
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalysis-free synthesis of thiazolidine–thiourea ligands for metal coordination (Au and Ag) and preliminary cytotoxic studies†
Daniel
Salvador-Gil
a,
Raquel P.
Herrera
b and
M. Concepción
Gimeno
 *a
*a
aDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: gimeno@unizar.es
bDepartamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, 50009 Zaragoza, Spain
First published on 24th February 2023
Abstract
The reaction of propargylamines with isothiocyanates results in the selective formation of iminothiazolidines, aminothiazolines or mixed thiazolidine–thiourea compounds under mild conditions. It has been observed that secondary propargylamines lead to the selective formation of cyclic 2-amino-2-thiazoline derivatives, while primary propargylamines form iminothiazoline species. In addition, these cyclic thiazoline derivatives can further react with an excess of isothiocyanate to give rise to thiazolidine–thiourea compounds. These species can also be achieved by reaction of propargylamines with isothiocynates in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Coordination studies of these heterocyclic species towards silver and gold with different stoichiometries have been carried out and complexes of the type [ML(PPh3)]OTf, [ML2]OTf (M = Ag, Au) or [Au(C6F5)L] have been synthesised. Preliminary studies of the cytotoxic activity in lung cancer cells have also been performed in both ligands and complexes, showing that although the ligands do not exhibit anticancer activity, their coordination to metals, especially silver, greatly enhances the cytotoxic activity.
:2. Coordination studies of these heterocyclic species towards silver and gold with different stoichiometries have been carried out and complexes of the type [ML(PPh3)]OTf, [ML2]OTf (M = Ag, Au) or [Au(C6F5)L] have been synthesised. Preliminary studies of the cytotoxic activity in lung cancer cells have also been performed in both ligands and complexes, showing that although the ligands do not exhibit anticancer activity, their coordination to metals, especially silver, greatly enhances the cytotoxic activity.
Introduction
In recent years, there has been an increased interest in the discovery and development of novel biologically active compounds as more selective agents for the treatment of different diseases. Cancer is a common illness in our society, the second leading cause of death in the world, responsible for important health costs associated with the treatment of patients and with a high level of mortality. As a result, numerous chemotherapeutic agents have been developed to avoid the progression of tumours or to prevent their recurrence, as well as with the main aim of avoiding many of the side effects of conventional antitumor drugs.1 In the continuous efforts to develop drugs with such anticancer properties, scientists have focused upon many different aspects of this illness. Among the antitumor organic compounds discovered in recent years, urea2 and thiourea3 derivatives have exhibited potent anticancer activity and have been reported as prodrugs. These structures have also been reported as important biologically active compounds in a variety of promising chemical prototypes for drug development. Additionally, thioureas could display different properties, such as anti-HIV,4 antiviral,5 antibacterial,6 antifungal,7 analgesic,8 or antithrombotic activity,9 among others (Fig. 1).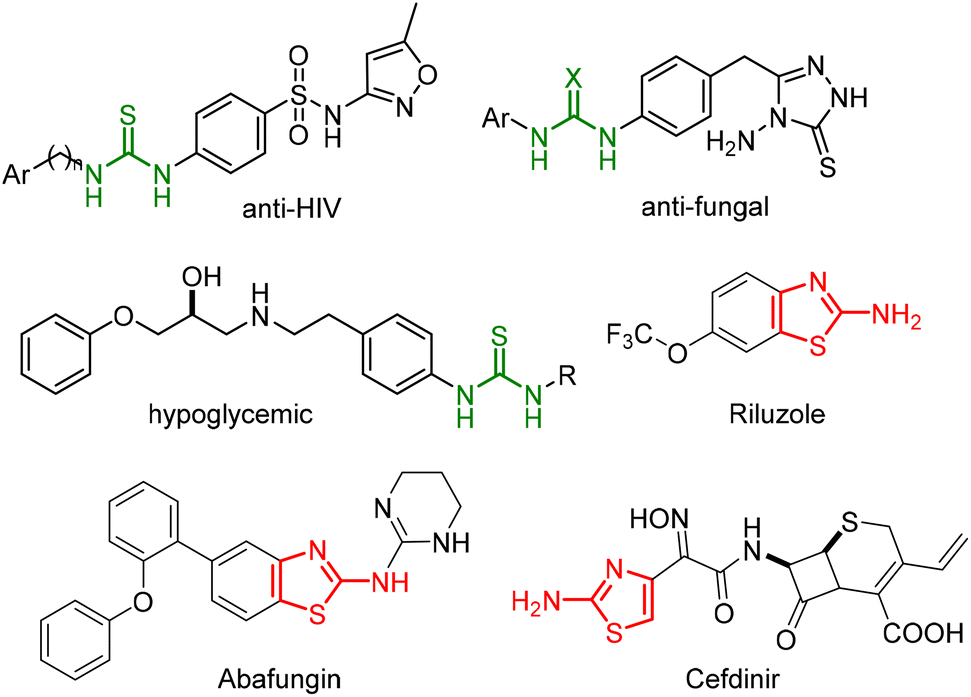 | ||
| Fig. 1 Biologically active thiourea and aminothiazole scaffolds. | ||
Heterocyclic compounds, such as thiazoline and thiazolidine scaffolds,10 have also been considered as privileged structural motifs in medicinal chemistry and of high importance in organic synthesis.11 These heterocycles provide the final drugs with improved pharmacokinetic properties and facilitate the construction of a large variety of biologically active molecules, such as anti-tumour,12 anti-bacterial,13 anti-viral,14 and anti-inflammatory compounds.15 Moreover, several aminothiazole derivatives are clinically approved drugs for the treatment of amyotrophic lateral sclerosis (Riluzone),16 or anti-fungals (Abafungin),17 or third generation cephalosporin antibiotics (Cefdinir) (Fig. 1).
Additionally, the development of new metallodrugs is currently becoming very important for cancer chemotherapy since they try to improve the activity of existing drugs such as cisplatin and to avoid its important side effects and resistance. In this field, improvements have been achieved with ruthenium18 and also with the design of gold and silver-based drugs,19 which present great potential as anticancer agents.
In this context, we have focused our efforts during the last decade on the search and development of new biologically active gold and silver20 complexes with new biological ligands21 in a synergic way as an excellent approach for discovering novel bioactive metallodrugs.
The search for new biological compounds goes in parallel with the development of new organic ligands or their corresponding metallodrug based compounds, because of the interesting properties that both parts could exhibit, thiourea or aminothiazoline derivatives and gold or silver complexes. We envisioned that coordination of these organic molecules with the respective group 11 metal centres should increase the biological activity of the resulting complexes in comparison with each part alone.
The conversion of propargylamines and isothiocyanates to afford the thiazolidine or thiazole moiety has been known long time ago; however, usually harsh conditions, microwaves or metal catalysts are used to achieve these heterocycles.
Results and discussion
Considering that heterocycles play an important role in many natural products and drug development, with a wide range of biological properties, we consider studying the formation of thiazolidines and thiazolidine–thiourea species under mild conditions. Additionally, their coordination to gold and silver centres will be explored because to the best of our knowledge the coordination of thiazoline containing heterocycles to gold or silver metal for biological purpose has been overlooked in the literature so far.To initiate the study, different model propargylamines 1a–c and isothiocyanates 2a–c were selected (Fig. 2).
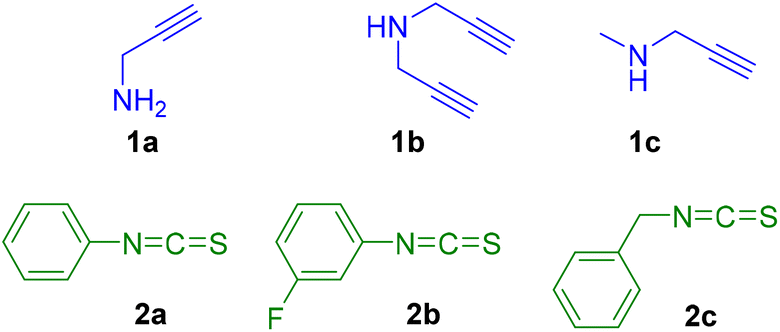 | ||
| Fig. 2 Propargylamines 1a–c and isothiocyanates 2a–c. | ||
For this purpose, the conditions for the formation of thiourea species 3 or aminothiazoline derivative 4 were investigated as disclosed in Scheme 1.
 | ||
| Scheme 1 Formation of thiourea 3 or aminothiazoline 4. | ||
Surprisingly, the normal reaction to afford thiourea 3 did not work between amine 1a and isothiocyanate 2a using EtOH as the solvent at room temperature. To our delight, after analysis of the 1H NMR spectrum of the reaction, it was observed that the signals of the obtained product were in agreement with the cyclic compound 4, the 2-aminothiazoline derivative.22 In fact, to obtain the desired thiourea 3, the solvent of the process was changed to a less polar solvent such as acetonitrile (Scheme 1). The reaction was carried out under the same conditions of time and temperature and finally the thiourea 3 was obtained.
However, in most of the assays the thiourea could not be isolated in the pure form, and a mixture of both thiazoline and thiourea derivatives was obtained, even if the reaction was carried out in acetonitrile, whereas pure thiazoline compounds are achieved in ethanol. Therefore, we continued working with EtOH. It is worth mentioning that when 2-thiazolines have an amino group at position 2, a tautomeric equilibrium is observed between the thiazoline and thiazolidine forms, that is 2-amino-2-thiazoline and 2-iminothiazolidine. Interestingly, we have observed that the use of secondary propargylamines 1b,c leads selectively to the formation of cyclic iminothiazolidine derivatives 5, 6, 8 and 9, while primary propargylamine 1a could form 2-aminothiazoline derivatives 4 and 7 (Scheme 2).
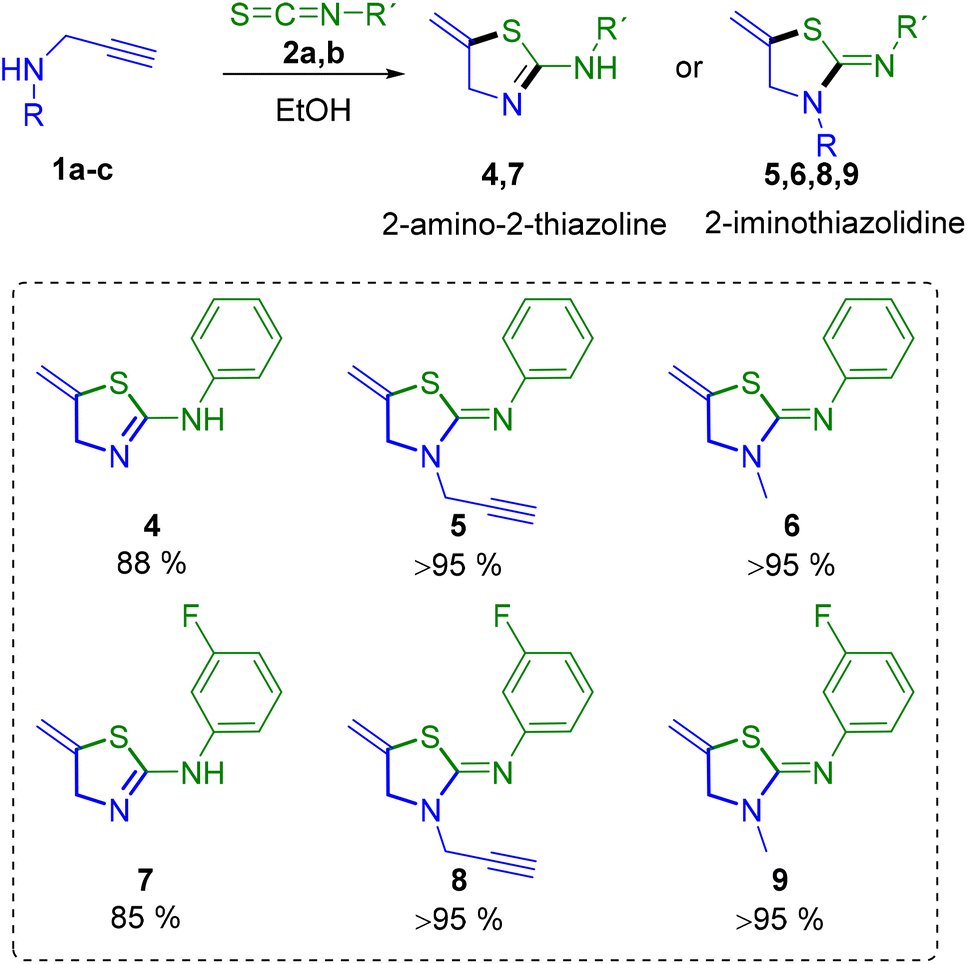 | ||
| Scheme 2 Synthesis of 2-amino-2-thiazolines and 2-iminothiazolidines. | ||
The easy formation of these thiazoline and thiazolidine derivatives was accomplished for all the alkynes and isothiocyanates assayed with very good yields. Thiazoline 4 crystallises with four independent molecules, with similar distances and bond angles in all the molecules (Fig. 3B).23 Moreover, it is possible to observe the existence of a double bond between the N1–C1 atoms of 1.278(2) Å, which corroborates the formation of the 2-amino-2-thiazoline 4 and also the presence of hydrogen bonds among the molecules that form a chain polymer. The shortest bonds correspond to the N7⋯H–N6 unit with a distance of 2.047 Å and an angle of 174° (Fig. 3B). In the structure of compound 6, the distances C5–N2 of 1.2759(16) and C5–N1 of 1.3590(15) support the formation of the iminothiazolidine (Fig. 3C). The crystal structure of compound 7 also supports the formation of the 2-amino-2-thiazoline derivative, with distances N1–C4 of 1.276(2) and N2–C4 of 1.357(2) Å (Fig. 3D).
