
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectroscopic and computational study of the interaction of Pt(II) pyrrole-imine chelates with human serum albumin†
Sheldon
Sookai
 a and
Orde Q.
Munro
*ab
a and
Orde Q.
Munro
*ab
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO WITS 2050, Johannesburg, South Africa. E-mail: S.Sookai92@gmail.com
bSchool of Chemistry University of Leeds, Woodhouse Lane, LS2 9JT, UK. E-mail: O.Munro@leeds.ac.uk
First published on 4th September 2023
Abstract
Three bis(pyrrolide-imine) Pt(II) chelates were synthesised and characterized with different bridging alkyl groups, specifically 2-hydroxypropyl (1), 2,2-dimethylpropyl (2), and 1,2-(S,S)-(+)-cyclohexyl (3). Novel compounds 1 and 2 were analysed by single-crystal X-ray diffraction (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ). The asymmetric unit of 1 comprises three independent molecules linked by hydrogen bonds involving the OH groups, forming a trimeric supramolecular structure. The Pt(II) chelates were reacted with human serum albumin (HSA) to investigate how the ligand bound to the Pt(II) ion influences the compound's affinity for HSA. Fluorescence quenching data obtained for native HSA and HSA bound to site-specific probes (warfarin, subdomain IIA; ibuprofen, subdomain IIIA) indicated that the three Pt(II) chelates bind close enough (within ∼30 Å) to Trp-214 to quench its intrinsic fluorescence. The bimolecular quenching constant (kq) was 103–104 -fold higher than the maximum diffusion-controlled collision constant in water (1010 M s−1) at 310 K, while the affinity constants, Ka, ranged from ∼5 × 103 to ∼5 × 105 at 310 K, and followed the order 1 > 3 > 2. The reactions of 1 and 3 with HSA were enthalpically driven, while that for 2 was entropically driven. Macromolecular docking simulations (Glide XP) and binding site specificity assays employing site-specific probes and UV-vis CD spectroscopy indicated that 1 and 2 target Sudlow's site II in subdomain IIIA, minimally perturbing the tertiary structure of the protein. Well-resolved induced CD signals from 1 and 2 bound to HSA in subdomain IIIA were adequately simulated by hybrid QM:MM TD-DFT methods. We conclude that the structure of the bis(pyrrolide-imine) Pt(II) chelate measurably affects its uptake by HSA without detectable decomposition or demetallation. Such compounds could thus serve as metallodrug candidates capable of utilising an HSA-mediated cellular uptake pathway.
). The asymmetric unit of 1 comprises three independent molecules linked by hydrogen bonds involving the OH groups, forming a trimeric supramolecular structure. The Pt(II) chelates were reacted with human serum albumin (HSA) to investigate how the ligand bound to the Pt(II) ion influences the compound's affinity for HSA. Fluorescence quenching data obtained for native HSA and HSA bound to site-specific probes (warfarin, subdomain IIA; ibuprofen, subdomain IIIA) indicated that the three Pt(II) chelates bind close enough (within ∼30 Å) to Trp-214 to quench its intrinsic fluorescence. The bimolecular quenching constant (kq) was 103–104 -fold higher than the maximum diffusion-controlled collision constant in water (1010 M s−1) at 310 K, while the affinity constants, Ka, ranged from ∼5 × 103 to ∼5 × 105 at 310 K, and followed the order 1 > 3 > 2. The reactions of 1 and 3 with HSA were enthalpically driven, while that for 2 was entropically driven. Macromolecular docking simulations (Glide XP) and binding site specificity assays employing site-specific probes and UV-vis CD spectroscopy indicated that 1 and 2 target Sudlow's site II in subdomain IIIA, minimally perturbing the tertiary structure of the protein. Well-resolved induced CD signals from 1 and 2 bound to HSA in subdomain IIIA were adequately simulated by hybrid QM:MM TD-DFT methods. We conclude that the structure of the bis(pyrrolide-imine) Pt(II) chelate measurably affects its uptake by HSA without detectable decomposition or demetallation. Such compounds could thus serve as metallodrug candidates capable of utilising an HSA-mediated cellular uptake pathway.
Introduction
Schiff base metal chelates have been extensively studied for their bioactivity and potential use in pharmaceuticals, with pyrrole-imine derivatives being among the most commonly synthesised due to their ease of preparation.1,2 These key ligands are formed by a condensation reaction between a primary aryl- or alkylamine and pyrrole-2-carboxaldehyde (or a pyrrole-2-ketone).3 Upon reaction with suitable metal ions, deprotonation of the pyrrole nitrogen occurs. Pyrrole-imine chelating agents are therefore strong σ-donor ligands with π-bonding character that dextrously stabilize metal ions in both low and high oxidation states, including Ni(II),4–6 Pd(II),6–8 Pt(II),9–11 Cu(II),12–15 Ag(I),16 and Au(III).17Pt(II) complexes are among the most widely studied metal complexes in medicine. Since the FDA approval of cisplatin in 1978 and its significant clinical success, work on a plethora of Pt(II) metallodrug candidates has flourished18 mainly to discover derivatives with reduced toxicity alongside responding to the emergence of cisplatin resistant tumours.19 It is postulated that one of the main reasons for the poor therapeutic index and toxic side effects of Pt(II) compounds is their interaction with serum proteins, in particular human serum albumin (HSA).20 For reference, after intravenous administration, between 68–98% of cisplatin is bound by serum proteins, mostly HSA, and up to five cisplatin molecules bind to a single HSA molecule.21 To maximize the activity and improve the safety of Pt(II) metallodrugs more broadly, it is essential to understand the binding mechanism of Pt(II) complexes to HSA (and other macromolecules) and to develop novel candidates with high biological stability and good solubility.
HSA is synthesised in the liver and is the most abundant serum protein with a concentration of 35–50 g L−1. The protein has three main functions in vivo: (i) aiding to maintain physiological pH, (ii) maintaining osmotic pressure, and (iii) the transport of endogenous and exogenous compounds.22 HSA comprises a single polypeptide chain with a heart-shaped tertiary structure consisting of 585 amino acids, culminating in a molar mass of 66.5 kDa (Fig. 1).23 The protein has three domains (I, II, and III), each made up of 10 α-helices and two subdomains (A and B).23,24 The two main drug binding sites are located in subdomains IIA and IIIA and are commonly known as Sudlow's site I and Sudlow's site II.25,26 Apart from Sudlow's sites I and II, HSA has several other binding sites including 4 thyroxine, 7 fatty acid, and several known metal ion binding sites.27 Delineating the mechanistic details of metal complex uptake by HSA is crucial to understanding in vivo pharmacokinetic and pharmacodynamic data of such systems, given the protein's role as the quintessential drug transporter.28
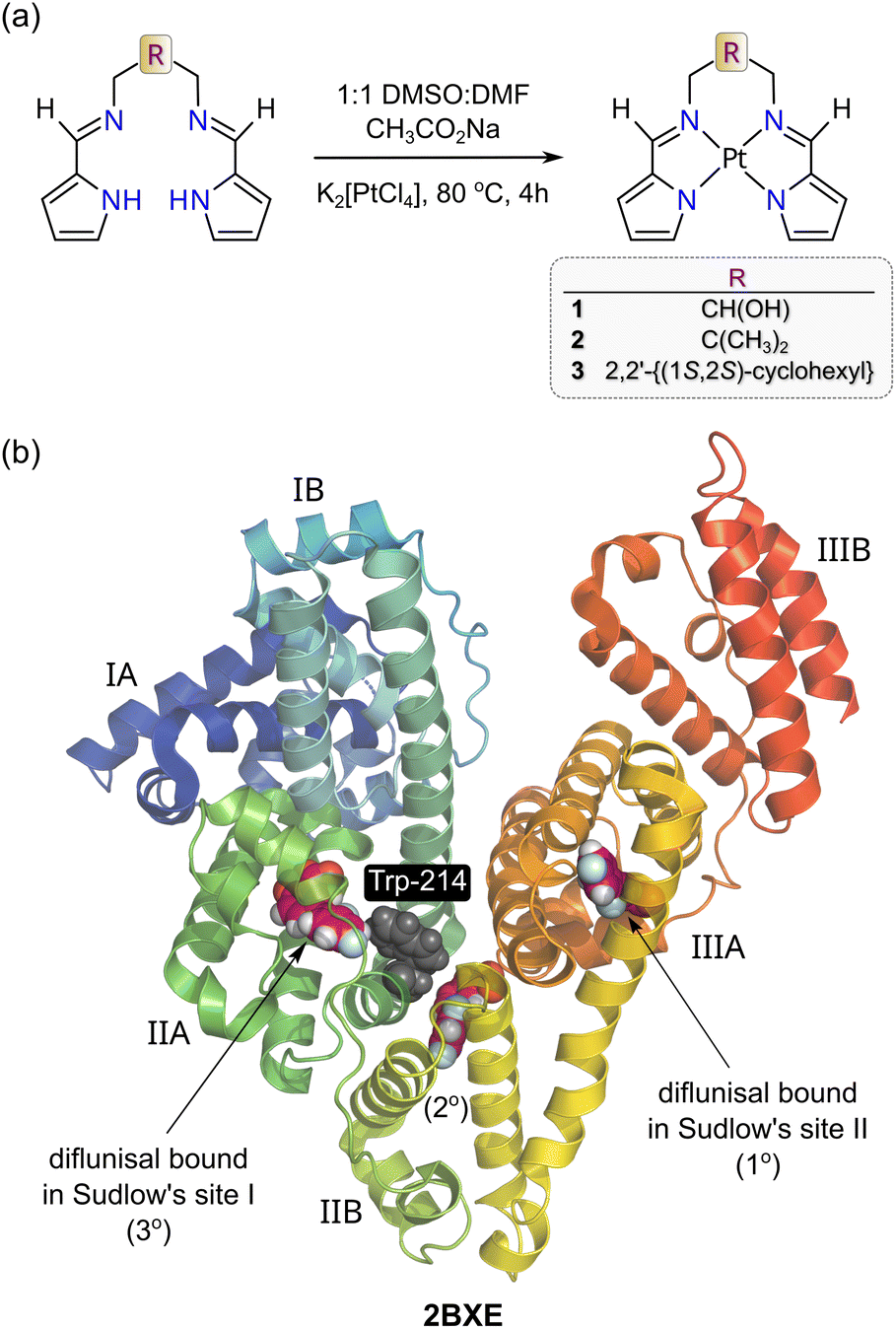 | ||
| Fig. 1 (a) Schematic synthesis procedure for the metalation of the three bis(pyrrole-imine) ligands with K2[PtCl4] relevant to this work. (b) X-ray structure of HSA bound to diflunisal (redrawn from PDB code 2BXE) illustrating the protein's two main drug binding sites, which are commonly called Sudlow's sites I and II. Site I (subdomain IIA) is larger than Site II (Subdomain IIIA). Diflunisal itself has three binding sites and the primary (1°) and tertiary (3°) locations are within Sudlow's sites II and I, respectively. The protein secondary structure elements are depicted schematically, coloured by domain, and labelled with Roman numerals and Arabic letters. | ||
By employing isoelectronic Ni(II), Pd(II), and Pt(II) (nd8) chelates of the propyl-bridged structural congener of 1 (R = CH2CH2CH2; H2PrPyrr), we recently showed that the thermodynamics governing complex uptake by HSA depended on the identity of the metal ion.29 Specifically, TΔS/ΔG and ΔH/ΔG together became increasingly positive (enthalpically driven) in the order Pt(II) < Pd(II) < Ni(II) < H2PrPyrr, thereby inversely tracking the metal ionic radii and polarizability order (5d > 4d > 3d).29 A key question emerging from this study was whether varying the ligand chelated to a given metal ion (e.g., Pt2+) affects its uptake by HSA and, if so, how? To answer this question, we synthesized three structurally-related, yet discrete, bis(pyrrole-imine) Schiff base ligands, namely 1,3-bis{[(1E)-1H-pyrrol-2-ylmethylene]amino}propan-2-ol, 2,2-dimethyl-N,N′-bis[(1E)-1H-pyrrol-2-ylmethylene]propane-1,3-diamine and (1S,2S)-N,N′-bis[(1E)-1H-pyrrol-2-ylmethylene]cyclohexane-1,2-diamine. The ligands were then metalated with Pt(II) to obtain the desired series of square planar Schiff base chelates 1–3 (Fig. 1a). Steady-state fluorescence and CD spectroscopy were used to investigate the binding of the complexes to HSA under physiological conditions, which revealed how the pyrrole-imine Schiff base ligand architecture influences the affinity and preferred binding site(s) targeted by the Pt(II) chelates. Finally, molecular docking (GLIDE XP) and TD-DFT simulations were employed to confirm the spectroscopically identified binding sites.
Results and discussion
Metal chelate synthesis and spectroscopic characterization
Chiral 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and the two novel derivatives 1 and 2 were straightforwardly synthesized as depicted in Fig. 1. Ligand metalation was achieved by reacting the Schiff bases with sodium acetate in a 50% (v/v) DMF-DMSO mixture, with acetate ions serving as a general base. Thereafter, K2[PtCl4] was added and the solutions heated under reflux for 4 hours. Purification by recrystallisation followed by X-ray crystallography afforded two novel crystal structures for 1 and 2; complex 3 had been previously reported by Shan et al.10 and was not studied by X-ray diffraction methods (vide infra).
