
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel design of dual-action Pt(IV) anticancer pro-drugs based on cisplatin and derivatives of the tyrosine kinase inhibitors imatinib and nilotinib†
Darren Fergal
Beirne
 a,
Barbara
Farkaš
b,
Chiara
Donati
c,
Valentina
Gandin
c,
Isabel
Rozas
b,
Trinidad
Velasco-Torrijos
*ad and
Diego
Montagner
*ad
a,
Barbara
Farkaš
b,
Chiara
Donati
c,
Valentina
Gandin
c,
Isabel
Rozas
b,
Trinidad
Velasco-Torrijos
*ad and
Diego
Montagner
*ad
aDepartment of Chemistry, Maynooth University, Ireland. E-mail: diego.montagner@mu.ie
bSchool of Chemistry, Trinity College Dublin, Ireland
cDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Italy
dKathleen Londsdale Institute for Human Health Research, Maynooth University, Ireland
First published on 7th September 2023
Abstract
Tyrosine kinases (TKs) are emerging as important targets in cancer therapy and some of their inhibitors, TKIs (e.g. imatinib and nilotinib), are FDA-approved drugs that are used as selective anti-cancer therapeutics against cell lines that overexpress TKs. Many examples of metal-based complexes functionalised with TKIs are reported in the literature but very few have been functionalised with platinum. Here we report the design, a detailed computational analysis/simulation, the complete chemical characterisation and the preliminary biological evaluation of two novel Pt(IV) anticancer pro-drugs based on cisplatin tethered with a derivative of either imatinib or nilotinib in the axial position. Pt(IV) complexes are a strategic scaffold in combination therapy due to their axial ligands that can be functionalised to form dual action drugs. The activation by reduction releases the Pt(II) core and the axial ligands upon cellular internalisation. The antiproliferative activity and the TK inhibition properties of the novel adducts are analysed with a theoretical approach and confirmed in vitro with preliminary biological assays.
Introduction
Tyrosine kinases (TKs) are enzymes whose action is to transfer a phosphate group from adenosine triphosphate (ATP) specifically to the tyrosine residues of cellular targets; they are often implicated in tumoral tissue formation as their overexpression and/or mutation is connected with increased cellular proliferation.1 There are several tyrosine kinase inhibitors (TKIs), some of which (such as imatinib and nilotinib) are FDA approved drugs that suppress signal transduction pathways by inhibiting the activity of TKs.2,3 Naturally, TKIs are used as selective anti-cancer therapeutics against cell lines that overexpress TKs. Four of these TKIs, namely imatinib, nilotinib, dasatinib and erlotinib, feature on the WHO List of Essential Medicines under “targeted therapies” (Fig. 1).4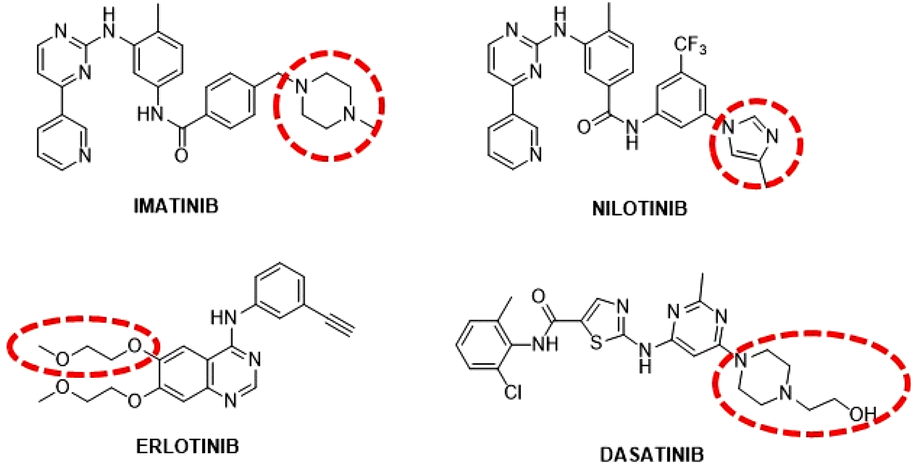 | ||
| Fig. 1 Structures of the FDA-approved TKIs. Red dotted circle: solvent exposed regions. | ||
Imatinib and Nilotinib are inhibitors targeting, among others, the receptors PDGFR (Platelet Derived Growth Factor Receptor) and c-KIT (Mast/stem cell growth factor receptor) and they act by binding the receptors intracellularly, via a reversible ATP-competitive mechanism.5–7 Resistance to imatinib motivated the development of new TKIs such as nilotinib, which is approximately 30 times more potent than imatinib and features a trifluoromethyl group that increases van der Waals (wdW) interactions with the active site.8 Nilotinib can circumvent 32 out of 33 clinically relevant imatinib resistant mutations. Considering that imatinib and nilotinib show synergistic anti-tumour effects when used in combination with cisplatin, the key concept behind the present work is to combine the TKIs’ inhibitory effect with the anticancer properties of cisplatin, by designing and synthesising Pt(IV) species containing in one unique molecule the Pt(II) scaffold (i.e. cisplatin) and the TKI (i.e. nilotinib and imatinib). The dual mode of action may have the benefit not only of circumventing resistance associated with cisplatin therapy, but also enhancing the selectivity towards cancer cells.
Pt(IV) species have become relevant in the last two decades with several examples described in the literature and in different reviews.9–13 The main advantages of this oxidation state are the higher stability under physiological conditions and the higher lipophilicity that enhances cellular uptake. These species are known as pro-drugs because they are intracellularly activated by reduction with the release of the Pt(II) scaffold and the axial ligands.14–17 Here, we describe the design, synthesis and characterisation of two novel dual-action Pt(IV)-complexes based on cisplatin and the two mentioned TKIs, nilotinib and imatinib (Fig. 2).
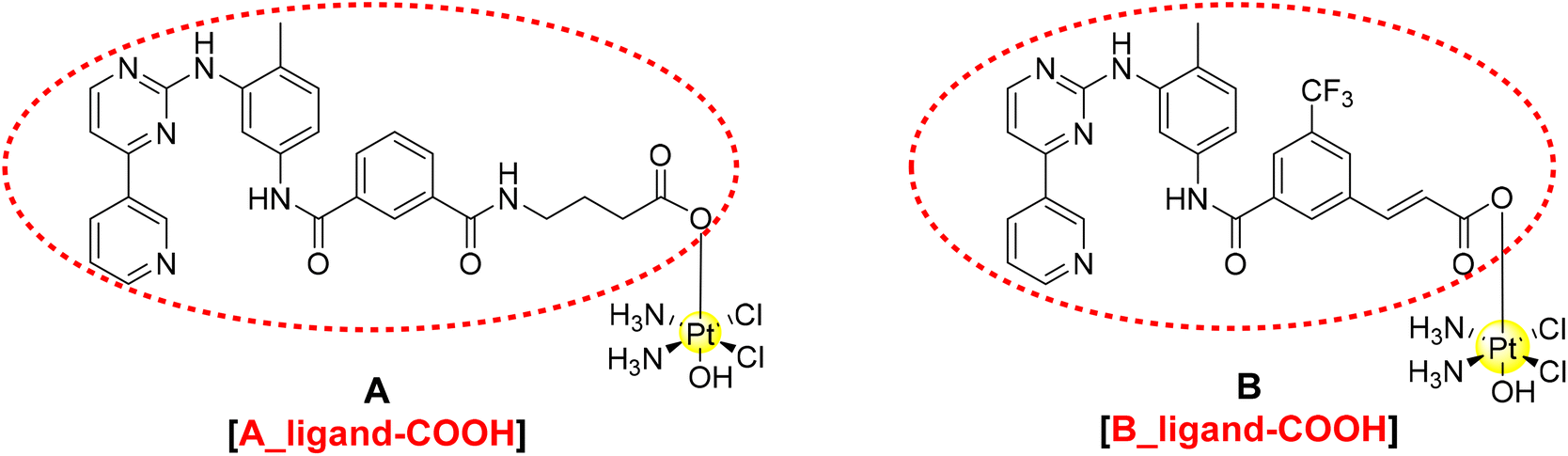 | ||
| Fig. 2 Proposed structure of the TKI–Pt(IV) complexes A and B and circled in red the corresponding carboxylic ligands A_ligand-COOH and B_ligand-COOH. | ||
While the idea of combining TKIs with different metals goes back a few years, as described by us in a recent review,18 examples containing platinum are very limited and mainly refer to Pt(II) species.19–25 To the best of our knowledge, this is the first time that a TKI such as nilotinib or imatinib is conjugated to a Pt(IV) scaffold and thus, in this paper, we report the strategic design of these novel pro-drugs supported by an extensive theoretical study on the mode of binding of these adducts to the TK targets. As described in our previous review, the functionalisation of the TKI with a metal complex should happen in a position that does not interfere with their binding to the receptors (Fig. 1).18 Therefore, the complexes are then synthesised and characterised with different techniques (multinuclear 1D and 2D NMR spectroscopy, mass spectrometry, IR spectroscopy, and elemental analyses), their stability under physiological conditions is assessed and a preliminary biological screening as anticancer agents and the TK inhibition activity on isolated enzymes are discussed.
