H), 2.23 (br q, J = 1.6 Hz, 3H), 2.31 (s, 3H), 2.40 (s, 3H), 3.49 (s, 2H), 3.51 (s, 2H), 3.77 (s, 6H); 13C NMR (101 MHz, chloroform-d) δ 7.6, 13.7, 15.8, 16.0, 39.3, 40.4, 53.2 (2C), 59.2, 125.2, 127.8, 130.9, 131.4, 139.8, 143.2, 144.9, 154.1, 172.2 (2C); 199.4; HRMS (ESI-MS) calcd for C20H23O5+ [M + H]+ 343.1540, found 343.1541; IR νmax/cm−1 1734, 1693, 1429, 1265, 1065, 1159, 1080, 1049.
:1 to 8:2). Mp 140 °C; 1H NMR (300 MHz, chloroform-d) δ 1.74 (br q, J = 1.3 Hz, 3H), 2.22 (br q, J = 1.3 Hz, 3H), 2.24 (s, 3H), 2.32 (s, 3H), 2.41 (s, 3H), 4.45–4.61 (m, 4H), 7.33 (d, J = 8.5 Hz, 2H), 7.78 (d, J = 8.5 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 7.5, 13.5, 15.6, 15.9, 21.7, 53.4, 54.1, 123.7, 127.7 (2C), 128.4, 129.4, 130.1 (2C), 132.0, 134.1, 136.3, 141.1, 143.8, 143.9, 153.8, 198.7; HRMS (ESI-MS) calcd for C22H24NO3S [M + H]+ 382.1471 found 382.1468; IR νmax/cm−1 2360, 1689, 1631, 1352, 1161, 1099.
:5 to 8:2). 1H NMR (300 MHz, chloroform-d) δ 1.76 (br q, J = 1.3 Hz, 3H), 2.25 (br q, J = 1.3 Hz, 3H), 2.27 (s, 3H), 2.37 (s, 3H), 5.07 (s, 4H); 13C NMR (75 MHz, chloroform-d) δ 7.5, 13.9, 15.9, 16.0, 73.9, 74.3, 122.7, 128.4, 128.5, 131.7, 139.1, 143.8, 144.2, 153.9, 199.1; HRMS (ESI-MS) calcd for C15H17O2+ [M + H]+ 229.1223, found 229.1223; IR νmax/cm−1 2922, 1685, 1471, 1375, 1055, 906.
:1 to 8:2) to give the corresponding hydroxylactam 7 (166 mg, 62% yield). 1H NMR (300 MHz, chloroform-d) δ 0.94 (t, J = 7.3 Hz, 3H), 1.24 (q, J = 7.0 Hz, 3H), 1.25 (q, J = 7.0 Hz, 3H), 1.35 (sixt, J = 7.5 Hz, 2H), 1.57–1.70 (m, 2H), 1.74 (br t, J = 2.5 Hz, 3H), 2.01 (br d, J = 1.6 Hz, 3H), 2.11–2.21 (m, 1H), 2.28–2.38 (m, 1H), 2.42 (s, 3H), 2.64–2.82 (m, 6H), 3.03–3.13 (m, 1H), 3.40–3.50 (m, 1H), 3.95–4.2 (m, 6H), 5.76 (br q, J = 1.6 Hz, 1H), 7.29 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), OH not observed; 13C NMR (75 MHz, CDCl3) δ 3.6, 12.1, 13.9, 14.2 (2C), 20.7, 21.7, 22.9, 23.1, 31.6, 32.5, 38.0, 38.8, 42.0, 56.6, 62.1 (2C), 73.0, 76.6, 79.3, 80.7, 91.9, 123.1, 127.5 (2C), 129.9 (2C), 135.9, 143.9, 158.8, 169.0 (2C), 169.8; HRMS (ESI-MS) calcd for C33H45N2O8S [M + H]+ 629.2891 found 629.2892; IR νmax/cm−1 2954, 1732, 1683, 1355, 1327, 1290, 1197, 1153, 1087, 1047, 723, 663, 576.
