
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interesting chemical and physical features of the products of the reactions between trivalent lanthanoids and a tetradentate Schiff base derived from cyclohexane-1,2-diamine†
Ioannis
Mylonas-Margaritis
 *a,
Zoi G.
Lada
b,
Alexandros A.
Kitos
a,
Diamantoula
Maniaki
a,
Katerina
Skordi
c,
Anastasios J.
Tasiopoulos
c,
Vlasoula
Bekiari
d,
Albert
Escuer
e,
Julia
Mayans
*e,
Vassilios
Nastopoulos
*a,
Evangelos G.
Bakalbassis
*f,
Dionissios
Papaioannou
*a and
Spyros P.
Perlepes
*ab
*a,
Zoi G.
Lada
b,
Alexandros A.
Kitos
a,
Diamantoula
Maniaki
a,
Katerina
Skordi
c,
Anastasios J.
Tasiopoulos
c,
Vlasoula
Bekiari
d,
Albert
Escuer
e,
Julia
Mayans
*e,
Vassilios
Nastopoulos
*a,
Evangelos G.
Bakalbassis
*f,
Dionissios
Papaioannou
*a and
Spyros P.
Perlepes
*ab
aDepartment of Chemistry, University of Patras, 26504 Patras, Greece. E-mail: ioanismylonasmargaritis@gmail.com; dapapaio@upatras.gr; nastopoulos@upatras.gr; perlepes@upatras.gr; bakalbas@chem.auth.gr
bInstitute of Chemical Engineering Sciences (ICE-HT), Foundation for Research and Technology-Hellas (FORTH), P.O. Box 1414, Platani, 26504 Patras, Greece
cDepartment of Chemistry, University of Cyprus, 1678 Nicosia, Cyprus
dDepartment of Agriculture, University of Patras, 26504 Patras, Greece
eDepartment de Quimica Inorganica i Organica, Secio Inorganica and Institute of Nanoscience (IN2UB) and Nanotechnology, Universitat de Barcelona, Marti i Franques 1-11, 08028 Barcelona, Spain. E-mail: julia.mayans@qi.ub.edu
fDepartment of Chemistry, Aristotle University of Thessaloniki, University Campus, 54124 Thessaloniki, Greece. E-mail: bakalbas@chem.auth.gr
First published on 15th May 2023
Abstract
The initial use of a tetradentate Schiff base (LH2) derived from the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 condensation between 2-hydroxyacetophenone and cyclohexane-1,2-diamine in 4f-metal chemistry is described. The 1:2 reaction of Ln(NO3)3·xH2O (Ln = lanthanoid or yttrium) and LH2 in MeOH/CH2Cl2 has provided access to isostructural complexes [Ln(NO3)3(L′H2)(MeOH)] in moderate to good yields. Surprisingly, the products contain the corresponding Schiff base ligand L′H2 possessing six aliphatic –CH2– groups instead of the –CH–(CH2)4–CH– unit of the cyclohexane ring, i.e. an unusual ring-opening of the latter has occurred. A mechanism for this LnIII-assisted/promoted LH2 → L′H2 transformation has been proposed assuming transient LnII species and a second LH2 molecule as the H2 source for the reduction of the cyclohexane moiety. DFT calculations provide strong evidence for the great thermodynamic stability of the products in comparison with analogous complexes containing the original intact ligand. The structures of the PrIII, SmIII, GdIII, TbIII, and HoIII complexes have been determined by single-crystal X-ray crystallography. The 9-coordinate LnIII centre in the molecules is bound to six oxygen atoms from the three bidentate chelating nitrato groups, two oxygen atoms that belong to the bidentate chelating organic ligand, and one oxygen atom from the coordinated MeOH group. In the overall neutral bis(zwitterionic) L′H2 ligand, the acidic H atoms are clearly located on the imino nitrogen atoms and this results in the formation of an unusual 16-membered chelating ring. The coordination polyhedra defined by the nine donor atoms around the 4f-metal-ion centres can be best described as distorted, spherical capped square antiprisms. The EuIII, TbIII, and DyIII complexes exhibit LnIII-based luminescence in the visible region, with the coordinated L′H2 molecule acting as the antenna. Ac magnetometry experiments show that the DyIII member of the family behaves as an SIM at zero field and under external dc fields of 0.1 and 0.2 T without the enhancement of the peaks’ maxima, suggesting that QTM is not the relaxation path. The GdIII complex behaves, rather unexpectedly, as a SIM with two different magnetic relaxation paths occurring at very close temperatures; this behaviour is tentatively attributed to a very small axial zero-field splitting (D ∼ 0.1 cm−1), which cannot be detected by magnetization or susceptibility experiments. The prospects of the present, first results in the lanthanoid(III)-LH2 chemistry are discussed.
:1 condensation between 2-hydroxyacetophenone and cyclohexane-1,2-diamine in 4f-metal chemistry is described. The 1:2 reaction of Ln(NO3)3·xH2O (Ln = lanthanoid or yttrium) and LH2 in MeOH/CH2Cl2 has provided access to isostructural complexes [Ln(NO3)3(L′H2)(MeOH)] in moderate to good yields. Surprisingly, the products contain the corresponding Schiff base ligand L′H2 possessing six aliphatic –CH2– groups instead of the –CH–(CH2)4–CH– unit of the cyclohexane ring, i.e. an unusual ring-opening of the latter has occurred. A mechanism for this LnIII-assisted/promoted LH2 → L′H2 transformation has been proposed assuming transient LnII species and a second LH2 molecule as the H2 source for the reduction of the cyclohexane moiety. DFT calculations provide strong evidence for the great thermodynamic stability of the products in comparison with analogous complexes containing the original intact ligand. The structures of the PrIII, SmIII, GdIII, TbIII, and HoIII complexes have been determined by single-crystal X-ray crystallography. The 9-coordinate LnIII centre in the molecules is bound to six oxygen atoms from the three bidentate chelating nitrato groups, two oxygen atoms that belong to the bidentate chelating organic ligand, and one oxygen atom from the coordinated MeOH group. In the overall neutral bis(zwitterionic) L′H2 ligand, the acidic H atoms are clearly located on the imino nitrogen atoms and this results in the formation of an unusual 16-membered chelating ring. The coordination polyhedra defined by the nine donor atoms around the 4f-metal-ion centres can be best described as distorted, spherical capped square antiprisms. The EuIII, TbIII, and DyIII complexes exhibit LnIII-based luminescence in the visible region, with the coordinated L′H2 molecule acting as the antenna. Ac magnetometry experiments show that the DyIII member of the family behaves as an SIM at zero field and under external dc fields of 0.1 and 0.2 T without the enhancement of the peaks’ maxima, suggesting that QTM is not the relaxation path. The GdIII complex behaves, rather unexpectedly, as a SIM with two different magnetic relaxation paths occurring at very close temperatures; this behaviour is tentatively attributed to a very small axial zero-field splitting (D ∼ 0.1 cm−1), which cannot be detected by magnetization or susceptibility experiments. The prospects of the present, first results in the lanthanoid(III)-LH2 chemistry are discussed.