10 and the two novel derivatives 1 and 2 were straightforwardly synthesized as depicted in Fig. 1. Ligand metalation was achieved by reacting the Schiff bases with sodium acetate in a 50% (v/v) DMF-DMSO mixture, with acetate ions serving as a general base. Thereafter, K2[PtCl4] was added and the solutions heated under reflux for 4 hours. Purification by recrystallisation followed by X-ray crystallography afforded two novel crystal structures for 1 and 2; complex 3 had been previously reported by Shan et al.10 and was not studied by X-ray diffraction methods (vide infra).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H) resonates as a singlet at 8.20 ppm (DMSO-d6), downfield from that of the Ni(II) congener (δH = 7.29 ppm, CDCl3)30 and both enantiomers of 3 in CDCl3 (δH = 7.61 ppm).10 Since 2 and 3 give similar imine CH signals to 1 (δH = 8.15 and 8.16 ppm, respectively), differences in solvent polarity lead to significant proton chemical shift variations for this class of Pt(II) chelates. Significantly, spin–spin coupling (3J{1H,195Pt}) of the imine CH proton to 195Pt (33.8%)31 culminates in discernible satellite peaks flanking the 8.20 ppm resonance. The coupling constants for 1, 2, and 3, 3JPt–H, are similar, measuring 72.14, 72.63, and 75.16 Hz, respectively. This exceeds the range reported for simple cis and trans square planar PtI2(NH2R)2 complexes (3JPt–H = 35–49 Hz)32 as well as the coupling constants for complexes with three-centre N–H⋯Pt(II) interactions (1JPt–H = 33–55 Hz).33 Such large spin–spin coupling undoubtedly reflects the rigid, planar structure of the bis(pyrrolide-imine) chelate, which enforces Pt–N–C–H torsion angles, ϕ, of ∼180° and thus optimal 1H–195Pt coupling (based on the Karplus equation).34 The strong 3JPt–H spin couplings for 1–3 are in fact comparable to the 2J{1H,195Pt} coupling (∼60 Hz) reported for trans-[Pt(ethene)(2-carboxypyridine)Cl]35 and the 58–70 Hz 2J{1H,195Pt} couplings for all geometric isomers of the former PtI2(NH2R)2 complexes.32 The 13C NMR spectrum of 1 was straightforwardly assigned (Fig. 2b). Close inspection of the signal for pyrrole α-carbon 1 reveals a pair of 195Pt satellites that reflect strong 13C–195Pt spin–spin coupling (2JPt–C = 76.3 Hz), commensurate with the structure of 1.
C–H) resonates as a singlet at 8.20 ppm (DMSO-d6), downfield from that of the Ni(II) congener (δH = 7.29 ppm, CDCl3)30 and both enantiomers of 3 in CDCl3 (δH = 7.61 ppm).10 Since 2 and 3 give similar imine CH signals to 1 (δH = 8.15 and 8.16 ppm, respectively), differences in solvent polarity lead to significant proton chemical shift variations for this class of Pt(II) chelates. Significantly, spin–spin coupling (3J{1H,195Pt}) of the imine CH proton to 195Pt (33.8%)31 culminates in discernible satellite peaks flanking the 8.20 ppm resonance. The coupling constants for 1, 2, and 3, 3JPt–H, are similar, measuring 72.14, 72.63, and 75.16 Hz, respectively. This exceeds the range reported for simple cis and trans square planar PtI2(NH2R)2 complexes (3JPt–H = 35–49 Hz)32 as well as the coupling constants for complexes with three-centre N–H⋯Pt(II) interactions (1JPt–H = 33–55 Hz).33 Such large spin–spin coupling undoubtedly reflects the rigid, planar structure of the bis(pyrrolide-imine) chelate, which enforces Pt–N–C–H torsion angles, ϕ, of ∼180° and thus optimal 1H–195Pt coupling (based on the Karplus equation).34 The strong 3JPt–H spin couplings for 1–3 are in fact comparable to the 2J{1H,195Pt} coupling (∼60 Hz) reported for trans-[Pt(ethene)(2-carboxypyridine)Cl]35 and the 58–70 Hz 2J{1H,195Pt} couplings for all geometric isomers of the former PtI2(NH2R)2 complexes.32 The 13C NMR spectrum of 1 was straightforwardly assigned (Fig. 2b). Close inspection of the signal for pyrrole α-carbon 1 reveals a pair of 195Pt satellites that reflect strong 13C–195Pt spin–spin coupling (2JPt–C = 76.3 Hz), commensurate with the structure of 1.
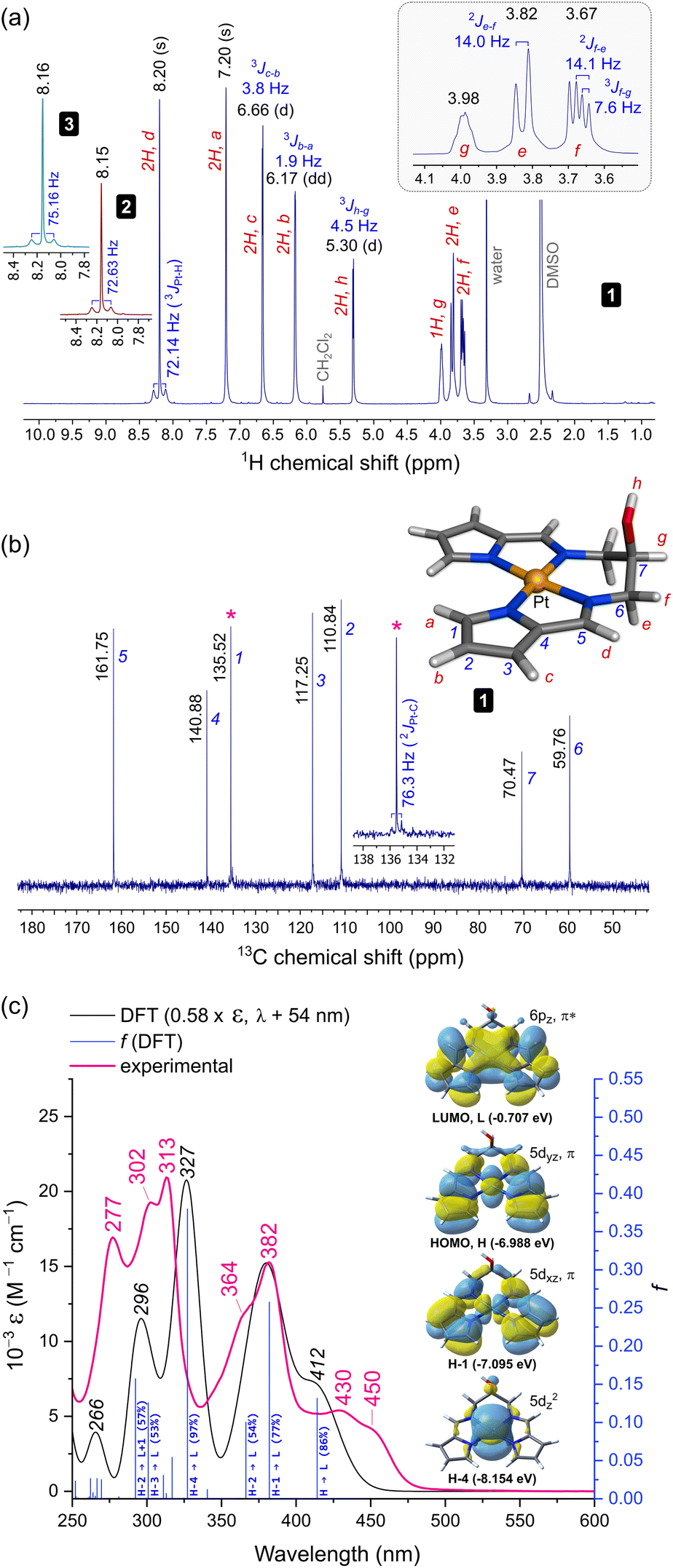 | ||
| Fig. 2 Spectroscopic characterization and structural assignments for 1. (a) Proton NMR spectrum (298 K, 400 MHz, DMSO-d6). The insets show the imine CH signals for 2 and 3. (b) Carbon NMR spectrum (298 K, 100 MHz) with the labelled and assigned DFT-calculated structure of 1. The inset highlights the 13C–195Pt spin–spin coupling for pyrrole carbon 1. (c) Electronic spectrum of 1 recorded in KH2PO4 buffer (50 mM, pH 7.5, 1% DMSO (v/v)). The TD-DFT calculated spectrum scaled (ε and λ) to best match the experimental spectrum is shown with selected transition assignments (bandwidth = 2500 cm−1, fwhm). The four key frontier MOs accounting for the bands at λ > 325 nm are shown. A full list of transition assignments is given in Table S9.† | ||
The 1H NMR spectrum for 1 exhibits two additional noteworthy features: (i) a sharp doublet for the hydroxyl proton resonance and (ii) resolvable splitting patterns for the methylene protons of the ligand's alkyl bridge. The former suggests that intermolecular H-bonding limits solvent-mediated exchange broadening of the hydroxyl proton resonance, allowing delineation of the J-coupling to methine proton g (3Jh–g = 4.5 Hz, Fig. 2). Regarding the methylene groups of the ligand's alkyl bridge, the axial (e) and equatorial (f) geminal protons resonate at 3.82 and 3.67 ppm, respectively. The resolved J-couplings for these signals indicate limited dynamic motion for the chelate ring (specifically its half-chair inversion). The experimental coupling constants 2Jf–e (14 Hz, geminal) and 3Jf–g (7.6 Hz) were confirmed by a static DFT calculation of the spin–spin coupling constants with the geometry-optimized structure (Fig. 2b, CAM-B3LYP36/def2-QZVP37/GD3BJ38 level of theory) in a DMSO solvent continuum (−16.1 Hz and 5.1 Hz, respectively). Formation of a supramolecular complex in DMSO (an oligomer), as seen in the asymmetric unit of the X-ray structure of 1 (vide infra), is both feasible and consistent with both hallmark features of the 1H NMR spectrum of 1. Interestingly, the methylene protons of 2 resonate as a broad singlet (Fig. S6†) wherein no geminal or 4J spin–spin coupling is evident, suggesting that dynamic/conformational exchange for the six-membered chelate ring is operative in this system.
The more intense band at 382 nm is similar with 77% 1[Pt(5dxz),π → Pt(6pz),π*] character and a prominent vibronic shoulder (364 nm) overlapping a weaker transition of mainly ligand π → π* character (HOMO−2 → LUMO, Table S9†). Finally, the far-UV bands are the most intense with the 313 nm band in the experimental spectrum (ε = 1.53 × 04 M−1 cm−1) correlating with the 327 nm band of the calculated spectrum. This interesting transition is almost pure MMLCT (metal-to-metal–ligand charge transfer) in character, i.e., 97% 1[Pt(5dz2) → Pt(6pz),π*] with a sharp vibronic satellite at 302 nm. The remaining band at 277 nm may be assigned from the DFT-calculated counterpart at 296 nm, which is based on two closely-spaced transitions: 57% HOMO−2 → LUMO+1 and 53% HOMO−3 → LUMO. Significantly, the latter transition, like the HOMO → LUMO transition, has part MLMLCT character (metal–ligand-to-metal–ligand charge transfer) as it originates from an MO which is a substantial admixture of the Pt 5dyz orbital with a bis(pyrrolide-imine) π MO, i.e., 1[Pt(5dyz),π → Pt(6pz),π*].
X-ray crystal structures
Both 1 and 2 crystallized in the triclinic space group P with three and four molecules in the asymmetric unit (ASU), respectively. The Pt(II) ion adopts the expected square planar geometry within the tetradentate chelate (Fig. 3). Typically, the sum of the angles around a square planar metal centre equates to 360°. The sum of the angles around Pt in complexes 1 and 2 were 350.38 ± 0.13° and 350.63 ± 0.14°, respectively, consistent with the constraints of the three chelate rings imparted by the ligands. The slightly non-planar conformations of the ligands mainly reflect non-bonded crystal packing interactions and the formation of discrete, stacked dimers.
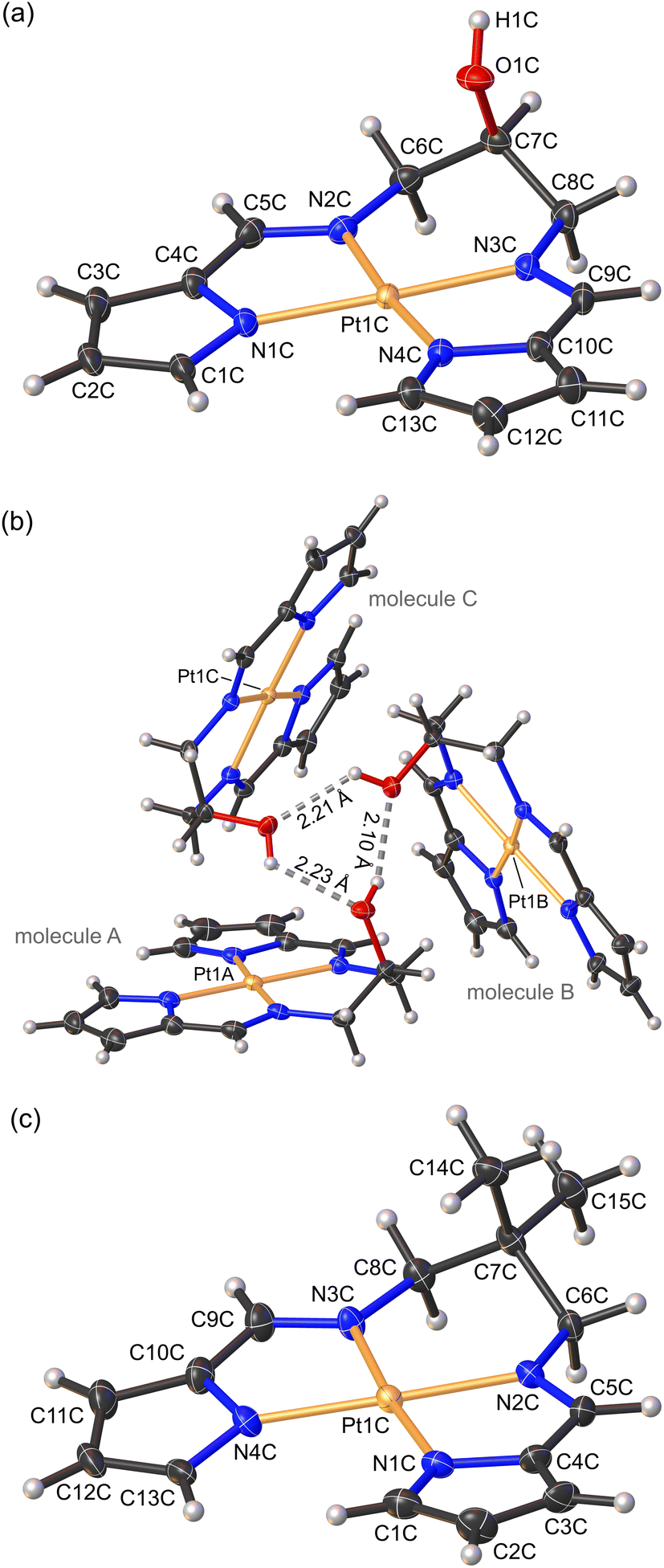 | ||
| Fig. 3 (a) Labelled view of the low-temperature X-ray structure of 1. One of the three independent molecules (molecule C) in the asymmetric unit is illustrated. (b) View of the asymmetric unit of the crystal structure of 1 with the three independent molecules forming an H-bonded trimer. (c) Labelled view of the low-temperature X-ray structure of 2. One of the four independent molecules in the asymmetric unit (molecule C) is illustrated. Thermal ellipsoids are rendered at 35% for 1 and 50% for 2. Hydrogen atoms are drawn as spheres with an arbitrary radius. | ||
Complexes 1 and 2 exhibit Pt–Npyrrole distances of 2.011 (3) and 2.013 (3) Å, respectively, while the Pt–Nimine distances were marginally shorter (∼0.4%), measuring 2.005 (3) and 2.002 (3) Å for complexes 1 and 2, respectively. Shorter Pt–Nimine bond lengths are typical for bis(pyrrolide-imine) chelates.10 The Pt–N bond distances of 1 and 2 are in agreement with the enantiomers of 3 reported by Shan et al.,10 with coordination group distances ranging from 1.936–2.026 Å.