:EtOAc 8:2) to give both enriched diastereomers (m ((Z)-8 + (E)-8) = 299 mg, 96% yield). HRMS (ESI-MS) calcd for C33H43N2O7S [M + H]+ 611.2786 found 611.2789.
(Z)-8: 1H NMR (300 MHz, chloroform-d) δ 0.91 (t, J = 7.3 Hz, 3H), 1.21 (t, J = 7.1 Hz, 6H), 1.25–1.37 (m, 2H), 1.45–1.56 (m, 2H), 1.72 (br t, J = 2.4 Hz, 3H), 1.97 (br d, J = 1.5 Hz, 3H), 2.43 (s, 3H), 2.67 (br q, J = 2.4 Hz, 2H), 2.73 (br t, J = 2.1 Hz, 2H), 3.69 (dd, J = 7.3 Hz, 2H), 4.08–4.26 (m, 8H), 4.98 (t, J = 7.0 Hz, 1H), 5.85 (br q, J = 1.5 Hz, 1H), 7.32 (d, J = 7.9 Hz, 2H), 7.70 (d, J = 7.9 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 3.6, 12.4, 13.9, 14.1 (2C), 20.0, 21.7, 22.8, 23.0, 32.0, 37.3, 40.7, 43.4, 56.6, 62.0 (2C), 73.0, 76.0, 79.2, 81.3, 104.8, 120.5, 127.8 (2C), 129.8 (2C), 136.0, 141.9, 144.0, 148.5, 168.9 (2C), 170.9; IR νmax/cm−1 1728, 1357, 1296, 1199, 1159, 1089, 1051, 894, 752, 655.
(E)-8: 1H NMR (300 MHz, chloroform-d) δ 0.89 (t, J = 7.2 Hz, 3H), 1.20 (t, J = 7.1 Hz, 6H), 1.18–1.30 (m, 2H), 1.34–1.47 (m, 2H), 1.72 (t, J = 2.5 Hz, 3H), 2.29 (br d, J = 1.6 Hz, 3H), 2.43 (s, 3H), 2.67 (br q, J = 2.5 Hz, 2H), 2.74 (br t, J = 2.3 Hz, 2H), 3.44 (t, J = 7.2 Hz, 2H), 4.05–4.19 (m, 6H), 4.23 (d, J = 7.6 Hz, 2H), 5.14 (t, J = 7.6 Hz, 1H), 5.93 (br q, J = 1.6 Hz, 1H), 7.28–7.37 (d, J = 7.9 Hz, 2H), 7.64–7.76 (d, J = 7.9 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 3.5, 13.9, 14.1 (2C), 16.8, 20.2, 21.7, 22.8, 23.0, 30.8, 36.9, 38.6, 43.2, 56.5, 61.9 (2C), 73.0, 76.1, 79.1, 81.1, 107.3, 124.7, 127.7 (2C), 129.8 (2C), 136.2, 142.2, 143.9, 145.7, 168.8 (2C), 168.9; IR νmax/cm−1 2960, 1743, 1689, 1352, 1205, 1159, 1085, 1057.
:0.07 inseparable mixture of 9-cis and 9-trans (16 mg, 58% yield, 62% yield, 71% of conversion). 1H NMR (300 MHz, chloroform-d) δ 0.69 (s, 3H), 1.17 (t, J = 6.9 Hz, 3H), 1.29 (t, J = 7.1 Hz, 3H), 1.32–1.36 (m, 7H), 1.41–1.70 (m, 4H), 2.20–2.35 (m, 1H), 2.45 (s, 3H), 3.10–3.66 (m, 9 H), 4.12 (s, 5H), 4.25 (q, J = 7.1 Hz, 2H), 4.35 (br q, J = 7.1 Hz, 2H), 5.59 (br q, J = 1.6 Hz, 1H), 7.42 (d, J = 8.1 Hz, 2H), 7.82 (d, J = 8.1 Hz, 2H); 13C NMR (75 MHz, chloroform-d) δ 12.6, 14.2, 14.3 (2C), 17.5, 21.6, 21.7, 30.2, 39.4, 40.1, 44.0, 48.7, 51.4, 53.6, 59.4, 60.5, 62.4, 62.6, 63.7, 84.0 (5C), 89.8, 95.2, 121.4, 127.9 (2 C), 130.0 (2 C), 134.0, 144.2, 160.6, 171.7, 172.2, 173.4 (one quaternary C carbon is in CDCl3peaks); HRMS (ESI-MS) calcd for C38H48N2O7SCo [M + H]+ 735.2509, found 735.2509; IR νmax/cm−1 2960, 2927, 1726, 1685, 1342, 1246, 1161, 1101, 1045, 1008, 802, 673.