Results and discussion
Computational studies
A detailed theoretical investigation was performed on the potential binding of imatinib and nilotinib (Fig. 1), the Pt(IV) complexes A and B and their corresponding ligands with the free carboxylic acids (the entities that will be released upon intracellular reduction, A_ligand-COOH and B_ligand-COOH, Fig. 2) to PDGFR, c-KIT and also to a mutation of c-KIT (c-KIT T670I) as targets.Since the X-ray crystal structures of PDGFR-α and c-KIT co-crystallised with imatinib (Fig. S14a/b in the ESI†) were previously resolved by Mol et al.,26 it was possible to identify the binding pocket for both targets and use them as templates for docking studies. Thus, molecular docking using both rigid and flexible protocols as implemented in Glide was performed and the results obtained for the six structures proposed and the corresponding targets are summarised in Table 1 and in Fig. S15–S20 (ESI†). The strength of binding is measured by the corresponding G-scores for the best poses obtained. Flexible docking provides better binding (more negative G-scores) than the rigid approach; for that reason, we will only discuss the flexible binding results hereafter. Both Pt(IV) complexes A and B as well as their corresponding free COOH ligands show stronger binding to PDGFR-α than the known TKIs, imatinib and nilotinib, for around 4 kcal mol−1 (Table 1).
| PDGFR-α | c-KIT | c-KIT T670I | ||||
|---|---|---|---|---|---|---|
| G-score, r | G-score, IFD | G-score, r | G-score, IFD | G-score, r | G-score, IFD | |
| Imatinib | −8.955 | −9.675 | −12.175 | −12.190 | −5.152 | −7.484 |
| A | −11.572 | −14.172 | −12.556 | −13.838 | −8.466 | −9.165 |
| A_ligand-COOH | −12.996 | −13.476 | −12.737 | −12.765 | −8.074 | −13.614 |
| Nilotinib | −7.690 | −10.115 | −10.478 | −10.790 | −5.818 | −8.169 |
| B | −7.248 | −14.025 | −13.306 | −15.135 | −6.493 | −10.653 |
| B_ligand-COOH | −12.520 | −13.991 | −12.588 | −12.907 | −7.112 | −11.153 |
In the case of the c-KIT target, the binding of imatinib, complex A and A_ligand-COOH is predicted to be of similar strength (Table 1).
However, after the c-KIT T670I mutation, which is known to be the culprit for the loss of imatinib's inhibitory activity, the improvement in the binding is almost twofold for A_ligand-COOH indicating the ability of this free ligand to inhibit c-KIT both prior and after the mentioned mutation with similar potency.
Complex B and its free carboxylic ligand follow a very similar trend, and both showed an improvement in their interactions with c-KIT compared to nilotinib. After the T670I mutation, binding of all three ligands weakens, but B_ligand-COOH maintains the best docking score. It is interesting that binding of both complexes’ ligands without the metal head in mutated c-KIT shows stronger interaction with the target than imatinib, nilotinib and their metal prodrugs A and B.
Looking at the interactions established between ligands and targets, the best binding poses of the six derivatives flexibly docked to PDGFR-α are shown in Fig. 3.
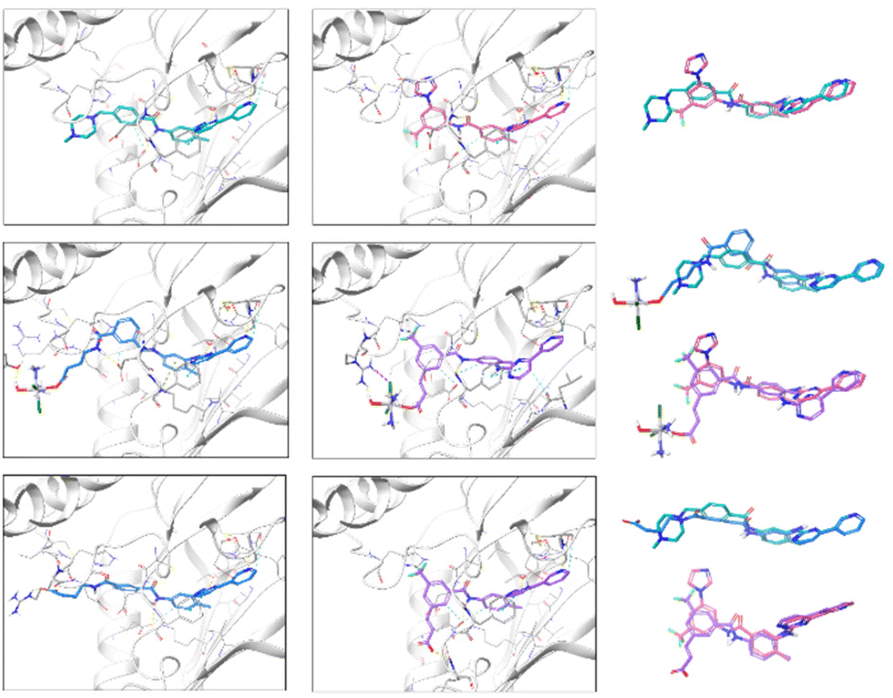 | ||
| Fig. 3 Best scoring poses of imatinib, A and A_ligand-COOH (left panel, top to bottom), and nilotinib, B and B_ligand-COOH (middle panel, top to bottom) after induced fit docking (IFD) in PDGFR-α with Glide. Alignment of ligand pairs is shown in the right panel (top to bottom), colouring matching the colouring in the ligand–receptor figures. HBs are represented by yellow dashed lines, π-cation interactions by green dashed lines, π–π stacking by cyan dashed lines connecting ring centres represented by cyan dots, salt bridges by pink dashed lines, XBs by purple dashed lines, and aromatic HBs by light blue dashed lines. Ribbon structure of the receptor has been intersected due to the enclosed nature of the binding pocket and amino acid residues far from the binding site have been hidden for clarity. | ||
Among the several hydrogen bonds (HBs) and other non-covalent interactions formed, the most remarkable are the HB formed by the pyridine N and the amino backbone group of Cys677 and the π–π stacking between the pyrimidine ring and Phe837, which are maintained in all the ligands. Except for the best scoring pose of complex B, a HB between the NH group connecting the pyrimidine and tri-substituted phenyl rings and Thr674 is also seen in all the systems. In the case of both imatinib and nilotinib, the amide functional group between aryl systems interacts with the target, where the NH acts as a HB-donor to Glu644 in both compounds, and the CO of nilotinib forms a HB with Asp836. Carboxylic groups in A_ligand-COOH and B_ligand-COOH and metal centres of Pt-prodrug complexes form HB and halogen bonds (XBs) outside the binding pocket. More details can be found in the ESI.†
From the two-dimensional interaction diagrams shown in Fig. S16,† it can be concluded that the 4-pyridyl-pyrimidine core, shared across all six ligands, remains locked in the hydrophobic end of the binding pocket containing a series of non-polar amino acids (Lue599, Val607, Ala625, Val658, Leu825, Phe837) and two aromatic residues (Tyr676 and Phe837) with consistent π–π bonding. The positively charged side-chain of Lys627 is conveniently positioned in the centre of the cavity followed by HB accepting carboxyl groups of Glu644 and Asp836 thus allowing for an efficient HB and π-cation network that secures ligand binding. The entrance to the cavity is highly hydrophobic, surrounded by side chains of Ile647, Leu651, and Leu809. As the surface of the protein opens up, more points for a strong HB and/or salt-based interaction arise, including Ser643 and Tyr872 or Lys646 and Arg817. To reach these reactive residues, long ligands whose fragments can extend beyond the enclosed cavity, such as A_ligand-COOH and B_ligand-COOH, are necessary.
Regarding the interactions observed between the six ligands and c-KIT, Fig. 4 captures their best binding poses as flexibly docked into c-KIT while Fig. S17 (ESI†) shows the corresponding two-dimensional interaction diagrams. It needs to be noticed that the binding cavity of c-KIT shows similar features to that of PDGFR-α, which, in principle, would allow for dual inhibition of the two proteins (Fig. S18, ESI†). In addition, the positioning and type of interaction established during the binding of the six derivatives in the two targets are also similar. The common features found are the position of the 4-pyridyl-pyrimidine core which sits comfortably in the hydrophobic cocoon enclosed by non-polar amino acids (i.e. Leu595, Val603, Ala621, Val654, and Leu799) and two aromatic residues (i.e. Phe811 and Tyr672). Besides, the pyridine ring, common to all ligands, is positioned to ensure HBs with the amino backbone group of Cys673, the phenyl ring forms π-cation interactions with the cationic side chain of Lys623 and the nearby Glu640 and Asp810 establish HBs with the amide and amine linkers of the ligands. Outside the binding cavity, Arg791 seems to be the favoured residue to interact with the complexes A and B. Finally, the interactions within the flexible docking poses with c-KIT with the T670I mutation were analysed and the corresponding flexible docked poses and two-dimensional interaction diagrams of the six compounds in c-KIT T670I are shown in Fig. S19 and S20 (ESI†). This crucial mutation of a polar residue in the centre of the binding pocket of c-KIT (i.e. Thr670 which seems to work as a HB acceptor for all compounds studied) into hydrophobic Ile with an ethyl side chain pointing into the binding pocket, is known to significantly hinder the inhibitory activity of imatinib and nilotinib. Loss of this HB interaction affects the ability of the 4-pyridyl-pyrimidine core to bury itself deeply into the binding site, causing a reduced depth of entry of the whole ligand into the binding cavity, as well as a less than ideal volume fit with a significant empty space (see the example of imatinib in Fig. 5, left).
 | ||
| Fig. 4 Best scoring poses of imatinib, A and A_ligand-COOH (left panel, top to bottom), and nilotinib, B and B_ligand-COOH (middle panel, top to bottom) after induced fit (IFD) in c-KIT docking using Glide. Alignment of ligand pairs is shown in the right panel (top to bottom), colouring matching the colouring in the ligand–receptor figures. For interaction colour scheme, follow the caption of Fig. 3. | ||
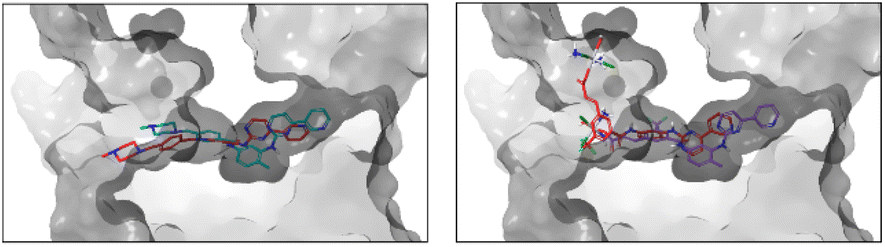 | ||
| Fig. 5 Shift in the imatinib binding pose (left) and B binding pose (right) upon the mutation of c-KIT in position 670 (T670I). Due to the enclosed binding cavity, the protein surface has been intersected and made transparent. Poses in the original colour scheme were obtained in non-mutated c-KIT, and those in red in T670I in mutated c-KIT. | ||
This, in turn, distances the pyridine ring of the ligands from the Cys673 residue which was also seen as a consistent HB donor for the six derivatives. Instead, interactions of the 4-pyridyl-pyrimidine core are established only through the pyrimidine ring by forming π–π stacking and HB with Phe811 and Lys623 for imatinib, and π-cationic interaction and HB with Lys623 and Asp810 for nilotinib.
