
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ruthenium(II) polypyridyl complexes with visible light-enhanced anticancer activity and multimodal cell imaging†
Yan
Kang
abc,
Yao
Zhao
 *ade,
Yuanyuan
Wei
ab,
Yang
Zhang
ad,
Zhaoying
Wang
ad,
Qun
Luo
ad,
Jun
Du
*b and
Fuyi
Wang
*ad
*ade,
Yuanyuan
Wei
ab,
Yang
Zhang
ad,
Zhaoying
Wang
ad,
Qun
Luo
ad,
Jun
Du
*b and
Fuyi
Wang
*ad
aBeijing National Laboratory for Molecular Sciences; CAS Key Laboratory of Analytical Chemistry for Living Biosystems; National Centre for Mass Spectrometry in Beijing; Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: yaozhao@iccas.ac.cn; fuyi.wang@iccas.ac.cn
bCollege of Chemistry and Materials Science, Key Laboratory of Functional Molecular Solids, the Ministry of Education, Anhui Laboratory of Molecular-Based Materials, Anhui Normal University, Wuhu 241000, P. R. China. E-mail: dujun@ahnu.edu.cn
cCollege of Advanced Interdisciplinary Studies, National University of Defense Technology, Changsha 410073, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
eState Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha 410082, P.R. China
First published on 8th August 2023
Abstract
Ruthenium(II) polypyridyl complexes have drawn growing attention due to their photophysical properties and anticancer activity. Herein we report four ruthenium(II) polypyridyl complexes [(N^N)2RuII(L)]2+ (1–4, L = 4-anilinoquinazoline derivatives, N^N = bidentate ligands with bis-nitrogen donors) as multi-functional anticancer agents. The epidermal growth factor receptor (EGFR) is overexpressed in a broad range of cancer cells and related to many kinds of malignance. EGFR inhibitors, such as gefitinib and erlotinib, have been approved as clinical anticancer drugs. The EGFR-inhibiting 4-anilinoquinazoline ligands greatly enhanced the in vitro anticancer activity of these ruthenium(II) polypyridyl complexes against a series of human cancer cell lines compared to [Ru(bpy)2(phen)], but interestingly, these complexes were actually not potent EGFR inhibitors. Further mechanism studies revealed that upon irradiation with visible light, complexes 3 and 4 generated a high level of singlet oxygen (1O2), and their in vitro anticancer activities against human non-small-cell lung (A549), cervical (HeLa) and squamous (A431) cancer cells were significantly improved. Specifically, complex 3 displayed potent phototoxicity upon irradiation with blue light, of which the photo-toxicity indexes (PIs) against HeLa and A431 cells were 11 and 8.3, respectively. These complexes exhibited strong fluorescence emission at ca. 600 nm upon excitation at about 450 nm. A subcellular distribution study by fluorescence microscopy imaging and secondary ion mass spectrometry imaging (ToF-SIMS) demonstrated that complex 3 mainly localized at the cytoplasm and complex 4 mainly localized in the nuclei of cells. Competitive binding with ctDNA showed that complex 4 was more favorable to bind to the DNA minor groove than complex 3. These differences support that complex 3 possibly exerts its anticancer activities majorly by photo-induced 1O2 generation and complex 4 by binding to DNA.
Introduction
Platinum-based anticancer drugs have been successful in the clinic in the past decades but suffer from various defects such as acquired resistance and severe side effects.1,2 In recent years, ruthenium anticancer complexes are regarded as promising alternatives to platinum drugs in cancer chemotherapies.3–7 One of the important merits is that ruthenium complexes show lower system toxicity and less cross-resistance towards platinum drugs.8 Remarkably, three ruthenium(III) complexes, NAMI-A,9 KP1019,10 and KP1339,11 and a ruthenium(II) complex TLD143312 entered clinical trials for various types of cancers.13 In the past few decades, Ru polypyridyl complexes have drawn considerable attention for their anticancer activities.7,14,15 Because of their tunable photo-physical/chemical properties,16 facile synthetic chemistry, redox activities and good water solubility, RuII polypyridyl complexes have been widely applied in immunogenic therapy17 and cell imaging,18,19 Some ruthenium polypyridyl complexes were found to inhibit thioredoxin reductase,20 up-regulate the ROS levels in cancer cells,21–23 bind to DNA,24 and induce DNA photocleavage25 and mitochondria-mediated cancer cell apoptosis.14,21 A series of drug delivery nanosystems have also been used to load ruthenium polypyridyl complexes for cancer treatment and bioimaging.26–28It is widely accepted that DNA is one of the major targets for most of the metal-based anticancer drugs. Specifically, the interactions of ruthenium-based anticancer complexes with DNA have been extensively studied. For example, the Barton group first discovered that two RuII polypyridyl complexes, [Ru(bpy)2(dppz)]2+ and [Ru(phen)2(dppz)]2+ (bpy = 2,2′-bipyridine, phen = 1,10-phenanthroline, dppz = dipyridophenazine), bind to DNA with high affinity.29 The [Ru(phen)3]2+ complex was also found to photocleave DNA30 or DNA-mediated photoelectron transfer.31 Ruthenium anticancer complexes may interact with DNA through covalent bonding32 and (partial) intercalative,33 electrostatic and major/minor-groove bindings.34
Apart from DNA, enzymes were also found to be important targets of anticancer agents. The epidermal growth factor receptor (EGFR) is believed to be overexpressed in a broad range of cancer cells and related to many kinds of malignance, such as non-small cell lung, ovarian, breast, and squamous cell cancers.35 By selectively inhibiting the phosphorylating site of EGFR, successful anticancer drugs have been developed and a number of them have been approved for clinical use.36 Among them, 4-anilinoquinazoline derivatives, such as gefitinib and erlotinib, are highly selective EGFR inhibitors and effective anticancer drugs in the clinic for the treatment of non-small-cell lung cancer and squamous carcinoma.37 A notable advantage of this kind of targeted anticancer drug is its lower toxicity towards normal tissues compared to that of traditional cytotoxic drugs.
Ruthenium(II) polypyridyl complexes also have tunable photochemical and photophysical properties,38 including high quantum yields, long lifetimes, and large Stokes shifts for photoluminescence.18,39–41 Ruthenium(II) polypyridyl complexes have been used in photodynamic therapy (PDT), where photosensitized complexes can transfer the energy from light to ground state oxygen molecules and excite them to the singlet state (1O2).201O2 is an important reactive oxygen species (ROS) which can react with various biomolecules within the cell and lead to cell apoptosis or necrosis. For example, a series of ruthenium(II) polypyridyl complexes exhibited visible light-enhanced in vitro anticancer activity.42–44
Cancer is a class of fatal disease featuring multiple gene mutations that lead to disordered cell proliferation and replication. Therefore, drugs that simultaneously act at multiple targets can achieve better efficacy and lower the chance of resistance.2,45,46 For instance, ruthenium complexes with both enzyme inhibiting activity and DNA interaction ability have been demonstrated to be efficient anticancer agents.1,47–49 In our group, a series of dual-targeting ruthenium arene anticancer complexes bearing EGFR inhibitory pharmacophores have been designed and synthesized.50,51 Ruthenium(II) polypyridyl subunits were conjugated with derived imidazole monodentate ligands containing EGFR-inhibiting 4-anilinoquinazoline pharmacophores, giving rise to a series of dual-targeting anticancer complexes that exhibit high antiproliferation activities against cancer cells.52 In this work, we endeavor to endow these complexes with fluorescence, so the distribution of the complexes in cells can be directly followed by fluorescence microscopy. Also, we tried to introduce photosensitizers into the complexes so as to further reinforce their anticancer activities with light. Moreover, an EGFR-inhibiting 4-anilinoquinazoline pharmacophore was employed to enhance the targeted accumulation in cancer cells. Therefore, in this work, we synthesized phenanthroline-based bidentate ligands containing 4-anilinoquinazoline pharmacophores, and further produced ruthenium polypyridyl complexes 1–4 (Scheme 1). As expected, complexes 1–4 showed strong fluorescent emission at about 610 nm with an excitation wavelength of about 460 nm. Also, they displayed antiproliferation activities against A431, HeLa and A549 cells. Moreover, complexes 3 and 4 generate 1O2 upon irradiation with visible light, and their cytotoxicity was enhanced up to 10-fold towards HeLa and A431 cells.
