
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and catalytic activity of well-defined Co(I) complexes based on NHC–phosphane pincer ligands†
Ana
Luque-Gómez
 ,
Pilar
García-Orduña
,
Fernando J.
Lahoz
and
Manuel
Iglesias
*
,
Pilar
García-Orduña
,
Fernando J.
Lahoz
and
Manuel
Iglesias
*
Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, C/Pedro Cerbuna 12, 50009-Zaragoza, Spain. E-mail: miglesia@unizar.es
First published on 15th August 2023
Abstract
A new methodology for the preparation of Co(I)–NHC (NHC = N-heterocyclic carbene) complexes, namely, [Co(PCNHCP)(CO)2][Co(CO)4] (1) and [Co(PCNHCP)(CO)2]BF4 (2), has been developed (PCNHCP = 1,3-bis(2-(diphenylphosphanyl)ethyl)-imidazol-2-ylidene). Both complexes can be straightforwardly prepared by direct reaction of their parent imidazolium salts with the Co(0) complex Co2(CO)8. Complex 1 efficiently catalyses the reductive amination of furfural and levulinic acid employing silanes as reducing agents under mild conditions. Furfural has been converted into a variety of secondary and tertiary amines employing dimethyl carbonate as the solvent, while levulinic acid has been converted into pyrrolidines under solventless conditions. Dehydrocoupling of the silane to give polysilanes has been observed to occur as a side reaction of the hydrosilylation process.
Introduction
The switch from petrochemicals to renewable bio-based products is pivotal for successful transition towards a circular economy.1 However, the transformation of biomass feedstocks into value-added products also requires the development of sustainable processes that avoid the generation of waste and the use of energy-extensive and cost-ineffective reactions.2 In this regard, catalysis is at the core of modern chemical synthesis, being a fundamental pillar of green and sustainable chemistry.3Earth-abundant metals (EAMs), compared to platinum group metals (PGMs), are readily available and inexpensive, and usually have a relatively lower environmental impact. In consequence, the development of homogeneous catalysts based on EAMs has undergone remarkable revitalization over the last decade.4 Among them, cobalt catalysts have shown excellent performances in a broad variety of reactions,5 for example, in the reduction of organic substrates via hydrogenation,6 transfer hydrogenation,7 hydrosilylation,8,9 reductive amination10 or hydroboration.11
Co(I) complexes have often been proposed as the active species in cobalt-catalysed processes;12,11b however, convenient synthetic methods for the preparation of well-defined Co(I) complexes are scarce.13 In particular, the synthesis of NHC–Co(I) complexes (NHC = N-heterocyclic carbene) has been described by the reaction of the free NHC ligand with [Co(PPh3)3Cl],14 by the reduction of NHC–Co(II) complexes,15 or by the oxidation of NHC–Co(0) complexes with 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO).16 The limitations of these synthetic methodologies have hampered the preparation of NHC–Co(I) organometallic complexes, which are promising homogeneous catalysts owing to the unique properties of NHCs and their success story as ancillary ligands in catalysis.17
Levulinic acid (LA) and furanics are key bio-based building blocks that can be easily obtained from the hexoses in lignocellulosic biomass.18 The catalytic reductive amination reaction shows promise as a sustainable method for the functionalization of biomass, which allows the synthesis of amines directly from carbonyl compounds (ketones, aldehydes or carboxylic acids).19 However, examples of the cobalt-catalyzed reductive amination of carbonyl compounds employing hydrosilanes as reducing agents are scarce.10 In fact, homogeneous cobalt catalysts have been extensively explored in the hydrosilylation of alkenes, alkynes and, to a lesser extent, carbonyl compounds;9 however, imines have been widely overlooked.20 An interesting example of the reductive amination of carbonyl compounds promoted by homogeneous cobalt catalysts has been reported by Beller and Jagadeesh.10c
The valorisation of levulinic acid via reductive amination may afford pyrrolidinones or pyrrolidines by the ensuing reduction of the carbonyl group. Most of the homogeneous catalysts reported for these transformations are based on PGMs.21 The only examples of EAM catalysts for the preparation of pyrrolidines are complexes [Fe(Cp*)(IMes)(CO)2]I and [Fe(IMes)(CO)4] (Cp* = 1,2,3,4,5-pentamethylcyclopentadiene; IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene), which operate in the presence of 6 equiv. of PhSiH3 and visible light irradiation at 100 °C.22 In this regard, it is worth mentioning that the N-alkylation of a variety of carboxylic acids—including LA—with silanes as reducing agents and B(C6F5)3 as a catalyst has been also described.23
We envisaged that the use of a PCP ligand containing a central NHC moiety (PCNHCP), recently employed by us for the synthesis of Ir(I) complexes,24 would provide an interesting new class of stable, well-defined NHC–Co(I) catalysts. The novel Co(I) complexes prepared herein were studied as catalysts for the reductive amination of bio-based platform chemicals, namely, furfural and levulinic acid.
Results and discussion
Synthetic procedures via free-NHC or transmetallation from silver–NHC complexes proved to be unsuccessful for the synthesis of cobalt(I) complexes with our PCNHCP ligand. Therefore, we explored the direct reaction of the parent imidazolium salt PCNHCP·HX (X = Cl or BF4) with 0.5 equiv. of [Co2(CO)8]. We reasoned that oxidative addition of the C2–H bond of the imidazolium salt would afford a transient PCNHCP–Co(II) hydride, which upon release of 0.5 equiv. of H2 would render the desired PCNHCP–Co(I) complex, analogously to the reductions of Co(II) halides with K(HBEt3) as reported by Gomes and co-workers.25Both imidazolium salts, PCNHCP·HX (X = Cl or BF4), were initially reacted with 0.5 equiv. of [Co2(CO)8] in THF at room temperature (Scheme 1). The reactions were monitored by 31P NMR spectroscopy, showing complete consumption of PCNHCP·HCl and intermediate species after 24 h. In contrast, in the case of the BF4 salt, the crude product needs to be refluxed in acetonitrile for the peaks of the intermediates to disappear in the 31P NMR spectrum. To our surprise, in the case of PCNHCP·HCl, the Co(I)/Co(–I) complex [Co(κ3-P,C,P′-PCNHCP)(CO)2][Co(CO)4] (1) was obtained, while the reaction with the BF4 salt afforded the Co(I) complex [Co(κ3-P,C,P′-PCNHCP)(CO)2]BF4 (2).