Regarding the coordination group bond angles for 1 and 2, the mean Npyrrole–Pt–Npyrrole bond angle (104.04 ± 0.14°) is ∼8% narrower, and the mean Nimine–Pt–Nimine bond angle (95.70 ± 0.14°) ∼13.9% wider compared with those reported by Shan et al.10 for the structurally-related enantiomers of 3. This reflects the 3-carbon chain linking the imine groups of 1 and 2, which is more flexible than the sterically strained 2-carbon chain of 3, and the resulting 6-membered chelate ring which substantially narrows and widens the Npyrrole–Pt–Npyrrole and Nimine–Pt–Nimine bond angles, respectively. Furthermore, the pyrrole groups are anionic σ-donors with more acute C–Npyrrole–C bond angles averaging 106.98 (3)° compared with the CNimine–C bonds which were more obtuse, averaging 120.87 (4)°. The CNimine bonds measured 1.299 (5) Å, reflecting the sp2 hybridized imine carbon atom. Selected bond lengths and bond angles are summarized in Table 1.
| 1 | 2 | |
|---|---|---|
| Bond distances (Å) | ||
| Pt–Npyrrole | 2.011 (3) | 2.013 (3) |
| Pt–Nimine | 2.005 (3) | 2.002 (3) |
| CN |
1.300 (5) | 1.298 (5) |
| Bond angles (°) | ||
| Npyrrole–Pt–Npyrrole | 103.82 (14) | 104.25 (14) |
| Nimine–Pt–Nimine | 95.65 (14) | 95.75 (14) |
| cis-Npyrrole–Pt–Nimine | 80.27 (14) | 79.96 (13) |
| trans-Npyrrole–Pt–Nimine | 175.50 (13) | 175.27 (14) |
| C–Npyrrole–C | 107.05 (3) | 106.91 (3) |
| C–Nimine–C | 120.78 (3) | 120.96 (4) |
The asymmetric unit (ASU) of complex 1 consists of three independent molecules that are linked to each other by hydrogen bonds (Fig. 3b), forming a trimeric supramolecular structure. A similar trimer was observed previously for the isoelectronic and isostructural Ni(II) derivative.30 The OH group acts as both a hydrogen bond acceptor and donor, which results in the formation of a six-membered hydrogen-bonded ring (Fig. 3b). Typically, short bond lengths indicate a stronger bond. However, in the case of hydrogen bonding the bond length does not always correlate with the bond strength.39 The trimeric supramolecular structure of 1 did exhibit significantly shorter (20%) hydrogen bond lengths, averaging 2.18 ± 0.01 Å, compared with the Ni(II) analogue (2.749 ± 0.002 Å). The ASU of 2 consisted of 4 independent molecules A–D (Fig. S13†). Further inspection of the crystal packing of both 1 and 2 indicated molecule A of one ASU forms an inverted face-to-face stacked interaction with molecule B′ from a second ASU (Fig. S12 and S13†). The symmetry-unique intermolecular Pt⋯Pt distances measure 3.723 and 3.771 Å and are greater than 3.5 Å, which is double the van der Waals radius of Pt,39 and thus fall outside the range for proper dz2(Pt)–dz2(Pt) orbital interactions.
HSA fluorescence quenching by 1–3
The interaction between a protein and a small molecule, located close to an aromatic amino acid residue such as Trp, Tyr, and to a lesser extent Phe,40,41 can result in the disruption of the protein's intrinsic fluorescence.41 Among the aromatic amino acids, Trp-214 situated in subdomain IIA is primarily responsible for the intrinsic fluorescence of HSA due to its higher quantum yield and more efficient resonance energy transfer compared to Tyr and Phe.42 In this study, we aimed to investigate the quenching of the intrinsic emission spectrum of HSA (λex, 295 nm) by titrating the protein with Pt(II) complexes 1–3 at varying concentrations with spectral acquisition from 310–400 nm (Fig. 4). The λemmax was observed at ∼343 nm in KH2PO4 buffer (50 mM, pH 7.5), and the intrinsic emission intensity of HSA was quenched monotonically with the addition of each aliquot of the respective Pt(II) chelate (Fig. 4 and S14†). Note that the stability of complexes 1–3 in the buffer system used for the spectroscopic titrations with HSA was independently established (Fig. S22 and S23†). Significantly, and in contrast to similar chelates of Pd(II),291–3 were substitution-inert and did not hydrolyse to form aqua or hydroxo species. The titrimetric data for 1–3 with HSA thus reflect reactions of the intact, tetradentate Pt(II) chelates with the protein target.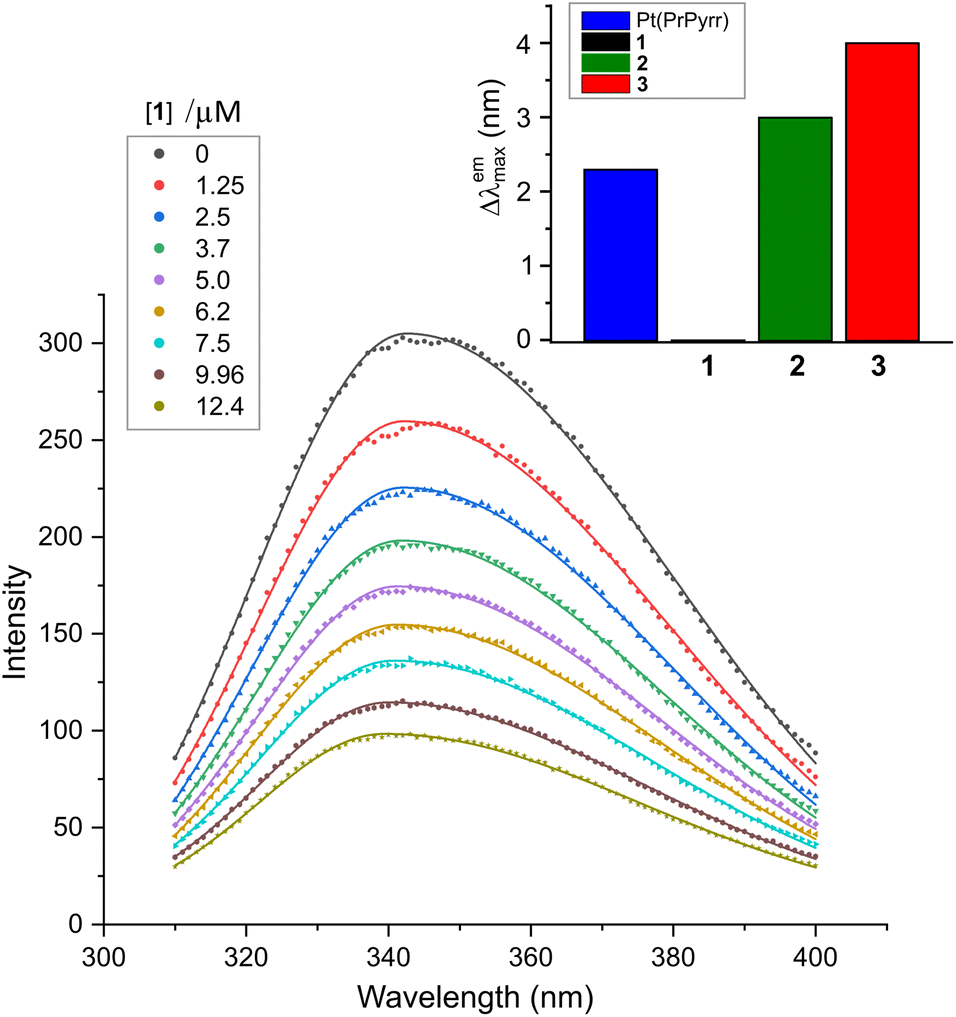 | ||
| Fig. 4 Emission spectra of human serum albumin (HSA, 5.0 μM) recorded as a function of [1] at 298 K in KH2PO4 (50 mM, pH 7.50). Solid lines are least squares fits of the data to the sum of two overlapping Gaussian bands. Analogous spectra for the reaction of HSA with 2 and 3 are given in Fig. S14 (ESI†). The wavelength shift accompanying ligand uptake (Δλemmax) is plotted as the inset to the main figure (upper right). | ||
Since protein fluorescence quenching is dependent on the overlap between the donor fluorophore (Trp-214) and acceptor's absorbance as well as the spatial proximity of the donor and quencher, we can deduce that the current Pt(II) chelates bind to HSA within range of Trp-214 to disrupt its microenvironment and quench its fluorescence.43,44 The mechanisms that may cause fluorescence quenching include ground state complex formation, collisional quenching, energy transfer, or molecular rearrangements.45,46 Binding of 2 and 3 to HSA induced band broadening and red shifts in λemmax of +3 and +4 nm, respectively, while 1 did not induce a spectral shift, as illustrated in Fig. 4.
The λemmax shift data suggest that the microenvironment surrounding Trp-214 in HSA has become more hydrophilic.47 The small red shifts in the emission maximum for 2 and 3 are comparable to that of Pt(PrPyrr).29 Two potential explanations accounting for the λemmax red shifts are: (i) the Pt(II) complexes bind to HSA sufficiently close to Trp-214 to induce direct electronic polarization of the indole ring in the residue, and/or (ii) the disruption of the ordered water molecules located within 15–25 Å of Trp-214 can lead to orientation-dependent polarization of the fluorophore. It is possible that both mechanisms impact the lowest energy 1A1 → 1La ground state transition of Trp's indole ring,43 thereby causing the λemmax shift. In the case of 1, no λemmax shift was observed, suggesting that 1 did not induce sufficient direct electronic polarization of Trp-214's indole ring to cause a wavelength shift in the protein's emission spectrum.
