
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaching strong absorption up to 700 nm with new benzo[g]quinoxaline-based heteroleptic copper(I) complexes for light-harvesting applications†
Cecilia
Bruschi
 a,
Xin
Gui
b,
Olaf
Fuhr
cd,
Wim
Klopper
bc and
Claudia
Bizzarri
*a
a,
Xin
Gui
b,
Olaf
Fuhr
cd,
Wim
Klopper
bc and
Claudia
Bizzarri
*a
aInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76137 Karlsruhe, Germany. E-mail: bizzarri@kit.edu
bInstitute of Physical Chemistry–Theoretical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany
cInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dKarlsruhe Nano Micro Facility (KNMFi), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 3rd May 2023
Abstract
Heteroleptic copper(I) complexes, with a diimine as a chromophoric unit and a bulky diphosphine as an ancillary ligand, have the advantage of a reduced pseudo Jahn–Teller effect in their excited state over the corresponding homoleptic bis(diimine) complexes. Nevertheless, their lowest absorption lies generally between 350 to 500 nm. Aiming at a strong absorption in the visible by stable heteroleptic Cu(I) complexes, we designed a novel diimine based on 4-(benzo[g]quinoxal-2′-yl)-1,2,3-triazole derivatives. The large π-conjugation of the benzoquinoxaline moiety shifted bathochromically the absorption with regard to other diimine-based Cu(I) complexes. Adding another Cu(I) core broadened the absorption and extended it to considerably longer wavelengths. Moreover, by fine-tuning the structure of the dichelating ligand, we achieved a panchromatic absorption up to 700 nm with a high molar extinction coefficient of 8000 M−1 cm−1 at maximum (λ = 570 nm), making this compound attractive for light-harvesting antennae.
Introduction
The economic evolution is deeply connected to the consumption of fossil fuels, which has rapidly increased over the years. However, this energy source is not limitless and is renewable only in an unsustainable time.1 Moreover, the combustion of fossil fuels leads to an increased CO2 concentration in the atmosphere, responsible for aggravating global warming. Therefore, moving our economy towards renewable energy sources is urgent. The most abundant, cleanest, and easily accessible energy is solar light.2 Nature can catch and use light, converting water and CO2 into organic compounds, like glucose, through a cascade of different processes.3 In particular, natural photosynthesis converts solar into chemical energy through photoinduced charge separation reactions, happening in complex reaction centres. Light-harvesting systems surrounding those reaction centres are composed of many chromophores and proteins to absorb and amplify the energy.4 To develop artificially solar-energy conversion schemes, many scientists have designed efficient light-absorbing compounds, which can be used in artificial light-harvesting arrays.5 As for natural photosynthetic systems, thus chromophores should absorb a large portion of the visible spectrum with high molar extinction coefficients.6,7 This purpose was achieved, for example, by Arrigo et al. building dendrimers based on multinuclear Ru(II) and Os(II) polypyridine.8 A panchromatic single photosensitizer based on Os(II) was also recently reported by Irikura et al. and successfully employed in photocatalytic CO2 reduction.9 The choice of these compounds is related to their appealing photophysical properties, such as absorption in the visible spectrum and long-lived excited states, and the possibility of tuning their absorptions by modulating their ligands. However, these compounds are based on rare and expensive metals. The element abundance on the earth crust should be considered for future scalability and application.10 Copper(I) complexes have emerged as a promising alternative to rare metal-based complexes.11 Indeed, not only copper(I) is an earth-abundant metal, but its 3d10 configuration does not allow any metal-centred (MC) excited states, known to preferentially deactivate through nonradiative pathways, enabling the population of metal-to-ligand charge-transfer (MLCT) states. However, the photophysical properties of copper(I) complexes are highly affected by the Jahn–Teller distortion of their excited state, favouring exciplex quenching processes and the formation of a low energy flattened state.12,13 Heteroleptic diimine–diphosphine copper complexes, (NN)Cu(PP), have superior photophysical properties than homoleptic Cu(NN)2 complexes,14,15 since the introduction of bulky and rigid phosphine reduces the flexibility of the system and enlarges the sterical hindrance in the excited state, disfavouring the formation of the flattened geometry.16 In heteroleptic (NN)Cu(PP) complexes, the lowest unoccupied molecular orbital (LUMO) is generally localized on the diimine ligand. The phosphine ligands contribute to the HOMO, mainly confined to the metal core. As the π-acidity of the phosphines lowers the HOMO level, the MLCT transitions in (NN)Cu(PP) generally lie between 360–450 nm; thus, they are hypsochromic shifted in comparison to the absorption of Cu(NN)2, usually covering the 450–700 nm range.13,17–19 This feature makes the synthesis of heteroleptic diimine–diphosphine complexes with a broader absorption in the visible spectrum challenging. By now, there are still a few cases of heteroleptic diimine–diphosphine copper(I) complexes showing an absorption at longer wavelengths than 450 nm.20–25 In this work, we aimed at new heteroleptic Cu(I) complexes of type (NN)Cu(PP) with a broad and intense absorption in the visible range. We used bis[(2-diphenylphosphino) phenyl] ether (DPEPhos) as chelating diphosphine and as diimine ligands, 4-(benzo[g]quinoxal-2′-yl)-1,2,3-triazole-derivatives. Benzoquinoxalines have been mainly synthesized for biological applications,26 as their scaffold is contained in numerous bioactive molecules. Even so, their luminescent properties made them also appealing as emitters in OLED devices.27 Concerning metal complexes, benzoquinoxaline-derivatives were used only in few cases, as ligands for bimetallic osmium complexes28 and bimetallic or trimetallic ruthenium complexes.29 As far as we know, this work is the first one where benzoquinoxaline-triazoles are used as chelating ligands for Cu(I). We synthesized three novel heteroleptic Cu(I) complexes: a mononuclear and two binuclear ones. The two binuclear copper complexes differ in the presence or absence of a methylene spacer between the benzoquinoxaline and triazole rings. This small diversity impacts the strength of the bond between the ligand and the copper centres, and in turn, on the photophysical properties. Their photophysical and electrochemical properties have been investigated and compared with the theoretical calculations. The π-extension of the benzoquinoxaline ring, combined with the presence of two copper cores, allows absorption at longer wavelengths than 450 nm and up to 700 nm, making them appealing for light-harvesting antennae applications.Synthesis and structure
The synthesis of the monochelating ligand 4 and of the corresponding dichelating ligand 5, both bearing a methylene spacer between the benzoquinoxaline ring and the triazole unit, was straightforward. It involves two simple and efficient steps: a fast condensation in MeOH, at room temperature, between 2,3-diaminonaphthalene and the corresponding diketone derivative (1-bromobutan-2,3-dione for 7 and 1,4-dibromobutane-2,3-dione for 9), followed by a Cu alkyne–azide cycloaddition (CuAAC) reaction, adding NaN3 and ethynylbenzene as reactants.24,30 The yields of the final ligands 4 and 5 are 54% and 44%, respectively. Differently, the synthesis of the dichelating ligand 6 required harsher conditions (Scheme 1). The first step is the condensation between 2,3-diaminonaphthalene and 1,6-bis(triisopropylsilyl)hexa-1,5-diyn-3,4-dione, previously prepared. This reaction needed higher temperature (80 °C) and longer time in respect to the other condensation reactions. Thus, it was performed in a mixture of ethanol and acetic acid (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), under reflux, overnight.27 For the deprotection of the so-formed 2,3-bis((triisopropylsilyl)ethynyl)benzo[g]quinoxaline 9 a solution of tetrabutylammonium fluoride (TBAF) (1 M, THF) was added dropwise at −78° C. Finally, 10 underwent a CuAAC reaction, employing benzylazide as a reactant. While for ligands 4 and 5, precipitation with pentane gave pure compounds, the CuAAC reaction of 6 required additional purification via column chromatography, affording product 3 with low yield (13%). A general procedure was followed for the coordination of the metal core.31 In an anhydrous dichloromethane solution of a Cu(I) precursor (Cu(CH3CN)4BF4) and the dichelating phosphine DPEPhos, at room temperature under Ar.