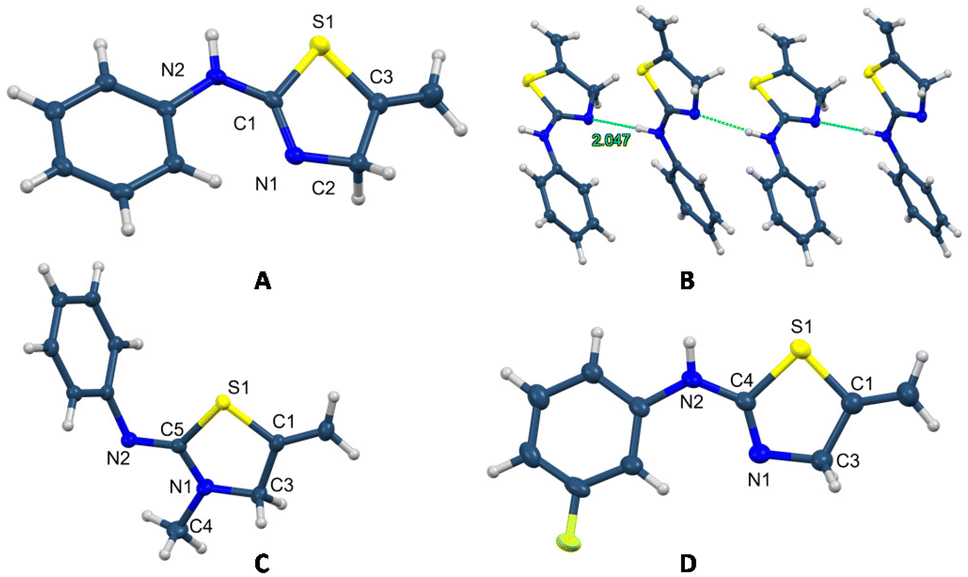 | ||
| Fig. 3 Crystal molecular structures of 4 (A), 6 (C), and 7 (D), and the chain formed by hydrogen bonding in 4 (B) showing ellipsoids at the 50% probability level. | ||
Encouraged with this reactivity, we tested the scope of this approach by adding 2 equivalents of isothiocyanate 2a–c to propargylamine 1a. Interestingly, new thiazolidine–thiourea derivatives 10–12 were prepared, incorporating three molecules of reagents in the final product in an atom-economical process (Scheme 3).
 | ||
| Scheme 3 Synthesis of thiazolidine–thiourea adducts 10–12. | ||
For the formation of these thiazolidine–thiourea species, the phenyl (2a), 3-fluorophenyl (2b) and benzyl (2c) isocyanates have been used. The reactions were carried out in ethanol and the compounds precipitated as a white solid, being obtained in the pure form and in very good yields. As a proof of the final structures of thiourea–thiazolidine derivatives 10 and 11, crystal structures were also corroborated by X-ray diffraction and are presented in Fig. 4.24 In both cases, we observe the presence of an intramolecular hydrogen bond between the amino group of the thiourea and the imino group of the iminothiazolidine. For compound 10 the hydrogen bond N1–H6⋯N3 has a donor⋯acceptor distance of 2.621 Å and an angle of 142.2°. In compound 11, the N1–H1⋯N3 bond has a donor⋯acceptor distance of 2.602 Å and an angle of 146.1°.
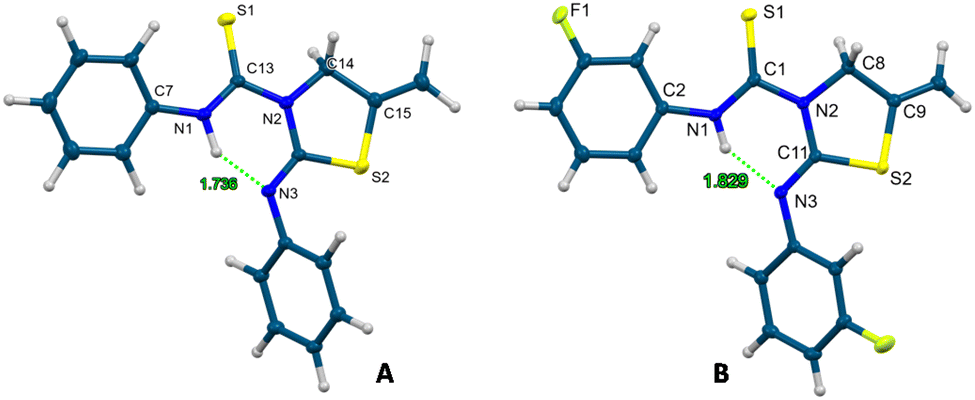 | ||
| Fig. 4 Crystal molecular structures of compounds 10 (A) and 11 (B) showing ellipsoids at the 50% probability level. | ||
Mechanism of the reaction
A plausible mechanism for the formation of thiazolines or thiazolidine–thiourea derivatives is proposed in Scheme 4. The first step would be the reaction of propargylamine 1a with the isothiocyanate 2, giving rise to the formation of the corresponding thiourea I. If two equivalents of isothiocyanate are used, thiourea III could be first generated. In this case, only thiourea I could be isolated in a pure form. The next step would be the generation of a more reactive allene intermediate II or IV, in equilibrium with its thiourea species, probably favoured by the use of a polar solvent.25 The formation of these intermediates would receive a posterior intramolecular nucleophilic attack by the sulfur atom to the quaternary carbon of the allene, giving rise to the cyclisation of the compound and the formation of the corresponding 2-iminothiazolidines V and VI. At this point, we cannot discard the direct attack of the sulfur atom over the triple bond as previously proposed by other authors.26 In the case of 2-iminothiazolidines V, the internal amino NH group could also produce a nucleophilic attack on another molecule of isothiocyanate giving rise to the same thiazolidine–thiourea VI. Alternatively, this route could also justify the obtainment of the same compound VI (Scheme 4).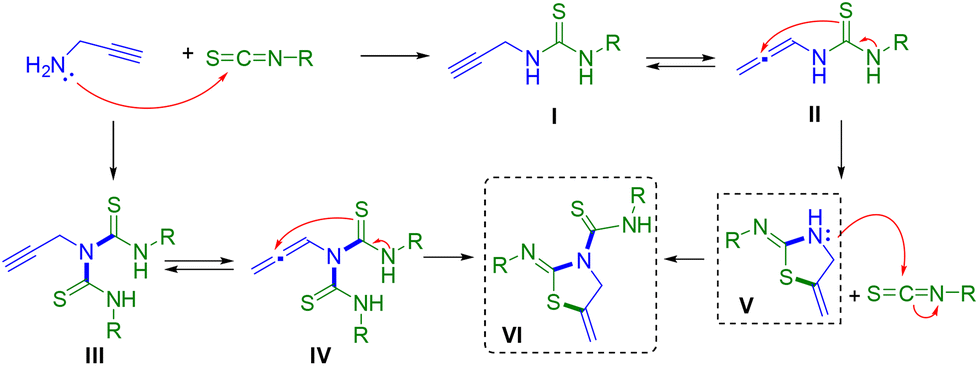 | ||
| Scheme 4 Proposed formation of the iminothiazolidine V and thiazolidine–thiourea VI. | ||
Metal coordination studies
It is known that both thioureas and thiazolidines alone may have some biological activity as described above. Therefore, we envisioned that the tandem of the two units in the same molecule could improve their activity. We also foresaw that coordinating these types of ligands to group 11 metals, with proven biological activity, probably would result in an enhancement of the potential biological activity.27 For this reason, the binding of thiazolidine–thiourea ligands to silver or gold fragments has been addressed in this section.First, the coordination of silver to 10–12 was explored, using [Ag(OTf)(PPh3)] or Ag(OTf) as the metallic precursor, to afford the heteroleptic 13–15 or the homoleptic 16–18 silver complexes (Scheme 5).
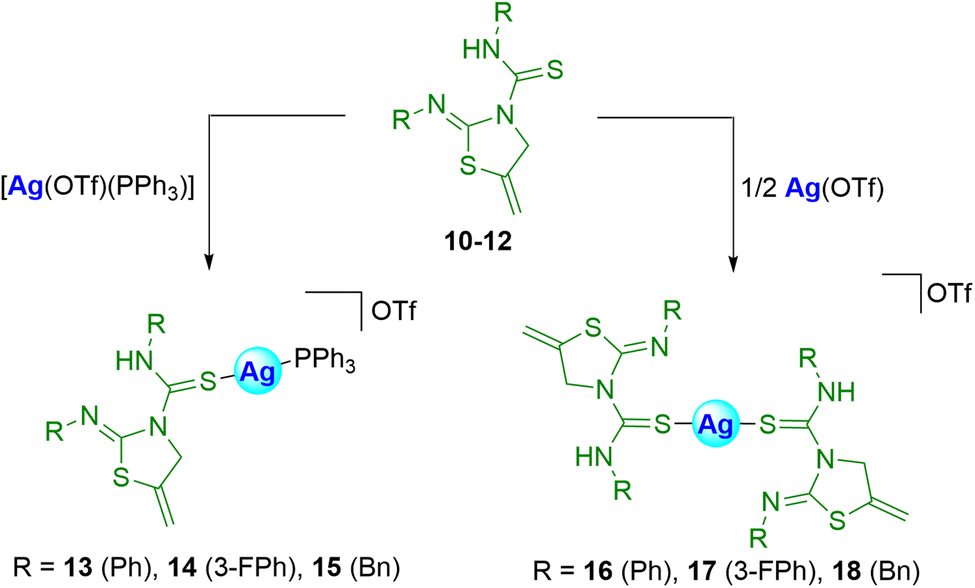 | ||
| Scheme 5 Preparation of silver complexes 13–18. | ||
In both cases, coordination of silver to the sulfur atoms of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S group of the ligands is proposed. The resonances of the complexes in the spectra appear shifted with respect to those of the ligands. In all cases, the new complexes were obtained in very good yields.