 | ||
| Scheme 1 Chemical structures of ruthenium complexes 1–4. | ||
Experimental section
Materials
RuCl3·xH2O (Ru > 36.7%) was purchased from Shenyang Jingke Reagent Co. (China); bipyridine, phenanthroline and NH4PF6 were purchased from Alfa Aesar; 5-amine-1,10-phenanthroline, trifluoroacetic acid (TFA), DMSO and cisplatin were purchased from Sigma Aldrich; 1,2-dibromoethane and ethyl bromoacetate were purchased from Beijing Ouhe Technology Co. (China); 4-(3′-chloro-4′-fluoroanilino)-6-hydroxy-7-methoxyquinazoline was purchased from Shanghai FWD Chemicals Co. (China). cis-[Ru(bpy)2Cl2]·2H2O53 and cis-[Ru(phen)2Cl2]·2H2O54 were synthesized following literature methods. Organic solvents including absolute methanol, absolute ethanol, absolute ether, acetonitrile, dichloromethane and THF were all analytical grade and used without further purification. Chromatographic grade acetonitrile was purchased from Fisher. DiD and NucRed Live 647 were purchased from Invitrogen.The protein tyrosine kinase, epidermal growth factor receptor (EGFR), and epidermal growth factor (EGF) were purchased from Sigma, and other biological agents including the ELISA kits for EGFR inhibitor screening were purchased from Cell Signaling Technology Inc. (USA). Sodium silicate nonahydrate (Na2SiO3·9H2O), cetyltrimethylammonium bromide (CTAB), iron nitrate nonahydrate (Fe(NO3)3·9H2O), ammonium hexafluorotitanate ((NH4)2TiF6), and boric acid (H3BO3) were purchased from Shanghai General Chemical Reagent Manufacture (China). The deionized water used in the experiments was prepared with a Milli-Q water purification system (Millipore, Milford, MA).
Elemental analysis was performed on a Flash EA 1112 element analysis instrument (Thermo Quest). NMR spectra were obtained using a Bruker Avance III HD 400 spectrometer (Germany). Electrospray ionization mass spectroscopy (ESI-MS) was carried out using a Q-TOF (Waters) mass spectrometer equipped with an Acquity (Waters) UHPLC system. The LC-MS data were processed using MassLynx (ver. 4.1, Waters). MALDI-TOF MS was carried out using a Bruker Daltonics Autoflex III.
Synthesis and characterization of ligands (Scheme S1†)
Intermediates a and b were synthesized according to a published method.50(a) 4-(3′-Chloro-4′-fluoroanilino)-6-hydroxy-7-methoxyquinazoline (6.0 g, 18.8 mmol) and potassium carbonate (12.0 g, 86.8 mmol) were mixed in DMF (250 mL). Then 1,2-dibromoethane (6 mL, 69.3 mmol) was added and the resulting mixture was heated at 80 °C for 8 h. After cooling to room temperature, the mixture was filtered in a vacuum and the filtrate was collected. Then the solvent was evaporated in a vacuum and the residue was recrystallized from ethanol. The yellow crude residue was further purified by flash chromatography on silica gel using ethyl acetate/petroleum (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as an eluent to give 4-(3′-chloro-4′-fluoroanilino)-6-(2-bromoethoxy)-7-methoxyquinazoline (a) as a white powder (3.5 g, 44%).
:2) as an eluent to give 4-(3′-chloro-4′-fluoroanilino)-6-(2-bromoethoxy)-7-methoxyquinazoline (a) as a white powder (3.5 g, 44%).
(b) Potassium phthalimide (4.6 g, 24.8 mmol) was added to DMF (80 mL). The reaction mixture was heated to 85 °C and refluxed for 0.5 h. DMF (50 mL) solution of the intermediate (a) (3.5 g, 8.2 mmol) was added to the reaction mixture, and refluxed for another 7 h. After cooling to room temperature, the mixture was filtered in a vacuum and the filtrate was collected. Then the solvent was evaporated in a vacuum and 50 mL of deionized water was added to the residue, then stirred at r.t. for 0.5 h. The mixture was filtered in a vacuum and washed with water to obtain a white powder. The white solid was dissolved in ethanol (80 mL) and THF (120 mL), and then hydrazine hydrate (4.5 mL) was added to the solution. The mixture was heated to 75 °C and refluxed for 4 h. After cooling to room temperature, the mixture was filtered in a vacuum and the filtrate was collected. Then the solvent was evaporated in a vacuum to obtain a yellow crude product. The yellow crude product was further purified by flash chromatography on silica gel using dichloromethane/methanol (15:1) as an eluent to give 4-(3′-chloro-4′-fluoroanilino)-6-(2-aminoethoxy)-7-methoxyquinazoline (b) as a white powder (1.19 g, 40%).
(c) A mixture of b (0.26 g, 0.72 mmol), succinic anhydride (0.36 g, 3.6 mmol) and pyridine (2 mL, 25 mmol) in THF (15 mL) and DMF (12 mL) was stirred at r.t. for 2 d. The mixture was filtered in a vacuum and washed with water, and a white solid (c) (0.183 g, 55%) was collected. 1H NMR (400 MHz, DMSO-d6, TMS) δH (ppm): 11.86 (s, 1H), 9.55 (s, 1H), 8.51 (s, 1H), 8.30 (t, J1 = J2 = 5.2 Hz, 1H), 8.13 (dd, J1 = J2 = 2.4 Hz, 1H), 7.84 (s, 1H), 7.82–7.78 (m, 1H), 7.44 (t, J1 = J2 = 9.2 Hz, 1H), 7.2 (s, 1H), 4.16 (t, J1 = 5.2 Hz, J2 = 5.6 Hz, 2H), 3.95 (s, 3H), 3.54–3.53 (d, 5.6H), 2.46 (t, J1 = 6.4 Hz, J2 = 6 Hz, 2H), 2.38 (t, J1 = 6 Hz, J2 = 6.4 Hz, 2H). Anal. calcd (%) for C21H22ClFN4O6 (F.W. 480.88, M + H2O): C, 52.45; H, 4.61; N, 11.65; found: C, 52.58; H, 4.98; N, 11.36.
(L1) A mixture of c (168.68 mg, 0.365 mmol), 5-amine-1,10-phenanthroline (71.80 mg, 0.368 mmol), EDCI (71.02 mg, 0.370 mmol) and pyridine (4 mL) in DMF (6.5 mL) was stirred at r.t. for 2 d. The mixture was poured into 50 mL of water, stirred for another 5 h, and centrifuged, and a yellow precipitate was collected. DMF (5 mL) was added to the precipitate and sonicated for 2 min, then water (10 mL) was added to the mixture, and it was sonicated for 2 min and centrifuged, and a pale yellow precipitate was collected. The precipitate was washed with DMF and water three times to collect a white solid (L1) (0.107 g, 46%).1H NMR (400 MHz, DMSO-d6, TMS) δH (ppm): 10.19 (s, 1H), 9.54 (s, 1H), 9.08 (dd, J = 4.1, 1.3 Hz, 1H), 9.05–8.97 (m, 1H), 8.65 (d, J = 8.4 Hz, 1H), 8.52 (s, 1H), 8.39 (d, J = 6.6 Hz, 2H), 8.17–8.08 (m, 2H), 7.86 (s, 1H), 7.82–7.74 (m, 2H), 7.71 (dd, J = 8.1, 4.3 Hz, 1H), 7.43 (t, J = 9.1 Hz, 1H), 7.23 (s, 1H), 4.21 (t, J = 5.4 Hz, 2H), 3.95 (s, 3H), 3.60 (d, J = 5.4 Hz, 2H), 2.81 (t, J = 6.5 Hz, 2H), 2.60 (t, J = 6.9 Hz, 2H). 13C NMR (DMSO-d6, 100 MHz, TMS) δC (ppm): 171.96, 171.73, 156.00, 154.38, 152.72, 149.73, 149.22, 148.10, 147.06, 145.81, 143.78, 135.66, 131.87, 131.74, 128.03, 123.47, 123.37, 122.71, 122.24, 122.17, 119.83, 118.83, 118.65, 116.60, 116.38, 108.71, 107.42, 102.74, 67.85, 55.84, 38.31, 31.18, 30.27. Anal. calcd (%) for C33H30ClFN7O5.5 (F.W. 667.09, M + 1.5H2O): C, 59.42; H, 4.53; N, 14.70; found: C, 59.86; H, 4.73; N, 14.65.
(d) A mixture of 4-(3′-chloro-4′-fluoroanilino)-6-hydroxy-7-methoxyquinazoline (2.02 g, 6.31 mmol), ethyl bromoacetate (1.4 mL, 12.66 mmol), and potassium carbonate (3.8 g, 27.54 mmol) in DMF (80 mL) was stirred at 80 °C for 4 h. After cooling to room temperature, the mixture was filtered in a vacuum and the filtrate was concentrated to 60 mL. After cooling, the filtrate was poured into water (250 mL), and the mixture was stirred at r.t. for 10 min, and then allowed to stand overnight. The next day, the mixture was filtered by vacuum, washed with water, and a yellow solid was collected. To a mixture of the yellow solid in water (80 mL) and ethanol (60 mL), NaOH:H2O (2.4 g:12 mL) was added. The mixture was refluxed at 75 °C for 4 h. Upon cooling, most of the ethanol was evaporated in a vacuum, and then water (45 mL) was added. After cooling, hydrochloric acid was added to regulate the pH to 2–3, and filtered in a vacuum to collect the precipitate (d) (1.21 g, 51%). 1H NMR (400 MHz, DMSO-d6, TMS) δH (ppm): 10.50 (s, 1H), 8.43 (s, 1H), 8.30 (dd, J1 = 6.4 Hz, J2 = 2.0 Hz, 1H), 7.98 (s, 1H), 7.96 (s, 1H), 7.32 (t, J1 = J2 = 9.2 Hz, 1H), 7.03 (s, 1H), 4.52 (s, 3H), 3.85 (s, 2H). Anal. calcd (%) for C29H22ClFN6O4 (F.W. 395.77, M + H2O): C, 51.59; H, 3.82; N, 10.62; found: C, 51.43; H, 3.69; N, 10.73.