The presence of the Pt-complex as in complex A causes an even more pronounced pull-out from the binding pocket to accommodate a HB between the OH group and the backbone of Ala599. The cost of the shift of B, shown as an example in Fig. 5 right, was compensated by HBs with Arg830 and Tyr846. Similarly, B_ligand-COOH forms three interactions through the carboxylic group (i.e. a salt bridge and HB with Arg791 and another HB with the neighbouring Ile789), thus allowing for maximised formation of HBs and π–π stacking of the 4-pyridyl-pyrimidine core with Lys623, Asp810, and Phe811.
Molecular dynamics (MD) simulation analysis is crucial for exploring the dynamic behaviour and stability of the ligand binding modes and interactions established through static docking. Thus, to explore the dynamics of the protein–ligand complexes obtained by docking, all-atom 50 ns MD simulations were carried out on each of the 18 systems. Stability was assessed by the time evolution of potential energy, RMSD and radius of gyration of the protein backbone, residue RMSF, RMSD of ligand heavy atoms, and principal component analysis and the corresponding results can be found in the ESI (Fig. S21–S27†). Additionally, quantitative prediction of the free energy of binding was obtained using the MMPBSA approach. All systems were found to be stable under the defined conditions of atmospheric pressure (1 bar) and at 310 K, with an example of potential energy and temperature–time evolution shown for the imatinib–PDGFR-α complex in Fig. S21 (ESI†).
Hydrogen bonding, a critical driving force behind the stability of the receptor–ligand complexes, was examined and the results are shown in the right panel of Fig. S22 (ESI†) for each of the imatinib, A and A_ligand-COOH complexes with PDGFR-α. The detected average number of HBs formed is 3.4, 2.8, and 4.8, respectively, showing the stability of the established HB network over time and corresponding well with the contacts predicted by docking studies. The average HB acceptor–donor distance captured for most of the bonds (>75%) was in the 2.8–3.2 Å range. To better understand which residues of the receptors are relevant for the binding process, trajectories were analysed in terms of contacts between ligands and single residues and the corresponding maps and contribution of each contact to the free binding energy are shown in Fig. S24.† Docked binding poses of imatinib, complex A and ligand A_ligand-COOH showed reoccurrence of HBs with specific residues as well as a consistent positioning of the 4-pyridyl-pyrimidine core in the hydrophobic cavity domain; the contact analysis carried out from the MD trajectory clearly exemplifies this behaviour capturing a constant interaction with residues such as Ala625, Val626, Lys627, Glu644, Thr674, Cys677, Cys835 or Asp836. The largest difference in contacts between imatinib, complex A and ligand A_ligand-COOH with PDGFR-α lay in those with residues farther away from the binding cavity entrance.
Structural and HB analysis for imatinib and complex A complexed with c-KIT and its T670I mutation is summarised in Fig. S25 (ESI†). Due to similarities with PDGFR-α ligand pairs, a detailed explanation is avoided. The biggest change in ligand orientation was observed for complex A, which was embedded in the c-KIT binding cavity with the Pt-metal centre moved from Arg791 to the positive side chain of Lys623 and backbone of Leu813, both situated on the opposite side of the cavity entrance; this coincides with the second most stable binding pose predicted through the flexible docking protocol.
Fig. S26 (ESI†) depicts residue contact frequency and decomposed contribution to free binding energies of imatinib, complex A, and ligand A_ligand-COOH binding with c-KIT before and after the T670I mutation. The major contact differences between the non-mutated and mutated protein can be seen with the residues of the hydrophobic domain (Leu595, Val603, Ala621, Val622) as well as other residues deeply embedded in the binding cavity (Glu671, Tyr672, Cys673, Gly676). This arises from the significant shift in the positioning of the 4-pyridyl-pyrimidine core upon the T670I mutation.
A detailed discussion of the analysis of MD trajectories generated with nilotinib and its analogues complex B and ligand B_ligand-COOH complexed with the three targets of interest is avoided here due to the repeating patterns already explained in the example of imatinib and its conjugated systems. The structural, energetic, and bonding descriptors analysed are shown fully in Fig. S27 and S28† for PDGFR-α, followed by Fig. S29 and S30 (ESI†) for c-KIT and its T670I mutation.
To more accurately quantify the free energy of binding of the ligands in a macromolecule–ligand complex, the MM/PBSA approach was used to calculate the favourable interactions, including solvent accessible surface area (SASA), as well as unfavourable polar solvation energy. The computed MM/PBSA free binding energies with decomposed energy terms for the 18 systems are listed in Table S1 (ESI†). A set of energies belonging to the three imatinib conjugates bonded to PDGFR-α indicates a similar binding strength. Contributions of vdW interactions as well as the non-polar solvation energy are also very much alike across the series. The binding strength is somewhat smaller for the Pt-complex A compared to imatinib due to higher polar solvation and dispersion energies. The acidic conjugate A_ligand-COOH showed not only an expected increase in the favourable electrostatic energy, but also a significant growth of the unfavourable polar solvation energy, resulting in a null effect overall. This pattern is also observed for the set of imatinib derivatives binding in mutated and unmutated c-KIT targets, with a more pronounced difference in the dispersion energies. The same applies to nilotinib and its derivatives. Altogether, the calculated binding free energies indicate that the conjugation of imatinib and nilotinib with Pt(IV)-carrying systems and the corresponding free carboxylic acid derivatives does not jeopardise their binding and inhibition of the studied targets.
Syntheses
Considering the promising results obtained in the molecular modelling study, we proceeded to prepare the ligands A_ligand-COOH and B_ligand-COOH as well as the corresponding Pt(IV) complexes A and B. As previously discussed, the imatinib and nilotinib derivatives should be functionalised at the solvent exposed region in order to minimise any possible negative effect on the TKI binding to the receptor. While the design of the synthesis of complex A with the imatinib derivative was easy, the synthetic strategy for the nilotinib derivative B showed several critical points.Accordingly, complex A has been obtained as shown in Scheme 1.
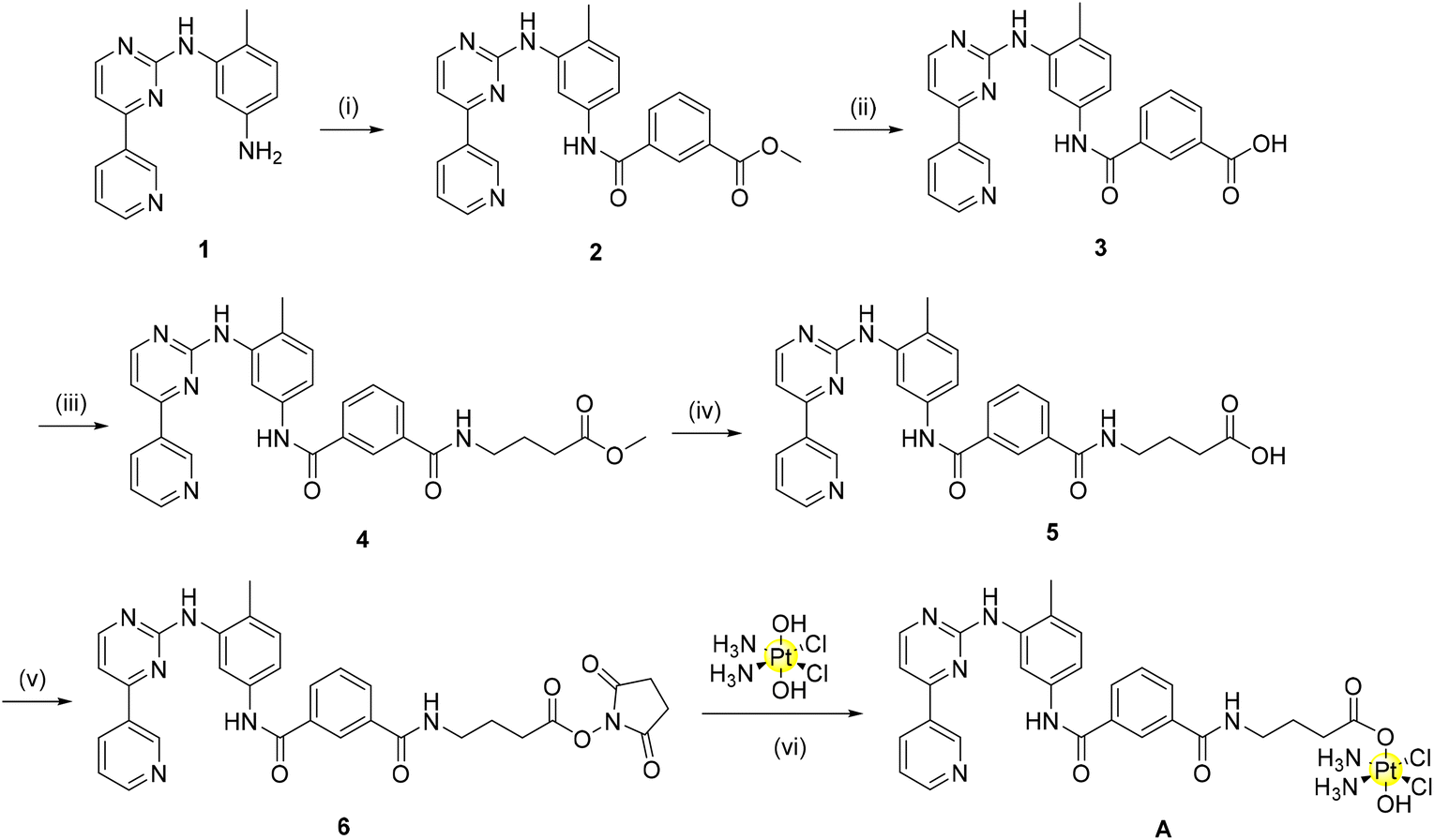 | ||
| Scheme 1 Synthesis of complex A: (i) mono-methyl isophthalate, BOP, TEA, DMF, r.t., 24 h, 88% (ii) NaOH, MeOH, 45 °C, 16 h, 90% (iii) 4-aminobutyric acid, TBTU, TEA, DMF, r.t., 24 h, 73% (iv) NaOH, MeOH, 45 °C, 16 h, 78% (v) TSTU, TEA, DMF, r.t., 20 min, 33% (vi) DMSO, 60 °C, 24 h, 64%. | ||
Briefly, four reactions were conducted as previously described by Dalla Via to synthesize the starting organic component of imatinib (1).27 From here, amide coupling with mono-methyl isophthalate using BOP as the coupling agent yields the methyl ester (2). Base hydrolysis deprotection of this methyl ester provides the carboxylic acid derivative (3) in excellent yield. Initially, 3 itself was converted to a N-hydroxysuccinimide-ester (NHS-ester) using NHS, TBTU and triethylamine. NHS-activation has previously been shown by our group to be a highly successful strategy to synthesize Pt(IV) pro-drugs.28 However, in this case, the reaction of the NHS-ester derivative of 3 with oxoplatin did not indicate the formation of a mono-axial functionalised Pt(IV) derivative as desired. As the reaction may not have progressed due to steric hindrance, we decided to introduce a linker to our compounds to alleviate this problem. This linker introduces an element for potential future optimization of the pharmacokinetic properties of the complexes (e.g. by using pegylated linkers). In the present case, a three carbon long alkyl linker was selected to provide sufficient distance from the Pt(IV) moiety, positioned in the solvent exposed region, to the main sites of enzyme interaction in the TKs’ binding sites.
