
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation on the predictive power of tolerance factor τ for A-site double perovskite oxides†
Elisabeth K.
Albrecht
and
Antti J.
Karttunen
 *
*
Department of Chemistry and Materials Science, Aalto University, P.O. Box 16100, FI-00076 Aalto, Finland. E-mail: antti.karttunen@aalto.fi
First published on 15th August 2023
Abstract
We have used a recently introduced new tolerance factor τ to create a stability map of all possible A-site double perovskite titanates AA′Ti2O6 and niobates AA′Nb2O6. The predictive power of τ is relatively good based on comparisons with available experimental data for A-site double perovskites. We carried out quantum chemical calculations on two hypothetical double perovskite compositions CsScTi2O6 and YRbTi2O6, where τ predicts high probability for their existence. In both cases, we found limits in the predictive power of the new tolerance factor for ion combinations on the A and A′ site which are very different in size. A difference in oxidation state may decrease the accuracy, as well. Overall, the A-site double perovskite stability mapping provides a starting point for the discovery of novel A-site double perovskites.
1. Introduction
Crystal structure prediction for compounds such as perovskites is still a challenging task due to the variety of compositions and structural modifications they can adapt. The general structural formula ABX3 with the space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m for ideal perovskites has the A cation on the corners of a primitive cubic unit cell, while the B cation sits in the center. The X anion occupies the face centers, leading to an octahedral coordination of the B cation. The BX6 octahedra are corner-sharing and can tilt and distort due to for example size mismatch in the cations.1 The size mismatches and resulting structural effects make it difficult to predict the existence and crystal structure of perovskites. Goldschmidt suggested a tolerance factor t
m for ideal perovskites has the A cation on the corners of a primitive cubic unit cell, while the B cation sits in the center. The X anion occupies the face centers, leading to an octahedral coordination of the B cation. The BX6 octahedra are corner-sharing and can tilt and distort due to for example size mismatch in the cations.1 The size mismatches and resulting structural effects make it difficult to predict the existence and crystal structure of perovskites. Goldschmidt suggested a tolerance factor t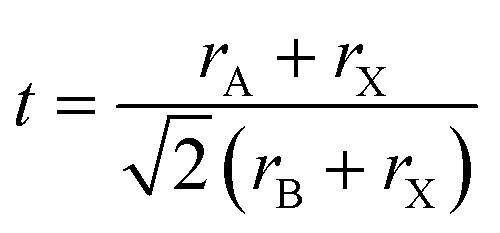 | (1) |
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 due tilting and distortion of octahedra as well as displacement of cations. For t > 1.059, a hexagonal perovskite-like structure is expected, and for t < 0.825 other structure types such as ilmenite are more likely. Overall, the tolerance factor t correctly predicts only about 74%4 of the materials as perovskites or non-perovskites. Therefore, Bartel et al.4 have suggested a new tolerance factor τ
4 due tilting and distortion of octahedra as well as displacement of cations. For t > 1.059, a hexagonal perovskite-like structure is expected, and for t < 0.825 other structure types such as ilmenite are more likely. Overall, the tolerance factor t correctly predicts only about 74%4 of the materials as perovskites or non-perovskites. Therefore, Bartel et al.4 have suggested a new tolerance factor τ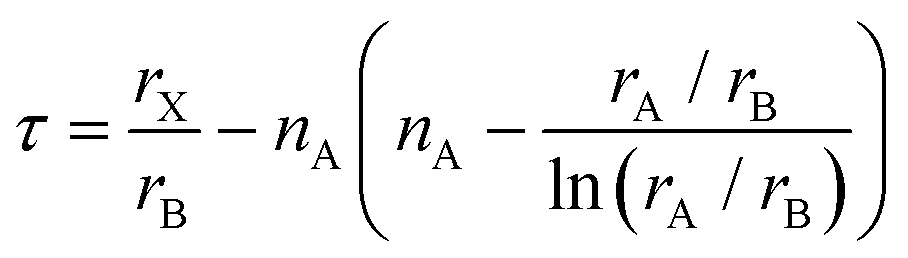 | (3) |
In 2018, Filip and Giustino7 screened ions in 2291 known compounds for their ability to form B-site ordered double perovskites using the average B-ion radius together with tolerance factor t. The screening was based on Goldschmidt's non-rattling principle which is also the basis of the Goldschmidt tolerance factor t. The octahedral factor μ was divided into the average octahedral factor 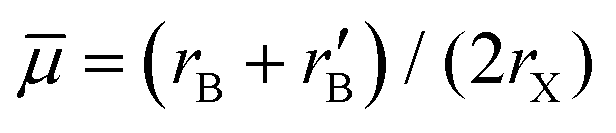 and the octahedral mismatch factor Δμ = |rB − r′B|/(2rX). These three parameters were then used to predict 94232 hitherto-unknown perovskites.
and the octahedral mismatch factor Δμ = |rB − r′B|/(2rX). These three parameters were then used to predict 94232 hitherto-unknown perovskites.
While B-site ordered double perovskites have been investigated in several tolerance factor-based high-throughput screening studies, A-site ordered double perovskites (Fig. 1) have received less attention. No high-throughput screening studies on the existence or crystal structure of A-site (ordered) double perovskites have been carried out, in particular with respect to the potential ordering of the A-site cations. It is relatively difficult to predict whether two different ions on the A-site will order or not. In fact, considering the relatively small number of A-site ordered double perovskites it is more likely that the A-site ions will not order. However, as an initial screening approach, compositions with two different ions on the A-site can in any case be considered. A screening of potential A-site double perovskites gives an idea about how well the new tolerance factor τ works for A-site double perovskites when comparing the predictions with known compounds. Furthermore, such screening can be used to predict potential compositions that might be worth investigating experimentally.