S group of the ligands is proposed. The resonances of the complexes in the spectra appear shifted with respect to those of the ligands. In all cases, the new complexes were obtained in very good yields.
Attempts were also made to synthesize the analogous cationic gold complexes, starting from [Au(PPh3)(MeCN)]SbF6 or [Au(tht)2]OTf precursors, or neutral derivatives with [Au(C6F5)(tht)] as the starting material (Scheme 6). The heteroleptic complexes 19–21 with the thiazolidine–thiourea and phosphine ligands were obtained with a very small amount of the homoleptic [Au(PPh3)2]OTf complex.
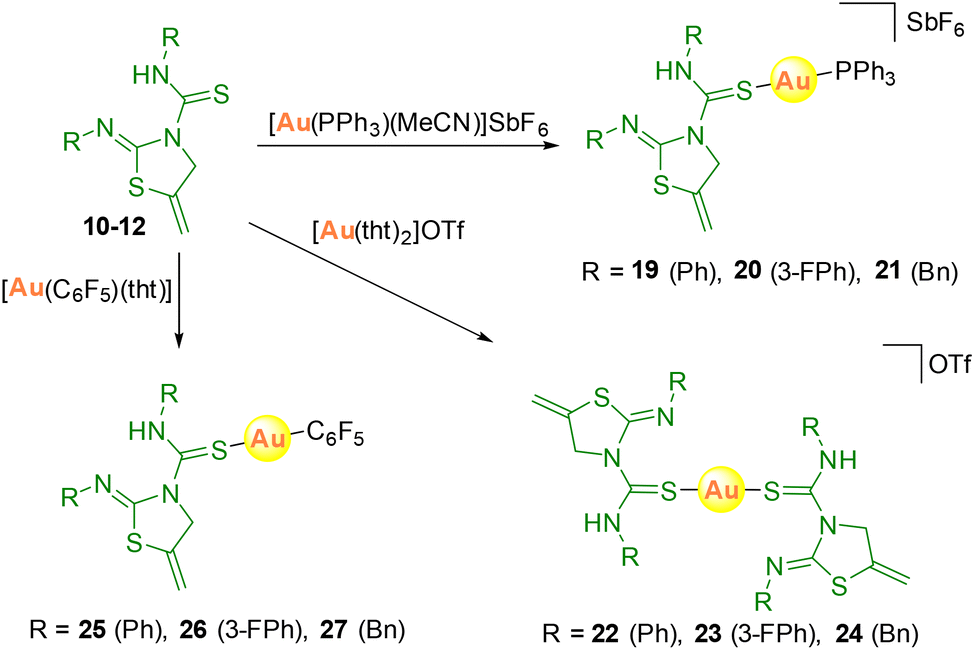 | ||
| Scheme 6 Preparation of gold complexes 19–27. | ||
The crystal structure of complex 20 has been established by X-ray diffraction studies. In this complex, the gold atom is in a linear environment bonded to the sulfur atom of thiourea with a distance Au1–S1 of 2.336(2) Å and to the phosphorus atom with a distance of Au1–P1 2.274(2) Å (Fig. 5).28 The angle around the gold centre is slightly distorted from linearity, P1–Au1–S1 175.34(9)°. The molecules associate in dimers through a short aurophilic interaction of Au1⋯Au1 3.0861(7) Å (Fig. 5).
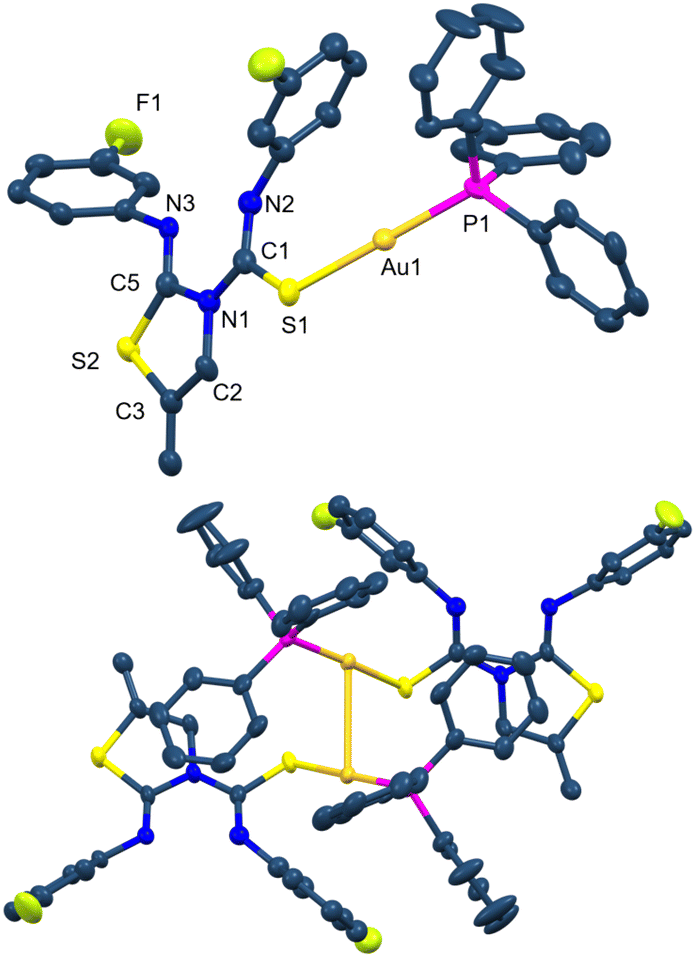 | ||
| Fig. 5 Molecular structure of complex 20 showing ellipsoids at the 30% probability level, and its association in dimers thought aurophilic interactions. Hydrogen atoms are omitted for clarity. | ||
In complexes 25–27, the 19F NMR spectra provide good information about the formation of the complexes, in which three different resonances are assigned to the pentafluorophenyl group, two multiplets for the ortho- and meta-fluoro and a triplet for the para-fluoro. In addition, for compound 26 two additional signals are observed, as expected, corresponding to the fluorine atoms of the thiazolidine–thiourea ligand.
The crystal structure of complex 27 has also been characterised by X-ray diffraction (Fig. 6).29 The geometry around the gold centre is also linear with a C1–Au1–S1 angle of 175.51(6)°. The main bond distances are Au1–C1 2.019(2) and Au1–S1 2.3069(6) Å. There is an intramolecular hydrogen bond as occurred in the ligands, with a donor⋯acceptor N3–H3⋯N2 distance of 2.626 Å and an angle of 146.2°.
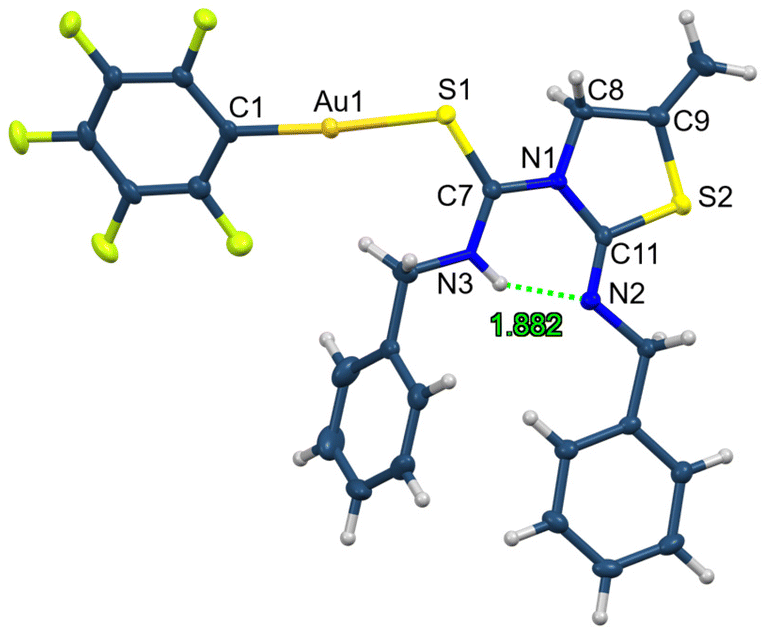 | ||
| Fig. 6 Molecular structure of complex 27 showing ellipsoids at the 50% probability level. | ||
Biological studies
Before the biological tests, analyses of solubility and stability were performed for our complexes. Hence, the ligands and the complexes were not soluble in DMSO and mixtures of DMSO/water were used to carry out the tests. Stability studies were performed in DMSO by 1H NMR studies and all compounds maintained stable unless for a period of 48 h, as no changes were observed between the initial 1H NMR spectra and the corresponding ones after 24 and 48 h, confirming the retention of the complexes in solution. Preliminary studies of the cytotoxic activity of the compounds were carried out using the MTT assay for the lung carcinoma cell line A549.30 The IC50 values for the complexes after 24 h of incubation are shown in Table 1.From these results some statements can be concluded. Interestingly, the thiazolidine–thiourea ligands 10–12 did not show any type of anticancer activity against this cancer line. In contrast, the silver complexes 13–15 that incorporate the triphenylphosphine ligand in their structure show excellent activity with IC50 values ranging from 3.97 ± 0.63 to 6.26 ± 0.29, respectively. Unfortunately, no comparison is possible with the equivalent gold species 19–21 because in these cases the complexes were not obtained completely pure and, consequently, the biological test has not been performed. It is possible to compare the cytotoxic activity of the silver phosphine complexes with other related complexes, such as [Ag(PPh3)(Py-CO-aaOMe)]OTf (aaOMe = amino acid ester), which exhibit much less activity in A549 cells (around 25 μM),20f or with thiourea silver species, which present similar activity in the same cells (around 7 μM).27c Additionally, those complexes bearing pentafluorophenyl groups, such as 25–27, do not show in our experience cytotoxic activity and they have not been measured. The homoleptic silver and gold complexes exhibit low activity. The best one corresponds to the silver complex 17 with an IC50 of 25.16 ± 1.8.
On comparison with the reference drug cisplatin, which exhibits a high value after 24 h (in water) and only a moderate value after 48 h, our silver derivatives present much higher activity at 24 h.
In summary, we can conclude that these types of organic ligands do not exhibit antiproliferative activity in cancer cells but their combination with metal compounds strongly enhance their activity, especially when a phosphine ancillary ligand is present.
Experimental
Experimental details
Mass spectra were recorded on a BRUKER ESQUIRE 3000 PLUS, with the electrospray (ESI) technique. 1H, 13C{1H}, 31P{1H} and 19F NMR spectra were recorded at room temperature on a BRUKER AVANCE 400 spectrometer (1H, 400 MHz, 13C{1H}, 101 MHz, 31P{1H}, 162 MHz and 19F 376.5 MHz) with chemical shifts (δ, ppm) reported relative to the solvent peaks of the deuterated solvent.Crystal structure determination
Crystals were mounted in inert oil on glass fibres and transferred to the cold gas stream of an Xcalibur Oxford diffraction or a Smart APEX CCD diffractometer equipped with a low-temperature attachment. Data were collected using monochromated MoKα radiation (λ = 0.71073 Å). Scan type ϖ. Absorption corrections based on multiple scans were applied using SADABS33 or spherical harmonics implemented in the SCALE3 ABSPACK scaling algorithm.34 The structures were solved by direct methods and refined on F2 using the program SHELXT-2016,35 or by using Olex2 as the graphical interface.36 All non-hydrogen atoms were refined anisotropically.Materials and procedures
The starting materials [AuCl(PPh3)], [Ag(OTf)(PPh3)], [Au(tht)2]OTf and [Au(C6F5)(tht)] were prepared according to published procedures.37 All other reagents were commercially available and used without further purification. Solvents were dried with a SPS solvent purification system.Cell culture
A549 (human lung carcinoma) cells were grown in DMEM medium supplemented with 5% fetal bovine serum (SFB), 100 mg mL−1 pyruvate, 2.2 g L−1 sodium bicarbonate, 5 mL non-essential amino acids (Invitrogen), 2 mM L-glutamine, and antibiotics. Cells were grown at 2 × 105 cells per mL, in 96-well flat-bottomed microplates and allowed to attach for 24 h prior to addition of compounds.Antiproliferative studies: MTT assay
The complexes were dissolved in DMSO and added at different concentrations between 1 and 50 μM in quadruplicate. Each complex was added in four different concentrations of the drugs obtained by different dilutions and with a column of negative controls (cells with a DMSO concentration similar to which contain the solutions of the complexes) and another column of blank (medium only).To make these dilutions, between 2 and 3 mg of the compound were weighed and dissolved in DMSO to obtain a stock solution of 0.1 M. It was then diluted to obtain a solution of 0.001 M in medium (DMEM) and finally from this, dilutions were made to obtain the different concentrations that would be added in the 96-well plate.