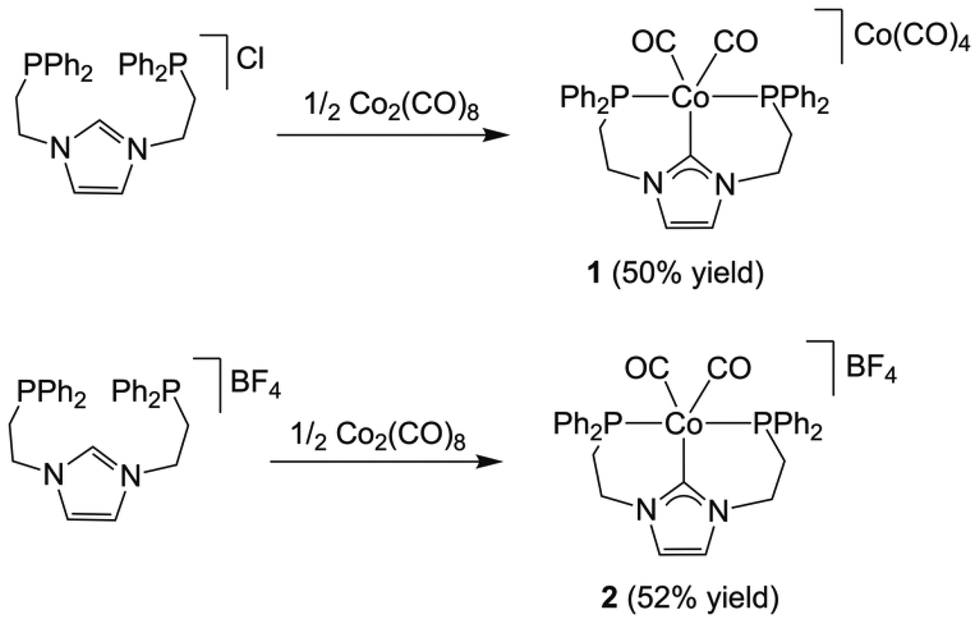 | ||
| Scheme 1 Synthetic route for the preparation of 1 and 2. Reaction conditions: X = Cl: THF, room temperature, 1 d. X = BF4: THF, room temperature, 1 d; followed by CH3CN, 80 °C, 3 d. | ||
The gradual diffusion of hexane into a solution containing complex 1 in THF allowed us to obtain crystals suitable for single-crystal X-ray diffraction. The cation displays a slightly distorted trigonal bipyramidal geometry with the κ3-P,C,P′-PCNHCP ligand showing a mer coordination with a P(1)–Co(1)–P(2) angle of 165.820(19)°. The two phosphane moieties are situated at the axial positions, while the NHC and carbonyl ligands occupy the equatorial plane. The NHC ring N(1)–C(1)–N(2)–C(17)–C(16) forms an angle of 39.46(7)° with the equatorial plane Co(1)–C(1)–C(33)–C(32), which situates the wingtip groups above and below the NHC plane. Both six-membered chelate rings, C(1)–Co(1)–P(2)–C(19)–C(18)–N(2) and C(1)–Co(1)–P(1)–C(14)–C(15)–N(1), present a highly deviated from planarity 1,4B boat conformation (Fig. 1).26
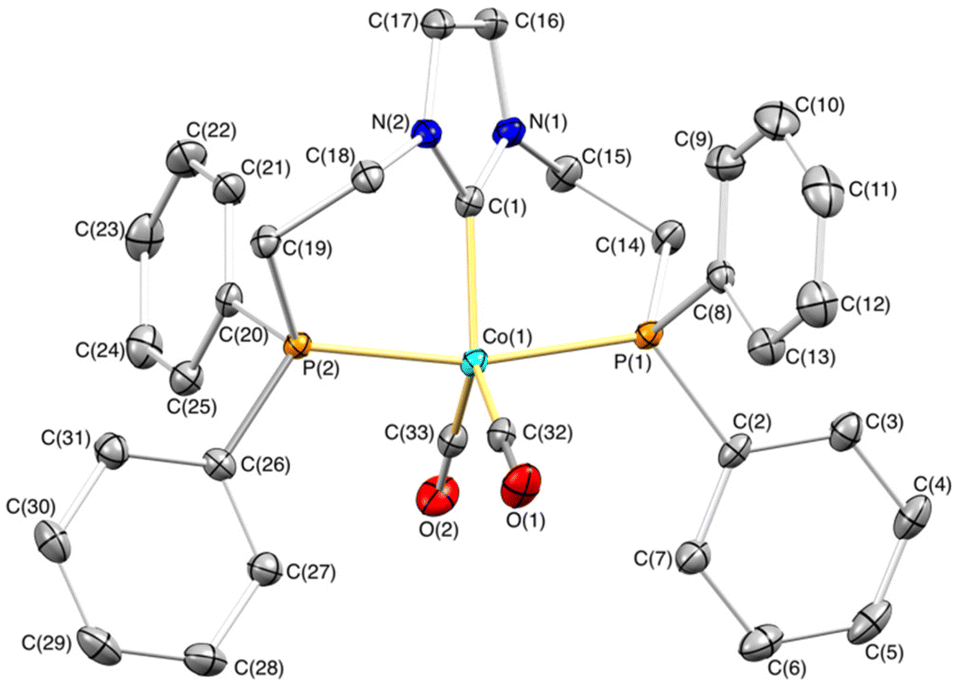 | ||
| Fig. 1 Molecular structure of cationic complex 1. Hydrogen atoms, solvent molecules and the [Co(CO)4]− counterion were omitted for clarity. Selected bond lengths (Å) and angles (°): Co(1)–P(1) 2.1868(5), Co(1)–P(2) 2.1796(5), Co(1)–C(1) 1.9502(17), Co(1)–C(32) 1.7724(18), Co(1)–C(33) 1.7738(18), P(1)–Co(1)–C(32) 92.11(6), P(1)–Co(1)–C(33) 95.45(5), P(2)–Co(1)–C(1) 82.78(5), P(2)–Co(1)–C(32) 94.21(6), P(2)–Co(1)–C(33) 92.83(5), P(1)–Co(1)–P(2) 165.820(19), C(1)–Co(1)–C(32) 121.56(7), P(1)–Co(1)–C(1) 83.09(5), C(1)–Co(1)–C(33) 120.78(8). | ||
Complexes 1 and 2 are both diamagnetic, which allowed full characterisation by NMR spectroscopy.
The 1H NMR spectrum of 1 shows the disappearance of the resonance that corresponds to imidazolium's C2 proton and the emergence of two multiplets at δ 4.26–4.11 and 3.05–2.93 ppm that correspond to the methylenic protons of the NCH2 and PCH2 moieties, respectively. The 13C{1H} NMR spectrum of 1 confirms the κ3-P,C,P′ coordination of the PCNHCP ligand to the cobalt centre, as well as the presence of a carbonyl ligand. The carbene carbon shows a triplet at δ 174.0 ppm (2JCP = 38.1 Hz) due to the coupling with the two 31P nuclei of the wingtip groups. The CO ligands also appear as a triplet at δ 198.6 ppm (2JCP = 23.6 Hz). The 31P{1H} NMR features a singlet resonance at δ 59.1 ppm, thus suggesting the presence of a symmetry plane that renders both P nuclei equivalent.
In order to gain mechanistic insight into the formation of 1, we monitored the reaction of PCNHCP·HCl with 0.5 equiv. of Co2(CO)8 in THF-d8 at 298 K by 31P{1H} NMR (Fig. 2). After 1 h, the 31P{1H} NMR spectrum shows a broad singlet at ca. δ −19 ppm, and two broad multiplets between δ 45 and 57 ppm. At this point, also the incipient formation of 1 is observed as a sharp singlet at δ 59.2 ppm. The broad resonances suggest that, initially, a mixture of complexes in equilibrium with each other is formed. These initial species can be assigned to a Co(I)/Co(–I) dinuclear complex ([Co(κ2-P,P′-PCNHCP·HCl)(CO)n]2) or a [Co(κ2-P,P′-PCNHCP·HCl)2][Co(CO)4] cationic complex.27 Subsequently, these species plausibly evolve to give the Co(I)/Co(–I) complex 1 and paramagnetic Co species.
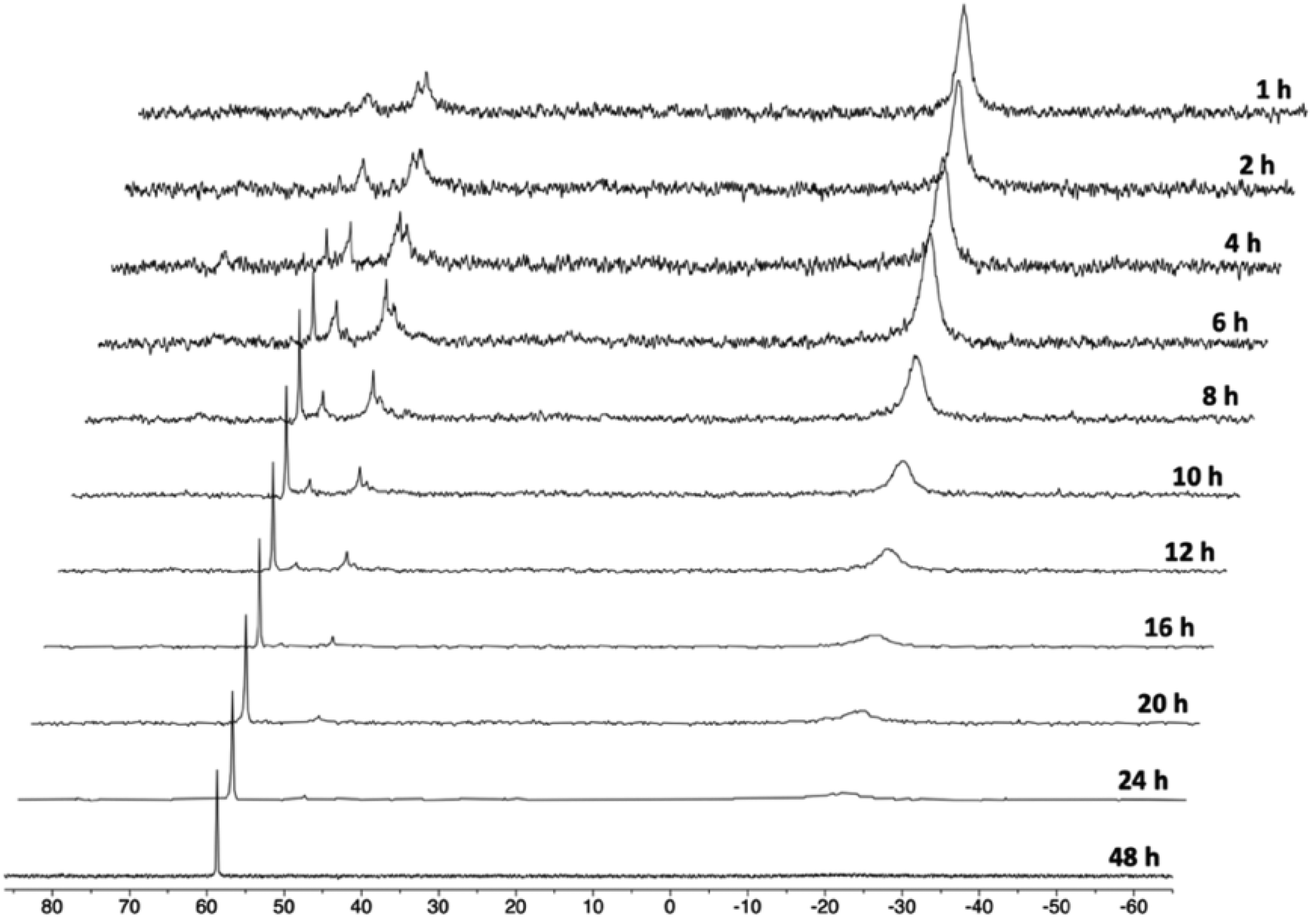 | ||
| Fig. 2 31P{1H} NMR monitoring of the formation of 1. | ||
The 1H, 13C{1H} and 31P{1H} NMR spectra of 2 in CD3CN show no remarkable differences compared with those described above for 1.
High-resolution electrospray ionization mass spectrometry (HR-ESI-MS) of 1 and 2 shows the cations [Co(PCNHCP)(CO)]+ and [Co(PCNHCP)]+ as main species. The FT-IR spectrum of 1 shows two strong bands at 1991 and 1935 cm−1, which are consistent with the presence of two CO ligands,28 and an intense band at 1882 cm−1 that can be attributed to the [Co(CO)4]− anion.13 In contrast, complex 2 shows only two strong bands at 1989 and 1930 cm−1 that correspond to the two carbonyl ligands of the Co(I) cation (Fig. 3). The presence of the BF4− counterion is further supported by a broad band at 1000–1100 cm−1 in the FTIR spectrum and a resonance at δ −151.2 ppm in the 19F NMR spectrum.
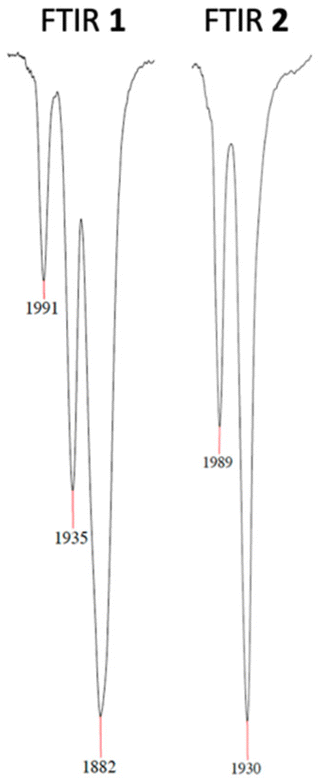 | ||
| Fig. 3 ν(CO) vibration bands in the FTIR spectra of complexes 1 and 2. | ||
Conductometric determinations of solutions of 1 and 2 in CH3CN (5 × 10−4 M) at room temperature revealed molar conductivities of 45 and 50 S mol−1 cm−1, respectively, which are compatible with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolyte.
:1 electrolyte.
The lability of the second carbonyl ligand has been reported for related complexes,28a,29 which may lead to the formation of an unsaturated species under catalytic conditions. In order to explore the catalytic activity of complexes 1 and 2, initially, they were tested as catalysts in the reductive amination of furfural (FAL). The reaction was performed in dimethylcarbonate (DMC), which is considered a green solvent, mainly due to its low volatility, good biodegradability and low toxicity (Scheme 2 and Table 1).30 The one-pot reaction of p-toluidine with FAL with PhSiH3 as the reducing agent and 5 mol% of 1 (entry 1) at 80 °C led to an 88% yield of silylamine. The yield was improved by using a sequential process that entails the reaction of p-toluidine with FAL in the presence of 1 (5 mol%) for 1 h at 80 °C followed by the addition of PhSiH3 (entry 2). It must be noted that the use of PhSiH3 leads to the formation of a mixture of aminosilanes, diaminosilanes, and oligomers, except in the case of secondary amines.