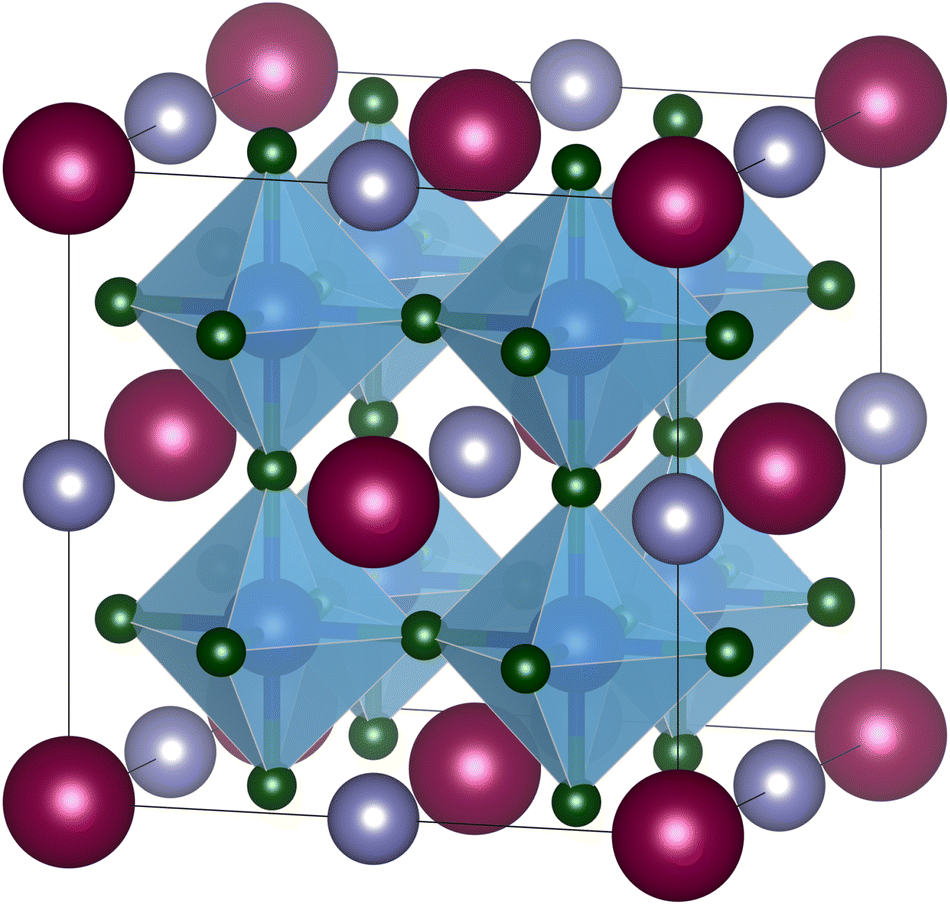 | ||
| Fig. 1 A-site ordered double perovskite AA′B2X6 with rock-salt ordering of the A-site cations. A cations colored in dark red, A′ cations in purple, B cations in blue, and X anions in green. Figure created with VESTA.8 | ||
Various functional properties such as ferroelectricity, pyroelectricity, superconductivity, and ferromagnetism make perovskites an important materials research platform due to high demand on materials with very specific functionalities.9–11 By extending the perovskite compositions from simple to double perovskites, their properties can be further fine-tuned by combining different ions. While there has been a significant research focus on B-site ordered double perovskites,12 A-site ordered double perovskites have been studied less.13–26 Here, we aim to provide a starting point to make predictions on the crystal structures of new A-site ordered double perovskites (AA′B2O6). For this purpose, we have compiled a database of possible A-site double perovskite compositions and applied the new tolerance factor τ of Bartel et al. to measure the probability of obtaining a double perovskite for each composition. We have also carried out quantum chemical calculations on several promising compositions based on τ to investigate the strengths and weaknesses of this tolerance factor when applied to A-site double perovskites.
2. Methods
2.1. Data acquisition
First, we built a database of possible compositions for A-site ordered double perovskites. For the A-site, we included all metals with an atomic number ≤83, except for technetium. For the B-site, we chose niobium (+V) and titanium (+IV), as they are common B-site ions in perovskites. They can also show second-order Jahn–Teller effect which supports layered ordering in A-site ordered double perovskites.13 From this set of elements, we removed all combinations for the A-site which did not lead to a zero net charge with the chosen B-ions. Similar to Bartel et al.4 in the case of A2BB′O6 double perovskites, we used the Shannon ionic radii for the calculation of τ.27 Since the ionic radii depend on the coordination number of the ion, we chose to use the 12-coordinated ionic radius for the A-site cations if available. In those cases where there was no 12-coordination available, we chose the ionic radius of the coordination closest to twelve, in line with Bartel et al. In reality, the ion may not form a structure with such high coordination number and this would be accounted for by a tilt of the oxygen octahedra, which may also change the coordination of the oxygen anions from six to a smaller number. However, the ionic radius of oxygen was kept the same for all evaluated compositions (1.4 Å, corresponding to coordination number six).In addition to the ionic radius choices discussed above, we also evaluated another approach: if a 12-coordinated ionic radius was not available for an ion, we estimated it from Bond Valence Sum (BVS) calculations using
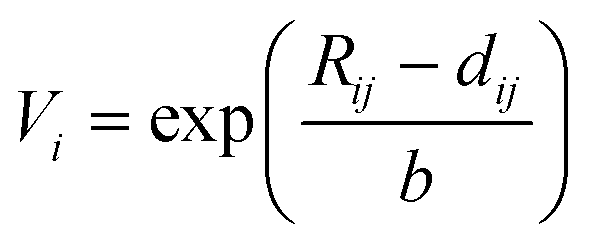 | (4) |
To get the probability of forming a perovskite with a certain composition, we fitted the calculated values of τ to the Platt-scaled classification probabilities of Bartel et al.4 Next, we translated the probabilities to a color scale and constructed a map out of them. In those cases where multiple ionic combinations with the same elements were possible, we used the average probability. For example in the case of AuTlTi2O6, both Au and Tl can have the oxidation state of +I or +III which leads to different average ionic radii and thus to different values of τ. The probabilities obtained for such combinations do not differ much, which is why we chose to use the average value. In the case of AuTlTi2O6, the probabilities are 58% for Au(III)/Tl(I) and 52% for Au(I) and Tl(III). This averaging applies to five combinations, CoCuTi2O6, CuEuTi2O6, MnCuTi2O6, TiCuTi2O6, and AuTlTi2O6.