(L2) A mixture of (d) (222.85 mg, 0.59 mmol), 5-amine-1,10-phenanthroline (116.4 mg, 0.60 mmol), EDCI (136 mg, 0.71 mmol) and pyridine (9 mL) in DMF (18 mL) was stirred at r.t. for 2 d. The mixture was poured into 80 mL of water, stirred for another 5 h, and centrifuged, and a yellow precipitate was collected. DMF (5 mL) was added to the precipitate and sonicated for 2 min, then methanol (10 mL) was added to the mixture and it was sonicated for 2 min and centrifuged, and a pale yellow precipitate was collected. The precipitate was washed with DMF and methanol three times to collect a white solid (L2) (0.147 g, 45%). 1H NMR (400 MHz, DMSO-d6, TMS) δH (ppm): 10.31 (s, 1H), 9.66 (s, 1H), 9.15 (d, J = 3.2 Hz, 1H), 9.06 (d, J = 2.9 Hz, 1H), 8.64 (d, J = 8.3 Hz, 1H), 8.56 (s, 1H), 8.48 (d, J = 7.9 Hz, 1H), 8.27 (s, 1H), 8.14 (d, J = 4.5 Hz, 1H), 8.04 (s, 1H), 7.83 (dd, J = 8.4, 4.2 Hz, 1H), 7.76 (dd, J = 8.0, 4.3 Hz, 1H), 7.47 (t, J = 9.1 Hz, 1H), 7.32 (s, 1H), 5.12 (s, 2H), 4.05 (s, 3H), 3.17 (d, J = 4.8 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz, TMS) δC(ppm): 167.20, 156.22, 154.62, 153.06, 150.00, 149.57, 147.47, 147.43, 145.85, 143.97, 135.93, 131.29, 130.88, 127.97, 124.56, 123.65, 123.45, 122.89, 122.33, 122.26, 120.41, 118.76, 116.74, 116.52, 108.63, 107.69, 104.10, 68.35, 56.13. Anal. calcd (%) for C29H22ClFN6O4 (F.W. 572.97, M + H2O): C, 60.79; H, 3.87; N, 14.67; found: C, 60.85; H, 3.59; N, 14.91.
Synthesis and characterization of [(N^N)2Ru(L)](PF6)2 (1–4)
Complex 1: MALDI-TOF-MS (m/z): 526.613 ((M − 2PF6 − 2H)2+, C53H41ClFN11O4Ru requires 526.611). 1H NMR (MeOD-d4, 400 MHz, TMS) δ (ppm): 8.79 (d, J = 8.4 Hz, 1H), 8.68 (dd, J = 15.9, 8.1 Hz, 4H), 8.34 (t, J = 6.0 Hz, 3H), 8.19–8.10 (m, 3H), 8.09–8.00 (m, 3H), 7.88 (t, J = 5.1 Hz, 2H), 7.85 (dd, J = 6.7, 2.6 Hz, 1H), 7.78 (s, 1H), 7.74 (dd, J = 8.5, 5.2 Hz, 1H), 7.68 (dd, J = 8.2, 5.3 Hz, 1H), 7.61 (d, J = 5.3 Hz, 1H), 7.54 (dt, J = 13.2, 6.2 Hz, 4H), 7.29 (dt, J = 14.0, 6.5 Hz, 2H), 7.17–7.07 (m, 2H), 4.29 (t, J = 5.5 Hz, 2H), 3.95 (s, 3H), 3.91 (s, 1H), 3.71 (t, J = 5.4 Hz, 2H), 3.04–2.93 (m, 2H), 2.82–2.72 (m, 2H). 13C NMR (DMSO-d6, 100 MHz, TMS) δ (ppm): 172.07, 171.85, 156.79, 156.56, 156.01, 154.37, 152.72, 152.22, 151.45, 151.37, 151.02, 148.08, 147.17, 147.05, 144.42, 137.94, 137.81, 136.77, 136.19, 133.94, 132.68, 130.24, 127.83, 127.78, 127.71, 126.47, 126.32, 125.66, 124.45, 124.38, 123.37, 122.26, 122.20, 118.94, 118.85, 118.66, 116.62, 116.40, 108.71, 107.43, 102.71, 67.76, 62.00, 55.85, 38.29, 31.32, 30.05. Anal. calcd (%) for C53H47ClF13N11O6P2Ru (F.W. 1379.46, M + 2H2O): C, 46.15; H, 3.43; N, 11.17; found: C, 46.07; H, 3.43; N, 11.37.
Complex 2: MALDI-TOF-MS (m/z): 550.610 ((M − 2PF6 − 2H)2+, C57H41ClFN11O4Ru requires 550.611).1H NMR (DMSO-d6, 400 MHz) δ (ppm): 10.52 (s, 1H), 9.56 (s, 1H), 8.94 (d, J = 8.3 Hz, 1H), 8.76 (d, J = 8.0 Hz, 4H), 8.69–8.57 (m, 2H), 8.47 (s, 1H), 8.39 (d, J = 8.9 Hz, 5H), 8.07 (q, J = 10.7 Hz, 6H), 7.87 (s, 1H), 7.83–7.70 (m, 6H), 7.70–7.62 (m, 1H), 7.40 (t, J = 9.1 Hz, 1H), 7.18 (s, 1H), 7.01 (s, 1H), 4.20 (s, 2H), 3.91 (s, 3H), 3.62–3.51 (m, 2H), 2.87 (d, J = 9.5 Hz, 3H), 2.60 (d, J = 6.4 Hz, 2H). 13C NMR (DMSO-d6, 100 MHz) δ (ppm): 172.09, 171.81, 156.00, 154.37, 152.70, 151.86, 151.48, 148.06, 147.53, 147.22, 147.18, 147.02, 144.81, 136.80, 136.11, 133.94, 132.72, 132.41, 130.43, 130.18, 128.03, 126.27, 125.53, 123.34, 122.28, 122.21, 118.79, 118.61, 116.54, 116.33, 108.74, 107.38, 102.82, 67.67, 55.82, 38.28, 31.41, 30.26. Anal. calcd (%) for C57H47ClF13N11O6P2Ru (F.W. 1427.50, M + 2H2O): C, 47.96; H, 3.32; N, 10.79; found: C, 47.74; H, 3.20; N, 10.58.
Complex 3: MALDI-TOF-MS (m/z): found 484.109 ((M − 2PF6 − 2H)2+, C49H34ClFN10O3Ru requires 484.084). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 10.86 (s, 1H), 9.82 (s, 1H), 9.04 (d, J = 8.6 Hz, 1H), 8.85 (dd, J = 16.3, 8.2 Hz, 4H), 8.75 (d, J = 8.0 Hz, 1H), 8.67 (s, 1H), 8.54 (s, 1H), 8.25–8.14 (m, 5H), 8.10 (q, J = 6.7 Hz, 2H), 8.04 (d, J = 4.7 Hz, 1H), 7.92–7.87 (m, 1H), 7.85–7.80 (m, 3H), 7.57 (dt, J = 11.9, 6.0 Hz, 4H), 7.43 (t, J = 9.1 Hz, 1H), 7.33 (d, J = 7.5 Hz, 2H), 7.30 (s, 1H), 5.34–5.24 (m, 2H), 4.01 (s, 3H). 13C-NMR (DMSO-d6, 100 MHz) δ (ppm): 167.70, 156.80, 156.54, 156.52, 156.19, 154.54, 153.00, 151.47, 151.39, 147.40, 147.22, 144.78, 137.96, 137.84, 136.40, 133.10, 130.10, 127.83, 127.79, 126.58, 126.48, 125.67, 124.46, 124.38, 123.25, 122.17, 122.10, 118.65, 116.62, 116.41, 108.64, 107.69, 104.06, 67.98, 56.09. Anal. calcd (%) for C49H40ClF13N10O5P2Ru (F.W. 1294.36, M + 2H2O): C, 45.47; H, 3.12; N, 10.82; found: C, 45.43; H, 3.33; N, 10.64.