:0.09 inseparable mixture of 9-cis and 9-trans (29 mg, 69% yield, 90% of conversion). 1H NMR (300 MHz, chloroform-d) δ 0.77 (s, 3H), 0.83 (t, J = 7.0 Hz, 3H), 1.09–1.20 (m, 3H), 1.20–1.27 (m, 1H), 1.30 (t, J = 7.1 Hz, 3H), 1.36 (t, J = 7.1 Hz, 3H), 1.41–1.51 (m, 1H), 2.26–2.38 (m, 1H), 2.43 (s, 3H), 2.53 (br d, J = 1.5 Hz, 3H), 2.60–2.73 (m, 1H), 3.08 (d, J = 17.0 Hz, 1H), 3.16 (br s, 2H), 3.34 (br dd, J = 7.8 and 8.6 Hz, 1H), 3.45 (d, J = 17.0 Hz, 1H), 3.55 (d, J = 17.0 Hz, 1H), 3.68–3.73 (m, 1H), 3.73 (d, J = 17.0 Hz, 1H), 4.23 (s, 5H), 4.23–4.39 (m, 4H), 5.82 (br q, J = 1.5 Hz, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.75 (d, J = 8.3 Hz, 2H); 13C NMR (75 MHz, chloroform-d) δ 13.9, 14.2, 14.3, 17.8, 17.9, 20.5, 21.7, 30.8, 39.3, 39.7, 39.8, 49.4, 50.9, 53.2, 56.5, 59.8, 60.1, 62.4, 62.6, 77.5, 84.0 (5C), 89.7, 95.2, 126.3, 127.7 (2 C), 130.0 (2C), 134.1, 144.1, 158.2, 169.6, 171.7, 171.9; HRMS (ESI-MS) calcd for C38H48N2O7SCo [M + H]+ 735.2509, found 735.2509; IR νmax/cm−1 1734, 1687, 1348, 1259, 1182, 1151, 1093, 1051, 1010, 823, 673.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. C and M. D. thanks the French Research Ministry, Aix-Marseille University and CNRS for financial support.References
- Selected references: (a) W. Reppe, O. Schichting, K. Klager and T. Toepel, Justus Liebigs Ann. Chem., 1948, 560, 1–92 CrossRef CAS; (b) J. M. Halford-McGuff, A. M. Z. Slawin and A. J. B. Watson, ACS Catal., 2023, 13, 3463–3470 CrossRef CAS; (c) P. Matton, S. Huvelle, M. Haddad, P. Phansavath and V. Ratovelomanana-Vidal, Synthesis, 2022, 54, 4–32 CrossRef CAS; (d) A. Lledo, A. Pla-Quintana and A. Roglans, Chem. Soc. Rev., 2016, 45, 2010–2023 RSC; (e) Y. Sato and Y. Obora, Eur. J. Org. Chem., 2015, 5041–5054 CrossRef; (f) S. Okamoto and Y. Sugiyama, Synlett, 2013, 1044–1060 CrossRef CAS; (g) Transition-Metal-Mediated Aromatic Ring Construction, ed. K. Tanaka, John Wiley & Sons, Hoboken, 2013 Search PubMed; (h) Y. Shibata and K. Tanaka, Synthesis, 2012, 323–350 CAS; (i) K. Tanaka, Heterocycles, 2012, 85, 1017–1043 CrossRef CAS; (j) D. L. J. Broere and E. Ruijter, Synthesis, 2012, 2639–2672 CAS; (k) G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC; (l) M. R. Shaaban, R. El-Sayed and A. H. M. Elwahy, Tetrahedron, 2011, 67, 6095–6130 CrossRef CAS; (m) N. Wedding and M. Hapke, Chem. Soc. Rev., 2011, 40, 4525–4538 RSC; (n) D. Leboeuf, V. Gandon and M. Malacria, in Handbook of Cyclization Reactions, ed. S. Ma, Wiley-VCH, Weinheim, 2009 Search PubMed; (o) N. Agenet, V. Gandon, O. Buisine, F. Slowinski and M. Malacria, Organic Reactions, ed. T. V. RajanBabu, Wiley, Hoboken, NJ, 2007 Search PubMed; (p) V. Gandon, C. Aubert and M. Malacria, Chem. Commun., 2006, 2209–2217 RSC.