Cells were incubated with our compounds for 24 h at 37 °C and with a 5% CO2 atmosphere. Then, 10 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazole) of concentration 5 mg mL−1 was added to each well and plates were incubated for 2 h under the same conditions as described above (37 °C and with a 5% CO2 atmosphere). The crystals of blue formazan that formed are adhered to the bottom of the well, so before dissolving them it is necessary to remove the supernatant by the “flipping” technique and then, dissolve them in DMSO (100 μL per well). The plates are shaken to help to dissolve the formazan precipitated. The optical density was measured at 550 nm using a 96-well multiscanner autoreader (ELISA). The IC50 was calculated by nonlinear regression analysis.
Synthesis of thioureas 3
S), 7.45 (m, 2H, Phortho), 7.33 (m, 1H, Phpara), 7.23 (m, 1H, Phmeta), 6.10 (br s, 1H, CS-NH-CH2), 4.46 (dd, 2H, 3JHH = 5.0 Hz, 4JHH = 2.6 Hz, NH-CH2-C), 2.26 (t, 1H, 4JHH = 2.6 Hz, CH). 13C{1H} APT NMR (ppm) (400 MHz, CDCl3): δ = 180.7 (s, 1C, CS), 135.8 (s, 1C, Phipso), 130.4 (s, 2C, Phortho), 127.7 (s, 1C, Phpara), 125.4 (s, 1C, Phmeta), 77.4 (s, 1C, C-CH2), 72.6 (s, 1C, CH), 35.2 (s, 1C, C-CH2-NH).
Synthesis of thiazolidines 4–9
CH2), 5.12 (dd, 1H, 2JHH = 4.3 Hz, 3JHH = 2.6 Hz, CCH2), 4.58 (t, 2H, 4JHH = 2.3 Hz, N-CH2). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 158.4 (s, 1C, N-CN), 145.1 (s, 1C, CipsoPh), 144.2 (s, 1C, CCH2), 129.1 (s, 2C, CorthoPh), 123.6 (s, 1C, CparaPh), 121.0 (s, 2C, CmetaPh), 104.2 (s, 1C, CCH2), 59.3 (s, 1C, NCH2).
CH2), 5.12 (dd, 1H, 2JHH = 4.0 Hz, 3JHH = 2.4 Hz, CCH2), 4.38 (d, 2H, 4JHH = 2.5 Hz, CH2-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 4.37 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 2.35 (t, 1H, 4JHH = 2.5 Hz, CCH). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 156.3 (s, 1C, N-CN), 151.2 (s, 1C, CipsoPh), 137.0 (s, 1C, CCH2), 129.0 (s, 2C, CorthoPh), 123.6 (s, 1C, CparaPh), 121.9 (s, 2C, CmetaPh), 106.0 (s, 1C, CCH2), 77.4 (s, 1C, CCH), 72.9 (s, 1C, CCH), 55.1 (s, 1C, N-CH2), 33.3 (s, 1C, CH2-CCH).
CH2), 5.07 (dd, 1H, 2JHH = 4.0 Hz, 3JHH = 2.4 Hz, CCH2), 4.28 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 3.09 (s, 3H, CH3). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 151.8 (s, 2C, CipsoPh, N-CN), 137.7 (s, 1C, CCH2), 129.0 (s, 2C, CorthoPh), 123.4 (s, 1C, CparaPh), 122.2 (s, 2C, CmetaPh), 105.3 (s, 1C, CCH2), 58.1 (s, 1C, N-CH2), 33.2 (s, 1C, CH3).
CH2), 5.14 (dd, 1H, 2JHH = 4.4 Hz, 3JHH = 2.6 Hz, CCH2), 4.56 (t, 2H, 3JHH = 2.4 Hz, N-CH2). 13C{1H} APT (ppm) (101 MHz, CDCl3): δ = 163.3 (d, 1C, CipsoPh, 1JCF = 243.7 Hz), 162.1 (s, 1C, N-CN), 149.8 (s, 1C), 130.2 (d, 1C, 3JCF = 9.6 Hz), 116.4 (d, 1C, 4JCF = 2.5 Hz), 110.2 (d, 1C, 2JCF = 21.12 Hz), 108.2 (d, 1C, 2JCF = 23.8 Hz), 104.8 (s, 1C, CH2), 60.44 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, CDCl3): δ = −108.5 (m, 1F, Ph-F).
CH2), 5.12 (m, 1H, CCH2), 4.38 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 4.36 (d, 2H, 4JHH = 2.5 Hz, CH2-CCH), 2.33 (t, 1H, 4JHH = 2.5 Hz, CH2-CCH). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 163.2 (d, 1C, 1JCF = 243.7 Hz, CFPh), 156.7 (s, 1C, N-CN), 151.9 (d, 1C, 3JCF = 9.6 Hz, CipsoPh), 136.7 (s, 1C, CCH2), 130.1 (d, 1C, 3JCF = 9.6 Hz, C-Ar), 117.7 (d, 1C, 4JCF = 2.7 Hz, C-Ar), 110.3 (d, 1C, 2JCF = 21.1 Hz, C-Ar), 109.3 (d, 1C, 2JCF = 22.0 Hz, C-Ar), 106.3 (s, 1C, CCH2), 77.7 (s, 1C, CCH), 73.1 (s, 1C, CCH), 55.2 (s, 1C, N-CH2), 33.3 (s, 1C, CH2-CCH). 19F{1H} NMR (ppm) (376.5 MHz, CDCl3): δ = −112.9 (m, 1F, Ph-F).
CH2), 5.09 (m, 1H, CCH2), 4.28 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 3.07 (s, 3H, CH3). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 163.8 (d, 1C, 1JCF = 245.3 Hz, CFPh), 157.8 (s, 1C, N-CN), 154.0 (d, 1C, 3JCF = 9.6 Hz, CipsoPh), 137.6 (s, 1C, CCH2), 130.3 (d, 1C, 3JCF = 9.7 Hz, C-Ar), 118.3 (d, 1C, 4JCF = 2.7 Hz, C-Ar), 110.2 (d, 1C, 2JCF = 21.3 Hz, C-Ar), 109.7 (d, 1C, 2JCF = 21.9 Hz, C-Ar), 105.9 (s, 1C, CCH2), 57.9 (s, 1C, N-CH2), 32.8 (s, 1C, CH3). 19F NMR (ppm) (376.5 MHz, CDCl3): δ = −113.0 (m, 1F, Ph-F).
CH2), 5.38 (t, 2H, 4JHH = 2.4 Hz, N-CH2), 5.24 (dt, 1H, 2JHH = 2.7 Hz, 4JHH = 2.2 Hz, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 178.6 (s, 1C, CS), 158.6 (s, 1C, NC-N), 149.2 (s, 1C, CipsoPh-N), 139.9 (s, 1C, CipsoPh-NH), 132.8 (s, 1C, CH2C), 130.3 (s, 2C, Ph), 129.4 (s, 2C, Ph), 126.8 (s, 2C, Ph), 126.3 (s, 1C, Ph), 125.2 (s, 1C, Ph), 122.3 (s, 2C, Ph), 107.9 (s, 1C, CCH2), 60.9 (s, 1C, N-CH2).
CH2), 5.38 (t, 2H, JHH = 2.4 Hz, N-CH2), 5.28 (m, 1H, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 165.5 (s, 1C, CS), 157.9 (s, 1C, NC-N), 150.0 (s, 1C, CipsoPh), 140.9 (s, 1C, CipsoPh), 131.2 (s, 1C, CH2C), 131.9 (d, 1C, 3JCF = 9.5 Hz, CmetaPh), 130.9 (d, 1C, 3JCF = 9.4 Hz, CmetaPh), 120.8 (d, 1C, 4JCF = 3.0 Hz, CparaPh,), 118.4 (d, 1C, 4JCF = 3.0 Hz, CparaPh), 113.3 (d, 1C, 2JCF = 21.4 Hz, CorthoPh), 112.9 (d, 1C, 2JCF = 21.3 Hz, CorthoPh), 112.0 (d, 1C, 2JCF = 25.6 Hz, CorthoPh), 109.8 (d, 1C, 2JCF = 23.3 Hz, CorthoPh), 108.3 (s, 1C, CCH2), 61.0 (s, 1C, N-CH2). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −111.0 (m, 1F, Ph-F), −111.5 (m, 1F, Ph-F).
CH2), 5.30 (m, 1H, CCH2), 5.26 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 4.81 (d, 2H, 3JHH = 4.9 Hz, NH-CH2), 4.41 (s, 2H, N-CH2-Ph). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 180.2 (s, 1C, CS), 156.9 (s, 1C, N-CN), 139.5 (s, 1C, CipsoPh-CH2-N), 138.2 (s, 1C, CipsoPh-CH2-NH), 133.2 (s, 1C, CCH2), 129.5–127.8 (s, 10C, Ph), 107.8 (s, 1C, CH2), 60.4 (s, 1C, CH2-N), 59.0 (s, 1C, N-CH2-Ph), 50.2 (s, 1C, NH-CH2).
CH2), 5.46 (br t, 2H, N-CH2), 5.35 (br s, 1H, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 164.8 (s, 1C), 156.5 (s, 1C), 151.2 (s, 1C), 134.8 (d, 6C, 3JH–P = 15.6 Hz, CorthoPPh3), 130.2 (d, 6C, 3JH–P = 9.6 Hz, CmetaPPh3), 132.2–130.4 (s, 14C, CparaPPh3 + Ph), 129.1 (s, 1C, Ph), 126.9 (s, 1C, Ph), 126.5 (s, 1C, Ph), 122.1 (s, 1C, Ph), 109.0 (s, 1C, CCH2), 60.8 (s, 1C, N-CH2). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 12.0 (br s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C35H30AgN3PS2]+ 694.0664, found 694.0647.
CH2), 5.46 (s, 2H, CCH2), 5.36 (m, 1H, N-CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 165.2 (s, 1C, CS), 162.7 (s, 1C, Cq), 151.1 (s, 1C), 149.5 (s, 1C), 134.7 (d, 6C, 2JCP = 15.3 Hz, CorthoPPh3), 132.1 (s, 3C, CparaPPh3), 131.4 (d, 3C, 1JCP = 35.1 Hz, CipsoPPh3), 130.2 (d, 6C, 3JCP = 8.6 Hz, CmetaPPh3), 118.3–109.6 (m, 10C, Ph-F), 109.5 (s, 2C, CCH2), 61.0 (s, 1C, N-CH2). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −79.9 (s, 1F, OTf), −113.8 (m, 1F, Ph-F), −114.3 (m, 1F, Ph-F). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 12.2 (s, 1P, PPh3).