Fluorescence quenching mechanism
The fluorescence quenching mechanism for a protein's intrinsic fluorescence can be described as static, dynamic, or mixed inhibition (a combination of both) and is typically analysed using the Stern–Volmer equation (eqn (1)),41| I0/I = 1 + KSV[Q] = 1 + kqτ0[Q] | (1) |
Stern–Volmer (SV) plots for the fluorescence emission quenching of HSA as a function of temperature (298 K) and [Pt(II) chelate] are shown in Fig. 5a. From the slope of the least squares fit of eqn (1) to the data, the bimolecular fluorescence quenching rate constant (kq) for the HSA-Pt(II) chelate interaction can be calculated (eqn (2)):
| kq = KSV/τ0 | (2) |
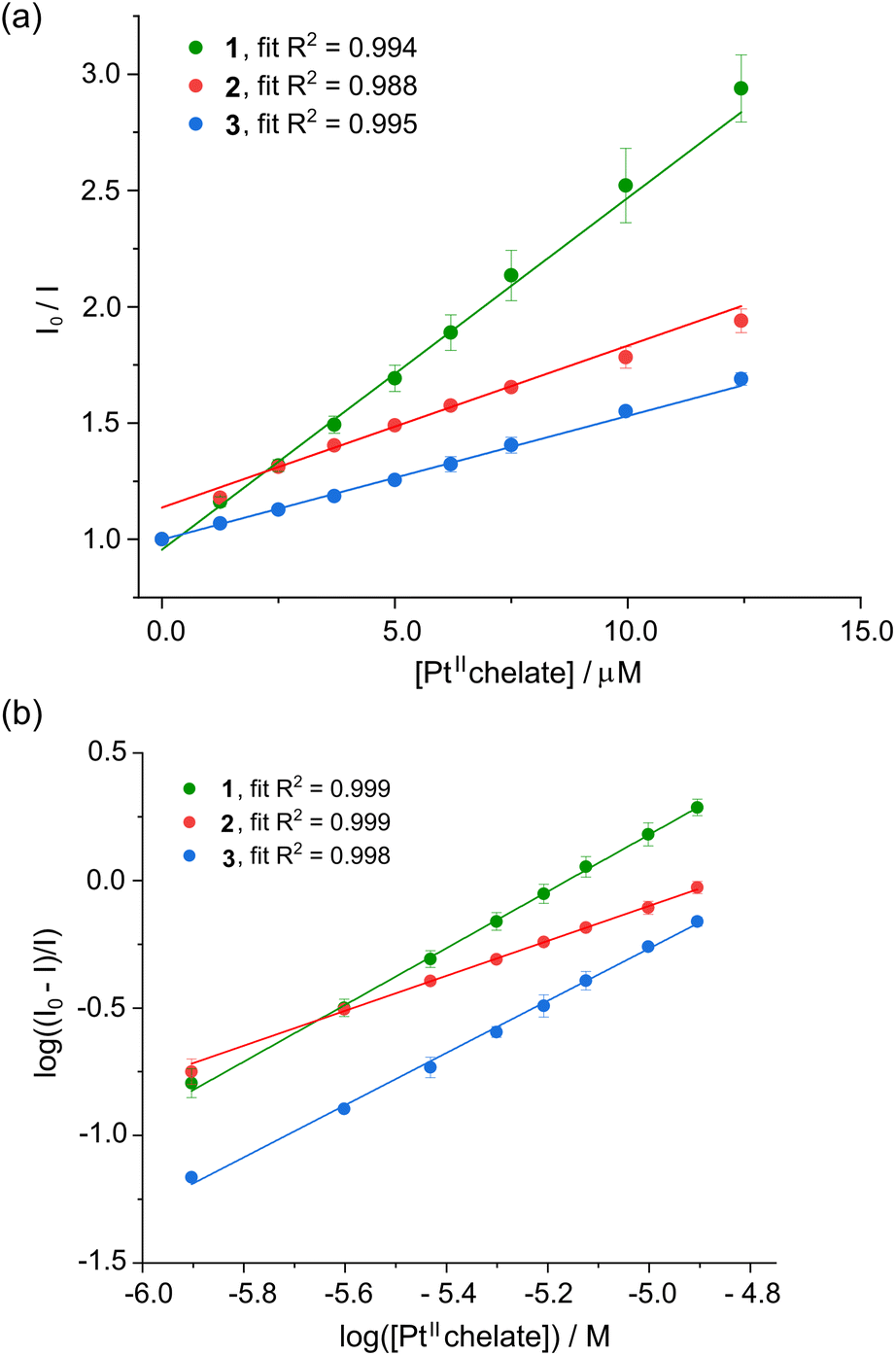 | ||
| Fig. 5 (a) Stern–Volmer plot for HSA (5 μM) in KH2PO4 buffer (50 mM, pH 7.5) recorded as the fluorescence intensity ratio (I0/I) vs. [1–3] at 298 K. Each plot represents the average of three independent experiments and the error bars represent ESD's. The data are well fitted to linear eqn (1), suggesting a single quenching mechanism is dominant. (b) Double logarithm plot of the fractional change in fluorescence intensity for human serum albumin (HSA, 5.0 μM) recorded as a function of the concentration of 1–3 (50 mM KH2PO4, pH 7.50, 298 K). Error bars are ESD's based on the average of three independent determinations. The data are described by eqn (3), which affords the affinity constant and stoichiometric coefficient for the reaction (Table 2). | ||
The KSV and kq values describing the interaction of the Pt(II) bis(pyrrolide-imine) chelates and HSA are summarised in Table 2. Linear Stern–Volmer plots typically indicate a single dominant quenching mechanism either static (binding-related) or dynamic (diffusion-limited collisional).40,41 The quenching mechanism may be differentiated by the KSV dependence on temperature. If KSV values decrease with increasing temperature, the dominant quenching mechanism is static because there is a decrease in the HSA·{ligand} complexes’ formation constant (the term “ligand” is used in a biochemical context; metal complexes 1–3 are the ligands or quenchers herein). However, if the reverse is observed the quenching mechanism is dynamic, which is largely due to an increase in the diffusion rate and collisional frequency.51
| Compound | Temp. T (K) | 10–5KSVa,c (M−1) |
10–13kqb (M−1 s−1) |
log(Ka/M−1)c | n |
|---|---|---|---|---|---|
| a K SV values were determined by fitting the data to linear eqn (1) to the data. b A mean excited state lifetime, τ0, of 5.87(76) ns for HSA was used to calculate the bimolecular quenching rate constant, kq. c The estimated standard deviations of the least significant digits are given in parentheses. d Ligand:HSA binding stoichiometry from the fit of the data to eqn (3). | |||||
| 1 | 288 | 1.89 (0.1) | 3.22 | 5.84 (0.04) | 1.13 (0.02) |
| 298 | 1.59 (0.2) | 2.46 | 5.67 (0.03) | 1.10 (0.01) | |
| 310 | 1.16 (0.1) | 2.27 | 5.50 (0.01) | 1.06 (0.02) | |
| 2 | 288 | 0.68 (0.04) | 1.16 | 3.25 (0.004) | 0.68 (0.01) |
| 298 | 0.71 (0.04) | 1.22 | 3.45 (0.05) | 0.71 (0.01) | |
| 310 | 0.95 (0.04) | 1.61 | 3.64 (0.04) | 0.73 (0.004) | |
| 3 | 288 | 0.54 (0.02) | 0.93 | 5.17 (0.04) | 1.10 (0.01) |
| 298 | 0.56 (0.01) | 0.95 | 4.81 (0.02) | 1.02 (0.01) | |
| 310 | 0.65 (0.1) | 1.11 | 4.67 (0.02) | 0.98 (0.01) | |
The Stern Volmer plots for 1–3 are linear, indicating that a single quenching mechanism is dominant—either static (binding-related) or dynamic (diffusion-limited collisional).40,41 Notably, the KSV values of 1 decreased with increasing temperature, consistent with a static quenching mechanism. In contrast, the KSV values of 2 and 3 increased with increasing temperature; therefore, a dynamic quenching mechanism is operative.40,41 The kq values for 1–3 all exceed the diffusion-controlled limit (1 × 1010 M−1 s−1)52 by 2 to 3 orders of magnitude, which is consistent with significant HSA⋯ligand binding interactions rather than non-specific collisional interactions.45 The KSV values followed the order 1 > 2 > 3 and ranged between 105–104 M−1, indicating a strong quenching phenomenon. The KSV values are consistent with those of other Pt(II) complexes interacting with HSA.29,53,54 A summary of KSV and kq values obtained for the interaction of the three Pt(II) complexes with HSA is reported in Table 2. The Stern–Volmer plots for 1, 2 and 3 over the full range of temperatures are provided in the ESI (Fig. S15†).
Ligand binding equilibrium constants
Quenching of intrinsic HSA fluorescence emission as a function of [PtII(L)], where L is the dianionic bis(pyrrolide-imine) chelating ligand, can be used to calculate the biophysical binding parameters for the interaction of 1–3 with HSA, i.e., the affinity constant (Ka) and the reaction stoichiometry (n). Both parameters can be obtained from a double log plot and least-squares fit of the emission quenching data as a function of increasing ligand concentration (eqn (3)),55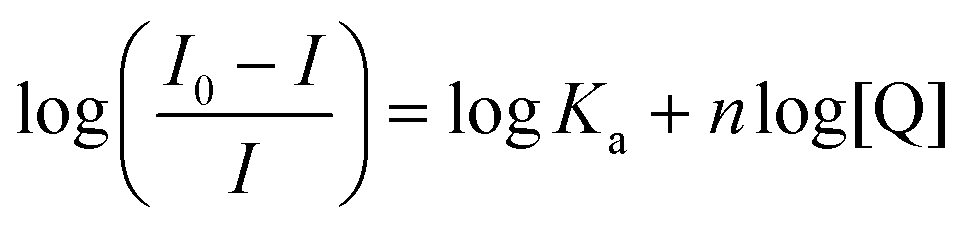 | (3) |
Ka and n, respectively. The data for the Pt(II) chelates are summarised in Table 2 and are plotted in Fig. 5b. (Similar plots for 1–3 at 288, 298, and 310 K are given in Fig. S16.†).
The affinity constant (Ka) for 1 and 3 decreases with increasing temperature, while Ka for 2 increases with increasing temperature. The reaction stoichiometry from the slope of eqn (3) (Fig. 5b and S16†) tended to deviate from unity (0.68 < n < 1.13). From Table 2, the Ka values follow the order 1 > 3 > 2. The Ka for the reaction of Pt(PrPyrr) with HSA was previously reported,29 covering a similar range to that found for 1 here. Of the three Pt(II) bis(pyrrolide-imine) chelates, 2 bound endothermically to HSA, while the binding of 1 and 3 to HSA was exothermic (see Table 3). All three Pt(II) bis(pyrrolide-imine) chelates had moderate binding affinities for HSA, suggesting non-covalent binding. Weaker ligand binding interactions are ideal since one potential application is to use HSA as a carrier protein for metal chelates. Moderate binding affinity could signify faster diffusion rates for a drug/metallodrug within the circulatory system.46
| Compound | T (K) | ΔGa (kJ mol−1) | ΔHa (kJ mol−1) | TΔSa (kJ mol−1) |
|---|---|---|---|---|
| a The estimated standard uncertainties of the least significant digits are given in parentheses. | ||||
| 1 | 288 | −32.1 (0.2) | ||
| 298 | −32.4 (0.2) | −26.2 (2.7) | 6.2 (2.6) | |
| 310 | −32.5 (0.1) | |||
| 2 | 288 | −19.3 (2.0) | ||
| 298 | −29.6 (2.0) | 30.3 (4.9) | 47.9 (4.9) | |
| 310 | −19.6 (1.6) | |||
| 3 | 288 | −27.8 (0.3) | ||
| 298 | −25.5 (0.4) | −37.4 (2.4) | –10.2 (1.5) | |
| 310 | −28.13 (0.4) | |||
Thermodynamics of Pt(II) chelate binding by HSA
The reactions of the three Pt(II) chelates follow a linear van't Hoff relationship; plots for triplicate measurements are represented in Fig. 6a. Under non-standard conditions the thermodynamic parameters, i.e., enthalpy change (ΔH), entropy change (ΔS), and Gibbs free energy change (ΔG), may be deduced from eqn (4) and (5).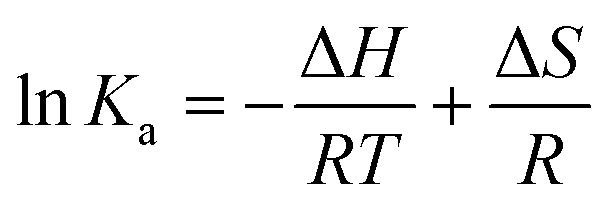 | (4) |
| ΔG = ΔH − TΔS | (5) |
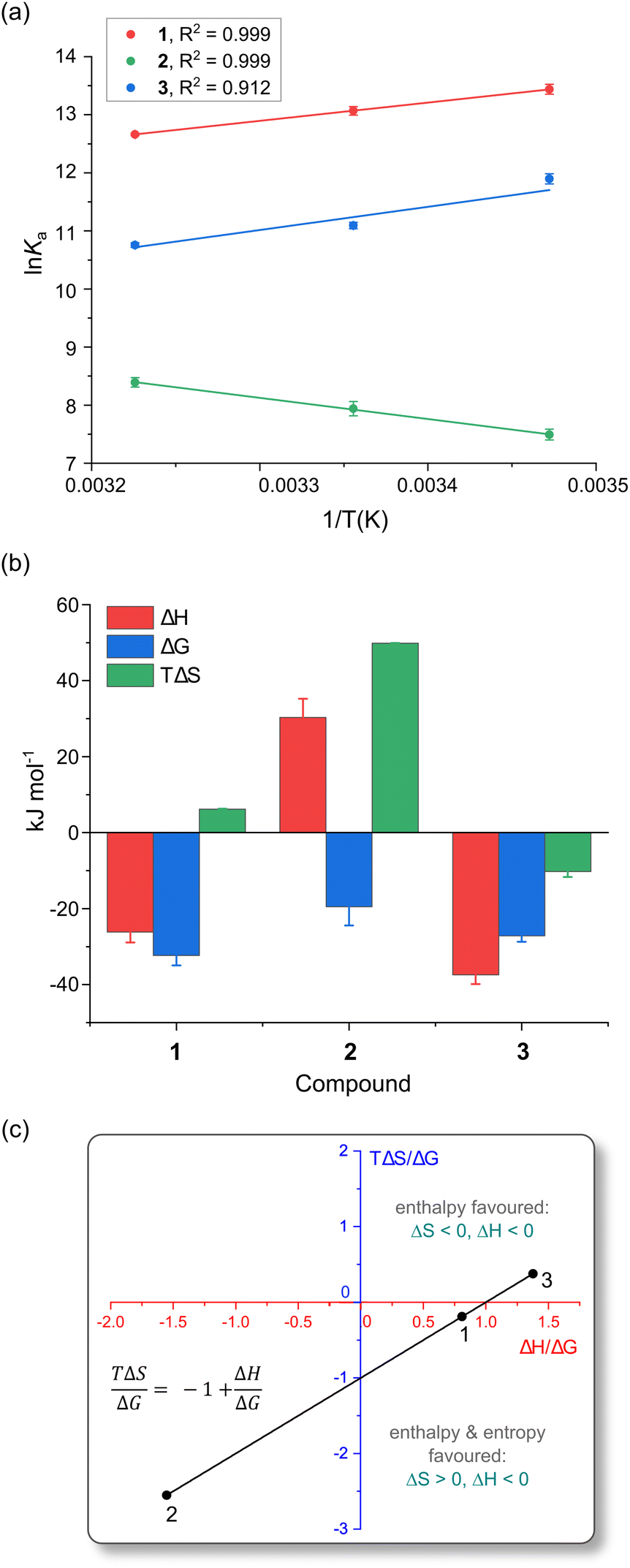 | ||
| Fig. 6 (a) Linear van't Hoff plots for the reactions of 1, 2, and 3 with HSA in 50 mM KH2PO4 buffer at pH 7.50. (b) Comparison of the thermodynamic parameters (298 K) governing the reactions of 1–3 with HSA. All measurements were done in triplicate, derived parameters were individually averaged, and error bars are standard deviations. (c) Plot of the Gibbs–Helmholtz relationship (eqn (5)) for the reaction of the three Pt(II) bis(pyrrolide-imine) chelates with HSA at 298 K in 50 mM KH2PO4 buffer at pH 7.50. The straight line fit of the data gives R2 = 0.9997 with a slope and intercept of −1. For all reactions, ΔG < 0. | ||
Table 3 summarises the thermodynamic parameters for the reaction of the current Pt(II) chelates with HSA in KH2PO4 buffer (50 mM, pH 7.5). All three reactions are exergonic,55 with ΔG values of −32.3 (±0.1), −19.5 (±0.08) and −27.2 (±1.4) kJ mol−1 for 1, 2 and 3, respectively at 298 K.