Introduction
Inorganic chemistry was dominated by transition metals (3d, 4d, 5d) in the second half of the 20th century.1 In the last 20 years or so, there has been a considerable shift of focus on the chemistry of f metals, mainly that of lanthanoids (Ln′s).2 When scientists, including S. P. P. (the co-author of this paper), started working in the Ln metal area in the early 1980s, they were told that this chemistry had no future.3 Today, the opposite is true! The Ln elements exhibit pronounced chemical similarities as a group near the bottom of the periodic table, but simultaneously they express distinctive and varied electronic properties. These atomistic properties are very useful and are the basis for application in many technological areas, including the construction of devices.4 The chemical (production of fine chemicals, catalytic activity, reactivity, bonding models), optical (optical fiber Er amplifiers, phosphors, luminescence thermometers, lasers, coloured ceramics, and glasses), quantum (atomic clocks, trapped 171Yb+ qubits, time crystals), mechanical (polishing powders, metallurgy, alloys with special characteristics), magnetic (GdIII-MRI agents, high Tc superconductors, hard magnets such as Nd2Fe14B, single-molecule magnets (SMMs), single-ion magnets), and biological (methanol dehydrogenase, Ln3+-dependent bacteria) properties of the metals and their ions are exploited in various areas of technology and fundamental research.3,4 The shielded nature of the 4f orbitals results in well-defined energy levels that are weakly perturbed by the coordination environment and are accompanied by large spin–orbit coupling; these characteristics enable the exploitation of Ln3+ ions in optical and magnetic applications. The magnetic and luminescence properties of some members of a new family of LnIII complexes are part of the present work.As far as the magnetic properties of molecular LnIII complexes are concerned, the discovery of slow relaxation of the magnetization and magnetic hysteresis in a bis(phthalocyaninato)terbium(III) complex in 20035 ignited an explosive growth of research interest in LnIII-based molecular nanomagnets, either coordination clusters (single-molecule magnets, SMMs) or mononuclear complexes (single-ion magnets, SIMs), because of their exciting physical phenomena and potential applications in magnetic memory storage, molecular spintronics, and quantum computing.6,7 Design principles developed by Rinehart and Long directed synthetic chemists and physicists towards longer relaxation times by means of extremely large increases in the energy barrier to magnetization reversal (Ueff).8 The massive increases did not lead to corresponding increases in the blocking temperature (TB) due to the vibronically-induced spin relaxation mechanisms – irrespective of the Ueff value – that can operate.9 A breakthrough happened in 2017 when it was shown10 that the cation [Dy(Cpttt)2]+, where Cpttt is C5H5tBu-1,2,4, exhibits magnetic hysteresis up to 60 K. Subsequent studies showed impressive results in this area.11a,b Removal of C–H groups from the C5 ring of Cpttt through the synthesis of peralkylated bis(cyclopentadienyl) DyIII complexes led to hysteresis temperatures TB around the ligand nitrogen temperature (∼80 K), which is the current record.11c
As far as the optical properties are concerned, most LnIII ions luminesce in the solid state. Unlike luminescence from organic compounds, most 4f-metal ion emissions consist of sharp lines. This property has been used in EuIII and TbIII phosphors, and in the NdIII YAG laser. EuIII and TbIII, and sometimes DyIII, display luminescence in the visible region.12 The 4f → 4f transitions are Laporte forbidden and thus excitation of LnIII to an emissive state by this route is not an efficient process. An alternative method of excitation is via an organic ligand, usually an aromatic system, which has an excited triplet state higher in energy than the LnIII emissive state, the so-named antenna effect.13
We are interested in mononuclear LnIII complexes that show magnetic relaxation (SIMs) and/or LnIII-based luminescence. For the realization of this general goal, the choice of the primary organic ligand is very important. For the preparation of SIMs, the ligand should behave as terminal (either monodentate or chelating). It is also crucial for the appearance of the required magnetic anisotropy of the molecule and a high separation between Mj and Mj ± 1 states (in order to obtain high Ueff values), and these imply a rational design of the ligand field. For oblate-type ions (e.g. TbIII and DyIII), the ligand donor atoms with the greatest electron density should bind at axial positions enhancing the required axial anisotropy, whereas for prolate-type ions (e.g. ErIII and YbIII), the ligand donor atoms with the largest electron density should coordinate to equatorial positions to achieve axial anisotropy. For the synthesis of luminescent LnIII complexes with metal ion-based emission, the ligands should bear chromophores which can facilitate efficient energy transfer “sensitization” of the LnIII ion's excited (i.e. emissive) levels from the ligand's triplet state. Schiff bases are often used for these purposes.14 We have been using Schiff bases of various types to achieve the above-mentioned objectives.15
We would also like to mention that the chemistry of mononuclear LnIII-Schiff base complexes is attracting the interest of inorganic chemists for two additional reasons. First, it has been shown that two and four electrons can be stored, respectively, in intramolecular and intermolecular C–C bonds formed by LnIII-assisted reduction of the imino group of Schiff-base ligands, providing new synthetic avenues to the reductive chemistry of lanthanoids.16 Second, complexes of YbIII with chelating Schiff bases are qubit candidates because of the huge splitting between the electronic ground doublet and the first excited crystal field state and their intrinsic slow paramagnetic relaxation,17 as well as candidates for novel coupled electronic qubit-nuclear qubit systems.18
We have recently embarked on a new subarea of LnIII-Schiff base chemistry by using N,N′-bis(salicylidene)ethylenediamine (salen)-type ligands, in which non-conjugated, e.g. cyclohexyl, bridges are linked with two salicylaldehyde moieties functionalised either on the aromatic rings and/or at the aliphatic carbon atoms. Our general goal is to compare the magnetic (e.g. SIM responses) and optical (e.g. aggregation-induced emission, AIE, or aggregation-caused quenching, ACQ, behaviour19) properties of the LnIII complexes containing functionalized Schiff bases with those of the corresponding complexes containing non-functionalized ligands. We plan to use various electron-accepting (–NO2, –F, –Cl, –CN, –Ph), electron-donating (–OMe, –OH, –NEt2, –Me) or bulky (e.g.tBu) substituents. This work started with the reactions of lanthanoid(III) nitrates with the Schiff base LH2 (Scheme 1), which possesses methyl groups at the aliphatic carbon atoms of the salicylaldehyde moiety. No LnIII complexes of the neutral or anionic forms of LH2 have been reported; in contrast, LnIII complexes of salcnH2 or salcn2−, i.e. the unsubstituted Schiff base, have been synthesized and structurally characterised, and their emission and magnetic properties were studied.20 However, we were surprised to discover that LH2 undergoes a LnIII-promoted/assisted cyclohexane ring opening and the isolated complexes contain a neutral L′H2 ligand (Scheme 1). The interesting structural features and properties (optical, magnetic) of the products are described in this article.
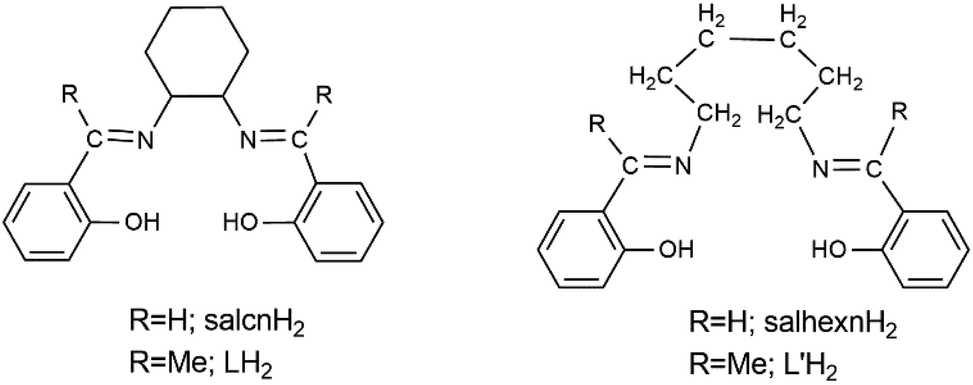 | ||
| Scheme 1 General structural formulae and abbreviations of the Schiff bases discussed in this work. In the complexes reported in this work, L′H2 exists in the overall neutral, bis(zwitterionic) form (vide infra). | ||
Experimental section
Chemicals and instrumentation
All manipulations were performed under aerobic conditions using materials and solvents (reagent grade) as received. The free organic ligand (E,E)-2,2′-[1,1′-(cyclohexane-1,2-diyldinitrilo)diethylidine]diphenol (LH2, Scheme 1)21 was synthesized by the 2:1 condensation reaction between 2-hydroxyacetophenone and (±)-trans-1,2-diaminocyclohexane in refluxing MeOH for 8 h; typical yields were in the 65–75% range. Its purity was checked by elemental analysis (C, H, N), and IR and 1H NMR (d6-DMSO) spectroscopy. Elemental microanalyses (C, H, N) were performed at the Instrumental Analysis Laboratory of the University of Patras. Conductivity measurements were performed at 25 ± 1 °C with a Metrohm-Herisau E-527 bridge and a cell of standard constant using DMSO as the solvent. FT-IR spectra (4000–400 cm−1) were recorded using a PerkinElmer spectrometer with the samples being in the form of KBr pellets. FT-Raman spectra were recorded using an EQUINOX spectrometer to which a Bruker (D) FRA-106/S component was attached. A R510 diode-pumped Nd:YAG laser was used for Raman excitation at 1064 nm with a maximum power of 500 mW on the samples, utilizing an average of 100 scans at 4 cm−1 resolution. 1H NMR and 13C NMR spectra were recorded at 600 and 150 MHz, respectively, on a Bruker Avance III HD spectrometer; the signal of the undeuteriated portion of d6-DMSO at δ 2.52 ppm was used as the 1H NMR reference. Excitation and emission spectra were recorded using a Cary Eclipse fluorescence spectrometer. Solid-state, variable-temperature and variable-field direct current (dc) magnetic data were collected on the powdered samples using a MPMS5 Quantum Design magnetometer operating at 0.03 T in the 300–2.0 K range for the magnetic susceptibility measurements and at 2.0 K in the 0–5 T range for the magnetization experiments. Diamagnetic corrections were applied to the observed susceptibilities using Pascal's constants. Alternating current (ac) magnetic susceptibility experiments were carried out in the 10–1488 Hz frequency range.