2.2. Computational details
For the quantum chemical calculations, CRYSTAL1731 program package was used. We applied hybrid PBE0 density functional method, (DFT-PBE0)32,33 in combination with all-electron, Gaussian-type basis sets based on Karlsruhe def2 sets.34 Triple-ζ-valence + polarization (TZVP) quality basis sets for O, Sc, Ti, Y, and Nb were taken from ref. 35. Split-valence + polarization(SVP) quality basis sets for K, Rb, and Cs were taken from ref. 36. The Monkhorst–Pack type k-meshes used to sample the reciprocal space are similar to our previous paper on CaMnTi2O6 double perovskite.37,38 In all calculations, default DFT integration grids and optimization convergence thresholds of CRYSTAL17 were applied. The calculations were carried out with Coulomb and exchange integral tolerance factors (TOLINTEG) set to tight values of 8, 8, 8, 8, and 16. All reported relative energies are based on electronic energies obtained at 0 K, Gibbs Free Energies have not been considered. Harmonic frequency calculations were carried out on selected crystal structures with the approach implemented in CRYSTAL.39,403. Results and discussion
3.1. Overview of the possible A-site double perovskites based on τ
The map in Fig. 2 provides an overview of possible A-site double perovskites and their probability of forming a perovskite titanate (upper triangle) or niobate (lower triangle). The probabilities are based on tolerance factor τ (eqn (3)). Since A-site double perovskites are less studied than B-site ordered double perovskites, the map gives a starting point for choosing potential new AA′ combinations in an AA′B2O6 double perovskite. The darker the field in Fig. 2, the higher the probability of forming a perovskite based on the tolerance factor τ. Due to the oxidation state of +V of niobium, there are only few possible combinations for A-site ions in the lower triangle. When the B-site is occupied by Nb, both the A and A′ ion must have an oxidation state of +I. When the B-site is occupied by Ti in oxidation state +IV, there is a larger variety of ionic combinations. Combinations of oxidation states II/II, I/III, and III/I are possible. We note that the map does not provide information of whether the A-site ions will order in a certain way or if they will be disordered, forming a solid solution.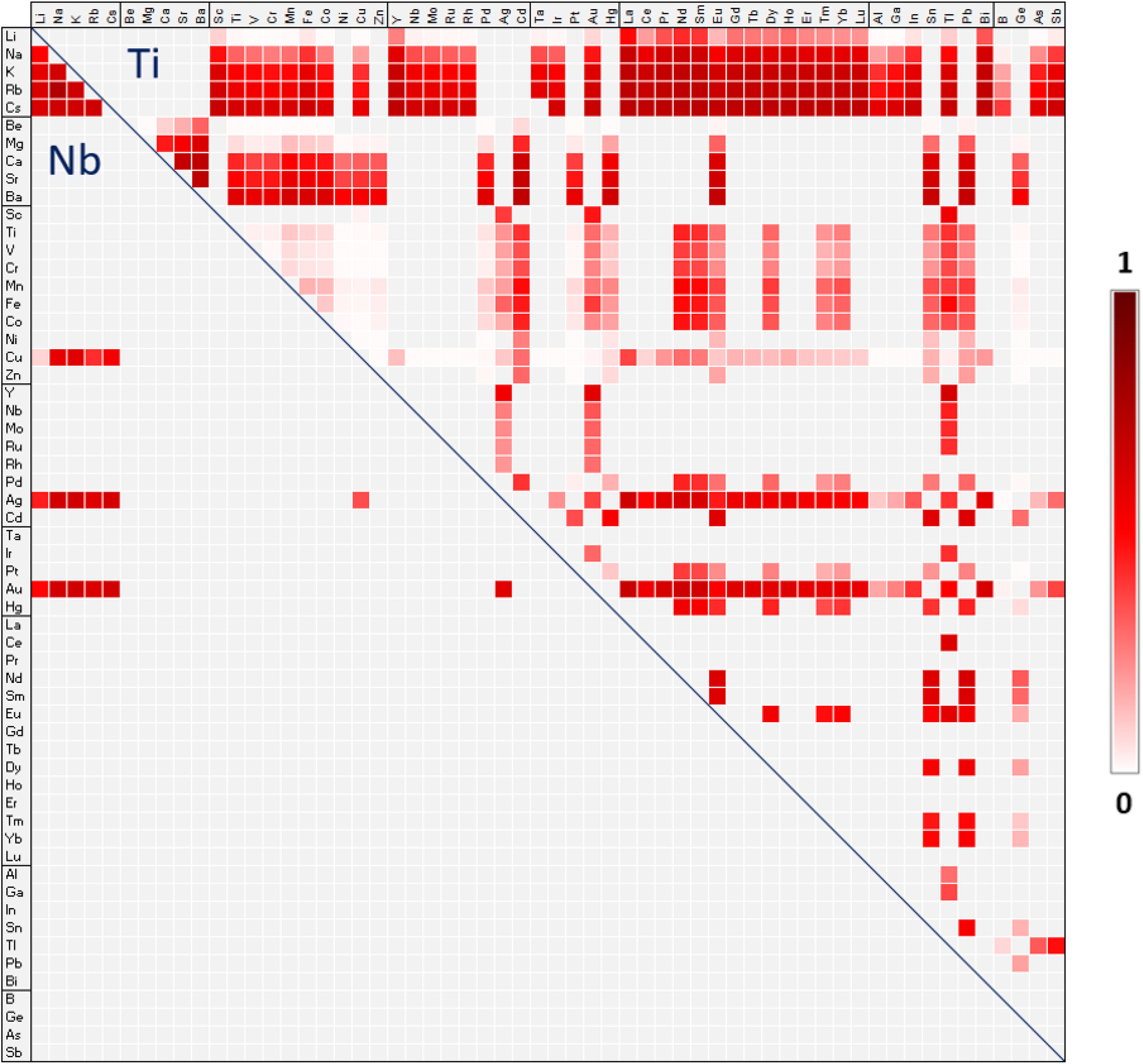 | ||
| Fig. 2 Map of the possible A-site double perovskite titanates (upper triangle) and niobates (lower triangle). The color scale gives the probability of forming a perovskite with the formula AA′(Ti/Nb)2O6. Probabilities have been obtained by fitting the calculated τ values to the Platt-scaled classification probabilities from Bartel et al.4 The combination of A and A′ ions must provide charge-balanced composition and per definition the average of the ionic radii of A and A′ must be larger than the ionic radius of B. | ||
Based on the tolerance factor τ, the most likely A-site double perovskites are expected for cases where the A- and A′-site are filled with an alkali metal in combination with a d-metal or a lanthanide. The larger the alkali metal cation, the more likely the perovskite, which makes compositions with Cs the most likely ones. According to Bartel et al.,4 a perovskite is expected if τ < 4.18. The most likely composition, with the smallest positive τ value of 3.751 is CsLaTi2O6, which has not been experimentally observed yet. In the case of CsLaTi2O6, the difference in oxidation state of cesium(I) and lanthanum(III), as well as the difference in ionic radii of about 0.52 Å suggests an A-site ordered perovskite.14 Since gold can show oxidation states I, II, and III, it forms many possible combinations. Here, again, those with the lanthanides and alkali metals are the most likely ones as in AuLaTi2O6 and CsAuTi2O6 with τ = 3.860.
The map in Fig. 2 has been drawn based on Shannon ionic radii only, but the use of BVS-derived radii for the ions that do not have a 12-coordinated Shannon radii changes most of the τ values close to 4.18 only by few percent, leading only in small changes in probability. A more significant change can be seen for six negative τ values, where the average ionic radius of A and A′ is smaller than the ionic radius of the B ion (Table S1 in ESI†). These are LiBTi2O6, BeZnTi2O6, BeCuTi2O6, BeGeTi2O6, BeNiTi2O6, and CuBTi2O6. The map in Fig. 2 does not take these compositions into account since they do not fulfil the definition of rA > rB. Geometrically, this definition makes sense and we do not expect those combinations to be likely. When using BVS-derived radii for Li, Be, and B, the τ values become large and positive, suggesting the compositions to be highly unlikely.