:3), under reflux, overnight.27 For the deprotection of the so-formed 2,3-bis((triisopropylsilyl)ethynyl)benzo[g]quinoxaline 9 a solution of tetrabutylammonium fluoride (TBAF) (1 M, THF) was added dropwise at −78° C. Finally, 10 underwent a CuAAC reaction, employing benzylazide as a reactant. While for ligands 4 and 5, precipitation with pentane gave pure compounds, the CuAAC reaction of 6 required additional purification via column chromatography, affording product 3 with low yield (13%). A general procedure was followed for the coordination of the metal core.31 In an anhydrous dichloromethane solution of a Cu(I) precursor (Cu(CH3CN)4BF4) and the dichelating phosphine DPEPhos, at room temperature under Ar.
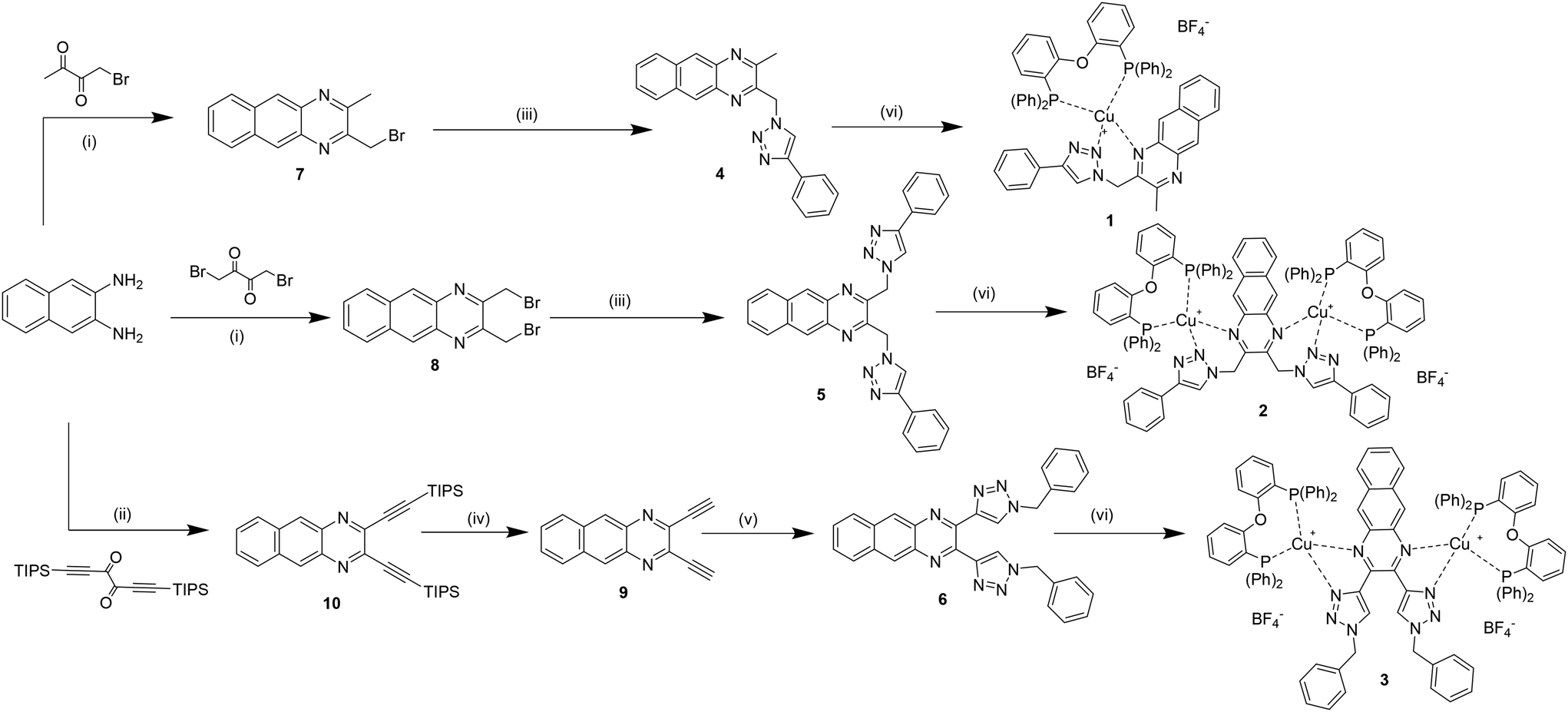 | ||
| Scheme 1 Synthesis of the benzoquinoxaline-based ligands (4–6) and their complexes (1–3). (i) NH4Cl, in MeOH, room T; (ii) in EtOH:AcOH (7:3, v/v), reflux; (iii) NaN3, phenylacetylene, CuSO4, NaCO3, in EtOH:H2O (7:3, v/v) at room T; (iv) TBAF in dry THF, −78 °C; (v) benzylazide, CuSO4, NaCO3, in EtOH:H2O (7:3, v/v) at room T; (vi) Cu(CH3CN)4BF4, DPEPhos, in dry DCM, Ar atmosphere, room T. | ||
The ratio between ligand and DPEPhosCu(CH3CN)2BF4 was 1:1 and 1:2 for the synthesis of 1 and 2, respectively. For the binuclear copper complex 3, DPEPhosCu(CH3CN)2BF4 was added in slight excess. Indeed, reacting 6 and the other starting materials in a precise ratio of 1:2, two species are formed: the desired binuclear copper complex and a not isolated species that is suspected to be the mononuclear copper complex, where the Cu(I) is coordinating with the nitrogen atoms of the two triazoles, which are then pointing internally. This mononuclear copper complex is probably the kinetic product of the coordination reaction, which reacts further with an excess of DPEPhosCu(CH3CN)2BF4 to give the desired binuclear copper complex 3 as the only species. The change from the copper complex mixture to the isolation of the binuclear complex was monitored by 1H NMR analysis, which shows the disappearance of the peaks attributed to the mononuclear species after the reaction (see Fig. S3.1 and S3.2†).