Synthetic procedures
Analytical data, calcd for C23H32LnN5O12 (found values are in parentheses): 2: C 38.64 (39.13), H 4.52 (4.47), N 9.80 (9.66); 3: C 38.31 (37.99), H 4.48 (4.59), N 9.72 (9.63); 4: C 38.24 (38.57), H 4.47 (4.56), N 9.69 (9.75); 5: C 37.95 (37.43), H 4.47 (4.41), N 9.62 (9.39); 6: C 37.86 (37.98), H 4.43 (4.52), N 9.60 (9.41); 7: C 37.68 (37.92), H 4.41 (4.32), N 9.56 (9.48); 8: C 37.56 (38.09), H 4.39 (4.28), N 9.52 (9.40); 9: C 37.44 (37.80), H 4.38 (4.49), N 9.49 (9.52); 10: C 37.15 (36.99), H 4.35 (4.38), N 9.42 (9.29); 11: C 41.88 (42.04), H 4.90 (4.81), N 10.62 (10.55)%.
Single-crystal X-ray crystallography
Single crystals of complexes 1, 3, 5, 6 and 8 were coated with paratone-N oil and attached to cryo-loops at the end of a copper pin. Diffraction data were collected by the ω-scan technique on a SuperNova Rigaku diffractometer under a stream of nitrogen gas at 100(2) K using Mo Kα radiation (λ = 0.7107 Å), except for compound 8 where Cu Kα radiation (λ = 1.5418 Å) was used. Data were collected and processed by the CRYSALIS CCD and RED software,22 respectively. An empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm, was applied to the intensities of the collected reflections. All structures were solved with SHELXT23a and refined by full-matrix least-squares techniques on F2 with SHELXL.23b All non-H atoms were refined anisotropically. Carbon-bound H atoms were included in the calculated positions (riding model). The H atoms on the protonated imino nitrogen atoms of L′H2, together with the hydroxyl H atoms of the coordinated MeOH molecule in all five structures, were located in difference Fourier maps and refined isotropically applying soft distance restraints (DFIX). Geometric/crystallographic calculations were carried out using PLATON,24 Olex25 and WINGX26 packages. Molecular/packing graphics were prepared with DIAMOND27 and MERCURY.28Important crystallographic data are listed in Table S1.† Full details can be found in the CIF files. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication with the deposition numbers 2246267–2246271.†
Computational details
DFT calculations for complex 1, employing the B3LYP functional with the 6-31+G (for the carbon, oxygen and nitrogen atoms) and 6-31G (for the hydrogen atoms) basis sets, were carried out with Gaussian09 (version B.01).29 Moreover, the effective core potential of the Stuttgart group,30 in which the 4f wave functions are imbedded in the core in an effective way – greatly simplifying the calculations without any loss of accuracy – was used for the PrIII ion. Analytical frequencies were calculated at the same level of theory, and the nature of the stationary points was determined according to the number of negative eigenvalues of the Hessian matrix (i.e., no imaginary frequency for the local minimum and one imaginary frequency for transition states). The relative stability of the two complexes (both the experimental structure and a similar one containing the initially used ligand) was calculated based on the free energy values (Gibbs).Results and discussion
Brief synthetic procedures
The reactions of Ln(NO3)3·xH2O (x = 5 or 6) or Y(NO3)3·6H2O and LH2 in a 1:2 molar ratio in CH2Cl2/MeOH gave yellow solutions that upon storage at room temperatures gave yellow crystals or crystalline powders of [Ln(NO3)3(L′H2)(MeOH)] (1–10) or [Y(NO3)3(L′H2)(MeOH)] (11) in moderate to good yields (40–65%). The crystals of the PrIII, SmIII, GdIII, TbIII and HoIII compounds (complexes 1, 3, 5, 6 and 8, respectively) were of X-ray quality and their structures were solved by X-ray crystallography. The other six complexes are proposed to be isostructural with 1, 3, 5, 6 and 8 based on elemental analyses and the identical IR spectra. Three synthetic points are of interest: (i) the experimental reaction ratio used (LnIII:LH2 = 1:2) is not the same with the stoichiometric ratio (LnIII:L′H2 = 1:1); the use of the stoichiometric molar ratio results in the isolation of the same products albeit with much lower yields. (ii) The molecules of the complexes do not contain the initial ligand LH2, but instead the transformed ligand L′H2 (Scheme 1; vide supra). (iii) Complex [Y(NO3)3(L′H2)(H2O)] (11a), i.e. the aquo analogue of 11, can be prepared by the 1:2 reaction between Y(NO3)3·6H2O and LH2 in MeCN or Me2CO–CH2Cl2. This fact has implications in the mechanistic proposal (see the ESI†). Since the slow evaporation (almost until dryness) of the solvents from a CH2Cl2/MeOH solution containing only LH2 results in the precipitation of this ligand (IR and 1H NMR evidence), i.e. the used ligand, we propose that the unique LH2 → L′H2 transformation is LnIII-assisted; our mechanistic proposal (ESI†) is based on this assumption. Synthetic organic chemistry has witnessed an explosive growth from the use of lanthanoid(III) reagents.31 This transformation (or a similar transformation involving salcnH2, Scheme 1) has never been observed in the coordination chemistry of LH2. DFT calculations (ESI†) reveal the greater thermodynamic stability of complex 1 compared with that of a structurally similar, hypothetical PrIII complex possessing the initially used LH2 ligand in its bis(zwitterionic) O,O′-bidentate chelating mode, three bidentate chelating nitrato groups and one MeOH ligand. This difference in stability explains the LnIII-assisted/promoted LH2 → L′H2 transformation.