CH2), 5.38 (t, 2H, N-CH2), 5.34 (m, 1H, CCH2), 4.95 (d, 2H, JHH = 4.8 Hz, NH-CH2), 4.44 (s, 2H, N-CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 177.9 (s, 1C, CS), 158.2 (s, 1C, N-CN), 138.8 (s, 1C, CipsoPh-CH2-N), 136.5 (s, 1C, CipsoPh-CH2-NH), 133.2 (s, 1C, CCH2), 134.7 (d, 6C, 2JHP = 15.3 Hz, CorthoPPh3), 132.1 (s, 3C, CparaPPh3), 131.6 (d, 3C, CipsoPPh3, 1JHP = 31.5 Hz), 130.2 (d, 6C, 3JHP = 8.4 Hz, CmetaPPh3), 129.8–128.1 (m, 10C, Ph), 108.9 (s, 1C, CCH2), 60.6 (s, 1C, N-CH2), 59.2 (s, 1C, Ph-CH2-NC), 51.3 (s, 1C, Ph-CH2-NH). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 11.5 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C37H34AgN3PS2]+ 724.0975, found 724.0977.
CH2), 5.42 (br s, 4H, N-CH2), 5.39 (br s, 2H, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 178.8 (s, 2C, CS), 160.1 (s, 2C, Cq), 148.1 (s, 2C, Cipso), 137.5 (s, 2C, Cipso), 132.0 (s, 2C, Cquaternary), 131.1–122.1 (s, 20C, Ph), 109.1 (s, 2C, CH2), 60.7 (s, 2C, N-CH2).
CH2), 5.44 (br s, 4H, N-CH2), 5.42 (br s, 2H, CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 168.1 (s, 2C, CS), 163.3 (s, 1C), 157.4 (s, 1C), 149.1 (s, 1C), 132.5 (d, 4C, 3JHF = 9.2 Hz, Ar), 132.2 (d, 4C, 3JHF = 9.3 Hz, Ar), 123.1 (s, 2C, Ar), 118.2 (s, 2C, Ar), 116.4 (d, 2C, 2JHF = 21.9 Hz, Ar), 114.6 (d, 2C, 2JHF = 24.8 Hz, Ar), 113.6 (d, 2C, 2JHF = 22.1 Hz, Ar), 109.7 (d, 2C, 2JHF = 23.9 Hz, Ar), 109.5 (s, 2C, CCH2), 60.8 (s, 2C, N-CH2). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −79.9 (s, 1F, OTf), −112.2 (m, 2F, Ph), −114.2 (m, 2F, Ph). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C34H26AgF4N6S4]+ 831.0079, found 831.0056.
CH2), 5.39 (m, 4H, N-CH2), 5.35 (m, 2H, CCH2), 4.98 (m, 4H, NH-CH2), 4.43 (s, 4H, N-CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 176.5 (s, 2C, CS), 156.5 (s, 2C, N-CN), 138.7 (s, 2C, CipsoPh-CH2-N), 136.0 (s, 2C, CipsoPh-CH2-NH), 132.0 (s, 2C, CCH2), 130.0–128.1 (s, 20C, Ph), 109.0 (s, 2C, CCH2), 60.5 (s, 2C, N-CH2), 59.2 (s, 1C, Ph-CH2-NC), 51.5 (s, 1C, Ph-CH2-NH). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C38H38AgN6S4]+ 815.1083, found 815.1099.
S), 160.8 (s, 1C, N-CN), 147.7 (s, 1C, CipsoPh-CH2-N), 138.1 (s, 1C, CipsoPh-CH2-NH), 135.3 (s, 2C, CorthoPh), 135.2 (d, 6C, CorthoPPh3, 2JC–P = 13.2 Hz), 133.4 (d, 3C, CparaPPh3, 4JC–P = 2.7 Hz), 131.6 (s, 1C, CCH2), 130.9 (s, 2C, CmetaPh), 130.5 (d, 6C, CmetaPPh3, 3JC–P = 11.8 Hz), 130.5 (s, 2C, CorthoPh), 128.7 (d, 3C, CipsoPPh3, 1JC–P = 63.2 Hz), 127.2 (s, 2C, CparaPh), 122.1 (s, 2C, CmetaPh), 109.5 (s, 2C, CH2), 60.5 (s, 1C, CH2-N). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 45.2 (s, 1P, Au(PPh3)2), 37.5 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C35H30AuN3PS2]+ 784.1279, found 784.1277.
S), 164.9 (d, 1C, CipsoPh-F-NH, 1JC–F = 247.7 Hz), 162.4 (d, 1C, CipsoPh-F-N, 1JC–F = 245.8 Hz), 161.4 (s, 1C, N-CN), 149.2 (d, 1C, CipsoPh-F-N, 3JC–F = 9.2 Hz), 139.5 (d, 1C, CipsoPh-F-NH, 3JC–F = 10.0 Hz), 134.9 (d, 6C, CorthoPPh3, 2JC–P = 13.8 Hz), 133.4 (d, 3C, CparaPPh3, 4JC–P = 2.6 Hz), 132.5 (d, 1C, C7, 3JC–F = 9.2 Hz), 132.1 (d, 1C, C3, 3JC–F = 9.4 Hz), 131.2 (s, 1C, CCH2), 130.4 (d, 6C, CmetaPPh3, 3JC–P = 11.9 Hz), 129.0 (d, 3C, CipsoPPh3, 1JC–P = 61.0 Hz), 123.4 (s, 1C, C8), 118.2 (d, 1C, C4, 4JC–F = 2.7 Hz), 116.6 (d, 1C, C6, 2JC–F = 21.0 Hz), 115.1 (d, 1C, C1, 2JC–F = 24.4 Hz), 113.9 (d, 1C, C2, 2JC–F = 21.1 Hz), 109.9 (s, 1C, CH2), 109.5 (d, 1C, C5, 2JC–F = 23.5 Hz), 60.7 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −118.8 (s, 1F, F), −113.9 (s, 1F, F), −127.5 (m, 6F, SbF6). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 45.2 (s, 1P, Au(PPh3)2), 37.4 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C35H28AuF2N3PS2]+ 820.1091, found 820.1088.
S), 158.9 (s, 1C, N-CN), 138.4 (s, 1C, CipsoPh-CH2-N), 136.0 (s, 1C, CipsoPh-CH2-NH), 135.0 (d, 6C, CorthoPPh3, 2JC–P = 13.8 Hz); 133.4 (d, 3C, CparaPPh3, 4JC–P = 2.7 Hz), 131.7 (s, 1C, CCH2), 130.6 (d, 6C, CmetaPPh3, 3JC–P = 11.9 Hz), 129.9 (s, 2C, CorthoPh), 129.2 (s, 2C, CmetaPh), 129.0 (s, 2C, CparaPh), 128.6 (s, 2C, CorthoPh), 128.4 (s, 2C, CmetaPh), 128.2 (s, 2C, CparaPh), 109.4 (s, 2C, CH2), 60.6 (s, 1C, CH2-N), 59.1 (s, 1C, Ph-CH2-N), 51.8 (s, 1C, CH2-NH). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 45.2 (s, 1P, Au(PPh3)2), 37.6 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C37H34AuN3PS2]+ 812.1592, found 812.1591.
CH2), 5.43 (br s, 6H, N-CH2 + CCH2). 13C{1H} APT NMR (ppm) (400 MHz, (CD3)2CO): δ = 160.1 (s, 1C), 154.1 (s, 1C), 146.4 (s, 1C), 138.8–131.8 (s, 6C, Ph), 130.6–122.1 (s, 20C, Ph), 109.5 (s, 2C, CCH2), 60.2 (s, 2C, N-CH2). 19F{1H} NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −80.1 (s, 3F, OTf).
CH2), 5.47 (br d, 6H, N-CH2 + CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 166.2 (s, 2C, CS), 162.8 (s, 1C, Cq), 132.1 (d, 4C, 3JCF = 9.3 Hz, Ar), 132.1 (d, 4C, 3JCF = 9.4 Hz, Ar), 118.3 (s, 4C, Ar), 113.4 (d, 4C, 2JCF = 21.2 Hz, Ar), 109.7 (d, 4C, 2JCF = 23.5 Hz, Ar), 109.2 (s, 2C, CCH2), 60.9 (s, 1C, N-CH2). 19F{1H} NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −73.5 (s, 1F, OTf), −106.4 (m, 2F, Ph-F), −107.5 (m, 2F, Ph-F).
CH2), 5.42 (m, 6H, N-CH2, CCH2), 5.08 (m, 2H, NH-CH2), 4.48 (s, 2H, N-CH2). 13C{1H} APT (ppm) (101 MHz, (CD3)2CO): δ = 176.1 (s, 2C, CS), 158.2 (s, 2C, N-CN), 138.9 (s, 2C, CipsoPh-CH2-N), 136.9 (s, 2C, CipsoPh-CH2-NH), 133.3 (s, 2C, CH2C), 129.7–128.1 (s, 20C, Ph), 108.9 (s, 2C, CCH2), 60.4 (s, 2C, CH2), 59.1 (s, 2C, Ph-CH2-NC), 51.2 (s, 2C, Ph-CH2-NH). 19F{1H} NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −79.8 (s, 1F, OTf).
C), 130.4 (s, 4C, CorthoPh), 129.3 (s, 1C, CparaPh), 127.4 (s, 2C, CmetaPh), 127.0 (s, 1C, CparaPh), 121.1 (s, 2C, CmetaPh), 109.2 (s, 1C, CH2), 60.5 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −116.4 (m, 2F, ForthoC6F5), −164.1 (t, 1F, FparaC6F5, 3JFF = 19.7 Hz), −166.3 (m, 2F, FmetaC6F5).
C), 123.8 (s, 1C), 118.2 (s, 1C), 116.2 (d, 1C, 2JCF = 21.3 Hz), 115.3 (d, 1C, 2JCF = 28.5 Hz), 113.6 (d, 1C, 2JCF = 21.3 Hz), 109.7 (d, 1C, 2JCF = 23.7 Hz), 109.6 (s, 1C, CH2), 60.6 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −114.0 (m, 1F, Ph-F), −114.1 (m, 1F, Ph-F), −116.6 (m, 2F, ForthoC6F5), −163.8 (t, 1F, FparaC6F5, 3JFF = 19.7 Hz), −166.2 (m, 2F, FmetaC6F5).
N), 138.5 (s, 1C, CipsoPh-F), 132.0 (s, 1C, CH2C), 129.8–128.1 (m, 10C, Ph), 109.3 (s, 1C, CH2), 60.5 (s, 1C, CH2-N), 59.1 (s, 1C, Ph-CH2-NC), 52.3 (s, 1C, Ph-CH2-NH). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −117.7 (m, 2F, ForthoC6F5), −163.4 (t, 1F, FparaC6F5, 3JFF = 19.6 Hz), −166.7 (m, 2F, FmetaC6F5).
CH), 4.37 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 2.35 (t, 1H, 4JHH = 2.5 Hz, CCH). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 156.3 (s, 1C, N-CN), 151.2 (s, 1C, CipsoPh), 137.0 (s, 1C, CCH2), 129.0 (s, 2C, CorthoPh), 123.6 (s, 1C, CparaPh), 121.9 (s, 2C, CmetaPh), 106.0 (s, 1C, CCH2), 77.4 (s, 1C, CCH), 72.9 (s, 1C, CCH), 55.1 (s, 1C, N-CH2), 33.3 (s, 1C, CH2-CCH).
CH2), 5.07 (dd, 1H, 2JHH = 4.0 Hz, 3JHH = 2.4 Hz, CCH2), 4.28 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 3.09 (s, 3H, CH3). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 151.8 (s, 2C, CipsoPh, N-CN), 137.7 (s, 1C, CCH2), 129.0 (s, 2C, CorthoPh), 123.4 (s, 1C, CparaPh), 122.2 (s, 2C, CmetaPh), 105.3 (s, 1C, CCH2), 58.1 (s, 1C, N-CH2), 33.2 (s, 1C, CH3).