As mentioned above, a perovskite is expected if τ < 4.18. Out of the 208 predicted A-site double perovskites with τ < 4.18, 26 have been synthesized and characterized as perovskites according to ICSD. Most of those, however, show tilting and/or distortion of the BO6 octahedra. So far, nine A-site double perovskites with τ > 4.18 have been synthesized, six of them under high pressure and high temperature. NaEuTi2O6 has been obtained at atmospheric pressure but high temperature.41 It is very close to the border of being determined a perovskite with τ = 4.182. The compound Li0.2Na0.8Nb2O342 is synthesized at atmospheric pressure and 1000 °C, the same as Li0.1Ag0.9Nb2O3.43 The small amount of lithium in the compounds explains the rather mild synthesis conditions. Of those combinations with τ < 4.18, we found 30 compounds which were synthesized and characterized but are not perovskites. However, in none of these cases the stoichiometry fits a perovskite-like stoichiometry. It is possible that with the correct stoichiometry and under other synthesis conditions a perovskite would form. Considering the relatively large number of correctly predicted A-site double perovskites, and the fact that perovskites with τ > 4.18 have been synthesized under high pressure, the predictive power of the tolerance factor τ appears to be rather good also for A-site double perovskites.
Most interesting A-site double perovskites are those where the A and A′ ions order in columnar, planar, or rock-salt ordering.14 Next, we will investigate how combining the map in Fig. 2 with quantum chemical methods can be used as a starting point for exploring new A-site ordered double perovskites. After choosing any potential composition, quantum chemical methods can be used to investigate the structural characteristics and energetics in more detail.
3.2. Quantum chemical calculations on potential A-site double perovskites
Looking at Fig. 2 for likely A-site ordered double perovskite compositions, we focus on titanates with alkali metals (+I) on the A-site because they pair with a +III cation on the A′-site, leading to a difference in the oxidation states. This difference enhances the likelyhood of ordering, in particular in combination with matching octahedral tilting, anion vacancies, or second-order Jahn–Teller distortions of B-site cations.14 Furthermore, we pay attention to the ionic radii. Alkali metals and lanthanides are close in ionic size and King et al. found several of them to form stable compounds in their 2007 paper44 on doubly cation ordered perovskites of the form AA′BWO6. In fact, King and Garcia-Martin provided in 2019 a list of all known doubly cation ordered perovskites of the form AA′BB′O6,21 and almost all compounds in the list have a combination of an alkali metal ion and a lanthanide ion on the A and A′ sites (few compounds with Y(III) cation are included). Perovskites with A-site cations of similar size (e.g. Na and La) thus appear to be easier to synthesize. However, in the abovementioned papers, the focus was on doubly cation ordered perovskites where the different B-site cations can also influence the ordering of the A-site. In their 2010 paper,14 King and Woodward note the A-site to be more likely to order in a layered fashion if the A-site cations are different in size. Layered ordering is also the most common type of ordering on the A-site. In 2017, Gou et al.45 also suggested that A-site ordering requires a difference in size for the ionic radii.For the +III cation, we decided to test the predictive power of the τ tolerance factor for d-metals instead of the more studied lanthanides. Combination of smaller d-metal cation and larger alkali metal cation should promote ordering on the A-site. It can be seen in Fig. 2 that of such combinations, Cs as the A ion and Sc as the A′ ion has one of the highest likelihoods with a τ value of 3.85. In this composition, there is a large size difference of 1.01 Å in the ionic radii (rCs = 1.88 Å, rSc = 0.87 Å). It needs to be mentioned here that these values belong to a twelve coordination of Cs but an eight-coordination of Sc since this is the highest coordination listed by Shannon. BVS-derived 12-coordinated radius for Sc would be 0.96 Å, which would lead to a difference of 0.92 Å and τ value of 3.82 Å (even higher likelyhood of existence). To put these values in perspective, the largest difference between A and A′ ionic radii in known double perovskite titanates is 0.63 Å in KCeTi2O6.46 CsScTi2O6 has yet not been synthesized according to ICSD.