Since the observed transformation was totally unexpected, we re-examined carefully the purity of LH2. Also we examined the purity of the 1,2-diaminocyclohexane precursor to investigate if it contained a portion of 1,6-diaminohexane whose condensation with 2-hydroxyacetophenone would have given L′H2 as a contaminant of LH2; in the case of contamination, the L′H2 component of the mixture with LH2 would result in the isolation of 1–11. Our studies proved the purity of LH2 and its 1,2-diaminocyclohexane precursor. Thus, (1) LH2 was obtained in the form of yellow single crystals; the unit cell determination of a few of them revealed that they represent21 the structurally characterised compound (E,E)-2,2′-[1,1′-(cyclohexane-1,2-diyldinitrilo)diethylidene]diphenol (Scheme 2); for the preparation of the complex, we used batches of single crystals which were subsequently powdered. (2) The 1H NMR spectrum of the used powder in d6-DMSO shows the expected32 signals for pure LH2 with the correct integration ratio; there are four signals in the aromatic region (δ 7.54–6.83 ppm) representing eight protons, a doublet of doublet signal at δ 4.02 ppm due to the two protons attached to positions 1 and 2 of the cyclohexane ring, a singlet signal at δ 2.38 ppm representing the six protons of the two methyl groups and a group of three multiplet signals at δ values of 2.07 (four protons), 1.84 (two protons) and 1.64 (two protons) ppm which represent the eight –CH2– protons of the cyclohexane ring. The broad singlet at δ ∼13.5 ppm (two protons) is assigned19 to the –OH hydrogen atoms. (3) The 1H NMR spectrum of (±)-trans-1,2-diaminocyclohexane (used for the synthesis of LH2) in d6-DMSO confirms its purity. The spectrum (Fig. S1†) shows five multiplet signals at δ 2.03, 1.70, 1.58, 1.17 and 0.96 ppm with an integration ratio of 1:1:1:1:1 (or 2:2:2:2:2). The signal at a lower field (δ 2.03 ppm) represents the two protons at the 1 and 2 positions of the cyclohexane ring (i.e. the protons belonging to the carbon atoms attached to the two nitrogen atoms), while the other signals correspond to the eight –CH2– protons. The –NH2 protons do not appear because of their exchangeable character. The spectrum is identical (as expected) to the spectra of the pure (+)-S,S-trans (Fig. S2†) and (−)-R,R-trans enantiomers of 1,2-diaminocyclohexane (provided by Fluorochem). Moreover, this spectrum is different from the recorded spectrum of 1,6-diaminohexane in d6-DMSO; the latter shows a triple signal at δ 2.68, a multiplet at δ 1.35 and a multiplet at δ 1.12 with an integration ratio of 1:1:1 (or 4:4:4) attributed to the twelve –(CH2)– protons. (4) The 13C NMR spectrum of (±)-trans-1,2-diaminocyclohexane, in d6-DMSO (Fig. S3†), which is again identical to the 13C NMR spectra of the pure (+)-S,S-trans and (−)-R,R-trans, (Fig. S4†) enantiomers, shows the three expected signals of the cyclohexane ring at δ values of 58.1, 35.4 and 25.7 ppm, the former being assigned to the carbons attached to the nitrogen atoms. Moreover, this spectrum is different from the recorded 13C spectrum of 1,6-diaminohexane, which displays the three expected signals at δ values of 41.8 (assigned to the C atoms attached to nitrogen), 32.8 (assigned to the C atoms at the 2 and 5 positions) and 26.8 (assigned to the C atoms at the 3 and 4 positions). All the above results indicate that our starting material (±)-trans-1,2-diaminocyclohexane is pure, and not contaminated with 1,6-diaminohexane.
 | ||
| Scheme 2 Schematic illustration of the E,E configuration of LH2 as observed in its single-crystal X-ray structure.21 | ||
The molar conductivity (ΛM) values of 10−3 M solutions of 1–11 are in the range of 85–110 S cm2 mol−1 indicating 1:3 electrolytes33 and the decomposition of the complexes in solution.
Description of the structures
The aspects of the molecular and crystal structures of complexes 3, 5, 6, and 8 are shown in Fig. 1, 2 and S5–S9.† The five complexes are isomorphous since (i) the structures all have the same space group and quite similar cell dimensions, and (ii) the types and the positions of atoms in the structures are the same except for a replacement of the LnIII atoms. To facilitate molecular comparison, the same numbering scheme has been assigned to all five complexes presented herein. Thus, only the molecular and crystal structures of compound 1 will be presented in detail. | ||
| Fig. 1 Partially labelled plot of the structure of the molecule [Pr(NO3)3(L′H2)(MeOH)] that is present in complex 1. Most H atoms are omitted for clarity. Selected bond lengths (Å): Pr–O1 2.334(2), Pr–O2 2.337(2), Pr–O12 2.501(2), Pr–O(nitrato) 2.534(2)–2.616(2). | ||
 | ||
| Fig. 2 The spherical capped square antiprismatic coordination polyhedron of PrIII in complex 1. The very small spheres define the vertices of the ideal polyhedron. | ||
The crystal structure of 1 consists of [Pr(NO3)3(L′H2)(MeOH)] molecules. The 9-coordinate PrIII centre is bound to six oxygen atoms from the three slightly anisobidentate chelating nitrato groups, two oxygen atoms that belong to the bidentate chelating L′H2 ligand and one oxygen atom from the coordinated MeOH molecule. The acidic H atoms, which are bound to the oxygen atoms of the hydroxyl groups in the structure of the free ligand,21 are clearly located on the imino nitrogen atoms (N1, N2); thus, the formally neutral ligand participates in its bis(zwitterionic) form leaving the oxygen atoms deprotonated, blocking the N coordination sites and forming an unusual 16-membered chelating ring. The two N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(imino)–C(aromatic)–C(aromatic)–O parts of the ligand and the PrIII centre are almost coplanar, with the largest deviation from their best mean plane being that of O2 (0.137(2) Å); the two aromatic rings are also coplanar and the angle between them is 1.2(2)°. The PrIII–O bond lengths fall in the range of 2.334(2)–2.616(2) Å and are typical of 9-coordinate PrIII complexes. The bond lengths of PrIII to deprotonated phenolato oxygens (2.334(2) and 2.337(2) Å) are shorter than the distances of the nitrato groups (2.534(2)–2.616(2) Å) and methanol (2.501(2) Å). There are two strong intramolecular H bonds in the complex molecule (Table S2†) with the protonated nitrogen atoms being the donors and the negatively charged coordinated oxygen atoms being the acceptors.
C(imino)–C(aromatic)–C(aromatic)–O parts of the ligand and the PrIII centre are almost coplanar, with the largest deviation from their best mean plane being that of O2 (0.137(2) Å); the two aromatic rings are also coplanar and the angle between them is 1.2(2)°. The PrIII–O bond lengths fall in the range of 2.334(2)–2.616(2) Å and are typical of 9-coordinate PrIII complexes. The bond lengths of PrIII to deprotonated phenolato oxygens (2.334(2) and 2.337(2) Å) are shorter than the distances of the nitrato groups (2.534(2)–2.616(2) Å) and methanol (2.501(2) Å). There are two strong intramolecular H bonds in the complex molecule (Table S2†) with the protonated nitrogen atoms being the donors and the negatively charged coordinated oxygen atoms being the acceptors.
There are no Platonic, Archimedean and Catalan polyhedra with nine vertices, and also this number of vertices cannot result in prisms or antiprisms. Thus, the only shapes that may be considered are those listed in Table S3.† With the help of program SHAPE,34 the best fit obtained for the PrIII coordination polyhedron is for the spherical capped square antiprism, with the nitrato atom O9 being the capping atom. Since the coordinated nitrato groups impose small coordination angles (∼50°), the polyhedron is distorted (Fig. 2).
A variety of intermolecular interactions stabilize the crystal structure of 1. The two molecules of the unit cell form a centrosymmetric dimer bridged by two relatively strong (Table S2†) O12–H12⋯O11 H bonds (Fig. 3) about the centre of symmetry located at ½, ½, ½ of the cell. The molecules are further packed in a 3D network through weak C–H⋯O interactions.
 | ||
| Fig. 3 The centrosymmetric dimeric species {[Pr(NO3)3(L′H2)(MeOH)]}2 formed through intermolecular O(methanol)–H⋯O(nitrato) H bonds in the crystal structure of 1. The intramolecular N–H⋯O H bonds, discussed in the text, are also shown. The PrIII atoms (large spheres) are not labelled for clarity. | ||
Each analogous Ln–O bond length in the five isomorphous complexes follows the order Pr > Sm > Gd > Tb > Ho, which is a consequence of the LnIII contraction.
Compounds 1, 3, 5, 6 and 8 are the first structurally characterized complexes of L′H2 or/and its anionic form with any metal. Homometallic LnIII complexes of the salicylaldehyde analogues of L′H2, i.e. salhexnH235 (Scheme 1), have been reported.36 They are: (i) {[Eu2(hfac)4(O2CMe)2(salhexnH2)2]}n,36a a 1D coordination polymer in which the neutral Schiff-base molecules behave as O,O′-bidentate bridging ligands with the acidic H atoms being located at the oxygen atoms (hfac− is the hexafluoroacetylacetato ligand); (ii) [Eu2(dbm)4(O2CMe)2(salhexnH2)2],36a where the two EuIII centres are linked by two O,O′-bidentate bridging ligands with the location of the acidic H atoms being not reported (dbm− is the dibenzoylmethanide ligand); (iii) {[Gd2(tta)4(O2CMe)2(salhexnH2)2]}n,36b a 1D polymer in which the neutral salhexnH2 molecules behave as O,O′-bidentate bridging ligands with the acidic H atoms being located at the imino nitrogen atoms (tta− is the 2-thenoyltrifluoroacetylacetonato ligand). Analogous O,O′-bidentate bridging coordination modes have been reported for homometallic LnIII polymers (1D, 2D)37,38 and dimers36a possessing the –(CH2)4– (i.e. N,N′-bis(salicylidene-1,4-butanediamine)36a,37 and –(CH2)5–38 (i.e. N,N′-bis(salicylidene-1,5-pentanediamine)38 analogues of salhexnH2. Thus, the O,O′-bidentate chelating mode of L′H2 in 1–11 is novel in the coordination chemistry of such ligands with aliphatic backbones containing four or more carbon atoms, not only with lanthanoids but also with other metal ions. All the above complexes of the salhexnH2, and –(CH2)4– and –(CH2)5– analogues were prepared using the open-chain Schiff base which was incorporated in the complexes; no transformation similar to the LH2 → L′H2 one reported in this work was noticed.