The enthalpy values (ΔH) for reactions of 1 and 3 with HSA are exothermic, measuring −26.2 kJ mol−1 and −37.4 kJ mol−1, respectively. Complex 2, in contrast, has an endothermic reaction with HSA (ΔH = +30.3 kJ mol−1). Fig. 6b highlights the dominance of the enthalpy term for the reactions of 1 and 3 with HSA, while the TΔS term is dominant for 2. It is well established that HSA has two main small molecule binding sites that are mainly hydrophobic,23 namely Sudlow's site I (subdomain IIA)55 and Sudlow's site II (subdomain IIIA).55 The favourable heats of reaction for 1 and 3 suggest that they cause minimal disruption of ordered water molecules within HSA. London dispersion forces (LDF) are likely the dominant binding forces for 1 and 3. In the case of complex 2, a positive enthalpy (ΔH > 0) is suggestive of hydrophobic interactions.55,56
In Fig. 6c we used the Gibbs–Helmholtz relationship (eqn (5)) and the experimental thermodynamic data (Table 3) to determine the influence that the ligand chelated to Pt(II) has on the thermodynamic parameters for complex formation with HSA. From Fig. 6c, when TΔS/ΔG is plotted against ΔH/ΔG, the resulting fit is linear with x- and y-intercepts of exactly 1. Reactions for 1 and 3 with HSA are enthalpically driven (ΔH < 0, upper and lower right quadrants) with the reaction for the hydroxy derivative 1 also being entropically favoured (ΔS > 0). Complex 2, in contrast, exhibits a purely entropically driven reaction with HSA (ΔH > 0, ΔS > 0; lower left quadrant). In all three quadrants, spontaneity is assured (ΔG < 0) since changes in ΔH/ΔG are compensated for by changes in TΔS/ΔG. Exact TΔS values for the three Pt(II) chelates are listed in Table 3. Both 1 and 2 have a positive entropy change, while 3 has a negative entropy change, with TΔS ranging from −10.2 kJ mol−1 to +49 kJ mol−1. Because the reaction for 2 is entropically driven, ordered water molecules are probably displaced from the ligand binding site into the buffer, suggesting hydrophobic and electrostatic interactions55,56 could play a key role in the binding of 2 by HSA.
The HSA binding thermodynamics of 1–3 reported here are similar to data for other Pt(II) chelates,53 which have moderately positive ΔH and ΔS values with both compounds binding specifically to Sudlow's site I. For further comparison, binding of the organic drugs ibuprofen57 and several benzodiazepines58 are characterized by moderately negative ΔH (−50 to −75 kJ mol−1) and ΔS values with Ka1 values ∼105 M−1.59 The latter drugs bind to Sudlow's site II initially, followed by secondary site occupation at higher doses (Ka2 values ∼103 M−1). The ΔH and ΔS values were, however, similar for both drug-binding steps.
Fluorescence-based probe displacement assays to determine the main Pt(II) chelate binding site(s)
Considering the binding constants and thermodynamic data for the uptake of 1–3 by HSA presented in Tables 2 and 3, it is likely that the unique steric and electronic properties of the bis(pyrrolide-imine) ligands coordinated to Pt(II) modulate the affinity and preferred binding site for each metal chelate in the protein. For 1–3, these binding sites are all in close enough proximity to Trp-214 to quench its fluorescence. This hypothesis was tested by carrying out a competitive site displacement assay using fluorescence spectroscopy. For the assay, 1–3 were titrated into a solution of native HSA and solutions of HSA that had been pre-equilibrated with the site-specific markers warfarin and ibuprofen. Both warfarin and ibuprofen have been shown to bind to subdomain IIA60 and IIIA,61 respectively, using X-ray crystallography. (Ibuprofen also has a secondary binding site located between subdomains IIA and IIB.61).One caveat concerning the assay is that HSA has several ligand-binding sites,61 which could make interpreting the data difficult. The idea here is that the displacement of either warfarin or ibuprofen by the incoming Pt(II) chelate would signal binding to either Sudlow's site I or II. However, if the presence of either warfarin or ibuprofen redirects the Pt(II) chelate to an alternate binding site within HSA (or prevents Pt(II) chelate uptake due to allosteric inhibition) the exact binding site may be difficult to elucidate. Although there may be certain limitations, examining changes in the logKa, n, and KSV values, as well as movements of the fluoroprobe, could reveal plausible binding locations for these metal complexes.
For 1–3, the assay for their uptake by HSA in the presence of key site-specific probes has been summarised in Fig. 7 (and Fig. S17 and S18†). Initially, when 1–3 react with native HSA, all ligand binding sites are available for complexation. The Stern–Volmer plots (Fig. 5 and Table 2) reveal 1 and 3 are dynamic quenchers, while 2 is a static quencher of HSA intrinsic fluorescence, while the double log plot (Fig. 5b) indicates a nominally 1:1 Pt(II) complex:HSA reaction stoichiometry.
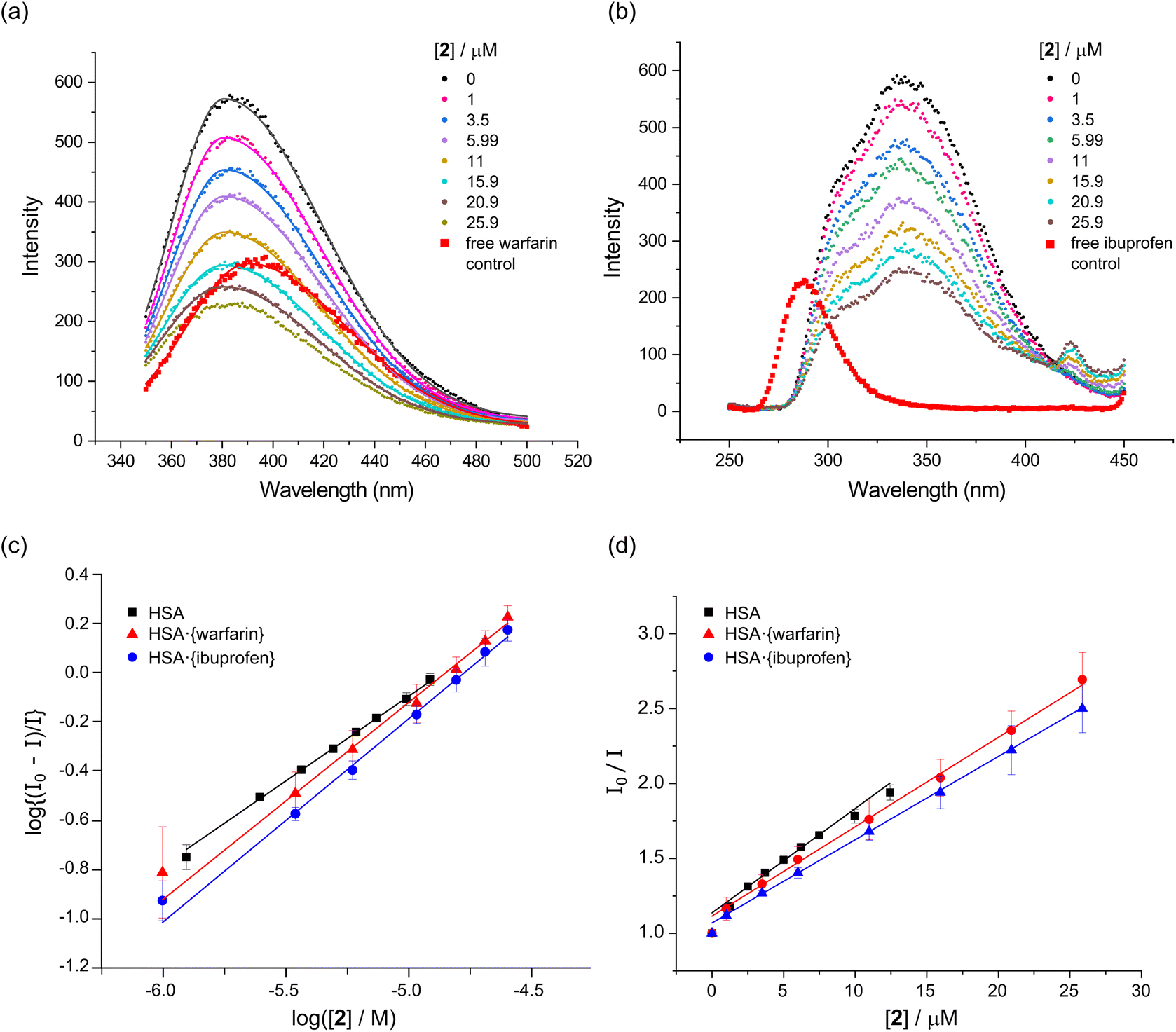 | ||
Fig. 7 Binding site determination for the reaction of 2 with HSA (5.0 μM protein, 50 mM KH2PO4, pH 7.50, 298 K). (a) Emission spectra of HSA·{warfarin} (5.0 μM warfarin, λex = 320 nm) recorded as a function of the concentration of 2 at 298 K. The spectra are fitted by single Gaussian functions to locate the emission maxima. Correlation coefficients, R2, ranged from 0.990 to 0.999. (b) Emission spectra of HSA·{ibuprofen} (5.0 μM ibuprofen, λex = 228 nm) recorded as a function of the concentration of 2 at 298 K. (c) Stern–Volmer (SV) plot for native HSA, HSA·{warfarin}, and HSA·{ibuprofen} as a function of the concentration of 2 at 298 K. The plots are linear with an intercept of 1.0 when static quenching is operative (i.e., the quencher binds to the target protein and co-bound fluorophore(s)). The excitation and emission wavelengths for the fluorophore probes were: (i) Trp-214, λex = 295 nm, λem = 340 nm (native HSA); (ii) warfarin, λex = 320 nm, λem = 382 nm (HSA·{warfarin}), and (iii) ibuprofen, λex = 228 nm, λem = 332 nm (HSA·{ibuprofen}). (d) Double-log plot (eqn (3)) of the fluorescence quenching data, 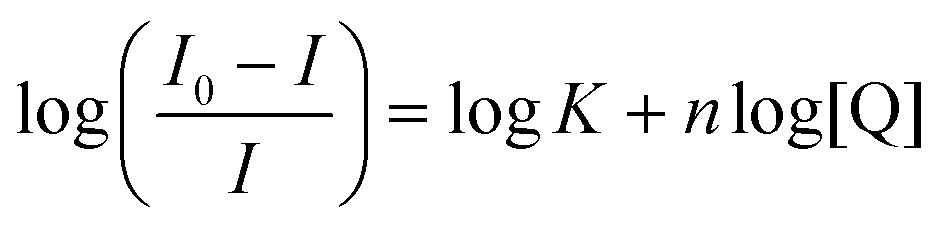 , to enable measurement of logKa and the reaction stoichiometry (n). , to enable measurement of logKa and the reaction stoichiometry (n). | ||
Titration of 1 into HSA·{warfarin} (λex = 320 nm, λem = 382 nm) and HSA·{ibuprofen}n (λex = 228 nm, λem = 332 nm) resulted in a decrease in the measured logKa values from 5.67 ± 0.03 (ntotal = 1.10 ± 0.01) in the native protein to 5.28 ± 0.05 (ntotal = 1.03 ± 0.03) and 5.38 ± 0.15 (ntotal = 1.06 ± 0.03) for the probe-bound targets HSA·{warfarin} and HSA·{ibuprofen}n, respectively. Similarly, for 3 the logKa values decreased from 4.85 ± 0.20 (ntotal = 1.02 ± 0.05) in the native protein to 3.88 ± 0.16 (ntotal = 0.85 ± 0.04) and 4.06 ± 0.15 (ntotal = 0.87 ± 0.03) for HSA·{warfarin} and HSA·{ibuprofen}n, respectively.
In contrast, the logKa values for the binding of 2 increased somewhat from 3.33 ± 0.20 (ntotal = 1.02 ± 0.05) in the native protein to 3.88 ± 0.16 (ntotal = 0.85 ± 0.04) and 4.06 ± 0.15 (ntotal = 0.87 ± 0.03) for the probe-bound targets HSA·{warfarin} and HSA·{ibuprofen}n, respectively. The Stern–Volmer plots for the binding of 1–3 by the probe-laden targets were linear throughout, consistent with a single mode of fluorescence quenching (Fig. 7d, S17 and S18†).
The foregoing results suggest that over the concentration range examined, neither warfarin nor ibuprofen are displaced to a substantial extent from HSA by the incoming Pt(II) chelate. However, the fluorescence from both site-specific probes is clearly quenched by 1–3, no doubt via a long-range FRET-based mechanism. Our interpretation is that all three Pt(II) complexes bind to HSA in the presence of warfarin or ibuprofen, without fully displacing either probe under the conditions employed for these experiments. Nevertheless, the probes can influence the binding affinity of 1–3. The observation that the probes are not fully displaced is not unexpected, considering that the logKa values for the binding of both warfarin (logKa ∼5.5)60 and ibuprofen (logKa ∼6.3)61 to native HSA are higher than those determined here for 2 and 3, while only the logKa value for ibuprofen uptake surpasses that of complex 1.
One interpretation of the data is that 1–3 can target both Sudlow's sites I and II. However, if either of the site-specific markers is already present in one of these sites, the unbound site is vacant and becomes fully saturated by the metal chelate. Alternatively, the presence of warfarin and ibuprofen may cause subtle changes in the tertiary structure of HSA, resulting in 1–3 favouring alternative binding sites in the protein. Such allosteric redirection of ligand uptake could explain why the logKa values for 1 and 3 decrease, while logKa for 2 increases for the probe-bound targets. While the current spectroscopic data cannot pinpoint the preferred binding site with absolute certainty, our in silico docking and CD studies (vide infra) suggest that while both sites are accessible, the ibuprofen binding site (Sudlow's site II) is preferred (Fig. 10 and Table S7;†vide infra). Because of the ambiguity of the fluorescence quenching assays in this case, we recommend that an ICD assay be conducted as a definitive cross-check for the binding site(s) favoured by a metal chelate (see Fig. 10).