In the transformation from 3 to 4, amide coupling was again employed to introduce the alkyl linker. Following ester hydrolysis to yield the carboxylic acid 5, TSTU (N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate) was employed as the optimal reagent for the synthesis of the desired NHS-ester (6) with 90% yield. This strategy as previously described by our group has the benefit of a short reaction time (20 min) with the formation of a pure product following column chromatography.29 Conjugation of the NHS-ester with oxoplatin afforded complex A in good yield (60%) without the need for chromatographic purification.
In a similar fashion, complex B was synthesised incorporating a linker to aid the conjugation to the platinum scaffold (Scheme 2). A two-carbon long alkene linker was selected for further renditions of Pt(IV)-imatinib/nilotinib conjugates for two main reasons: (i) development of a more convergent synthesis relative to complex A and (ii) early indications that a change in the linker may allow complexes previously unobtainable due to purification issues to be obtained now. Briefly, commercially bought 7 was brominated using N-bromosuccinimide via an electrophilic aromatic substitution reaction in excellent yield. A Heck reaction was employed to afford the linker 9; it was important to conduct the reaction in a sealed vessel as various attempts screened without it proved unsuccessful (including when conducted under an inert atmosphere, addition of dry-solvents and when the mol% of the catalyst used was varied). The methyl ester (10) was obtained as above via coupling with TBTU, deprotected to 11 using basic ester hydrolysis and converted to the NHS-ester, 12, using TSTU. The complexation reaction to produce B proceeded smoothly with little requirement for optimisation.
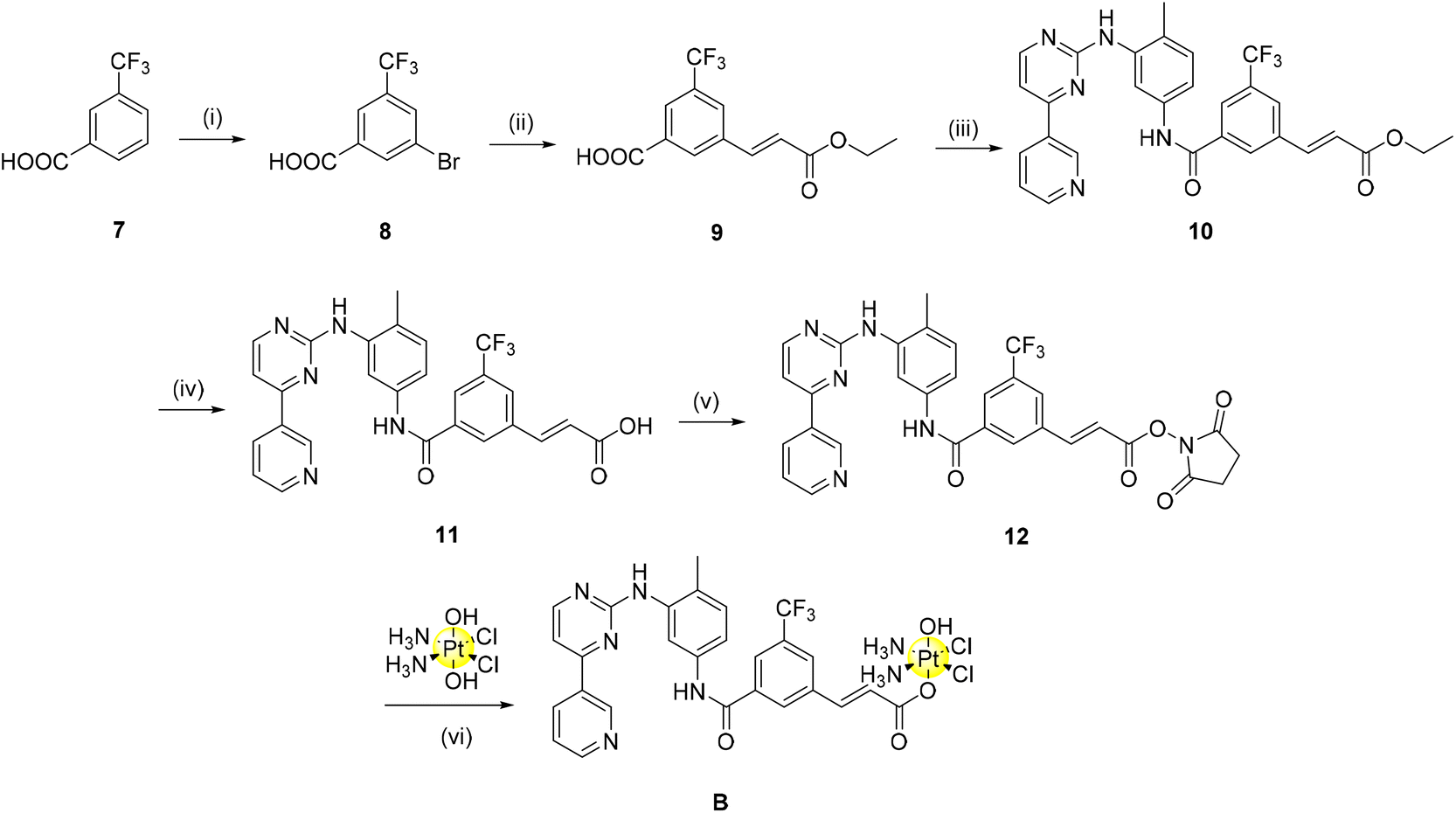 | ||
| Scheme 2 Synthesis of the alkene linker Pt(IV)-nilotinib based pro-drug. (i) TFA, NBS, H2SO4, 60 °C, 5 h, 82% (ii) ethyl acrylate, Pd(PPh3)4, TEA, sealed tube, 100 °C, 18 h, 70% s (iii) imatinib scaffold (1), TBTU, TEA, DMF, r.t., 24 h, 100% (iv) NaOH, MeOH/H2O, 70 °C, 16 h, 100% (v) TSTU, TEA, DMF, r.t., 20 min, 53% (vi) oxoplatin, DMSO, 60 °C, 24 h, 64%. | ||
The final complexes were characterised by 1D and 2D multinuclear NMR (1H, 13C, 19F, 195Pt) spectroscopy, IR spectroscopy, and mass spectrometry (Fig. S1–S11, ESI†) and the purity was assessed with elemental analysis. The complexes are stable under physiological conditions as seen from the UV spectra obtained in a solution of DMSO/PBS buffer 1/9 (Fig. S12†). The pro-drug nature of these complexes was assessed by 1H (Fig. S13a/b†) and 195Pt NMR spectroscopy through the addition of an excess of ascorbic acid to DMSO solutions of A and B.29 Both complexes are fully reduced by ascorbic acid after 24 hours, as observed by the complete disappearance of the triplet peaks at ∼6 ppm, corresponding to the NH3 protons of the equatorial amines in a Pt(IV) scaffold. Additionally, concomitant emergence of peaks assigned to the released carboxylic acid axial ligand can be observed (for example, at δ 6.87 ppm the alkene proton corresponding to the carboxylic acid 11, Fig. S12b†). 195Pt NMR of A and B with 10 equivalents of ascorbic acid after 24 hours confirmed the appearance of the cisplatin signal at −2080 ppm.
