
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the spin-crossover properties of FeII4L6 cages via the interplay of coordination motif and linker modifications†
Tobias
Paschelke
a,
Eicke
Trumpf
a,
David
Grantz
 a,
Malte
Pankau
a,
Niclas
Grocholski
a,
Christian
Näther
b,
Frank D.
Sönnichsen
a and
Anna J.
McConnell‡
*a
a,
Malte
Pankau
a,
Niclas
Grocholski
a,
Christian
Näther
b,
Frank D.
Sönnichsen
a and
Anna J.
McConnell‡
*a
aOtto Diels Institute of Organic Chemistry, Kiel University, Otto-Hahn-Platz 4, Kiel 24098, Germany
bInstitute of Inorganic Chemistry, Kiel University, Max-Eyth-Straße 2, Kiel 24118, Germany
First published on 24th August 2023
Abstract
Despite the increasing number of spin-crossover FeII-based cages, the interplay between ligand modifications (e.g. coordination motif substituents and linker) is not well-understood in these multinuclear systems, limiting rational design. Here, we report a family of FeII4L6 spin-crossover cages based on 2,2′-pyridylbenzimidazoles where subtle ligand modifications lowered the spin crossover temperature in CD3CN by up to 186 K. Comparing pairs of cages, CH3 substituents on either the coordination motif or phenylene linker lowered the spin-crossover temperature by 48 K, 91 K or 186 K, attributed to electronic effects, steric effects and a combination of both, respectively. The understanding of the interplay between ligand modifications gained from this study could be harnessed on the path towards the improved rational design of spin-crossover cages.
Introduction
Most known spin-crossover complexes are based on FeII ions.1–5 These complexes can be switched between paramagnetic high- and diamagnetic low-spin states by external stimuli like light,6–12 pressure6,13,14 or temperature6,10,15–20 if their spin-pairing and ligand field splitting energies are similar.2,3,21 Despite extensive studies, there are still limitations to the prediction of spin-crossover properties22 but methods have been established for predicting the spin state23,24 and tuning the spin-crossover properties.9,25 For example, steric bulk proximal to the coordination site is known to stabilize the high-spin state through lengthening the metal–ligands bonds.26,27 However, rationalisation of electronic effects on the spin-crossover temperature (T1/2) is more difficult, especially since most studies were performed in the solid state where intermolecular interactions also need to be considered.18,28 Halcrow's systematic solution study on 2,6-di(pyrazol-1-yl)pyridine-based complexes revealed that electron-donating p-pyridine substituents lower T1/2 while m-pyrazole substituents raise it,18 attributed to differing σ and π bonding contributions.18,28Spin-crossover cages29–39 consist of multiple FeII ions and organic ligands with two or more coordination motifs connected via linkers. They are an interesting emerging class of spin-crossover materials2,3 since multiple avenues exist for tuning the spin-crossover properties. These include modifications to the coordination motif,31,36 guest encapsulation,30,33,35 linker modifications30 and cooperative effects. Furthermore, the potential to access multiple spin-states with distinct optical and magnetic properties makes cages appealing for applications as sensors, electronic switches and in information storage.1–5
However, spin-crossover cage examples, especially those studied in solution,29,30,33,36,38 are limited and they are usually comprising imidazolimine ligands that form cubic33,35,36,38,39 or tetrahedral29–32,38 cages. The rational design of a cage with predictable spin-crossover properties is also challenging and the effect of modifications to the coordination motif and linker has not been extensively studied in FeII-based cages.