Far-UV CD spectroscopy
Far-UV circular dichroism (CD) spectroscopy is a highly effective technique for analysing changes in a protein's secondary structure (i.e., α-helices, β-sheets, and random coils) upon ligand binding.62,63 The impact of Pt(II) chelates 1 and 2 on HSA's secondary structure was measured over the 200–260 nm range (Fig. 8). The CD spectrum of native HSA displayed two minima at 208 (±1 nm) and 222 nm (±1 nm), which is typical of an α-helical rich protein (Fig. 8a and b). The electronic transitions for both minima are attributed to π → π* (208 nm) and n → π* (222 nm) excitations,62 respectively, with the transitions originating within the amide bonds of the peptide backbone.62,63 The far-UV CD spectrum of HSA in the presence of 1 and 2 was similar to that of native HSA, with only a minimal decrease in ellipticity (Fig. 8a and b). Both 1 and 2 exhibited moderately high binding affinities for HSA (Table 2), effecting only small CD intensity changes without a wavelength shift, even at concentrations as high as 40 μM (8-fold higher than the HSA concentration). Collectively, the data indicate that the Pt(II) chelates negligibly impact the secondary structure of HSA.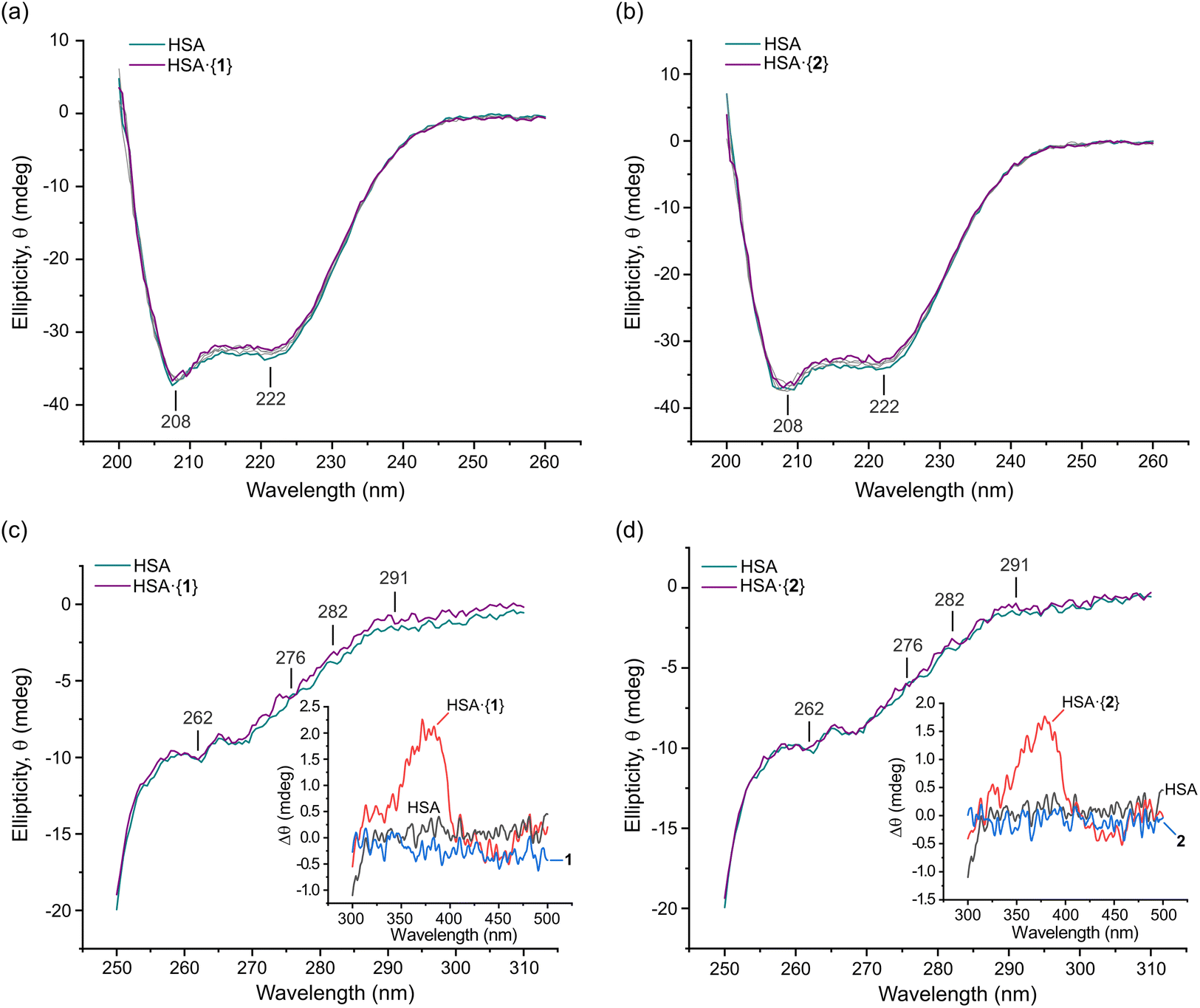 | ||
| Fig. 8 Plots of the far-UV CD spectra of native HSA and the protein incubated with saturating doses of (a) 1 and (b) 2 recorded at 298 K in 50 mM KH2PO4 buffer at pH 7.50. The plots are unsmoothed spectra at Pt(II) bis(pyrrolide-imine) chelate doses ranging from 0 to 40 μM. Grey spectra indicate concentrations of 5, 10, and 20 μM of 1 and 2. Band minima are indicated. Plots of the UV-visible CD spectra of native HSA (15 μM) and the protein incubated with a saturating dose (40 μM) of (c) 1 and (d) 2 were recorded at 298 K in 50 mM KH2PO4 buffer at pH 7.50. The graphical insets are plots of the HSA·{PtII(L)} complex; they represent the induced CD (ICD) spectrum of each achiral protein-bound ligand. Perturbations in the protein structure around Phe (250–270 nm), Tyr (∼280 nm), and Trp (285–300 nm) residues may also be resolved in some difference spectra. Selected wavelengths in the UV CD spectrum are marked. | ||
The far-UV CD data were further analysed using JASCO Spectra Manager™ to calculate the percentage composition of α-helices, turns, and other secondary structure elements present for each HSA·{PtII(L)} adduct (Table 4). The dominant secondary structure domains are α-helices (∼56%), turns (∼11%), and unordered coils (∼32%). In solution, the secondary structure composition of HSA differs from that of native HSA in the solid state23 (68.5% α-helix, 0% β-sheet, 9.6% turns, and 21.9% unordered coils; PDB code 1BM0 analysed with BeStSel64). However, our CD data are consistent with solution state spectral decompositions reported previously by others.65–67 It is accepted that enhanced subdomain mobility and general thermal motion/disorder account for the decrease of α helicity.50,63
| Compound | Conc. (μM) | Helix (%) | Turn (%) | Unordered (%) |
|---|---|---|---|---|
| 1 | 0 | 56.39 | 11.03 | 32.58 |
| 5 | 55.15 | 10.43 | 32.42 | |
| 10 | 54.31 | 11.10 | 34.59 | |
| 20 | 54.04 | 10.78 | 35.18 | |
| 40 | 53.94 | 11.37 | 34.69 | |
| 2 | 0 | 57.21 | 10.88 | 31.91 |
| 5 | 55.72 | 10.67 | 33.61 | |
| 10 | 55.93 | 11.08 | 32.99 | |
| 20 | 55.19 | 11.05 | 33.76 | |
| 40 | 55.14 | 10.85 | 34.01 | |
Near-UV and visible region CD spectroscopy
To investigate the impact of 1 and 2 on the tertiary structure of HSA, we utilised near-UV CD spectroscopy. This technique probes the electronic structure of the three aromatic amino acid residues, Trp (285–300 nm), Tyr (275–285 nm), and Phe (250–270 nm), and allows for the examination of conformational changes in the protein's tertiary structure. Moreover, induced circular dichroism (ICD) bands from the bound metal chelates were observed; these can be structurally informative when an achiral small molecule binds to a chiral host macromolecule such as a protein.62 Here, we present the experimental near-UV CD and ICD spectra for HSA bound to the Pt(II) chelates of interest, and we complement the experimental data interpretation with results from our Glide XP68,69 (Schrödinger Release 2022-3) molecular docking and TD-DFT simulations (ONIOM70 CAM-B3LYP36/SDD71/GD3BJ:38 UFF72) of the electronic structures of the protein-bound metal chelates (TD-DFT; time-dependent density functional theory).
Fig. 8 shows the spectra of HSA in its native form and in the presence of identical saturating doses (40 μM) of 1 and 2. Through difference spectroscopy, we were able to resolve the ICD spectra for both Pt(II) chelates. Notably, the intensity of the ICD response for 1 was greater than that of 2, in accord with the affinity constants (logKa, Table 2) for uptake of the two Pt(II) chelates by HSA. A stronger ligand–HSA interaction results in a stronger ICD signal from the optically inactive ligand. This phenomenon was previously observed for related metal chelates by Sookai and Munro.29 The presence of the ICD band in the UV-visible region suggests that the Pt(II) chelates do not undergo decomposition, demetallation, imine hydrolysis, or ligand dissociation upon binding to HSA in solution. Unlike simple metal complexes such as cisplatin73 and NAMI-A (trans-[RuCl4(1H-indazole)2]−),74 whose ligands can dissociate from the metal, the present class of bis(pyrrolide-imine) Pt(II) chelates exhibit the potential for reversible transport in vivo via an HSA-mediated cellular uptake pathway.75 This finding highlights the possibility that other polydentate chelates and macrocyclic metal complexes could be similarly transported.
The near-UV CD spectrum of native HSA typically shows two minima at 262 and 280 nm and a maximum at 290 nm due to disulphide bonds and aromatic amino acids.76 Perturbations in this region provide insights into the conformational changes occurring in HSA. Phenylalanine and tyrosine residues have low quantum yields and extinction coefficients but high symmetry, making them less sensitive to changes in the residue's microenvironment. Tryptophan residues, in contrast, have high quantum yields and extinction coefficients but possess low symmetry. Therefore, they are most sensitive to changes in their immediate microenvironment.77 All three amino acid residues have π → π* transitions (1La and 1Lb) and have the potential to be directly involved in π bonding. Importantly, the π → π* transitions have charge transfer character and thus respond to dielectric changes within the chromophore microenvironment such that solvent changes may affect the transition.78 Thus, changes in the near-UV CD spectrum can be correlated to tertiary structure changes in the protein due to its interaction with other molecules or due to solvent exposure perturbing the aromatic residue's microenvironment.79
Near-UV CD spectra were recorded from 250–310 nm for HSA·{1} and HSA·{2} (Fig. 8c and d). The 291 nm Trp 1Lb (0,0) band and Tyr and Trp overlap bands at 282 and ∼276 nm (1La; solvated Trp),80,81 respectively, are all present. When 1 and 2 bind to HSA, these marker bands exhibit slight positive changes in ellipticity with a negligible wavelength shift. The change in ellipticity suggests that both Tyr and Trp are more exposed to the solvent and/or affected by the electrostatic field of the bound metal chelates. The former would occur for minor alterations in the secondary structure of HSA (Fig. 8a and b), i.e., the decrease in α-helicity and partial unfolding upon ligand uptake noted earlier. Partial unfolding of HSA is expected to alter both the number and arrangement of ordered water molecules around Trp and Tyr, culminating in the observed, subtle perturbations for these chromophores.