Biological studies
The Pt(IV) complexes A and B together with the free carboxylic acid derivatives 5 (A_ligand-COOH, in the computational studies) and 11 (B_ligand-COOH, in the computational studies) were tested on two cancer cell lines: neuroblastoma (SH-SY5Y) and ovarian (2008) cancer. The cells were incubated with the compounds for 72 h and the IC50 values obtained from the MTT assays are reported in Table 2.| SH-SY5Y | 2008 | |
|---|---|---|
| 5 (A_ligand-COOH) | >50 | >50 |
| 11 (B_ligand-COOH) | >50 | >50 |
| Complex A | 6.8 ± 0.9 | 4.4 ± 0.2 |
| Complex B | 5.7 ± 0.5 | 5.3 ± 0.8 |
| Cisplatin | 15.8 ± 2.3 | 2.2 ± 1.4 |
The neuroblastoma cell line SH-SY5Y was chosen for in vitro testing as it has been previously shown that a high proportion of primary neuroblastoma tumours express PDGFR and c-KIT receptors.30,31 In particular, the expression of c-KIT in neuroblastoma tumours has been demonstrated to aid tumour cell survival by suppressing apoptosis,32 while the role of PDGFR has yet to be elucidated.33 Specifically, the SH-SY5Y cell line was chosen as it expresses c-KIT, PDGFR-α and PDGFR-β.34 Against SH-SY5Y, the two complexes A and B displayed promising activity (IC50: A = 6.8 μM, B = 5.7 μM) as they were shown to be ∼3 times more active than the cisplatin reference drug (IC50: 15.8 μM). The potential of these complexes is enhanced when the activity of complexes A or B is compared to conventional combination chemotherapy of imatinib and cisplatin. Previously, it has been shown that combination chemotherapy of cisplatin with imatinib against SH-SY5Y in vitro has either an antagonistic effect or negligible benefit (depending on the concentration of imatinib) on cytotoxicity when compared to cisplatin alone.34 The complexes in this study display a clear benefit of using the Pt(IV)–imatinib conjugate approach over conventional combination chemotherapy, as activity of complexes A and B have outperformed that of cisplatin, instead of having antagonistic/negligible impact.
The ovarian cancer cell line 2008 (derived from epithelial cells) was chosen for in vitro testing as, in a similar fashion to above, numerous pre-clinical studies have indicated that both c-KIT and PDGFR may have a role in ovarian tumorigenesis, with a high frequency of PDGFR expression observed in higher-grade ovarian cancer tumours.35 Previous Phase II clinical trials of imatinib against epithelial ovarian cancer suggested that monotherapy of imatinib against platinum and taxane-resistant ovarian cancer had little activity.36 The two complexes A and B displayed similar activity (IC50: A = 4.4 μM, B = 5.3 μM) to the cisplatin reference drug (IC50: 2.2 μM) against the ovarian cancer cell line 2008. This itself is a promising preliminary result as cisplatin is the current clinically approved therapy for ovarian cancer37 and the complexes could potentially possess the ability to overcome platinum-resistance. Additionally, appropriately functionalised metal–tyrosine conjugates appear to demonstrate selectivity towards cancer cells over non-malignant human cell lines.18 Against both cell lines, complexes A and B were shown to have similar activity.
The negligible activity exhibited by the two ligands against both cell lines can be explained due to their decreased lipophilicity resulting from the presence of the carboxylic acid that negatively impacts the cellular uptake. To confirm the theoretical studies that showed both the complexes and the ligands to be effective binders to PDGFR and c-KIT receptors due to appropriately conducted functionalisation, the inhibitory activity of these compounds was evaluated on the isolated enzymes and the corresponding IC50 values are reported in Table 3. As visible from the heat chart in Fig. 6, both complexes are strong inhibitors of both receptors, with ligand 5 being very effective against PDGFRα.
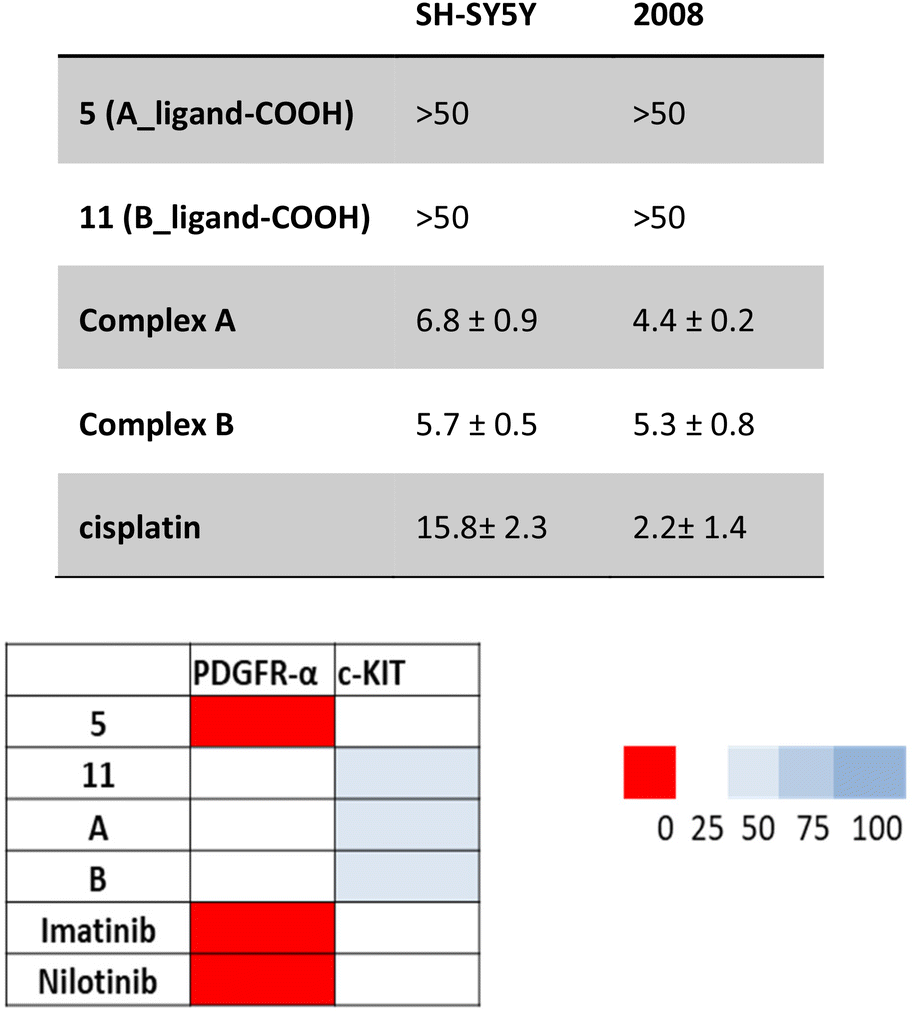 | ||
| Fig. 6 Inhibition of isolated enzyme isoforms. Heat-map showing the relative inhibition of tested compounds (at 0.2 μM) on PDGFRα and c-KIT enzymes. Results are expressed as enzyme residual activity. Data are the means of three independent experiments. | ||
| PDGR-α (μM) | c-KIT (μM) | |
|---|---|---|
| 5 (A_ligand-COOH) | 0.08 ± 0.02 | 0.15 ± 0.04 |
| 11 (B_ligand-COOH) | 0.11 ± 0.03 | 0.23 ± 0.08 |
| A | 0.12 ± 0.02 | 0.31 ± 0.09 |
| B | 0.15 ± 0.03 | 0.33 ± 0.06 |
| Imatinib | 0.07 ± 0.01 | 0.10 ± 0.03 |
| Nilotinib | 0.09 ± 0.02 | 0.15 ± 0.04 |
All IC50 values obtained were in the low micromolar concentrations, confirming strong inhibition properties for the two complexes A and B and the two carboxylic acids 5 (A_ligand-COOH) and 11 (B_ligand-COOH). These results have enabled us to confirm the theoretical approach.
Overall, these preliminary results are interesting and seem to confirm the hypothesis that the combination of the two biologically active moieties released upon intracellular reduction (cisplatin scaffold and TKIs imatinib and nilotinib) outperform conventional combination chemotherapy of imatinib/nilotinib with cisplatin. More detailed biological analyses (i.e. drug uptake, cellular TK inhibition, in vivo studies, etc.) are planned to better understand the mechanism of action and fully determine their potential in a clinical setting.
Experimental section
Detailed material and methods are reported in the ESI.† The authors have cited additional references within the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41–65
41–65
Syntheses
Compound 1, cisplatin and oxoplatin were synthesised as previously reported.27,38,39:MeOH = 9:1, Rf = 0.6). Using a pipette, the solution was added dropwise to saturated NH4Cl aqueous solution (90 mL). The precipitate was collected via vacuum filtration, washed with abundant H2O, and dried using a Schlenk line. The solid was used without further purification (yield: 0.25 g, 88%). 1H NMR (500 MHz, CDCl3): δ 9.25 (dd, J = 0.7, 2.27 Hz, 1H); 8.71 (dd, J = 1.7, 4.80 Hz, 1H); 8.64 (d, J = 1.71 Hz, 1H); 8.54 (m, 2H); 8.53 (m, 1H); 8.51 (t, J = 1.6 Hz, 1H); 8.22 (m, 1H); 8.17 (m, 1H); 7.94 (s, 1H); 7.60 (t, J = 7.8 Hz, 1H); 7.45 (ddd, J = 0.8, 4.8, 7.9 Hz, 1H); 7.34 (dd, J = 5.1, 11.1 Hz); 7.24 (d, J = 8.4 Hz, 1H); 7.21 (d, J = 5.2 Hz, 1H); 7.04 (s, 1H); 3.97 (s, 3H); 2.37 (s, 3H). 13C NMR (126 MHz, CDCl3): δ 183.77, 175.46, 166.31, 162.81, 160.53, 159.06, 151.53, 148.53, 136.27, 134.97, 132.67, 132.06, 130.87, 130.63, 129.19, 127.47, 123.77, 115.27, 112.98, 108.49, 52.49, 17.72. IR (ATR): 2953, 1721, 1646, 1578, 1525, 1444, 1245, 796, 607 cm−1. LC-MS: calculated mass for (M + H)+: 439.475, found: 440 (93.4%). HR-MS: calculated mass for (M + H)+: 440.1723. Found: 440.1721.
:MeOH = 9:1, Rf = 0.475). 0.036 g (0.91 mmol, 2 eq.) of NaOH was dissolved in 2 ml of deionised H2O and added to the MeOH solution. The solution was heated at 45 °C for a further 4 hours (TLC, CHCl3:MeOH = 9:1). The solution was allowed to cool to room temperature prior to removal of the organic solvent in vacuo. ∼40 ml of 0.5 M aqueous HCl was added to the remaining aqueous solvent until complete precipitation. The precipitate was collected via vacuum filtration, washed with abundant H2O, and dried using a Schlenk line (yield: 0.173 g, 90%). 1H NMR (500 MHz, DMSO-d6): δ 10.44 (s, 1H); 9.41 (s, 1H); 9.14 (s, 1H); 8.86 (t, J = 5.8, 6.5 Hz, 2H); 8.60 (d, J = 4.9 Hz, 1H); 8.53 (s, 1H); 8.21 (d, J = 7.8 Hz, 1H); 8.13 (d, J = 7.2 Hz, 1H); 7.86 (m, 1H); 7.66 (t, J = 7.7, 7.73 Hz, 1H); 7.54 (d, J = 5.1 Hz, 1H); 7.48 (d, J = 8.3 Hz, 1H); 7.22 (m, 1H); 2.24 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6): δ 167.33, 165.12, 161.44, 160.18, 138.02, 137.49, 135.69, 134.16, 132.56, 131.47, 130.62, 129.31, 128.94, 128.27, 125.86, 117.78, 117.46, 108.24, 104.15, 103.81, 103.32. IR (ATR): 2953, 2551, 2023, 1657, 1411, 1248, 989, 804, 666 cm−1. LC-MS: calculated mass for (M + H)+: 426.448, found: 426.1 (100%). Rf (CHCl3:MeOH, 9:1): 0.1. HR-MS: calculated mass for (M + H)+: 426.1566. Found: 426.1562. Characterisation data were analogous to a previously synthesised TFA salt of 3.40
:MeOH = 9:1, Rf = 0.45). The solvent was removed in vacuo at 60 °C. 3 ml of DMF was added to the residue and transferred into a Falcon tube. 33 ml of saturated NH4Cl aqueous solution was added to the tube. The resulting precipitate was collected via centrifugation, washed with 20 ml of deionised and then dried using a Schlenk line (yield: 0.090 g, 73%).1H NMR (500 MHz, DMSO-d6): δ 10.36 (s, 1H, amide–NH); 9.27 (d, J = 1.7 Hz, 1H); 8.99 (s, 1H, NH); 8.68 (m, 2H); 8.51 (d, J = 5.1 Hz, 1H); 8.48 (m, 1H); 8.40 (s, 1H); 8.09 (d, J = 1.9 Hz, 1H); 8.06 (d, J = 7.8 Hz, 1H); 8.01 (m, 1H); 7.61 (t, J = 7.8 Hz, 1H); 7.52 (m, 2H); 7.43 (d, J = 5.2 Hz, 1H); 7.22 (d, 8.5 Hz, 1H); 3.58 (s, 3H); 3.29 (m, 2H overlapped); 2.38 (m, 3H); 2.24 (s, 3H); 1.79 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 173.15, 165.81, 165.03, 161.62, 161.18, 159.48, 151.39, 148.19, 135.27, 134.75, 134.42, 130.15, 130.08, 128.43, 126.54, 123.81, 117.21, 116.74, 107.54, 51.27, 38.61, 30.80, 24.43, 17.67. LC-MS: calculated mass for (M + H)+: 523, found: 523 (95.7%). IR (ATR): 3284, 2928, 1731, 1658, 1579, 1526, 1478, 1442, 1415, 1296, 1252, 1171, 1090, 1025, 800, 688 cm−1. HR-MS: calculated mass for (M + Na)+: 547.2064. Found: 547.2063 (M + Na)+.
:MeOH = 9:1, Rf = 0.44). A further 0.03 g (0.7624 mmol, 2 eq.) of NaOH was dissolved in 2 ml of deionised H2O and added to the MeOH/H2O solution. The solution was heated at 45 °C for 4 hours (TLC, DCM:MeOH = 9:1). The solution was allowed to cool to room temperature prior to removal of the organic solvent in vacuo. The solution was neutralised using a minimum amount of 0.1 M HCl (and saturated NaHCO3 aqueous solution if a pH ∼7 was surpassed), until a light precipitate was visible. The mixture was kept on an ice bath for 15 minutes. The pale precipitate was collected via vacuum filtration, washed with abundant H2O, and dried using a Schlenk line (yield: 0.153 g, 78%). 1H NMR (500 MHz, DMSO-d6): δ 10.46 (s, 1H, amide-NH); 9.45 (s, 1H); 9.28 (s, 1H); 8.99 (s, 1H); 8.69 (d, J = 4.6 Hz, 1H); 8.52 (d, J = 5.1 Hz, 1H); 8.49 (d, J = 1.6 Hz, 1H); 8.48 (m, 1H); 8.13 (d, J = 1.8 Hz, 1H); 8.05 (t, J = 8.3 Hz, 2H); 7.60 (t, J = 7.7 Hz, 1H); 7.56 (m, 1H); 7.53 (m, 1H); 7.43 (d, J = 5.1 Hz, 1H); 7.22 (d, J = 8.4 Hz, 1H); 3.29 (unknown, 2H overlapped); 2.23 (s, 3H); 2.18 (t, J = 6.7 Hz, 2H); 1.76 (s, 2H). 13C NMR (126 MHz, DMSO-d6): δ 165.96, 165.48, 162.08, 161.66, 159.95, 151.85, 148.65, 138.26, 137.64, 135.61, 135.45, 134.91, 130.52, 130.50, 128.91, 126.98, 124.29, 117.72, 117.32, 107.99, 40.57, 34.88, 25.19, 18.15. LC-MS: calculated mass for (M + H)+: 511, found: 511.5. (95.9%). IR (ATR): 3281, 1644, 1579, 1555, 1527, 1447, 1399, 1286, 1254, 1093, 1024, 1006, 796, 688 cm−1. Rf (DCM:MeOH, 9:1) = 0.44. HR-MS: calculated mass for (M + H)+: 511.2088. Found: (M + H)+ 511.2084.
:MeOH = 9:1, Rf = 0.33). The solution was then removed in vacuo at 45 °C. 30 ml of ice cold deionised water was added. The resulting precipitate was collected via centrifugation, washed with 20 ml of deionised water and dried using a Schlenk line. The crude was purified using silica gel column chromatography (CHCl3: acetone, 2:8) (yield: 0.06 g, 33%). Rf (CHCl3:MeOH, 9:1) = 0.33. 1H NMR (500 MHz, DMSO-d6): δ 10.36 (d, J = 4.3 Hz, 1H); 9.29 (s, 1H); 9.00 (s, 1H); 8.73 (t, J = 5.5 Hz, 1H); 8.70 (s, 1H); 8.52 (d, J = 5.1 Hz, 1H); 8.49 (d, J = 8.0 Hz, 1H); 8.41 (d, J = 6.9 Hz, 1H); 8.11–8.00 (m, 3H); 7.62 (td, J = 7.8, 3.8 Hz, 1H); 7.56–7.49 (m, 2H); 7.44 (d, J = 5.2 Hz, 1H); 7.23 (d, J = 8.4 Hz, 1H); 3.31–3.21 (m, 2H); 2.85–2.75 (m, 4H); 2.24 (s, 2H); 1.96–1.87 (m, 2H). 13C NMR (126 MHz, DMSO-d6): δ 170.71, 169.35, 166.42, 165.52, 162.10, 161.66, 159.96, 151.86, 148.66, 138.32, 137.54, 135.78, 135.14, 134.90, 130.68, 130.57, 128.91, 128.25, 127.05, 124.30, 117.68, 117.21, 108.02. IR (ATR): 3273, 2927, 1813, 1733, 1643, 1578, 1526, 1447, 1412, 1299, 1253, 1204, 1138, 1069, 877, 801, 704 cm−1. HRMS: calculated mass for (M + H)+: 608.2252. Found: 608.2213. LC-MS: calculated mass for (M + H)+: 608, found: 608.4 (92.3%).
:MeOH = 9:1, Rf = 0.675). No colour change was observed. The solution was allowed to cool to room temperature and was then added to 40 ml of a water/ice mixture. The white precipitate was collected via vacuum filtration, washed with a large amount of H2O and residual solvent was removed in vacuo using a Schlenk line (yield: 1.1 g, 82%). 1H NMR (500 MHz, CDCl3): δ 8.42 (s, 1H); 8.30 (d, 1H); 8.01 (s, 1H), 2.45 (s, 1H). 13C NMR (126 MHz, CDCl3): δ 168.26, 136.29, 133.31, 132.89, 132.69, 131.61, 125.51, 123.00. 19F NMR (470 MHz, CDCl3): δ 62.95 (s, 3F).
:EtOAc = 8:2, Rf = 0.125). A dark black colour was observed. The mixture was allowed to cool prior to adding to 40 ml of 0.1M HCl by using a minimum amount of acetone was to aid with mass transfer. The resulting precipitate was collected via vacuum filtration, washed with deionised water (30 ml) and n-hexane (50 ml). The crude solid was filtered through Celite by dissolving in ∼400 mL of DCM, washed with 2 × 30 ml of Brine, dried using anhydrous Na2SO4, concentrated in vacuo, dried using a Schlenk line, transferred into a vial using 25 ml of diethyl ether and dried again (yield: 0.36 g, 70%). 1H NMR (500 MHz, DMSO-d6): δ 13.70 (broad s, 1H, COO-H); 8.49 (s, 1H); 8.44 (s, 1H); 8.17 (d, J = 7.5 Hz, 1H); 7.84 (dd, J = 7.0, 16.14 Hz, 1H); 6.94 (dd J = 6.1, 16.1 Hz, 1H); 4.22 (q, J = 7.1, 2H); 1.28 (t, J = 7.1, 3H). 13C NMR (126 MHz, DMSO-d6): δ 166.28, 166.15, 142.24, 136.64, 133.35, 133.11, 130.81, 130.57, 129.13, 127.05, 125.10, 122.93, 122.07, 60.82, 14.63. 19F NMR (470 MHz, CDCl3): δ 61.35 (s, 3F). IR (ATR): 2990, 2599, 1716, 1664, 1646, 1449, 1274, 1187, 1125, 869, 686 cm−1. HR-MS: calculated mass for (M − H)−: 287.0537. Found: 287.0535 (M − H)−.
:MeOH = 9:1, Rf = 0.9). The solvent was removed in vacuo. The residue was triturated in 50 ml of H2O for 15 minutes. The resulting precipitate was collected via centrifugation, washed with 2 × 30 ml of deionised water and dried using a Schlenk line (yield: 0.88 g, 100%). 1H NMR (500 MHz, DMSO-d6): δ 10.45 (s, 1H), 9.31 (d, J = 1.6 Hz, 1H), 9.00 (s, 1H), 8.70 (dd, J = 4.7, 1.4 Hz, 1H), 8.61 (s, 1H), 8.53 (d, J = 5.1 Hz, 1H), 8.49 (d, J = 8.0 Hz, 1H), 8.36 (s, 1H), 8.27 (s, 1H), 8.12 (s, 1H), 7.85 (d, J = 16.1 Hz, 1H), 7.54 (dd, J = 7.8, 4.7 Hz, 1H), 7.51–7.47 (m, 1H), 7.45 (d, J = 5.1 Hz, 1H), 7.26 (d, J = 8.2 Hz, 1H), 6.97 (d, J = 16.1 Hz, 1H), 4.23 (q, J = 7.1 Hz, 2H), 2.25 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6): 166.31, 163.83, 162.09, 161.59, 159.99, 151.88, 148.71, 142.49, 138.41, 137.08, 137.03, 136.20, 134.91, 132.64, 130.95, 130.67, 130.41, 128.47, 128.40, 126.07, 124.28, 121.91, 117.69, 117.27, 108.12, 60.82, 18.14, 14.62. 19F NMR (470 MHz, DMSO-d6): δ −61.157 (s, 3F). HRMS: calculated mass for (M + Na)+: 570.1723. Found: 570.1722 (M + Na)+. Difference −0.09 ppm (99.07% IMS). IR (ATR): 3286, 2994, 1768, 1653, 1600, 1574, 1534, 1360, 1290, 1170, 997, 795, 691 cm−1. LC-MS: calculated mass for (M + H)+: 548.6, found: 548.6 (98.4%). 1H NMR data was as previously reported.27
0.5 g (0.91 mmol, 1 eq.) of 10 was suspended in 90 ml of MeOH. The mixture was heated at 45 °C to allow the ester to dissolve. 0.18 g (4.57 mmol, 5 eq.) of NaOH was dissolved in 0.5 ml of deionised H2O and added to the MeOH solution. The solution was heated at 70 °C for 4 hours (TLC, CHCl3:MeOH = 9:1, Rf = 0.55). The organic solvent was removed in vacuo. 0.1 M HCl was added to the remaining aqueous solvent until complete precipitation (∼pH = 4). The precipitate was collected via vacuum filtration, washed with abundant H2O, 1 × 10 ml of diethyl ether and dried using a Schlenk line. The solid was triturated with 30 ml of CHCl3, collected via vacuum filtration as before, washed with 2 × 20 ml of CHCl3 and dried using a Schlenk line (yield: 0.29 g, 61%). 1H NMR (500 MHz, DMSO-d6): δ 10.46 (s, 1H), 9.31 (s, 1H), 9.00 (s, 1H), 8.70 (d, J = 3.7 Hz, 1H), 8.58 (s, 1H), 8.53 (d, J = 5.1 Hz, 1H), 8.49 (d, J = 8.0 Hz, 1H), 8.28 (d, J = 18.1 Hz, 2H), 8.12 (s, 1H), 7.76 (d, J = 16.0 Hz, 1H), 7.52 (ddd, J = 10.2, 8.0, 3.4 Hz, 2H), 7.45 (d, J = 5.1 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 16.1 Hz, 1H), 2.26 (s, 3H). 13C NMR (126 MHz, DMSO-d6): 167.77, 167.50, 163.61, 161.86, 161.34, 159.98, 159.72, 151.63, 148.72, 148.45, 138.16, 136.76, 136.27, 134.66, 130.50, 130.38, 128.20, 124.01, 123.19, 117.45, 117.02, 107.86, 17.89. 19F NMR (470 MHz, DMSO-d6): δ −61.147 (s, 3F). IR (ATR): 3276, 1645, 1581, 1529, 1448, 1280, 1120, 1046, 795, 689 cm−1. LC-MS: calculated mass for (M + H)+: 520, found: 520.2 (100%). HR-MS: calculated mass for (M + H)+: 520.1591. Found: 520.1550 (M + H)+. 1H NMR data was as previously reported.27
:MeOH = 9:1, Rf = 0.5). The solution was then removed in vacuo at 45 °C. 33 ml of cold deionised water was added to the residue. The resulting precipitate was collected via centrifugation, washed with 20 ml of deionised water and dried using a Schlenk line. The crude was purified using silica gel column chromatography (CHCl3:acetone, 1:1, Rf = 0.45) (yield: 0.047 g, 53%). 1H NMR (500 MHz, DMSO-d6): δ 10.48 (s, 1H), 9.31 (d, J = 1.7 Hz, 1H), 9.00 (s, 1H), 8.72 (s, 1H), 8.70 (dd, J = 4.7, 1.5 Hz, 1H), 8.54 (d, J = 5.1 Hz, 1H), 8.52–8.46 (m, 2H), 8.34 (s, 1H), 8.19 (d, J = 16.1 Hz, 1H), 8.12 (d, J = 1.5 Hz, 1H), 7.52 (ddd, J = 10.4, 8.1, 3.4 Hz, 2H), 7.45 (d, J = 5.2 Hz, 1H), 7.33 (d, J = 16.2 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 2.88 (s, 4H), 2.26 (s, 3H). 13C NMR (126 MHz, DMSO-d6): 170.78, 163.80, 162.51, 162.11, 161.60, 159.98, 151.89, 148.72, 147.97, 138.44, 135.40, 134.90, 132.64, 132.02, 130.68, 128.49, 124.27, 117.66, 117.22, 115.67, 108.12, 26.01, 18.16. LC-MS: calculated mass for (M + H)+: 617, found: 617 (100%). 19F NMR (470 MHz, DMSO-d6): δ −61.143 (s, 3F). IR (ATR): 3278, 1758, 1601, 1406, 1178, 1046, 1008, 872, 621 cm−1. HR-MS: calculated mass for (M + H)+: 617.1755. Found: 617.1746 (M + H)+.
Conclusion
For the first time, two novel Pt(IV) complexes based on cisplatin with the tyrosine kinase inhibitors imatinib and nilotinib have been designed, synthesized, characterised and their preliminary anticancer activity and enzymatic inhibition properties studied. A detailed theoretical study, comprising DFT optimisation, docking and molecular dynamics, was used to analyse whether or not the functionalisation of the two inhibitors (imatinib and nilotinib) with the platinum centre could jeopardize the inhibitory activity on three TK targets, PDGFR-α, c-KIT and the T670I mutated c-KIT. Based on the promising results obtained from the calculations, the two ligands and the corresponding Pt(IV) complexes were prepared and characterised and the inhibitory properties of the complexes and of the ligands were confirmed on the isolated enzymes. Additionally, the preliminary anticancer activity of the novel platinum(IV) pro-drugs, which are stable under physiological conditions, demonstrated that this novel approach is highly promising, with strong activity against neuroblastoma (SH-SY5Y) and ovarian (2008) cancer cell lines.As aforementioned, these preliminary results are very intriguing and seem to confirm the hypothesis that the combination of the two biologically active moieties released upon intracellular reduction (cisplatin scaffold and TKIs imatinib and nilotinib) outperform conventional combination chemotherapy of imatinib/nilotinib with cisplatin. More detailed biological analyses (i.e. drug uptake, cellular TK inhibition, in vivo studies, etc.) are planned to better understand the mechanism of action and fully determine their potential in a clinical setting.
Author contributions
D. F. B.: Investigation, formal analysis, funding acquisition, visualization, writing – original draft, writing – review & editing. B. F.: investigation, formal analysis. C. D.: investigation. V. G.: data curation, supervision, visualization. I. R.: data curation, funding acquisition, visualization, writing. T. V. T.: conceptualization, data curation, supervision, visualization, writing – review & editing. D. M.: conceptualization, data curation, funding acquisition, project administration, supervision, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. F. B. would like to thank the Irish Research Council for funding the research via a Government of Ireland Postgraduate Scholarship (GOIPG/2020/55). D. M. and T. V. T. thankfully acknowledge support from Science Foundation Ireland (SFI): SFI 2012 Strategic Opportunity Fund (Infrastructure award 12/RI/2346/SOF) for NMR facilities. SFI Opportunistic Infrastructure Fund 2016 (16/RI/3399) for LC-MS. B. F. and I. R. gratefully acknowledge financial support from Science Foundation Ireland (20/FFP-P/8760). Thanks to Johnson Matthey that provided the catalyst [Pd(PPh3)4] through the JM PGM Award Scheme PGMAS54.References
- M. K. Paul and A. K. Mukhopadhyay, Int. J. Med. Sci., 2012, 101–115 Search PubMed.
- C. B. Gambacorti-Passerini, R. H. Gunby, R. Piazza, A. Galietta, R. Rostagno and L. Scapozza, Lancet Oncol., 2003, 75–85 CrossRef PubMed.
- C. Gambacorti-Passerini, L. Antolini, F. X. Mahon, F. Guilhot, M. Deininger, C. Fava, A. Nagler, C. M. Della Casa, E. Morra, E. Abruzzese, A. D'Emilio, F. Stagno, P. Le Coutre, R. Hurtado-Monroy, V. Santini, B. Martino, F. Pane, A. Piccin, P. Giraldo, S. Assouline, M. A. Durosinmi, O. Leeksma, E. M. Pogliani, M. Puttini, E. Jang, J. Reiffers, M. G. Valsecchi and D. W. Kim, J. Natl. Cancer Inst., 2011, 103, 553–561 CrossRef CAS PubMed.
- World Health Organization Model List of Essential Medicines, World Health Organization, Geneva, 22nd List, 2021. https://iris.who.int/bitstream/handle/10665/325771/WHO-MVP-EMP-IAU-2019.06-eng.pdf Search PubMed.
- M. A. Babaei, B. Kamalidehghan, M. Saleem, H. Z. Huri and F. Ahmadipour, Drug Des., Dev. Ther., 2016, 10, 2443–2459 CrossRef CAS PubMed.
- S. Kaulfuß, H. Seemann, R. Kampe, J. Meyer, R. Dressel, B. König, J. G. Scharf and P. Burfeind, Oncotarget, 2013, 4, 1037–1049 CrossRef PubMed.
- R. Roskoski, Pharmacol. Res., 2018, 129, 65–83 CrossRef CAS PubMed.
- E. P. Reddy and A. K. Aggarwal, Genes Cancer, 2012, 3, 447–454 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Eur. J. Inorg. Chem., 2017, 2017, 1596–1612 CrossRef CAS.
- D. Gibson, J. Inorg. Biochem., 2021, 217, 111353 CrossRef CAS PubMed.
- Z. Xu, Z. Wang, Z. Deng and G. Zhu, Coord. Chem. Rev., 2021, 442, 213991 CrossRef CAS.
- C. Marotta, E. Giorgi, F. Binacchi, D. Cirri, C. Gabbiani and A. Pratesi, Inorg. Chim. Acta, 2023, 548, 121388 CrossRef CAS.
- E. Wexselblatt and D. Gibson, J. Inorg. Biochem., 2012, 220–229 CrossRef CAS PubMed.
- L. Fang, X. Qin, J. Zhao and S. Gou, Inorg. Chem., 2019, 58, 2191–2200 CrossRef CAS PubMed.
- X. Q. Song, Z. Y. Ma, Y. G. Wu, M. L. Dai, D. B. Wang, J. Y. Xu and Y. Liu, Eur. J. Med. Chem., 2019, 167, 377–387 CrossRef CAS PubMed.
- T. Babu, H. Ghareeb, U. Basu, H. Schueffl, S. Theiner, P. Heffeter, G. Koellensperger, N. Metanis, V. Gandin, I. Ott, C. Schmidt and D. Gibson, Angew. Chem., Int. Ed., 2023, 62, e202217233 CrossRef CAS PubMed.
- D. F. Beirne, M. Dalla Via, T. Velasco-Torrijos and D. Montagner, Coord. Chem. Rev., 2022, 469, 214655 CrossRef CAS.
- R. Kok, K. Temming and M. Fretz, Curr. Mol. Pharmacol., 2010, 1–12 Search PubMed.
- Y. Wei, D. C. Poon, R. Fei, A. S. M. Lam, S. C. F. Au-Yeung and K. K. W. To, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- Y. Zhang, Q. Luo, W. Zheng, Z. Wang, Y. Lin, E. Zhang, S. Lü, J. Xiang, Y. Zhao and F. Wang, Inorg. Chem. Front., 2018, 5, 413–424 RSC.
- C. Li, F. Xu, Y. Zhao, W. Zheng, W. Zeng, Q. Luo, Z. Wang, K. Wu, J. Du and F. Wang, Front. Chem., 2020, 8, 1–14 CrossRef PubMed.
- M. Yang, H. Wu, J. Chu, L. A. Gabriel, Y. Kim, K. S. Anderson, C. M. Furdui and U. Bierbach, Chem. Commun., 2018, 54, 7479–7482 RSC.
- M. Yang, PhD Thesis, Wake Forest University, North Carolina, United States of America, 2017. https://wakespace.lib.wfu.edu/handle/10339/89873.
- S. Harmsen, M. E. M. Dolman, Z. Nemes, M. Lacombe, B. Szokol, J. Pató, G. Kéri, L. Öfi, G. Storm, W. E. Hennink and R. J. Kok, Bioconjugate Chem., 2011, 22, 540–545 CrossRef CAS PubMed.
- C. D. Mol, D. R. Dougan, T. R. Schneider, R. J. Skene, M. L. Kraus, D. N. Scheibe, G. P. Snell, H. Zou, B. C. Sang and K. P. Wilson, J. Biol. Chem., 2004, 279, 31655–31663 CrossRef CAS PubMed.
- M. Dalla Via, PhD Thesis, Università degli Studi di Padova, 2018. https://www.research.unipd.it/handle/11577/3421807.
- E. Moynihan, G. Bassi, A. Ruffini, S. Panseri, M. Montesi, T. Velasco-Torrijos and D. Montagner, Front. Chem., 2021, 9, 795997 CrossRef CAS PubMed.
- E. Moynihan, S. Panseri, G. Bassi, A. Rossi, E. Campodoni, E. Dempsey, M. Montesi, T. Velasco-Torrijos and D. Montagner, Int. J. Mol. Sci., 2023, 24, 6028 CrossRef CAS PubMed.
- P. S. Cohen, J. P. Chan, M. Lipkunskaya, J. L. Biedler and R. C. Seeger, Blood, 1994, 84, 3465–3472 CrossRef CAS PubMed.
- T. Matsui, K. Sano, T. Tsukamoto, M. Ito, T. Takaishi, H. Nakata, H. Nakamura and K. Chihara, J. Clin. Invest., 1993, 92, 1153–1160 CrossRef CAS PubMed.
- F. Timeus, N. Crescenzio, P. Valle, P. Pistamiglio, M. Piglione, E. Garelli, E. Ricotti, P. Rocchi, P. Strippoli, L. Cordero di Montezemolo, E. Madon, U. Ramenghi and G. Basso, Exp. Hematol., 1997, 25, 1253–1260 CAS.
- A. Shimada, J. Hirato, M. Kuroiwa, A. Kikuchi, R. Hanada, K. Wakai and Y. Hayashi, Pediatr. Blood Cancer, 2008, 50, 213–217 CrossRef PubMed.
- E. Palmberg, J. I. Johnsen, J. Paulsson, H. Gleissman, M. Wickström, M. Edgren, A. Östman, P. Kogmer and M. Lindskog, Int. J. Cancer, 2009, 124, 1227–1234 CrossRef CAS PubMed.
- R. E. Schmandt, R. Broaddus, K. H. Lu, H. Shvartsman, A. Thornton, A. Malpica, C. Sun, D. C. Bodurka and D. M. Gershenson, Cancer, 2003, 98, 758–764 CrossRef CAS PubMed.
- R. L. Coleman, R. R. Broaddus, D. C. Bodurka, J. K. Wolf, T. W. Burke, J. J. Kavanagh, C. F. Levenback and D. M. Gershenson, Gynecol. Oncol., 2006, 101, 126–131 CrossRef CAS PubMed.
- S. Dasari and P. Bernard Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- S. Dhara, Indian J. Chem., 1970, 8, 193–194 Search PubMed.
- R. J. Brandon and J. C. Dabrowiak, J. Med. Chem., 1984, 27, 861–865 CrossRef CAS PubMed.
- P. Finkbeiner, J. P. Hehn and C. Gnamm, J. Med. Chem., 2020, 63, 7081–7107 CrossRef CAS PubMed.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS.
- G. Madhavi Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput. Aided Mol. Des., 2013, 27, 221–234 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- W. Sherman, H. S. Beard and R. Farid, Chem. Biol. Drug Des., 2006, 67, 83–84 CrossRef CAS PubMed.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Curr. Comput.-Aided Drug Des., 2011, 7, 145–157 Search PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154 CrossRef CAS PubMed.
- J. M. Seminario, Int. J. Quantum Chem., 1996, 60, 1271–1277 CrossRef.
- S. K. Burger, M. Lacasse, T. Verstraelen, J. Drewry, P. Gunning and P. W. Ayers, J. Chem. Theory Comput., 2012, 8, 554–562 CrossRef CAS PubMed.
- S. Zheng, Q. Tang, J. He, S. Du, S. Xu, C. Wang, Y. Xu and F. Lin, J. Chem. Inf. Model., 2016, 56, 811–818 CrossRef CAS PubMed.
- S. Boonstra, P. R. Onck and E. Van Der Giessen, J. Phys. Chem. B, 2016, 120, 3692–3698 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. Van Der Spoel and R. Van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- S. Páll, A. Zhmurov, P. Bauer, M. Abraham, M. Lundborg, A. Gray, B. Hess and E. Lindahl, J. Chem. Phys., 2020, 153, 134110 CrossRef PubMed.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- J. Srinivasan, T. E. Cheatham, P. Cieplak, P. A. Kollman and D. A. Case, J. Am. Chem. Soc., 1998, 120, 9401–9409 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- C. Wang, D. Greene, L. Xiao, R. Qi and R. Luo, Front. Mol. Biosci., 2018, 4, 87 CrossRef PubMed.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theor. Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theor. Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- Available online: https://www.discoverx.com/products-applications/kinase-solutions?gclid=EAIaIQobChMIs-3mrL2V_gIVlM13Ch0RrAK8EAAYAiAAEgJEsPD_BwER.J.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt02030d |
| This journal is © The Royal Society of Chemistry 2023 |