- G. Domínguez and J. Pérez-Castells, Chem. – Eur. J., 2016, 22, 6720–6739 CrossRef PubMed.
- (a) D. Leboeuf, L. Iannazzo, A. Geny, M. Malacria, K. P. C. Vollhardt, C. Aubert and V. Gandon, Chem. – Eur. J., 2010, 16, 8904–8913 CrossRef CAS PubMed; (b) V. Gandon, N. Agenet, K. P. C. Vollhardt, M. Malacria and C. Aubert, J. Am. Chem. Soc., 2006, 128, 8509–8520 CrossRef CAS PubMed; (c) N. Agenet, V. Gandon, K. P. C. Vollhardt, M. Malacria and C. Aubert, J. Am. Chem. Soc., 2007, 129, 8860–8871 CrossRef CAS PubMed.
- A. Roglans, A. Pla-Quintana and M. Solà, Chem. Rev., 2021, 121, 1894–1979 CrossRef CAS PubMed.
- L. Chaussy, D. Hagebaum-Reignier, S. Humbel and P. Nava, Phys. Chem. Chem. Phys., 2022, 24, 21841–21852 RSC.
- M. Delorme, A. Punter, R. Oliveira, C. Aubert, Y. Carissan, J.-L. Parrain, M. Amatore, P. Nava and L. Commeiras, Dalton Trans., 2019, 48, 15767–15771 RSC.
- (a) A. A. Bengali, R. G. Bergman and C. B. Moore, J. Am. Chem. Soc., 1995, 117, 3879–3880 CrossRef CAS; (b) P. E. M. Siegbahn, J. Am. Chem. Soc., 1996, 118, 1487–1496 CrossRef CAS; (c) A. A. Dahy and N. Koga, Bull. Chem. Soc. Jpn., 2005, 78, 781–791 CrossRef CAS; (d) A. A. Dahy, C. H. Suresh and N. Koga, Bull. Chem. Soc. Jpn., 2005, 78, 792–803 CrossRef CAS.
- M. I. D. Mardjan, A. Mayooufi, J.-L. Parrain, J. Thibonnet and L. Commeiras, Org. Process Res. Dev., 2020, 24, 606–614 CrossRef CAS.
- TURBOMOLE V7.5 2020, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from https://www.turbomole.com.
- (a) P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1929, 123, 714–733 CrossRef CAS; (b) J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS; (c) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed; (d) J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed; (e) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (f) F. Furche and J. P. Perdew, J. Chem. Phys., 2006, 124, 044103 CrossRef PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1992, 799–805 Search PubMed.
- (a) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (c) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed . Erratum: J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 Search PubMed.
- (a) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed; (b) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (c) J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional computations, computational details, 1H and 13C NMR spectra of each compound, computed coordinates. See DOI: https://doi.org/10.1039/d3dt02291a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |