DOI:
10.1039/D3DT01018J
(Paper)
Dalton Trans., 2023,
52, 7336-7351
Tuning the optical and magnetic properties of lanthanide single-ion magnets using nitro-functionalized trispyrazolylborate ligands†
Received
4th April 2023
, Accepted 4th May 2023
First published on 15th May 2023
Abstract
We report the synthesis, crystal structures, photophysical and magnetic properties of 11 novel lanthanide complexes with the asymmetrically functionalized trispyrazolylborate ligand 4-nitrotrispyrazolylborate, 4-NO2Tp−: [Ln(4-NO2Tp)3] (Ln = La–Dy, except Pm). In-depth photophysical characterization of the ligands via luminescence, reflectance and absorption spectroscopic techniques, decay lifetimes, quantum yields supported by time-dependent density functional theory (TD-DFT) and natural bond order (NBO) analysis reveal that n-NO2Tp− ligands are dominated by intra-ligand charge transfer (ILCT) transitions and that second-sphere interactions are critical to the stabilization of the T1 state of n-NO2Tp− ligands and hence their ability to sensitize Ln3+ emission. The luminescence properties of the complexes indicate that 4-NO2Tp− is a poor sensitizer of Ln3+ emission, unlike 3-NO2Tp−. Moreover, [Nd(4-NO2Tp)3] (crystallized as a hexane solvate) displays single-molecule magnet (SMM) properties, with longer relaxation times and larger barrier than the non-functionalized [NdTp3], attributed to the addition of the NO2-group and subsequent rigidification of the molecular structure.
Introduction
Lanthanide (Ln) hybrid materials with tunable optical and magnetic properties have been attractive targets in fields such as opto-electronics and molecular magnetism.1–3 While there has been great success with tailoring the optical properties of the lanthanides towards specific applications such as metal ion and explosives sensors,4,5 thermometers,6 information storage devices7etc., the ability to tune the molecular magnetic properties of the lanthanide ions remains quite elusive.2,8 The intrinsic, magnetic properties of 4f ions, namely, their large single-ion anisotropy and high degree of spin–orbit coupling allow for Ln-materials to have applications such as high density, molecule-based information storage, spintronic devices and quantum computing.9 Recent reports on lanthanide molecular magnets that seek to harness the large single-ion anisotropy of the lanthanides, do so by engineering a wide variety of chemical environments including radical-bridged complexes to promote superexchange interactions,10–15 low coordination number (CN < 8) complexes,16–19 and many di-cyclopentadienyl (metallocene) complexes.20–22
There has, however, been comparatively very little work done on optimizing both the optical and magnetic properties of the lanthanides simultaneously, or using the lessons learned from harnessing the optical properties to inform design of lanthanide molecular magnets.23 Bi-functional Ln-materials that display useful photophysical and magnetic properties are advantageous because they could display unusual properties, e.g. optical polarization of nuclear spin,24 and yield insights into the fundamental properties of f-electrons by studying excited state dynamics in a magnetic field. Such work would require a ligand platform that can: (i) exert a large degree of control over the primary coordination sphere of the lanthanides, (ii) sensitize lanthanide luminescence and (iii) accommodate a wide variety of ring substituents to facilitate tunability of properties.23
Pyrazolylborate ligands (H4−xBPzx) meet all these criteria. They are widely used to make lanthanide chelates owing to their ability to control the spherical coordination environment of f-block elements via steric bulk/chelate effect,25,26 and can accommodate a very diverse array of ring substituents.27–29 Moreover, lanthanide pyrazolylborates, [Ln(H4−xBPzx)y], have been characterized as light-emitting devices (LEDs),30,31 single-molecule magnets (SMMs)32,33 and even magneto-luminescent materials that display both sensitized luminescence and SMM properties.34 Despite the breadth of work done on harnessing the desirable, opto-electronic and magnetic properties of the f-elements using pyrazolylborate ligands, our understanding of how ring substituents affect the properties of [Ln(H4−xBPzx)y] materials is lacking, especially when compared to Ln-benzoate or Ln-β-diketonate systems.35
Latva et al.,36 Tsaryuk et al.,37,38 de Bettencourt-Dias et al.,39,40 Raymond et al.41 among others, have detailed descriptions on how to tune the optical properties of Ln hybrid materials using aromatic ring substituents to control the antenna effect. The antenna effect describes the energy transfer from the S1 or T1 states of coordinated “antenna” ligands (organic chromophores) to lanthanide ions’ 2S+1LJ states by the Förster or Dexter energy transfer mechanisms. The influence of both electron-donating and electron-withdrawing groups, as well as the impact of the number of substituents and ring position on the photophysical properties of lanthanide complexes is well understood and has been implemented with great success.35 Conversely, less explored is the influence of said substituent effects on slow magnetic relaxation dynamics (SMM properties), considering both structural changes (intermolecular interactions) and perturbations of the crystal field experience by the Ln3+ ion. We are aware of only two reports, by Sushila et al. and Gálico et al, on the influence of halo-substitution on the optical and magnetic properties of lanthanide amino‑bisphenol and β-diketonate complexes respectively, that addresses the impact of substituent (halogen) effects on optical and magnetic properties of Ln-materials.42,43 Given the variety of lanthanide molecular magnets with pyrazolylborates and our lab's recent work with selective functionalization of trispyrazolylborate (Tp−) ligands,29,44 we postulate that asymmetrically functionalized Tp− ligands (Fig. 1) are an ideal platform for establishing how aromatic ring substituents influence the properties of lanthanide molecular magnets.
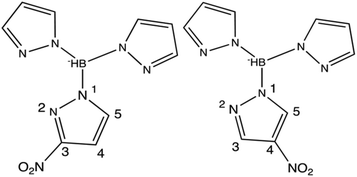 |
| | Fig. 1 Diagrams of asymmetric trispyrazolylborate ligands, left – 3-nitrotrispyrazolylborate (3-NO2Tp−) and right – 4-nitrotrispyrazolylborate (4-NO2Tp−). | |
We previously reported five families of lanthanide 3-nitrotripyrazolylborates, Ln(3-NO2Tp)x complexes that displayed tunable nuclearity and charge transfer mediated optical properties, and we elucidated the impact of strongly electron-withdrawing substituents on the coordination chemistry and photophysical properties of Ln(H4−xPzx)y complexes.45 We noted that the drastic difference in structural types and photophysical properties, when compared to other homoleptic Ln(H4−xBPzx)y complexes was primarily due to the presence of new binding site via the oxygen of the nitro-group (Ln-ONO2) on the 3-NO2Tp−. With this in mind, we substituted 3-NO2Tp− for the 4-nitrotrispyrazolylborate ligand, 4-NO2Tp−, to investigate the influence of the nitro-group on lanthanide pyrazolylborates without Ln-ONO2 coordination. Herein, we report that the coordination compounds of 4-NO2Tp− with Ln3+ ions are indeed 9-coordinate homoleptic complexes, [Ln(4-NO2Tp)3] (Ln = La–Dy, except Pm) analogous to the previously reported [LnTp3]. Moreover, the inclusion of the non-coordinating nitro-group imparts control over the crystal packing and crystallographic symmetry of [Ln(4-NO2Tp)3] which may be used as a tool to influence the slow magnetic relaxation of [Nd(4-NO2Tp)3]. The photophysics of [Ln(4-NO2Tp)3] feature Ln3+-selective singlet sensitization of Eu3+ and Tb3+ and various intra-ligand charge transfer (ILCT) transitions between the Sn and Tn states of the boron-bridged pyrazole and 4-nitropyrazole rings. We also provide a comparative analysis of the performance of Tp−vs. n-NO2Tp− (n = 3 or 4) as Ln3+ sensitizers and SMM-generating ligands (Tp−vs. 4-NO2Tp−).
Results
Synthesis of Ln(4-NO2Tp)3·solvent
The reported complexes, [Ln(4-NO2Tp)3] (Ln = La–Dy, except Pm, 1–9) were synthesized by combining methanolic solutions of LnCl3 salts with [TBA][4-NO2Tp]. The solid crude product may be readily isolated by removing the methanol in vacuo and washing away tert-butylammonium chloride in isopropanol. The complexes are stable in acetonitrile (as indicated by their well-defined NMR spectra, see Fig. S7–S14†). They are also soluble in common solvents such as dicholoromethane and acetone, but only sparingly soluble in methanol and ethanol and insoluble in benzene and isopropanol. This differs drastically from the non-functionalized [LnTp3] complexes which are insoluble in all solvents when Ln = La–Gd and only sparingly soluble in benzene and dicholoromethane for Ln = Dy and Tb.46 Crude [Ln(4-NO2Tp)3] can be recrystallized as one of two polymorphs, a hexane solvate, [Ln(4-NO2Tp)3]·C6H14 (a) and a benzene solvate, [Ln(4-NO2Tp)3]·7C6H6 (b).
Crystal structures
Structural description of [Ln(4-NO2Tp)3]·C6H14.
Compounds 1a–9a are all isomorphous and hence only the crystal structure of 4a will be discussed in detail. 4a contains one crystallographically unique Nd3+ ion coordinated to 9 nitrogen atoms from three unique 4-NO2Tp− ligands, with the Nd3+ center exhibiting a tricapped trigonal prismatic coordination geometry of approximately D3h site symmetry. Ln–N bonds lengths range from 2.565(2) to 2.579(3) Å for the nitrogen atoms of the trigonal prism and 2.739(3) to 2.753(2) Å for the capping nitrogen atoms. The mononuclear [Ln(4-NO2Tp)3] complex displays an approximate C3h point symmetry, analogous to that of the 9-coordinate [LnTp3] complexes,46 with a C3 axis passing through the Nd3+ center perpendicular to the plane defined by the three nitropyrazoles. However, unlike in [LnTp3], there are three unique NO2⋯H–C hydrogen bonds between adjacent [Ln(4-NO2Tp)3] molecules, whereas [LnTp3] does not feature any significant intermolecular interactions. The NO2⋯H–C hydrogen bonds as shown Fig. 2(b) range from 3.1916(1) Å to 3.4382(1) Å, where two of the three H bonds occur between pyrazole and 4-nitropyrazole rings and the last between two 4-nitropyrazole rings. Compound 4a crystallizes in the C2/c space group and it contains two highly disordered hexane sites in the lattice. The hexane molecule does not significantly separate the [Ln(4-NO2Tp)3] molecules, resulting in fairly dense crystal packing in 4a with the shortest intermolecular Nd3+-to-Nd3+ distance being 9.471(6) Å, similar to [LnTp3] at 9.686(1) Å. Interestingly this packing features sheets of [Ln(4-NO2Tp)3] molecules, along the [001] direction, such that a given sheet is sandwiched between a parallel one and another that is slighted canted by 19° (Fig. S1†).
 |
| | Fig. 2 (a) Asymmetric unit of 4a (with the hexane omitted for clarity) featuring the [Ln(4-NO2Tp)3] complex. (b) NO2⋯H–C hydrogen bonding between [Ln(4-NO2Tp)3] complexes forming a 2-D layer in the xy plane. | |
Structural description of [Ln(4-NO2Tp)3]·7C6H6 (5b, Ln = Sm and 8b, Ln = Tb).
Compounds 5b and 8b constitute a second solvate polymorph of [Ln(4-NO2Tp)3], crystalized with 7 solvent benzene molecules per molecule of [Ln(4-NO2Tp)3]. Since 5b and 8b are isostructural and only the structure of 5b will be discussed. The [Ln(4-NO2Tp)3] complex of 5b (Ln = Sm) is the same as 4a (Ln = Nd) (Fig. S2a†), with Sm–N bond lengths that range from 2.5427(11)–2.5440(13) Å for the trigonal prism nitrogrens and 2.7408(11) Å for the capping nitrogens, however it crystallizes in the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group. Additionally, the crystallographic 3-fold rotoinversion axis overlaps with the molecular C3 that through Sm3+, resulting in only a third of the [Ln(4-NO2Tp)3] complex being present in the asymmetric unit of 5b, while the entire molecule is contained with the asymmetric unit of 4a. There are 7 lattice benzene molecules per molecule of [Ln(4-NO2Tp)3], resulting in significantly less dense crystal packing of the [Ln(4-NO2Tp)3] units (Fig. S2b†), with fairly long intermolecular Sm3+-to-Sm3+ distances, the shortest being 12.667(1) Å. All of the [Sm(4-NO2Tp)3] complexes lie perfectly parallel to each other and there are no significant non-covalent interactions between the monomers (Fig. 3).
space group. Additionally, the crystallographic 3-fold rotoinversion axis overlaps with the molecular C3 that through Sm3+, resulting in only a third of the [Ln(4-NO2Tp)3] complex being present in the asymmetric unit of 5b, while the entire molecule is contained with the asymmetric unit of 4a. There are 7 lattice benzene molecules per molecule of [Ln(4-NO2Tp)3], resulting in significantly less dense crystal packing of the [Ln(4-NO2Tp)3] units (Fig. S2b†), with fairly long intermolecular Sm3+-to-Sm3+ distances, the shortest being 12.667(1) Å. All of the [Sm(4-NO2Tp)3] complexes lie perfectly parallel to each other and there are no significant non-covalent interactions between the monomers (Fig. 3).
 |
| | Fig. 3 Packing diagram of 5b (solvent benzene molecules omitted for clarity) where the [Sm(4-NO2Tp)3] molecules align collinearly in parallel planes. | |
Photophysical properties of 4-NO2Tp− and [Ln(4-NO2Tp)3]·C6H14
Optical properties of 4-NO2Tp−.
We utilized diffuse reflectance and luminescence spectroscopy techniques, in addition to time-dependent density functional theory (TD-DFT) to fully characterize the photophysical properties of 4-NO2Tp−. Diffuse reflectance measurements for K[4-NO2Tp] (Fig. S16†) reveal a broad absorption band, spanning the UV to blue region, between 225 and 430 nm. The solid-state luminescence spectra of K[4-NO2Tp], 1a (fluorescence) and 7a (phosphorescence) spectra respectively, for 4-NO2Tp−, are summarized in Fig. 4. La and Gd complexes are well known to yield emission spectra that display decay of Sn and Tn states almost exclusively45,47,48 and are included to aid in characterization of those transitions/states with respect to 4-NO2Tp−. The similar profiles for all three (emission) spectra, even 7a at 77 K, indicate that the decay of the Tn states of 4-NO2Tp− is non-radiative (NR) and/or that T1 state is unstable and excitation energy is readily lost. The most probable cause would be the NO2-group intra-ligand charge transfer (ICLT) quenching pathway, described by Tsaryuk et al., where excitation energy is lost due to the non-radiative π*–nNO2 transition.37,38
 |
| | Fig. 4 Top: Calculated S0 → S1 transition of Calc-1 as predicted by TD-DFT. Bottom: Room temperature luminescence excitation (left, broken lines) and emission (right, solid lines) of K[4-NO2Tp] (λexc = 393 nm, λem = 490 nm), 1a (λexc = 400 nm, λem = 481 nm) and 7a (at 77 K, λexc = 394 nm, λem = 481 nm). The excitation peak at 400 nm is assigned as S0 → S1 of 4-NO2Tp−. * – background signal from the Xe arc lamp observable owing to very weak emission intensity of 1a and 7a. | |
To assign the S0 → S1 and T1 → S0 transitions of 4-NO2Tp− associated with the antenna effect, we carried out TD-DFT calculations on a geometry optimized model of 1, Calc-1 (Fig. 4 and Fig. S26†). The population analysis of Calc-1 indicates that the HOMOs and LUMOs of the ligand are localized at the π systems of aromatic pyrazole (Pz) and 4-nitropyrazole (4-NO2Pz) rings. There is good agreement between the calculated absorption spectrum of Calc-1 and the solution absorption spectrum of crude 1 in acetonitrile (Fig. 5). However, the low energy absorptions in the excitation spectra of K[4-NO2Tp] and 1a at ∼400 nm, that lead to radiative decay (ca. 485 nm) are not predicted by TD-DFT nor are they present in the solution absorption spectrum. We attribute this to the nature of collecting luminescence excitation spectra where only transitions that lead to emission can be observed and not necessarily the transitions with large ε values, which is what is typically observed in an absorption spectrum. Since reflectance spectroscopy is sensitive to transitions with very low molar absorptivity/f-oscillator strengths (see Ln3+ absorptions in Fig. S19–S25†), we note that the 400 nm transition of 4-NO2Tp− (which has a very low ε); is present in the reflectance spectrum of 1a. Given that the higher energy transitions with larger oscillator strengths do not lead to emission from 4-NO2Tp−, assignment of the peaks in the excitation spectra is quite difficult. However, based on the TD-DFT calculations (Fig. 5) and our previous work with the 3-NO2Tp− ligand and we assign the 4-NO2Tp− transitions as:
excitation peak at 400 nm: S0 → S1, nNO2 → π*4-NO2Pz ICLT,
emission at 485 nm: S1 → S0, π*4-NO2Pz → nNO2 ICLT,
theoretical emission of T1 → S0: π*4-NO2Pz → nNO2 ILCT.
 |
| | Fig. 5 Left – Electronic absorption spectrum (solid line) of crude 1 in acetonitrile (1.03 × 10−5 M) and calculated UV-VIS of Calc-1 in the gas phase. Right – TD-DFT output of the assigned S0 → S7 as an ILCT πPz → π*4-NO2Pz transition and the calculated S0 → T1 transition associated with the antenna effect. | |
Luminescence properties of [Ln(4-NO2Tp)3]·C6H14.
All of the reported complexes (except 2a and 7a) display characteristic Ln3+ emission in the UV-visible region (Fig. 6). The profiles and relative intensities for 3a (Pr), 4a (Nd), 5a (Sm), 8a (Tb) and 9a (Dy) emission spectra are typical for the line-like, f–f transitions of the trivalent lanthanides.34,45 The europium analogue, 6a, features an emission spectrum with the expected 5D0 → 7FJ (J = 1–4) transitions. Induced electric dipole (5D0 → 7F2, 5D0 → 7F4) transitions are allowed in 6a, given the lack of a center of symmetry, while fully forbidden transitions, 5D0 → 7F0 and 5D0 → 7F3 are either not observable (5D0 → 7F0) or very weak (5D0 → 7F3). The intensity of magnetic dipole (5D0 → 7F1) transition is independent on any Eu3+–ligand interactions,49 and just happens to coincide with 5D0 → 7F2. The difference in intensity between J = 2 and J = 4 is somewhat unexpected, as the former transition is usually considered hypersensitive and thus expected to be more intense.49 This can be attributed to two factors: (i) lack of electric quadrupole contributions to J = 2 and (ii) a static coupling mechanism between Eu3+ and 4-NO2Tp−, i.e. a weakly covalent Eu3+-4-NO2Tp− bond.50,51 A preliminary Judd–Ofelt analysis of the luminescence data performed on 6a (Table S2†) provides some evidence for the latter with a low value for Ω2.50 Finally, the crystal field splitting observed for the 5D0 → 7FJ (J = 1–4) transitions is in agreement with the high local symmetry (D3h) of the europium center.52
 |
| | Fig. 6 298 K temperature solid-state luminescence excitation (left, broken lines) and emission (right, solid lines) of 1a (λexc = 400 nm, λem = 481 nm), 3a (λexc = 446 nm, λem = 602 nm), 4a (λexc = 350 nm), 5a (λexc = 405 nm, λem = 600 nm), 6a (λexc = 394 nm, λem = 700 nm), 8a (λexc = 486 nm, λem = 544 nm) and 9a (λexc = 450 nm, λem = 575 nm) with assignments of the various 4-NO2Tp− or direct f-to-f Ln3+ transitions. The emission spectrum of 4a collected using 350 nm may be a 4-NO2Tp− transition or Nd3+ absorption at ∼350 nm (2I11/2).55 | |
The observed emission spectra in Fig. 6 is due primarily to direct f–f transitions and not sensitization via the 4-NO2Tp− ligand. The excitation spectra of 1a–9a (except 2a and 7a) and the weak (or in some cases absent) absorption associated with 4-NO2Tp−, highlights that 4-NO2Tp− is an inefficient sensitizer for Ln3+ emission. Interestingly, the presence of several direct f–f absorption peaks in Fig. 6 reveal that 4-NO2Tp− does not directly quench Ln3+ luminescence via Ln3+-to-4-NO2Tp− back-energy transfer and that direct f–f absorption and/or Ln3+-to-Ln3+ energy transfer (energy migration) are fairly efficient sensitization mechanisms. 2 likely does not display room temperature luminescence due to a fast dCe → π*4-NO2Pz metal-to-ligand charge transfer (MLCT) and subsequent non-radiative π*4-NO2Pz → nNO2 transition, analogous to monomeric, [Ce(3-NO2Tp)2(NO3)].45
6a and 8a are the only compounds that feature a significant absorption band ca. 330–340 nm that could be attributed to ligand absorption and sensitization via 4-NO2Tp−. This band coincides somewhat with the absorption at 284 nm in the absorption spectrum of crude 1 and a πPz → π*4-NO2Pz transition predicted at 276 nm for Calc-1 and therefore, this peak is assigned as the S0 → S7, πPz → π*4-NO2Pz ICLT transition. The disparity between the transitions observed in the absorption spectrum versus the excitation and emission spectra may be attributed to two distinct pathways associated with luminescence vs. energy transfer to Eu3+/Tb3+; usually these pathways are identical (to be discussed later, see Fig. 10). The ability of 4-NO2Tp− to weakly sensitize Tb3+ and Eu3+ emission suggests that the T1 state of 4-NO2Tp− (which we could not determine experimentally), may have enough energy to sensitize Eu3+ (5D0, 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1) and Tb (5D4, 21500 cm−1) emission, yet may be too short lived to participate in energy transfer for the lanthanide ions studied, and that the rate of non-radiative decay via a fast π*4-NO2Pz → nNO2 transition is large (knonrad ≫ krad). Another explanation for the sensitized emission in 6a and 8a and not the other lanthanides, may be a singlet energy transfer pathway53,54via the S0 → S7, πPz → π*4-NO2Pz ICLT transition that typically only Eu3+ and Tb3+ can participate in, whereas triplet sensitization is usually necessary for more weakly luminescent lanthanides (Pr3+, Sm3+, Dy3+).
500 cm−1) and Tb (5D4, 21500 cm−1) emission, yet may be too short lived to participate in energy transfer for the lanthanide ions studied, and that the rate of non-radiative decay via a fast π*4-NO2Pz → nNO2 transition is large (knonrad ≫ krad). Another explanation for the sensitized emission in 6a and 8a and not the other lanthanides, may be a singlet energy transfer pathway53,54via the S0 → S7, πPz → π*4-NO2Pz ICLT transition that typically only Eu3+ and Tb3+ can participate in, whereas triplet sensitization is usually necessary for more weakly luminescent lanthanides (Pr3+, Sm3+, Dy3+).
Magnetic properties of [Ln(4-NO2Tp)3]·C6H14
Static magnetic measurements.
The magnetic susceptibility of 4a, 8a and 9a were studied under a static field of 1000 Oe (0.1 T) as shown in Fig. 7. The room temperature χmT values were 1.65 cm3 K mol−1, 11.85 cm3 K mol−1 and 13.93 cm3 K mol−1 for 4a, 8a and 9a respectively. These values agree well with those anticipated for free uncoupled Ln3+ ions at 1.64 cm3 K mol−1 (Ln = Nd, 4I9/2, S = 3/2, L = 6, g = 8/11), 11.82 cm3 K mol−1 (Ln = Tb, 7F6, S = 3, L = 3, g = 3/2) and 14.17 cm3 K mol−1 (Ln = Dy, 6H15/2, S = 5/2, L = 5, g = 4/3). For all three compounds, χ=T decreases with T, owing to the depopulation of higher energy mJ states generated by crystal field splitting of the ground state.34 Assuming a perfect D3h coordination sphere for the lanthanide center, the crystal field Hamiltonian can be expressed as:
| ĤCF = B02θ2Ô02 + B04θ4Ô04 + B06θ6Ô06 + B66θ6Ô66 |
where Bqk are the crystal field parameters (including orbital reduction parameters), θk, are the operator equivalent factors and Ôqk the Stevens operator equivalents.56,57 A survey of the crystal field parameters between −5000 and +5000 cm−1 indicated that the best fits of the experimental data around B06 = B66 = 0 for all three cations. While the negligible value of the sole equatorial term B66 is in agreement with the substantially longer Ln—NNO₂Pz distances in the plane of the tricapped trigonal prismatic coordination sphere, the B06 value is more surprising for 4f centers. Attempts to determine values for the B02 and B04 parameters were unsuccessful owing to overparameterization of the systems (Fig. S27–S29†). Even when further narrowing the possibilities by assuming a linear dependence of the crystal field parameters on the f-electron count of the metal center, two sets of parameters remained equally suited to account for the DC magnetization data (Table 1, Fig. 7 and S30†).
 |
| | Fig. 7 Temperature dependence of χmT product at 1000 Oe for 4a (Nd,  ), 8a (Tb, ), 8a (Tb,  ) and 9a (Dy, Δ). Solid lines correspond to simulations using the crystal field parameters of set 1 in Table 1, dashed lines to those using set 2. ) and 9a (Dy, Δ). Solid lines correspond to simulations using the crystal field parameters of set 1 in Table 1, dashed lines to those using set 2. | |
Table 1 Crystal field parameters suitable for simulations of the static magnetization
| Crystal field parameters (cm−1) |
4a
|
8a
|
9a
|
|
B
02/B04 (set 1) |
960/−300 |
−220/−70 |
−460/−20 |
|
B
02/B04 (set 2) |
−560/340 |
−610/10 |
−620/−50 |
Dynamic magnetic measurements.
Dynamic (ac) magnetic susceptibility experiments were collected on 4a, 8a and 9a in the 0–1000 Hz frequency range between 1.8 and 9.8 K. None of the complexes displayed an out-of-phase signal in the absence of a magnetic field bias, but the Nd3+ complex, 4a did so, under a small applied field (H = 400 Oe). χ′mvs. f and χ″mvs. f isotherms are presented in Fig. 8(a) and (b) respectively and the frequency dependence of χ″m confirm that 4a is indeed a single-molecule magnet. The time dependence of the relaxation times, extracted using the CCFit2 program,58 can be modelled using eqn (1), which contains a direct term (AT) and a two phonons Raman term.59| |  | (1) |
 |
| | Fig. 8 (a) In-phase molar susceptibility (χ′m) vs. frequency and (b) out-of-phase molar susceptibility (χ″m) vs. frequency of 4a between 2 and 9 K at H = 400 Oe. | |
Since 4a is a field-induced SMM, no quantum tunneling parameter was included when modelling the relaxation data. Full details on all of the terms and parameters used for the fit are outlined in Fig. 11 and S30.† Initial inclusion of a Raman term (CTn) yielded a power dependence n term of ∼3.5; since this is a significant departure from the idealized Raman relaxation mechanism, we excluded any direct Raman contribution. Moreover, the use of a model with an Orbach 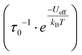 and a direct term (AT) yielded a sufficient model, however the Ueff was significantly smaller than what is expected for a true Orbach process at 14 cm−1. We therefore turned to a model with direct and two-phonons Raman terms, which yielded a nearly identical Ueff, or rather Evib at 13 cm−1 (Fig. S30†), corresponding to phonon-coupled under-barrier relaxation process rather than a true ΔmJ transition/Orbach process.
and a direct term (AT) yielded a sufficient model, however the Ueff was significantly smaller than what is expected for a true Orbach process at 14 cm−1. We therefore turned to a model with direct and two-phonons Raman terms, which yielded a nearly identical Ueff, or rather Evib at 13 cm−1 (Fig. S30†), corresponding to phonon-coupled under-barrier relaxation process rather than a true ΔmJ transition/Orbach process.
Discussion
Ligand design for Tp−-based antenna ligands
Comparison of the optical properties of the [Ln(4-NO2Tp)3] complexes and those of previously published complexes [LnTp3] and [Ln(3-NO2Tp)2(NO3)] shows the markedly inferior sensitizing efficiency of the 4-NO2Tp− ligand. Given the similarity of 3-NO2Tp− and 4-NO2Tp−, including the prevalence in both ligands of πPz → π*n-NO2Pz charge transfer transitions, predicted by TD-DFT and confirmed experimentally, this difference is rather surprising. To elucidate the origin of the disparity between the n-NO2Tp− (n = 3 or 4) ligands, we determined the quantum yield (Φexp), emission lifetime (τEu) and derived their intrinsic quantum yield (ΦEuEu), the rates of radiative (krad) and nonradiative decay (knonrad) as well as their efficiency of sensitization (ηsens) (Table 2). The radiative lifetime for Eu3+ is similar for all three complexes, with a slight reduction in [Eu(3-NO2Tp)2(NO3)]. Expectedly, 10-coordinate [Eu(3-NO2Tp)2(NO3)] has a lower intrinsic quantum yield than the two 9-coordinate complexes, which show nearly identical ΦEuEu. The primary difference between the EuTp-complexes is their quantum yields. Consistent with the reduction in sensitization efficiency often associated with nitrated antenna ligands,38 the quantum yield and radiative decay rate of 6a are about half of those of [EuTp3] (∼2.4% vs. 4.6% for Φexp and ∼13 s−1vs. 25 s−1 for krad), however the quantum yield and radiative decay rate for [Eu(3-NO2Tp)2(NO3)] are 4% and 21 s−1 respectively, approximately equal to those of [EuTp3]. The reduction in the radiative decay of 6a as compared to [EuTp3] can be attributed to the fast decay/short lifetime of the T1 state of 4-NO2Tp− and inefficient energy transfer to Eu3+ owing to the presence of the nitro-group. However, the nitro group appears to have no impact in [Eu(3-NO2Tp)2(NO3)].
Table 2 Photophysical data comparing [EuTp3], [Eu(3-NO2Tp)2(NO3)] and 6a
Examination of the differences between [Eu(3-NO2Tp)2(NO3)] and 6a seem to indicate that the triplet state of 3-NO2Tp− is more stable/longer-lived than that of 4-NO2Tp−, given that the emission from 4-NO2Tp− could not be observed at even 77 K, while we recorded room temperature T1 emission from 3-NO2Tp− in [Gd(3-NO2Tp)2(NO3)].45 TD-DFT calculations predict that both 3-NO2Tp− and 4-NO2Tp− should have similar triplet energies (422 and 404 nm respectively), indicating that the enhanced stability of 3-NO2Tp− must be due to external factors, outside of the energy of the molecular orbitals. One possible explanation is revealed by a comparison of the non-covalent interactions in the crystal structures of [Eu(3-NO2Tp)2(NO3)] and 6a. These interactions are π–π stacking and NO2⋯H–C hydrogen bonding in [Eu(3-NO2Tp)2(NO3)] and 6a respectively, both interactions involving the n-NO2Pz (n = 3 or 4) rings. Natural bond order (NBO) derived stabilization energies associated with each NCI (π–π stacking or NO2⋯H–C hydrogen bonding) (Fig. 9) and indicates that the π–π stacking interactions are significantly stronger than NO2⋯H–C hydrogen bonding. We postulate that the stronger π–π stacking NCIs of [Eu(3-NO2Tp)2(NO3)] increase the rigidity of 3-NO2Tp−. The coordination of the 3-NO2 group to the Ln3+ centers is likely to also contribute to an increased rigidity, accounting overall for the higher sensitization efficiency observed in [Eu(3-NO2Tp)2(NO3)] vs.6a. This is consistent with recent reports on NCIs stabilizing the triplet state of aromatic ligands.62–65 The enhanced stability afforded to the T1 state of 3-NO2Tp− in [Eu(3-NO2Tp)2(NO3)] likely increased its lifetime enough that it could undergo substantial energy transfer with Eu3+ (Fig. 10), while the T1 state of 4-NO2Tp− undergoes a rapid non-radiative decay. By changing the position of the ring substituent in n-NO2Tp− (n = 3 or 4) ligands, we can control the type and strength of NCIs in Ln(n-NO2Tp)x complexes and consequently tune the efficiency of n-NO2Tp-to-Ln3+ energy transfer(s) using second-sphere interactions.
 |
| | Fig. 9 Top: Representative natural bond orbitals (NBOs) of the π–π stacking interactions in [Eu(3-NO2Tp)2(NO3)] and the NO2⋯H–C hydrogen bonding in 6a. Bottom: Table summarizing the donor and acceptor pairs and associated contribution to the stabilization energy in kJ mol−1. | |
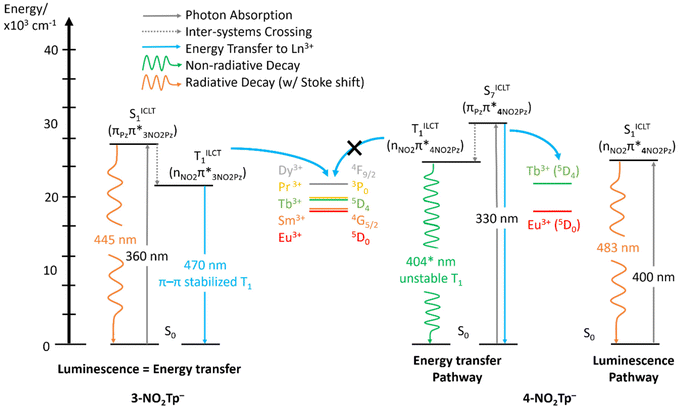 |
| | Fig. 10 Jablonski diagram comparing the luminescence and energy transfer pathways in 3-NO2Tp− and 4-NO2Tp−. | |
Ligand design for Tp−-based single-molecule magnets
The capped trigonal prismatic coordination sphere provided by the 4-NO2Tp− ligands corresponds to a relatively strongly axial crystal. Therefore, single molecule magnet properties are more likely to be obtained with lanthanide ions with an oblate electronic density along with a relatively high J value.8 Those include neodymium (J = 9/2), terbium (J = 6) and dysprosium (J = 15/2). Magnetic studies on 4a (Nd), 8a (Tb) and 9a (Dy) offer a preliminary confirmation of the axiality of the crystal field, as no Bqk term with q ≠ 0 appears in the modelisation of the dc magnetization data. However, either set of crystal field parameters (Table 1) obtained from those fits indicate ground states with mJ = 0 for 8a and mJ = ½ for 9a, in agreement with the absence of single molecule magnetism observed for those complexes. Of the two sets obtained for 4a, the first corresponds to a ground state mJ = 3/2 compatible with the observed slow magnetic relaxation and should therefore be preferred to the second set which would indicate a mJ = ½ ground state.
In 4a, as in the unsubstituted analogue [NdTp3],32 slow relaxation can only be observed under a small external magnetic field. This indicates substantial quantum tunneling of the magnetization at zero field, and in turn the presence of some small equatorial contribution to the crystal field, albeit one that could not be quantified from the dc data. Interestingly, relaxation is about 5 times slower in 4a than in [NdTp3] at the same temperature (Fig. 11) despite the virtually identical geometry of the two complexes (and higher crystallographic symmetry in [NdTp3]). Moreover, 4a's relaxation barrier, Evib is about 6 times larger than NdTp3's “Orbach” barrier (12.7 vs. 2.8 cm−1). [NdTp3] was previously reported to have a small relaxation barrier (Ueff = 2.84 cm−1, τ0 = 4.2 × 10−5 s),32 likely not a “true” Orbach relaxation barrier but rather a value corresponding to under barrier relaxation due to vibronic coupling. Given that dilution of NdTp3 with diamagnetic La3+ ions, did not significantly affect the relaxation behavior,32 the slight increase in intermolecular Ln3+-to-Ln3+ distances in 4a is likely not responsible for the improvement either, nor is the change from the parallel anisotropy axes in [NdTp3]66 to the slightly canted layers of 4a, which one would usually assume to be detrimental to slow relaxation. A change in vibrational levels is therefore the most likely mechanism for the observed difference, linked to the lattice rigidifying effect of the NO2⋯H–C hydrogen bonding in 4a. This marked increase in relaxation times and under-barrier in 4a over [NdTp3], signify that the former is a better SMM than the latter owing to the nitro-functionalization. The apparent similarity in the primary coordination sphere of 4a and [NdTp3] facilitates the first example of the influence of a strongly electron-withdrawing group on relaxation dynamics via a direct comparison of the two Nd3+ chelates.
 |
| | Fig. 11 Temperature dependence of the magnetic relaxation times, under a small bias field, of [NdTp3] (black triangles, 100 Oe) with the reported Orbach fit (black line),32 and 4a (blue squares, 400 Oe) with the fit (blue broken line) generated using eqn (1). From the fitted AC data (solid lines), [NdTp3]: Ueff = 2.84(2) cm−1, τ0 = 4.2(2) × 10−4 s; 4a: A = 318 (8) K, Ueff = 12.7(1.6) cm−1 and τ0 = 4.1(1.6) × 10−5 s. | |
Conclusion
Eleven new lanthanide complexes with an asymmetric nitro-functionalized trispyrazolylborate ligand, 4-NO2Tp− were synthesized and their optical and magnetic properties are presented. The [Ln(4-NO2Tp)3] complexes display crystalline polymorphism based on the solvent used for recrystallization. The choice of lattice solvent can be used as a method of controlling crystal packing and supramolecular assembly (or lack thereof) via NO2⋯H–C hydrogen bonding. An in-depth analysis of the optical properties of 4-NO2Tp− revealed two distinct pathways for luminescence and energy transfer comprised of several ILCT transitions such as nNO2 → π*4-NO2Pz and πPz → π*4-NO2Pz absorptions and π*4-NO2Pz → nNO2 and π*4-NO2Pz → πPz emissions. Moreover, 4-NO2Tp− is a poor sensitizer for Ln3+ emission (vs. Tp− and 3-NO2Tp−), as a direct result of adding a non-coordinating nitro-group. With regard to the magnetic properties of the reported complexes, the opposite is true, whereby adding a nitro-group increased the relaxation times by a factor of 5 as well as the under-barrier of [Nd(4-NO2Tp)3] over [NdTp3] by one order of magnitude. Our findings on the ability to control the crystal packing/non-covalent interactions, diverse charge transfer optical properties and enhancement of relaxation barriers using 4-NO2Tp− over Tp−, in context with our previous work on [Ln(3-NO2Tp)x] complexes, highlights the utility of asymmetric Tp− ligands for tuning the properties of the lanthanides to generate and study magneto-luminescent materials and design better molecular magnets.
Experimental
Materials
Lanthanide chloride salts, LnCl3·7H2O (Ln = La3+, Ce3+, Fisher Scientific, 99.9%), PrCl3·7H2O (Strem Chemicals, 99.9%), LnCl3·6H2O (Ln = Nd3+, Sm3+–Dy3+, Aldrich, 99.9%) are commercially available and were used as received.
Synthesis of tetrabutylammonium 4-nitrotrispyrazolylborate, [Bu4N][4-NO2Tp]
[Bu4N][4-NO2Tp] was prepared as described in the literature.29,44 After each synthesis of [Bu4N][4-NO2Tp], a standard solution, typically 0.3 M, was prepared by dissolving the reaction product in methanol.
Synthesis of potassium 4-nitrotrispyrazolylborate, K[4-NO2Tp]
K[4-NO2Tp] was prepared by dissolving 4-nitropyrazole (0.4485 g, 4 mmol) and potassium trispyrazolylborate (1.000 g, 4 mmol) in dry ethyl acetate and heating under reflux for 2 hours. Pentane (50 mL) was added to the cloudy mixture which was then placed in a refrigerator at 5 °C overnight. Two distinct phases were always isolated from this reaction, a white powder of K[4-NO2Tp] in very low yield (>5%) and a yellow powdery mixture of K[4-NO2Tp] and 4-nitropyrazole. Owing to the low yields, the solvent-free synthesis with the tetrabutylammonium salt was always used to generate the 4-NO2Tp−.
Synthesis of Ln(4-NO2Tp)3·solvent (1–9)
Complexes of 1–9 were synthesized using the same procedure differing only in the recrystallization conditions. A standard solution of LnCl3 was generated by dissolving an appropriate amount of the salt, e.g. LaCl3 (0.4910 g, 2.002 mmol) in methanol (20 mL, 0.1 M). Solutions of LaCl3 (1 mL, 0.1 M, 1 mmol) and [Bu4N][4-NO2Tp] (1 mL, 0.3 M, 3 mmol) were combined in a scintillation vial. The solvent was removed in vacuo and the oil-like residue was suspended in 3 mL 1:1 cyclohexane:isopropanol; vacuum filtration and washing with of isopropanol (3 × 3 mL) yielded dry, crude La(4-NO2Tp)3 as a white powder.
Recrystallized solvates were prepared by dissolving the crude product in of methanol (3 mL, La–Eu) or 1:1 methanol:ethanol (3 mL, Gd–Dy). Hexanes or benzene for (6 mL) was layered on top of the solution. Single crystals of hexane (1a–9a) or benzene (5b, 8b) solvates were observed after 1 day and the final product was isolated and collected after 3 days via vacuum filtration and washing with hexanes (3 × 3 mL). Crystals of the hexane polymorph had a tendency to re-dissolve in the mother liquor after 5 or 6 days. X-ray diffraction quality single crystals were collected when the recrystallization was carried out in a sealed scintillation vial, while a microcrystalline powder was obtained when the synthesis was done in a parafilmed 25 mL Erlenmeyer flask.
[La(4-NO2Tp)3]·C6H14 (1a).
White single crystals were isolated from methanol/hexanes. Yield (recrystallized): 51%. Elemental Anal. Calc. for LaC33H41O6N21B3: C 39.67, H 4.11, N 29.45. Found: C 39.40, H 3.59, N 29.05.
[Ce(4-NO2Tp)3]·C6H14 (2a).
Pale yellow single crystals were isolated from methanol/hexanes. Yield (recrystallized): 52%. Elemental Anal. Calc. for CeC33H41O6N21B3: C 39.62, H 4.10, N 29.41. Found: C 39.21, H 3.88, N 29.55.
[Pr(4-NO2Tp)3]·C6H14 (3a).
Green single crystals were isolated from methanol/hexanes. Yield (recrystallized): 45%. Elemental Anal. Calc. for PrC33H41O6N21B3: C 39.60, H 4.10, N 29.39. Found: C 39.45, H 3.94, N 29.59.
[Nd(4-NO2Tp)3]·C6H14 (4a).
Lilac single crystals were isolated from methanol/hexanes. Yield (recrystallized): 51%. Elemental Anal. Calc. for NdC33H41O6N21B3: C 39.47, H 4.09, N 29.29. Found: C 39.29, H 3.94, N 29.13.
[Sm(4-NO2Tp)3]·C6H14 (5a).
Pale yellow single crystals were isolated from methanol/hexanes. Yield (recrystallized): 36%. Elemental Anal. Calc. for SmC33H41O6N21B3: C 39.22, H 4.06, N 29.11. Found: C 39.11, H 3.91, N 29.30. τ = 0.116 ms (λexc = 340 nm (6P3/2), λem = 604 nm).
[Eu(4-NO2Tp)3]·C6H14 (6a).
White single crystals were isolated from methanol/hexanes. Yield (recrystallized): 30%. Elemental Anal. Calc. for EuC33H41O6N21B3: C 39.16, H 4.05, N 29.07. Found: C 38.87, H 3.87, N 29.07. τ = 1.80 ms (λexc = 394 nm (5L6), λem = 700 nm).
[Gd(4-NO2Tp)3]·C6H14 (7a).
White single crystals were isolated from methanol/hexanes. Yield (recrystallized): 33%. Elemental Anal. Calc. for GdC33H41O6N21B3: C 38.96, H 4.03, N 28.92. Found: C 38.91, H 3.97, N 29.04.
[Tb(4-NO2Tp)3]·C6H14 (8a).
White single crystals were isolated from methanol/hexanes. Yield (recrystallized): 50%. Elemental Anal. Calc. for TbC33H41O6N21B3: C 38.90, H 4.03, N 28.87. Found: C 38.71, H 4.00, N 28.92. τ = 1.87 ms (λexc = 486 nm (5D4), λem = 542 nm).
[Dy(4-NO2Tp)3]·C6H14 (9a).
White single crystals were isolated from methanol/hexanes. Yield (recrystallized): 36%. Elemental Anal. Calc. for DyC33H41O6N21B3: C 38.76, H 4.01, N 28.77. Found: C 38.78, H 3.92, N 28.66.
[Sm(4-NO2Tp)3]·7C6H6 (5b).
Pale yellow single crystals were isolated from methanol/benzene. Yield (recrystallized): 44%. Elemental Anal. Calc. for SmC69H69O6N21B3: C 56.33, H 4.69, N 20.00. Found: C 55.41, H 4.66, N 20.61.
[Tb(4-NO2Tp)3]·7C6H6 (8b).
White single crystals were isolated from methanol/benzene. Yield (recrystallized): 40%. Elemental Anal. Calc. for TbC69H69O6N21B3: C 56.01, H 4.67, N 19.88. Found: C 55.40, H 4.70, N 20.43.
X-ray structure determination
Crystals of 1a–9a, 5b and 8b, were harvested from mother liquors and mounted on 50 µm MiTeGen mounts. All measurements were made using monochromated microfocus Mo Kα (λ = 0.71073) radiation on a Bruker D8 Quest, equipped with a Photon II detector. All reflection data were collected at 100(2) K with 0.5° φ and ω scans. The data were reduced using SAINT,67 and empirical absorption corrections were applied using SADABS,68 for 1a–9a, 5b and 8b. Structure solutions solved using intrinsic phasing were performed using the ShelXT package69 in APEX III. All data were subsequently refined using SHELXL-2014 in the program SHELXle.70 All atoms were refined anisotropically. The reported CIFs for 1a–9a, feature structural models that were refined with scattering contributions from the disordered hexane sites removed from the diffraction data using the bypass procedure in PLATON.71 The electron count from the “squeezed” model converged in good agreement with a single hexane molecule. Aromatic hydrogen atoms were placed in idealized positions and allowed to ride on the coordinates of the parent atom with isotropic thermal parameters (Uiso) fixed at 1.2Ueq for all carbon atoms and at 1.5Ueq for all boron atoms. Details of the X-ray diffraction experiments and crystal data are summarized in Table 3.
Table 3 Crystallographic data for 1a–9a, 5b and 8b
| |
1a
|
2a
|
3a
|
4a
|
5a
|
5b
|
| CCDC no. |
2226885 |
2226886 |
2226887 |
2226888 |
2226889 |
2226890 |
| Formula |
LaN21O6B3C27H27 |
CeN21O6B3C27H27 |
PrN21O6B3C27H27 |
NdN21O6B3C27H27 |
SmN21O6B3C27H27 |
SmN21O6B3C69H69 |
| Formula weight |
913.04 |
914.25 |
915.04 |
918.37 |
924.49 |
1471.24 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Trigonal |
| Space group |
C2/c |
C2/c |
C2/c |
C2/c |
C2/c |
R |
|
a, Å |
24.473(5) |
24.400(8) |
24.369(6) |
24.2443(7) |
24.324(15) |
21.6878(4) |
|
b, Å |
13.830(3) |
13.801(3) |
13.783(3) |
13.7711(3) |
13.756(10) |
21.6878(4) |
|
c, Å |
29.778(4) |
28.778(10) |
28.792(7) |
28.8113(11) |
28.833(15) |
25.7723(8) |
|
α, ° |
90 |
90 |
90 |
90 |
90 |
90 |
|
β, ° |
116.898(5) |
116.759(12) |
112.453(8) |
112.537(1) |
112.49(2) |
90 |
|
γ, ° |
90 |
90 |
90 |
90 |
90 |
90 |
|
V, Å3 |
8988(3) |
8954(5) |
8937(4) |
8921.3(5) |
8913(10) |
10498.2(5) |
|
Z
|
8 |
8 |
8 |
8 |
8 |
6 |
|
T, K |
100 |
100 |
100 |
100 |
100 |
100 |
|
ρ
calc, g cm−3 |
1.349 |
1.356 |
1.360 |
1.367 |
1.378 |
1.396 |
|
μ, mm−1 |
1.011 |
1.077 |
1.150 |
1.224 |
1.378 |
0.907 |
|
λ, Mo Kα |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
|
R
int
|
0.0483 |
0.0590 |
0.0455 |
0.0639 |
0.0673 |
0.0403 |
| Residuals:aR; Rw |
0.0219; 0.0477 |
0.0236; 0.0546 |
0.0252; 0.0638 |
0.0295; 0.0651 |
0.0268; 0.0613 |
0.0175; 0.0426 |
| Goodness of fit |
1.022 |
1.042 |
1.049 |
1.007 |
0.996 |
1.071 |
| |
6a
|
7a
|
8a
|
8b
|
9a
|
|
R = R1 = ∑|Fo| − |Fc||/∑|Fo| for observed data only. Rw = wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2 for all data.
|
| CCDC no. |
2226891 |
2226892 |
2226893 |
2227039 |
2226894 |
| Formula |
EuN21O6B3C27H27 |
GdN21O6B3C27H27 |
TbN21O6B3C27H27 |
TbN21O6B3C69H69 |
DyN21O6B3C27H27 |
| Formula weight |
926.10 |
931.38 |
933.06 |
1479.80 |
936.63 |
| Crystal System |
Monoclinic |
Monoclinic |
Monoclinic |
Trigonal |
Monoclinic |
| Space group |
C2/c |
C2/c |
C2/c |
R |
C2/c |
|
a, Å |
24.313(16) |
24.296(6) |
24.248(2) |
21.6423(9) |
24.291(5) |
|
b, Å |
13.724(10) |
13.742(2) |
13.7248(17) |
21.6423(9) |
13.695(3) |
|
c, Å |
29.722(15) |
29.768(4) |
28.843(4) |
25.7873(17) |
29.707(8) |
|
α, ° |
90 |
90 |
90 |
90 |
90 |
|
β, ° |
115.893(13) |
116.414(8) |
112.506(4) |
90 |
115.032(5) |
|
γ, ° |
90 |
90 |
90 |
90 |
90 |
|
V, Å3 |
8922(10) |
8901(3) |
8867.9(18) |
10460.3(11) |
8954(4) |
|
Z
|
8 |
8 |
8 |
6 |
8 |
|
T, K |
100 |
100 |
100 |
100 |
100 |
|
ρ
calc, g cm−3 |
1.379 |
1.390 |
1.398 |
1.409 |
1.390 |
|
μ, mm−1 |
1.466 |
1.551 |
1.656 |
1.082 |
1.729 |
|
λ, Mo Kα |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
|
R
int
|
0.0643 |
0.0608 |
0.0394 |
0.0800 |
0.0477 |
| Residuals:aR; Rw |
0.0430; 0.0872 |
0.0251; 0.0629 |
0.0323; 0.0713 |
0.0355; 0.0689 |
0.0426; 0.0852 |
| Goodness of fit |
0.975 |
1.020 |
1.003 |
1.118 |
1.163 |
Powder X-ray diffraction
Powder X-ray diffraction (PXRD) data on the bulk recrystallization products from each sample were collected on a Rigaku Miniflex (Cu Kα 2θ = 5–60) and analyzed using the Match! software program. The PXRD patterns of the bulk products for 1a–9a, 5b and 8b were used to check purity and reproducibility and are provided in ESI Fig. S3 and S4.†
Infrared spectroscopy
Infrared spectra were collected from 650 to 4000 cm−1 using a PerkinElmer Frontier FT-IR spectrophotometer with a diamond attenuated total reflectance (ATR) sample holder. The IR spectra of the bulk, recrystallized products for 1a–9a, 5b and 8b were used to check purity and reproducibility and are provided in ESI Fig. S5 and S6.†
1H nuclear magnetic resonance spectroscopy
NMR spectra of crude 1–9 (no solvates) were recorded on a Varian 400 MHz spectrometer. Spectra of crude 1–9 (except 7) are provided in the ESI Fig. S4–S14.†
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was performed on a PerkinElmer Pyris 1 TGA. Samples were heated from 30–500 °C at 20 °C min−1 in air. TGA curves of 5a and 5b are given in Fig. S15† indicating thermal stability of [Ln(4-NO2Tp)3] up to ∼240 K, with solvent loss at 185 °C for a (hexanes) solvate and 77 °C for b (benzene) solvate.
Elemental analysis
Elemental analyses were performed by ALS Environment for 1a and 9a, and Atlantic Microlab for 2a–8a, 5b and 8b.
Photophysical measurements
Visible and NIR solid-state luminescence measurements were obtained at room temperature for 1a–9a (and 77 K for 6). Luminescence spectra were collected with a Horiba Jobin Yvon Fluorolog-3 spectrophotometer using a 450 W xenon arc lamp combined with a double excitation monochromator and double emission monochromator. For spectra in the visible region, a photomultiplier tube at 950 V was used as the emission detector, whereas for spectra in the near-IR region, a liquid nitrogen cooled, Symphony II NIR InGaAs diode array detector was used as the emission detector. Data were collected and analyzed using the FluorEssence software package. The solid samples were mounted on a J1933 Solid Sample Holder using non-emitting high vacuum grease for room temperature scans. Low temperature luminescence measurements were collected on solid samples under vacuum using a Janis VPF-100 cryostat equipped with UV-grade fused silica windows coupled with a Lakshore model 325 temperature controller. Lifetime measurements were collected with a Horiba Jobin Yvon Flurolog-3 spectrophotometer adapted for time-correlated single photon counting (TCSPC) measurements using a xenon flash lamp as the light source. Lifetime profiles for 5a, 6a and 8a were obtained using the TCSPC module and the data were fit using DAS6 software. Quantum yields measurements were collected in duplicate using a Horiba PTI QM-400 fluorometer using a PTFE powder holder under ambient conditions and an 8.9 cm integrating sphere with a Spectralon fluropolymer coating. The samples were crushed with a mortar and pestle, diluted with KBr in a 1:50 ratio of sample:KBr prior to data collection. Diffuse reflectance spectra were collected on solid samples at 298 K. The light source was a Mikropack DH-2000-BAL deuterium and halogen light source coupled with an Ocean Optics Flame detector. Scattered light was collected with a fiber-optic cable. Spectra were referenced with BaSO4. Data were processed using OceanView spectroscopy software. The electronic absorption spectrum of 1 was collected on a SPECORD 600 UV-VIS diode array spectrophotometer.
Magnetic SQUID measurements
Crystalline samples for static SQUID magnetometry were prepared inside a glovebox where a measured amount of sample was added to a half-sealed quartz tube using a glass pipet followed by a measured amount of eicosane. The top of the tube was then fitted to an Ultra Torr Swagelok adapter. This was taken out of the glovebox and the eicosane was melted using hot water to fix the sample. The assembly was then attached to a Schlenk line, and the top of the tube was sealed using an H2/O2 torch while the sample was under vacuum. The sealed tube was taped to a straw using Kapton tape and loaded onto the instrument. For the dynamic measurements, the samples were enclosed in a copper foil pouch and mounted on a quartz holder with Kapton tape. The molar diamagnetic susceptibilities of the compounds were estimated from their molar mass (χdia (cm3 mol−1) = −[MW (g mol−1) × 10−6]/2) and subtracted from the experimental value. Data was collected at 0.1 T for static measurements and 0.04 T for dynamic measurements for using a Quantum Design SQUID MPMS3 magnetometer.
Computational methods
Computational studies were conducted using the high-performance computing cluster at the George Washington University. The input structure for Calc-1 was derived from the crystal structure of 1a. The frontier molecular orbitals and the UV/VIS spectrum of Calc-1 were computed using density functional theory (DFT) in the Gaussian 16 software (Gaussian Inc.).72 A ground state optimization was performed on Calc-1, using the B3LYP73,74 level of theory with the modified scalar-relativistic effective core potential (ECP) basis set def2-TZVP as implemented in the software with the def2-TZVP pseudopotential applied to La3+.75–78 Geometry optimizations were performed on the input structure without symmetry constraints in the gas phase. Subsequent frequency calculations were performed on Calc-1 to confirm that the optimized structure was its global minimum. No imaginary frequencies were present in the calculated IR and Raman spectra. Time-dependent DFT calculations were then conducted to determine the expected absorption spectra of Calc-1 considering singlet (S0 → Sn) and triplet (S0 → Tn) transitions. 100 singlet and triplet states each were identified for Calc-1. NBO calculations were performed using NBO7 on two models. Model A consisted of two molecules of [La(3-NO2Tp)2(NO3)] (generated from crystallographically determined atomic coordinates of [Gd(3-NO2Tp)2(NO3)]·¼H2O) participating in a π–π stacking interaction. Model B comprised two [La(4-NO2Tp)]2+ units participating in a NO2⋯H–C hydrogen bond (generated from crystallographically determined atomic coordinates in 1a). NBO second-order perturbation theory was applied to quantify the magnitude of the donor–acceptor interaction between adjacent complexes as well as to identify the atomic and molecular orbitals involved with Jmol as the visualization software.79
Author contributions
The manuscript was written through contributions of all authors.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
This material is based upon work supported by the Department of Energy National Nuclear Security Administration through the Nuclear Science and Security Consortium under award number DE-NA0003180. This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or limited, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof. This study was also supported by the Mary Hopkinson Shepard Endowed Graduate Fellowship for Science Award through the Columbian College of Arts and Sciences (CCAS) at the George Washington University (CHH). This work was completed in part with resources provided by the High Performance Computing Cluster at the George Washington University, Research Technology Services. CHH acknowledges Alexander Marwitz (Georgetown University) for his help with determination of quantum yields, Grant Wilkinson (Georgia Institute of Technology) for collection of the static SQUID measurements and Michael Womble (The George Washington University) for collection of the TGA data.
References
- K. Binnemans, Lanthanide-Based Luminescent Hybrid Materials, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- A. Dey, P. Kalita and V. Chandrasekhar, Lanthanide(III)-Based Single-Ion Magnets, ACS Omega, 2018, 3, 9462–9475 CrossRef CAS PubMed.
- C. A. P. Goodwin, Blocking like it's hot: a synthetic chemists’ path to high-temperature lanthanide single molecule magnets, Dalton Trans., 2020, 49, 14320–14337 RSC.
- J. A. Smith, M. A. Singh-Wilmot, K. P. Carter, C. L. Cahill and J. A. Ridenour, Lanthanide-2,3,5,6-Tetrabromoterephthalic Acid Metal–Organic Frameworks: Evolution of Halogen⋯Halogen Interactions across the Lanthanide Series and Their Potential as Selective Bifunctional Sensors for the Detection of Fe3+, Cu2+, and Nitroaromatics, Cryst. Growth Des., 2019, 19, 305–319 CrossRef CAS.
- J. A. Smith, M. A. Singh-Wilmot, K. P. Carter, C. L. Cahill and J. A. Ridenour, Supramolecular assembly of lanthanide-2,3,5,6-tetrafluoroterephthalic acid coordination polymers via fluorine⋯fluorine interactions: a platform for luminescent detection of Fe3+ and nitroaromatic compounds, New J. Chem., 2020, 44, 12317–12330 RSC.
- J. H. S. K. Monteiro, F. A. Sigoli and A. D. Bettencourt-Dias, A water-soluble TbIII complex as a temperature-sensitive luminescent probe, Can. J. Chem., 2018, 96, 859–864 CrossRef CAS.
- J. F. C. B. Ramalho, S. F. H. Correia, L. Fu, L. M. S. Dias, P. Adão, P. Mateus, R. A. S. Ferreira and P. S. André, Super modules-based active QR codes for smart trackability and IoT: a responsive-banknotes case study, npj Flexible Electron., 2020, 4, 11 CrossRef.
- J. D. Rinehart and J. R. Long, Exploiting single-ion anisotropy in the design of f-element single-molecule magnets, Chem. Sci., 2011, 2, 2078–2085 RSC.
- S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Molecular single-ion magnets based on lanthanides and actinides: Design considerations and new advances in the context of quantum technologies, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS.
- D. I. Alexandropoulos, K. R. Vignesh, B. S. Dolinar and K. R. Dunbar, End-to-end azides as bridging ligands in lanthanide coordination chemistry: Magnetic and magnetocaloric properties of tetranuclear Ln4 (Ln = Gd, Dy) complexes exhibiting a rare rhombus topology, Polyhedron, 2018, 151, 255–263 CrossRef CAS.
- S. Demir, I.-R. Jeon, J. R. Long and T. D. Harris, Radical ligand-containing single-molecule magnets, Coord. Chem. Rev., 2015, 289, 149–176 CrossRef.
- S. Demir, M. I. Gonzalez, L. E. Darago, W. J. Evans and J. R. Long, Giant coercivity and high magnetic blocking temperatures for N23− radical-bridged dilanthanide complexes upon ligand dissociation, Nat. Commun., 2017, 8, 2144 CrossRef PubMed.
- C. A. Gould, E. Mu, V. Vieru, L. E. Darago, K. Chakarawet, M. I. Gonzalez, S. Demir and J. R. Long, Substituent Effects on Exchange Coupling and Magnetic Relaxation in 2,2′-Bipyrimidine Radical-Bridged Dilanthanide Complexes, J. Am. Chem. Soc., 2020, 142, 21197–21209 CrossRef CAS PubMed.
- R. Ishikawa, S. Michiwaki, T. Noda, K. Katoh, M. Yamashita, K. Matsubara and S. Kawata, Field-Induced Slow Magnetic Relaxation of Mono- and Dinuclear Dysprosium(III) Complexes Coordinated by a Chloranilate with Different Resonance Forms, Inorganics, 2018, 6, 7 CrossRef.
- R. Ishikawa, S. Michiwaki, T. Noda, K. Katoh, M. Yamashita and S. Kawata, Series of Chloranilate-Bridged Dinuclear Lanthanide Complexes: Kramers Systems Showing Field-Induced Slow Magnetic Relaxation, Magnetochemistry, 2019, 5(2), 30 CrossRef CAS.
- T. P. Latendresse, V. Vieru, A. Upadhyay, N. S. Bhuvanesh, L. F. Chibotaru and M. Nippe, Trends in trigonal prismatic Ln-[1]ferrocenophane complexes and discovery of a Ho3+ single-molecule magnet, Chem. Sci., 2020, 11, 3936–3951 RSC.
- K. R. Meihaus, J. D. Rinehart and J. R. Long, Dilution-Induced Slow Magnetic Relaxation and Anomalous Hysteresis in Trigonal Prismatic Dysprosium(III) and Uranium(III) Complexes, Inorg. Chem., 2011, 50, 8484–8489 CrossRef CAS PubMed.
- K. R. Meihaus, S. G. Minasian, W. W. Lukens, S. A. Kozimor, D. K. Shuh, T. Tyliszczak and J. R. Long, Influence of Pyrazolate vs N-Heterocyclic Carbene Ligands on the Slow Magnetic Relaxation of Homoleptic Trischelate Lanthanide(III) and Uranium(III) Complexes, J. Am. Chem. Soc., 2014, 136, 6056–6068 CrossRef CAS PubMed.
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, On Approaching the Limit of Molecular Magnetic Anisotropy: A Near-Perfect Pentagonal Bipyramidal Dysprosium(III) Single-Molecule Magnet, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed.
- K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, High-temperature magnetic blocking and magneto-structural correlations in a series of dysprosium(III) metallocenium single-molecule magnets, Chem. Sci., 2018, 9, 8492–8503 RSC.
- C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II), J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS PubMed.
- C. A. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Molecular magnetic hysteresis at 60 kelvin in dysprosocenium, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- J. Long, Y. Guari, R. A. S. Ferreira, L. D. Carlos and J. Larionova, Recent advances in luminescent lanthanide based Single-Molecule Magnets, Coord. Chem. Rev., 2018, 363, 57–70 CrossRef CAS.
- K. S. Kumar, D. Serrano, A. M. Nonat, B. Heinrich, L. Karmazin, L. J. Charbonnière, P. Goldner and M. Ruben, Optical spin-state polarization in a binuclear europium complex towards molecule-based coherent light-spin interfaces, Nat. Commun., 2021, 12, 2152 CrossRef CAS PubMed.
- E. M. Matson, J. J. Kiernicki, N. H. Anderson, P. E. Fanwick and S. C. Bart, Isolation of a uranium(III) benzophenone ketyl radical that displays redox-active ligand behaviour, Dalton Trans., 2014, 43, 17885–17888 RSC.
- S. Isabel and N. Marques, Recent advances in the chemistry of f-element poly(pyrazolyl)borate complexes, New J. Chem., 1995, 19, 55–571 Search PubMed.
- A. J. Amoroso, A. M. C. Thompson, J. C. Jeffery, P. L. Jones, J. A. McCleverty and M. D. Ward, Synthesis of the new tripodal ligand tris-[3-(2′-pyridyl)pyrazol-1-yl]hydroborate, and the crystal structure of its europium(III) complex, J. Chem. Soc., Chem. Commun., 1994, 2751–2752, 10.1039/C39940002751.
- J. Fleming, E. Psillakis, S. Couchman, J. Jeffery, J. McCleverty and M. Ward, Complexes of the potentially hexadentate ligand bis{3-[6-(2,2′-bipyridyl)]pyrazol-1-yl}hydroborate with representative s-, p-, d- and f-block metal ions: factors promoting formation of mononuclear or double-helical dinuclear complexes, Dalton Trans., 1998, 537–544, 10.1039/a707936b.
- H. Flötotto, T. Secker, P. Kögerler and C. Besson, Amine-Functionalized Spin Crossover Building Blocks, Eur. J. Inorg. Chem., 2019, 2019, 4621–4624 CrossRef PubMed.
- P. Fang, L. Wang, G. Zhan, W. Yan, P. Huo, A. Ying, Y. Zhang, Z. Zhao, G. Yu, Y. Huang, S. Gong, L. Duan, Z. Liu, Z. Bian and C. Huang, Lanthanide Cerium(III) Tris(pyrazolyl)borate Complexes: Efficient Blue Emitters for Doublet Organic Light-Emitting Diodes, ACS Appl. Mater. Interfaces, 2021, 13, 45686–45695 CrossRef CAS PubMed.
- W. Yan, L. Wang, H. Qi, G. Zhan, P. Fang, Z. Liu and Z. Bian, Highly Efficient Heteroleptic Cerium(III) Complexes with a Substituted Pyrazole Ancillary Ligand and Their Application in Blue Organic Light-Emitting Diodes, Inorg. Chem., 2021, 60, 18103–18111 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Slow magnetic relaxation in homoleptic trispyrazolylborate complexes of neodymium(III) and uranium(III), Dalton Trans., 2012, 41, 13572–13574 RSC.
- J. D. Rinehart, K. R. Meihaus and J. R. Long, Observation of a Secondary Slow Relaxation Process for the Field-Induced Single-Molecule Magnet U(H2BPz2)3, J. Am. Chem. Soc., 2010, 132, 7572–7573 CrossRef CAS PubMed.
- E. A. Mikhalyova, M. Zeller, J. P. Jasinski, R. J. Butcher, L. M. Carrella, A. E. Sedykh, K. S. Gavrilenko, S. S. Smola, M. Frasso, S. C. Cazorla, K. Perera, A. Shi, H. G. Ranjbar, C. Smith, A. Deac, Y. Liu, S. M. McGee, V. P. Dotsenko, M. U. Kumke, K. Müller-Buschbaum, E. Rentschler, A. W. Addison and V. V. Pavlishchuk, Combination of single-molecule magnet behaviour and luminescence properties in a new series of lanthanide complexes with tris(pyrazolyl)borate and oligo(β-diketonate) ligands, Dalton Trans., 2020, 49, 7774–7789 RSC.
- M. L. P. Reddy and S. Sivakumar, Lanthanide benzoates: a versatile building block for the construction of efficient light emitting materials, Dalton Trans., 2013, 42, 2663–2678 RSC.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, Correlation between the lowest triplet state energy level of the ligand and lanthanide(III) luminescence quantum yield, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- V. Tsaryuk, K. Zhuravlev, V. Zolin, P. Gawryszewska, J. Legendziewicz, V. Kudryashova and I. Pekareva, Regulation of excitation and luminescence efficiencies of europium and terbium benzoates and 8-oxyquinolinates by modification of ligands, J. Photochem. Photobiol., A, 2006, 177, 314–323 CrossRef CAS.
- V. Tsaryuk, V. Kudryashova, P. Gawryszewska, R. Szostak, A. Vologzhanina, K. Zhuravlev, Z. Klemenkova, J. Legendziewicz and V. Zolin, Structures, luminescence and vibrational spectroscopy of europium and terbium nitro- and dinitro-substituted benzoates. Nitro groups as quenchers of Ln3+ luminescence, J. Photochem. Photobiol., A, 2012, 239, 37–46 CrossRef CAS.
- S. Viswanathan and A. De Bettencourt-Dias, Eu(III) and Tb(III) Luminescence Sensitized by Thiophenyl-Derivatized Nitrobenzoato Antennas, Inorg. Chem., 2006, 45, 10138–10146 CrossRef CAS PubMed.
- A. D. Bettencourt-Dias and S. Viswanathan, Nitro-functionalization and luminescence quantum yield of Eu(III) and Tb(III) benzoic acid complexes, Dalton Trans., 2006, 4093–4103, 10.1039/B606970C.
- A. P. S. Samuel, J. Xu and K. N. Raymond, Predicting Efficient Antenna Ligands for Tb(III) Emission, Inorg. Chem., 2009, 48, 687–698 CrossRef CAS PubMed.
- Sushila, R. Siddiqui, S. Patra, K. Shivam, A. Sil, B. Guchhait, H. Tian, R. Kataria, S. Goswami, P. Venugopalan and R. Patra, Halogen Bond Mediated Self-Assembly of Mononuclear Lanthanide Complexes: Perception of Supramolecular Interactions, Slow Magnetic Relaxation, and Photoluminescence Properties, Inorg. Chem., 2022, 61, 11484–11496 CrossRef CAS PubMed.
- D. A. Gálico, R. Marin, G. Brunet, D. Errulat, E. Hemmer, F. A. Sigoli, J. O. Moilanen and M. Murugesu, Triplet-State Position and Crystal-Field Tuning in Opto-Magnetic Lanthanide Complexes: Two Sides of the Same Coin, Chem. Eur. J., 2019, 25(64), 14625–14637 CrossRef PubMed.
- C. Ma and C. Besson, Precise control of the degree and regioselectivity of functionalization in nitro- and amino-functionalized di(trispyrazolylborato)iron(II) spin crossover complexes, Dalton Trans., 2021, 50, 18077–18088 RSC.
- C. H. Hossack, R. J. Butcher, C. L. Cahill and C. Besson, Structural Diversity of Lanthanide 3-Nitrotrispyrazolylborates: Tunable Nuclearity and Intra-Ligand Charge Transfer Sensitization of Visible and NIR Ln3+ Emission, Inorg. Chem., 2021, 60, 15724–15743 CrossRef CAS PubMed.
- C. Apostolidis, J. Rebizant, B. Kanellakopulos, R. von Ammon, E. Dornberger, J. Müller, B. Powietzka and B. Nuber, Homoscorpionates (hydridotris(1-pyrazolyl)borato complexes) of the trivalent 4fions. The crystal and molecular structure of [(HB(N2C3H3)3]3LnIII, (Ln = Pr, Nd), Polyhedron, 1997, 16, 1057–1068 CrossRef CAS.
- G. Crosby, R. Whan and R. Alire, Intramolecular energy transfer in rare earth chelates. Role of the triplet state, J. Chem. Phys., 1961, 34, 743–748 CrossRef CAS.
- W. Sager, N. Filipescu and F. Serafin, Substituent effects on intramolecular energy transfer. I. Absorption and phosphorescence spectra of rare earth β-diketone chelates, J. Phys. Chem., 1965, 69, 1092–1100 CrossRef CAS.
- K. Binnemans, Interpretation of europium(III) spectra, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- J. G. Santos, J. D. L. Dutra, S. A. Junior, R. O. Freire and N. B. da Costa Junior, Theoretical Spectroscopic Study of Europium Tris(bipyridine) Cryptates, J. Phys. Chem. A, 2012, 116, 4318–4322 CrossRef CAS PubMed.
-
A. De Bettencourt-Dias, in Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, 2014, pp. 1–48, DOI:10.1002/9781118682760.ch01.
- P. A. Tanner, Some misconceptions concerning the electronic spectra of tri-positive europium and cerium, Chem. Soc. Rev., 2013, 42, 5090–5101 RSC.
- C.-L. Liu, R.-L. Zhang, C.-S. Lin, L.-P. Zhou, L.-X. Cai, J.-T. Kong, S.-Q. Yang, K.-L. Han and Q.-F. Sun, Intraligand Charge Transfer Sensitization on Self-Assembled Europium Tetrahedral Cage Leads to Dual-Selective Luminescent Sensing toward Anion and Cation, J. Am. Chem. Soc., 2017, 139, 12474–12479 CrossRef CAS PubMed.
- K. D. Matthews, S. A. Bailey-Folkes, I. A. Kahwa, G. L. McPherson, C. A. O'Mahoney, S. V. Ley, D. J. Williams, C. J. Groombridge and C. A. O'Connor, Luminescence Dynamics and 13C NMR Characteristics of Dlnuclear Complexes Exhibiting Coupled Lanthanide(III) Cation Pairs, J. Phys. Chem., 1992, 96, 7021–7027 CrossRef CAS.
- K. Binnemans and C. Görller-Walrand, On the color of the trivalent lanthanide ions, Chem. Phys. Lett., 1995, 235, 163–174 CrossRef CAS.
-
E. Bauer and M. Rotter, in Properties and Applications of Complex Intermetallics, 2009, pp. 183–248, DOI:10.1142/9789814261647_0005.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, PHI: A powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- D. Reta and N. F. Chilton, Uncertainty estimates for magnetic relaxation times and magnetic relaxation parameters, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
- A. Lunghi and S. Sanvito, The Limit of Spin Lifetime in Solid-State Electronic Spins, J. Phys. Chem. Lett., 2020, 11(15), 6273–6278 CrossRef CAS PubMed.
- M. Bortoluzzi, G. Paolucci, S. Polizzi, L. Bellotto, F. Enrichi, S. Ciorba and B. S. Richards, Photoluminescence studies on europium-based scorpionate-complex, Inorg. Chem. Commun., 2011, 14, 1762–1766 CrossRef CAS.
- G. F. de Sá, O. L. Malta, C. de Mello Donegá, A. M. Simas, R. L. Longo, P. A. Santa-Cruz and E. F. da Silva, Spectroscopic properties and design of highly luminescent lanthanide coordination complexes, Coord. Chem. Rev., 2000, 196, 165–195 CrossRef.
- S.-N. Liu, K.-N. Tong, Y. Zhao, J.-F. Cheng, M.-K. Fung and J. Fan, Efficient red phosphorescent Ir(III) complexes based on rigid ligands with high external quantum efficiency and low efficiency roll-off, J. Mater. Chem. C, 2020, 8, 6168–6175 RSC.
- J.-C. Jin, N.-N. Shen, Y.-P. Lin, L.-K. Gong, H.-Y. Tong, K.-Z. Du and X.-Y. Huang, Modulation of the Structure and Photoluminescence of Bismuth(III) Chloride Hybrids by Altering the Ionic-Liquid Cations, Inorg. Chem., 2020, 59, 13465–13472 CrossRef CAS PubMed.
- J.-C. Jin, Y.-P. Lin, Y.-H. Wu, L.-K. Gong, N.-N. Shen, Y. Song, W. Ma, Z.-Z. Zhang, K.-Z. Du and X.-Y. Huang, Long lifetime phosphorescence and X-ray scintillation of chlorobismuthate hybrids incorporating ionic liquid cations, J. Mater. Chem. C, 2021, 9, 1814–1821 RSC.
- A. C. Marwitz, A. D. Nicholas, L. M. Breuer, J. A. Bertke and K. E. Knope, Harnessing Bismuth Coordination Chemistry to Achieve Bright, Long-Lived Organic Phosphorescence, Inorg. Chem., 2021, 60, 16840–16851 CrossRef CAS PubMed.
- C. Apostolidis, A. Kovács, A. Morgenstern, J. Rebizant and O. Walter, Tris-{Hydridotris(1-pyrazolyl)borato}lanthanide Complexes: Synthesis, Spectroscopy, Crystal Structure and Bonding Properties, Inorganics, 2021, 9(6), 44 CrossRef CAS.
-
SAINT, version 8.34a
Bruker AXS Inc.: Madison, WI
2013
Search PubMed.
-
G. Sheldrick, Bruker AXS Inc.: Madison, WI, 2005.
- G. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. B. Hubschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- A. Spek, Platon/Squeeze, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Density–functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. Dolg, H. Stoll, A. Savin and H. Preuss, Energy-adjusted pseudopotentials for the rare earth elements, Theor. Chim. Acta, 1989, 75, 173–194 CrossRef CAS.
- R. Gulde, P. Pollak and F. Weigend, Error-Balanced Segmented Contracted Basis Sets of Double-ζ to Quadruple-ζ Valence Quality for the Lanthanides, J. Chem. Theory Comput., 2012, 8, 4062–4068 CrossRef CAS PubMed.
- M. Dolg, H. Stoll and H. Preuss, A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds, Theor. Chim. Acta, 1993, 85, 441–450 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Jmol: an open-source Java viewer for chemical structures in 3D, https://www.jmol.org/.Journal.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, IR, NMR and reflectance spectra, TGA curves, computational details, Judd–Ofelt analysis, DC and AC SQUID data fits and parameters (PDF). CCDC 2226885–2226894 and 2227039. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01018j |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Folasade
Abdul
,
Christopher
Cahill
,
Folasade
Abdul
,
Christopher
Cahill







 ), 8a (Tb,
), 8a (Tb,  ) and 9a (Dy, Δ). Solid lines correspond to simulations using the crystal field parameters of set 1 in Table 1, dashed lines to those using set 2.
) and 9a (Dy, Δ). Solid lines correspond to simulations using the crystal field parameters of set 1 in Table 1, dashed lines to those using set 2.

 and a direct term (AT) yielded a sufficient model, however the Ueff was significantly smaller than what is expected for a true Orbach process at 14 cm−1. We therefore turned to a model with direct and two-phonons Raman terms, which yielded a nearly identical Ueff, or rather Evib at 13 cm−1 (Fig. S30†), corresponding to phonon-coupled under-barrier relaxation process rather than a true ΔmJ transition/Orbach process.
and a direct term (AT) yielded a sufficient model, however the Ueff was significantly smaller than what is expected for a true Orbach process at 14 cm−1. We therefore turned to a model with direct and two-phonons Raman terms, which yielded a nearly identical Ueff, or rather Evib at 13 cm−1 (Fig. S30†), corresponding to phonon-coupled under-barrier relaxation process rather than a true ΔmJ transition/Orbach process.