Spectroscopic discussion in brief
Representative data are presented in Fig. 4–6 and S10–S12.† The IR spectra of 1–11 are almost identical. The weak-to-medium intensity band at ∼3450 cm−1 with two submaxima at lower wavenumbers is due to the ν(OH) vibration of coordinated MeOH39 and the ν(NH+) vibration15a of the coordinated L′H2 ligand; the relatively low wavenumber and broadness of the band are both indicative of H bonding, which has been crystallographically confirmed (vide supra). The strong band at ∼1610 cm−1 can be safely assigned to the ν(CN) vibration of the protonated imine group. The bands at ∼1460, 1300 cm−1 and ∼1025 cm−1 are assigned40 to the vibrational modes ν1(A1)[ν(NO)], ν2(B2)[νas(NO2)] and ν2(A1)[ν5(NO2)], respectively, of the bidentate chelating nitrato groups, with the former being possibly overlapped with an aromatic stretch. The separation of the two highest-wavenumber bands is ∼160 cm−1, which is a typical value of bidentate nitrato ligands. The Raman peaks at 3067, 2953 and 2942, 1611, 1477 and 1267 cm−1 (Fig. 4) are due40,41 to the ν(CH)aromatic, ν(CH)aliphatic, ν(CN), ν1(A1)[ν(NO)] and ν5(B2)[νas(NO2)] modes, respectively.
 | ||
| Fig. 4 The Raman spectrum of solid [Pr(NO3)3(L′H2)(MeOH)] (1). | ||
 | ||
| Fig. 5 Solid-state, room-temperature excitation (curve 1; maximum emission at 616 nm) and emission (curve 2; maximum excitation at 399 nm) spectra of solid [Eu(NO3)3(L′H2)(MeOH)] (4). | ||
 | ||
| Fig. 6 Solid-state, room-temperature excitation (curve 1; maximum emission at 488 nm) and emission (curve 2; maximum excitation at 405 nm) spectra of solid [Tb (NO3)3(L′H2)(MeOH)] (6). | ||
The solid-state, room-temperature luminescence spectra of the EuIII (4), TbIII (6) and DyIII (7) complexes, which are candidates for LnIII-based emission in the visible region,12,13 were recorded. Upon maximum excitation at 399 nm, a solid sample of 4 displays photoluminescence with maxima at 595, 616, 652 and 695 nm due to EuIII.13a,b,42 These peaks are assigned to the characteristic 5D0 → 7Fj (j = 0–4) transitions. Specific assignments are as follows: 5D0 → 7F1,2 (595 nm), 5D0 → 7F2 (616 nm), 5D0 → 7F3 (652 nm) and 5D0 → 7F4 (695 nm). The dominant peak at 616 nm is due to the hypersensitive 5D0 → 7F2 transition. The higher intensity of this transition compared with that of the magnetic-dipole allowed 5D0 → 7F1 transition indicates that this complex has no imposed symmetry at EuIII, in accordance with our belief that 4 is isomorphous with the structurally characterized complexes 1, 3, 5, 6 and 8.
Upon maximum excitation at 405 nm, solid 6 displays photoluminescence with maxima at 488, 543, 588, 621 (shoulder) and 648 nm due to TbIII.42c,43 These emission maxima are assigned to the transitions from the 5D4 state to its 7F6 (488 nm), 7F5 (543 nm), 7F4 (588 nm), 7F3 (621 nm) and 7F2 (648 nm) states.
The excitation spectrum of complex 7 (Fig. S12†) shows medium-to-strong intensity bands which are attributed44 to the appropriate transitions of DyIII: 6H15/2 → 4M17/2, 6P7/2 (330 nm), 6H15/2 → 2K17/2 (380 nm) and 6H15/2 → 4I15/2 (418 nm). Upon maximum excitation at 380 nm, solid 7 displays photoluminescence with maxima at 486 and 519 nm being attributed to the intraconfigurational 4f–4f transitions of the metal ion (Fig. S12†).44 The maximum located at 486 nm can be safely assigned to the magnetic dipole transition 4F9/2 → 6H15/2, which generally is not sensitive to the crystal field. The expected “yellow” 4F9/2 → 6H13/2 around 575 nm is not observed in our spectrum; this emission is related to the forced electric dipole transition-type and its intensity is strongly influenced (and often strongly quenched) by the crystal-filed environment. Thus, the origin of the 519 nm maximum is not clear.
Magnetic studies
Direct current (dc) magnetic susceptibility data on the powdered samples of complexes 5, 6, 7 and 8 were collected in the temperature range of 300–2.0 K under an applied field of 0.03 T (Fig. 7 and S13†). The room-temperature χMT values are 7.7 for 5, 11.9 for 6, 14.2 for 7 and 13.5 for 8 cm3 K mol−1. These values are very close to those expected for the isolated GdIII (8S7/2, 7.88 cm3 K mol−1), TbIII (7F6, 11.82 cm3 K mol−1), DyIII (6H15/2, 14.17 cm3 K mol−1) and HoIII (5I8, 14.07 cm3 K mol−1) centres, respectively. | ||
| Fig. 7 Temperature dependence of the XMT product at 0.03 T for compounds 6 (squares), 7 (diamonds) and 8 (circles) (main curves). The χMT vs. T (100–2.0 K) variation for complex 5 is shown in the inset. Solid lines show the best fit of the experimental data. | ||
Upon cooling, the value of the χMT product for the isotropic (4f7) GdIII complex 5 is constant down to 25 K and then decreases slowly to a value of 6.0 cm3 K mol−1 at 2.0 K. The low-temperature decrease of the product should be attributed to weak intermolecular antiferromagnetic exchange interactions promoted by the two intermolecular Omethanol–H⋯“free” Onitrato H bonds (vide supra, Table S2†) which form {GdIII}2 dimers. The fit of the experimental data, applying the spin Hamiltonian H = −2J(S1·S2), give a satisfactory simulation for J = −0.03 cm−1 with a g value of 1.98. Magnetization measurements at 2.0 K show a saturation value of 7.03Nμβ in agreement with the expected 7/2 spin. Reduced magnetization data indicate a quasi-negligible anisotropy (Fig. 8, right).
 | ||
| Fig. 8 (Left) Magnetization plots for complexes 6–8; squares, diamonds and circles represent the data for compounds 6, 7 and 8, respectively. (Right) Reduced magnetization for GdIII complex 5 between 1.8 and 6.8 K with increments of 1 K. | ||
For the TbIII (6), DyIII (7) and HoIII (8) complexes, the values of the χMT product decrease slowly when lowering T due to the progressive depopulation of the mj levels of the ground state J, with a more pronounced decay below 50 K; the final values at 2.0 K are 7.01 (6), 9.21 (7) and 5.07 (8) cm3 K mol−1. The sharp decay at low temperatures might also be due to weak antiferromagnetic LnIII⋯LnIII interactions within the H-bonded dimers. The susceptibility data were fitted assuming implicitly a regular distribution of the mj states. The Hamiltonian used is represented by eqn (1), where Ŝ is the spin operator, L is the orbit operator,45,46λ is the spin–orbit coupling and Δ describes the energy gap between mL components. The value for the orbital reduction parameter k was assumed as 1 (the LnIII ions behave as purely ionic). Thus, the first term of the Hamiltonian describes the spin–orbit coupling, the second is related to the ligand field around the lanthanoid cation, and the third term describes the Zeeman effect.
| H = (λŜL) + Δ[LZ2 − L(L + 1)/3] + [βH(−kL + 2Ŝ)] | (1) |
In spite of the low symmetry of the LnIII environment in 6–8, the χMT simulations were accurate with Δ parameters of −37, −10 and −14 cm−1 for 6, 7 and 8, respectively. The negative values of Δ indicate that the highest mj value corresponds to the ground state. The magnetization plots of 6–8 at 2.0 K show a fast increase at a low field with quasi saturation (Fig. 8), confirming the negative Δ values for these complexes.