It is important to underline that this binuclear copper complex 3, compared to the binuclear copper complex 2, does not present the flexible methylene spacer between the benzoquinoxaline and the triazole ring. Moreover, the triazoles coordinate the metal centres with the N atoms in position 3, that were already proved to be higher σ-donor24,32–35 than the N atoms in position 2 of the 1,2,3-triazoles, involved in the coordination bonds of complex 2, instead (Scheme 1).
The mononuclear copper complex 1 gave crystals suitable for X-ray analysis, by diffusion of a cyclohexane layer to a concentrated dichloromethane solution of the compound. The complex crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two formula units per unit cell. As expected, the cation in 1 (Fig. 1) shows a pseudotetrahedral geometry with a bite angle degree of 90.2° for the N(2)–Cu–N(4) angle and of 113.90° for the P(1)–Cu–P(2). The distances between the copper centre and the nitrogen atoms have a value of 2.091 Å, 2.117 Å; while the value for the distances between the copper and the phosphorous atoms are higher and equal to 2.244 Å, 2.295 Å. These results are consistent with the ones obtained for quinoxaline-based copper complexes, previously published by our group.24 As for this latter class of compounds, the presence of the methylene spacer induces a higher flexibility of the diimine bite angle that is wider compared to those in similar copper complexes, where the methylene spacer was absent.24,31,36
with two formula units per unit cell. As expected, the cation in 1 (Fig. 1) shows a pseudotetrahedral geometry with a bite angle degree of 90.2° for the N(2)–Cu–N(4) angle and of 113.90° for the P(1)–Cu–P(2). The distances between the copper centre and the nitrogen atoms have a value of 2.091 Å, 2.117 Å; while the value for the distances between the copper and the phosphorous atoms are higher and equal to 2.244 Å, 2.295 Å. These results are consistent with the ones obtained for quinoxaline-based copper complexes, previously published by our group.24 As for this latter class of compounds, the presence of the methylene spacer induces a higher flexibility of the diimine bite angle that is wider compared to those in similar copper complexes, where the methylene spacer was absent.24,31,36
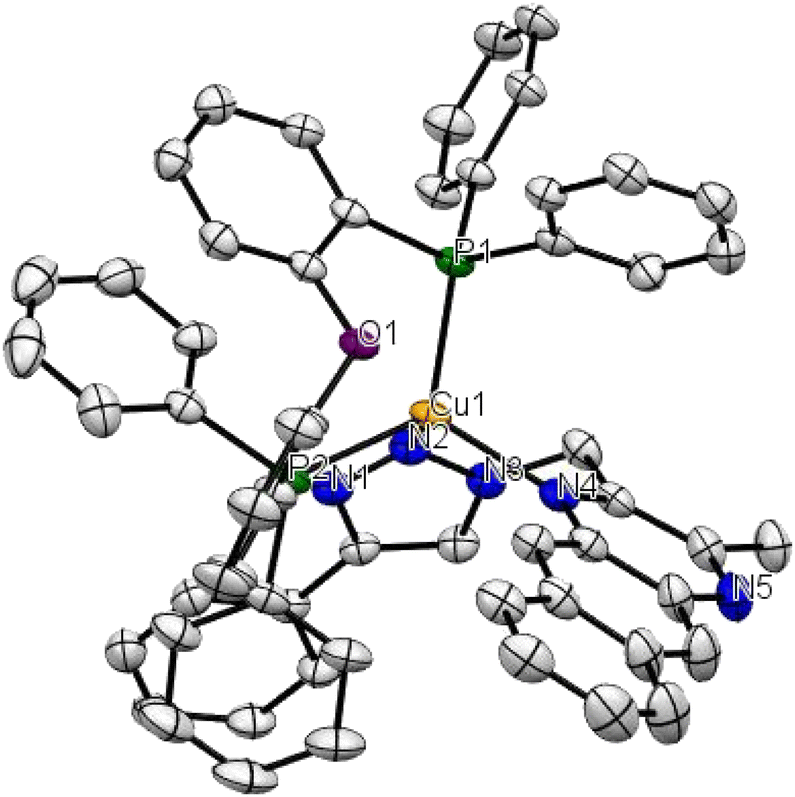 | ||
| Fig. 1 ORTEP drawing of the molecular structure of 1. Hydrogen atoms, counterion, and solvent molecules were omitted for clarity. | ||
Photophysical characterization
UV-vis absorption spectra were recorded for all compounds in dichloromethane (DCM). To understand the nature of the recorded transition bands, and to investigate thoroughly the strength of the coordination bonds, a comparison between the absorption spectra of the copper complexes 1–3 and their corresponding ligands 4–6 was done (Fig. 2a–c). For an easy comparison, the absorption spectra of the three complexes, are reported together (Fig. 2d and Table 1). | ||
| Fig. 2 Absorption spectra recorded in dichloromethane solution: (a) comparison between the absorption spectra of the copper complex 1 and its corresponding ligand 4; (b) comparison between the absorption spectra of the copper complex 2 and its corresponding ligand 5; (c) comparison between the absorption spectra of the copper complex 3 and its corresponding ligand 6; (d) comparison between the absorption spectra of the three new copper complexes 1. | ||
| Compound | λ abs/nm (ε/104 M−1 cm−1) |
|---|---|
| 1 | 272 (9.99), 357 (0.67), 371 (0.83), 415 (0.28), 470 (0.16) |
| 2 | 279 (9.27), 359 (0.43), 377 (0.63), 480 (0.19) |
| 3 | 275 (5.79), 330 (3.62), 405 (0.79), 424 (0.99), 568 (0.81) |
The absorption spectra of the mononuclear copper complex 1 and of its corresponding binuclear copper complex 2 show in the UV region an intense band (ε ≅ 1 × 105 M−1 cm−1) around 270 nm and much less intense peaks (ε ≅ 4–8 × 103 M−1 cm−1) in the 300–400 nm range. Since their corresponding ligands 4 and 5 show similar features, these bands could be attributed to ligand centred (1LC) transitions on the diimine ligands. On the other hand, the increased absorption of the copper complexes at around 300 nm could be attributed to the 1LC localized on the phosphine ligand. These results are consistent with the ones obtained for previously published heteroleptic copper complexes, bearing a DPEPhos ligand, showing a 1LC peak in the same region.24,36–38 The copper complexes 1 and 2 present in addition a red-shifted band in the visible region, extended from 400 nm to 550 nm for 1 and to 650 nm for 2. These bands could be assigned to the population of a metal-to-ligand charge transfer state 1MLCT. According to theoretical calculations (vide infra), a contribution of a 1MLLCT (metal–ligand-to-ligand charge transfer) cannot be excluded. Comparing the absorption of the binuclear copper complex 2 with that one of its corresponding mononuclear complex 1, there is a red-shift (≅0.05 eV) of the visible band (1MLCT/1MLLCT) in 2, which could be attributed to a cooperative effect between the metal centres.24 The moderate change in the absorption of the complexes concerning their ligands might be caused by a weak coordination of the Cu(I) centre, as previously discussed, affecting their photophysical properties.
The binuclear copper complex 3 shows intense peaks in the UV region at about 270 nm (ε ≅ 6 × 104 M−1 cm−1) and 330 nm (ε ≅ 4 × 104 M−1 cm−1), and both can be assigned to 1LC transitions, as for complexes 1 and 2. Therefore, the same attribution can be done for the peaks lying in the visible range, between 400 and 450 nm (ε ≅ 5–10 × 103 M−1 cm−1), which presents a similar profile to the ones of the 1 and 2, although slightly red-shifted and more intense. In the visible region, the absorption spectrum of the copper complex 3 shows an extended band, going from 450 nm to 700 nm, with a maximum peak at about 570 nm. The oscillator strength of this transition increased significantly, having an absorptivity coefficient of almost 8000 cm−1 M−1. To the best of our knowledge, this copper complex presents one of the most extended absorption with a considerable extinction coefficient (ε > 200 M−1 cm−1 up to 650 nm) compared with other heteroleptic Cu(I) complexes of type (NN)Cu(PP). The binuclear DPEPhosCu(I) complex with a quinolyl–triazole from our previous work presents an absorption up to 800 nm; nevertheless, the extinction coefficient was lower than 250 M−1 cm−1 at longer wavelengths than 600 nm.24 So far, among the (NN)Cu(PP) complexes with long absorption, there are the 4H-imidazolate-based complexes, with a 1MLCT maximum of 520 nm in DCM.21,22 4H-Imidazolate ligands were also used in homoleptic Cu(I) complexes, reaching a panchromatic light absorption up to 900 nm.19 However, we have already discussed that the substitution of a chelating phosphine with a diimine ligand leads to a bathochromic shift.13,18,39 Moreover, the 1MLCT of the binuclear copper complex 3 is much more intense compared to that of the binuclear copper complex 2 (ε ≅ 8 × 103 M−1 cm−1 for 3 and ε ≅ 2 × 103 M−1 for 2). Lastly, the much more significative change between the absorption spectra of the copper complex 3 and of its corresponding ligand 6, leads to conclude that the absence of a flexible methylene spacer, added to the higher sigma donation of the coordinating N-atom in position 3 of the 1,2,3-triazole, makes the coordination bonds in complex 3 stronger, and its effect on the photophysical properties of the copper complex higher.
The emission was measured for all the compounds in dichloromethane solution at room temperature. The mono- and the bis-chelating ligands 4–6 are fluorescent and emit in the green-cyano region (Fig. 3 and Table 2). The ligand 4 is the most blue-shifted, with an emission maximum of circa 480 nm and a photoluminescence quantum yield (PLQY) of 6% and a lifetime (τ) of 6 ns. Similarly, ligand 6 emits a bathochromic shifted wavelength of 500 nm circa, with 5% PLQY and 6 ns lifetime. The emission of ligand 5 is brighter and longer-lived (PLQY: 15%, τ: 26 ns). Interestingly, none of the Cu(I) complexes reported in this study emits when excited at its corresponding 1MLCT/1MLLCT bands. Probably, they deactivate through radiative pathways in the infrared region or only through nonradiative pathways.
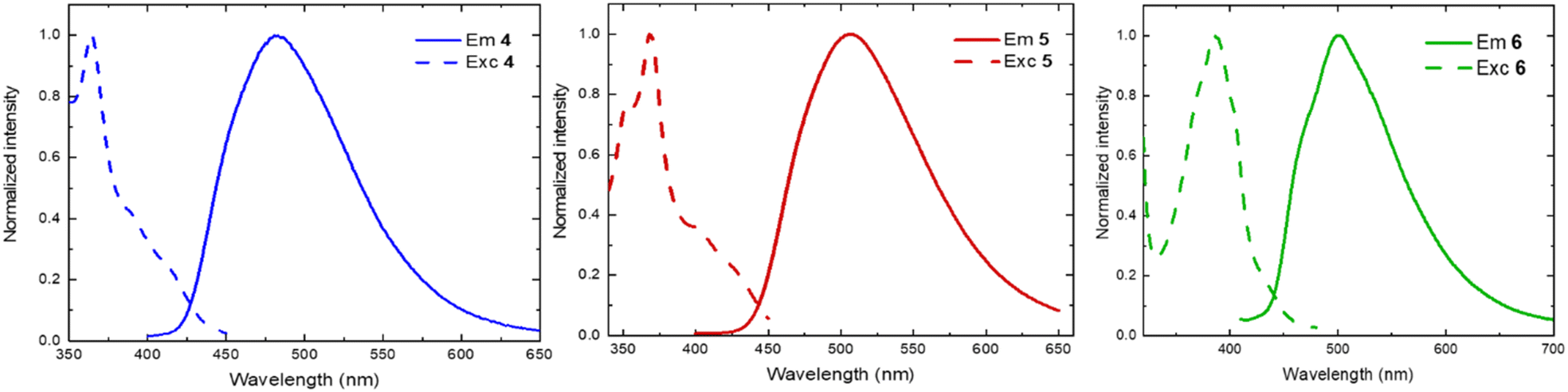 | ||
| Fig. 3 Emission (solid lines) and excitation (dashed lines) spectra of ligand: 4 (left), 5 (middle), 6 (right), recorded in dichloromethane solution at room temperature (λexc: 350 nm; λem: 500 nm). | ||
| Compound | λ abs/nm | λ em/nm | PLQYa | τ /ns | k r/s−1 | k nr/s−1 |
|---|---|---|---|---|---|---|
| a PLQY values in the solution were estimated using coumarin153 in methanol solution as reference (PLQY = 0.38).40,41 b Lifetimes in solution were recorded with a time-correlated single photon counting and with NanoLED as the excitation source (λexc = 366 nm). | ||||||
| 4 | 271, 350, 366, ∼392, ∼415 | 481 | 0.06 | 6.0 | 1.02 × 108 | 1.57 × 109 |
| 5 | 276, 354, 370, ∼410 | 507 | 0.15 | 26 | 5.77 × 107 | 3.27 × 108 |
| 6 | 274, 300, 375, 389, ∼435 | 501 | 0.05 | 6.0 | 8.17 × 107 | 1.59 × 109 |
On the contrary, when exciting at shorter wavelengths (up to 420 nm), the emission spectra of 1 and 2 are superimposable with those of their corresponding ligands 4 and 5, with a maximum peak at 486 nm and 510 nm, respectively. In order to exclude the emission coming from the free ligand, the stability of the complexes was tested in deuterated DCM by 1H NMR. The complexes are stable in solution for over a week (Fig. S2.17 and S2.18†); nevertheless, a small (less than 5%) de-coordination of the ligand could not be ruled out. The weak metal coordination in 1 and 2 and the dominant LC character were observed already in the UV-vis absorption. Instead, the binuclear copper complex 3 gave no emission, even exciting at high energy. This observation agrees with the superior chelating features of the corresponding ligand 6, achieving a stable Cu(I) complex, as already noted in the UV-vis absorption data.
Electrochemical characterization
Cyclic voltammetry (CV) of complexes 1–3 was recorded in dichloromethane, under inert atmosphere (Ar). Tetrabutylamonium hexafluorophosphate (TBAPF6) was employed as supporting electrolyte and ferrocene as the internal standard (Fig. 4; Table 3).42 The cathodic waves of their corresponding ligands 4–6 were measured for comparison (see Fig. S4.1–S4.3†). Due to the low solubility of the ligand 5 in DCM, a solution in N,N-dimethylformamide (DMF) was used. The copper complexes showed an irreversible oxidation, attributed to the oxidation of the copper centre Cu(I)/Cu(II). This oxidation process lies at about 0.85 V for the complexes 1 and 2 and at a slightly higher potential (1.01 V) for 3. In the cathodic scan, all the reduction can be attributed to processes centred on the diimine ligands. Indeed, the CV of the ligands shows similar peaks, lying at lower potentials, compared to those of their complexes. The mononuclear copper complex 1 shows an irreversible reduction peak at −1.83 V. In contrast, its corresponding binuclear copper complex 2 presents a higher number of irreversible peaks, recorded at −1.20 V, −1.84 V, and −2.08 V. Differently, the reversibility of the reduction processes is improved in the binuclear copper complex 3 bearing the highest reduction potential, among these three compounds, at −1.12 V and a second peak at −2.05 V. The ability of 3 to be more easily reduced agrees with the observation that this complex has the lowest energy absorption among the three complexes herein presented. It is relevant to mention that for the two binuclear copper complexes 2 and 3, the current associated with the oxidation process is circa the double intensity of those relative to the reduction processes, while for the mononuclear copper complex 1, their intensities are similar. Thus, the two copper metals in the two binuclear complexes 2 and 3 are equivalent and oxidize simultaneously. | ||
| Fig. 4 Cyclic voltammetry recorded for the copper complexes: 1 (left), 2 (middle), and 3 (right) at scan rate 100 mV s−1 in Ar-saturated DCM solutions (0.1 M TBAPF6). Potentials are reported versus ferrocene/ferrocenium redox couple (Fc/Fc+). | ||
| Compound | E ox/V | E red/V | ΔE/V |
|---|---|---|---|
| a Obtained in deaerated dichloromethane solutions at scan rate 100 mV s−1 and reported vs. Fc/Fc+. b in DMF. | |||
| 1 | 0.85 | −1.83 (n.r.) | 2.78 |
| 2 | 0.88 | −1.20, −1.84, −2.08 (n.r.) | 2.1 |
| 3 | 0.95 | −1.12, −2.05 | 2.13 |
| 4 | n.d. | −2.00; −2.14 | — |
| 5b | n.d. | −1.65; −2.03; −2.46; −2.68 | — |
| 6 | n.d. | −1.80; −1.52 | — |
Quantum chemical calculations
To further investigate the photophysical properties of the heteroleptic copper(I) complexes, quantum chemical calculations were performed with the TURBOMOLE program package.43,44 The equilibrium geometries were optimised at PBE0-D3(BJ)45–48 level of theory, and the electronic excitations were calculated utilising the GW approximation and Bethe–Salpeter equation (GW/BSE)49,50 at both one component (1c, scalar-relativistic) and quasirelativistic two-component (2c, including spin–orbit coupling) levels. As in our previous works,24,51 we prefer to apply the GW/BSE method over time-dependent density-functional theory (TDDFT) to avoid problems with computations of charge-transfer states due to the self-interaction error in the TDDFT approach. The simulated absorption spectra of the ligands show a good overlay with experimental data (Fig. S5.1†). This is also true for the calculated emission spectra of the benzoquinoxaline–triazole diimines (Fig. S5.3†). Regarding the complexes, a natural transition orbital (NTO) analysis confirms the strong LC contribution in all Cu(I) complexes, so that it is reasonable to associate the lowest absorption band to a mixture of MLCT and LC character of the transitions in the visible region (Fig. S5.2†). The electronic transition involves charge transfer from the copper atom to the benzoquinoxaline moiety of the diimine ligand, while the triazole moiety is not concerned. Interestingly, calculations suggest relative absorption maxima (MLLCT) at longer wavelengths than observed in the DCM experiments. This is particularly evident for compound 2, where the calculated absorption goes up to 800 nm. This different behaviour between the theory and the experiments might be rationalised with a distinct oscillator strength between complexes 2 and 3, due to the more rigid structure in 3.Conclusion
We presented one mononuclear and two dinuclear heteroleptic Cu(I) complexes based on novel benzoquinoxaline–triazole diimine ligands. Thanks to their extensive π-conjugation, the three complexes present absorption in the visible region of the electromagnetic spectrum, up to 700 nm. This feature is not typical for heteroleptic Cu(I) complexes, bearing chelating phosphines. Electrochemical and photophysical analyses suggested a metal–ligand-to-ligand charge transfer as the lowest excited state, confirmed by theoretical calculations. Due to the overall geometry of the complex, the dinuclear complex 3 is the most stable and presents an extended visible light absorption with significant oscillator strength, providing the necessary characteristics to perform in light-harvesting arrays.Experimental
Experimental details and general remarks
All starting materials were purchased from commercial suppliers and used as received. For the synthesis of 1,6-bis(triisopropylsilyl)hexa-1,5-diyne-3,4-dione a previously published procedure was followed.52,53Caution: azides can generate N2 with a very exothermic reaction. Explosion hazards.54
Synthesis
:7) solution at 80° for 15 h. After this time, the reaction mixture was quenched with water. The organic product was extracted three times with DCM. The collected organic phase was washed with water (three times), brine and then dried over Na2SO4 and filtered. The solvent was reduced under vacuum. The crude was purified via a silica gel column chromatography using cyclohexane/DCM (50:50) as eluent to obtain a yellow solid (0.620 g, 1.14 mmol). Yield: 62%. 1H NMR (400 MHz, CDCl3) δ = 8.61 (s, 2H, H), 8.07 (dt, J = 6.5, 3.2 Hz, 2H), 7.57 (dt, J = 6.5, 3.2 Hz, 2H), 1.25–1.18 (m, 42H) ppm. 13C NMR (101 MHz, CDCl3) δ = 140.14, 137.18, 134.46, 128.84, 127.52, 127.38, 104.05, 99.41, 18.95, 11.63 ppm. HRMS (ESI) m/z ([M + H]+, C34H48N2Si2): 541.3434 (calc.), 541.3418 (found).
:3). Then the other reactants were added in the following order: sodium azide (0.285 g, 4.38 mmol, 1.20 equiv.), sodium ascorbate (0.407 g, 2.05 mmol, 0.56 equiv.), copper sulfate pentahydrate (0.119 g, 0.75 mmol, 0.20 equiv.), sodium carbonate (0.181 g, 2.18 mmol, 0.59 equiv.), ethynylbenzene (0.373 g, 3.65 mmol, 1.00 equiv.). The reaction mixture was left under stirring at room temperature for two days. A solution of NH4OH (10%) was subsequently added for the quenching. The organic product was extracted three times with dichloromethane, then it was washed with water (three times) and brine, dried over Na2SO4 and filtered. The crude is purified dissolving the compound in a minimum amount of dichloromethane and adding an excess of pentane for the precipitation. The precipitate was filtered, dissolved in dichloromethane and lastly the solvent was removed under reduced pressure to obtain the product as a light brown solid (0.692 g, 1.97 mmol). Yield: 54%. 1H NMR (400 MHz, CD2Cl2) δ = 8.58 (d, J = 3.9 Hz, 2H), 8.14–8.07 (m, 2H), 8.02 (s, 1H, H), 7.87–7.82 (m, 2H), 7.62–7.55 (m, 2H), 7.46–7.39 (m, 2H), 7.37–7.31 (m, 1H), 5.97 (s, 2H), 2.84 (s, 3H) ppm. 13C NMR (126 MHz, CD2Cl2) δ = 157.87, 154.16, 149.16, 148.62, 139.08, 136.79, 134.69, 134.53, 133.86, 133.80, 133.73, 133.61, 132.38, 130.79, 130.65, 130.56, 129.80, 129.17, 129.11, 129.07, 129.03, 128.51, 128.44, 127.93, 127.84, 127.49, 127.27, 126.14, 125.51, 124.68, 124.56, 124.45, 123.37, 120.44, 52.32, 23.76 ppm. HRMS m/z ([M + H]+, C22H17N5): 352.1562 (calc.), 352.1554 (found).
:3) and 20 mL acetonitrile. Then the other reactants were added in the following order: sodium azide (0.302 g, 4.65 mmol, 2.40 equiv.), sodium ascorbate (0.438 g, 2.21 mmol, 1.14 equiv.), copper sulfate pentahydrate (0.128 g, 0.80 mmol, 0.42 equiv.), sodium carbonate (0.195 g, 2.36 mmol, 1.20 equiv.), ethynylbenzene (0.396 g, 3.87 mmol, 2.00 equiv.). The reaction mixture was left under stirring for two days. Then a NH4OH (10%) solution was added for quenching the reaction and the organic product was extracted with dichloromethane (3 times). The organic phase collected was washed with water (three times) and brine, dried over Na2SO4, filtered. The crude was purified via a precipitation adding an excess of pentane to a few mL solution of the compound in dichloromethane to obtain a yellow solid. The precipitate was filtered, dissolved in dichloromethane and lastly the solvent was removed under reduced pressure to obtain the product as a yellow solid (424 mg, 0.85 mmol). Yield: 44%. 1H NMR (400 MHz, C2D6OS) δ = 8.73 (s, 2H), 8.63 (s, 2H), 8.22–8.16 (m, 2H), 7.95 (d, J = 7.6 Hz, 4H), 7.60 (dt, J = 6.7, 3.4 Hz, 2H), 7.48 (t, J = 7.6 Hz, 4H), 7.37 (t, J = 7.4 Hz, 2H), 6.35 (s, 4H) ppm. 13C NMR (101 MHz, DMSO) δ 149.74, 146.53, 136.80, 133.47, 130.80, 128.95, 128.30, 127.92, 127.16, 127.10, 125.25, 123.00, 52.10 ppm. HRMS m/z (C30H23N8): 495.2046 (calc.), 495.2044 (found).
:3). Then the following reactants were added: sodium ascorbate (0.102 g, 0.51 mmol, 1.12 equiv.), copper sulphate penta-hydrate (0.048 g, 0.19 mmol, 0.42 equiv.), sodium carbonate (0.058 mg, 0.55 mmol, 1.20 equiv.), benzylazide (0.122 g, 0.92 mmol, 2.00 equiv.). The mixture of reaction was left under stirring for two days. After this period time, a solution of NH4OH (10%) was added. The organic product was extracted three times with dichloromethane, then it was washed with water (three times) and brine, dried over Na2SO4 and filtered. The solvent was removed under vacuum. The product was purified by a silica gel column chromatography using a solution of DCM/MeOH (95:5) as eluent that was performed twice. The product was obtained as a brownish powder (36 mg, 0.07 mmol). Yield: 16%. 1H NMR (400 MHz, CDCl3) δ = 8.70 (s, 1H), 8.10 (d, J = 8.3 Hz, 2H), 7.58 (dd, J = 6.6, 3.2 Hz, 1H), 7.46–7.33 (m, 5H), 5.61 (s, 2H). 13C NMR (101 MHz, CDCl3) δ = 146.42, 145.03, 137.65, 134.47, 134.43, 129.33, 129.00, 128.72, 128.54, 128.42, 127.81, 127.21, 125.47 ppm. HRMS (ESI) m/z ([M + H]+, C30H22N8): 495.2045 (calc.), 495.2044 (found).
General procedure for the synthesis of the copper complexes
Following a previously published procedure,24 the synthesis was performed as follows. The precursor, Cu(CH3CN)4BF4 (1.00 equiv.), was dissolved together with DPEPhos (1.00 equiv.) in a solution of dry dichloromethane, under an Ar atmosphere. After circa 30 minutes, the chosen benzoquinoxaline-based ligand (1.00 equiv. for the mononuclear, 0.5 equiv. for the binuclear complexes) was added to the solution, still under inert atmosphere. The reaction stirred for four hours. Lastly, the solvent was removed under reduced pressure in a rotary evaporator. The crude product was dissolved in a minimum amount of dichloromethane and crystallized by a slow diffusion of cyclohexane.Computational details
The GW/BSE computations were carried out at CD-ev GW(10)/BSE level of theory, i.e. eigenvalue-only self-consistent GW (evGW)55 employing contour deformation (CD)56 for the highest 10 occupied and lowest 10 unoccupied orbitals followed by the Bethe–Salpeter equation (BSE). For non- and scalar-relativistic one-component (1c) calculations, the def2-TZVP basis set57 was taken for Cu, P, N and C atoms in triazole and benzoquinoxaline moieties, and the def2-SV(P) basis set57 was taken for the rest of the atoms. For quasirelativistic two-component (2c) calculations, the all-electron x2c-TZVPall/SV(P)all-2c basis set58 was used. The resolution-of-the-identity (RI) approximation was used for all two-electron integrals.All orbital and auxiliary basis sets were taken from the TURBOMOLE basis-set library.43,44 The “Coulomb-fitting” auxiliary basis sets (denoted jbas) were used in the ground-state density functional theory (DFT) computations, and the “MP2-fitting” auxiliary basis sets (denoted cbas) were used in the GW/BSE computations. The ground-state DFT computations were carried out with the modules DSCF and RIDFT, and the self-consistent field convergence criterion scfconv = 8 and DFT grid 4 were used. The geometry optimization was considered converged when the change in energy and cartesian gradients reached thresholds of 10−7 and 10−4 hartree, respectively. The GW/BSE computations were carried out with the ESCF module, and the convergence criterion rpaconv = 6 was used. Furthermore, in evGW, the damping parameter was set to η = 0.001 in order to achieve rapid convergence. In CD-GW, 128 grid points were used, also 128 parameters were taken in the Padé approximant.
Author contributions
C. Bruschi performed the synthesis, the electrochemical and photophysical characterization, guided by C. Bizzarri. X. Gui performed the theoretical calculations, supervised by Prof. W. Klopper. O. Fuhr solved and refined the X-ray structure. C. Bizzarri ideated the project. C. Bizzarri and C. Bruschi wrote the manuscript. All authors revised the draft and gave the final approval.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the German Research Foundation (DFG) for the financial support through the transregional collaborative research centre SFB/TRR 88 (3MET) (Projects B9 and C1). Bizzarri also acknowledges the support of the Klaus Tschira Boost Fund and the German Scholar Organization. Open access supported by KIT through Publish & Read Agreements.References
- N. Armaroli and V. Balzani, Chem. – Asian J., 2011, 6, 768–784 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Chem. – Eur. J., 2016, 22, 32–57 CrossRef CAS PubMed.
- V. Krewald, M. Retegan and D. A. Pantazis, in Solar Energy for Fuels, ed. H. Tüysüz and C. K. Chan, Springer International Publishing, Cham, 2016, pp. 23–48, DOI:10.1007/128_2015_645.
- A. Harriman, Chem. Commun., 2015, 51, 11745–11756 RSC.
- P. Ceroni, A. Credi, M. Venturi and V. Balzani, Photochem. Photobiol. Sci., 2010, 9, 1561–1573 CrossRef CAS PubMed.
- S. Campagna, F. Nastasi, G. La Ganga, S. Serroni, A. Santoro, A. Arrigo and F. Puntoriero, Phys. Chem. Chem. Phys., 2023, 25, 1504–1512 RSC.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- A. Arrigo, G. La Ganga, F. Nastasi, S. Serroni, A. Santoro, M.-P. Santoni, M. Galletta, S. Campagna and F. Puntoriero, C. R. Chim., 2017, 20, 209–220 CrossRef CAS.
- M. Irikura, Y. Tamaki and O. Ishitani, Chem. Sci., 2021, 12, 13888–13896 RSC.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933–7947 RSC.
- D. R. McMillin, M. T. Buckner and B. T. Ahn, Inorg. Chem., 1977, 16, 943–945 CrossRef CAS.
- A. Lavie-Cambot, M. Cantuel, Y. Leydet, G. Jonusauskas, D. M. Bassani and N. D. McClenaghan, Coord. Chem. Rev., 2008, 252, 2572–2584 CrossRef CAS.
- M. Sandroni, Y. Pellegrin and F. Odobel, C. R. Chim., 2016, 19, 79–93 CrossRef CAS.
- M. T. Buckner and D. R. McMillin, J. Chem. Soc., Chem. Commun., 1978, 759–761, 10.1039/C39780000759.
- S.-M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313–3322 CrossRef CAS PubMed.
- Y. Zhang, M. Schulz, M. Wächtler, M. Karnahl and B. Dietzek, Coord. Chem. Rev., 2018, 356, 127–146 CrossRef CAS.
- J. Beaudelot, S. Oger, S. Peruško, T.-A. Phan, T. Teunens, C. Moucheron and G. Evano, Chem. Rev., 2022, 122(22), 16365–16609 CrossRef CAS PubMed.
- M. Sandroni, L. Favereau, A. Planchat, H. Akdas-Kilig, N. Szuwarski, Y. Pellegrin, E. Blart, H. Le Bozec, M. Boujtita and F. Odobel, J. Mater. Chem. A, 2014, 2, 9944–9947 RSC.
- J. H. Tran, P. Traber, B. Seidler, H. Görls, S. Gräfe and M. Schulz, Chem. – Eur. J., 2022, 28, e202200121 CrossRef CAS PubMed.
- C. Müller, M. Schulz, M. Obst, L. Zedler, S. Gräfe, S. Kupfer and B. Dietzek, J. Phys. Chem. A, 2020, 124, 6607–6616 CrossRef PubMed.
- M. Schulz, N. Hagmeyer, F. Wehmeyer, G. Lowe, M. Rosenkranz, B. Seidler, A. Popov, C. Streb, J. G. Vos and B. Dietzek, J. Am. Chem. Soc., 2020, 142, 15722–15728 CrossRef CAS PubMed.
- B. Seidler, M. Sittig, C. Zens, J. H. Tran, C. Müller, Y. Zhang, K. R. A. Schneider, H. Görls, A. Schubert, S. Gräfe, M. Schulz and B. Dietzek, J. Phys. Chem. B, 2021, 125, 11498–11511 CrossRef CAS PubMed.
- A. Listorti, G. Accorsi, Y. Rio, N. Armaroli, O. Moudam, A. Gégout, B. Delavaux-Nicot, M. Holler and J.-F. Nierengarten, Inorg. Chem., 2008, 47, 6254–6261 CrossRef CAS PubMed.
- C. Bruschi, X. Gui, N. Salaeh-arae, T. Barchi, O. Fuhr, S. Lebedkin, W. Klopper and C. Bizzarri, Eur. J. Inorg. Chem., 2021, 2021, 4074–4084 CrossRef CAS.
- M. Rentschler, P. J. Boden, M. A. Argüello Cordero, S. T. Steiger, M.-A. Schmid, Y. Yang, G. Niedner-Schatteburg, M. Karnahl, S. Lochbrunner and S. Tschierlei, Inorg. Chem., 2022, 61, 12249–12261 CrossRef CAS PubMed.
- R. P. Pandit, S. H. Kim and Y. R. Lee, Adv. Synth. Catal., 2016, 358, 3586–3599 CrossRef CAS.
- B. D. Lindner, Y. Zhang, S. Höfle, N. Berger, C. Teusch, M. Jesper, K. I. Hardcastle, X. Qian, U. Lemmer, A. Colsmann, U. H. F. Bunz and M. Hamburger, J. Mater. Chem. C, 2013, 1, 5718–5724 RSC.
- M. Milkevitch, E. Brauns and K. J. Brewer, Inorg. Chem., 1996, 35, 1737–1739 CrossRef CAS PubMed.
- G. N. A. Nallas and K. J. Brewer, Inorg. Chim. Acta, 1996, 253, 7–13 CrossRef CAS.
- F. K. Merkt, K. Pieper, M. Klopotowski, C. Janiak and T. J. J. Müller, Chem. – Eur. J., 2019, 25, 9447–9455 CrossRef CAS PubMed.
- C. Bizzarri, A. P. Arndt, S. Kohaut, K. Fink and M. Nieger, J. Organomet. Chem., 2018, 871, 140–149 CrossRef CAS.
- T. U. Connell, J. M. White, T. A. Smith and P. S. Donnelly, Inorg. Chem., 2016, 55, 2776–2790 CrossRef CAS PubMed.
- C. B. Anderson, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Organometallics, 2013, 32, 788–797 CrossRef CAS.
- K. J. Kilpin, E. L. Gavey, C. J. McAdam, C. B. Anderson, S. J. Lind, C. C. Keep, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2011, 50, 6334–6346 CrossRef CAS PubMed.
- P. M. Guha, H. Phan, J. S. Kinyon, W. S. Brotherton, K. Sreenath, J. T. Simmons, Z. Wang, R. J. Clark, N. S. Dalal, M. Shatruk and L. Zhu, Inorg. Chem., 2012, 51, 3465–3477 CrossRef CAS PubMed.
- L.-L. Gracia, L. Luci, C. Bruschi, L. Sambri, P. Weis, O. Fuhr and C. Bizzarri, Chem. – Eur. J., 2020, 26, 9929–9937 CrossRef CAS PubMed.
- E. Leoni, J. Mohanraj, M. Holler, M. Mohankumar, I. Nierengarten, F. Monti, A. Sournia-Saquet, B. Delavaux-Nicot, J.-F. I. Nierengarten and N. Armaroli, Inorg. Chem., 2018, 57, 15537–15549 CrossRef CAS PubMed.
- M. Mohankumar, M. Holler, E. Meichsner, J.-F. Nierengarten, F. Niess, J.-P. Sauvage, B. Delavaux-Nicot, E. Leoni, F. Monti, J. M. Malicka, M. Cocchi, E. Bandini and N. Armaroli, J. Am. Chem. Soc., 2018, 140, 2336–2347 CrossRef CAS PubMed.
- M. Sandroni, M. Kayanuma, M. Rebarz, H. Akdas-Kilig, Y. Pellegrin, E. Blart, H. Le Bozec, C. Daniel and F. Odobel, Dalton Trans., 2013, 42, 14628–14638 RSC.
- A. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- G. Jones, W. R. Jackson, C. Y. Choi and W. R. Bergmark, J. Phys. Chem., 1985, 89, 294–300 CrossRef CAS.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56(4), 461–466 CrossRef.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- TURBOMOLE V7.4 2019, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, https://www.turbomole.com.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- K. Krause and W. Klopper, J. Comput. Chem., 2017, 38, 383–388 CrossRef CAS PubMed.
- X. Gui, C. Holzer and W. Klopper, J. Chem. Theory Comput., 2018, 14, 2127–2136 CrossRef CAS PubMed.
- M. Grupe, P. Boden, P. Di Martino-Fumo, X. Gui, C. Bruschi, R. Israil, M. Schmitt, M. Nieger, M. Gerhards, W. Klopper, C. Riehn, C. Bizzarri and R. Diller, Chem. – Eur. J., 2021, 27, 15252–15271 CrossRef PubMed.
- R. Faust, C. Weber, V. Fiandanese, G. Marchese and A. Punzi, Tetrahedron, 1997, 53, 14655–14670 CrossRef CAS.
- G. Li, S. Wang, S. Yang, G. Liu, P. Hao, Y. Zheng, G. Long, D. Li, Y. Zhang, W. Yang, L. Xu, W. Gao, Q. Zhang, G. Cui and B. Tang, Chem. – Asian J., 2019, 14, 1807–1813 CrossRef CAS PubMed.
- D. S. Treitler and S. Leung, J. Org. Chem., 2022, 87, 11293–11295 CrossRef CAS PubMed.
- X. Blase, C. Attaccalite and V. Olevano, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115103 CrossRef.
- C. Holzer and W. Klopper, J. Chem. Phys., 2019, 150, 204116 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Y. J. Franzke, R. Treß, T. M. Pazdera and F. Weigend, Phys. Chem. Chem. Phys., 2019, 21, 16658–16664 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2074014. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00902e |
| This journal is © The Royal Society of Chemistry 2023 |