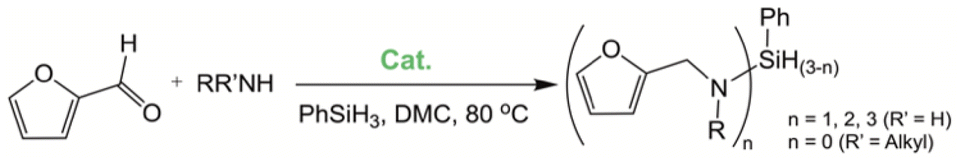 | ||
| Scheme 2 Reductive amination of furfural in DMC employing PhSiH3 as a reducing agent. | ||
| Entry | Amine | Time (h) | Conversione (%) |
|---|---|---|---|
| a Reaction conditions: 0.125 mmol of amine, 0.125 mmol of furfural and 0.25 mmol of PhSiH3, 5 mol% of 1, at 80 °C in DMC. b One pot reaction. c 5 mol% of 2. d 1 mol% of 1. e Conversions were determined by 1H NMR spectroscopy of the crude material. | |||
| 1b |
|
23 | 88 |
| 2 |
|
20 | >99 |
| 3c |
|
20 | 53 |
| 4d |
|
20 | 56 |
| 5 |

|
20 | 82 |
| 6 |
|
20 | 80 |
| 7 |
|
20 | 64 |
| 8 |
|
20 | 50 |
| 9 |
|
20 | 73 |
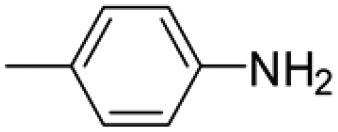
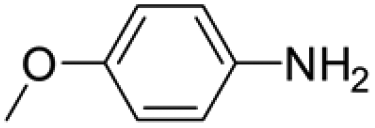
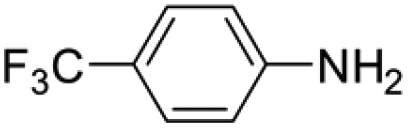
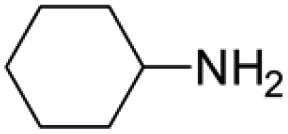
Under analogous conditions, catalyst 2 leads to a noticeably lower yield (entry 3). The higher activity of 1 can be ascribed to the presence of the Co(CO)4− counterion since this is the only structural difference between both complexes. A decrease in the loading of 1 to 1 mol% also results in a significant drop in the catalytic activity (entry 4). Therefore, other amines were also tested under the conditions described above for entry 2. The use of aniline or p-anisidine results in yields of ca. 80% (entries 5 and 6), while in the case of 4-(trifluoromethyl)aniline, the yield drops to 64% (entry 7). Longer reaction times for the formation of the imine led to similar yields. Cyclohexylamine was explored as a substrate in order to expand the scope of the reaction to aliphatic amines, resulting in a 50% yield (entry 8). It is noteworthy that secondary aliphatic amines readily react under analogous conditions to afford the corresponding tertiary amines in good yields (entry 9). In order to probe the synthetic utility of this methodology, the reaction of furfural with p-toluidine (under conditions analogous to those described in entry 2, Table 1) was scaled up to 2 mmol of the substrate. After purification, a 79% yield was obtained for the synthesis of N-(furan-2-ylmethyl)-4-methylaniline.
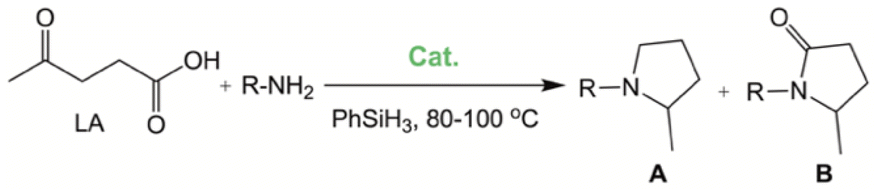
The results described above and the dearth of suitable EAM catalysts for the synthesis of pyrrolidines by reductive amination of LA prompted us to study the catalytic activity of complexes 1 and 2 in these reactions (Scheme 3). The optimization of reaction conditions was explored by employing p-toluidine under solventless conditions and 1 as the catalyst. First, reaction conditions similar to those reported by Darcel and co-workers were used,22i.e., 6 equiv. of PhSiH3, 5 mol% of 1, 100 °C for 20 h; however, in our case, visible light irradiation was not required. Quantitative formation of the corresponding pyrrolidine was observed, which prompted us to evaluate the activity of 1 under milder conditions. Quantitative yields of pyrrolidine (A) were also obtained at shorter reaction times, namely, after 5 h (Table 2; entry 1). The reduction of the amount of PhSiH3 to 2 equiv. also leads to the exclusive formation of A in quantitative yields at 80 °C (Table 2; entry 2). It is worth mentioning that, under analogous conditions, the use of 2 as a catalyst leads to a 42% yield, which is consistent with the reactivity trend described previously for the reductive amination of furfural. Finally, we evaluated the influence of catalyst loading. The use of 1 mol% of 1 also allows quantitative yields of A at 80 °C in 5 h, but 6 equiv. of PhSiH3 is required (Table 2; entry 3). Under these conditions, 4-methoxyaniline also brings about quantitative yields (Table 2; entry 4). However, aniline gives rise to lower conversions and selectivities (Table 2; entry 5), but total conversion was achieved by increasing the reaction time to 20 h (Table 2; entry 6). The presence of an electron-attracting group in aniline, such as CF3, results in a significant drop in the catalytic activity in the case of 4-trifluoromethylaniline (Table 2; entry 7). In this case, a mixture of A, B and other reaction intermediates is observed after 20 h at 100 °C. The reaction was scaled up to 1.35 mmol of levulinic acid to probe the synthetic value of this reaction, employing 1 equivalent of p-toluidine (under conditions analogous to those described in entry 1, Table 2), leading to an 87% yield of 2-methyl-1-(p-tolyl)pyrrolidine after purification.
 | ||
| Scheme 3 Solventless reductive amination of levulinic acid to pyrrolidine or pyrrolidinone with PhSiH3 as a reducing agent. | ||
| Entry | Amine | Equiv. PhSiH3 | Loading (mol %) | T (°C) | Time (h) | Conversion % (A/B ratio)b |
|---|---|---|---|---|---|---|
| a Reaction conditions: 0.25 mmol of levulinic acid, 0.25 mmol of amine, 1 mol% of 1, at 80 °C neat. b Conversions were determined by 1H NMR spectroscopy of the crude material. | ||||||
| 1 |
|
6 | 5 | 100 | 5 | >99 (100/0) |
| 2 |
|
2 | 5 | 80 | 5 | >99 (100/0) |
| 3 |
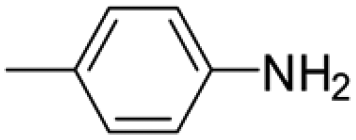
|
6 | 1 | 80 | 5 | >99 (100/0) |
| 4 |
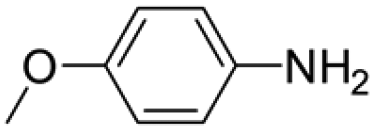
|
6 | 1 | 80 | 5 | >99 (100/0) |
| 5 |
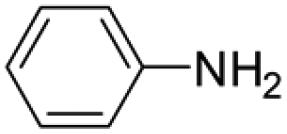
|
6 | 1 | 80 | 5 | 83 (70/30) |
| 6 |
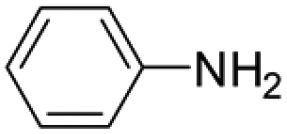
|
6 | 1 | 80 | 20 | >99 (100/0) |
| 7 |
|
6 | 5 | 100 | 20 | 12 (—) |
To explore the stability of the catalyst (1), the crude mixtures of all the catalytic experiments were studied by 31P{1H} NMR spectroscopy at the end of the reaction, showing exclusively one peak at δ 59.2 ppm in all cases, which corresponds to the shift observed for 1 in acetonitrile. However, this resonance could be attributed to the presence of an unreacted catalyst at the end of the reaction. Consequently, stoichiometric experiments between 1 and PhSiH3 were carried out in a Young NMR tube to shed light on the nature of the active species. Complex 1 was reacted with 2 equivalents of PhSiH3 in THF-d8, showing moderate gas evolution at 60 °C. After 24 h at 60 °C, the 31P{1H} NMR spectrum (Fig. S32†) shows the peak characteristic of 1 at δ 59.2 ppm, together with two broad peaks in a 1:1 ratio at δ 36.2 and 33.2 ppm, as the main species in solution. These two resonances plausibly correspond to a complex where the PCNHCP ligand adopts a facial geometry,31 which makes both phosphorous nuclei inequivalent.32 A sharp singlet at δ 26.2 ppm and a broad multiplet at δ −20.3 ppm are also observed as minor products. The 1H NMR spectrum shows no hydride peak attributable to the oxidative addition of the Si–H bond, but it confirms the presence of 1 accompanied by a mixture of other diamagnetic species. Remarkably, a sharp singlet due to H2 formation appears at δ 4.57 ppm, which suggests that acceptorless dehydrogenative coupling of the silane is taking place (Fig. S33†).33 This reactivity agrees with the fact that the 1H NMR spectra of the catalytic reactions show the formation of polysilanes via dehydrocoupling of PhSiH3. This was confirmed by the 19Si–1H HMBC spectrum of the reaction mixture of the reductive amination of levulinic acid with p-toluidine (Table 2, entry 3), which shows a correlation between the resonances at 5.37–5.21 in the 1H NMR spectrum and a peak at ca. δ −28 ppm in the 19Si NMR spectrum (Fig. S36†). The 29Si DEPT spectrum shows two groups of resonances, from δ −20 to −29 ppm and from δ −40 to −52 ppm, that correspond to the different dehydrocoupling products formed as a by-product of the hydrosilylation reaction (Fig. S37†). Moreover, it is noteworthy that a white precipitate, attributable to the formation of a polymer, forms at long reaction times. An analogous experiment was performed in CD3CN at 80 °C, showing comparable results. However, in this case, the 1H and 31P{1H} NMR spectra show 1 as the main species throughout the reaction, which allows the sharp singlet of H2 at δ 4.55 ppm to be observed in a cleaner region (Fig. S34 and S35†). Homogeneous catalysts based on other metal centres have been reported for the dehydrocoupling of PhSiH3 to give polysilanes;34 however, to the best of our knowledge, this is the first example of a cobalt catalyst for this transformation.