Another example for a potential A-site ordered double perovskite is YRbTi2O6, which also has a difference of two in the oxidation states of Y(III) and Rb(I). It has not yet been synthesized according to ICSD. The difference in ionic radius is smaller than in CsScTi2O6: with rY = 1.08 Å and rRb = 1.72 Å, the difference is only 0.64 Å which is in the range of the ionic size difference in KCeTi2O6 (0.63 Å, the largest difference between A and A′ ionic radii in known double perovskite titanates). Again, the ionic radius for Rb is given for twelve coordination while the one for Y is in nine coordination. The BVS-derived 12-coordinated ionic radius of Y would be 1.13 Å which leads to a difference of 0.59 Å. For the two radii choices, the τ values are essentially identical with 3.83 and 3.84, both values suggesting that a perovskite should form.
To sum up, the tolerance factor τ together with other structural characteristics would suggest CsSCTi2O6 and YRbTi2O6 to be A-site ordered double perovskites. We now follow our previous work38 where we systematically investigated the ordering and Glazer tilt system47 of A-site ordered double perovskites. We take into account 11 Glazer tilt systems together with columnar, planar, or rock-salt ordering which leaves us with 33 possible crystal structures for each composition. We optimize each crystal structure with DFT-PBE0 method and analyze the resulting structures and energetics.
3.3. CsScTi2O6
In CsScTi2O6, the A and A′ ion are very different in size, and as a result the TiO6 octahedra are mildly to strongly distorted in most of the 33 crystal structures studied, within the constraints of the space group symmetry. Typically, Glazer tilt systems are built from rigid B-site coordination octahedra (Ti in this case) that can be tilted but not distorted. In some of the optimized crystal structures, the octahedra distort to the point where no octahedra are formed anymore. At the same time in the A-site, the coordination number of Sc can go down to five or even smaller, which is too small for scandium.27 The lowest-energy crystal structure optimized for CsScTi2O6 is in space group Pmc21 in planar A-site ordering, where a a−b+a− tilt system was expected. Fig. 3 shows the calculated structure. It is obvious that the perovskite structure is not the preferred structure for this compound since the lowest energy system does not even show the typical octahedra. In fact, the six lowest-energy structures, planar a−b+a−, a−b−b−, a−a−a− and a+b0c− and rock salt a−b+a− and a+b0c− do not show octahedral coordination of titanium ions. According to Shannon,27 scandium prefers a coordination of at least six which also is unavailable in many of those low-energy structures. The lowest-energy perovskite-like structure with octahedrally coordinated titanium is planar a0b−b− which can be seen in Fig. 4. It is 114 kJ mol−1 higher in energy than the lowest energy structure (Fig. 3) which is a significant difference. A large ionic size difference of the A-site cations might thus undermine the predictive value of the tolerance factor τ in its current form. Eqn (3) only uses the average ionic radius. There is a possibility that ions which are too different in size will not form a perovskite despite τ predicting it to be likely.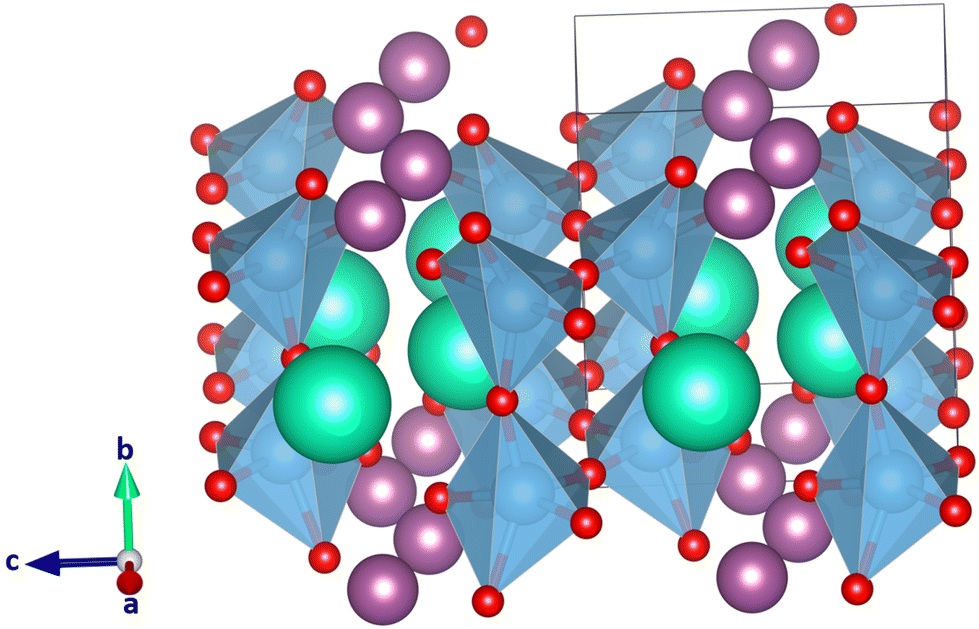 | ||
| Fig. 3 Lowest-energy structure of the 33 optimized CsScTi2O6 crystal structures. Cs(I) cations colored in green, Sc(III) cations in purple, Ti(IV) cations in blue, and O(−II) anions in red. The starting point of the structural optimization was the a−b+a− tilt system in planar A-site ordering with the space group Pmc21. Lattice parameters a, b, and c have not been constrained during the optimization even though the tilt system would a−b+a− require two lattice parameters to be the same. | ||
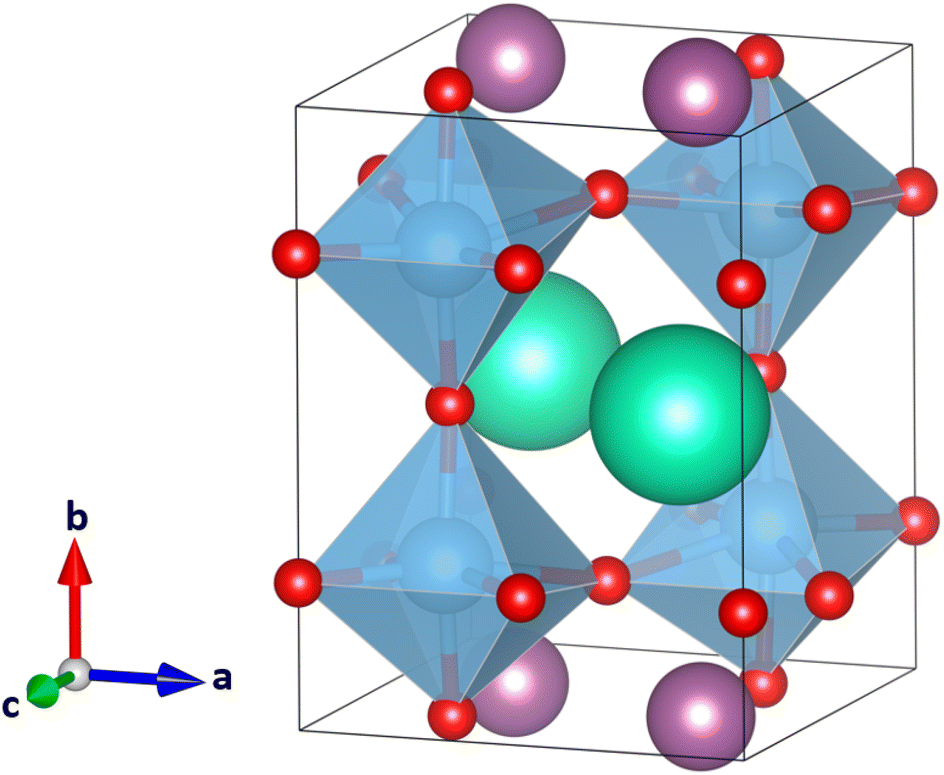 | ||
| Fig. 4 Lowest-energy perovskite-like structure of the 33 optimized CsScTi2O6 crystal structures, retaining the TiO6 octahedra. Cs(I) cations colored in green, Sc(III) cations in purple, Ti(IV) cations in blue, and O(−II) anions in red. In planar A-site ordering and with the tilt system a0b−b− it has the space group Pmma. Lattice parameters were not constrained in any way, although in principle the tilt system a0b−b− would require two lattice parameters to be the same. | ||
All in all, the compound CsScTi2O6 shows the limits of the new tolerance factor τ for A-site ordered double perovskites for A-site cations with large size difference. It remains to be tested whether CsScTi2O6 can form a perovskite or not. According to the map in Fig. 2 the compound should form a double perovskite while quantum chemical calculations do not support this prediction.