Motivated to improve the rational design of spin-crossover cages, we studied a family of FeII4L6 cages (1–11, Fig. 1) and varied the coordination motif and linker to determine their influence on T1/2. These subtle ligand modifications tuned the T1/2 values, determined using the ideal solution model, from >430 K to 244 K in CD3CN; T1/2 decreased by 48 K from introducing CH3 groups on the pyridine motifs (cage 6vs.10), 91 K from increasing the steric bulk of the linker with CH3 groups (cage 7vs.9) and 186 K for a combination of electronic and steric effects (cage 7vs.11).
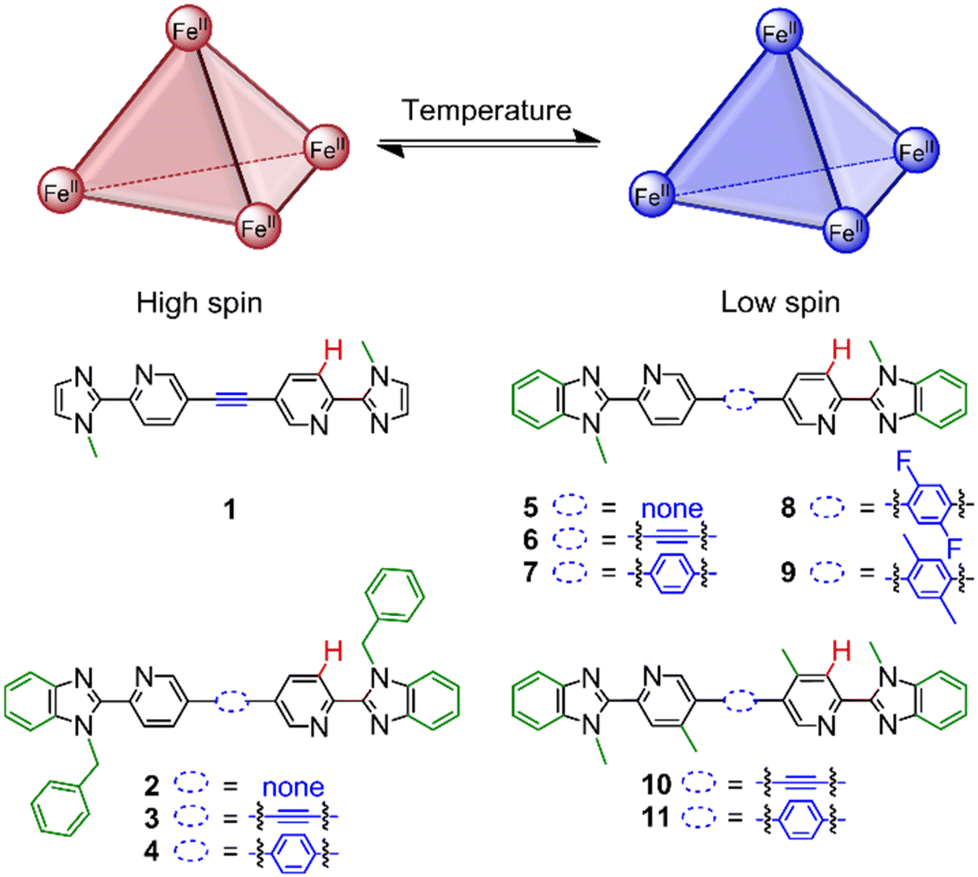 | ||
| Fig. 1 FeII4L6 cages 1–11 for investigating the effect of ligand modifications within the coordination motif (represented by the changes in green) and linker (represented in blue) on the spin-crossover properties. The chemical shift changes of the proton in red were used for fitting to the ideal solution model. | ||
Results and discussion
Complexes and helicates based on 2,2′-pyridyl(benz)imidazole ligands exhibit a range of ligand field strengths, resulting in low-spin40 complexes and those showing spin-crossover41,42 depending on the ligand substitution. We decided to focus on FeII4L6 cages to probe the interplay of coordination site modifications and linker on T1/2 since: (i) there are relatively few spin-crossover examples;31,34,37 (ii) a variety of linkers can be introduced into the bis-bidentate ligand design with relative synthetic ease; (iii) the guest binding (e.g. solvent or counteranion) influence on T1/2 is likely minimised by the more open faces of the cages as compared to the more enclosed faces of an FeII4L4 cage. The studies were performed in solution rather than the solid state to disentangle these intramolecular ligand field strength changes from intermolecular interactions. Furthermore, the structure and spin-state changes of individual species with temperature could be probed by NMR spectroscopy.A family of FeII4L6 cages was designed based on four related coordination motifs with the modifications highlighted in green (Fig. 1): (i) imidazole-based 1 and benzimidazole-based (ii) 2–4 with N-benzyl substituents; (iii) 5–9 with N-methyl substituents; (iv) 10–11 with N-methyl substituents and CH3 groups on the pyridine ring. Furthermore, the linker (Fig. 1, blue) was also varied from no spacer to an alkyne and phenylene derivatives. Edge-bridged FeII4L6 cages 1–11 (ESI, section 4†) were self-assembled at room temperature in anhydrous acetonitrile in a glovebox using four equivalents of Fe(OTf)2 and six equivalents of the appropriate ligand (preparations in ESI, section 2† using our recently reported one-pot Sonogashira-type procedure43 or via Suzuki couplings). The corresponding ZnII4L6 cages were also prepared using Zn(OTf)2 to serve as diamagnetic analogues. However, a discrete cage species did not form with the imidazole ligand (ESI, section 3.1†).
The formation of M4L6 cages was confirmed by ESI mass spectrometry, the observation of one set of signals in the NMR spectra and in the case of cages 5 and 7, by X-ray crystallography (Fig. 2 and ESI, section 6†). Although the diffraction data were of limited quality due to poor diffraction and disorder, as is commonly observed for metallosupramolecular cages,44 they were sufficient to establish the cages’ connectivity. In both cages, an anti ligand conformation was adopted resulting in a T-symmetric cage with the same stereochemistry at each metal center (Fig. 2). Cage 5 was also observed to bind a triflate counterion in its cavity.
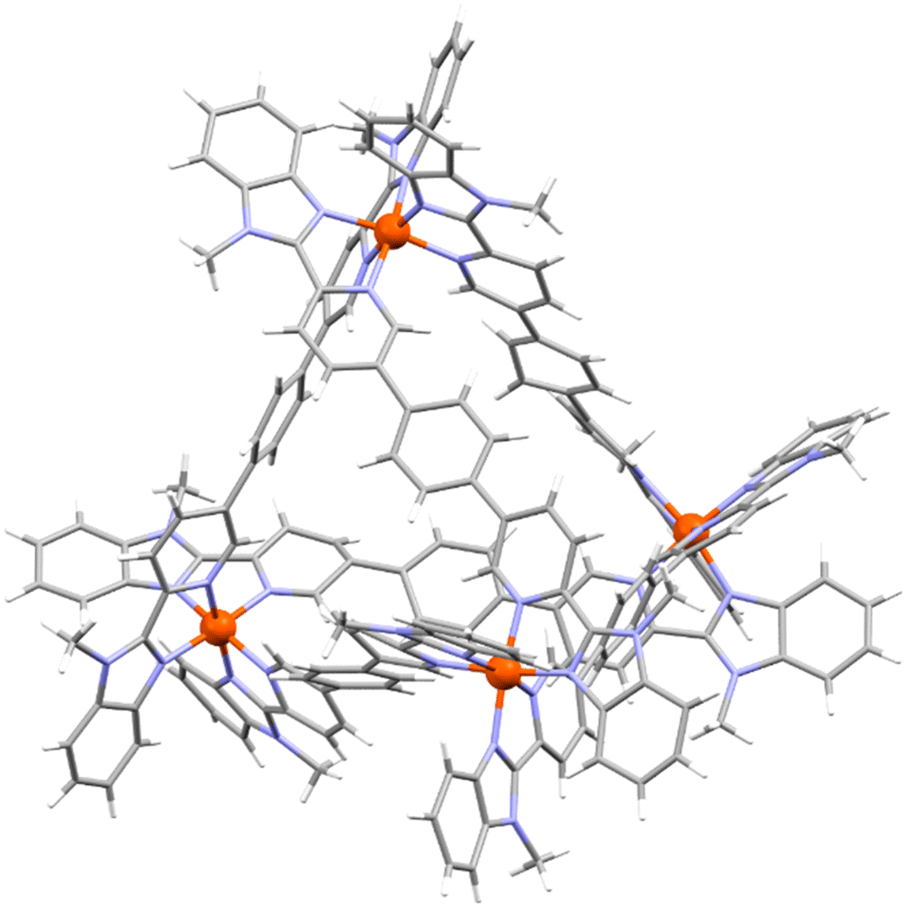 | ||
| Fig. 2 X-ray crystal structure of cage 7. | ||