Hydride resonances are not observed by 1H NMR spectroscopy, but it is conceivable that transient hydride species are formed during the reaction. In this regard, the most widely accepted mechanism for the dehydrocoupling of PhSiH3 has been reported to require the formation of a metal hydride, which reacts with the silane via σ-bond metathesis to give the silyl intermediate and H2. Subsequently, Si–Si bond formation occurs by reaction of the silyl intermediate with a new molecule of silane, again via a σ-bond metathesis step.35 However, it must be noted that this mechanism has been proposed for Ti and Zr catalysts.
Ionic outer-sphere mechanisms are frequently observed in silane-mediated reductions and do not entail the oxidative addition of the Si–H bond. Consequently, it can be postulated that the aforementioned reactions may proceed via this class of mechanisms. The seminal work on these types of mechanisms by Piers,36 Brookhart37 and Bullock38 has triggered the development of a myriad of new hydrosilylation catalysts. Noteworthy precedents of this type of catalyst have been reported by the groups of Brookhart,39 Nikonov,40 Oestreich41 and others.42 A tentative reaction mechanism for the hydrosilylation of imines with 1 would entail the end-on coordination of PhSiH3 to the cation [Co(κ3-P,C,P′-PCNHCP)(CO)]+, followed by heterolytic splitting of the Si–H bond assisted by the imine or the [Co(CO)4]− ion. Bifunctional catalysts based on the Lewis base characteristics of the latter have been reported; namely, the cooperative ring opening of epoxides by a Lewis acid metal centre and a [Co(CO)4]− counterion acting as a Lewis base has been proposed as a key step in epoxide functionalization.43 The heterolytic splitting of the Si–H bond results in the formation of a Co-hydride species and a silyl iminium cation, either directly or via a [Co(CO)4]− shuttle. Finally, hydride transfer furnishes the hydrosilylation product, regenerating the active species. However, detailed mechanistic studies are required to confirm this postulation.
Conclusions
In summary, we have developed a new methodology for the preparation of NHC–Co(I) complexes directly from their parent imidazolium salts. Remarkably, the nature of the counterion of the imidazolium salt plays an important role in the selectivity of the reaction, with PCNHCP·HCl affording the Co(I)/Co(–I) complex [Co(κ3-P,C,P′-PCNHCP)(CO)2][Co(CO)4] (1), and PCNHCP·HBF4 affording the Co(I) complex [Co(κ3-P,C,P′-PCNHCP)(CO)2]BF4 (2). Complex 1 shows promise as a catalyst for reductive amination reactions employing PhSiH3 as a reducing agent; however, the use of 2 results in markedly worse performances under analogous conditions. The higher activity of 1 seems to be originated by the presence of the [Co(CO)4]− counterion. Furfural was converted into a variety of secondary and tertiary amines, in DMC, by reaction with aromatic and aliphatic primary amines, and aliphatic secondary amines. Moreover, levulinic acid was converted into pyrrolidines under mild, solventless conditions using a 1 mol% loading of 1, this being the first example of a cobalt complex to catalyse this transformation. Moreover, it is remarkable that 1 also catalyses the dehydrocoupling of PhSiH3, with polysilanes being observed as reaction by-products. Finally, we believe that the synthetic methodology described here provides useful information for the preparation of novel, well-defined NHC–Co(I) catalysts.Experimental
General methods
All experiments and manipulations were carried out under an argon atmosphere using standard Schlenk techniques or in an argon-filled glove box. The solvents were dried by known procedures and distilled under argon prior to use or obtained oxygen- and water-free from a solvent purification system (Innovative Technologies). Deuterated solvents were dried over molecular sieves or calcium hydride (THF-d8, CDCl3 and CD3CN). All other commercially available starting materials were used without further purification. 1H, 13C{1H}, 31P{1H} and 19F spectra were recorded either on a Bruker ARX 300 MHz or a Bruker Avance 400 MHz instrument. Chemical shifts (expressed in parts per million) are referenced to residual solvent peaks (1H, 13C{1H}). Coupling constants, J, are given in Hz. Spectral assignments were achieved by a combination of 1H–1H COSY, 13C APT and 1H–13C HSQC/HMBC experiments. High-resolution electrospray mass spectra (HRMS) were acquired using a MicroTOF-Q hybrid quadrupole time-of-flight spectrometer (Bruker Daltonics, Bremen, Germany). Attenuated total reflection infrared spectra (ATR-IR) of solid samples were recorded on a PerkinElmer Spectrum 100 FT-IR spectrometer.General procedure for the catalytic reductive amination of furfural
To a Schlenk flask equipped with a magnetic stir bar, furfural (10 μL, 0.125 mmol), p-toluidine (13.4 mg, 0.125 mmol), 5 mol% of the catalyst 1 (3.9 mg, 0.0063 mmol) and 1 mL of DMC as solvent were added. The resulting solution was stirred at 80 °C for 1 h. Afterwards, 2 equivalents of PhSiH3 (31 μL, 0.25 mmol) was added under argon and stirred again at 80 °C for 20 h. The resulting solution was evaporated under reduced pressure and the conversion was determined by 1H NMR spectroscopy of the crude material in CDCl3.44General procedure for the catalytic reductive amination of levulinic acid
To a Schlenk flask equipped with a magnetic stir bar, levulinic acid (26 μL, 0.25 mmol), p-toluidine (26.8 mg, 0.25 mmol), 1 mol% of the catalyst 1 (3.9 mg, 0.0026 mmol) and 6 equivalents of PhSiH3 (185 μL, 1.5 mmol) were added under argon. To the mixture thus obtained, mesitylene (6 μL, 0.04 mmol) was added as an internal standard and the solution was stirred at 80 °C for 5 h. CDCl3 was added to an aliquot and measured by 1H NMR to determine the yield of the reaction.45:90) mixture. After the collection and concentration under reduced pressure, the resulting amber-yellow oil was obtained in 79% yield (296 mg, 1.58 mmol). The NMR spectra matched those previously reported in the literature.42a
:90) mixture. After the collection and concentration under reduced pressure, the resulting amber oil was obtained in 87% yield (207 mg, 1.18 mmol). The NMR spectra matched those previously reported in the literature.45a
620 reflections measured, 10813 unique; Rint = 0.0516; number of data/restraint/parameters 10813/0/532; R1 = 0.0361 [9320 ref. with I > 2σ(I)], wR(F2) = 0.0954 (all data); largest difference peak 0.646 e·Å−3.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grants RTI2018-099136-A-I00 and PID2021-126212OB-I00 MCIN/AEI/10.13039/501100011033 funded by “ERDF A way of making Europe”, as well as E42_23R, are gratefully acknowledged. The authors would like to acknowledge the use of “Servicio General de Apoyo a la Investigación-SAI” at the Universidad de Zaragoza and at the ISQCH/CEQMA (CSIC).References
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- H. Clark, T. J. Farmer, L. Herrero-Davila and J. Sherwood, Green Chem., 2016, 18, 3914–3934 RSC.