Insights from DFT simulations
Based on our in silico docking data obtained with published protein X-ray structures and grids centred on probe ligands (warfarin, diazepam, and ibuprofen), we used hybrid QM:MM TD-DFT simulations (ONIOM method82) to calculate the electronic structures of HSA·{1} and HSA·{2}. The best pose taken directly from the optimized Glide XP structure (Fig. S21†) was assigned to the quantum mechanics (QM) layer, and five amino acid residues (Ile-388, Leu-407, Phe-403, Tyr-411, and Arg-485) interacting with 2 in Sudlow's site II were added to the QM layer for the most accurate ICD spectrum calculation. The protein amino acid residues were simulated using molecular mechanics (UFF72). The TD-DFT calculated ICD spectrum of HSA·{1} broadly matches the key spectral features of the experimental ICD spectrum (Fig. 9a). The simulations indicate that for both ibuprofen and aromatic amino acid residues within Sudlow's sites I and II (vide infra, Tables S7 and S8†), there are several possible binding sites within 9.6–30 Å of Trp-214 that can account for (i) FRET-based quenching of this fluorophore and (ii) location of the ligand within 5.9–12 Å of at least one Tyr residue. Interestingly, none of the binding sites are directly within Sudlow's site I; instead, the in silico evidence suggests that both 1 and 2 target Sudlow's site II.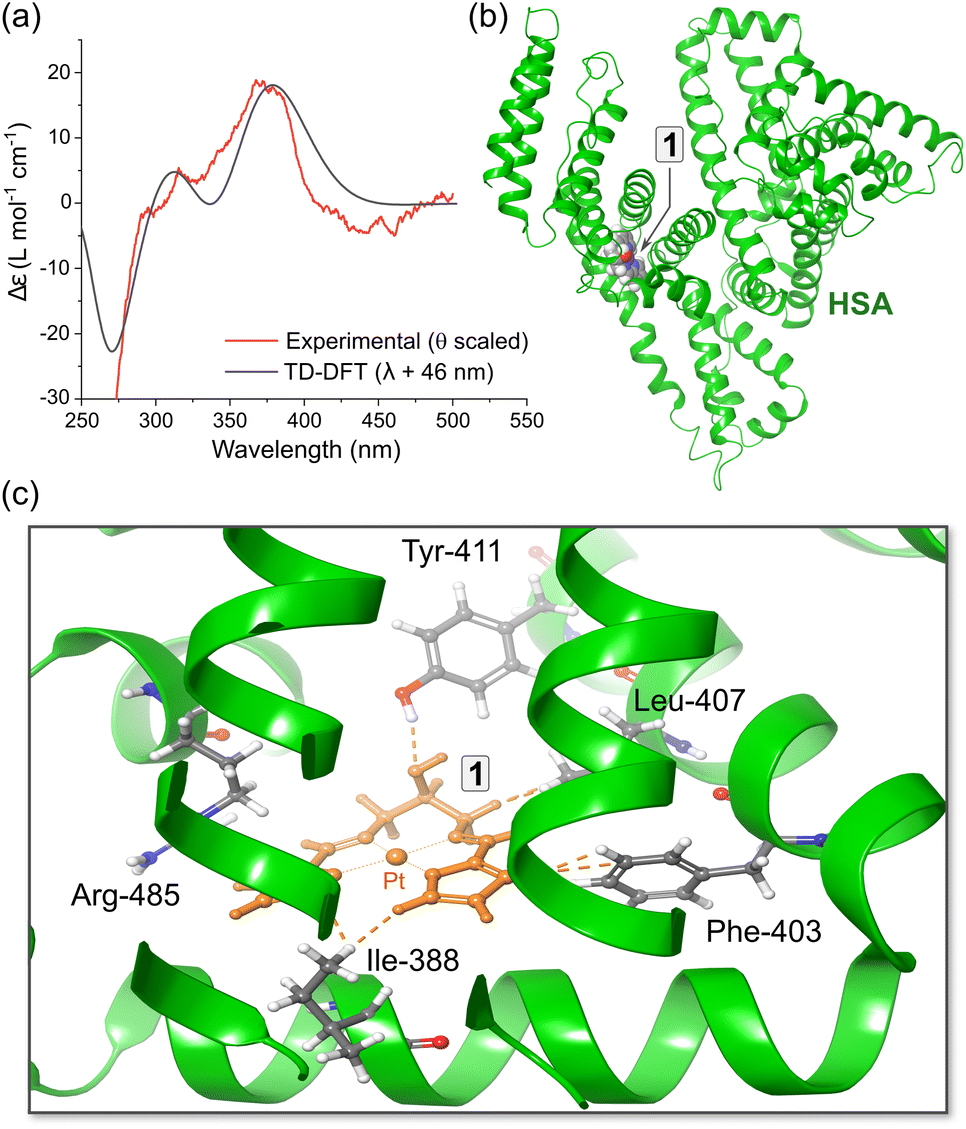 | ||
| Fig. 9 (a) Comparison of the experimental ICD spectrum (Δθ) recorded for HSA·{1} at pH 7.5 in 50 mM (KH2PO4) and the CD spectrum calculated using hybrid QM:MM TD-DFT simulations (CAM-B3LYP/SDD/GD3BJ:UFF) for the top-scoring docked pose of 1 bound in subdomain IIIA (PDB code: 2BXF). The DFT-calculated spectrum of the best pose matches the experimental spectrum reasonably well; a bandwidth of 1580 cm−1 (hwhm) was used for the TD-DFT data. (b) Structural model used for the TD-DFT simulations showing the location of 1 within subdomain IIIA. (c) View of the metal chelate binding site containing the best pose of 1 and the closest amino acid residues. | ||
Regarding the assignment of the near-UV CD and visible ICD spectra of HSA·{1} and HSA·{2}, the fine structure perturbations for the TD-DFT ICD spectra matched those observed in the experimental spectra with an amplitude of 1 > 2, which was mainly due to the larger Ka of HSA·{2} compared to HSA·{1}. The TD-DFT data for unbound 1 (Fig. 2c, Tables S5 and S9†) suggest that the UV-vis absorption bands at 350–410 nm involve 1[π → Pt(6pz),π*] and 1[Pt(5dπ),π → Pt(6pz),π*] transitions of the bis(pyrrolide-imine) chelate, while the visible region absorption bands from 410–500 nm have somewhat more metal-rich charge transfer character (MLMLCT, 1[Pt(5dπ),π → Pt(6pz),π*]). The absorption bands peaking at ∼380 nm exhibit a slight blue shift of ∼3 nm upon uptake of the Pt(II) chelates by HSA, giving rise to the positive ICD bands at ∼378 nm (Fig. 9a).
Site displacement assay employing CD spectroscopy proves Sudlow's site II is targeted by Pt(II) chelates 1 and 2
To investigate the site-specific binding of 1 and 2 to HSA, an induced circular dichroism (ICD) site displacement assay was performed by incubating either complex in KH2PO4 buffer (50 mM, pH 7.5) with HSA and titrating in either warfarin (Sudlow's site I probe) or ibuprofen (Sudlow's site II probe). The Pt(II) chelates themselves are achiral and therefore do not produce a CD signal unless they are bound to a chiral host such as HSA. If the incoming site-specific probe ligand can displace the Pt(II) chelate, the ICD signal is lost due to the displaced metal chelate returning to an achiral protein-free state in solution.As shown in Fig. 10a, for HSA·{2} the ICD spectrum in the near-UV region was only weakly perturbed after adding a low dose of warfarin (4.2 μM); subsequent higher doses of the probe failed to displace 2 from the protein. Importantly, the logKa value of 2 for HSA (3.45) is significantly lower than that of warfarin for the protein (5.5),60 which would allow complete substitution of 2 by warfarin if the Pt(II) chelate occupied Sudlow's site I. The results clearly demonstrate that Sudlow's site I is not the main binding site of 2 within HSA. For HSA·{1} (Fig. S19†), there was a similarly negligible change in the ICD spectrum upon titration of warfarin into the solution of the protein complex, suggesting that warfarin did not displace 1. This could be due to the logKa of warfarin (logKa = 5.560) being slightly lower than that of 1 (logKa = 5.67) or indicate that 1 (like 2) does not bind within Sudlow's site I. At the highest concentration of warfarin (54 μM), the emergence of the warfarin-induced CD peak at 306 nm was observed.
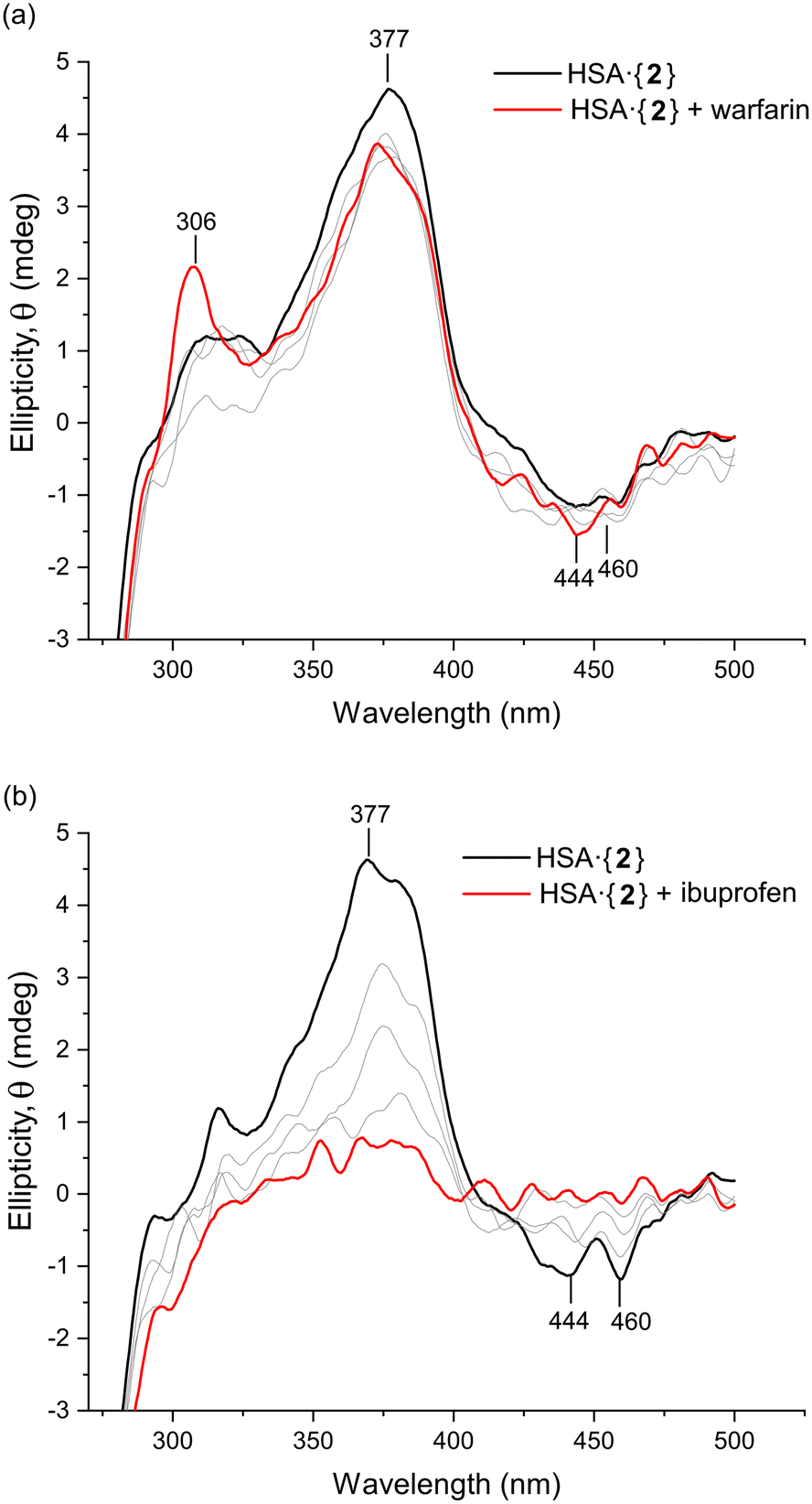 | ||
| Fig. 10 Induced CD site displacement assay carried out by measuring the UV-vis CD spectrum of HSA (15 μM in 50 mM KH2PO4 buffer, pH 7.5) bound to 2 (15 μM). The site-specific markers warfarin (Sudlow's site I) and ibuprofen (Sudlow's site II) were titrated into the solution of HSA·{2} in Parts (a) and (b), respectively, at concentrations of 4.1, 8.3, 12.5, and 20.1 μM (the grey lines represent doses <20.1 μM). The assay identifies which probe drug or marker can displace the Pt(II) chelate from its target binding site and thus delineates the true location of the metal chelate within the protein. The spectra were smoothed using a Lowess function (0.07 span) for clarity. | ||
When ibuprofen was titrated into HSA·{2}, there was a monotonic decrease in the ICD signal from 2 within the protein (Fig. 10b), consistent with displacement of 2 by ibuprofen into the bulk solution and confirming that 2 bound in either one or both ibuprofen binding sites (subunits IIIA or between IIA and IIIA).27 The evidence collectively demonstrates that both Pt(II) chelates preferentially bind in Sudlow's site II (subdomain IIIA). Notably, in silico data (TD-DFT simulations) endorsed the binding of 2 within Sudlow's site II of HSA by matching the key features of the experimental CD spectrum (Fig. 9a). Despite compelling spectroscopic evidence, however, only X-ray data can delineate the precise binding site(s) of 1 and 2 with absolute certainty.
Macromolecular simulations – general perspectives
Docking studies were carried out using Glide XP (Schrödinger Suite 2022-3) to predict potential binding sites in HSA for 1–3 and were compared to previously docked control compounds (warfarin, diazepam, ibuprofen).29 It is important to note that rigid X-ray structures of proteins are used to dock flexible ligands, and these methods provide an approximate insight into how a group of compounds may bind and interact with the target. Furthermore, docking scores usually have a poor quantitative relationship with experimental thermodynamic data (ΔG values),83,84 because a docking experiment does not replicate a physical reaction between the macromolecule and incoming ligand in solution. Therefore, only qualitative conclusions may be drawn between the thermodynamic data (Tables 1 and 2) and docking scores (Table S7†). Consequently, we have focussed on in silico data that confirms the experimental results (CD spectra, Ka values, and fluorescence quenching data). The key trends are discussed below.From Table S7,† HSA has several binding sites for 1 and 2, with docking scores (ΔGdock) ranging from −1.4 to − 5.2 kcal mol−1. Both Pt(II) chelates repeatedly favoured similar binding locations (∼3 in total including Sudlow's I and II) and a third site between subdomain IA–IIA, consistent with X-ray data for diverse drug and ligand types bound by HSA61 and recent data reported for d8 metal ions chelated by a related bis(pyrrolide-imine) ligand system.29
The binding sites targeted by the Pt(II) chelates were all located between 5–30 Å of Trp-214. Distances longer than 20 Å are less likely to cause quenching of Trp-214 fluorescence. In such instances, these may be secondary, less-favourable Pt(II) chelate binding sites. Regarding Tyr and Phe (Table S8†), both amino acids were within 6–19 Å from the docked Pt(II) chelate which afforded observable perturbations in the near-UV CD spectrum between 250 and 295 nm (Fig. 8c and d).
The presence of warfarin and diazepam bound within HSA subdomains IIA and IIIA, respectively, minimally influenced the docking scores for the uptake of 2, while there was a decrease in the docking score for 1 (Table S7†). Thus, ΔGdock for 1 is reduced from −4.62 to −1.39 kcal mol−1 when warfarin is present (Table S7†) and from −5.15 to −2.02 kcal mol−1 when diazepam is present (Table S7†). In both instances, 1 is redirected to less favourable binding sites. Ibuprofen bound within Sudlow's site II (subdomain IIIA, as well as at its secondary site, IIA-IIB) did not affect the docking scores of either 1 or 2. This in silico result accords with the site displacement data for HSA·{ibuprofen} (Fig. 7), since neither 1 nor 2 have sufficient affinity (high enough logKa values) to displace ibuprofen. Consequently, it seems likely that the Pt(II) chelates were redirected to alternate binding site(s) in the fluorescence quenching experiments depicted in Fig. 7.
Conclusions
The interaction of Pt(II) chelates 1–3 with HSA was investigated using complementary spectroscopic techniques to understand how the identity of the bis(pyrrole-imine) Schiff base ligand coordinated to the Pt(II) ion affects uptake of the complexes by the protein. All three Pt(II) chelates quenched the intrinsic Trp-214 fluorescence of HSA, with 1 exerting the effect via a static quenching mechanism, while 2 and 3 favoured a dynamic quenching mechanism. The three Pt(II) chelates had HSA affinity constants (Ka values) that were typical of many small molecule ligands for the protein (103–105 M−1).85,86 The Stern–Volmer quenching constants (Ksv) followed the order 1 > 2 > 3, while the Ka values followed the order 1 > 3 > 2. The reaction stoichiometry for all three Pt(II) chelates was 1:1 (Pt:HSA), with possible partial occupation of a second, lower affinity site occurring at higher concentrations.