Complex 4: MALDI-TOF-MS (m/z): 508.085 ((M − 2PF6 − 2H)2+, C53H34ClFN10O3Ru requires 508.084). 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 10.86 (s, 1H), 9.77 (s, 1H), 9.01 (d, J = 8.5 Hz, 1H), 8.78 (dd, J = 8.2, 3.4 Hz, 4H), 8.72 (d, J = 8.2 Hz, 1H), 8.68 (s, 1H), 8.54 (s, 1H), 8.39 (s, 4H), 8.22–8.16 (m, 2H), 8.12 (t, J = 5.8 Hz, 2H), 8.07 (dd, J = 9.3, 5.3 Hz, 3H), 8.00 (d, J = 5.0 Hz, 1H), 7.89 (dt, J = 6.8, 3.8 Hz, 1H), 7.81–7.69 (m, 6H), 7.40 (t, J = 9.0 Hz, 1H), 7.30 (s, 1H), 5.34–5.21 (m, 2H), 4.01 (s, 3H). 13C NMR (DMSO-d6, 100 MHz, TMS) δ (ppm): 167.66, 156.15, 154.50, 152.99, 152.72, 151.84, 147.61, 147.45, 147.37, 147.18, 145.17, 136.83, 136.30, 133.08, 132.54, 130.43, 130.05, 128.03, 126.43, 126.29, 125.53, 123.18, 122.10, 122.03, 119.95, 118.82, 118.64, 116.58, 116.36, 108.64, 107.70, 104.00, 84.24, 68.01, 56.06. Anal. calcd (%) for C53H40ClF13N10O5P2Ru (F.W. 1342.40, M + 2H2O): C, 47.42; H, 3.00; N, 10.43; found: C, 47.26; H, 3.17; N, 10.60.
Absorption, fluorescence and emission titration studies
Complexes 1–4 were dissolved in DMSO, respectively, to yield 2 mM stock solutions, and then diluted using PBS buffer (pH 7.4) to 10 μM. UV-vis absorption spectroscopy of complexes 1–4 (10 μM) was performed on a SHIMADZU UV-2550 spectrophotometer (Japan). Fluorescence spectroscopy was carried out using a HITACHI F-4600 fluorescence spectrophotometer (Japan). Emission titration was carried out for complex 3 by adding various amounts of ctDNA in Tris-HCl buffer (pH 7.4) after 24 h of incubation at 370 K.Fluorescence confocal microscopy imaging
5 × 103 A549 cells per well were plated in a laser scanning confocal Petri dish and cultured for 12 h. After removing the medium, the cells were incubated in the absence or presence of 3 or 4 (10 μM) at 310 K for 3 h and 24 h. The controls groups were incubated in 0.5% DMSO medium. After incubation in 3 or 4 for a certain time at 310 K, the supernatant was removed, and stained with DiD (10 μM) or NucRed Live 647 (1.5 drops per mL medium) for 20 min at 310 K. After staining, the cells were washed three times with 1 mL of PBS. The cells were maintained in colourless minimal medium. Fluorescence images were obtained using an FV1000-IX81 confocal laser scanning microscope (OLYMPUS); for complexes 3 and 4, a 405 nm or 488 nm laser and emission wavelengths of 570–600 nm were used. For DiD, excitation at 633 nm and emission at 650–680 nm were used; for NucRed Live 647, excitation at 633 nm and emission at 655–755 nm were used.Generation of singlet oxygen
The generation of 1O2 of complexes 1, 3 and 4 was investigated using a fluorescence probe for singlet oxygen, Singlet Oxygen Sensor Green (SOSG®, Invitrogen), following the instruction manual.Enzyme-linked immunosorbent assay (ELISA)
ELISA screening was performed following the instructions provided by the supplier of the assay kits (no. 7909, Cell Signalling Technology, Inc.).In vitro anti-proliferation assays
The human lung adenocarcinoma cell line A549, the human cervical cancer cell line HeLa, and squamous cell carcinoma A431 were obtained from the Centre for Cell Resource of Peking Union Medical College Hospital and maintained in complete medium [90% DMEM (Invitrogen, USA) +10% fetal bovine serum (Invitrogen, USA) +1% penicillin–streptomycin (Invitrogen, USA)]. On request, an aliquot of 100 ng mL−1 epidermal growth factor (Sigma, USA) was added to the media. The cells were grown at 310 K under a humidified atmosphere containing 5% CO2 for 2–3 days prior to screening experiments.The in vitro anti-proliferation activities of complexes 1–4 were evaluated using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Cells were plated at a density of 3000 cells per well (A549), 6500 cells per well (HeLa), and 8000 cells per well squamous cell carcinoma (A431) (Luna™ Automated Counter, Logos Biosystems, Korea) in 100 μL of media in 96-well plates and cultured for 16 h. The stock solutions [10 mM, except for cisplatin (1 mM)] of all tested compounds were made up fresh in DMSO before dilution in media to give the required concentration for addition to the cells. For each ruthenium complex, eight different concentrations were prepared from the stock solution by diluting with the cell culture medium prior to use and the concentration of DMSO was less than 1.5% in all dilutions. Cells were then exposed to each tested compound at various concentrations for 48 h in the dark. The phototoxicity of 3 and 4 was investigated, and the cells treated with various concentrations of 3 or 4 were first incubated for 24 h in the dark at 310 K, and then irradiated at λ 460 nm or 520 nm for 1 h, followed by 23 h in the dark at 310 K. The cell growth was measured using MTT assay following the reported procedure. After incubation for 48 h, the drug solution was aspirated and washed three times with PBS, then 100 μL of complete medium containing MTT (0.5 mg ml−1) was added to each well and incubated at 310 K for 4 h. The MTT medium was removed, and 100 μL of DMSO was added to each well to dissolve the formazan crystals at room temperature for 10 min. The optical density (OD) value for each well was measured using a microplate reader (SpectraMax M5 Molecular Devices Corporation) at a wavelength of 570 nm. The inhibition rate (IR) was calculated based on the following equation:
| IR (%) = [1 − (ODcompound − ODblank)/(ODcontrol − ODblank)] × 100%. |
Secondary ion mass spectrometry (ToF-SIMS) imaging
A549 cells were seeded at a density of 1 × 104 ml−1 in a cell culture dish containing a 1 × 1 cm silicon wafer and incubated for 24 h where the cells were allowed to attach. Then the cells were incubated in the absence or presence of complex 3 or 4 (10 μM) for 24 h at 310 K. The supernatant culture medium was removed, and the cells were washed six times with ammonium acetate (150 mM, pH = 7.4). Then the cells were lyophilized for 16 h using an LGJ-12 lyophilizer (Beijing Songyuan Huaxing Technology Develop Co., Ltd). ToF-SIMS analysis and imaging were conducted using a ToF-SIMS V spectrometer (IONTOF GmbH, Munster, Germany). Dual-beam experiments were performed using a 10 keV argon cluster ion beam (Arn+) as the sputtering beam and a 30.0 keV Bi3+ beam as the analysis beam. High spatial resolution images were collected at 256 × 256 pixels with the highest resolution of 300 nm over a 100 × 100 μm2 area using a pulsed analysis beam (DC current = 200 pA, pulse width = 23 ns and repetition rate = 5 kHz) at the center of a 300 × 300 μm crater eroded by an Arn+ sputtering source. The current of Arn+ was ∼2 nA with a lead-off time of 60 μs. Positive ion spectra were recorded and calibrated by CH3+, C2H3+, C2H5+, C3H5+ and C3H7+. Signals were collected layer by layer, and images were processed using SurfaceLab software (version 6.4, ION-TOF, Münster, Germany). The signal intensities were directly related to the level of detected ions of interest.Competitive binding with DNA
Emission measurements were carried out using a JASCO FP-6600 fluorospectrometer. Tris buffer was used as a blank to make preliminary adjustments. The excitation wavelength was fixed, and the emission range was adjusted before measurements. All measurements were made at 25 °C. For emission spectral titrations, the complex concentration was maintained constant at 10 μM and the concentration of DNA was varied from 0.05 to 0.5 μM. The emission enhancement factors were measured by comparing the intensities at the emission spectral maxima under similar conditions.Hoechst 33342® displacement assay was performed in Tris buffer solution (5 mM, pH = 7.4). The concentrations of CT-DNA and Hoechst 33342 were kept constant at 20 μM and 200 μM, respectively, and they were titrated with different concentrations of competing complexes from 0 to 100 μM. The CT-DNA-Hoechst 33342 complex was excited at 370 nm and emission spectra were recorded from 400 nm to 680 nm. Measurement parameters: PMT voltage, 700 V; EX slit, 5.0 nm; EM slit, 5.0 nm.