While the FeII4L6 cages were obtained as the only species, a second minor species was also observed in some self-assemblies with ZnII (ESI, sections 3.7, 3.8 and 3.10†). Analysis by 1H and DOSY NMR spectroscopy and ESI mass spectrometry revealed the formation of Zn2L3 helicates. Lusby and co-workers have reported a helicate/tetrahedron equilibrium with the Co(II) and Co(III) analogues of cage 7.45 We attribute the formation of Zn2L3 but not Fe2L3 helicates to a combination of the metal's ionic radius and ligand's steric bulk increasing the energy of the Fe2L3 relative to the Fe4L6 self-assembly, as has been observed in related systems.46,47
The spin-crossover properties of cages 1–11 were investigated in solution between 248 K and 348 K in CD3CN. While the Evans method48 can determine the magnetic susceptibility from variable-temperature (VT) NMR experiments, preliminary studies with cage 6 showed the NMR signals shifted upon addition of the p-xylene standard, suggesting possible guest binding (Fig. S239†). In addition, the magnetic susceptibility is dependent on accurate determination of the paramagnetic species concentration and influenced by the presence of other paramagnetic species, leading to large sources of error and inaccurate T1/2 values.49 Therefore, the ideal solution model50 was used since the spin-crossover properties of a single species can be determined even in the presence of other paramagnetic impurities. In addition, our recent report of a paramagnetic NMR toolbox would enable the detailed characterisation of the cages in the high-spin state.51 The chemical shift change of proton H (Fig. 1, red) was fitted using Origin (eqn (1)) to obtain ΔH, ΔS and T1/2 values.§
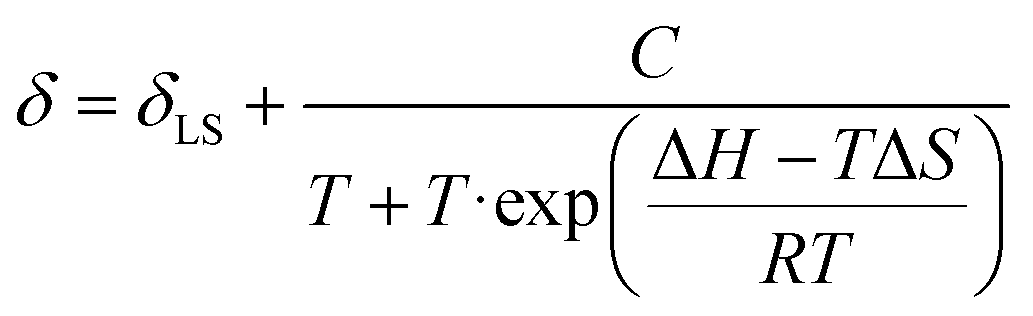 | (1) |
Only small chemical shift changes were observed for cages 1 and 2, indicating that these cages are predominantly low spin over the measured temperature range (Fig. 3). For the other cages, initial fitting of all four parameters (C, ΔH, ΔS, δLS) resulted in large errors or the fitting did not converge (ESI, section 5†). Therefore, δLS was fixed to the chemical shift of the diamagnetic ZnII4L6 cage analogue, yielding ΔH, ΔS and T1/2 values for cages 6–11 (Table 1). The fitting for cages 3–5 still did not converge, likely attributable to an insufficient change in the spin-state populations within the temperature range available for the solvent CD3CN.¶ Since cages 1–7 were predominantly low spin at 248 K, the magnitude of Δδ was used as a measure for T1/2 in comparisons between cages where thermodynamic data could not be obtained.
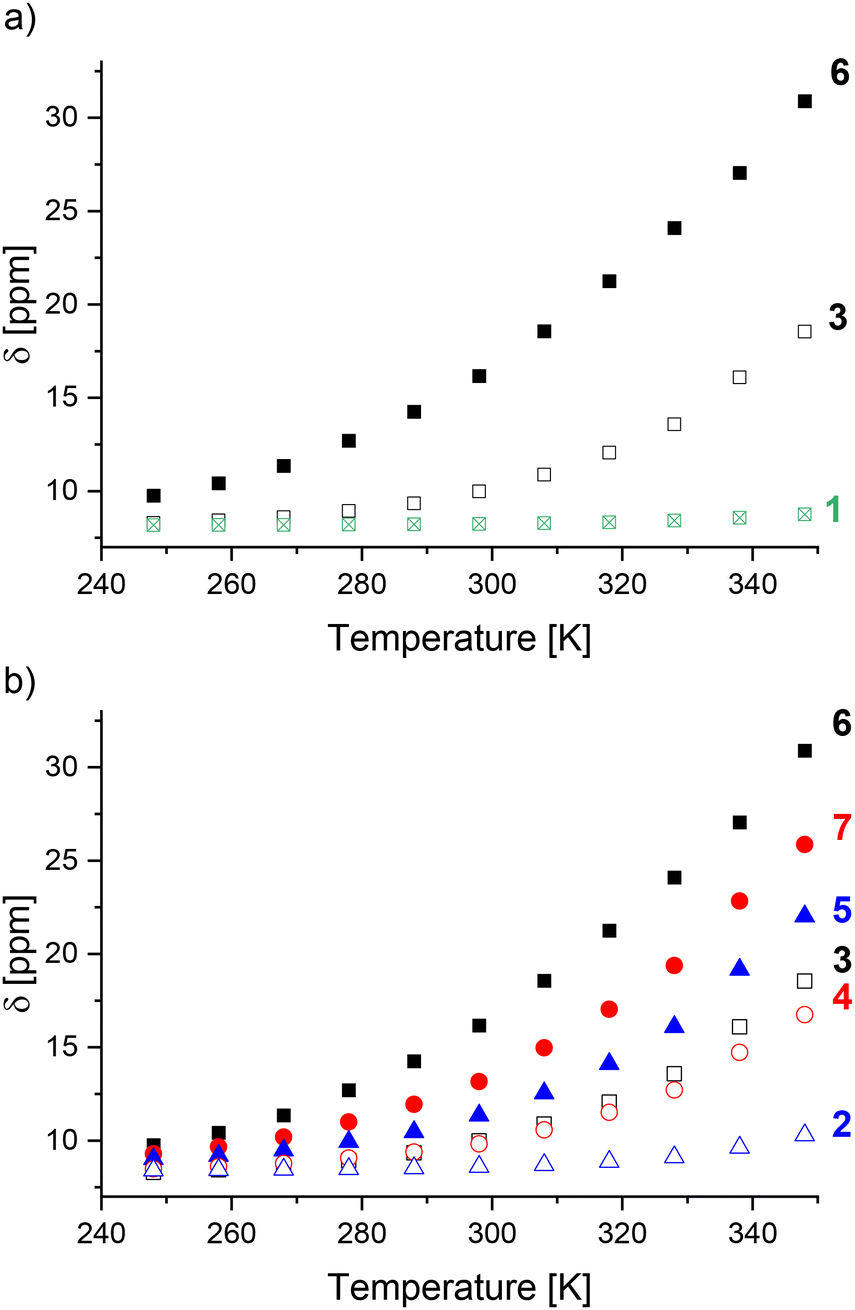 | ||
| Fig. 3 Comparison of the chemical shift changes between 248 K and 348 K for cages: (a) 1, 3 and 6; (b) 2–7. The symbol shading represents the change to the coordination motif: imidazole (⊠); N-benzylated benzimidazole (unfilled); N-methylated benzimidazole (filled). The symbol color and shape represent the linker: alkyne linker (black square); phenylene linker (red circle); no linker (blue triangle). | ||
| Cage | Δδa (ppm) | ΔH (kJ mol−1) | ΔS (J mol−1 K−1) | T 1/2 (K) |
|---|---|---|---|---|
| a Based on the chemical shift change of proton H (red, Fig. 1) between 248 K and 348 K. b Could not be determined since fitting did not converge. c Low spin cage. d Proposed to be >430 K. | ||||
| 1 | 0.58 | |||
| 2 | 1.89 | |||
| 3 | 10.2 | |||
| 4 | 8.26 | |||
| 5 | 13.0 | |||
| 6 | 21.1 | 24.46 ± 0.58 | 60.94 ± 3.00 | 401 |
| 7 | 17.7 | 27.71 ± 0.91 | 64.48 ± 5.78 | 430 |
| 8 | 30.1 | 27.22 ± 0.93 | 72.61 ± 4.00 | 375 |
| 9 | 31.4 | 21.79 ± 0.48 | 64.32 ± 2.00 | 339 |
| 10 | 27.8 | 27.44 ± 1.12 | 77.65 ± 4.40 | 353 |
| 11 | 12.5 | 20.78 ± 0.22 | 85.05 ± 0.93 | 244 |