CH2), 5.14 (dd, 1H, 2JHH = 4.4 Hz, 3JHH = 2.6 Hz, CCH2), 4.56 (t, 2H, 3JHH = 2.4 Hz, N-CH2). 13C{1H} APT (ppm) (101 MHz, CDCl3): δ = 163.3 (d, 1C, CipsoPh, 1JCF = 243.7 Hz), 162.1 (s, 1C, N-CN), 149.8 (s, 1C), 130.2 (d, 1C, 3JCF = 9.6 Hz), 116.4 (d, 1C, 4JCF = 2.5 Hz), 110.2 (d, 1C, 2JCF = 21.12 Hz), 108.2 (d, 1C, 2JCF = 23.8 Hz), 104.8 (s, 1C, CH2), 60.44 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, CDCl3): δ = −108.5 (m, 1F, Ph-F).
CH2), 5.12 (m, 1H, CCH2), 4.38 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 4.36 (d, 2H, 4JHH = 2.5 Hz, CH2-CCH), 2.33 (t, 1H, 4JHH = 2.5 Hz, CH2-CCH). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 163.2 (d, 1C, 1JCF = 243.7 Hz, CFPh), 156.7 (s, 1C, N-CN), 151.9 (d, 1C, 3JCF = 9.6 Hz, CipsoPh), 136.7 (s, 1C, CCH2), 130.1 (d, 1C, 3JCF = 9.6 Hz, C-Ar), 117.7 (d, 1C, 4JCF = 2.7 Hz, C-Ar), 110.3 (d, 1C, 2JCF = 21.1 Hz, C-Ar), 109.3 (d, 1C, 2JCF = 22.0 Hz, C-Ar), 106.3 (s, 1C, CCH2), 77.7 (s, 1C, CCH), 73.1 (s, 1C, CCH), 55.2 (s, 1C, N-CH2), 33.3 (s, 1C, CH2-CCH). 19F{1H} NMR (ppm) (376.5 MHz, CDCl3): δ = −112.9 (m, 1F, Ph-F).
CH2), 5.09 (m, 1H, CCH2), 4.28 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 3.07 (s, 3H, CH3). 13C{1H} APT NMR (ppm) (101 MHz, CDCl3): δ = 163.8 (d, 1C, 1JCF = 245.3 Hz, CFPh), 157.8 (s, 1C, N-CN), 154.0 (d, 1C, 3JCF = 9.6 Hz, CipsoPh), 137.6 (s, 1C, CCH2), 130.3 (d, 1C, 3JCF = 9.7 Hz, C-Ar), 118.3 (d, 1C, 4JCF = 2.7 Hz, C-Ar), 110.2 (d, 1C, 2JCF = 21.3 Hz, C-Ar), 109.7 (d, 1C, 2JCF = 21.9 Hz, C-Ar), 105.9 (s, 1C, CCH2), 57.9 (s, 1C, N-CH2), 32.8 (s, 1C, CH3). 19F NMR (ppm) (376.5 MHz, CDCl3): δ = −113.0 (m, 1F, Ph-F).
CH2), 5.38 (t, 2H, 4JHH = 2.4 Hz, N-CH2), 5.24 (dt, 1H, 2JHH = 2.7 Hz, 4JHH = 2.2 Hz, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 178.6 (s, 1C, CS), 158.6 (s, 1C, NC-N), 149.2 (s, 1C, CipsoPh-N), 139.9 (s, 1C, CipsoPh-NH), 132.8 (s, 1C, CH2C), 130.3 (s, 2C, Ph), 129.4 (s, 2C, Ph), 126.8 (s, 2C, Ph), 126.3 (s, 1C, Ph), 125.2 (s, 1C, Ph), 122.3 (s, 2C, Ph), 107.9 (s, 1C, CCH2), 60.9 (s, 1C, N-CH2).
CH2), 5.38 (t, 2H, JHH = 2.4 Hz, N-CH2), 5.28 (m, 1H, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 165.5 (s, 1C, CS), 157.9 (s, 1C, NC-N), 150.0 (s, 1C, CipsoPh), 140.9 (s, 1C, CipsoPh), 131.2 (s, 1C, CH2C), 131.9 (d, 1C, 3JCF = 9.5 Hz, CmetaPh), 130.9 (d, 1C, 3JCF = 9.4 Hz, CmetaPh), 120.8 (d, 1C, 4JCF = 3.0 Hz, CparaPh,), 118.4 (d, 1C, 4JCF = 3.0 Hz, CparaPh), 113.3 (d, 1C, 2JCF = 21.4 Hz, CorthoPh), 112.9 (d, 1C, 2JCF = 21.3 Hz, CorthoPh), 112.0 (d, 1C, 2JCF = 25.6 Hz, CorthoPh), 109.8 (d, 1C, 2JCF = 23.3 Hz, CorthoPh), 108.3 (s, 1C, CCH2), 61.0 (s, 1C, N-CH2). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −111.0 (m, 1F, Ph-F), −111.5 (m, 1F, Ph-F).
CH2), 5.30 (m, 1H, CCH2), 5.26 (t, 2H, 4JHH = 2.3 Hz, N-CH2), 4.81 (d, 2H, 3JHH = 4.9 Hz, NH-CH2), 4.41 (s, 2H, N-CH2-Ph). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 180.2 (s, 1C, CS), 156.9 (s, 1C, N-CN), 139.5 (s, 1C, CipsoPh-CH2-N), 138.2 (s, 1C, CipsoPh-CH2-NH), 133.2 (s, 1C, CCH2), 129.5–127.8 (s, 10C, Ph), 107.8 (s, 1C, CH2), 60.4 (s, 1C, CH2-N), 59.0 (s, 1C, N-CH2-Ph), 50.2 (s, 1C, NH-CH2).
CH2), 5.46 (br t, 2H, N-CH2), 5.35 (br s, 1H, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 164.8 (s, 1C), 156.5 (s, 1C), 151.2 (s, 1C), 134.8 (d, 6C, 3JH–P = 15.6 Hz, CorthoPPh3), 130.2 (d, 6C, 3JH–P = 9.6 Hz, CmetaPPh3), 132.2–130.4 (s, 14C, CparaPPh3 + Ph), 129.1 (s, 1C, Ph), 126.9 (s, 1C, Ph), 126.5 (s, 1C, Ph), 122.1 (s, 1C, Ph), 109.0 (s, 1C, CCH2), 60.8 (s, 1C, N-CH2). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 12.0 (br s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C35H30AgN3PS2]+ 694.0664, found 694.0647.
CH2), 5.46 (s, 2H, CCH2), 5.36 (m, 1H, N-CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 165.2 (s, 1C, CS), 162.7 (s, 1C, Cq), 151.1 (s, 1C), 149.5 (s, 1C), 134.7 (d, 6C, 2JCP = 15.3 Hz, CorthoPPh3), 132.1 (s, 3C, CparaPPh3), 131.4 (d, 3C, 1JCP = 35.1 Hz, CipsoPPh3), 130.2 (d, 6C, 3JCP = 8.6 Hz, CmetaPPh3), 118.3–109.6 (m, 10C, Ph-F), 109.5 (s, 2C, CCH2), 61.0 (s, 1C, N-CH2). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −79.9 (s, 1F, OTf), −113.8 (m, 1F, Ph-F), −114.3 (m, 1F, Ph-F). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 12.2 (s, 1P, PPh3).
CH2), 5.38 (t, 2H, N-CH2), 5.34 (m, 1H, CCH2), 4.95 (d, 2H, JHH = 4.8 Hz, NH-CH2), 4.44 (s, 2H, N-CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 177.9 (s, 1C, CS), 158.2 (s, 1C, N-CN), 138.8 (s, 1C, CipsoPh-CH2-N), 136.5 (s, 1C, CipsoPh-CH2-NH), 133.2 (s, 1C, CCH2), 134.7 (d, 6C, 2JHP = 15.3 Hz, CorthoPPh3), 132.1 (s, 3C, CparaPPh3), 131.6 (d, 3C, CipsoPPh3, 1JHP = 31.5 Hz), 130.2 (d, 6C, 3JHP = 8.4 Hz, CmetaPPh3), 129.8–128.1 (m, 10C, Ph), 108.9 (s, 1C, CCH2), 60.6 (s, 1C, N-CH2), 59.2 (s, 1C, Ph-CH2-NC), 51.3 (s, 1C, Ph-CH2-NH). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 11.5 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C37H34AgN3PS2]+ 724.0975, found 724.0977.
CH2), 5.42 (br s, 4H, N-CH2), 5.39 (br s, 2H, CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 178.8 (s, 2C, CS), 160.1 (s, 2C, Cq), 148.1 (s, 2C, Cipso), 137.5 (s, 2C, Cipso), 132.0 (s, 2C, Cquaternary), 131.1–122.1 (s, 20C, Ph), 109.1 (s, 2C, CH2), 60.7 (s, 2C, N-CH2).
CH2), 5.44 (br s, 4H, N-CH2), 5.42 (br s, 2H, CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 168.1 (s, 2C, CS), 163.3 (s, 1C), 157.4 (s, 1C), 149.1 (s, 1C), 132.5 (d, 4C, 3JHF = 9.2 Hz, Ar), 132.2 (d, 4C, 3JHF = 9.3 Hz, Ar), 123.1 (s, 2C, Ar), 118.2 (s, 2C, Ar), 116.4 (d, 2C, 2JHF = 21.9 Hz, Ar), 114.6 (d, 2C, 2JHF = 24.8 Hz, Ar), 113.6 (d, 2C, 2JHF = 22.1 Hz, Ar), 109.7 (d, 2C, 2JHF = 23.9 Hz, Ar), 109.5 (s, 2C, CCH2), 60.8 (s, 2C, N-CH2). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −79.9 (s, 1F, OTf), −112.2 (m, 2F, Ph), −114.2 (m, 2F, Ph). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C34H26AgF4N6S4]+ 831.0079, found 831.0056.
CH2), 5.39 (m, 4H, N-CH2), 5.35 (m, 2H, CCH2), 4.98 (m, 4H, NH-CH2), 4.43 (s, 4H, N-CH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 176.5 (s, 2C, CS), 156.5 (s, 2C, N-CN), 138.7 (s, 2C, CipsoPh-CH2-N), 136.0 (s, 2C, CipsoPh-CH2-NH), 132.0 (s, 2C, CCH2), 130.0–128.1 (s, 20C, Ph), 109.0 (s, 2C, CCH2), 60.5 (s, 2C, N-CH2), 59.2 (s, 1C, Ph-CH2-NC), 51.5 (s, 1C, Ph-CH2-NH). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C38H38AgN6S4]+ 815.1083, found 815.1099.
S), 160.8 (s, 1C, N-CN), 147.7 (s, 1C, CipsoPh-CH2-N), 138.1 (s, 1C, CipsoPh-CH2-NH), 135.3 (s, 2C, CorthoPh), 135.2 (d, 6C, CorthoPPh3, 2JC–P = 13.2 Hz), 133.4 (d, 3C, CparaPPh3, 4JC–P = 2.7 Hz), 131.6 (s, 1C, CCH2), 130.9 (s, 2C, CmetaPh), 130.5 (d, 6C, CmetaPPh3, 3JC–P = 11.8 Hz), 130.5 (s, 2C, CorthoPh), 128.7 (d, 3C, CipsoPPh3, 1JC–P = 63.2 Hz), 127.2 (s, 2C, CparaPh), 122.1 (s, 2C, CmetaPh), 109.5 (s, 2C, CH2), 60.5 (s, 1C, CH2-N). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 45.2 (s, 1P, Au(PPh3)2), 37.5 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C35H30AuN3PS2]+ 784.1279, found 784.1277.