- (a) R. A. Sheldon, J. R. Soc., Interface, 2016, 13, 20160087 CrossRef PubMed; (b) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- (a) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed; (b) M. Albrecht, R. Bedford and B. Plietker, Organometallics, 2014, 33, 5619–5621 CrossRef CAS; (c) P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS PubMed.
- (a) P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194–1206 CrossRef CAS PubMed; (b) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed; (c) B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS.
- (a) S. W. Anferov, A. S. Filatov and J. S. Anderson, ACS Catal., 2022, 12(16), 9933–9943 CrossRef CAS PubMed; (b) L. N. Mendelsohn, C. S. MacNeil, L. Tian, Y. Park, G. D. Scholes and P. J. Chirik, ACS Catal., 2021, 11, 1351–1360 CrossRef CAS; (c) P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS; (d) V. Papa, J. R. Cabrero-Antonino, A. Spannenberg, K. Junge and M. Beller, Catal. Sci. Technol., 2020, 10, 6125–6137 RSC; (e) S. Lapointe, D. K. Pandey, J. M. Gallagher, J. Osborne, R. R. Fayzullin, E. Khaskin and J. R. Khusnutdinova, Organometallics, 2021, 40(21), 3617–3626 CrossRef CAS; (f) H. Alawisi, H. D. Arman and Z. J. Tonzetich, Organometallics, 2021, 40(8), 1062–1070 CrossRef CAS; (g) T. P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS PubMed.
- (a) G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC; (b) S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS; (c) M. Pang, J.-Y. Chen, S. Zhang, R.-Z. Liao, C.-H. Tung and W. Wang, Nat. Commun., 2020, 11, 1249 CrossRef CAS PubMed; (d) R. V. Jagadeesh, D. Banerjee, P. B. Arockiam, H. Junge, K. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, Green Chem., 2015, 17, 898–902 RSC; (e) W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2018, 119, 2876–2953 CrossRef PubMed.
- (a) C. Chen, M. B. Hecht, A. Kavara, W. W. Brennessel, B. Q. Mercado, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13244–13247 CrossRef CAS PubMed; (b) W. J. Teo, C. Wang, Y. W. Tan and S. Ge, Angew. Chem., Int. Ed., 2017, 56, 4328–4332 CrossRef CAS; (c) B. Raya, S. Biswas and T. V. RajanBabu, ACS Catal., 2016, 6, 6318–6323 CrossRef CAS PubMed; (d) Y. Gao, L. Wang and L. Deng, ACS Catal., 2018, 8, 9637–9646 CrossRef CAS.
- Examples for the hydrosilylation of carbonyl compounds: (a) T. W. Lyons and M. Brookhart, Chem. – Eur. J., 2013, 19, 10124–10127 CrossRef CAS PubMed; (b) Q. Niu, H. Sun, X. Li, H.-F. Klein and U. Flörke, Organometallics, 2013, 32, 5235–5238 CrossRef CAS; (c) Y. Li, J. A. Krause and H. Guan, Organometallics, 2018, 37, 2147–2158 CrossRef CAS; (d) Y. Li, J. A. Krause and H. Guan, Organometallics, 2020, 39, 3721–3730 CrossRef CAS; (e) K. Matsubara, T. Mitsuyama, S. Shin, M. Hori, R. Ishikawa and Y. Koga, Organometallics, 2021, 40, 1379–1387 CrossRef CAS.
- (a) V. Kumar, U. Sharma, P. K. Verma, N. Kumar and B. Singh, Adv. Synth. Catal., 2012, 354, 870–878 CrossRef CAS; (b) C. C. Bories, M. Barbazanges, E. Derat and M. Petit, ACS Catal., 2021, 11, 14262–14273 CrossRef CAS; (c) K. Murugesan, Z. H. Wei, V. G. Chandrashekhar, H. Neumann, A. Spannenberg, H. J. Jiao, M. Beller and R. V. Jagadeesh, Nat. Commun., 2019, 10, 5443 CrossRef PubMed.
- (a) L. Zhang, Z. Zuo, X. Leng and Z. A. Huang, Angew. Chem., Int. Ed., 2014, 53, 2696–2700 CrossRef CAS PubMed; (b) L. Zhang, Z. Zuo, X. Wan and Z. Huang, J. Am. Chem. Soc., 2014, 136, 15501–15504 CrossRef CAS PubMed; (c) J. Guo, B. Cheng, X. Shen and Z. Lu, J. Am. Chem. Soc., 2017, 139, 15316–15319 CrossRef CAS PubMed.
- (a) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef CAS; (b) A. D. Ibrahim, S. W. Entsminger, L. Zhu and A. R. Fout, ACS Catal., 2016, 6, 3589–3593 CrossRef CAS; (c) B. Raya, S. Biswas and T. V. Rajanbabu, ACS Catal., 2016, 6, 6318–6323 CrossRef CAS PubMed; (d) B. Raya, S. Jing, V. Balasanthiran and T. V. Rajanbabu, ACS Catal., 2017, 7, 2275–2283 CrossRef CAS; (e) M. L. Scheuermann, E. J. Johnson and P. J. Chirik, Org. Lett., 2015, 17, 2716–2719 CrossRef CAS PubMed; (f) W. N. Palmer, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2015, 5(2), 622–626 CrossRef CAS; (g) J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135(51), 19107–19110 CrossRef CAS PubMed.
- C. M. Boudreaux, D. Nugegoda, W. Yao, N. Le, N. C. Frey, Q. Li, F. Qu, M. Zeller, C. E. Webster, J. H. Delcamp and E. T. Papish, ACS Catal., 2022, 12, 8718–8728 CrossRef CAS.
- (a) Y. Gao, Q. Chen, X. Leng and L. Deng, Dalton Trans., 2019, 48, 9676–9683 RSC; (b) Z. Mo, Y. Li, H. Lee and L. Deng, Organometallics, 2011, 30, 4687–4694 CrossRef CAS; (c) Z. Mo, J. Xiao, Y. Gao and L. Deng, J. Am. Chem. Soc., 2014, 136, 17414–17417 CrossRef CAS; (d) Y. Meng, Z. Mo, B. Wang, Y. Zhang, L. Deng and S. Gao, Chem. Sci., 2015, 6, 7156–7162 RSC; (e) Z. Mo, D. Chen, X. Leng and L. Deng, Organometallics, 2012, 31, 7040–7043 CrossRef CAS.