One influence of the coordination motif on the spin-state can be delineated by comparing cages 1, 3 and 6 (Fig. 3a). The increased Δδ values for benzimidazole-based cages 3 and 6vs. diamagnetic imidazole-based cage 1 indicated the stabilization of the high-spin state, attributed to lengthening of the M–L bonds by the benzimidazole's steric bulk in 3 and 6 (Table 1).40
For benzimidazole-based cages 2–7 (Fig. 3b), the effect of N-benzylation was compared to N-methylation for three different linkers (alkyne, phenylene or no linker). Comparing cages with the same linker (e.g.2 and 5), larger Δδ values were observed for the N-methylated cages 5–7 than the N-benzylated analogues 2–4, suggesting cages 5–7 have lower T1/2 values.
The linker's influence on the spin-crossover properties was also investigated. For both the N-methylated (5–7) and N-benzylated series (2–4), the largest Δδ values were observed for an alkyne followed by the phenylene and no linker (Table 1). This was also reflected by the increasing T1/2 values for the N-methylated cages: 401 K for cage 6 (alkyne linker), 430 K for cage 7 (phenylene linker) and >430 K for cage 5 (no linker).
Different factors were considered to explain the influence of the linker on T1/2. First, the electronic effect of the linker was estimated using the Hammett parameter σm for C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (0.21), C6H5 (0.06) and 3-pyridine (0.23).52 However, their trend does not correlate with the observed T1/2 trend. The Hammett parameters do not take into account electronic effects from metal complexation and these will be particularly significant for the cages without a linker as the 3-pyridine is part of the coordination motif.
CH (0.21), C6H5 (0.06) and 3-pyridine (0.23).52 However, their trend does not correlate with the observed T1/2 trend. The Hammett parameters do not take into account electronic effects from metal complexation and these will be particularly significant for the cages without a linker as the 3-pyridine is part of the coordination motif.
The ligand conformation and mechanical coupling between the FeII centers could also play a role since the linkers’ steric bulk dictates twisting within the ligand in order to adopt the required anti ligand conformation. Dihedral angles of approx. 10–22° for the phenylene linker and 50–65° for no linker were observed in the X-ray structures of cages 5, 7 and related cages.45,53 No cooperativity and weaker mechanical coupling is likely in cages with alkyne and phenylene linkers (3–4, 6–7) given the large FeII–FeII distances (>13 Å in cage 7) and ligand twisting, thus making spin-crossover more favorable.38 In contrast, cages with no linkers (2, 5) likely exhibit stronger mechanical coupling and cooperative effects cannot be excluded.
Finally, the cavity size as dictated by the ligand length could also influence T1/2via host–guest chemistry. While the same counteranion OTf− was used for all cages to minimize guest binding effects, the smallest cages 3 and 5 were observed to bind the counterion in slow exchange by 19F NMR spectroscopy (Fig. S174 and S195†) and in the case of 5, in the X-ray crystal structure (Fig. S293†).
While these different factors were considered, it was not possible to rationalize the observed T1/2 trend as a function of the linker. Therefore, to gain further insight into coordination site and linker modifications on T1/2, sets of cages (6 and 10, 7–9, 7 and 11) with the same ligand scaffold but minor modifications (e.g. a F or CH3 substituent) were compared. Given the larger size of cages 7–11 and the observation of fast exchange of the counterion OTf− in the 19F NMR spectra at room temperature (Fig. S208, S215, S223, S230 and S236†), it was hypothesised that ligand field strength effects would dominate over solvent, guest binding and ion-pairing effects enabling the determination of trends based on steric and electronic effects.