The Stern–Volmer constant (Ksv) was used to evaluate the fluorescence quenching efficiency. The classic Stern–Volmer equation: F0/F = 1 + KSV [Q] where F0 and F are the fluorescence intensities before and after addition of the quencher, respectively. [Q] is the concentration of the quencher and Ksv is the quenching constant.
Results and discussion
Synthesis and characterization
Two linkers with different lengths and flexibilities were used for the conjugation of a phenanthroline bidentate donor and a 4-anilinoquinazoline derivative (Scheme S1, ESI†). The precursor compound 4-(3′-chloro-4′-fluoro)-6-hydroxy-7-methoxyquinazoline reacted with 1,2-dibromoethane or ethyl bromoacetate in the presence of potassium carbonate at 353 K, giving rise to modified 4-anilinoquinazoline intermediates 4-(3′-chloro-4′-fluoroanilino)-6-(2-bromoethoxy)-7-methoxyquinazoline (a) and 4-(3′-chloro-4′-fluoroanilino)-6-(carboxylmethoxy)-7-methoxyquinazoline (d), respectively. The bromo group of compound a was substituted with an amine group to give compound b, and then b and 5-amine-1,10-phenanthroline were linked with succinic anhydride to give L1. L2 was synthesized by a one-step condensation reaction between compound d and 5-amine-1,10-phenanthroline.cis-[Ru(bpy)2Cl2]53 and cis-[Ru(phen)2Cl2]54 were synthesized following literature methods. The reactions between the 4-anilinoquinazoline-derived bidentate ligand L1 or L2 with the ruthenium polypyridyl complexes cis-[Ru(bpy)2Cl2] or cis-[Ru(phen)2Cl2] gave rise to complexes [Ru(bpy)2(L1)](PF6)2 (1), [Ru(bpy)2(L2)](PF6)2 (2), [Ru(phen)2(L1)](PF6)2 (3), and [Ru(phen)2(L2)](PF6)2 (4), respectively. Complexes 1–4 and ligands L1 and L2 were characterized by ESI-MS, 1H NMR spectroscopy, and 13C NMR spectroscopy. The NMR spectra are shown in Fig. S1–S12.† From the NMR analysis, residual DMF was found in L1–L2 and complexes 1–4, so the samples were washed with cold methanol several times, and then washed with cold diethyl ether. The resulting solid complexes were left in a vacuum drying oven overnight at 60 °C before being analyzed by elemental analysis. The data are listed in the Experimental section above. The elemental analysis data are satisfactory except for some water of crystallization with the complexes.
Photophysical properties of the ruthenium complexes
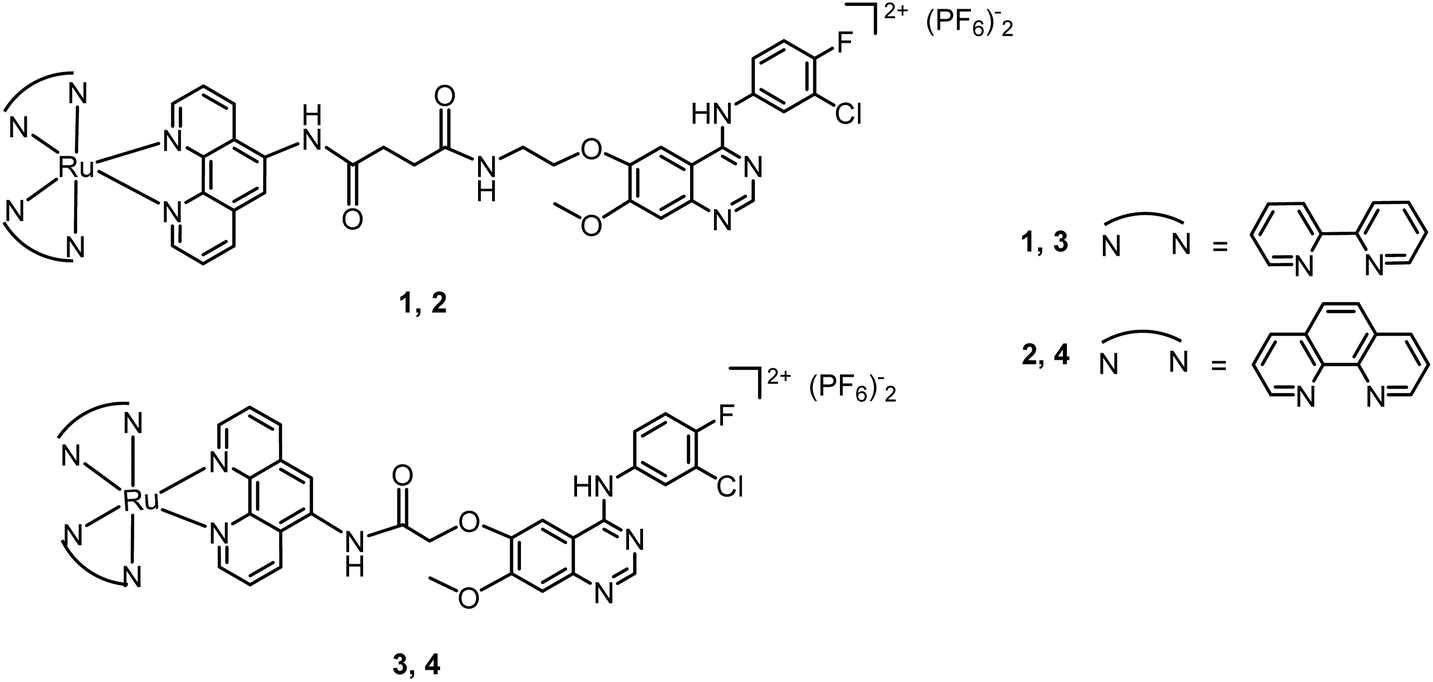
The electronic absorption spectra of the complexes in phosphate-buffered saline (PBS) at 298K are shown in Fig. 1a. Complexes 1–4 show intense spin-allowed intra ligand (1IL) absorption bands in the UV region at approximately 230–375 nm and less intense spin-allowed ligand-to-metal charge-transfer (LMCT) absorption bands at approximately 380–500 nm; the latter bands indicate the promotion of an electron from the metal t2g orbital to the π* antibonding orbital of the ligand. The maximum absorption bands (λmax) of complexes 1 and 3 are longer than those of complexes 2 and 4, respectively. By substituting phenanthroline (phen) with bipyridine (bpy), there is a small bathochromic shift in the 1IL absorption bands. These are typical absorption properties of RuII polypyridyl complexes.40 The fluorescence emission spectra of the complexes are shown in Fig. 1b. The maximum excitation wavelengths of complexes 1–4 are λ 453 nm, 443 nm, 456 nm and 453 nm, respectively, and the maximum emission bands are λ 605 nm, 602 nm, 601 nm and 602 nm, respectively. Complexes 1–4 exhibit strong fluorescence in the visible region, and broad Stokes shifts (ca. 150 nm) in PBS. These results are consistent with other RuII-polypyridyl complexes with similar structures.14,18,20,52 | ||
| Fig. 1 (a) UV-visible absorption spectra of complexes 1–4 (10 μM) in PBS (10 mM, pH = 7.4) at 298 K. (b) Fluorescence emission spectra of complexes 1–4 (10 μM) in PBS (10 mM, pH = 7.4). The λex values of complexes 1–4 are 453 nm, 443 nm, 456 nm and 453 nm, respectively. (c) Fluorescence spectra (λex = 504 nm for SOSG) of ruthenium complexes 1, 3 and 4 (50 μM) in the presence/absence of SOSG (1 μM) in H2O (3% acetone) upon irradiation with different wavelengths for 10 min. Complexes 1, 3 and 4 were irradiated with light at λirr = 445, 465 and 550 nm, respectively. | ||
Enzyme inhibition activity
Enzyme-linked immunosorbent assay (ELISA) was employed to evaluate the inhibitory activities of ruthenium complexes 1–4 against EGFR. The clinically available targeted anticancer drug gefitinib as an EGFR inhibitor was applied as a reference. Unfortunately, these complexes did not exhibit potent EGFR inhibitory activity, and their half-maximal enzyme activity inhibitory concentrations (IC50) were >500 nM. The lack of the EGFR inhibitory activities of complexes 1–4 may be influenced by the sterically hindered ruthenium(II) polypyridyl moiety and the short linker with the 4-anilinoquinazoline moiety. A longer and more flexible linker between the ruthenium moiety and the EGFR-inhibiting pharmacophore as well as a smaller Ru moiety can lower the steric hindrance for the binding affinity to EGFR, which affords complexes with high inhibition activity against EGFR, as shown in our previous work.50–52,55Generation of singlet oxygen
As the ruthenium(II) polypyridyl complexes usually exhibit activity in the photo-induced generation of singlet oxygen (1O2),21 in this work, the level of 1O2 by the photoactivation of Ru complexes was measured using a fluorescence probe named Singlet Oxygen Sensor Green (SOSG®).56 In the absence of 1O2, SOSG exhibits weak blue fluorescence, but in the presence of 1O2, SOSG emits strong green fluorescence with the excitation maximum at λ = 504 nm and the emission maximum at λ = 525 nm. Samples of complexes 1, 3 or 4 (50 μM) and SOSG (1 μM) were prepared in deionized water with 3% acetone. Control experiments were carried out under identical conditions without SOSG. All the samples were irradiated with visible light for 10 min, and for the groups with the addition of SOSG, the intensity of fluorescence at 525 nm (λex = 504 nm) increased rapidly (Fig. 1c). By comparison, the control groups showed very weak emission of the fluorescence. The wavelength-dependent activation efficiency for the generation of 1O2 by complexes 1, 3 and 4 was evaluated and their corresponding wavelengths that could induce the highest level of 1O2 were λ = 445, 465 and 550 nm, respectively, as shown in Fig. S13.† These results suggest that complexes 1, 3 and 4 produce 1O2 upon irradiation with visible light. As depicted in Fig. 2, the fluorescence intensity of SOSG with 3 or 4 increased more than that of complex 1, which suggests that, upon irradiation, 3 and 4 generated more 1O2 than complex 1. | ||