S), 164.9 (d, 1C, CipsoPh-F-NH, 1JC–F = 247.7 Hz), 162.4 (d, 1C, CipsoPh-F-N, 1JC–F = 245.8 Hz), 161.4 (s, 1C, N-CN), 149.2 (d, 1C, CipsoPh-F-N, 3JC–F = 9.2 Hz), 139.5 (d, 1C, CipsoPh-F-NH, 3JC–F = 10.0 Hz), 134.9 (d, 6C, CorthoPPh3, 2JC–P = 13.8 Hz), 133.4 (d, 3C, CparaPPh3, 4JC–P = 2.6 Hz), 132.5 (d, 1C, C7, 3JC–F = 9.2 Hz), 132.1 (d, 1C, C3, 3JC–F = 9.4 Hz), 131.2 (s, 1C, CCH2), 130.4 (d, 6C, CmetaPPh3, 3JC–P = 11.9 Hz), 129.0 (d, 3C, CipsoPPh3, 1JC–P = 61.0 Hz), 123.4 (s, 1C, C8), 118.2 (d, 1C, C4, 4JC–F = 2.7 Hz), 116.6 (d, 1C, C6, 2JC–F = 21.0 Hz), 115.1 (d, 1C, C1, 2JC–F = 24.4 Hz), 113.9 (d, 1C, C2, 2JC–F = 21.1 Hz), 109.9 (s, 1C, CH2), 109.5 (d, 1C, C5, 2JC–F = 23.5 Hz), 60.7 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −118.8 (s, 1F, F), −113.9 (s, 1F, F), −127.5 (m, 6F, SbF6). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 45.2 (s, 1P, Au(PPh3)2), 37.4 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C35H28AuF2N3PS2]+ 820.1091, found 820.1088.
S), 158.9 (s, 1C, N-CN), 138.4 (s, 1C, CipsoPh-CH2-N), 136.0 (s, 1C, CipsoPh-CH2-NH), 135.0 (d, 6C, CorthoPPh3, 2JC–P = 13.8 Hz); 133.4 (d, 3C, CparaPPh3, 4JC–P = 2.7 Hz), 131.7 (s, 1C, CCH2), 130.6 (d, 6C, CmetaPPh3, 3JC–P = 11.9 Hz), 129.9 (s, 2C, CorthoPh), 129.2 (s, 2C, CmetaPh), 129.0 (s, 2C, CparaPh), 128.6 (s, 2C, CorthoPh), 128.4 (s, 2C, CmetaPh), 128.2 (s, 2C, CparaPh), 109.4 (s, 2C, CH2), 60.6 (s, 1C, CH2-N), 59.1 (s, 1C, Ph-CH2-N), 51.8 (s, 1C, CH2-NH). 31P{1H} NMR (ppm) (162 MHz, (CD3)2CO): δ = 45.2 (s, 1P, Au(PPh3)2), 37.6 (s, 1P, PPh3). HRMS (ESI+ μ-TOF): m/z (%) = [M]+ calcd for [C37H34AuN3PS2]+ 812.1592, found 812.1591.
CH2), 5.43 (br s, 6H, N-CH2 + CCH2). 13C{1H} APT NMR (ppm) (400 MHz, (CD3)2CO): δ = 160.1 (s, 1C), 154.1 (s, 1C), 146.4 (s, 1C), 138.8–131.8 (s, 6C, Ph), 130.6–122.1 (s, 20C, Ph), 109.5 (s, 2C, CCH2), 60.2 (s, 2C, N-CH2). 19F{1H} NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −80.1 (s, 3F, OTf).
CH2), 5.47 (br d, 6H, N-CH2 + CCH2). 13C{1H} APT NMR (ppm) (101 MHz, (CD3)2CO): δ = 166.2 (s, 2C, CS), 162.8 (s, 1C, Cq), 132.1 (d, 4C, 3JCF = 9.3 Hz, Ar), 132.1 (d, 4C, 3JCF = 9.4 Hz, Ar), 118.3 (s, 4C, Ar), 113.4 (d, 4C, 2JCF = 21.2 Hz, Ar), 109.7 (d, 4C, 2JCF = 23.5 Hz, Ar), 109.2 (s, 2C, CCH2), 60.9 (s, 1C, N-CH2). 19F{1H} NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −73.5 (s, 1F, OTf), −106.4 (m, 2F, Ph-F), −107.5 (m, 2F, Ph-F).
CH2), 5.42 (m, 6H, N-CH2, CCH2), 5.08 (m, 2H, NH-CH2), 4.48 (s, 2H, N-CH2). 13C{1H} APT (ppm) (101 MHz, (CD3)2CO): δ = 176.1 (s, 2C, CS), 158.2 (s, 2C, N-CN), 138.9 (s, 2C, CipsoPh-CH2-N), 136.9 (s, 2C, CipsoPh-CH2-NH), 133.3 (s, 2C, CH2C), 129.7–128.1 (s, 20C, Ph), 108.9 (s, 2C, CCH2), 60.4 (s, 2C, CH2), 59.1 (s, 2C, Ph-CH2-NC), 51.2 (s, 2C, Ph-CH2-NH). 19F{1H} NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −79.8 (s, 1F, OTf).
C), 130.4 (s, 4C, CorthoPh), 129.3 (s, 1C, CparaPh), 127.4 (s, 2C, CmetaPh), 127.0 (s, 1C, CparaPh), 121.1 (s, 2C, CmetaPh), 109.2 (s, 1C, CH2), 60.5 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −116.4 (m, 2F, ForthoC6F5), −164.1 (t, 1F, FparaC6F5, 3JFF = 19.7 Hz), −166.3 (m, 2F, FmetaC6F5).
C), 123.8 (s, 1C), 118.2 (s, 1C), 116.2 (d, 1C, 2JCF = 21.3 Hz), 115.3 (d, 1C, 2JCF = 28.5 Hz), 113.6 (d, 1C, 2JCF = 21.3 Hz), 109.7 (d, 1C, 2JCF = 23.7 Hz), 109.6 (s, 1C, CH2), 60.6 (s, 1C, CH2-N). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −114.0 (m, 1F, Ph-F), −114.1 (m, 1F, Ph-F), −116.6 (m, 2F, ForthoC6F5), −163.8 (t, 1F, FparaC6F5, 3JFF = 19.7 Hz), −166.2 (m, 2F, FmetaC6F5).
N), 138.5 (s, 1C, CipsoPh-F), 132.0 (s, 1C, CH2C), 129.8–128.1 (m, 10C, Ph), 109.3 (s, 1C, CH2), 60.5 (s, 1C, CH2-N), 59.1 (s, 1C, Ph-CH2-NC), 52.3 (s, 1C, Ph-CH2-NH). 19F NMR (ppm) (376.5 MHz, (CD3)2CO): δ = −117.7 (m, 2F, ForthoC6F5), −163.4 (t, 1F, FparaC6F5, 3JFF = 19.6 Hz), −166.7 (m, 2F, FmetaC6F5).
Conclusions
Several thiazole-based heterocycles have been achieved by the reaction of propargylamines with isothiocyanates. The selective formation of thioureas, iminothiazolidines, aminothiazolines or mixed thiazolidine–thiourea compounds has been accomplished depending upon the reaction conditions. The use of secondary propargylamines leads selectively to the formation of cyclic iminothiazolidine derivatives, while primary propargylamines form iminothiazoline species. Further reaction of the cyclic thiazoline derivatives with isothiocyanate or the reaction of the starting compounds in a 1:2 molar ratio affords the thiazolidine–thiourea derivatives. The proposed mechanism for the formation of these heterocycles starts with the formation of the thiourea species following two possible pathways. The first one is the formation of the thiourea, cyclisation to form the iminothiazolidine and further attack of the isothiocyanate to produce the thiazolidine–thiourea. Alternatively, the generation of bis(thiourea) and cyclisation produce the same compounds. Coordination studies of these heterocyclic species towards silver and gold with different stoichiometries have been carried out to afford cationic [ML(PPh3)]OTf, [ML2]OTf (M = Ag, Au) or neutral [Au(C6F5)L] complexes. Preliminary studies of the cytotoxic activity in lung cancer cells have been carried out in both ligand and complexes, showing that although the ligands do not exhibit anticancer activity, the coordination of the metals, especially silver, greatly enhances the cytotoxic activity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors also thank Agencia Estatal de Investigación (PID2019-104379RB-C21 and PID2020-117455GB-I00/AEI/10.13039/501100011033; and RED2018-102471-T (MCIN/AEI/10.13039/501100011033)) and Gobierno de Aragón-Fondo Social Europeo (research group E07_20R) for financial support of our research.References
- N. Aydemir and R. Bilaloğlu, Mutat. Res., 2003, 537, 43–51 CAS.
- For selected examples, see: (a) C. Sanmartín, M. Echeverría, B. Mendívil, L. Cordeu, E. Cubedo, J. García-Foncillas, M. Font and J. A. Palop, Bioorg. Med. Chem., 2005, 13, 2031–2044 CrossRef PubMed; (b) E. Moreau, S. Fortin, M. Desjardins, J. L. C. Rousseau, E. Petitclerc and R. C. -Gaudreault, Bioorg. Med. Chem., 2005, 13, 6703–6712 CrossRef CAS PubMed; (c) G. Hallur, A. Jimeno, S. Dalrymple, T. Zhu, M. K. Jung, M. Hidalgo, J. T. Isaacs, S. Sukumar, E. Hamel and S. R. Khan, J. Med. Chem., 2006, 49, 2357–2360 CrossRef CAS PubMed; (d) H.-Q. Li, P.-C. Lv, T. Yan and H.-L. Zhu, Anti-Cancer Agents Med. Chem., 2009, 9, 471–480 CrossRef CAS PubMed; (e) H.-Q. Li, T.-T. Zhu, T. Yan, Y. Luo and H.-L. Zhu, Eur. J. Med. Chem., 2009, 44, 453–459 CrossRef CAS PubMed; (f) L. Garuti, M. Roberti, G. Bottegoni and M. Ferraro, Curr. Med. Chem., 2016, 23, 1528–1548 CrossRef CAS PubMed; (g) F. Auria-Luna, E. Marqués-López, E. Romanos, V. Fernández-Moreira, M. C. Gimeno, I. Marzo and R. P. Herrera, Bioorg. Chem., 2020, 105, 104364 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Esteves-Souza, K. Pissinate, M. G. Nascimento, N. F. Grynberg and E. Aurea, Bioorg. Med. Chem., 2006, 14, 492–499 CrossRef CAS PubMed; (b) S. N. Manjula, N. M. Noolvi, K. V. Parihar, S. A. M. Reddy, V. Ramani, A. K. Gadad, G. Singh, N. G. Kutty and C. M. Rao, Eur. J. Med. Chem., 2009, 44, 2923–2929 CrossRef CAS PubMed; (c) S. Saeed, N. Rashid, P. G. Jones, M. Ali and R. Hussain, Eur. J. Med. Chem., 2010, 45, 1323–1331 CrossRef CAS PubMed; (d) J. Yao, J. Chen, Z. He, W. Sun and W. Xu, Bioorg. Med. Chem., 2012, 20, 2923–2929 CrossRef CAS PubMed; (e) C. Ning, Y. Bi, Y. He, W. Y. Huang, L. Liu, Y. Li, S. Zhang, X. Liu and N. Yu, Bioorg. Med. Chem. Lett., 2013, 23, 6432–6435 CrossRef CAS PubMed.