Pt(II) chelates 1 and 3 bind to HSA with negative ΔH, ΔG, and positive ΔS values, reflecting a spontaneous enthalpy-driven process governed by van der Waals (London dispersion) forces, while 2 had a positive ΔH, negative ΔG and a strongly positive ΔS, consistent with an entropically driven reaction with HSA. Glide XP docking simulations in conjunction with fluorescence and induced CD site specificity assays employing probe ligands showed that 1 and 2 bind preferentially in Sudlow's site II, with possible additional binding sites being accessible. Far- and near-UV CD spectroscopy confirmed that the binding of 1 and 2 minimally perturbs the protein's secondary structure. Finally, the induced CD spectra recorded for HSA·{1} and HSA·{2} distinctly confirm uptake of the intact complexes without imine group hydrolysis or demetallation.
Author contributions
O. Q. M. and S. S. co-designed the study. S. S. synthesised and characterized the compounds, carried out the spectroscopy, collected and analysed the thermodynamic data, and performed most of the molecular simulations. O. Q. M. recorded the X-ray data for 1, solved and refined the structures of 1 and 2, and performed selected DFT simulations for spectroscopic data analysis. The initial manuscript was written by S. S.; the final manuscript and figures were reviewed, edited, and co-written/created in parts by O. Q. M.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work is based on research supported by the South African Research Chairs Initiative of the Department of Science and Innovation (DSI) and National Research Foundation (NRF) of South Africa (Grant No. 64799, OQM). The authors thank WITS University and the NRF for funding to purchase a JASCO J-1500 MCD spectrometer (Grant No. 116177, OQM) and a dual-wavelength Bruker D8 Venture X-ray diffractometer (Grant No. 129920, OQM). We also thank the Centre for High Performance Computing (Project CHEM1065, CHPC, Cape Town) for both the CPU time and resources needed for the DFT simulations. Some DFT simulations were undertaken on ARC4, part of the High-Performance Computing facilities at the University of Leeds, UK. We thank Prof. Manuel Fernandes (WITS University) for assistance with collecting the X-ray data for 2.References
- R. U. Braun, K. Zeitler and T. J. J. Müller, Org. Lett., 2001, 3, 3297–3300 CrossRef CAS PubMed.
- C. E. Hewton, M. C. Kimber and D. K. Taylor, Tetrahedron Lett., 2002, 43, 3199–3201 CrossRef CAS.
- M. P. Akerman and V. A. Chiazzari, J. Mol. Struct., 2014, 1058, 22–30 CrossRef CAS.
- C. H. Wei and J. R. Einstein, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 2591–2598 CrossRef CAS.
- Y. Wang, H. Fu, F. Shen, X. Sheng, A. Peng, Z. Gu, H. Ma, J. S. Ma and J. Yao, Inorg. Chem., 2007, 46, 3548–3556 CrossRef CAS PubMed.
- S. Bhikraj, O. Q. Munro and M. P. Akerman, Polyhedron, 2017, 124, 22–29 CrossRef CAS.
- J. S. Hart, G. S. Nichol and J. B. Love, Dalton Trans., 2012, 41, 5785–5788 RSC.
- H. Liang, J. Liu, X. Li and Y. Li, Polyhedron, 2004, 23, 1619–1627 CrossRef CAS.
- C. N. Iverson, C. A. G. Carter, R. T. Baker, J. D. Scollard, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2003, 125, 12674–12675 CrossRef CAS PubMed.
- X.-F. Shan, D.-H. Wang, C.-H. Tung and L.-Z. Wu, Tetrahedron, 2008, 64, 5577–5582 CrossRef CAS.
- Y. Kim, J. Lee, Y.-H. Son, S.-U. Choi, M. Alam and S. Park, J. Inorg. Biochem., 2020, 205, 111015 CrossRef CAS PubMed.
- C. H. Wei, Inorg. Chem., 1972, 11, 2315–2321 CrossRef CAS.
- N. A. Bailey, A. Barrass, D. E. Fenton, M. S. L. Gonzalez, R. Moody and C. O. R. de Barbarin, J. Chem. Soc., Dalton Trans., 1984, 2741–2746 RSC.
- F. Z. C. Fellah, J.-P. Costes, F. Dahan, C. Duhayon, G. Novitchi, J.-P. Tuchagues and L. Vendier, Inorg. Chem., 2008, 47, 6444–6451 CrossRef CAS PubMed.
- Y. Hu, Q. Li, H. Li, Q. Guo, Y. Lu and Z. Li, Dalton Trans., 2010, 39, 11344–11352 RSC.
- H. Adams, M. R. J. Elsegood, D. E. Fenton, S. L. Heath and S. J. Ryan, J. Chem. Soc., Dalton Trans., 1999, 2031–2038 RSC.
- K. J. Akerman, A. M. Fagenson, V. Cyril, M. Taylor, M. T. Muller, M. P. Akerman and O. Q. Munro, J. Am. Chem. Soc., 2014, 136, 5670–5682 CrossRef CAS PubMed.
- F. M. Muggia, A. Bonetti, J. D. Hoeschele, M. Rozencweig and S. B. Howell, J. Clin. Oncol., 2015, 33, 4219–4226 CrossRef CAS PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481–489 CrossRef CAS PubMed.
- T. Boulikas, A. Pantos, E. Bellis and P. Christofis, Cancer Ther., 2007, 5, 537–583 Search PubMed.
- T. Makovec, Radiol. Oncol., 2019, 53, 148–158 CrossRef CAS PubMed.
- T. Peters Jr., All About Albumin: Biochemistry, Genetics, and Medical Applications, Academic Press, 1995 Search PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- P. A. Zunszain, J. Ghuman, T. Komatsu, E. Tsuchida and S. Curry, BMC Struct. Biol., 2003, 3, 6 CrossRef PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1975, 11, 824–832 CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 CAS.
- M. Fasano, S. Curry, E. Terreno, M. Galliano, G. Fanali, P. Narciso, S. Notari and P. Ascenzi, IUBMB Life, 2005, 57, 787–796 CrossRef CAS PubMed.
- G. Rabbani and S. N. Ahn, Int. J. Biol. Macromol., 2019, 123, 979–990 CrossRef CAS PubMed.
- S. Sookai and O. Q. Munro, ChemistryEurope, 2023, 1, e202300012 CrossRef.
- M. P. Akerman, T. Chatturgoon and O. Q. Munro, Inorg. Chim. Acta, 2014, 421, 292–299 CrossRef CAS.
- J. R. L. Priqueler, I. S. Butler and F. D. Rochon, Appl. Spectrosc. Rev., 2006, 41, 185–226 CrossRef CAS.
- B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- A. Albinati, F. Lianza, P. S. Pregosin and B. Mueller, Inorg. Chem., 1994, 33, 2522–2526 CrossRef CAS.
- M. Karplus, J. Chem. Phys., 2004, 30, 11–15 CrossRef.
- I. M. Ismail, S. J. S. Kerrison and P. J. Sadler, Polyhedron, 1982, 1, 57–59 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- S. Soares, N. Mateus and V. de Freitas, J. Agric. Food Chem., 2007, 55, 6726–6735 CrossRef CAS PubMed.
- M. H. Gehlen, J. Photochem. Photobiol., C, 2020, 42, 100338 CrossRef CAS.
- O. K. Abou-Zied and O. I. K. Al-Shihi, J. Am. Chem. Soc., 2008, 130, 10793–10801 CrossRef CAS PubMed.
- P. R. Callis, J. Mol. Struct., 2014, 1077, 14–21 CrossRef CAS.
- Y. Yue, J. Liu, M. Yao, X. Yao, J. Fan and H. Ji, Spectrochim. Acta, Part A, 2012, 96, 316–323 CrossRef CAS PubMed.
- A. Hussain, M. F. AlAjmi, M. T. Rehman, S. Amir, F. M. Husain, A. Alsalme, M. A. Siddiqui, A. A. AlKhedhairy and R. A. Khan, Sci. Rep., 2019, 9, 5237 CrossRef PubMed.
- S. R. Feroz, S. B. Mohamad, N. Bujang, S. N. A. Malek and S. Tayyab, J. Agric. Food Chem., 2012, 60, 5899–5908 CrossRef CAS PubMed.
- A. Divsalar, M. J. Bagheri, A. A. Saboury, H. Mansoori-Torshizi and M. Amani, J. Phys. Chem. B, 2009, 113, 14035–14042 CrossRef CAS PubMed.
- M. K. Helms, C. E. Petersen, N. V. Bhagavan and D. M. Jameson, FEBS Lett., 1997, 408, 67–70 CrossRef CAS PubMed.
- K. Flora, J. D. Brennan, G. A. Baker, M. A. Doody and F. V. Bright, Biophys. J., 1998, 75, 1084–1096 CrossRef CAS PubMed.
- P. Alam, S. K. Chaturvedi, T. Anwar, M. K. Siddiqi, M. R. Ajmal, G. Badr, M. H. Mahmoud and R. H. Khan, J. Lumin., 2015, 164, 123–130 CrossRef CAS.
- G. G. Ariga, P. N. Naik, S. A. Chimatadar and S. T. Nandibewoor, J. Mol. Struct., 2017, 1137, 485–494 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, Boston MA, 3rd edn, 2006 Search PubMed.
- F. Samari, B. Hemmateenejad, M. Shamsipur, M. Rashidi and H. Samouei, Inorg. Chem., 2012, 51, 3454–3464 CrossRef CAS PubMed.
- Z. Chen, S. Zhang, Z. Zhu and Y. Zhang, New J. Chem., 2017, 41, 6340–6348 RSC.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed.
- A. Samanta, S. Jana, D. Ray and N. Guchhait, Spectrochim. Acta, Part A, 2014, 121, 23–34 CrossRef CAS PubMed.
- H. Aki, M. Goto and M. Yamamoto, Thermochim. Acta, 1995, 251, 379–388 CrossRef CAS.
- R. E. Olson and D. D. Christ, in Annual Reports in Medicinal Chemistry, ed. J. A. Bristol, Academic Press, 1996, vol. 31, pp. 327–336 Search PubMed.
- H. Aki and M. Yamamoto, J. Pharm. Sci., 1994, 83, 1712–1716 CrossRef CAS PubMed.
- I. Petitpas, A. A. Bhattacharya, S. Twine, M. East and S. Curry, J. Biol. Chem., 2001, 276, 22804–22809 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, in Methods in Enzymology, Academic Press, 2004, vol. 383, pp. 318–351 Search PubMed.
- A. Micsonai, F. Wien, É. Bulyáki, J. Kun, É. Moussong, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- F. Mohammadi, A.-K. Bordbar, A. Divsalar, K. Mohammadi and A. A. Saboury, Protein J., 2009, 28, 189–196 CrossRef CAS PubMed.
- H. A. Tajmir-Riahi, C. N. N'Soukpoe-Kossi and D. Joly, Spectroscopy, 2009, 23, 81–101 CrossRef CAS.
- A. Khammari, A. A. Saboury, M. H. Karimi-Jafari, M. Khoobi, A. Ghasemi, S. Yousefinejad and O. K. Abou-Zied, Phys. Chem. Chem. Phys., 2017, 19, 10099–10115 RSC.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- F. R. Clemente, T. Vreven and M. J. Frisch, Quantum Biochemistry, John Wiley & Sons, Ltd, 2010, pp. 61–83 Search PubMed.
- P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- G. Ferraro, L. Massai, L. Messori and A. Merlino, Chem. Commun., 2015, 51, 9436–9439 RSC.
- A. Bijelic, S. Theiner, B. K. Keppler and A. Rompel, J. Med. Chem., 2016, 59, 5894–5903 CrossRef CAS PubMed.
- H. Yin, L. Liao and J. Fang, JSM Clin. Oncol. Res., 2014, 2, 1010 Search PubMed.
- D. Lafitte, P. O. Tsvetkov, F. Devred, R. Toci, F. Barras, C. Briand, A. A. Makarov and J. Haiech, Biochim. Biophys. Acta, Proteins Proteomics, 2002, 1600, 105–110 CrossRef CAS PubMed.
- O. K. Abou-Zied and O. I. K. Al-Shihi, J. Am. Chem. Soc., 2008, 130, 10793–10801 CrossRef CAS PubMed.
- S. M. Kelly and N. C. Price, Curr. Protein Pept. Sci., 2000, 1, 349–384 CrossRef CAS PubMed.
- X.-X. Cheng, X.-Y. Fan, F.-L. Jiang, Y. Liu and K.-L. Lei, Luminescence, 2015, 30, 1026–1033 CrossRef CAS PubMed.
- A. Barth, S. R. Martin and P. M. Bayley, Biopolym. Orig. Res. Biomol., 1998, 45, 493–501 CAS.
- B. Valeur and G. Weber, Photochem. Photobiol., 1977, 25, 441–444 CrossRef CAS PubMed.
- T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815–826 CrossRef CAS PubMed.
- A. R. Leach, B. K. Shoichet and C. E. Peishoff, J. Med. Chem., 2006, 49, 5851–5855 CrossRef CAS PubMed.
- G. L. Warren, C. W. Andrews, A.-M. Capelli, B. Clarke, J. LaLonde, M. H. Lambert, M. Lindvall, N. Nevins, S. F. Semus, S. Senger, G. Tedesco, I. D. Wall, J. M. Woolven, C. E. Peishoff and M. S. Head, J. Med. Chem., 2006, 49, 5912–5931 CrossRef CAS PubMed.
- C. Dong, S. Ma and Y. Liu, Spectrochim. Acta, Part A, 2013, 103, 179–186 CrossRef CAS PubMed.
- G. Wang, X. Li, X. Ding, D. Wang, C. Yan and Y. Lu, J. Pharm. Biomed. Anal., 2011, 55, 1223–1226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details and ESI tables and figures in PDF format, and X-ray crystal structures. CCDC 2271558 and 2271559. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02039h |
| This journal is © The Royal Society of Chemistry 2023 |