3.4. YRbTi2O6
Since the large size difference of the A and A′ cations in CsScTi2O6 caused strong distortion of the octahedra and too small coordination number of the Sc ions in some of the structures, we now look at YRbTi2O6 (rY = 1.08 Å, nine-coordinated radius and rRb = 1.72 Å, 12-coordinated radius with a difference of 0.64 Å). The results of the calculations can be found in Table 1. The lowest-energy ordering-tilt combination, columnar a+b0c− (space group Pmm2), has a long Ti–O distance of 2.99 Å (Fig. 5). Harmonic frequency calculations show the optimized structure to be a true global minimum. The second-lowest energy structure, planar a−b+a−, is 35 kJ mol−1 higher in energy and exhibits slightly distorted but still perovskite-like TiO6 octahedra (Fig. 6). Table 1 shows that the planar ordering seems to be the lowest-energy ordering for the A-site cations, which is plausible since planar ordering is the most likely for A-site ordered double perovskites.14 Further research is necessary to determine whether YRbTi2O6 can form a perovskite structure or not.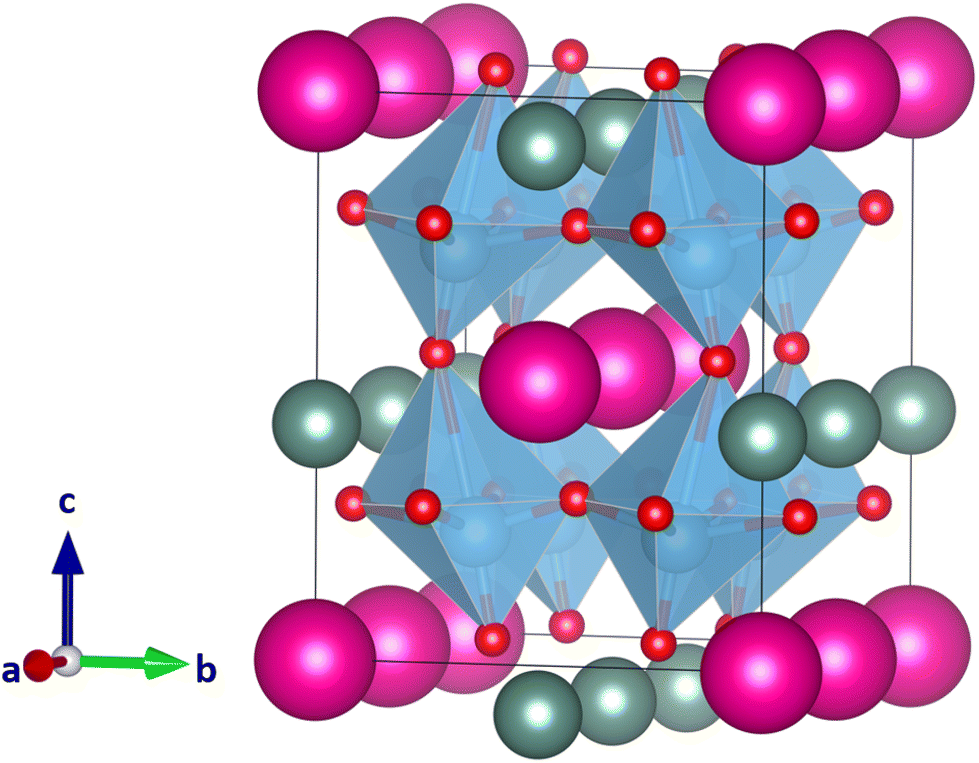 | ||
| Fig. 5 Lowest-energy perovskite-like structure of the 33 calculated YRbTi2O6 structures with Y(III) cations colored in grey, Rb(I) cations in pink, Ti(IV) cations in blue, and O(−II) anions in red. In columnar A-site ordering and with the tilt system a+b0c− the space group is Pmm2. | ||
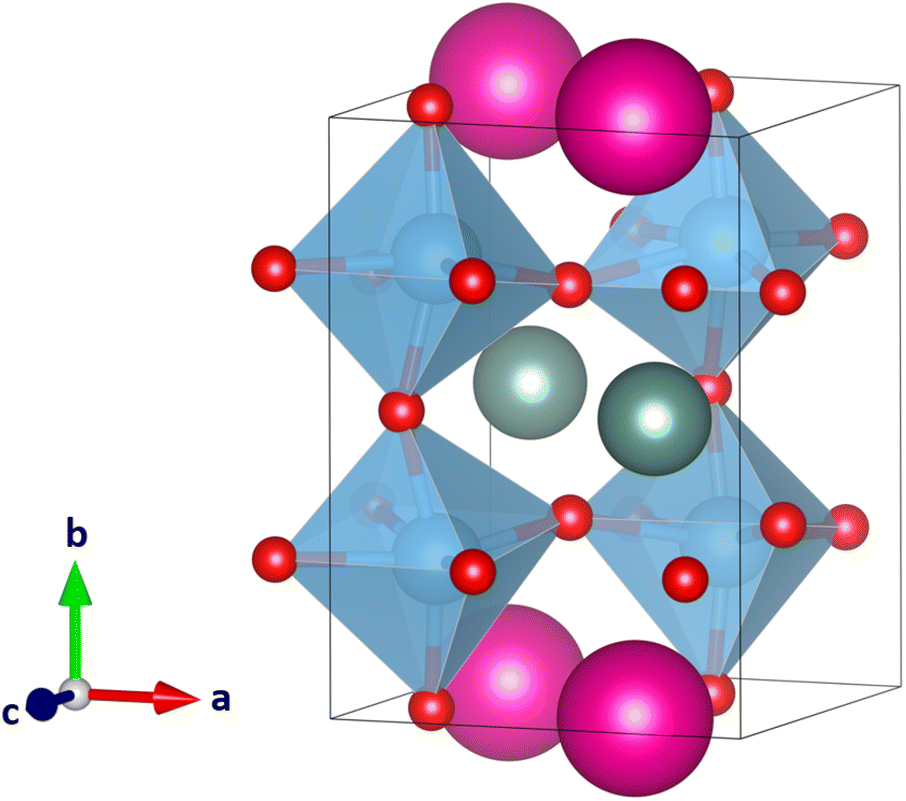 | ||
| Fig. 6 Second-lowest energy perovskite-like structure of the 33 calculated YRbTi2O6 structures with Y(III) cations colored in grey, Rb(I) cations in pink, Ti(IV) cations in blue, and O(−II) anions in red. In planar A-site ordering and with the tilt system a−b+a− the space group is Pmc21. The structure is 35 kJ mol−1 higher in energy compared to the lowest-energy structure in Fig. 5. | ||
| Tilt system | Glazer space group | Double perovskite space group | E/Z [Hartree] | E rel [kJ mol−1] |
|---|---|---|---|---|
| Columnar A-site ordering | ||||
| a + a + a + | 204 Im |
71 Immm | −2212.815026 | 74 |
| a + a + c − | 137 P42/nmc | 137 P42/nmc | −2212.814905 | 74 |
| a − b + a − | 62 Pnma | 11 P21/m | −2212.810291 | 86 |
| a − b − b − | 15 C2/c | 13 P2/c | −2212.807763 | 93 |
| a − a − a − | 167 Rc |
13 P2/c | −2212.805262 | 100 |
| a 0 b + b + | 139 I4/mmm | 139 I4/mmm | −2212.814899 | 74 |
| a + b 0 c − | 63 Cmcm | 25 Pmm2 | −2212.843203 | 0 |
| a 0 b − b − | 74 Imma | 51 Pmma | −2212.807772 | 93 |
| a 0 a 0 c + | 127 P4/mbm | 65 Cmmm | −2212.802939 | 106 |
| a 0 a 0 c − | 140 I4/mcm | 132 P42/mcm | −2212.803378 | 105 |
| a 0 a 0 a 0 | 221 Pmm |
123 P4/mmm | −2212.797404 | 120 |
| Planar A-site ordering | ||||
| a + a + a + | 204 Im |
47 Pmmm | −2212.823192 | 53 |
| a + a + c − | 137 P42/nmc | 115 P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) m2 m2 |
−2212.823046 | 53 |
| a − b + a − | 62 Pnma | 26 Pmc21 | −2212.830042 | 35 |
| a − b − b − | 15 C2/c | 13 P2/c/3 P2 | −2212.823397 | 52 |
| a − a − a − | 167 Rc |
13 P2/c | −2212.822985 | 53 |
| a 0 b + b + | 139 I4/mmm | 123 P4/mmm | −2212.821915 | 56 |
| a + b 0 c − | 63 Cmcm | 38 Amm2 | −2212.826897 | 43 |
| a 0 b − b − | 74 Imma | 51 Pmma | −2212.823317 | 52 |
| a 0 a 0 c + | 127 P4/mbm | 65 Cmmm | −2212.815461 | 73 |
| a 0 a 0 c − | 140 I4/mcm | 125 P4/nbm | −2212.821135 | 58 |
| a 0 a 0 a 0 | 221 Pmm |
123 P4/mmm | −2212.820837 | 59 |
| Rock salt A-site ordering | ||||
| a + a + a + | 204 Im |
200 Pm |
−2212.783521 | 157 |
| a + a + c − | 137 P42/nmc | 115 Pm2 |
−2212.795828 | 124 |
| a − b + a − | 62 Pnma | 31 Pmn21 | −2212.802313 | 107 |
| a − b − b − | 15 C2/c | 5 C2 | −2212.798448 | 118 |
| a − a − a − | 167 Rc |
5 C2 | −2212.801118 | 110 |
| a 0 b + b + | 139 I4/mmm | 123 P4/mmm | −2212.786199 | 150 |
| a + b 0 c − | 63 Cmcm | 38 Amm2 | −2212.803817 | 103 |
| a 0 b − b − | 74 Imma | 44 Imm2 | −2212.798429 | 118 |
| a 0 a 0 c + | 127 P4/mbm | 136 P42/mnm | −2212.785915 | 150 |
| a 0 a 0 c − | 140 I4/mcm | 121 I2m |
−2212.795261 | 126 |
| a 0 a 0 a 0 | 221 Pmm |
225 Fmm |
−2212.766282 | 202 |
3.5. YKTi2O6