| Fig. 2 (a–f) Fluorescence laser scan confocal microscopy images of A549 cells incubated with complex 3 or 4 (10 μM) at 310 K. The complex was added and further incubated with cells for 24 h before NucRed Live 647, a cell nucleus dye, was incubated for 20 min. Wavelengths for complexes 3 and 4: λex = 488 nm and λem = 600–630 nm; for NucRed Live 647: λex = 633 nm and λem = 655–755 nm. (c) Merged image of (a and b); (f) merged image of (d and e). | ||
Cancer cell antiproliferation activity
The in vitro antiproliferation activities of ruthenium(II) polypyridyl complexes 1–4 in non-small-cell lung (A549), cervical (HeLa) and squamous (A431) human cancer cell lines were evaluated by means of a colorimetric microculture MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-tetrazolium bromide]) assay. The cells were incubated in the presence of the corresponding complex in the dark for 48 h, and the results are shown in Table 1. As EGFR inhibition is not the principal mechanism for the anticancer activities of complexes 1–4, EGF stimulation57 was not involved. The antiproliferation activities of the classic cytotoxic metallodrug cisplatin and some ruthenium polypyridyl anticancer complexes were cited as a comparison. Against the tested tumor cell lines, all the synthesized ruthenium complexes showed potent antiproliferation activity. The antiproliferation activities of complexes 1–4 in the dark were much more potent than those of cis-[Ru(bpy)2Cl2],22cis-[Ru(phen)2Cl2]22 and [Ru(bpy)2(phen)],43 but the complexes were not more active than cisplatin.| IC50 against cancer cellsa (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell line | A549 | HeLa | A431 | ||||||
| Light conditions | Dark | Lightb | PIc | Dark | Light | PI | Dark | Light | PI |
| a The IC50 values were determined as the average of six independent experiments expressed as the mean ± SD. b The lights used were blue light (460 nm) for complex 3, green light (520 nm) for complex 4, and a >450 nm light for [Ru(bpy)2(phen)].43 c PI (phototoxicity index) is the ratio of the dark and light IC50 values. d — = not available. | |||||||||
| 1 | 27 ± 3 | —d | — | 27 ± 4 | — | — | 7 ± 2 | — | — |
| 2 | 35 ± 2 | — | — | 67 ± 10 | — | — | 36 ± 2 | — | — |
| 3 | 19 ± 2 | 10 ± 1 | 1.9 | 18 ± 4 | 1.7 ± 0.5 | 11 | 15 ± 2 | 1.8 ± 0.6 | 8.3 |
| 4 | 21 ± 2 | 12 ± 2 | 1.8 | 22 ± 1 | 13 ± 2 | 1.7 | 12 ± 1 | 7 ± 2 | 1.7 |
| Cisplatin | 10 ± 1 | — | — | 12 ± 1 | — | — | 5.9 ± 0.4 | — | — |
| cis-[Ru(bpy)2Cl2]22 | — | — | — | >200 | — | — | — | — | — |
| cis-[Ru(phen)2Cl2]22 | — | — | — | >200 | — | — | — | — | — |
| [Ru(bpy)2(phen)]43 | 250 ± 5 | 40 ± 4 | 6.3 | — | — | — | — | — | — |
As O21 can be efficiently generated by the photoactivation of complexes 3 and 4, the in vitro cytotoxicities of complexes 3 and 4 were also examined in the presence of a certain wavelength of light. The irradiation sources for 3 and 4 in MTT assay were blue (460 nm) and green (520 nm) LEDs respectively, which were chosen according to their optimal wavelength for generating 1O2. In the MTT assay, the cells were incubated with complex 3 or 4 for 24 h in the dark, and then irradiated with light of the corresponding wavelength for 1 h followed by incubation for another 23 h in the dark. As shown in Table 1, upon irradiation, the IC50 of complexes 3 and 4 against the tested tumor cell lines significantly decreased. Complexes 3 and 4 displayed much more potent anticancer activities against HeLa and A431 cells upon irradiation with light of the corresponding wavelengths, compared to cisplatin and gefitinib and also to their precursor Ru complexes, such as cis-[Ru(bpy)2Cl2],22cis-[Ru(phen)2Cl2]22 and [Ru(bpy)2(phen)].43 The phototoxic indexes42,43 (PI, the ratio of the IC50 of a complex in the dark to its IC50 upon irradiation) of complexes 3 and 4 were calculated to evaluate their light-induced toxicity. Complex 4 displayed moderate phototoxicity, with PI values for three cancer cell lines between 1.7 and 1.8. In contrast, complex 3 exhibited very potent phototoxicity, and its PI values for HeLa and A431 cells were 11 and 8.3, respectively. These results indicate that complex 3 exerts its anticancer activities majorly by photo-induced 1O2 generation. [Ru(bpy)2(phen)] is also phototoxic, and its PI was 6.3 for A549 cells,43 but its photoactivated IC50 value against A549 cells was ca. 40 μM, making it much less active compared to complexes 3 and 4. Upon irradiation with light, complexes 3 and 4 were more cytotoxic against HeLa and A431 cells than a series of structurally similar RuII-polypyridyl complexes containing EGFR-inhibiting groups developed by us.52
Fluorescence confocal microscopic imaging
The fluorescence emission activity of complexes 3 and 4 facilitates the observation of their intracellular uptake and distribution in living cancer cells. A549 cells were co-incubated in the presence of complexes 3 and 4, respectively, for 3 h, before DiD, a cell membrane marker, was added to stain the cell membrane. Fluorescence laser scan confocal microscopy (FLSCM) was employed to image the cells and the results are shown in Fig. S14.† Complexes 3 and 4 did not enter or accumulate in the cell membrane after the initial three hours of incubation.Then the incubation of complexes with A549 cells was extended to 24 h, and NucRed Live 647, as a cell nuclear marker, was added to the media and incubated for a further 20 min. As depicted in Fig. 2, complex 3 concentrated in the cytoplasm and complex 4 was distributed mainly in the nucleus and less in the cytoplasm. Remarkably, A549 cell nucleus swell after incubation with complex 3 and shrinks after incubation with complex 4, indicating the distinct behavior of cellular accumulation and the type of damage to the cell. Interestingly, the signal of NucRed Live 647 was very strong with complex 3 but weak with complex 4. We speculate that the high uptake of complex 4 in the cell nucleus is competing with the dye and preventing it from entering the cells.