As a control, anion binding experiments (ESI, section 7†) were carried out with cages 7 and 11 since this set of cages displayed the largest T1/2 change within this study (Table 1). To probe guest binding within the cavity, 8 equivalents (relative to the cage) of the smaller counterion BF4− were added to each cage and the broad 19F signals for BF4− indicated fast guest exchange (Fig. S296 and S299†). Small chemical shift changes (Δδ < 0.2 ppm) were observed in the 1H NMR spectra and the insignificant changes of the phenylene protons (Δδ < 0.05 ppm) suggested counteranion binding in the cavity has a minimal effect (Fig. S295 and S297†).
Further experiments with cage 7 focused on ion-pairing. NTf2− was chosen as a counterion since the sharp signal in the 19F NMR spectrum suggested that it is too large to fit in the cavity (Fig. S303†). Again, small 1H chemical shift changes were observed, meaning ion-pairing likely has an insignificant effect on T1/2. Finally, VT NMR experiments with cages 7 and 11 in the presence and absence of competing counteranions showed no evidence of a change from fast to slow counteranion exchange upon cooling to 248 K due to increased population of the low spin state with a smaller cavity (Fig. S297–S304†). Based on these control experiment results, the effect of ion-pairing or guest binding on the spin-crossover properties is proposed to be negligible for comparisons within a set of cages.
Cages 7–9 were compared where F or CH3 substituents were introduced on the phenylene linker to increase the steric bulk without changing the ligand length. T1/2 decreased from 430 K (cage 7) to 375 K for cage 8 with F substituents and to 339 K for cage 9 with CH3 substituents (Table 1). While the high-spin fraction of cage 7 at 298 K is 3%, increasing high-spin-state populations of 10% and 26% were obtained for cages 8–9, respectively (Fig. 4). Thus, T1/2 decreases with increasing substituent size on the linker, attributed to increased dihedral angles between the coordination motif and the linker, as observed in crystal structures of related cages.45 However, contributions from electronic effects cannot be completely neglected since CH3 groups are electron-donating, while F is electron-withdrawing.
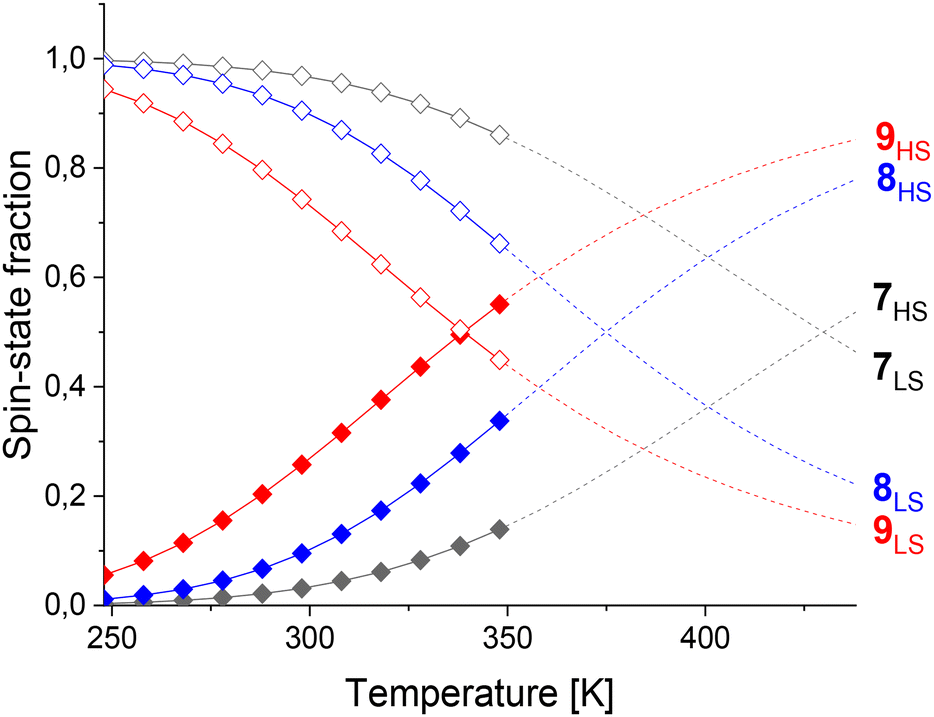 | ||
| Fig. 4 High (filled diamonds) and low (unfilled diamonds) spin-state fractions of cages 7 (black), 8 (blue) and 9 (red) from VT NMR measurements between 248 K and 348 K. Data above 348 K (dashed lines) was extrapolated. | ||
Thermodynamic data analysis revealed cages 7 and 8 have similar enthalpies but cage 8 has an increased entropy (Table 1). In contrast, cage 9 had a lower enthalpy compared to cage 7, but the entropy values were similar. Thus, the spin-transition of cage 9 is enthalpically driven, while for cage 8 it is entropically controlled. It is difficult to draw conclusions from these data since the relationship between both energies on spin-crossover properties has not been systematically studied.54 However, we attribute stabilization of the high-spin state for cage 9vs. cage 7 to the increased dihedral angles from the steric bulk of the CH3 groups and the required adoption of an anti-conformation, leading to weakened M–L bonds. Thus, modifications through CH3 substituents on the phenylene linker tuned T1/2 by 91 K.
Further coordination site modifications were introduced to investigate the influence of interactions between these and the linker on T1/2. For example, Halcrow and co-workers have shown that electron-donating groups on para-substituted pyridines in FeII mononuclear complexes based on 2,6-bis(pyrazol-1-yl)pyridines can stabilize the high-spin state.18 Therefore, CH3 groups were introduced on the pyridine motif in cages 10 and 11 in an attempt to decrease T1/2 towards room temperature. Comparing cages 6 and 10, the introduction of the CH3 groups decreased T1/2 from 401 K to 353 K (Table 1), resulting in an increase of the high-spin state fraction at 298 K from 7% to 15% (Fig. 5). We propose the stabilization of the high-spin state by 48 K is largely due to electronic effects since steric effects from the alkyne linker are minimized.