- (a) T. K. Venkatachalam, C. Mao and F. M. Uckun, Bioorg. Med. Chem., 2004, 12, 4275–4284 CrossRef CAS PubMed; (b) R. Hunter, Y. Younis, C. I. Muhanji, T.-l. Curtin, K. J. Naidoo, M. Petersen, C. M. Bailey, A. Basavapathruni and K. S. Anderson, Bioorg. Med. Chem., 2008, 16, 10270–10280 CrossRef CAS PubMed; (c) K. Chen, Z. Tan, M. He, J. Li, S. Tang, I. Hewlett, F. Yu, Y. Jin and M. Yang, Chem. Biol. Drug Des., 2010, 76, 25–33 CrossRef CAS PubMed.
- J. D. Bloom, R. G. Dushin, K. J. Curran, F. Donahue, E. B. Norton, E. Terefenko, T. R. Jones, A. A. Ross, B. Feld, S. A. Lang and M. J. DiGrandi, Bioorg. Med. Chem., 2004, 14, 3401–3406 CrossRef CAS PubMed.
- S. A. Khan, N. Singh and K. Saleem, Eur. J. Med. Chem., 2008, 43, 2272–2277 CrossRef CAS PubMed.
- (a) J. Stefanska, D. Szulczyk, A. E. Koziol, B. Miroslaw, E. Kedzierska, S. Fidecka, B. Busonera, G. Sanna, G. Giliberti, P. La Colla and M. Struga, Eur. J. Med. Chem., 2012, 55, 205–213 CrossRef CAS PubMed; (b) B. Kocyigit-Kaymakcioglu, A. O. Celen, N. Tabanca, A. Ali, S. I. Khan, I. A. Khan and D. E. Wedge, Molecules, 2013, 18, 3562–3567 CrossRef CAS PubMed.
- L. dos Santos, L. A. Lima, V. Cechinel-Filho, R. Corrêa, F. C. Buzzi and R. J. Nunes, Bioorg. Med. Chem., 2008, 16, 8526–8534 CrossRef CAS PubMed.
- A. L. Lourenço, M. S. Saito, L. E. G. Dorneles, G. M. Viana, P. C. Sathler, L. C. S. Aguiar, M. de Pádula, T. F. S. Domingos, A. G. M. Fraga, C. R. Rodrigues, V. P. de Sousa, H. C. Castro and L. M. Cabral, Molecules, 2015, 20, 7174–7200 CrossRef PubMed.
- (a) N. Sahiba, A. Sethiya, J. Soni, D. K. Agarwal and S. Agarwal, Top. Curr. Chem., 2020, 378, 34 CrossRef CAS PubMed; (b) X. Just-Baringo, F. Albericio and M. Alvarez, Curr. Top. Med. Chem., 2014, 14, 1244–1256 CrossRef CAS PubMed; (c) A.-C. Gaumont, M. Gulea and J. Levillain, Chem. Rev., 2009, 109, 1371–1401 CrossRef CAS PubMed.
- (a) D. Das, P. Sikdar and M. Baigari, Eur. J. Med. Chem., 2016, 109, 89–98 CrossRef CAS PubMed; (b) R. P. Singh, M. N. Aziz, D. Gout, W. Fayad, M. A. El-Manawaty and C. J. Lovely, Bioorg. Med. Chem., 2019, 27, 115047 CrossRef CAS PubMed; (c) S. A. Shehzadi, I. Khan, A. Saeed, F. A. Larik, P. A. Channar, M. Hassan, H. Raza, Q. Abbas and S.-Y. Seo, Bioorg. Chem., 2019, 84, 518–528 CrossRef CAS PubMed; (d) A. Singh, D. Malhotra, K. Singh, R. Chadha, P. Mohinder and S. Bedi, J. Mol. Chem., 2022, 1266, 133479 CAS.
- (a) S. R. Alizdeh and S. M. Hashemi, Med. Chem. Res., 2021, 30, 771–806 CrossRef PubMed; (b) Y. Wan, J. Long, H. Gao and Z. Tang, Eur. J. Med. Chem., 2021, 210, 112953 CrossRef CAS PubMed.
- (a) S. K. Bharti, G. Nath, R. Tilak and S. K. Singh, Eur. J. Med. Chem., 2010, 45, 651–660 CrossRef CAS PubMed; (b) A.-M. Borcea, I. Ionuţ, O. Crişan and O. Oniga, Molecules, 2021, 26, 624 CrossRef CAS PubMed.
- I. P. Singh, S. Gupta and S. Kumar, Med. Chem., 2020, 16, 4–23 CrossRef CAS PubMed.
- K. Liaras, M. Fesatidou and A. Geronikaki, Molecules, 2018, 23, 685 CrossRef PubMed.
- R. G. Miller, J. D. Mitchell and D. H. Moore, Cochrane Database Syst. Rev., 2012, 3, CD001447 Search PubMed.
- C. Borelli, M. Schaller, M. Niewerth, K. Nocker, B. Baasner, D. Berg, R. Tiemann, K. Tietjen, B. Fugmann, S. Lang-Fugmann and H. C. Korting, Chemotherapy, 2008, 54, 245–259 CrossRef CAS PubMed.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- (a) I. Ott, Coord. Chem. Rev., 2009, 153, 1670–1681 CrossRef; (b) S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550–580 CrossRef CAS PubMed; (c) C.-M. Che and R. W.-Y. Sun, Chem. Commun., 2011, 47, 9554–9560 RSC; (d) A. Casini and L. Messori, Curr. Top. Med. Chem., 2011, 11, 2647–2660 CrossRef CAS PubMed; (e) C. N. Banti and S. K. Hadjikakou, Metallomics, 2013, 5, 569–596 CrossRef CAS PubMed; (f) B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209–4219 RSC; (g) S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS; (h) T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC; (i) C. I. Yeo, K. K. Ooi and E. R. T. Tiekink, Molecules, 2018, 23, 1410 CrossRef PubMed; (j) V. Fernández-Moreira, R. P. Herrera and M. C. Gimeno, Pure Appl. Chem., 2019, 91, 247 CrossRef.
- For selected examples, see: (a) M. C. Gimeno, H. Goitia, A. Laguna, M. E. Luque, M. D. Villacampa, C. Sepúlveda and M. Meireles, J. Inorg. Biochem., 2011, 105, 1373–1382 CrossRef CAS PubMed; (b) H. Goitia, Y. Nieto, M. D. Villacampa, C. Kasper, A. Laguna and M. C. Gimeno, Organometallics, 2013, 32, 6069–6078 CrossRef CAS; (c) A. Gutiérrez, L. Gracia-Fleta, I. Marzo, C. Cativiela, A. Laguna and M. C. Gimeno, Dalton Trans., 2014, 43, 17054–17066 RSC; (d) L. Ortego, F. Cardoso, S. Martins, M. F. Fillat, A. Laguna, M. Meireles, M. D. Villacampa and M. C. Gimeno, J. Inorg. Biochem., 2014, 130, 32–37 CrossRef CAS PubMed; (e) A. Gutiérrez, I. Marzo, C. Cativiela, A. Laguna and M. C. Gimeno, Chem. – Eur. J., 2015, 21, 11088–11095 CrossRef PubMed; (f) L. Ortego, M. Meireles, C. Kasper, A. Laguna, M. D. Villacampa and M. C. Gimeno, J. Inorg. Biochem., 2016, 156, 133–144 CrossRef CAS PubMed; (g) D. Salvador-Gil, L. Ortego, R. P. Herrera, I. Marzo and M. C. Gimeno, Dalton Trans., 2017, 46, 13745–13755 RSC; (h) V. Fernández-Moreira, C. Val-Campillo, I. Ospino, R. P. Herrera, I. Marzo, A. Laguna and M. C. Gimeno, Dalton Trans., 2019, 48, 3098–3108 RSC.
- (a) I. Mármol, S. Montanel-Perez, J. C. Royo, M. C. Gimeno, M. D. Villacampa, M. J. Rodríguez-Yoldi and E. Cerrada, Inorg. Chem., 2020, 59, 17732–17745 CrossRef PubMed; (b) A. Luengo, I. Marzo, M. Reback, I. M. Daubit, V. Fernández-Moreira, N. Metzler-Nolte and M. C. Gimeno, Chem. – Eur. J., 2020, 26, 12158–12167 CrossRef CAS PubMed; (c) I. Mármol, P. Castellnou, R. Alvarez, M. C. Gimeno, M. J. Rodriguez-Yoldi and E. Cerrada, Eur. J. Med. Chem., 2019, 183, 111661 CrossRef PubMed; (d) H. Goitia, A. Laguna and M. C. Gimeno, Inorg. Chim. Acta, 2018, 24, 53–58 CrossRef.
- For a few precedents of this type of reaction under other conditions, see: (a) H. M.-F. Viart, T. S. Larsen, C. Tassone, T. L. Andresen and M. H. Clausen, Chem. Commun., 2014, 50, 7800–7802 RSC; (b) N. Scalacci, C. Pellajo, M. Radi and D. Castagnolo, Synlett, 2016, 27, 1883–1887 CrossRef CAS; (c) R. P. Singh, D. Gout and C. J. Lovely, Eur. J. Org. Chem., 2019, 1726–1740 CrossRef CAS.
- CCDC 2234522 (4), 2234523 (6) and 2234524 (7)† contain the supplementary crystallographic data for this paper.
- CCDC 2234525 (10) and 2234526 (11)† contain the supplementary crystallographic data for this paper.
- A. Johnson, A. Laguna and M. C. Gimeno, J. Am. Chem. Soc., 2014, 136, 12812–12815 CrossRef CAS PubMed.
- (a) A. Ranjan, R. Yerande, P. B. Wakchaure, S. G. Yerande and D. H. Dethe, Org. Lett., 2014, 16, 5788–5791 CrossRef CAS PubMed; (b) A. Ranjan, A. S. Deore, S. G. Yerande and D. H. Dethe, Eur. J. Org. Chem., 2017, 4130–4139 CrossRef CAS.
- (a) K. Yan, C.-N. Lok, K. Bierla and C.-M. Che, Chem. Commun., 2010, 46, 7691–7693 RSC; (b) S. Adhikari, O. Hussain, R. M. Phillips, W. Kaminsky and M. R. Kollipara, Appl. Organomet. Chem., 2018, 32, e4476G CrossRef; (c) G. Canudo-Barreras, L. Ortego, A. Izaga, I. Marzo, R. P. Herrera and M. C. Gimeno, Molecules, 2021, 26, 6891 CrossRef CAS PubMed.
- CCDC 2234527 (20)† contains the supplementary crystallographic data for this paper.
- CCDC 2234528 (27)† contains the supplementary crystallographic data for this paper.
- J. van Meerloo, G. J. Kaspers and J. Cloos, Methods Mol. Biol., 2011, 731, 237–245 CrossRef CAS PubMed.
- C. Marzano, V. Gandin, M. Pellei, D. Colavito, G. Papini, G. G. Lobbia, E. Del Giudice, M. Porchia, F. Tisato and C. Santi, J. Med. Chem., 2008, 51, 798–808 CrossRef CAS PubMed.
- M. Frik, J. Fernández-Gallardo, O. Gonzalo, V. Mangas-Sanjuan, M. González-Álvarez, A. Serrano del Valle, C. Hu, I. González-Álvarez, M. Bermejo, I. Marzo and M. Contel, J. Med. Chem., 2015, 58, 5825–5841 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Program for adsorption correction, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- CysAlisPro, Version 1.171.35.11; Agilent Technologies. Multiscan absorption correction with SCALE3 ABSPACK scaling algorithm.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (b) L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–71 CrossRef CAS PubMed.
- (a) R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, in Inorg. Synth, John Wiley & Sons, Inc., 1989, pp. 85–91 Search PubMed; (b) L. Malatesta, L. Naldini, G. Simonetta and F. Cariati, Coord. Chem. Rev., 1966, 1, 255–262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2234522–2234528. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00079f |
| This journal is © The Royal Society of Chemistry 2023 |