ToF-SIMS single-cell imaging
Time of flight secondary ion mass spectrometry (ToF-SIMS) is a powerful label-free high-resolution imaging tool to analyze the distribution of chemicals in thin surface layers, and is more and more frequently used in the analysis of biological samples in recent years.58,59 Ruthenium complexes in this work are perfect probes for both fluorescence imaging and ToF-SIMS imaging. Herein, ToF-SIMS single-cell imaging was employed to further verify the distribution of ruthenium complexes in cancer cells. As shown in Fig. 3, the distribution of ruthenium polypyridyl complexes 3 or 4 in a single A549 cell was imaged with ToF-SIMS. The red color shows the signals of Ru-containing fragments of complexes 3 or 4. The green color depicts the signals of the phosphocholine fragments at m/z 184 (C5H15NPO4+), which are regarded as markers of the phospholipid membrane and thus profile the whole cell. The blue color corresponds to the deoxyribose fragments at m/z 81 (C5H5O+), which only exist in the cell nucleus. When A549 cells were incubated with the corresponding complex for 24 h, complex 3 accumulated largely in the membrane and cytoplasm, but complex 4 was found to accumulate more in the cell nucleus. This result is consistent with the results of confocal fluorescence microscopy images. These ruthenium anticancer complexes were ideal for multimodal cell imaging, which in turn profiled their intracellular distribution very well. | ||
| Fig. 3 TOF-SIMS images obtained from A549 cells treated with 10 μM complex 3 (a–e) or 4 (f–j). In the first column, the red color indicates the total signal intensity of the Ru-containing fragments for 3 (m/z 257 for [Ru(bpy)–H]+ and m/z 281 for [Ru(phen)–H]+) or 4 (m/z 281 for [Ru(phen)–H]+ and m/z 461 for [Ru(phen)2–H]+). In the second column, the green color shows the signals of the phosphocholine fragment, C5H15NPO4+, at m/z 184 as the marker of the cell membrane that depicts the cell profile. In the third column, the blue color corresponds to the deoxyribose fragments, C5H5O+, at m/z 81 as the marker of the cell nucleus. Merged images: c = a + b; e = a + d; h = f + g; j = f + i. | ||
DNA binding
To further explore the interaction of the ruthenium(II) polypyridyl complexes 3 and 4 with DNA, a competitive binding assay with a fluorescent probe, Hoechst 33342® (Hoechst), was carried out. Hoechst 33342 shows weak fluorescence in Tris buffer (pH = 7.4) due to the solvent quenching effect. However, upon binding to the DNA minor groove, the fluorescence emission at ca. 488 nm can be significantly enhanced.60 Therefore, compounds which can decrease the fluorescence intensity of the DNA–Hoechst complex could be regarded as competitively binding to DNA at the minor groove. As shown in Fig. 4a and c, the addition of complexes 3 or 4 substantially quenched the fluorescence intensity of the ctDNA–Hoechst mixture at ca. 488 nm, suggesting that 3 could replace Hoechst and bind to DNA at the minor groove. The quenching constant (Ksv) for the fluorescence intensity of Hoechst bound to ctDNA by complexes 3 and 4 was calculated from the Stern–Volmer plot. As shown in Fig. 4b, for the competitive replacement of Hoechst from ctDNA, the Ksv value of complex 3 was 7.75 × 104 M−1, and that for complex 4 was 1.37 × 105 M−1. The higher Ksv value suggests higher affinity between the complex and the DNA minor groove, so these results demonstrate that complex 4 bound much more easily to DNA. This is consistent with the multimodal imaging results that more complex 4 was found to accumulate in the DNA, which also suggests that the anticancer mechanism of complex 4 also involved the interaction with DNA.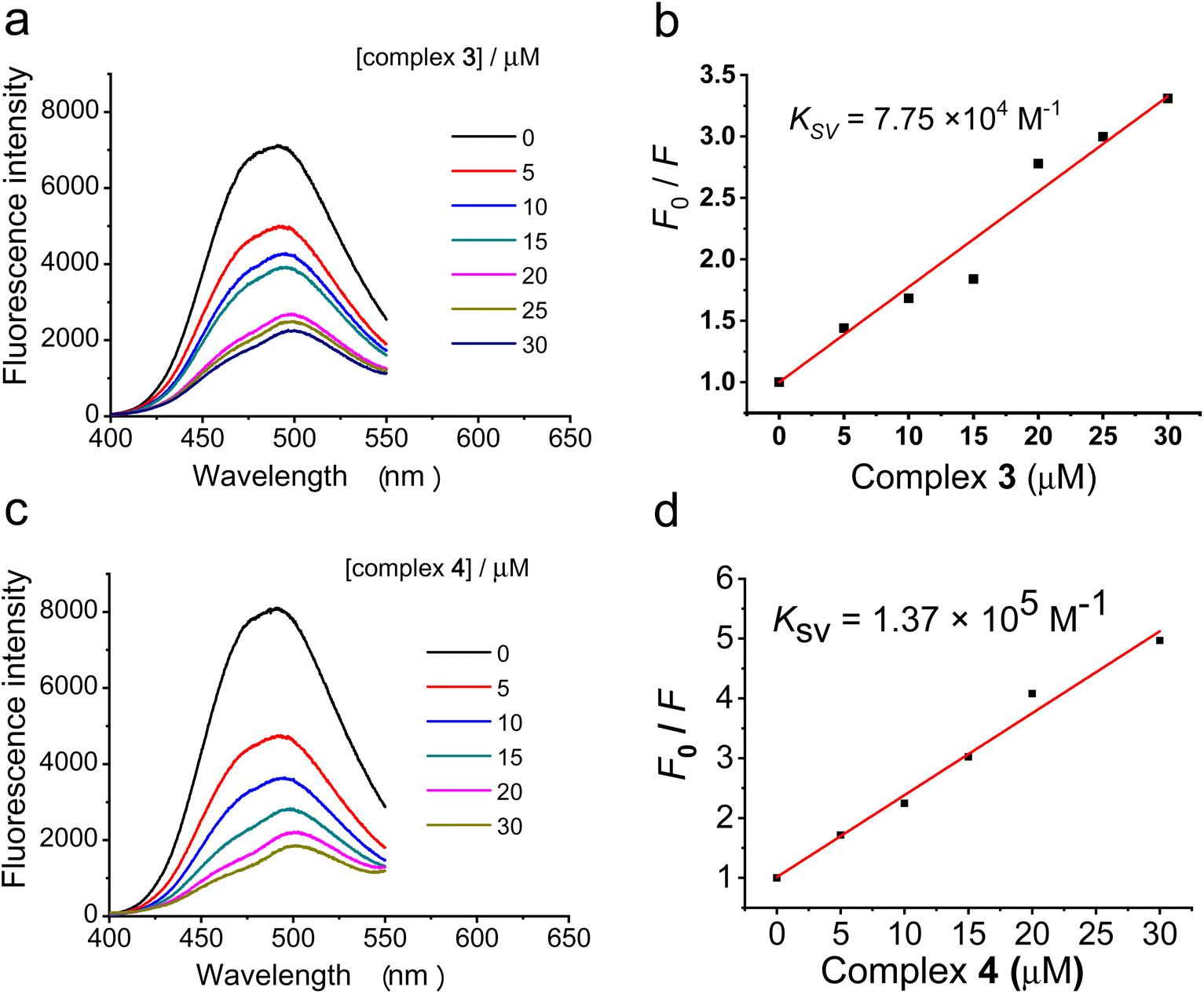 | ||
| Fig. 4 Competitive displacement assay. (a and c) Fluorescence titration of the Hoechst–ctDNA complex with complexes 3 and 4, respectively, in Tris buffer (Hoechst:ctDNA = 20:200 μM) at λex = 370 nm. (b and d) The corresponding Stern–Volmer plot for the quenching of fluorescence intensity at 488 nm upon the addition of complexes 3 and 4, respectively. | ||
Conclusion
In conclusion, we developed a series of multi-functional ruthenium(II) polypyridyl complexes [(N^N)2RuII(L)]2+ (1–4, L = 4-anilinoquinazoline derivative) that have visible light-enhanced in vitro anticancer activity and can be used as fluorescence and mass spectrometry cell imaging probes. These complexes exhibited strong red fluorescence emitting with large Stokes shifts that facilitated single-cell imaging by confocal scanning microscopy (LSCM). Complexes 3 and 4 were potent cytotoxic agents against A549, HeLa and A431 cancer cell lines. Upon irradiation with visible light, complex 3 displayed significantly enhanced toxicity against the HeLa and A431 cell lines. It is much more potent than cisplatin as well as its precursor Ru complexes, such as cis-[Ru(bpy)2Cl2], cis-[Ru(phen)2Cl2] and [Ru(bpy)2(phen)]. Single-cell fluorescence imaging and high-resolution mass spectrometry (ToF-SIMS) imaging were performed, showing that complex 3 largely accumulated in the cytoplasm and 4 tended to enter the cell nucleus. Interestingly, EGFR inhibition seemed not to be a key factor in their anticancer mechanisms. More research suggested that the visible light enhancement of the antiproliferation activity of complex 3 may be associated with the high level generation of singlet oxygen. In comparison, complex 4 exerted its anticancer activity largely by interaction with the DNA minor groove, and the photo-induced generation of singlet oxygen seemed to be not crucial.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Beijing Natural Science Foundation (No. 2232034) and the National Natural Science Foundation of China (No. 21927804, 22077003). Y. Z. is alsothankful for the financial support from the State Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, China.References
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- I. N. Stepanenko, M. S. Novak, G. Muhlgassner, A. Roller, M. Hejl, V. B. Arion, M. A. Jakupec and B. K. Keppler, Inorg. Chem., 2011, 50, 11715–11728 CrossRef CAS PubMed.
- A. A. Nazarov, J. Risse, W. H. Ang, F. Schmitt, O. Zava, A. Ruggi, M. Groessl, R. Scopelitti, L. Juillerat-Jeanneret, C. G. Hartinger and P. J. Dyson, Inorg. Chem., 2012, 51, 3633–3639 CrossRef CAS PubMed.