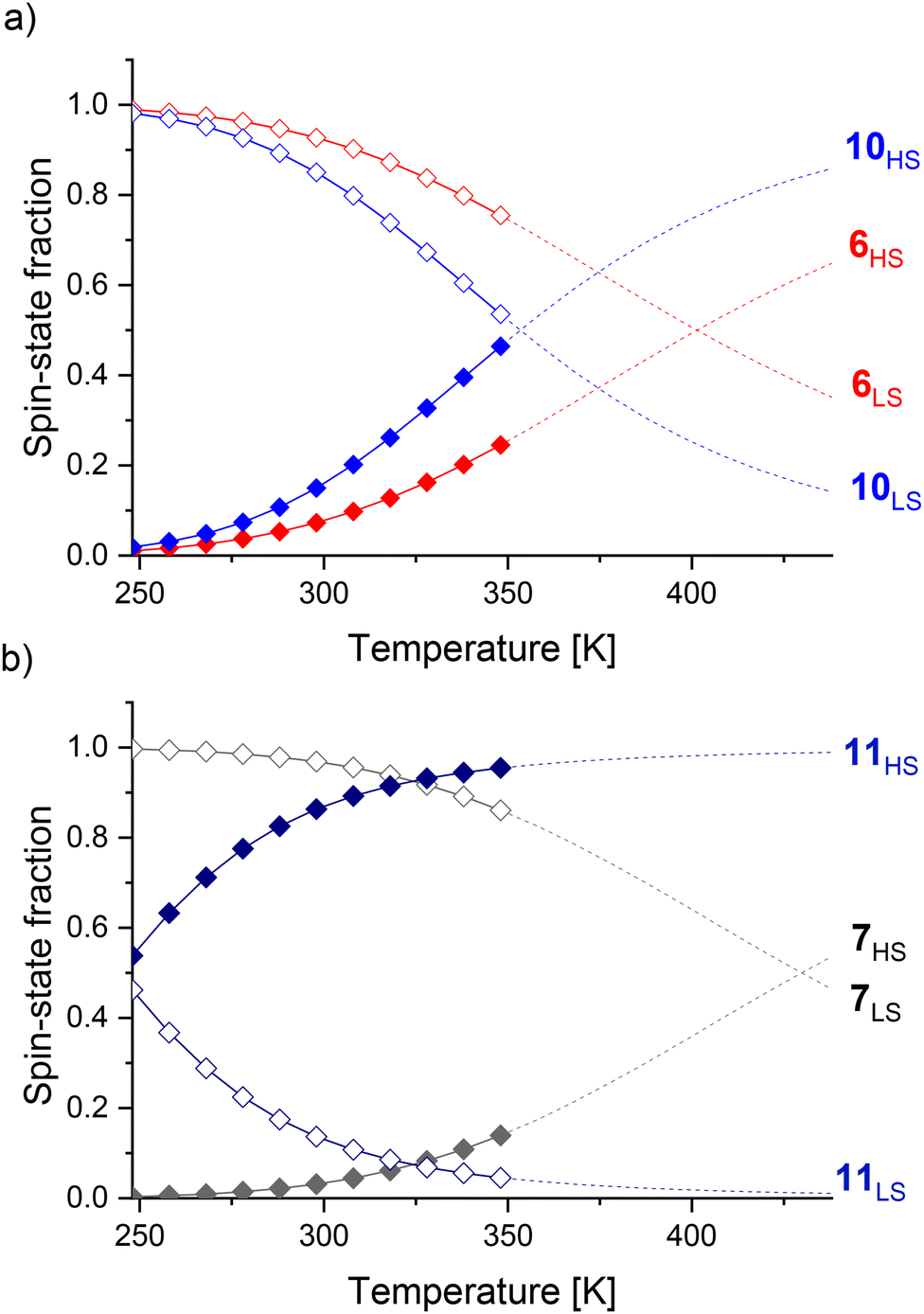 | ||
| Fig. 5 High (filled diamonds) and low (unfilled diamonds) spin-state fractions of cages: (a) 6 (red) and 10 (blue); (b) 7 (grey) and 11 (blue) from VT NMR measurements between 248 K and 348 K. Data above 348 K (dashed lines) was extrapolated. | ||
Surprisingly, T1/2 decreased by 186 K from 430 K to 244 K for cage 7vs.11 (Table 1). The introduction of a CH3 group in cage 11 yields a largely high-spin cage (86% high-spin state fraction) at 298 K, while cage 7 is almost completely low-spin (3% high-spin state fraction) (Fig. 5). We attribute this large change to electronic effects, and moreover, to strong steric clashes between the CH3 substituents and the phenylene linker in cage 11, increasing the dihedral angles between the linker and the pyridine rings as observed in crystal structures of related cages.55 Comparing thermodynamic data (Table 1), both the enthalpy and entropy increased for cage 10vs.6. Although the increased entropy favors the high-spin state, this is partly counteracted by the increased enthalpy stabilizing the low-spin state, resulting in a smaller T1/2 difference. In contrast, the decrease of the enthalpy and increase of the entropy for cage 11vs.7 both favor the high-spin state, leading to the larger T1/2 change of 186 K.
Conclusions
This study demonstrates the power of solution-based spin-crossover studies using the ideal solution model for quantifying ligand field strength modifications in the absence of intermolecular interactions, as exemplified in this family of FeII4L6 cages. Furthermore, information about the cage's structure and spin-state can be obtained from VT NMR experiments independently of other paramagnetic species.Imidazole-based cage 1 was low-spin over the measured temperature range. However, the steric bulk of the benzimidazole motif in cages 2–11 stabilized the high-spin state.
While substitution of the coordination motif is known to influence T1/2 in spin-crossover complexes, this systematic study reveals that the linker can also have a profound effect, despite the increased distance from the metal centers. Importantly, the interplay of coordination site and linker modification effects was quantified and minor modifications like the introduction of CH3 groups tuned T1/2 by up to 186 K. p-CH3 substituents on the pyridine motif decreased T1/2 by 48 K for cage 10vs.6 with alkyne linkers, attributed to predominantly electronic effects. CH3 substituents on the phenylene linker decreased T1/2 by 91 K for cage 9vs.7, largely attributed to steric clashes. Finally, a combination of both effects is proposed to result in the 186 K T1/2 decrease in cage 11vs.7 due to both electronic effects from the p-CH3 substituents on the pyridine and a steric clash with the phenylene linker.