It seems that despite the clear indication of the low τ value (Fig. 2), neither CsScTi2O6 nor YRbTi2O6 can be expected to be a double perovskite with high confidence. To study whether the difference in size between the two A-site ions causes the strongly distorted structures, we next studied YKTi2O6. Potassium cations have similar chemical properties to rubidium cations, but are smaller in size (rK = 1.64 Å, 12-coordinated radius) and therefore closer to yttrium (rY = 1.08 Å, nine-coordinated radius). The combination of Y and K even has already been found in the doubly cation ordered perovkite KYCaWO6.21 There have, however, not yet been found any compounds with Y and Rb or Y and Cs on the A-site. For comparison, Fig. 7 shows YKTi2O6 in space group Pmm2, the same space group as the lowest-energy system of YRbTi2O6 (Fig. 5). Again, frequency calculations show the structure to be true local minimum. The oxygen octahedra are still distorted but the maximum Ti–O distance decreases to 2.78 Å. This composition might not be any more likely to exist as a double perovskite, but it is another indicator that ionic sizes play a large role in A-site double perovskites. According to τ, YRbTi2O6 (τ = 3.83), is more likely to form a double perovskite than YKTi2O6 (τ = 3.87), but according to quantum chemical calculations, YKTi2O6 would seem somewhat more likely to form a double perovskite. Another point to consider is that the probability of A-site ordering decreases with the smaller size difference of the A-site cations.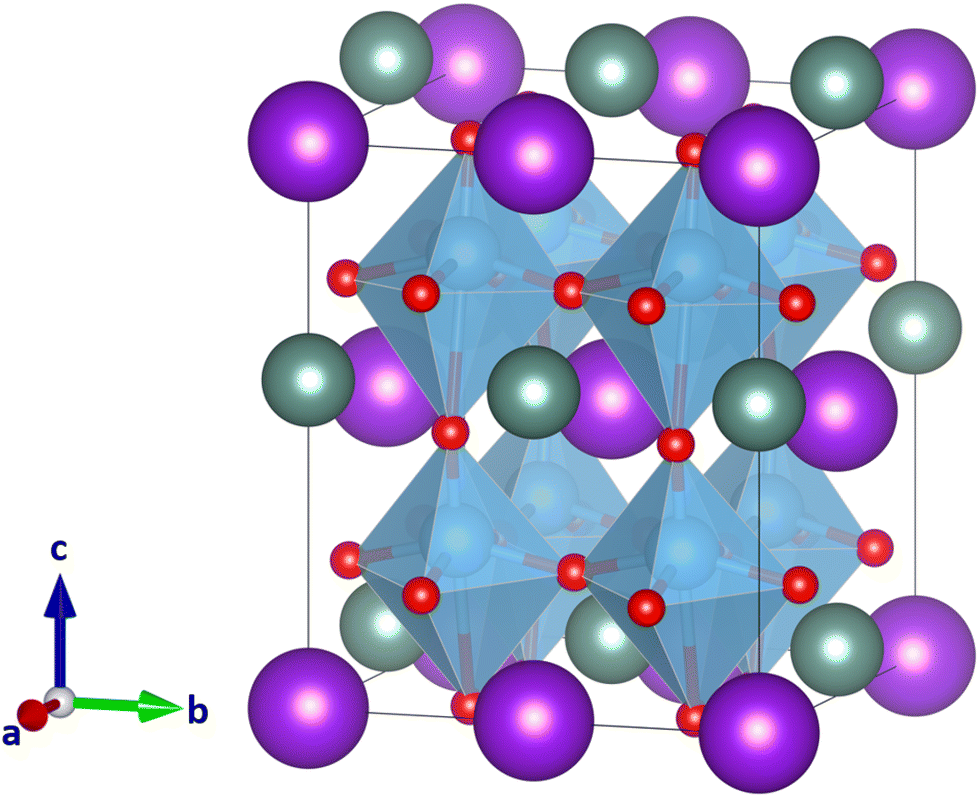 | ||
| Fig. 7 YKTi2O6 structure with Y(III) cations in grey, K(I) cations in dark purple, Ti(IV) cations in blue, and O(−II) anions red. In columnar A-site ordering and with the tilt system a+b0c− it has the space group Pmm2, the same space group as the lowest-energy structure of YRbTi2O6. | ||
The reason why the new tolerance factor τ works fairly well on B-site ordered double perovskites4 but needs the support of quantum chemical calculations for A-site double perovskites can be found in the class of elements which are typically found on these sites. The B-site typically is occupied by d-metal ions, which do not vary much in size from one element to another and are overall on the smaller side. On the A-site, however, large number of elements around the periodic table can be found. Especially the large alkali metal ions paired with small d-metal ions come out as medium ionic sizes when the radii are averaged which for the calculation of τ. These medium ionic sizes then lead to high probabilities of double perovskite, even though the combination of A-site ions is not actually likely to lead in a double perovskite structure.
4. Conclusions
The new tolerance factor τ can be used to screen all possible and feasible combination for A-site double perovskite oxides. Here the screening was carried out for double perovskite titanates and niobates The predictive power of τ is relatively good based on comparisons with available experimental data for A-site double perovskites. However, we found limitations for the use of the new tolerance factor for A-site double perovskites where the A and A′ ions are very different in size. Quantum chemical calculations on CsScTi2O6 have shown that the huge size difference of Cs and Sc leads to strongly distorted oxygen octahedra and the resulting lowest-energy structures are no longer perovskite-like. According to τ, YRbTi2O6 also had a rather high probability to form a perovskite, but with a smaller difference in ionic radii of A-site cations compared to CsScTi2O6. Consequently, quantum chemical calculations suggest a somewhat distorted, but still perovskite-like structure as the lowest-energy structure. The distortion further decreases when moving to YKTi2O6. This finding is in line with experimental observation of doubly cation ordered perovskite KYCaWO6.Overall, the new tolerance factor τ provides a reasonable starting point for the search of new A-site (ordered) double perovskites. However, consideration of tilt systems with the help of quantum chemical calculations is still necessary to make more robust predictions. In the future, the predictive power of τ can be investigated more extensively when more experimental data on novel A-site double perovskites becomes available.