- (a) Y. Liu and L. Deng, J. Am. Chem. Soc., 2017, 139, 1798–1801 CrossRef CAS PubMed; (b) A. D. Ibrahim, K. Tokmic, M. R. Brennan, D. Kim, E. M. Matson, M. J. Nilges, J. A. Bertke and A. R. Fout, Dalton Trans., 2016, 45, 9805–9811 RSC.
- S. Takebayashi and R. R. Fayzullin, Organometallics, 2021, 40, 500–507 CrossRef CAS.
- (a) S. P. Nolan, N-Heterocyclic, Carbenes in Synthesis, Wiley-VCH, Weinheim, 2006 CrossRef; (b) S. S. Bera and M. Szostak, ACS Catal., 2022, 12, 3111–3137 CrossRef CAS PubMed.
- (a) R. Weingarten, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2012, 5, 7559–7574 RSC; (b) P. J. Deuss, K. Barta and J. G. de Vries, Catal. Sci. Technol., 2014, 4, 1174–1196 RSC; (c) J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC; (d) Y. W. Tiong, C. L. Yap, S. Gan and W. S. P. Yap, Ind. Eng. Chem. Res., 2018, 57, 4749–4766 CrossRef CAS; (e) Z. Sun and K. Barta, Chem. Commun., 2018, 54, 7725–7745 RSC; (f) P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, ACS Catal., 2012, 2, 1487–1499 CrossRef CAS; (g) R. J. Van Putten, J. C. van der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed; (h) T. Werpy and G. R. Petersen, Top Value Added Chemicals from Biomass, Volume I. Results of Screening for Potential Candidates from Sugars and Synthesis Gas, U.S. Department of Energy; U.S. Government Printing Office, Washington, DC, 2004 Search PubMed.
- (a) J. He, L. Chen, S. Liu, K. Song, S. Yang and A. Riisager, Green Chem., 2020, 22, 6714–6747 RSC; (b) G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050–3054 CrossRef CAS; (c) J. D. Vidal, M. J. Climent, P. Concepcion, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2015, 5(10), 5812–5821 CrossRef CAS; (d) J. J. Roylance and K.-S. Choi, Green Chem., 2016, 18, 5412–5417 RSC; (e) A. Cukalovic and C. V. Stevens, Green Chem., 2010, 12, 1201–1206 RSC; (f) G. Chieffi, M. Braun and D. Esposito, ChemSusChem, 2015, 8, 3590–3594 CrossRef CAS PubMed; (g) M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC; (h) P. D. Pham, P. Bertus and S. Legoupy, Chem. Commun., 2009, 6207–6209 RSC.
- W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2019, 119, 2876–2953 CrossRef CAS PubMed.
- (a) Y. Wei, C. Wang, X. Jiang, D. Xue, J. Li and J. Xiao, Chem. Commun., 2013, 49, 5408–5410 RSC; (b) Z. Sun, J. Chen and T. Tu, Green Chem., 2017, 19, 789–794 RSC; (c) C. Wu, X. Luo, H. Zhang, X. Liu, G. Ji, Z. Liu and Z. Liu, Green Chem., 2017, 19, 3525–3529 RSC; (d) Y. B. Huang, J. J. Dai, X. J. Deng, Y. C. Qu, Q. X. Guo and Y. Fu, ChemSusChem, 2011, 4, 1578–1581 CrossRef CAS PubMed; (e) Z. Xu, P. Yan, H. Jiang, K. Liu and Z. C. Zhang, Chin. J. Chem., 2017, 35, 581–585 CrossRef CAS; (f) S. Wang, H. Huang, C. Bruneau and C. Fischmeister, ChemSusChem, 2017, 10, 4150–4154 CrossRef CAS PubMed.
- D. Wei, C. Netkaew and C. Darcel, Adv. Synth. Catal., 2019, 361, 1781–1786 CrossRef CAS.
- M. C. Fu, R. Shang, W. M. Cheng and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 9042–9046 CrossRef CAS PubMed.
- A. Luque-Gómez, S. García-Abellán, J. Munarriz, V. Polo, V. Passarelli and M. Iglesias, Inorg. Chem., 2021, 60, 15497–15508 CrossRef.
- (a) T. F. C. Cruz, L. F. Veiros and P. T. Gomes, Inorg. Chem., 2018, 57, 14671–14685 CrossRef CAS PubMed; (b) T. F. C. Cruz, P. S. Lopes, L. C. J. Pereira, L. F. Veiros and P. T. Gomes, Inorg. Chem., 2018, 57, 8146–8159 CrossRef CAS PubMed.
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS . Puckering parameters for the metallacycle Co(1)–P(1)–C(14)–C(15)–N(1)–C(1): q = 0.9340(14) Å, θ = 80.45(9)°, φ = 4.47(8)°, for Co(1)–P(2)–C(19)–C(18)–N(2)–C(1): q = 0.9433(13) Å, θ = 78.63(10)°, φ = 8.78(10)°.
- (a) R. L. Hollingsworth, J. W. Beattie, A. Grass, P. D. Martin, S. Groysman and R. L. Lord, Dalton Trans., 2018, 47, 15353–15363 RSC; (b) H. Van Rensburg, R. P. Tooze, D. F. Foster and A. M. Z. Slawin, Inorg. Chem., 2004, 43, 2468–2470 CrossRef CAS PubMed; (c) A. R. Manning, J. Chem. Soc. A, 1968, 1135 RSC.
- (a) L. M. Guard, T. J. Hebden, D. E. Linn and D. M. Heinekey, Organometallics, 2017, 36, 3104–3109 CrossRef CAS; (b) V. M. Krishnan, H. D. Arman and Z. J. Tonzetich, Dalton Trans., 2018, 47, 1435–1441 RSC.
- M. A. Kent, C. H. Woodall, M. F. Haddow, C. L. McMullin, P. G. Pringle and D. F. Wass, Organometallics, 2014, 33(20), 5686–5692 CrossRef CAS.
- M. Azam Rasool, P. P. Pescarmona and I. F. J. Vankelecom, ACS Sustainable Chem. Eng., 2019, 7(16), 13774–13785 CrossRef.
- Related complexes have been reported to adopt facial geometries: (a) P. L. Chiu and H. M. Lee, Organometallics, 2005, 24, 1692–1702 CrossRef CAS; (b) V. Tegethoff, T. Lübbering, C. Schulte to Brinke, B. Schirmer, J. Neugebauer and F. E. Hahn, Organometallics, 2021, 40(5), 606–617 CrossRef CAS.
- A similar pattern has been described by H. M. Lee for the same ligand fac-Ru complex (ref. 31a) and by us for a similar PCP ligand that coordinates to Ir adopting a facial geometry: M. Iglesias, A. Iturmendi, P. J. Sanz Miguel, V. Polo, J. J. Pérez-Torrente and L. A. Oro, Chem. Commun., 2015, 51, 12431–12434 RSC.
- For examples of catalysed acceptorless dehydrogenative coupling of the silanes see: (a) L. Rosenberg, C. W. Davis and J. Yao, J. Am. Chem. Soc., 2001, 123, 5120–5121 CrossRef CAS PubMed; (b) D. Schmidt, T. Zell, T. Schaubc and U. Radius, Dalton Trans., 2014, 43, 10816–10827 RSC; (c) E. E. Smith, G. Du, P. E. Fanwick and M. M. Abu-Omar, Organometallics, 2010, 29, 6527–6533 CrossRef CAS; (d) F. G. Fontaine and D. Zargarian, J. Am. Chem. Soc., 2004, 126, 8786–8794 CrossRef CAS PubMed; (e) M. Tanabe, A. Takahashi, T. Fukuta and K. Osakada, Organometallics, 2013, 32, 1037–1043 CrossRef CAS; (f) M. D. Spencer, Q. D. Shelby and G. S. Girolami, J. Am. Chem. Soc., 2007, 129, 1860–1861 CrossRef CAS PubMed; (g) T. D. Tilley, Acc. Chem. Res., 1993, 26, 22–29 CrossRef CAS; (h) J. Y. Corey, X. H. Zhu, T. C. Bedard and L. D. Lange, Organometallics, 1991, 10, 924–935 CrossRef CAS.