Ac magnetic susceptibility measurements were performed using a 4.0 G ac field on the powdered samples of 6–8 under different static fields in order to explore the magnetization dynamics of the three complexes. No out-of-phase (imaginary) components of the ac susceptibility, χ′′M, were detected in the TbIII and HoIII complexes at frequencies between 10 and 1488 Hz, either at zero field or under external applied fields. In contrast, DyIII complex 7 exhibits tails of peaks below 5 K as expected for a Kramers’ ion with mj = ±15/2 ground doublet (Fig. 9, left and S15†). The χ′′M response was measured at zero dc field, and also under applied external (bias) dc fields of 0.1 and 0.2 T without observing any enhancement of the peaks’ maxima; this indicates that the quantum tunnelling of the magnetization (QTM) is not the relaxation path in this case despite the distorted environment around the DyIII centre. Due to the lack of maxima in the 10–1480 Hz frequency range and in the measured temperature range, the system was fitted (Fig. 9, right and S14†) according to the Debye eqn (2),47 giving the mean values of 4.6 cm−1 and 7.6 × 10−7 s for the effective barrier for the magnetization reversal (Ueff) and pre-experimental factor (τ0), respectively. The experimental data were not enough for a complete Cole–Cole plot to investigate the magnetic relaxation mechanism; however, due to the Ueff value which is smaller than the energy of the first excited state, the Orbach relaxation is discarded as the main relaxation path.
| ln(χ′′M/χ′M) = ln(ωτ0) − Ueff/kBT | (2) |
 | ||
| Fig. 9 Ac data for complex 7. (Left) Out-of-phase magnetic susceptibility (χ′′M) versus T in a 4.0 G ac field oscillating between 1488 and 10 Hz at zero static (dc) field. (Right) Natural logarithm of χ′′M/χ′Mversus 1/T at different ac frequencies (1488–200 Hz) in zero static (dc) field. | ||
Given the recent discovery of and great interest on GdIII SIMs/SMMs,48,49 we investigated the dynamic properties of 5. Out-of-phase magnetic susceptibility signals were not observed at zero field, but upon increasing the static (dc) field tails of signals appeared with a maximum intensity around 0.4 T. The χ′′Mversus T measurements were carried out at this field in the 10–1488 Hz range and a clear frequency dependence was observed, Fig. 10. The χ′′M(T) plot shows two processes, one as a shoulder signal at higher frequencies around 2.5 K and a second one below 1.8 K, which are almost completely overlapped showing that the two different magnetic relaxation paths are occurring at very close temperatures (Fig. 10, top). A fit of the Argand plot performed by the generalized Debye model shows an α parameter with values between 0.06 and 0.16 indicating a narrow distribution of relaxation times and short τ values in the 10−5–10−6 s interval which are shown as ln(τ) vs. the inverse of temperature in Fig. 10, bottom and S14.† Fitting of the ln(τ) plot can be performed with similar quality factors combining several relaxation mechanisms such as Orbach plus Raman, Orbach plus direct or direct plus Raman. However, taking into account that the conventional Ueff barrier derived from a double-well potential cannot be applied for this kind of quasi-isotropic system (DS2 ≪ kT), the only realistic fit is from the direct (low temperature) plus Raman (high temperature, via lattice vibrations) relaxation combination applying the expression:
| τ−1 = AT + CTn | (3) |
 | ||
| Fig. 10 Top, temperature dependence (a) and frequency dependence (b) of χ′′M for complex 5 under a 0.4 T static field. Bottom, Argand plot (c) and fit of the ln(τ) vs. inverse of temperature (d). | ||
The best fit values were A = 9136, C = 6.85 and n = 4.90, Fig. 10 bottom. The n value is lower than the theoretical n = 9 value for the Kramers ions but is commonly attributed to the participation of optical lattice vibrations, which has been previously reported.50
Complex 5 joins a limited number of GdIII complexes exhibiting slow relaxation of the magnetization in spite of the isotropic character of the cation, which contrasts with the classical interpretation for the slow relaxation of the magnetization usually explained as an over barrier process depending on the axial zero field splitting. The mostly accepted explanation48a,g,h,49 is the presence of a weak axial zero-field splitting (D ∼ 0.1 cm−1), which cannot be detected by magnetization or susceptibility studies. These small values, which result from molecular distortion, allow very small double-well energy barriers around ∼1 cm−1 (calculated by the formula DS2 − 1/4), which are in the orders of magnitude lower than the experimentally calculated barriers (Ueff). Such low D values, however, imply that the relaxation phenomenon cannot be explained in terms of conventional over-barriers or QTM above 2 K. The weak anisotropy breaks the degeneration at the S = 7/2 level, mixing the mS sublevels under moderate (∼0.4 T) external magnetic fields and resulting in SIM/SMM behaviour.
Concluding comments and perspectives
It is rather difficult to conclude on a research project which is still at its infancy. The most salient features of this work are: (1) the first use of the tetradentate Schiff base LH2 (Schemes 1 and 2) has provided access to an interesting family of mononuclear LnIII complexes. (2) The original ligand LH2 has undergone a novel LnIII-assisted LH2 → L′H2 transformation and the resulting complexes contain the latter as the ligand; a mechanistic scheme for this transformation has been proposed (ESI†). These preliminary results may give “food for thought” concerning the potential use of LnIII ions as catalysts in ring-opening reactions of cyclohexane-containing substrates in MeOH under mild conditions. (3) The L′H2 ligand is incorporated in its bis(zwitterionic) form and exhibits a new coordination mode (O,O′-chelating) for this type of Schiff base. (4) The EuIII, TbIII and DyIII complexes display LnIII-based emission in the visible region proving that the coordinated L′H2 molecule can effectively transfer energy to the excited, emissive levels of these LnIII ions; (5) The DyIII member of the family is a weak SMM at zero external field, and the QTM or Orbach processes are not the relaxation paths; the GdIII compound exhibits SIM properties under a moderate external dc field, thus joining a small group of such complexes.Our future directions in the project include: (a) Reactions of LH2 with 3d and 5f (uranyl, thorium(IV)) ions to see if the LH2 → L′H2 transformation can be observed again; preliminary experiments indicate that 3d-metal complexes containing either LH2 or L′H2 can be isolated. For example, compounds {FeIIICl3(LH2)·Et2O}n, {[FeIIICl3(LH2)·2Me2CO}n and {[MnIICl2(LH2)2]·3MeOH}n have been recently structurally characterized, depending on the reaction conditions. Complex (Et3NH)[MnIII2(L)2(L′H2)](ClO4)3·CHCl3, which contains both the original and transformed ligands, can be isolated from the 1:1:2 Mn(ClO4)2·6H2O/LH2/Et3N reaction mixture in CHCl3–MeOH (Fig. S16–S19†) (b) Synthesis and full characterization of L′H2 (this compound is not known), followed by its reaction with lanthanoid(III) nitrates to investigate if the same complexes (1–11) with those described here can be obtained. (c) Reactions of ligands analogous to LH2, but possessing various substituents (–Cl, –Br, –Me,⋯) on the aromatic rings, with LnIII ions to investigate if the identity of the products depends on the nature of the substituent and if similar metal ion-assisted transformations are operative; (d) reactions of Schiff bases derived from the 2:1 condensation between 2-hydroxyacetophenone and 1,2-diaminocyclopentane or 1,2-diaminocyclobutane with LnIII ions; the ring-opening process might be easier with such Schiff bases given the larger strain of these rings than that in cyclohexane (cyclobutane 27 kcal mol−1, cyclopentane 5 kcal mol−1, and cyclohexane 1 kcal mol−1).