Thus, this study demonstrates how seemingly subtle modifications (i.e. substitution of H for CH3) can significantly impact the spin-crossover properties of multinuclear FeII-based cages in solution, resulting in large T1/2 changes of almost 200 K and altering the spin state from predominantly low spin to high spin at room temperature. Future work will investigate how this understanding of the interplay between ligand modifications can be applied to the design of spin-crossover cages with more predictable spin-crossover properties. This would facilitate the tailored application of spin-crossover cages, e.g. as sensors.
Author contributions
T. P.: conceptualisation (lead), data curation (lead), formal analysis (lead), investigation (lead), resources (lead), supervision (supporting), visualisation (lead), writing-original draft (lead), writing-review and editing (equal). E. T.: conceptualisation (lead), formal analysis (supporting), investigation (supporting), resources (supporting), visualisation (lead), writing-original draft (supporting), writing-review and editing (equal). D. G.: formal analysis (supporting), investigation (supporting), resources (supporting), writing-review and editing (supporting). M. P.: formal analysis (supporting), investigation (supporting), resources (supporting), visualisation (supporting), writing-review and editing (supporting). N. G.: formal analysis (supporting), investigation (supporting), resources (supporting), writing-review and editing (supporting). C. N.: formal analysis (supporting), investigation (supporting), resources (supporting), writing-review and editing (supporting). F. D. S.: investigation (supporting), methodology (lead), resources (supporting), writing-review and editing (equal). A. J. M.: conceptualisation (lead), formal analysis (supporting), funding acquisition (lead), project administration (lead), supervision (lead), writing-original draft (supporting), writing-review and editing (equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG, project number 429518153) for financial support. We thank Etienne Rommens for preliminary studies and the spectroscopy department (including Marion Höftmann and Gitta Kohlmeyer-Yilmaz for VT NMR measurements and Johanna Baum and Dr Claus Bier for high-resolution ESI measurements) for NMR and mass spectral data collection. We also thank Prof. Dr Felix Tuczek for useful discussions.References
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- A. J. McConnell, Supramol. Chem., 2018, 30, 858–868 CrossRef CAS.
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303–7338 RSC.
- H. S. Scott, R. W. Staniland and P. E. Kruger, Coord. Chem. Rev., 2018, 362, 24–43 CrossRef CAS.
- W. Huang, X. Ma, O. Sato and D. Wu, Chem. Soc. Rev., 2021, 50, 6832–6870 RSC.
- E. Breuning, M. Ruben, J.-M. Lehn, F. Renz, Y. Garcia, V. Ksenofontov, P. Gütlich, E. Wegelius and K. Rissanen, Angew. Chem., Int. Ed., 2000, 39, 2504–2507 CrossRef CAS PubMed.
- T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 CrossRef CAS PubMed.
- B. Rösner, M. Milek, A. Witt, B. Gobaut, P. Torelli, R. H. Fink and M. M. Khusniyarov, Angew. Chem., Int. Ed., 2015, 54, 12976–12980 CrossRef PubMed.
- M. Mörtel, A. Witt, F. W. Heinemann, S. Bochmann, J. Bachmann and M. M. Khusniyarov, Inorg. Chem., 2017, 56, 13174–13186 CrossRef PubMed.
- T. Shiga, G. N. Newton and H. Oshio, Dalton Trans., 2018, 47, 7384–7394 RSC.
- H.-Y. Sun, Y.-S. Meng and T. Liu, Chem. Commun., 2019, 55, 8359–8373 RSC.
- M. Oppermann, F. Zinna, J. Lacour and M. Chergui, Nat. Chem., 2022, 14, 739–745 CrossRef CAS PubMed.
- D. C. Fisher and H. G. Drickamer, J. Chem. Phys., 1971, 54, 4825–4837 CrossRef CAS.
- R. J. Butcher, J. R. Ferraro and E. Sinn, Inorg. Chem., 1976, 15, 2077–2079 CrossRef CAS.
- J. Elhaïk, V. A. Money, S. A. Barrett, C. A. Kilner, I. R. Evans and M. A. Halcrow, Dalton Trans., 2003, 2053–2060 RSC.
- B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2010, 49, 9274–9277 CrossRef CAS PubMed.
- S. Chorazy, R. Podgajny, K. Nakabayashi, J. Stanek, M. Rams, B. Sieklucka and S.-i. Ohkoshi, Angew. Chem., Int. Ed., 2015, 54, 5093–5097 CrossRef CAS PubMed.
- L. J. K. Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 55, 4327–4331 CrossRef PubMed.
- A. A. Pavlov, G. L. Denisov, M. A. Kiskin, Y. V. Nelyubina and V. V. Novikov, Inorg. Chem., 2017, 56, 14759–14762 CrossRef CAS PubMed.
- S. Chorazy, J. J. Stanek, J. Kobylarczyk, S.-i. Ohkoshi, B. Sieklucka and R. Podgajny, Dalton Trans., 2017, 46, 8027–8036 RSC.
- C.-M. Jureschi, J. Linares, A. Boulmaali, P. R. Dahoo, A. Rotaru and Y. Garcia, Sensors, 2016, 16, 187 CrossRef PubMed.
- N. Deorukhkar, C. Besnard, L. Guénée and C. Piguet, Dalton Trans., 2021, 50, 1206–1223 RSC.
- H. Phan, J. J. Hrudka, D. Igimbayeva, L. M. L. Daku and M. Shatruk, J. Am. Chem. Soc., 2017, 139, 6437–6447 CrossRef CAS PubMed.
- S. Rodríguez-Jiménez, M. Yang, I. Stewart, A. L. Garden and S. Brooker, J. Am. Chem. Soc., 2017, 139, 18392–18396 CrossRef PubMed.
- D. Y. Aleshin, I. Nikovskiy, V. V. Novikov, A. V. Polezhaev, E. K. Melnikova and Y. V. Nelyubina, ACS Omega, 2021, 6, 33111–33121 CrossRef CAS PubMed.
- H. A. Goodwin, E. S. Kucharski and A. H. White, Aust. J. Chem., 1983, 36, 1115–1124 CrossRef CAS.
- C. Bartual-Murgui, S. Vela, M. Darawsheh, R. Diego, S. J. Teat, O. Roubeau and G. Aromí, Inorg. Chem. Front., 2017, 4, 1374–1383 RSC.