- F. Y. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- A. Bergamo and G. Sava, Chem. Soc. Rev., 2015, 44, 8818–8835 RSC.
- M. Groessl, E. Reisner, C. G. Hartinger, R. Eichinger, O. Semenova, A. R. Timerbaev, M. A. Jakupec, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 2185–2193 CrossRef CAS PubMed.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 2006, 4003–4018 CrossRef.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- S. Monro, K. L. Colón, H. Yin, J. Roque, III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- C. G. Hartinger, S. Zorbas-Seifried, M. A. Jakupec, B. Kynast, H. Zorbas and B. K. Keppler, J. Inorg. Biochem., 2006, 100, 891–904 CrossRef CAS PubMed.
- T. Chen, Y. Liu, W.-J. Zheng, J. Liu and Y.-S. Wong, Inorg. Chem., 2010, 49, 6366–6368 CrossRef CAS PubMed.
- U. Schatzschneider, J. Niesel, I. Ott, R. Gust, H. Alborzinia and S. Wölfl, ChemMedChem, 2008, 3, 1104–1109 CrossRef CAS PubMed.
- J. Karges, T. Yempala, M. Tharaud, D. Gibson and G. Gasser, Angew. Chem., Int. Ed., 2020, 59, 7069–7075 CrossRef CAS PubMed.
- L. M. Lifshits, J. A. Roque Iii, P. Konda, S. Monro, H. D. Cole, D. von Dohlen, S. Kim, G. Deep, R. P. Thummel, C. G. Cameron, S. Gujar and S. A. McFarland, Chem. Sci., 2020, 11, 11740–11762 RSC.
- W. Xu, J. Zuo, L. Wang, L. Ji and H. Chao, Chem. Commun., 2014, 50, 2123–2125 RSC.
- S. Thota, S. Vallala, R. Yerra and E. J. Barreiro, Chin. Chem. Lett., 2015, 26, 721–726 CrossRef CAS.
- Z. Luo, L. Yu, F. Yang, Z. Zhao, B. Yu, H. Lai, K.-H. Wong, S.-M. Ngai, W. Zheng and T. Chen, Metallomics, 2014, 6, 1480–1490 CrossRef CAS PubMed.
- S. Qi, Z. Jin, Y. Jian, Y. Hou, C. Li, Y. Zhao, X. Wang and Q. Zhou, Chem. Commun., 2021, 57, 4162–4165 RSC.
- C. Tan, S. Wu, S. Lai, M. Wang, Y. Chen, L. Zhou, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, Dalton Trans., 2011, 40, 8611–8621 RSC.
- B.-J. Han, G.-B. Jiang, J. Wang, W. Li, H.-L. Huang and Y.-J. Liu, RSC Adv., 2014, 4, 40899–40906 RSC.
- G.-L. Liao, X. Chen, L.-N. Ji and H. Chao, Chem. Commun., 2012, 48, 10781–10783 RSC.
- Y. Zhang, Q. Zhou, Y. Zheng, K. Li, G. Jiang, Y. Hou, B. Zhang and X. Wang, Inorg. Chem., 2016, 55, 4296–4300 CrossRef CAS PubMed.
- N. Soliman, G. Gasser and C. M. Thomas, Adv. Mater., 2020, 32, 2003294 CrossRef CAS PubMed.
- J. Shen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2021, 443, 214016 CrossRef CAS.
- M. He, Z. Zhang, Z. Jiao, M. Yan, P. Miao, Z. Wei, X. Leng, Y. Li, J. Fan, W. Sun and X. Peng, Chin. Chem. Lett., 2023, 34, 107574 CrossRef CAS.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- M. B. Fleisher, K. C. Waterman, N. J. Turro and J. K. Barton, Inorg. Chem., 1986, 25, 3549–3551 CrossRef CAS.
- J. K. Barton, C. V. Kumar and N. J. Turro, J. Am. Chem. Soc., 1986, 108, 6391–6393 CrossRef CAS.
- A. Notaro, M. Jakubaszek, N. Rotthowe, F. Maschietto, R. Vinck, P. S. Felder, B. Goud, M. Tharaud, I. Ciofini, F. Bedioui, R. F. Winter and G. Gasser, J. Am. Chem. Soc., 2020, 142, 6066–6084 CrossRef CAS PubMed.
- S. Mardanya, S. Karmakar, D. Maity and S. Baitalik, Inorg. Chem., 2015, 54, 513–526 CrossRef CAS PubMed.
- A. Ghosh, P. Das, M. R. Gill, P. Kar, M. G. Walker, J. A. Thomas and A. Das, Chem. – Eur. J., 2011, 17, 2089–2098 CrossRef CAS PubMed.
- D. K. Moscatello, M. Holgado-Madruga, A. K. Godwin, G. Ramirez, G. Gunn, P. W. Zoltick, J. A. Biegel, R. L. Hayes and A. J. Wong, Cancer Res., 1995, 55, 5536–5539 CAS.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS PubMed.
- M. Muhsin, J. Graham and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2003, 2, 515–516 CrossRef CAS PubMed.
- M. Martínez-Alonso and G. Gasser, Coord. Chem. Rev., 2021, 434, 213736 CrossRef.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- L. O'Neill, L. Perdisatt and C. O'Connor, J. Phys. Chem. A, 2012, 116, 10728–10735 CrossRef PubMed.
- C.-P. Tan, Y.-M. Zhong, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2021, 12, 2357–2367 RSC.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- T. Sainuddin, J. McCain, M. Pinto, H. Yin, J. Gibson, M. Hetu and S. A. McFarland, Inorg. Chem., 2016, 55, 83–95 CrossRef CAS PubMed.
- Z. Zhao, X. Tao, Y. Xie, Q. Lai, W. Lin, K. Lu, J. Wang, W. Xia and Z.-W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202202855 CAS.
- S. He, G. Dong, Y. Li, S. Wu, W. Wang and C. Sheng, Angew. Chem., Int. Ed., 2020, 59, 3028–3032 CrossRef CAS PubMed.
- L. Feng, Y. Geisselbrecht, S. Blanck, A. Wilbuer, G. E. Atilla-Gokcumen, P. Filippakopoulos, K. Kraling, M. A. Celik, K. Harms, J. Maksimoska, R. Marmorstein, G. Frenking, S. Knapp, L. O. Essen and E. Meggers, J. Am. Chem. Soc., 2011, 133, 5976–5986 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bachler, R. Trondl, G. Muhlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 4839–4841 RSC.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- W. Zheng, Q. Luo, Y. Lin, Y. Zhao, X. Wang, Z. Du, X. Hao, Y. Yu, S. Lu, L. Ji, X. Li, L. Yang and F. Wang, Chem. Commun., 2013, 49, 10224–10226 RSC.
- Y. Zhang, W. Zheng, Q. Luo, Y. Zhao, E. Zhang, S. Liu and F. Wang, Dalton Trans., 2015, 44, 13100–13111 RSC.
- J. Du, Y. Kang, Y. Zhao, W. Zheng, Y. Zhang, Y. Lin, Z. Wang, Y. Wang, Q. Luo, K. Wu and F. Wang, Inorg. Chem., 2016, 55, 4595–4605 CrossRef CAS PubMed.
- B. P. Sullivan, D. J. Salmon and T. J. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS.
- R. M. Hartshorn and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5919–5925 CrossRef CAS.
- J. Du, E. Zhang, Y. Zhao, W. Zheng, Y. Zhang, Y. Lin, Z. Wang, Q. Luo, K. Wu and F. Wang, Metallomics, 2015, 7, 1573–1583 CrossRef CAS PubMed.
- A. Gollmer, J. Arnbjerg, F. H. Blaikie, B. W. Pedersen, T. Breitenbach, K. Daasbjerg, M. Glasius and P. R. Ogilby, Photochem. Photobiol., 2011, 87, 671–679 CrossRef CAS PubMed.
- H. Ogiso, R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J.-H. Kim, K. Saito, A. Sakamoto, M. Inoue, M. Shirouzu and S. Yokoyama, Cell, 2002, 110, 775–787 CrossRef CAS PubMed.
- C. Li, F. Xu, Y. Zhao, W. Zheng, W. Zeng, Q. Luo, Z. Wang, K. Wu, J. Du and F. Wang, Front. Chem., 2020, 8, 210 CrossRef PubMed.
- S. Liu, W. Zheng, K. Wu, Y. Lin, F. Jia, Y. Zhang, Z. Wang, Q. Luo, Y. Zhao and F. Wang, Chem. Commun., 2017, 53, 4136–4139 RSC.
- Y. Guan, W. Zhou, X. H. Yao, M. P. Zhao and Y. Z. Li, Anal. Chim. Acta, 2006, 570, 21–28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt01661g |
| This journal is © The Royal Society of Chemistry 2023 |