Author contributions
I. M.-M.: conceptualization, synthesis, and data analysis; Z. L.: Raman spectroscopy and data analysis; A. K.: synthesis and conventional characterization; D. M.: synthesis and conventional characterization; K. Skordi: collection of single-crystal crystallographic data; A. T.: supervision of the experimental work on single-crystal crystallography; V. B.: luminescence spectra and data analysis; A. E.: supervision of the experimental magnetic work; J. M. magnetic measurements, data analysis, and writing of the magnetic part of the manuscript; V. N.: solution and refinement of the single-crystal X-ray structures and creation of structural plots; E. B.: theoretical calculations and writing of the relevant part of the manuscript; D. P.: mechanistic proposals and writing of the relevant part of the manuscript; S. P.: conceptualization, supervision, project management, and final manuscript writing and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Research Director George Voyiatzis for permitting us to use the Raman facilities at ICE-HT/FORTH. AE and JM are grateful for the funding from MICINN (Project PGC2018-094031-B-100). We also thank the undergraduate student Chrysanthi Panteli for preliminary synthetic work and the PhD candidate Eleanna Vahlioti for her assistance to obtain the NMR spectra.References
- J. Ribas Gispert, Coordination Chemistry, Wiley-VCH, Weinheim, Germany, 2008, pp. XXIV–XXXVII Search PubMed.
- J.-P. Launay and M. Verdaguer, Electrons in Molecules, Oxford University Press, Oxford, UK, revised edition, 2018 Search PubMed.
- J. C. Wedal and W. J. Evans, J. Am. Chem. Soc., 2021, 143, 18354–18367 CrossRef CAS PubMed (perspective).
- For an excellent essay, see: T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- J. Tang and P. Zhang, Lanthanide Single Molecule Magnets, Springer-Verlag, Berlin, Germany, 2015 Search PubMed.
- For representative review-type articles, see: (a) N. F. Chilton, Ann. Rev., 2022, 52, 79–101 Search PubMed; (b) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (c) A. Zabala-Lekuona, J. M. Seco and E. Colacio, Coord. Chem. Rev., 2021, 441, 213984 CrossRef CAS; (d) P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923–3929 RSC (frontier); (e) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655–6669 RSC; (f) F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150–175 CrossRef CAS; (g) J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC; (h) S. K. Gupta and R. Murugavel, Chem. Commun., 2018, 54, 3685–3696 RSC (feature article); (i) K. L. M. Harriman, D. Errulat and M. Murugesu, Trends Chem., 2019, 1, 425–439 CrossRef CAS; (j) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS; (k) F. Habib and M. Murugesu, Chem. Soc. Rev., 2013, 42, 3278–3288 RSC; (l) K. Katoh, T. Komeda and M. Yamashita, Chem. Rec., 2016, 16, 987–1016 CrossRef CAS PubMed.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC (perspective).
- (a) T. Fukuda, N. Shigeyoshi, T. Yamamura and N. Ishikawa, Inorg. Chem., 2014, 53, 9080–9086 CrossRef CAS PubMed; (b) A. Lunghi, F. Totti, R. Sessoli and S. Sanvito, Nat. Commun., 2017, 8, 14620 CrossRef PubMed.
- (a) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed; (b) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- (a) K. R. McClain, C. A. Gould, K. Chakarawet, S. J. Teat, T. J. Groshens, J. R. Long and B. G. Harvey, Chem. Sci., 2018, 9, 8492–8503 RSC; (b) C. A. Gould, K. R. McClain, J. M. Yu, T. J. Groshens, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2019, 141, 12967–12973 CrossRef CAS PubMed; (c) F.-S. Guo, D. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- H. C. Aspinall, f-Block Chemistry, Oxford University Press, Oxford, UK, 2020, pp. 13–14, 28–32 Search PubMed.
- (a) J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef; (b) L. Armelao, S. Quici, F. Barigelletti, G. Accorsi, G. Bottaro, M. Cavazzini and E. Tondello, Coord. Chem. Rev., 2010, 254, 487–505 CrossRef CAS; (c) J. Andres and K. E. Borbas, Inorg. Chem., 2015, 54, 8174–8176 CrossRef CAS PubMed; (d) J. Vuojola and T. Soukka, Methods Appl. Fluoresc., 2014, 2, 1–27 Search PubMed.
- For example, see: (a) J. Long, I. V. Basalov, N. V. Forosenko, K. A. Lyssenko, E. Mamantova, A. V. Cherkasov, M. Damjanović, L. F. Chibotaru, Y. Guari, J. Larionova and A. A. Trifonov, Chem. – Eur. J., 2019, 25, 474–478 CrossRef CAS PubMed; (b) K. Kobayashi, Y. Harada, K. Ikenaga, Y. Kitagawa, M. Nakano and T. Kajiwara, Magnetochemistry, 2019, 5, 27 CrossRef CAS; (c) Y. Yao, H.-Y. Yin, Y. Ning, J. Wang, Y.-S. Meng, X. Huang, W. Zhang, L. Kang and J.-L. Zhang, Inorg. Chem., 2019, 58, 1806–1814 CrossRef CAS PubMed.
- For example see: (a) D. Maniaki, I. Mylonas-Margaritis, J. Mayans, A. Savvidou, C. P. Raptopoulou, V. Bekiari, V. Psycharis, A. Escuer and S. P. Perlepes, Dalton Trans., 2018, 47, 11859–11872 RSC; (b) I. Mylonas-Margaritis, D. Maniaki, J. Mayans, L. Ciammaruchi, V. Bekiari, C. P. Raptopoulou, V. Psycharis, S. Christodoulou, A. Escuer and S. P. Perlepes, Magnetochemistry, 2018, 4, 45 CrossRef CAS.
- C. Camp, V. Guidal, B. Biswas, J. Pécaut, L. Dubois and M. Mazzanti, Chem. Sci., 2012, 3, 2433–2448 RSC.
- K. S. Pedersen, A.-M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed.
- R. Hussain, G. Allodi, A. Chiesa, E. Garlatti, D. Mitcov, A. Konstantatos, K. S. Pedersen, R. De Renzi, S. Piligkos and S. Carretta, J. Am. Chem. Soc., 2018, 140, 9814–9818 CrossRef CAS PubMed.
- J. Cheng, Y. Li, R. Sun, J. Liu, F. Gou, X. Zhou, H. Xiang and J. Liu, J. Mater. Chem. C, 2015, 3, 11099–11110 RSC.
- (a) G.-L. Wang, Y.-M. Tian, D.-X. Cao, Y.-S. Yu and W.-B. Sun, Z. Anorg. Allg. Chem., 2011, 637, 583–588 CrossRef CAS; (b) G.-L. Wang, Y.-M. Tian, W.-B. Sun, B.-L. Han, M.-F. Yu, H. Xu and T. Gao, Z. Anorg. Allg. Chem., 2011, 637, 1616–1621 CrossRef CAS; (c) J. Zhu, H.-F. Song, P.-F. Yan, G.-F. Hou and G.-M. Li, Cryst. Eng. Commun., 2013, 15, 1747–1752 RSC; (d) Q. Liu, M. Ding, Y. Lin and Y. Xing, Polyhedron, 1998, 17, 555–559 CrossRef CAS; (e) Q. Liu and M. Ding, J. Organomet. Chem., 1998, 553, 179–181 CrossRef CAS; (f) Q. Liu, J. Huang, Y. Qian and A. S. C. Chan, Polyhedron, 1999, 18, 2345–2350 CrossRef CAS; (g) J. Zhu, H. Song, J. Sun, P. Yan, G. Hou and G. Li, Synth. Met., 2014, 192, 29–36 CrossRef CAS; (h) P.-F. Yan, P.-H. Lin, F. Habib, T. Aharen, M. Murugesu, Z.-P. Deng, G.-M. Li and W.-B. Sun, Inorg. Chem., 2011, 50, 7059–7065 CrossRef CAS PubMed; (i) J.-W. Sun, J. Zhu, H.-F. Song, G.-M. Li, X. Yao and P.-F. Yan, Cryst. Growth Des., 2014, 14, 5356–5360 CrossRef CAS; (j) X. Zou, C. Du, Y. Dong and G. Li, Inorg. Chim. Acta, 2020, 507, 119455 CrossRef CAS.
- F. Chen and H.-Y. Ye, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, o1757 CrossRef CAS PubMed.