- (a) L. Rosenberg, C. W. Davis and J. Yaob, J. Am. Chem. Soc., 2001, 123, 5120–5121 CrossRef CAS PubMed; (b) F.-G. Fontaine and D. Zargarian, J. Am. Chem. Soc., 2004, 126, 8786–8794 CrossRef CAS PubMed; (c) E. E. Smith, G. Du, P. E. Fanwick and M. M. Abu-Omar, Organometallics, 2010, 29, 6527–6533 CrossRef CAS; (d) D. Schmidt, T. Zell, T. Schaub and U. Radius, Dalton Trans., 2014, 43, 10816–10827 RSC; (e) M. Tanabe, A. Takahashi, T. Fukuta and K. Osakada, Organometallics, 2013, 32, 1037–1043 CrossRef CAS.
- (a) T. J. Clark, K. Lee and I. Manners, Chem. – Eur. J., 2006, 12, 8634–8648 CrossRef CAS PubMed; (b) R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC; (c) T. D. Tilley, Acc. Chem. Res., 1993, 26, 22–29 CrossRef CAS.
- (a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS; (b) D. J. Parks, J. M. Blackwell and W. E. Piers, J. Org. Chem., 2000, 65, 3090–3098 CrossRef CAS PubMed.
- S. Park and M. Brookhart, Organometallics, 2010, 29, 6057–6064 CrossRef CAS PubMed.
- V. K. Dioumaev and R. M. Bullock, Nature, 2003, 424, 530–5352 CrossRef CAS PubMed.
- (a) J. Yang and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 12656–12657 CrossRef CAS PubMed; (b) S. Park and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 640–653 CrossRef CAS PubMed; (c) J. Yang, P. S. White, C. K. Schauer and M. Brookhart, Angew. Chem., Int. Ed., 2008, 47, 4141–4143 CrossRef CAS PubMed.
- (a) D. V. Gutsulyak, S. F. Vyboishchikov and G. I. Nikonov, J. Am. Chem. Soc., 2010, 132, 5950–5951 CrossRef CAS PubMed; (b) D. V. Gutsulyak and G. I. Nikonov, Angew. Chem., Int. Ed., 2010, 49, 7553–7556 CrossRef CAS PubMed; (c) D. V. Gutsulyak, A. van der Est and G. I. Nikonov, Angew. Chem., Int. Ed., 2011, 50, 1384–1387 CrossRef CAS PubMed; (d) S.-H. Lee, D. V. Gutsulyak and G. I. Nikonov, Organometallics, 2013, 32, 4457–4464 CrossRef CAS.
- (a) T. T. Metsänen, D. Gallego, T. Szilvási, M. Driess and M. Oestreich, Chem. Sci., 2015, 6, 7143–7149 RSC; (b) J. Fuchs, H. F. T. Klare and M. Oestreich, ACS Catal., 2017, 7, 8338–8342 CrossRef CAS; (c) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed.
- (a) M. Iglesias, P. J. Sanz Miguel, V. Polo, F. J. Fernández-Alvarez, J. J. Pérez-Torrente and L. A. Oro, Chem. – Eur. J., 2013, 19, 17559–17566 CrossRef CAS PubMed; (b) R. Lalrempuia, M. Iglesias, V. Polo, P. J. Sanz Miguel, F. J. Fernández-Alvarez, J. J. Pérez-Torrente and L. A. Oro, Angew. Chem., Int. Ed., 2012, 51, 12824–12827 CrossRef CAS PubMed; (c) R. Puerta-Oteo, J. Munarriz, V. Polo, M. V. Jiménez and J. J. Pérez-Torrente, ACS Catal., 2020, 10(13), 7367–7380 CrossRef CAS.
- (a) Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS PubMed; (b) J. A. R. Schmidt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11426–11435 CrossRef CAS PubMed; (c) T. L. Church, Y. D. Y. L. Getzler and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 10125–10133 CrossRef CAS PubMed; (d) J. M. Rowley, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 4948–4960 CrossRef CAS PubMed; (e) Y. Wen, R. Nie, B. Li and S. Li, ACS Catal., 2023, 13, 3317–3322 CrossRef CAS.
- The products of the reductive amination of furfural have been previously reported in the literature. (a) N-(Furan-2-ylmethyl)-4-methylaniline: D. Weickmann, W. Frey and B. Plietker, Chem. – Eur. J., 2013, 19, 2741–2748 CrossRef CAS PubMed; (b) N-(Furan-2-ylmethyl)aniline: P. D. Pham, P. Bertus and S. Legoupy, Chem. Commun., 2009, 6207–6209 RSC; (c) N-(Furan-2-ylmethyl)-4-methoxyaniline: J. S. K. Clark, C. M. Lavoie, P. M. MacQueen, M. J. Ferguson and M. Stradiotto, Organometallics, 2016, 35, 3248–3254 CrossRef CAS; (d) N-(Furan-2-ylmethyl)-4-(trifluoromethyl)aniline: Y. Du, R. M. Pearson, C.-H. Lim, S. M. Sartor, M. D. Ryan, H. Yang, N. H. Damrauer and G. M. Miyake, Chem. – Eur. J., 2017, 23, 10962–10968 CrossRef CAS PubMed; (e) N-(Furan-2-ylmethyl)cyclohexanamine: C. D. Matier, J. Schwaben, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 17707–17710 CrossRef CAS PubMed; (f) 4-(Furan-2-ylmethyl)morpholine: Z. Şahin, N. Gürbüz, İ. özdemir, O. Şahin, O. Büyükgüngör, M. Achard and C. Bruneau, Organometallics, 2015, 34, 2296–2304 CrossRef.
- The products of the reductive amination of levulinic acid. 2-methyl-1-(p-tolyl)pyrrolidine, phenylpyrrolidine: (a) 2-Methyl-1- has been previously reported in the literature Y. Ogiwara, T. Uchiyama and N. Sakai, Angew. Chem., Int. Ed., 2016, 55, 1864–1867 CrossRef CAS PubMed; (b) 1-(4-Methoxyphenyl)-2-methylpyrrolidine: Y. Zhang, X. Yang, Q. Yao and D. Ma, Org. Lett., 2012, 14, 3056–3059 CrossRef CAS PubMed; (c) 2-Methyl-1-(4-(trifluoromethyl)phenyl)pyrrolidine: C. Li, Y. Kawamata, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 13088–13093 CrossRef CAS PubMed; (d) C. Wu, X. Luo, H. Zhang, X. Liu, G. Ji, Z. Liu and Z. Liu, Green Chem., 2017, 19, 3525–3529 RSC.
- SAINT+, version 6.01: Area-Detector Integration Software, Bruker AXS, Madison, WI, 2001 Search PubMed.
- SADABS, Area-Detector Absorption Program, Bruker AXS, Madison, WI, 1996 CrossRef CAS PubMed; L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2279813 for (1). See DOI: https://doi.org/10.1039/d3dt00463e |
| This journal is © The Royal Society of Chemistry 2023 |