- L. Bondì, A. L. Garden, F. Totti, P. Jerabek and S. Brooker, Chem. – Eur. J., 2022, 28, e202104314 CrossRef PubMed.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597–1599 RSC.
- R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, D. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem. – Eur. J., 2013, 19, 8058–8062 CrossRef CAS PubMed.
- D.-H. Ren, D. Qiu, C.-Y. Pang, Z. Li and Z.-G. Gu, Chem. Commun., 2015, 51, 788–791 RSC.
- L. Li, N. Saigo, Y. Zhang, D. J. Fanna, N. D. Shepherd, J. K. Clegg, R. Zheng, S. Hayami, L. F. Lindoy, J. R. Aldrich-Wright, C.-G. Li, J. K. Reynolds, D. G. Harman and F. Li, J. Mater. Chem. C, 2015, 3, 7878–7882 RSC.
- N. Struch, C. Bannwarth, T. K. Ronson, Y. Lorenz, B. Mienert, N. Wagner, M. Engeser, E. Bill, R. Puttreddy, K. Rissanen, J. Beck, S. Grimme, J. R. Nitschke and A. Lützen, Angew. Chem., Int. Ed., 2017, 56, 4930–4935 CrossRef CAS PubMed.
- L. Li, A. R. Craze, O. Mustonen, H. Zenno, J. J. Whittaker, S. Hayami, L. F. Lindoy, C. E. Marjo, J. K. Clegg, J. R. Aldrich-Wright and F. Li, Dalton Trans., 2019, 48, 9935–9938 RSC.
- H.-S. Lu, W.-K. Han, X. Yan, Y.-X. Xu, H.-X. Zhang, T. Li, Y. Gong, Q.-T. Hu and Z.-G. Gu, Dalton Trans., 2020, 49, 4220–4224 RSC.
- M. Hardy, J. Tessarolo, J. J. Holstein, N. Struch, N. Wagner, R. Weisbarth, M. Engeser, J. Beck, S. Horiuchi, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2021, 60, 22562–22569 CrossRef CAS PubMed.
- W. Li, C. Liu, J. Kfoury, J. Oláh, K. Robeyns, M. L. Singleton, S. Demeshko, F. Meyer and Y. Garcia, Chem. Commun., 2022, 58, 11653–11656 RSC.
- J. Zheng, L. K. S. von Krbek, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2022, 61, e202212634 CrossRef CAS PubMed.
- H. Min, A. R. Craze, M. J. Wallis, R. Tokunaga, T. Taira, Y. Hirai, M. M. Bhadbhade, D. J. Fanna, C. E. Marjo, S. Hayami, L. F. Lindoy and F. Li, Chem. – Eur. J., 2023, 29, e202203742 CrossRef CAS PubMed.
- K. H. Sugiyarto and H. A. Goodwin, Aust. J. Chem., 1987, 40, 775–783 CrossRef CAS.
- S. G. Telfer, B. Bocquet and A. F. Williams, Inorg. Chem., 2001, 40, 4818–4820 CrossRef CAS PubMed.
- T. Lathion, L. Guénée, C. Besnard, A. Bousseksou and C. Piguet, Chem. – Eur. J., 2018, 24, 16873–16888 CrossRef CAS PubMed.
- M. Lehr, T. Paschelke, V. Bendt, A. Petersen, L. Pietsch, P. Harders and A. J. McConnell, Eur. J. Org. Chem., 2021, 2728–2735 CrossRef CAS.
- K. Rissanen, Chem. Soc. Rev., 2017, 46, 2638–2648 RSC.
- M. J. Burke, G. S. Nichol and P. J. Lusby, J. Am. Chem. Soc., 2016, 138, 9308–9315 CrossRef CAS PubMed.
- R. G. Siddique, K. S. A. Arachchige, H. A. Al-Fayaad, J. C. McMurtrie and J. K. Clegg, Dalton Trans., 2022, 51, 12704–12708 RSC.
- Q. Shi, X. Zhou, W. Yuan, X. Su, A. Neniškis, X. Wei, L. Taujenis, G. Snarskis, J. S. Ward, K. Rissanen, J. de Mendoza and E. Orentas, J. Am. Chem. Soc., 2020, 142, 3658–3670 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- S. De, S. Tewary, D. Garnier, Y. Li, G. Gontard, L. Lisnard, A. Flambard, F. Breher, M.-L. Boillot, G. Rajaraman and R. Lescouëzec, Eur. J. Inorg. Chem., 2018, 2018, 414–428 CrossRef CAS.
- W. Kläui, W. Eberspach and P. Gütlich, Inorg. Chem., 1987, 26, 3977–3982 CrossRef.
- M. Lehr, T. Paschelke, E. Trumpf, A.-M. Vogt, C. Näther, F. D. Sönnichsen and A. J. McConnell, Angew. Chem., Int. Ed., 2020, 59, 19344–19351 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- N. Ousaka, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 1464–1468 CrossRef CAS PubMed.
- K. P. Kepp, Inorg. Chem., 2016, 55, 2717–2727 CrossRef CAS PubMed.
- W. Meng, J. K. Clegg, J. D. Thoburn and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 13652–13660 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2207255 (for 18), 2207256 (for cage 5) and 2207257 (for cage 7). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01569f |
| ‡ Current address: Department of Chemistry and Biology, University of Siegen, Adolf-Reichwein-Strasse 2, Siegen 57068, Germany. E-mail: anna.mcconnell@uni-siegen.de |
| § This proton was chosen to allow comparison over the family of cages and for its proximity to the paramagnetic FeII centres, resulting in large chemical shift changes without significant line broadening (relative to other protons) over the temperature range. For cages 9 and 11, similar T1/2 values were determined from fitting the chemical shift data for other protons close to the metal centre (ESI, sections 5.11 & 5.13†). |
| ¶ In the Evans method, it is often possible to obtain T1/2 values from data where there is a relatively small change of the spin-state fraction within the measured temperature range. This is because the maximum χmT value can be fixed to values reported in the literature for Fe(II) complexes during the fitting to the regular solution model. This is not possible in fitting data to the ideal solution model since the chemical shift of the high spin state (δHS) is not known. |
| This journal is © The Royal Society of Chemistry 2023 |