- Crysalis PRO (version 1.171.93), Rigauk Oxford Diffraction, Yarnton Oxfordshire, UK, 2018 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. Spek, Acta Crystallogr., Sect. D: Struct. Biol., 2009, 65, 148–155 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- K. Branderburg, DIAMOND: Program for Crystal and Molecular Structure Visualization, Crystal Impact Gbr, Bonn, Germany, 2014 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1993, 85, 441–450 CrossRef CAS.
- For example, see: (a) G. A. Molander, Chem. Rev., 1992, 92, 29–68 CrossRef CAS; (b) W. S. Trahanovsky, L. H. Young and M. H. Bierman, J. Org. Chem., 1969, 34, 869–871 CrossRef CAS.
- E. Szłyk, A. Wojtczak, E. Larsen, A. Surdykowski and J. Neumann, Inorg. Chim. Acta, 1999, 293, 239–244 CrossRef.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81–122 CrossRef CAS.
- M. Llunell, D. Casanova, J. Girera, P. Alemany and S. Alvarez, SHAPE, version 2.0, Barcelona, Spain, 2010 Search PubMed.
- I. Sheikhshoaie and M. A. Sharif, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o3563–o3565 CrossRef CAS.
- (a) T. Koizuka, K. Yanagisawa, Y. Hirai, Y. Kitagawa, T. Nakanishi, K. Fushimi and Y. Hasegawa, Inorg. Chem., 2018, 57, 7097–7103 CrossRef CAS PubMed; (b) Y.-Y. Xu, O. Sun, Y. Qi, B.-Y. Xie and T. Gao, New J. Chem., 2019, 43, 16706–16713 RSC; (c) Y.-Y. Xu, P. Chen, T. Gao, H.-F. Li and P.-F. Yan, Cryst. Eng. Commun., 2019, 21, 5965–5972 RSC.
- (a) Y. Yue, G. Hou, X. Yao and G. Li, Polyhedron, 2017, 129, 157–163 CrossRef CAS; (b) Y. Yue, P. Yan, J. Sun, G. Hou and G. Li, Polyhedron, 2015, 94, 90–95 CrossRef CAS; (c) Y. Jia, H. Li, P. Chen, T. Gao, W. Sun and P. Yan, Aust. J. Chem., 2018, 71, 527–533 CrossRef CAS; (d) W. Radecka-Paryzek, I. Pospieszna-Markiewicz and M. Kubicki, Inorg. Chim. Acta, 2007, 360, 488–496 CrossRef CAS; (e) Y. Yue, P. Yan, J. Sun and G. Li, Inorg. Chem. Commun., 2015, 54, 5–8 CrossRef CAS; (f) Y. Yue, J. Sun, P. Yan and G. Li, Inorg. Chem. Commun., 2015, 51, 42–45 CrossRef CAS; (g) I. Pospieszna-Markiewicz, M. T. Kaczmarek, M. Kubicki and W. Radecka-Paryzek, J. Alloys Compd., 2008, 451, 403–405 CrossRef CAS.
- W. Radecka-Paryzek, I. Pospieszna-Markiewicz and M. Kubicki, J. Rare Earths, 2010, 28, 51–55 CrossRef.
- C. Stamou, Z. G. Lada, C. T. Chasapis, D. Papaioannou, P. Dechambenoit and S. P. Perlepes, Dalton Trans., 2022, 51, 15771–15782 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, USA, 4th edn, 1986, pp. 254–257 Search PubMed.
- F. R. Dollish, W. G. Fateley and F. F. Bentley, Characteristic Raman Frequencies of Organic Compounds, Wiley, New York, USA, 1974, pp. 1–4, 136, 175–179 Search PubMed.
- (a) C. D. Polyzou, H. Nikolaou, C. P. Raptopoulou, K. F. Konidaris, V. Bekiari, V. Psycharis and S. P. Perlepes, Molecules, 2021, 26, 1622 CrossRef CAS PubMed; (b) H. Nikolaou, A. Terzis, C. P. Raptopoulou, V. Psycharis, V. Bekiari and S. P. Perlepes, J. Surf. Interf. Mater., 2014, 2, 311–318 Search PubMed; (c) V. Bekiari, K. A. Thiakou, C. P. Raptopoulou, S. P. Perlepes and P. Lianos, J. Lumin., 2008, 128, 481–488 CrossRef CAS.
- S. Kalusniak, E. Castellano-Hernández, H. Yalçinoğlu, H. Tanaka and C. Kränkel, Appl. Phys. B, 2022, 128, 33 CrossRef CAS.
- S. Chemingui, M. Ferhi, K. Horchani-Naifer and M. Férid, J. Lumin., 2015, 166, 82–87 CrossRef CAS.
- M. V. Marinho, D. O. Reis, W. X. C. Oliveira, L. F. Marques, H. O. Stumpf, M. del Déniz, J. Pasán, C. Ruiz-Pérez, J. Cano, F. Lloret and M. Julve, Inorg. Chem., 2017, 56, 2108–2123 CrossRef CAS PubMed.
- E. Pilichos, À. Tubau, S. Speed, M. Font-Bardia, A. Escuer, A. Grabulosa and J. Mayans, Dalton Trans., 2023, 52, 2485–2494 RSC.
- J. Bartolomé, G. Filoti, V. Kuncser, G. Schienteie, V. Mereacre, C. E. Anson, A. K. Powell, D. Prodius and C. Turta, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 014430 CrossRef.
- For example, see: (a) M. Orendáč, L. Sedláková, E. Čižmár, A. Orendáčová, A. Feher, S. A. Zvyagin, J. Wosnitza, W. H. Zhu, Z. M. Wang and S. Gao, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 214410 CrossRef; (b) A. Arauzo, A. Lazarescu, S. Shova, E. Bartolomé, R. Cases, J. Luzón, J. Bartolomé and C. Turta, Dalton Trans., 2014, 43, 12342–12356 RSC; (c) M. J. Martínez-Pérez, S. Carbona-Serra, C. Schlegel, F. Moro, P. J. Alonso, H. Prima-García, J. M. Clemente-Juan, M. Evangelisti, A. Gaita-Ariño, J. Sesé, J. van Slageren, E. Coronado and F. Luis, Phys. Rev. Lett., 2012, 108, 247213 CrossRef PubMed; (d) A. Vráblová, M. Tomás, L. R. Falvello, Ľ. Dlháň, J. Titiš, J. Černák and R. Boča, Dalton Trans., 2019, 48, 13943–13952 RSC; (e) T. K. Ghosh, S. Maity, J. Mayans and A. Ghosh, Inorg. Chem., 2021, 60, 438–448 CrossRef CAS PubMed; (f) M. Orts-Arroyo, R. Rabelo, A. Carrasco-Berlanga, N. Moliner, J. Cano, M. Julve, F. Lloret, G. De Munno, R. Ruiz-García, J. Mayans, J. Martínez-Lillo and I. Castro, Dalton Trans., 2021, 50, 3801–3805 RSC; (g) Y. Horii, K. Katoh, Y. Miyazaki, M. Damjanovic, T. Sato, L. Ungur, L. F. Chibotaru, B. K. Breedlove, M. Nakano, W. Wernsdorfer and M. Yamashita, Chem. – Eur. J., 2020, 26, 8076–8082 CrossRef CAS PubMed; (h) E. Pilichos, M. Font-Bardia, A. Escuer and J. Mayans, Dalton Trans., 2021, 50, 1746–1753 RSC; (i) R. J. Holmberg, L. T. A. Ho, L. Ungur, I. Korobkov, L. F. Chibotaru and M. Murugesu, Dalton Trans., 2015, 44, 20321–20325 RSC.
- J. Mayans and A. Escuer, Chem. Commun., 2021, 57, 721–724 RSC.
- L. Gu and R. Wu, Phys. Rev. B: Condens. Matter Mater. Phys., 2021, 103, 014401 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic (Table S1) and structural data (Tables S2 and S13), various spectra (Fig. S1–S4, S10–S12), structural plots (Fig. S5–S9 and S16–S19), magnetic plots (Fig. S13–S15), mechanistic schemes (Schemes S1–S6), and computational drawings (Schemes S7 and S8). CCDC 2246267–2246271. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00817g |
| This journal is © The Royal Society of Chemistry 2023 |