Author contributions
Elisabeth K. Albrecht: Conceptualization; investigation; visualization; writing – original draft preparation; writing – review and editing. Antti. J. Karttunen: Conceptualization; writing – review and editing; supervision; funding acquisition. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dawid Stryszyk for his data mining work, the Academy of Finland for funding (grant no. 317273), and CSC – The Finnish IT Center for Science for computational resources.References
- R. J. D. Tilley, Perovskites: structure-property relationships, Wiley, Chichester, West Sussex, United Kingdom, 1st edn, 2016 Search PubMed.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- C. Li, K. C. K. Soh and P. Wu, J. Alloys Compd., 2004, 372, 40–48 CrossRef CAS.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, eaav0693 CrossRef CAS PubMed.
- R. Ouyang, S. Curtarolo, E. Ahmetcik, M. Scheffler and L. M. Ghiringhelli, Phys. Rev. Mater., 2018, 2, 083802 CrossRef CAS.
- J. C. Platt, Advances in Large-Margin Classifiers, The MIT Press, 1999, vol. 10, pp. 61–74 Search PubMed.
- M. R. Filip and F. Giustino, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5397–5402 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- L. Schneemeyer, J. Waszczak, S. Zahorak, R. van Dover and T. Siegrist, Mater. Res. Bull., 1987, 22, 1467–1473 CrossRef CAS.
- B. Raveau, A. Maignan, C. Martin and M. Hervieu, Chem. Mater., 1998, 10, 2641–2652 CrossRef CAS.
- G. Hodes, Science, 2013, 342, 317–318 CrossRef CAS PubMed.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef CAS.
- M. C. Knapp and P. M. Woodward, J. Solid State Chem., 2006, 179, 1076–1085 CrossRef CAS.
- G. King and P. M. Woodward, J. Mater. Chem., 2010, 20, 5785 RSC.
- M. W. Lufaso, P. W. Barnes and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 397–410 CrossRef PubMed.
- C. Bernuy-Lopez, K. Høydalsvik, M.-A. Einarsrud and T. Grande, Materials, 2016, 9, 154 CrossRef PubMed.
- F. J. Garcia-Garcia, M. J. Sayagués and F. J. Gotor, Nanomaterials, 2021, 11, 380 CrossRef CAS PubMed.
- R. Ghosh, A. Barik, M. R. Sahoo, S. Tiwary, P. D. Babu, S. D. Kaushik and P. N. Vishwakarma, J. Appl. Phys., 2022, 132, 224107 CrossRef CAS.
- M. Goto, T. Saito and Y. Shimakawa, Chem. Mater., 2018, 30, 8702–8706 CrossRef CAS.
- B. Jiang, T. M. Raeder, D.-Y. Lin, T. Grande and S. M. Selbach, Chem. Mater., 2018, 30, 2631–2640 CrossRef CAS.
- G. King and S. Garcia-Martin, Inorg. Chem., 2019, 58, 14058–14067 CrossRef CAS PubMed.
- G. King, S. Garcia-Martin, E. Urones-Garrote, G. Nenert and P. M. Woodward, Additional Conferences (Device Packaging, HiTEC, HiTEN, and CICMT), 2013, 2013, 000006–000013.
- S. S. Pramana, A. Cavallaro, C. Li, A. D. Handoko, K. W. Chan, R. J. Walker, A. Regoutz, J. S. Herrin, B. S. Yeo, D. J. Payne, J. A. Kilner, M. P. Ryan and S. J. Skinner, J. Mater. Chem. A, 2018, 6, 5335–5345 RSC.
- K. Świerczek, A. Klimkowicz, K. Zheng and B. Dabrowski, J. Solid State Chem., 2013, 203, 68–73 CrossRef.
- J. Young and J. M. Rondinelli, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 174110 CrossRef.
- Y. Zhang, H. Zhao, Z. Du, K. Świerczek and Y. Li, Chem. Mater., 2019, 31, 3784–3793 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS PubMed.
- D. Brown and M. W. Lufaso, Bond valence parameters (bvparm2020.cif, accessed 2023-08-07), https://www.iucr.org/resources/data/datasets/bond-valence-parameters.
- D. Zagorac, H. Müller, S. Ruehl, J. Zagorac and S. Rehme, J. Appl. Crystallogr., 2019, 52, 918–925 CrossRef CAS PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–36 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. S. Kuklin, K. Eklund, J. Linnera, A. Ropponen, N. Tolvanen and A. J. Karttunen, Molecules, 2022, 27, 874 CrossRef CAS PubMed.
- R. E. Stene, B. Scheibe, A. J. Karttunen, W. Petry and F. Kraus, Eur. J. Inorg. Chem., 2019, 2019, 3672–3682 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- E. K. Albrecht and A. J. Karttunen, Dalton Trans., 2022, 51, 16508–16516 RSC.
- F. Pascale, C. M. Zicovich-Wilson, F. López Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 888–897 CrossRef CAS PubMed.
- C. M. Zicovich-Wilson, F. Pascale, C. Roetti, V. R. Saunders, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25, 1873–1881 CrossRef CAS PubMed.
- R. Ranjan, A. Senyshyn, H. Boysen, C. Baehtz and F. Frey, J. Solid State Chem., 2007, 180, 995–1001 CrossRef CAS.
- C. A. L. Dixon, J. A. McNulty, S. Huband, P. A. Thomas and P. Lightfoot, IUCrJ, 2017, 4, 215–222 CrossRef CAS PubMed.
- U. Farid, H. U. Khan, M. Avdeev, T. A. Whittle and B. J. Kennedy, J. Solid State Chem., 2019, 269, 401–408 CrossRef CAS.
- G. King, S. Thimmaiah, A. Dwivedi and P. M. Woodward, Chem. Mater., 2007, 19, 6451–6458 CrossRef CAS.
- G. Gou, N. Charles, J. Shi and J. M. Rondinelli, Inorg. Chem., 2017, 56, 11854–11861 CrossRef CAS PubMed.
- J. Brous, I. Fankuchen and E. Banks, Acta Crystallogr., 1953, 6, 67–70 CrossRef CAS.
- A. M. Glazer, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Database of investigated double perovskite oxide compositions in Excel format, optimized structures in CIF format, and absolute electronic energies of the studied systems. See DOI: https://doi.org/10.1039/d3dt01990j |
| This journal is © The Royal Society of Chemistry 2023 |